
Abstract
Tetraploid oysters are used to cross with diploids to produce triploid oysters that have become an important part of the oyster aquaculture industry worldwide. Although most tetraploid oysters are artificially induced autotetraploids, allotetraploids can be produced between closely related species, providing new opportunities for polyploid breeding and studying genome interactions. Using PacBio sequencing, Illumina sequencing, and high-throughput chromosome conformation capture scaffolding, we produced a high-quality genome assembly of an artificially induced allotetraploid between the Pacific oyster Crassostrea gigas and Portuguese oyster Crassostrea angulata. The assembled genome is 1.23 Gb, with a contig N50 of 2.56 Mb and a scaffold N50 of 57.22 Mb, and anchored to 20 chromosomes. The assembly contains 58,330 protein-coding genes, 98.34% of which are functionally annotated. The heterozygosity and the ratio of repetitive sequences is 5.50% and 46.43%, respectively. This chromosomal-level genome assembly of an allotetraploid oyster provides a valuable genetic resource for studying genome biology, especially genome reorganization after hybridization and duplication, and for advanced breeding of polyploids that are critical for the oyster aquaculture industry.
Similar content being viewed by others


Background & Summary
Oysters are among the most important aquaculture species worldwide, accounting for an annual production of ~7 million metric tons (World Food and Agriculture – Statistical Yearbook 2024 (fao.org)). Genetic improvement including selective breeding, hybridization and polyploidization, plays an important role in supporting oyster aquaculture1. One of the most significant advances over the last four decades is the development of triploid oysters. Triploid oysters have three sets of chromosomes and grow significantly faster than diploids due to their increased heterozygosity, polyploid gigantism and sterility1,2. Sterility is desired for aquaculture as it prevents uncontrolled reproduction of cultured stocks. Although incomplete, sterility in oysters inhibits excessive gonad development and improves meat quality during the reproductive season when mature diploids are undesirable. Because of their sterility, superior growth and improved meat quality, triploids have become one of the most popular stocks for oyster aquaculture1,2,3. The commercialization of triploid oysters has contributed significantly to oyster aquaculture, especially for the Pacific oyster Crassostrea gigas and Eastern oyster Crassostrea virginica, in meeting the market demand around the world4,5. Triploid oysters now account for 30–70% of the cultured oysters in major producing countries such as France, Australia, USA and China1. Originally, triploids were induced by retaining the second polar body in newly fertilized eggs with chemicals such as cytochalasin B (CB) or 6-dimethylaminopurine (6-DMAP)6,7. However, chemical induction had low efficiency, which hindered commercial production. The successful development of tetraploid oysters by Guo and Allen8 (1994) made it possible to produce mated triploids by mating diploids x tetraploids, which is 100% effective without any use of toxic chemicals5. Nowadays, triploid oysters are commercially produced through diploid x tetraploid crosses. Thus, the successful production and breeding of tetraploids are critical for oyster aquaculture that is heavily dependent on triploids.
The Guo and Allen method for tetraploid induction involves blocking the release of polar body I in eggs from triploid Pacific oysters fertilized by haploid sperm (3n♀ × 2n♂), which successfully introduced the first autotetraploid Pacific oyster8,9. While it is challenging and difficult to replicate, tetraploids can also be obtained using normal diploid eggs (2n × 2n)10. Induction of tetraploids has also been reported in several oyster species including C. gigas, C. virginica, C. angulata, Crassostrea hongkongensis, Crassostrea sikamea, and tropical oysters Crassostrea belcheri (Sowerby) and Crassostrea iredalei (Faustino)11,12,13,14, although it is not clear whether breeding populations of tetraploids have been established in the latter two species. Most of the tetraploid oysters produced so far were autotetraploids. Allotetraploids can also be produced between species that can hybridize. Tetraploid genomes represent a new state of whole genome duplication that may be unstable and go through rapid reorganization and evolution. With two different genomes, allotetraploids may be more stable because of the preferential pairing of homologs that reduces multivalent formation. The presence of two genomes provides a rare opportunity for studying genome interaction. They may also generate new genotypes by combining characteristics of two species and produce superior triploids for aquaculture. High-quality assemblies of diploid genomes have been produced for oyster species and led to advances in our understanding of oyster biology and environmental adaptation15,16,17,18,19,20,21,22. The sequencing and analysis of tetraploid genomes may provide insights into the biology and evolutionary potential of tetraploids.
We previously produced allotetraploid oysters between the Pacific oyster C. gigas and Portuguese oyster C. angulata, two closely related species that dominate oyster aquaculture production23. In this study, we used long reads generated by PacBio sequencing, short reads generated by Illumina sequencing, and high-throughput chromosomal conformation capture (Hi-C) analysis to construct a high-quality chromosomal-level genome assembly of the allotetraploid oyster. The final genome size is 1,230.39 Mb in 717 contigs, with a contig N50 length of 2.56 Mb and a scaffold N50 length of 57.22 Mb. More than 90% of contigs (1,108.13 Mb) were anchored on 20 chromosomes. The assembly contains 571.24 Mb (46.43%) of repetitive sequences and 7,961 noncoding RNAs. Using de novo prediction, mRNA transcripts and homolog-based strategies, a total of 58,330 protein-coding genes were predicted, and 98.34% of which (57,360) were annotated in the publicly available NCBI RefSeq non-redundant protein, eggNOG, KEGG, SWISS-PROT, Pfam, TrEMBL, GO, and KOG databases. This allotetraploid oyster genome assembly provides a valuable resource for studying interactions between two genomes after duplication and hybridization, which are important for our understanding of the evolutionary biology of polyploids. The interaction or reorganization of the two genomes will likely create novel genotypic combinations or structural variations that affect the phenotype and can be used to study genetic control of production traits and improve the aquaculture performance of polyploid oysters.
Methods
Sample and sequencing
The allotetraploid oyster was artificially induced between the Portuguese oyster C. angulata and the Pacific oyster C. gigas. First, allotriploids were produced in 2015 by mating diploid C. angulata females and autotetraploid C. gigas males; the latter was produced by blocking the release of polar body I in eggs from triploid Pacific oysters fertilized by haploid sperm, which then experienced several generations of random 4n × 4n mating from 2009 to 2015 involving several lines or populations. Second, allotetraploids were produced in 2018 with the Guo and Allen method8 using eggs from the allotriploids and sperm from diploid C. angulata. Subsequently, allotetraploids were reproduced by 4n × 4n crosses for three generations. For this study, one allotetraploid oyster was sampled on 05/27/2024 from the F3 allotetraploids that were produced in 2022 (Fig. 1). The tetraploidy of the sampled oyster was confirmed by flow cytometry. Adductor muscle was collected and flash-frozen in liquid nitrogen, and then used for genomic DNA extraction (with ~30 mg tissue) using the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany). Agarose (1.0%) gel electrophoresis, Qubit (Invitrogen, QubitTM3Flurometer) and NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) were used to determine DNA concentration and quality. The genomic DNA was used to build sequencing libraries, including 15-kb insert PacBio HiFi library and 150-bp insert Illumina paired-end library.

Breeding history leading to the sequenced allotetraploid oyster in this study.
High-molecular weight (HMW) gDNA was prepared for PacBio HiFi read production, and libraries were constructed using the PacBio Template Prep Kit 1.0 according to the standard protocol of Template Preparation using BluePippin size selection (Pacific Biosciences, USA). Sequencing of genomic libraries was performed on two cells using the self-testing high-precision CCS mode on the PacBio Sequel II system. A total of 34.37 Gb of HiFi long-read data with a read N50 length of 16.59 Kb (average read length of 16.18 Kb) was obtained, resulting in 27.94-fold coverage of the allotetraploid oyster genome size.
The short-insert library was constructed using the NR604-VAHTS Universal V6 RNA-seq Library Prep Kit (Vazyme), and then was sequenced by the Illumina NovaSeq. 6000 platform using the paired-end model (PE 150) following the standard protocol (Illumina Inc., San Diego, CA, USA). A total of 156.82 Gb (127.50-fold coverage) of clear reads with a Q30 of 93.52% were obtained to assess allotetraploid oyster genome size.
The Hi-C libraries were also constructed for genome assembly24,25. The same fresh adductor muscle was crosslinked with 1.0% formaldehyde and then was terminated with 0.2 M glycine. Libraries were generated according to the manufacturer’s instructions: (1) digestion with HindIII restriction enzyme, (2) labeling using Biotin-14-dATP (Thermo Fisher Scientific, USA), (3) ligation with T4 DNA ligase, (4) physically shearing into 300–700 bp fragments, (5) selectively capture using streptavidin magnetic beads. Illumina HiSeq 6000 platform was used for sequencing. We obtained 147.05 Gb (119.55-fold coverage) of clean data.
For genome annotation, we collected tissues from four organs (gill, mantle, adductor muscle and labial palp) for RNA-seq. Total RNA was extracted from tissues of each organ and then equally mixed into 1 sample. The RNA mixture was used for library construction and sequencing by the Illumina NovaSeq. 6000 platform following the standard protocol (Illumina Inc., San Diego, CA, USA). A total of 7.17 Gb of clear data was obtained.
Genome assessment and assembly
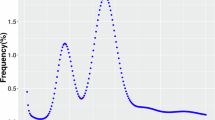
Illumina paired-end clear reads (156.82 Gb) were used to survey the genome features of the allotetraploid oyster via the k-mer method. GenomeScope v2.0026 (parameters: -k 19 -p 4 -m 1000000000) and Jellyfish v2.1.427 (parameter: -h 1000000000) were used for k-mer count histogram (k = 19) (Fig. 2). Estimation of genome size followed the formula of G = N k-mer/Daverage k-mer, where N k-mer is the total number of k-mers, Daverage k-mer is the average depth of k-mers, G is genome size. The survey results showed that the haploid genome size of allotetraploid oyster was estimated to be 544.56 Mb with the heterozygosity, repetitive sequence ratio and GC content of 5.50%, 46.69% and 34.61%, respectively (Table 1). In total, 34.37 Gb of HiFi long-reads were used for assembly using the Hifiasm v0.19 software28 with default parameters, resulting in a total length of 1,815.36 Mb comprising 1,610 contigs with a contig N50 length of 2.29 Mb for the allotetraploid oyster (Table 2).

The 19-mer frequency distribution in the allotetraploid oyster genome. The percentages of different genotypes are: aaaa [94.5%], aaab [3.15%], aabb[1.99%], aabc [0.001%] and abcd [0.361%]. The X-axis is the k-mer coverage, and Y-axis represents the product of frequency by coverage of the k-mer.
Chromosomal-level assembly with Hi-C
To anchor contigs, 491,784,314 clean reads generated from the Hi-C data were mapped to the assembly using BWA v0.7.17-r118829 with default parameters. Valid interaction pairs (125,471,951 pairs) were defined as paired reads with mate mapped to a different contig and then were used to do the Hi-C associated scaffolding using HiC-Pro v2.10.030 that can also filter out invalid interaction pairs including self-ligation, non-ligation, PCR amplification, random break, and extreme fragments. The LACHESIS v2.0.131 was used for agglomerative hierarchical clustering, sorting and orientation (cluster_min_re_sites = 544; cluster_max_link_density = 2; order_min_n_res_in_trunk = 908; order_min_n_res_in_shreds = 870). All of 1,610 contigs were clustered into 717 groups (contigs after Hi-C) with a contig N50 length of 2.56 Mb and a scaffold N50 length of 57.22 Mb (Table 2), and 91.35% (655) were anchored on 20 chromosomes. Finally, 583 contigs were successfully sorted and oriented with a total length of 1,108.13 Mb for the allotetraploid oyster (Table 3). Chromatin contact matrix was built by Juicebox v1.532, and the 20 chromosomes show clearly distribution in the heatmap, with distinct interaction signal around the diagonal within chromosome and between adjacent chromosomes (Fig. 3). Moreover, we carried out collinearity analysis of the assembled allotetraploid genome with the original diploid C. gigas (GCF_963853765.1) and C. angulata (GCA_025765675.3) reference genome using Diamond v0.9.29.13033 (e < 1e−5, C score > 0.5) and MCScanX34 (MCScanX -s 5 -m 5). The pronounced co-linearity relationships indicated highly conserved gene blocks among allotetraploid oyster, diploid C. gigas and C. angulata (Fig. 4).

Genome assembly of the allotetraploid oyster. (a) Chromosome interactive heat map of the Hi-C assembly. Color block indicates intensity of interaction from yellow (low) to dark-red (high). (b) Circos plot of the assembly. The a, b, c, d and e indicate chromosome ideograms, TE density, SSR density, gene density and GC content.

Collinearity analysis among genomes of the allotetraploid oyster, diploid C. gigas and C. angulata.
Repeat sequences annotation
Whole-genome repeat sequences, including tandem repeats and transposable elements (TEs), were annotated using the combined strategy of ab initio prediction and homology alignment. The MIcroSAtellite identification tool (MISA v2.135) and Tandem Repeat Finder (TRF v4.0936, 2 7 7 80 10 50 500 -d -h) were used to predict tandem repeats, which yielded a total of 83.22 Mb of tandem repeats (6.76% of the genome assembly) (Table 4). For TEs, a customized repeat library was built using RepeatModeler v2.0.137 (BuildDatabase -name; RepeatModeler -pa 12), which can initiate two de novo repeat finding programs of RECON v1.0.838 and RepeatScout v1.0.639. The library was then classified by RepeatClassifier with default parameters according to the public databases of Dfam v3.540 and Repbase v19.0641. The LTRharvest v1.5.1042 and LTR_finder v2.843 (ltr_finder -w 2 -C -D) were used to identify full-length long terminal repeat retrotransposons (fl-LTR-RTs). High-quality intact fl-LTR-RTs and non-redundant LTR library were then generated by LTR_retriever v2.9.044. We combined the above de novo TE sequences libraries with public databases to construct non-redundant species-specific TE library, which was then used to identify and classify the final TE sequences using homology search of RepeatMasker v4.1.245 (repeatmasker -nolow -no_is -norna -engine wublast -parallel 8 -qq). A total of 488.03 Mb of TEs were identified, accounting for 39.67% of genome assembly. Among TEs, DNA transposons and retroelements accounted for 26.85% (330.34 Mb) and 12.82% (157.68 Mb) of the genome assembly, respectively (Table 4).
Noncoding RNAs and pseudogene annotation
For noncoding RNAs annotation, miRNA, rRNA, tRNA, snoRNA and snRNA were identified by specific approaches. The miRNA was identified against miRBase database46. Based on the Rfam v14.547 database, rRNA and tRNA were identified by tRNAscan-SE v1.3.148 and barrnap v0.949 (barrnap–kingdom euk–threads 1) respectively, and snoRNA and snRNA were identified by Infernal v1.150 (cmscan–cpu 3–rfam). In total, 7,710 tRNA, 179 rRNA and 72 miRNA were predicted (Table 5).
The GenBlastA v1.0.451 and GeneWise v2.4.152 were used to identify homologous pseudogenes after excluding functional genes (genblasta -P wublast -pg tblastn) and to search for immature stop codons and frameshift mutations (genewise -both -pseudo), respectively. We obtained 362 pseudogenes with an average length of 5.64 Kb (Table 5).
Protein-coding gene prediction and functional annotation
Three approaches, de novo prediction, homology-based prediction, and mRNA-based prediction, were applied for protein-coding gene prediction in the allotetraploid genome. Two ab initio gene-prediction software, Augustus v3.1.053 and SNAP v2006-07-2854, were used for de novo gene model prediction in the repeat-masked assembly (hard-masking). For homology-based prediction, protein sequences of four well-annotated species (C. angulata (GCA_025765675.3), C. ariakensis (GCA_020567875.1), C. virginica (GCF_002022765.2) and Danio rerio (GCA_049306965.1)) were downloaded and aligned to the repeat-masked genome assembly. Then, the GeMoMa v1.755 (run.sh mmseqs) was used to predict gene model based on sequence alignment. The 7.17 Gb clean data from RNA-seq was used for mRNA-based prediction. The Hisat2 v2.1.056 (hisat2–dta -p 10) and StringTie v2.1.457 (stringtie -p 2) were used to assemble transcripts. The GeneMarkS-T v5.158 was used to predict genes based on transcripts. Finally, the EVidenceModeler (EVM) v1.1.159 was used to integrate all gene models predicted by the above methods, which was then modified by PASA v2.4.160 to generate a weighted and non-redundant gene set. A total of 58,330 protein-coding genes (Table 6) were predicted with an average exon number of 7.84 per gene and an average gene length of 8.27 Kb (Table 7).
For functional annotation of gene models, we searched against public biological functional databases, including Non-Redundant (NR), Evolutionary Genealogy of Genes: Non-supervised Orthologous Groups (eggNOG)61, Gene Ontology (GO), TrEMBL, Gene Ontology (GO), EuKaryotic Orthologous Groups (KOG), Kyoto Encyclopedia of Genes and Genomes (KEGG)62, SWISS-PROT63 and Pfam64, using Diamond blastp (Diamond v0.9.29.13033, diamond blastp–masking 0 -e 0.001). A total of 57,360 genes (98.34% of the total predicted genes) were functionally annotated (Table 8).
Data Records
The raw PacBio, Hi-C, and Illumina sequencing data are deposited in the NCBI Sequence Read Archive database under the accession numbers: SRR32607952, SRR32607953, SRR32459897, SRR32456008 and SRR3245587665. The genome assembly has been deposited on the NCBI GenBank database under the accession number JBPJCZ00000000066. Moreover, the genomic annotation results have been deposited in the figshare database67.
Technical Validation
Four methods were used to evaluate the genome assembly: the mapping of Illumina reads, PacBio HiFi reads, BUSCO assessment, and core gene integrity. The Illumina short-reads and PacBio HiFi-reads were mapped to the assembly using BWA v0.7.17-r118829 and Minimap2 v2.2868 to assess the quality, respectively. As shown in Table 9, 99.09% and 99.95% of short-reads and HiFi-reads were mapped to the allotetraploid genome, respectively. The completeness of the assembly was evaluated by the Core Eukaryotic Genes Mapping Approach (CEGMA) v2.569 database and Benchmarking Universal Single-Copy Orthologs (BUSCO) v2.070 against the metazoa_odb10. A total of 447 (97.60%) out of 458 conserved eukaryotic core genes from the CEGMA database and 938 (98.32%) out of the complete 954 BUSCO orthologous groups were identified in the assembled genome (Table 10). All single-copy genes are expected to be duplicated in the allotetraploid, and the fact that 12.4% of BUSCO orthologs are present in single copies indicates significant gene loss after the whole genome duplication. Moreover, we randomly selected 36 genes from the allotetraploid oyster and aligned with genome assemblies of C. gigas and C. angulata. A total of 33 genes showed high identity (å 90%) and 1 gene showed low identity (~80%) with both species. Two genes were aligned with one of the two species (Table 12). These findings indicate that some genome reorganization has occurred, which may alter the fitness and aquaculture performance of allotetraploids. The Hi-C heatmap shows strong interactions within intra-chromosomal regions and between paired inter-chromosomes (Fig. 3). Taken together, these results confirm that the allotetraploid oyster genome assembly is of high quality considering its high heterozygosity and repeat content.
Alignment of 4 randomly selected genes confirms the presence of both C. gigas and C. angulata alleles (Table 11). Some heterozygous alleles between A and B subgenomes clearly originated from Pacific oyster and Portuguese oyster genomes, respectively (black boxes in Fig. 5), confirming allotetraploidy of the sequenced oyster. Some homozygous alleles between A and B subgenomes originated from single parental genotype (C. gigas or C. angulata), and some loci had alleles absent in the reference genomes of both parental species. In addition, we used species-specific COI sequences (C. gigas: TAGTAGCAGACATGCAATTTCCTCGA; C. angulata: CGTGATAATTGGGGGGTTTGGTAACT) to align with 156.82 Gb Illumina short-read data. A total of 20,006 reads were mapped with C. angulata specific COI sequence, while only 3 reads were mapped with C. gigas specific COI sequence. This result indicates that the mitochondrial genome is from C. angulata, consistent with the known pedigree of the allotetraploid oyster.

Sequence alignment of 4 randomly selected gene segments among of C. gigas, C. angulata, A and B subgenome of allotetraploid oyster in this study.
Code availability
No custom code was used during this study for the curation and validation of the dataset. All commands and pipelines used in data processing were executed according to the manual and protocols of the corresponding bioinformatics software.
References
Guo, X. Genetics in shellfish culture. 393–413 (5m Books Ltd, 2021).
Guo, X., Wang, Y., Xu, Z. & Yang, H. Chromosome set manipulation in shellfish. 165–195 (Woodhead Publishing, 2009).
Nell, J. A. Farming triploid oysters. Aquaculture 210, 69–88 (2002).
Matt, J. L. & Allen, S. K. A classification system for gonad development in triploid Crassostrea virginica. Aquaculture 532, https://doi.org/10.1016/j.aquaculture.2020.735994 (2021).
Guo, X., DeBrosse, G. A. & Allen, S. K. Jr. All-triploid Pacific oysters (Crassostrea gigas Thunberg) produced by mating tetraploids and diploids. Aquaculture 142, 149–161 (1996).
J. Brianik, C. & Allam, B. The need for more information on the resistance to biological and environmental stressors in triploid oysters. Aquaculture 577, https://doi.org/10.1016/j.aquaculture.2023.739913 (2023).
Peachey, B. L. & Allen, S. K. Evaluation of cytochalasin B and 6-dimethylaminopurine for tetraploidy induction in the Eastern oyster, Crassostrea virginica. Aquaculture 450, 199–205, https://doi.org/10.1016/j.aquaculture.2015.07.034 (2016).
Guo, X. & Allen, S. K. Jr. Viable tetraploids in the Pacific oyster (Crassostrea gigas Thunberg) produced by inhibiting polar body 1 in eggs from triploids. Molecular Marine Biology & Biotechnology 3, 42–50 (1994).
Guo, X. & Allen, S. K. Jr. The successful induction of tetraploidy in the Pacific oyster Crassostrea gigas (Thunberg). Aquaculture 137, 152–153 (1995).
Benabdelmouna, A. & Ledu, C. Autotetraploid Pacific oysters (Crassostrea gigas) obtained using normal diploid eggs: induction and impact on cytogenetic stability. Genome 58, 333–348, https://doi.org/10.1139/gen-2015-0014 (2015).
Aileen Tan, S. H., Teh, C. P., Chang, G. O. & Yasin, Z. Tetraploid induction in tropical oysters, Crassostrea belcheri (Sowerby) and Crassostrea iredalei (Faustino). Aquaculture Research 48, 1406–1412, https://doi.org/10.1111/are.12976 (2017).
Qin, Y. et al. Tetraploid induction of Crassostrea hongkongensis and C. sikamea by inhibiting the polar body 1 release in diploid fertilized eggs. Marine Life Science & Technology 3, 463–473, https://doi.org/10.1007/s42995-021-00107-w (2021).
Guo, X. M. Production and breeding of tetraploid eastern oyster Crassostrea virginica. Journal of Shellfish Research 31, 292–292 (2012).
Zhang, Y., Qin, Y. & Yu, Z. Comparative study of tetraploid-based reciprocal triploid Portuguese oysters, Crassostrea angulata, from seed to marketsize. Aquaculture 547, https://doi.org/10.1016/j.aquaculture.2021.737523 (2022).
Li, A. et al. Chromosome-level genome assembly of the Suminoe oyster Crassostrea ariakensis in south China. Scientific Data 11, https://doi.org/10.1038/s41597-024-04145-8 (2024).
Li, A. et al. Genome of the estuarine oyster provides insights into climate impact and adaptive plasticity. Communications Biology 4, 1287, https://doi.org/10.1038/s42003-021-02823-6 (2021).
Zhang, G. et al. The oyster genome reveals stress adaptation and complexity of shell formation. Nature 490, 49–54, https://doi.org/10.1038/nature11413 (2012).
Qi, H., Cong, R., Wang, Y., Li, L. & Zhang, G. Construction and analysis of the chromosome-level haplotype-resolved genomes of two Crassostrea oyster congeners: Crassostrea angulata and Crassostrea gigas. GigaScience 12, https://doi.org/10.1093/gigascience/giad077 (2022).
Teng, W. et al. Parallel evolution in Crassostrea oysters along the latitudinal gradient is associated with variation in multiple genes involved in adipogenesis. Molecular ecology, https://doi.org/10.1111/mec.17108 (2023).
Liu, F. et al. MolluscDB 2.0: a comprehensive functional and evolutionary genomics database for over 1400 molluscan species. Nucleic acids research 53, D1075–D1086, https://doi.org/10.1093/nar/gkae1026 (2025).
Zhang, Y. et al. Comparative genomics reveals evolutionary drivers of sessile life and left-right shell asymmetry in bivalves. Genomics, proteomics & bioinformatics https://doi.org/10.1016/j.gpb.2021.10.005 (2022).
Zhang, X. et al. Whole genome sequencing of Crassostrea ariakensis (Mollusca: Ostreidae) and C. hongkongensis expands understandings of stress resistance in sessile oysters. Genomics 116, 110757, https://doi.org/10.1016/j.ygeno.2023.110757 (2024).
Wang, H., Qian, L., Liu, X., Zhang, G. & Guo, X. Classification of a common cupped oyster from southern China. Journal of Shellfish Research 29, 857–866, https://doi.org/10.2983/035.029.0420 (2010).
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009).
Rao, S. S. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680, https://doi.org/10.1016/j.cell.2014.11.021 (2014).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204, https://doi.org/10.1093/bioinformatics/btx153 (2017).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly with phased assembly graphs. arXiv, 2008.01237v01231 (2020).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome biology 16, 259, https://doi.org/10.1186/s13059-015-0831-x (2015).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nature biotechnology 31, 1119–1125, https://doi.org/10.1038/nbt.2727 (2013).
Durand, N. C. et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell systems 3, 99–101, https://doi.org/10.1016/j.cels.2015.07.012 (2016).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12, 59–60, https://doi.org/10.1038/nmeth.3176 (2015).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic acids research 40, e49, https://doi.org/10.1093/nar/gkr1293 (2012).
Beier, S. et al. MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585, https://doi.org/10.1093/bioinformatics/btx198 (2017).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic acids research 27, 573–580 (1999).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Bao, Z. & Eddy, S. R. Automated De Novo Identification of Repeat Sequence Families in Sequenced Genomes. Genome research 12, 1269–1276, https://doi.org/10.1101/gr.88502 (2002).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358, https://doi.org/10.1093/bioinformatics/bti1018 (2005).
Wheeler, T. J. et al. Dfam: a database of repetitive DNA based on profile hidden Markov models. Nucleic acids research 41, D70–D82, https://doi.org/10.1093/nar/gks1265 (2012).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob DNA 6, 11, https://doi.org/10.1186/s13100-015-0041-9 (2015).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. Bmc Bioinformatics 9, https://doi.org/10.1186/1471-2105-9-18 (2008).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic acids research 35, W265–268, https://doi.org/10.1093/nar/gkm286 (2007).
Ou, S. & Jiang, N. LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons. Plant Physiology 176, 1410–1422, https://doi.org/10.1104/pp.17.01310 (2018).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics Chapter 4, Unit 4 10, https://doi.org/10.1002/0471250953.bi0410s25 (2009).
Griffiths-Jones, S., Grocock, R. J., van Dongen, S., Bateman, A. & Enright, A. J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic acids research 34, D140–144, https://doi.org/10.1093/nar/gkj112 (2006).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic acids research 33, D121–124, https://doi.org/10.1093/nar/gki081 (2005).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic acids research 25, 955–964 (1997).
Loman, T. A Novel Method for Predicting Ribosomal RNA Genes in Prokaryotic Genomes Degree Projects in Bioinformatics thesis, Lund University, (2017).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935, https://doi.org/10.1093/bioinformatics/btt509 (2013).
She, R., Chu, J. S., Wang, K., Pei, J. & Chen, N. GenBlastA: enabling BLAST to identify homologous gene sequences. Genome research 19, 143–149, https://doi.org/10.1101/gr.082081.108 (2009).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome research 14, 10.1101/ (2004).
Stanke, M. & Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19(Suppl 2), ii215–225, https://doi.org/10.1093/bioinformatics/btg1080 (2003).
Korf, I. Gene finding in novel genomes. Bmc Bioinformatics 5, 59 (2004).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic acids research 44, e89, https://doi.org/10.1093/nar/gkw092 (2016).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360, https://doi.org/10.1038/nmeth.3317 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature biotechnology 33, 290–295, https://doi.org/10.1038/nbt.3122 (2015).
Tang, S., Lomsadze, A. & Borodovsky, M. Identification of protein coding regions in RNA transcripts. Nucleic acids research 43, e78, https://doi.org/10.1093/nar/gkv227 (2015).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome biology 9, R7, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic acids research 31, 5654–5666, https://doi.org/10.1093/nar/gkg770 (2003).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic acids research 47, D309–D314, https://doi.org/10.1093/nar/gky1085 (2019).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic acids research 44, D457–D462, https://doi.org/10.1093/nar/gkv1070 (2016).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic acids research 31, 365–370, https://doi.org/10.1093/nar/gkg095 (2003).
Finn, R. D. et al. Pfam: clans, web tools and services. Nucleic acids research 34, D247–251, https://doi.org/10.1093/nar/gkj149 (2006).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP565690 (2025).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_051201775.1 (2025).
Li, A., Guo, X., Li, L. & Zhang, G. The first chromosomal-level genome assembly of an allotetraploid oyster. figshare https://doi.org/10.6084/m9.figshare.28448168.v2 (2025).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100, https://doi.org/10.1093/bioinformatics/bty191 (2018).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067, https://doi.org/10.1093/bioinformatics/btm071 (2007).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212, https://doi.org/10.1093/bioinformatics/btv351 (2015).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (32471687 to A.L.), the Key Research and Development Program of Shandong (ZFJH202309 to G.Z.), the Youth Innovation Promotion Association, Chinese Academy of Sciences (2023215 to A.L.), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0730300 to A.L.), the Taishan Scholars Program (tsqn202312267 to A.L.), the Key Research and Development Program of Shandong (2022LZGC015 to L.L.), and the China Agriculture Research System of MOF and MARA (CARS-49 to L.L.).
Author information
Authors and Affiliations
Contributions
G.Z., Xm.G. and L.L. conceived the study. Y.L., X.L., J.F., Xr.G. and Z.X. produced allotetraploids and conducted flow cytometry. A.L., M.J.Z., J.Z., M.S.Z., M.H., J.D., L.W., H.Q. and W.W. collected the samples, extracted the genomic DNA, and conducted sequencing. M.J.Z., J.Z. and A.L. performed bioinformatics analysis. A.L., M.J.Z., J.Z. and Xm.G. wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, A., Zhao, M., Zhao, J. et al. Chromosomal-level genome assembly of an allotetraploid oyster. Sci Data 12, 1492 (2025). https://doi.org/10.1038/s41597-025-05775-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05775-2
