
Abstract
Extensive divergence of protein sequences from those of model eukaryotes makes recognition of well-studied signaling pathways impossible in Trypanosoma brucei. Consequently, substrates and effectors of most protein kinases are unknown. We addressed these general problems by developing workflows to (i) identify effectors for protein kinases, (ii) discover new effectors and substrates of CK1.2 as proof of principle, and (iii) decode linear motifs in CK1.2 substrates. Casein kinase CK1.2 promotes cytokinesis and division of mitochondrial DNA (kinetoplast) (kDNA). Applying our principles to CK1.2 we identified four new substrates, BBP59, LRRP1, CAP64, and BBP268, that harbored two unique linear motifs, S-x(1,3)-T-x(1,4)-S-x(2)-A-x(2)-[AIV] and S-x(1,3)-T-x(1,4)-S. Ectopic expression of BBP59 inhibited cytokinesis and kinetoplast DNA division, like knockdown of CK1.2. Phosphorylation of BBP59 was blocked after knockdown of CK1.2 or treatment with SB-431542 an inhibitor of CK1.2 (IC50 49.2 nM). Highlighting phospho-regulation of its activities, Ser72 substitution with Ala72 or Asp72 in BBP59 caused preferential blockage of cytokinesis or kDNA division. The sequence-agnostic workflow used to identify effectors of CK1.2 can be deployed for other protein kinases, and to assist functional annotation of hypothetical proteins.
Similar content being viewed by others


Introduction
Trypanosoma brucei sp. cause Human African Trypanosomiasis (HAT) and nagana, a wasting disease in cattle. Availability of the genome sequence1 has advanced biological studies of the trypanosome. Most predicted trypanosome proteins lack homologs in extensively studied (i.e., model) eukaryotes2 earning them classification as “hypothetical proteins” because no functions can be assigned to them.
T. brucei has approximately 180 protein kinases, of which 40 are essential for viability at the blood stream stage3,4. Thirty percent of the proteome, including 1491 hypothetical proteins5,6, is phosphorylated6. From experimental data, protein kinases regulate endocytosis7, kinetoplast (mitochondrial nucleoid) division8, basal body (centriole) separation, cell cycle transitions, RNA synthesis, and chromosome segregation (reviewed in8,9,10): Contributions of hypothetical phosphoproteins to these physiologic processes remains largely unexplored.
Casein kinase CK1.2 is essential for viability of T. brucei11, and it regulates kinetoplast division, basal body separation, and cytokinesis3,8. Despite its involvement in multiple biological systems each of which requires a cast of polypeptides only one substrate of CK1.2 is known12. In a search for possible new substrates of CK1.2 that might contribute to physiologic pathways affected by the enzyme, we used a bottom-up gene knockdown/phosphoproteomics strategy (described in a previous study8): we sought polypeptides whose phosphorylation changed after knockdown of CK1.2 by performing three independent phospho-proteomics studies: two label-free shotgun experiments13,14 and one SILAC “target list” study15,16.
After knockdown of CK1.2, abundance of 113 phospho-peptides decreased at least two-fold and abundance of 257 phospho-peptides increased at least two-fold, compared to the proteome of uninduced control trypanosomes. Phospho-peptides that changed in abundance after knockdown are considered “CK1.2-pathway proteins”; those that decreased in abundance are potential substrates of the enzyme. Potential CK1.2 substrates include four basal body proteins, BBP110, BB268, BB59, and BBP59017, one bi-lobe protein, LRRP1, and one flagellum protein, FAZ2, as well as two mitochondrial proteins, POMP12 and POMP2518. These data are consistent with detection of CK1.2 at basal bodies, bi-lobe and flagella8.
All genomes contain hypothetical proteins (including those with domains of unknown function) whose functions cannot be predicted19,20,21,22, because annotation of proteomes relies heavily on computational alignment of sequences and statistics23,24,25. These bioinformatic approaches may be supplemented with data from experiment-based approaches including genome-wide gene knockout or RNA interference, or polypeptide interactome mapping to predict protein functions.
To gain insight on roles of hypothetical proteins and how they impact biological processes, a systematic “divide and conquer” strategy which limits the number of candidates studied per experiment may be needed, due to the large number per genome. Major biological pathways are organized as “protein machines” or conformational ensembles (including networks of interacting proteins26,27) each comprised of at least 10 proteins28,29,30; examples of such macromolecular complexes include DNA replisomes, ribosomes, endosomes, proteasomes, anaphase promoting complex, and chromosome separation, chromatin remodeling, and cytokinesis complexes29,31,32,33,34,35,36. These protein assemblies (stable or transient) are controlled in response to environmental cues by signaling pathways containing enzymes that modify polypeptides post translationally. One “divide and conquer” strategy to identify hypothetical proteins in these complexes is to position them in pathways that are influenced by enzymes that regulate the macromolecular complexes. Enzymes that modify polypeptides post-translationally (i.e., post-translational modification (PTM) enzymes) such as protein kinases, peptidases/proteases, deacetylases/acetylases are a good starting point for this strategy; they could serve as in vivo specificity agents to identify biological processes in which hypothetical proteins participate.
To better understand how CK1.2 affects kinetoplast DNA (kDNA) division and cytokinesis in T. brucei we implemented a six-step strategy to find effectors of signaling by the protein kinase, bearing in mind that hypothetical proteins comprise a major fraction of the trypanosome proteome. Steps in the workflow can be applied to any protein kinase in T. brucei or other organisms. We present direct evidence for four new CK1.2 substrates, including hypothetical protein Tb927.10.350 (BBP59). Focusing on BBP59, because it met criteria for a CK1.2 effector protein, we identify residues that influence its phosphorylation by that protein kinase, and we use that information to produce phospho-regulatory versions of BBP59 whose biological roles were studied using a “dominant-negative” strategy. Our work with CK1.2 is ‘proof-of-concept’ for a bottom-up strategy to discover effectors of protein kinase functions. Due to their large number in most organisms our workflow invariably reveals kinase-directed functions of many “hypothetical” proteins, advancing studies of signaling pathways in the African trypanosome.
Results
SB-431542 inhibits enzyme activity of CK1.2 and blocks cytokinesis
In G1 a trypanosome has one kinetoplast (K) and one nucleus (1K1N) (the trypanosome division cycle is reviewed in37). In S-phase, kinetoplast DNA (kDNA) is synthesized and the kinetoplast divides, producing a trypanosome with two kinetoplasts and one nucleus (2K1N). After mitosis the trypanosome has two kinetoplasts and two nuclei (2K2N) and executes cytokinesis to produce two daughter cells each with one kinetoplast and one nucleus (1K1N). We reported earlier that after knockdown of CK1.2 for 24 h cytokinesis was blocked along with inhibition of kDNA division8. To check whether the cytokinesis block could be kinetically separated from the failure of kDNA division, we induced RNAi against the gene for 12 h, fixed trypanosomes using revised conditions (Materials and Methods) and enumerated cells with different kinetoplast and nucleus configurations.
Knockdown of CK1.2 for 12 h reduced amount of the protein by 40% (Fig. S1) and resulted in a 160% increase in the percentage of 2K2N trypanosomes, up from 10.4% of the total population in un-induced cells to 27.4% of the total population in induced cells (Fig. 1A). This data is consistent with failure of cytokinesis after CK1.2 knockdown8. Concurrently, there was a decrease in the number of 2K1N cells, from 14.6% of un-induced cells to 5.5% of induced trypanosomes (Fig. 1A). A reduction (14%) in the abundance of 1K1N trypanosomes occurred (Fig. 1A). A small proportion of trypanosomes with abnormal kinetoplast and nucleus configurations (2.5% 1K2N and 2.5% “other” cells, comprised of 1K0N, 0K0N, xKxN (x > 2) was detected (Fig. 1A)). Accumulation of 1K2N trypanosomes may be due to a failure of kinetoplast division8. The change in cell type distribution between un-induced (N = 468 cells from three biological replicates) and induced (N = 477 cells from three biological replicates) trypanosome populations was statistically significant (p = 2.8 × 10–14, χ2 test). We conclude that an early (i.e. 12 h) effect of CK1.2 knockdown is the inhibition of cytokinesis, accompanied by a minor effect on kinetoplast division that is pronounced at later time points8. Compared to results of a previous study, the block in cytokinesis after CK1.2 knockdown emerged at 12 h here instead of 18 h reported earlier8. Two factors may have contributed to the change in kinetics for detection of the cytokinesis block. First, in the current study trypanosomes are cultured regularly in tetracycline-free medium before inducing RNAi; that protocol was not used in the earlier work8,38. Second, conditions for aldehyde fixation and fluorescence detection of trypanosomes were different. So, it is understandable that kinetics of K/N phenotypes in this study is similar but not identical to that reported earlier.

Reduction of CK1.2 activity genetically or with SB-431542 prevents cytokinesis. (A) RNAi against CK1.2 was induced with tetracycline for 12 h. Kinetoplasts (K) and nuclei (N) were stained with DAPI and enumerated. At least 150 trypanosomes were examined in each of three independent experiments. Lines represent average percentages. Statistical significance of the differences in distribution of K/N per cell between RNAi and uninduced controls was determined with a χ2 test (p = 2.8 × 10–14). Inset: Image of a 2K2N trypanosome stained with DAPI (magenta) and mCLING (green) (K = kinetoplast, N = nucleus). (B) Effect of SB-431542 on the activity of recombinant CK1.2. Kinase assays were performed in triplicate with two technical replicates each time. IC50 of SB-431542 (49.2 nM ± 13.2 nM) was determined by non-linear regression analysis of data obtained. Error bars represent standard error of the mean. Inset: chemical structure of SB-431542. (C) Trypanosomes were treated with DMSO or SB-431542 for 4 h. Kinetoplasts (K) and nuclei (N) were stained with DAPI and detected by fluorescence microscopy (N > 150 for each experiment). “Other” contains trypanosomes with > 2 K or N or < 1 K or N. Lines represent average percentages. Statistical significance of the change in distribution of number of kinetoplast and nuclei between DMSO and SB-431542-treated trypanosomes was assessed with a χ2 test (p = 2.3 × 10–17). Inset: Image of a 2K2N trypanosome stained with DAPI (magenta) and mCLING (green). K = kinetoplast, N = nucleus.
We sought small molecule enzyme inhibitors as orthogonal tools to study the biology of CK1.2 on a shorter time scale. In this context an orthogonal strategy involves using multiple analytical techniques that operate on different principles to measure the same biological property (e.g., using small molecule inhibitors of CK1.2 or RNA interference of CK1.2 protein synthesis to reduce enzyme activity). That approach provides independent confirmation of a result, by avoiding the limitations of a single method. Towards that goal, we screened a Kinase Inhibitor Library (Selleck Chemicals) for inhibition (at 20 µM) of purified recombinant CK1.2 (Fig. S2). SB-43154239,40 and PF-47067241,42 inhibited CK1.2. Potency of the small molecules was confirmed in a dose–response study; IC50 of SB-431542 was 49.2 nM ± 13.2 nM (Fig. 1B), and that of PF-670462 was 29.1 nM ± 2.0 nM (Fig. S3).
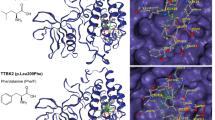
SB-431542 is not known to block human CK1 activity; instead, it is widely used as an inhibitor of TGF receptor kinase43,44,45,46, so our results (above) were surprising. Consequently, we sought supporting evidence for SB-431542 activity against CK1.2 (Fig. 1B) by performing structure modeling and molecular docking (Fig. S4) (see details in Supplemental Materials). Comparative analysis of trypanosome CK1.2 and orthologous CK1 structures complexed with inhibitors provided insight into kinase binding of SB-431542, confirming it as a novel inhibitor of CK1.2 (Fig. 1B). The data are consistent with SB-431542 inhibiting human CK147,48. Lastly, comparing the phenotypic effects of trypanosome treatment with SB-431542 to knockdown of CK1.2 (presented below) shows that in vivo the enzyme is a major target of that inhibitor.
Given that knockdown of CK1.2 prevents cytokinesis (Fig. 1A), and SB-431542 inhibits enzyme activity of CK1.2 (Fig. 1B), we speculated that treatment of T. brucei with SB-431542 might interfere with cytokinesis. To test this hypothesis, trypanosomes were treated with DMSO (vehicle) or SB-431542 (8 μM) for 4 h, and the number of kinetoplasts and nuclei were counted (Fig. 1C). The amount of inhibitor used for the in vivo study deserves explanation when compared to the IC50 reported earlier. As an ATP-competitive inhibitor, the amount of SB-431542 used (8 μM) for the whole trypanosome is in the range expected, compared to the in vitro of IC50 (49 nM). Concentration of ATP used for in vitro kinase assays was 20 µM whereas ATP concentration in T. brucei approximates 2.4 mM49. Used at 8 µM there is a 300-fold less SB-431542 compared to ATP concentration in trypanosomes. In the in vitro experiment (Fig. 1B) there is a 400-fold excess of ATP, compared to SB-431542, at IC50 of CK1.2. That said several caveats are worth noting. First, the amount of CK1.2 present affects IC50 or EC50 values; we do not know the in vivo concentration of CK1.2 compared to the quantity used for in vitro assay of IC50. Second, other proteins may bind to SB-431542 in vivo, causing a reduction of the amount of SB-431542 complexed to CK1.2. Third, some ATP in trypanosomes may not be accessible to CK1.2 due to sequestration from the cytoplasm where the enzyme is found. How these variables impact inhibition of CK1.2 in vivo by SB-431542 is unknown.
We observed a 250% increase in abundance of 2K2N cells following treatment with SB-431542, from 8.2% in DMSO-treated cells to 30.9% for SB-431542-treated trypanosomes (Fig. 1C). The proportion of 2K1N cells decreased from 15.8% in control cells to 8.3% after treatment with SB-431542 (Fig. 1C), and the fraction of 1K1N cells decreased from 74.4% (DMSO control) to 59.1% in SB-431542-treated trypanosomes (Fig. 1C). A minor (i.e., less than 5%) population of “Other” (i.e., with abnormal K and N configurations) trypanosomes was present (0.9% 1K2N, 0.6% xKxN, (x > 2)) (Fig. 1C). The difference in distribution of cells with different K and N configurations between DMSO- (N = 474 cells from three biological replicates) or SB-431542-treated (N = 469, three biological replicates) trypanosomes was statistically significant (p = 2.3 × 10–17, χ2 test). We conclude that SB-431542 inhibits cytokinesis in T. brucei.
Tb927.10.350, Tb927.11.8950, Tb927.3.1010, and Tb927.10.10280 are substrates for CK1.2
Since CK1.2 is protein kinase we speculated that its effect on cytokinesis (Fig. 1A) could be mediated by substrates acting as “effectors”8. In search of new CK1.2 substrates, we hypothesized that some proteins that were dephosphorylated after CK1.2 knockdown8 are substrates in T. brucei. To test this possibility, peptides from several CK1.2 “pathway proteins”8 (Fig. 2A) were evaluated as substrates, with proteins that localized to regions where CK1.2 was detected in T. brucei8 of highest interest. Six polypeptides meeting these criteria, namely, BBP59 (Tb927.10.350)17, LRRP1 (Tb927.11.8950), Tb11.01.273050, FAZ2 (Tb927.1.4310), CAP64 (Tb927.3.1010)50 and MARP2/BBP268 (Tb927.10.10280)17,51 were tested (Fig. 2A). Peptides corresponding to phosphorylation sites (Fig. 2A) were synthesized with at least 6 native residues flanking each predicted phosphosite and used as substrates for protein kinase assays (Fig. 2B). Peptides were tested in technical replicates in three independent biological experiments as a set of five, with “no substrate” controls. The experiment was designed to provide a “yes” or “no” answer rather than to compare efficiency of each peptide as a substrate for CK1.2, since in vivo protein kinases are best viewed as “molecular switches” instead of prolific enzymes (reviewed in52).

BBP59, LRRP1, BBP268, and CAP64 are phosphorylated by CK1.2 in vitro. (A) Peptides from CK1.2-pathway proteins. Six residues flanking predicted phosphosites (in red) were included in synthesized peptides. (B) Peptide phosphorylation by CK1.2. Peptides were tested as sets in three independent assays. Each peptide was tested in two technical replicates per (independent) experiment. Symbols represent mean counts per minute (CPM) of the technical replicates and are color and shape-coded according to the experimental replicate. Blue circles were measurements from the first experiment, red squares from the second, and black triangles from the third. Lines represent mean CPM values. A repeated measures one-way ANOVA with Dunnett’s multiple comparisons test was used to test for significance between each peptide and a “No Substrate” control. BBP59, p = 0.0001; LRRP1, p = 0.0077; Tb11.01.2730, p = 0.9996; FAZ2, p = 0.0548; BBP268, p < 0.0001, CAP64, p = 3.3 × 10–4.
Peptides from four proteins (BBP59, LRRP1, CAP64, and MARP2/BBP268) were phosphorylated by CK1.2 (Fig. 2B). To test for statistical significance of differences in phosphorylation, the natural log-transformed mean counts per minute (CPM) of each peptide and the control were compared by repeated measures one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparisons test (P-values were: BBP59, p = 1 × 10–4; LRRP1, p = 8 × 10–3; CAP64, p = 3.3 × 10–4 and MARP2/BBP268, p < 1 × 10–4). We conclude that BBP59, LRRP1, CAP64, and MARP2/BBP268 are substrates of CK1.2, whereas FAZ2 and Tb11.01.2730 are not.
BBP59 is cytoplasmic protein in bloodstream T. brucei
We determined whether BBP59 was near kDNA perhaps at the basal body or at sites for ongoing cytokinesis (e.g., cleavage furrow53) by fluorescence microscopy after tagging the protein in situ at its C-terminus with a 3X HA epitope54 or a “Spaghetti monster”-Myc tag. Following immunostaining of trypanosomes with anti-Myc or anti-HA antibodies, BBP59 was detected in puncta throughout the cytoplasm (Fig. 3A), like CK1.28. Potential colocalization of BBP59 with CK1.2 was assessed in a trypanosome line expressing V5- tagged CK1.2 and Myc-tagged BBP59 (Fig. 3B). Trypanosomes were labeled with both anti-Myc and anti-V5 antibodies and the degree of signal overlap between CK1.2 and BBP59 was determined with Manders’ overlap coefficient55 (Fig. 3C). At least 75 trypanosomes were analyzed in each of three biological replicates. Mean Manders’ overlap coefficient scores were low56; 0.343 for BBP59 with CK1.2, and 0.269 for CK1.2 with BBP59.

BBP59 is not a basal body protein in bloodstream T. brucei. (A) BBP59 was endogenously tagged at its C-terminus with either a 3 × HA epitope (upper row) or a “Spaghetti monster”(SpMon)-Myc tag (lower row). Trypanosomes were fixed and labeled with an anti-Myc or anti-HA primary antibody followed by fluorescent secondary antibody, then stained with DAPI and analyzed by fluorescence microscopy. Trypanosome cell bodies are outlined in the “Merge” column. Scale bar = 20 μm. (B) Trypanosomes containing both V5-CK1.2 and BBP59-Myc epitope tags were labeled with anti-V5 and anti-Myc antibodies. Scale bar = 5 μm. (C) Fluorescence signal overlap between V5-CK1.2 and BBP59-myc was determined using Mander’s coefficient. M1 = Overlap coefficient of BBP59 with CK1.2. M2 = Overlap coefficient of CK1.2 with BBP59. N > 75 trypanosomes in each of 3 biological replicates. Lines represent mean Mander’s overlap coefficient scores.
We conclude that BBP59 is not concentrated near basal bodies or at membrane sites for cytokinesis; it is a cytoplasmic protein in bloodstream T. brucei.
Electrophoretic mobility of BBP59 is shifted by phosphatase treatment in vitro or by SB-431542 treatment in vivo
BBP59 is dephosphorylated after CK1.2 knockdown8. In western blotting of lysates from trypanosomes expressing Myc-tagged BBP59, the protein migrates as a doublet close to the expected size of 59 kDa (Fig. 4A) (Images of uncropped representative western blots are presented in Fig. S5). Since phosphorylation alters electrophoretic mobility of some proteins in SDS–polyacrylamide gel, we hypothesized that the BBP59 doublet may be caused by phosphorylation of a fraction of the protein. To examine this possibility, we treated lysates of trypanosomes containing BBP59-Myc with protein phosphatase, and we quantified the intensity of the two BBP59-Myc bands in western blots. Phosphatase treatment decreased the intensity of the upper band by 97.2% (average ratio of total-protein normalized upper:lower band signal intensity decreased from 0.76 to 0.02 (p = 5 × 10–3, Student’s T-test)), (Fig. 4B). We then investigated the effect of CK1.2 knockdown on electrophoretic mobility of BBP59-Myc.

Electrophoretic mobility of BBP59-myc is affected by phosphatase treatment or down-regulation of CK1.2 enzyme activity. (A) Upper panel: Representative western blot of lysate from BBP59-myc trypanosomes treated with λ-protein phosphatase for 15 min. Arrows indicate two BBP59-myc bands observed in lysates not treated with phosphatase. Lower panel shows total protein stain. (B) Signal intensity of the upper and lower BBP59-myc bands was measured, normalized to total protein level per lane, and plotted as the ratio of upper:lower band intensity. Three independent experiments were performed. Statistical significance of the difference in upper band intensity with and without phosphatase treatment was determined using Student’s t-test (p = 5.0 × 10–3). (C) Upper panel: Representative western blot of CK1.2 RNAi/BBP59-myc trypanosomes. Knockdown of CK1.2 was induced for 12 h with tetracycline (1 μg/mL). Arrows indicate the two BBP59-myc bands observed in control cells. Lower panel shows total protein stain. (D) Signal intensity of the upper and lower BBP59-myc bands, analyzed as presented as in (B). Statistical significance of the difference in BBP59-myc band ratios between—Tet and + Tet samples was determined using Student’s t-test (p = 0.019). (E) Representative western blot of BBP59-myc T. brucei lysate following treatment with DMSO or SB-431542 for 4 h. Arrows indicate two BBP59-myc bands observed in control cells. Lower panel shows total protein stain. (F) Signal intensity of the upper and lower BBP59-myc bands, analyzed as presented as in panels B and D. Three independent experiments were performed. Statistical significance of the differences in BBP59-myc band ratios between DMSO and SB-431542 or PF-670462 treatments was determined using Student’s t-test (For SB-431542, p = 2.2 × 10–3. For PF-670462, p = 1.8 × 10–3).
After CK1.2 knockdown, the upper band of the BBP59-Myc doublet decreased in intensity (Fig. 4C) by 69% (average ratio of total-protein normalized upper:lower band signal intensity decreased from 0.36 to 0.11 (p = 0.02, Student’s T-test)) (Fig. 4D).
We then determined whether SB-431542 or PF-670462 affected electrophoretic mobility of BBP59-Myc. Here, we treated trypanosomes expressing BBP59-Myc with SB-431542 (8 µM) or PF-670462 (4 µM) for 4 h and evaluated the change in the ratio of BBP59 upper:lower band (normalized signal intensity) (Fig. 4E). Compared to DMSO, PF-670462 treatment decreased the upper:lower band ratio by 65%, from 0.51 to 0.18 (p = 1.8 × 10–3, Student’s T-test) and SB-431542 treatment similarly decreased the upper:lower band ratio 61%, from 0.51 to 0.20 (p = 2.2 × 10–3, Student’s T-test) (Fig. 4F).
Together the three sets of data indicate that BBP59 is phosphorylated in vivo (Fig. 4), confirming data from mass spectrometry analysis8. Further, genetic knockdown, and small-molecule inhibitor analyses show that CK1.2 contributes to in vivo phosphorylation of BBP59 (Fig. 4).
Ser72 is important for phosphorylation of BBP59 by CK1.2
Three sets of data support BBP59 phosphorylation by CK1.2. First, BBP59 is dephosphorylated following CK1.2 knockdown in vivo8. Second, phosphorylation of BBP59-Myc is affected in vivo by CK1.2 (Fig. 4). Third, a BBP59 peptide is phosphorylated by CK1.2 in vitro (Fig. 2B). To extend these observations, we sought to (i) test whether a predicted phosphosite8 could be verified using purified enzyme in vitro, and (ii) identify amino acids governing phosphorylation of BBP59 by CK1.2. Whereas the phosphopeptide had seven Ser/Thr residues only three of them were predicted to be phosphorylated (Fig. 2A): An amino acid pattern (i.e. linear motif57,58,59) recognized by T. brucei CK1.2 has not been described. When peptides contain multiple Tyr, Ser or Thr residues PhosphoRS or other computational approaches are used to assign probabilities of phosphosites60,61,62 (see Fig. 2 for examples). A protein kinase substrate contains two sets of information; (i) a pattern of amino acids recognized by the kinase (i.e., the motif), and (ii) an acceptor site for the phosphoryl group that is transferred from ATP to the peptide.
Variants of a BBP59 peptide with Ala replacements of single Ser or Thr residues were synthesized (Fig. 5A) to help determine residues important for CK1.2 phosphorylation of BBP59. A “silenced” version of the peptide in which all Ser or Thr residues were replaced by Ala served as a negative control. These peptides were tested as substrates of CK1.2 (Fig. 5B). To determine statistical significance of differences in mean phosphorylation, the signal (natural log-transformed mean counts per minute) for each peptide was compared to the un-modified “Original” peptide using a repeated measures ANOVA and Dunnet’s multiple comparison’s test.

Substitution of alanine for serine 72 decreases CK1.2 phosphorylation of a BBP59 peptide. (A) Alanine scanning mutants of the BBP59 peptide substrate of CK1.2. (B) Peptides were tested as substrates as sets in three independent experiments. Each peptide was tested in two technical replicates per independent experiment. Symbols represent mean counts per minute of technical replicates and are color and shape-coded according to the experimental replicate: blue triangles, red diamonds, and black squares represent mean counts per minute (CPM) from independent replicate sets. Lines represent mean CPM values. A repeated measures one-way ANOVA with Dunnett’s multiple comparisons test was used to test for significance between each peptide and the control (“Original”) (“Silenced”, p = < 0.0001, “No Substrate,” p = < 0.0001, S66A, p = 3.5 × 10–3; S69A, p = 2.8 × 10–3; T70A, p = 0.0993; S72A, p = < 0.0001). (C) Linear peptide motifs within CK1.2 substrates. Amino acids encompassing substrates of CK1.2 were analyzed for patterns using PRATT. Search parameters were C = 100, PL = 20, PN = 20, PX = 5, FN = 3, FL = 3, E = 3. Two motifs predicting the Ser72 residue of BBP59 are presented along with alignment of substrates for CK1.2 (from Fig. 2). The sequence of ZC3H11 protein was searched using ScanProsite, with CK1.2 target motifs as queries. Scan was run in match mode (greedy, overlaps, no includes).
Replacement of Ser with Ala at position 66 or 69 increased phosphorylation of the BBP59 peptide compared to the normal sequence (p = 3.5 × 10–3, and p = 2.8 × 10–3, respectively) (Fig. 5B). Substitution of Thr70 with Ala decreased phosphorylation of the peptide, but not to a statistically significant level (p = 0.1) (Fig. 5B). At position 72, substitution of Ala for Ser decreased phosphorylation of the peptide to background levels (p < 1 × 10–4) (Fig. 5B). Phosphorylation of the control “Silenced” peptide was very low compared to the normal peptide (p < 1 × 10–4) (Fig. 5B). Phosphorylation of the “No Substrate” control reaction was reduced compared to the “Original” peptide (p < 1 × 10–4) (Fig. 5B). We conclude that Ser72 is required for CK1.2 phosphorylation of a BBP59 peptide.
Two linear motifs are present in substrates of CK1.2 from T. brucei
Substrate specificity of trypanosome protein kinases has not been studied experimentally, although the enzymes have diverged from human or yeast homologs63. So, it is unclear that one can identify new substrates for T. brucei CK1.2 using linear motifs from human CK1s. Having experimentally documented four new substrates of CK1.2 (Fig. 2B) and knowing their predicted phosphosites (Fig. 2A) we extracted linear motifs in substrates of trypanosome CK1.2 using PRATT64.
All four substrates of CK1.2 contained two motifs that had three or four invariant residues, S-x(1,3)-T-x(1,4)-S (single letter amino acid code) and S-x(1,3)-T-x(1,4)-S-x(2)-A-x(2)-[AIV] (Fig. 5C). Ser72, which is essential for CK1.2 phosphorylation of BBP59 (Fig. 5A), was part of both motifs. Presence of Thr or Ala as invariant amino acids in the motifs is unique to trypanosome CK1.2, compared with human CK1 phosphosites (see below). Prior to our work (Fig. 2), only one substrate, ZC3H11, for CK1.2 was proposed12: It has a CK1.2 motif in residues 294 to 302.
Human substrates of CK1 were analyzed similarly. For twenty peptides studied three motifs together accounted for their phosphorylation; (i) S-x(2)-S-x(0,2)-S, (ii) S-x(3,4)-S-x(2)-[EST], and (iii) S-x(4,5)-S-x(2)-[EPST] (Fig. S6). A single motif with three invariant positions (e.g., S-x(2)-S-x(0,2)-S) was not found in all twenty peptides. Similarly, the widely publicized human CK1 motif S-x(2)-S may not be universal since it was not found in some substrates in an Eukaryotic Linear Motif (ELM) dataset65. We surmise that human CK1 recognizes a set of motifs, none of which is identical to the trypanosome pattern (Fig. 5C).
Ser72 mutants alter BBP59 effects on cytokinesis and kDNA division
In (non-differentiating) bloodstream T. brucei we obtained inconclusive data, using RNAi to determine functions of BBP59. After RNAi, BBP59 expression was reduced in six independent clonal lines but only three displayed loss-of-fitness while the other three proliferated normally and divided their kinetoplasts (Fig. S7). These observations suggest that some clonal lines adapt to loss of BBP59. This data is not surprising, because in Saccharomyces cerevisiae 4699 genes can be deleted without loss of cell viability, like loss of BBP59 in T. brucei. However, the S. cerevisiae knockout lines adapted to gene loss by changing the proteome; 84% of proteins changed two-fold in many knockout lines66. Given the complexity of data from BBP59 knockdown studies, we used ectopic expression of wild-type and mutant forms of the protein (i.e., a dominant-negative strategy67,68,69) to gain insight into biologic functions of BBP59 in T. brucei (Fig. 6).

Phosphomimetic residues at Ser72 alter BBP59 effects on kDNA division and cytokinesis. T. brucei expressing of ectopic BBP59 was induced for 4 h with tetracycline. Kinetoplasts (K) and nuclei (N) were stained with DAPI, visualized by fluorescence microscopy and enumerated. Statistical significance of the differences in cell types between parental single marker (SM) controls and induced trypanosomes expressing variants of BBP59 was determined with a χ2 test. (A) S72_BBP59 (unmutated) and SM T. brucei (p = 1.4 × 10–10). (B) A72_BBP59 (p = 3.3 × 10–8) and D72_BBP59 (p = 4.2 × 10–4) were compared to unmutated S72_BBP59. (C) Plots of KinD:Cyto ratios in cells with different genetic backgrounds. Cartoon shows major effects of D72_BBP59 and A72_BBP59 on cytokinesis and kDNA division. (D) Model (inferred from panel C) of effect of Ser72 phosphorylation on BBP59 regulation of cytokinesis and kDNA division.
Use of inducible ectopic overexpression to study T. brucei protein kinases is well-established. Protein overexpression in a wild-type (WT) background, exemplified in work with AUK170, PLK71, NRKC72, and DYRK, allows rapid detection of phenotypes caused by mutant proteins and enables studies that may not be possible when trypanosomes are not viable after gene knockout.
We hypothesized that BBP59 contributed to physiologic effects of CK1.2 (i.e., cytokinesis or kinetoplast division (Fig. 1A)8) since BBP59 is an in vivo substrate of CK1.2 (Fig. 4)8. Further, with Ser72 affecting phosphorylation of BBP59 by CK1.2 (Fig. 5) we predicted that mutations that prevent or mimic phosphorylation of BBP59 might affect cytokinesis or kDNA division. (BBP59 is a predicted protein kinase (Fig. S8) with S72 outside the kinase domain.) To test these ideas, we expressed ectopic copies of Myc-tagged BBP59 containing one of three forms of the protein; (i), normal WT-BBP59 (i.e., S72_BBP59), or (ii) a nonphosphorylatable A72_BBP59, or (iii) a phospho-mimetic D72_BBP59. Protein expression was induced for 4 h and quantitated by western blotting. For unmutated (WT) S72_BBP59, mean ectopic expression was 5510 arbitrary units (AU), twenty-one times background levels (251 AU) (Fig. S9B); A72_BBP59 mean ectopic expression was 5363 AU, twenty-eight times the control (185 AU) (Fig. S9D), and D72_BBP59 mean ectopic expression was 16,067 AU, thirty-five times background level (459 AU) (Fig. S9F).
To investigate the effects of Ser72 phosphosite mutants on cytokinesis or kinetoplast division, expression of each BBP59 was induced for 4 h, trypanosomes were stained with mCLING73 and DAPI, and the number of kinetoplasts and nuclei per cell was quantitated. Compared to parental single marker (SM) cells74, expression of S72_BBP59 (i.e., unmutated) decreased 1K1N abundance from 73.3 to 56.8%, increased 2K2N from 11.1 to 16.8% of cells, and produced 1K2N cells (6.6% of total population) (Fig. 6A). S72_BBP59 had a statistically significant effect on the distribution of K/N trypanosome populations (p = 1.4 × 10–10, χ2) (N = 468 for SM cells and 484 for S72_BBP59 cells). We calculated the relative contribution of BBP59 to kDNA division or cytokinesis in single cells (termed the KinD:Cyto ratio) by dividing the proportion 1K2N cells by the fraction of 2K2N (i.e., 6.6/16.8 for S72_BBP59). The resulting ratios compare the effects of S72 substitutions on cytokinesis or kDNA division. For S72_BB59 KinD:Cyto was 0.4 (Fig. 6C).
A72_BBP59 expression, compared to S72_BBP59, increased abundance of 2K2N to 24.6% of the population. Concomitantly, only 2.4% of trypanosomes were 1K2N (Fig. 6B) (N = 443 cells from three biological replicates) (p = 3.3 × 10–6, χ2). KinD:Cyto dropped from 0.40 for S72_BBP59 to 0.11 for A72_BBP59 (Fig. 6C) implying that A72_BBP59 drives division of kDNA while reducing cytokinesis (Fig. 6B and C). Since A72 lacks a phosphorylatable hydroxyl, we infer that dephosphorylation of S72 (mimicked by A72_BBP59) promotes division of kDNA and prevents cytokinesis (Fig. 6D). (Interestingly, after knockdown of CK1.2 KinD:Cyto (0.10) was similar to that of A72_BBP59 (Fig. 6C), suggesting that effects of knockdown of the kinase on kinetoplasts and cytokinesis is phenocopied by A72_BBP59.)
Expression of D72_BBP59, compared to S72_BBP59, produced 1K2N (7.6%) like the effect of S72_BBP59, and reduced amount of 2K2N to 11.9% of trypanosomes (Fig. 6B). The difference in K/N distribution caused by D72_BBP59, compared to S72_BBP59, was statistically significant (p = 4.2 × 10–4, χ2) (N = 353 D72_BBP59 trypanosomes from three biological replicates). KinD:Cyto ratio of D72_BBP59 rose to 0.85 from 0.40 for S72_BBP59 (Fig. 6C). Together the data imply that D72_BBP59 prevents kinetoplast division, like S72_BBP59, and facilitates cytokinesis (Fig. 6C). D72_BBP59 mimics persistent phosphorylation of S72, so we deduce that phospho-S72-BBP59 preferentially drives cytokinesis (Fig. 6D). In summary, phospho-mimetic mutants reveal the role of S72 phosphorylation on biological functions of BBP59 (Fig. 6D).
A72_BBP59 expression changes phosphorylation of cytokinesis proteins
Expression of A72_BBP59 blocks cytokinesis preferentially (Fig. 6B and C). BBP59 is a predicted protein kinase, and S72 is not within the kinase domain (Fig. S8). For these reasons, we reasoned that production of A72_BBP59 (functionally representing dephosphorylated BBP59) might change the global phosphoproteome in ways that provide clues about cytokinesis proteins affected by BBP59 (i.e., BBP59 “pathway proteins”). To test this hypothesis, we performed quantitative phosphoproteomics to identify phosphopeptides whose stability was perturbed by A72_BBP59 expression. Towards that goal, A72_BBP59 expression was induced for 4 h in cells grown in heavy SILAC medium75 while uninduced A72_BBP59 trypanosomes were cultured in light medium. Trypanosomes (1 × 108) from each group were combined and phosphopeptides analyzed by LC–MS/MS8,76. Phosphopeptides were filtered using a 1% FDR threshold77, and those with changes in abundance greater than two-fold (in two out of three biological replicates) (Fig. 7) were compiled (Table S1).

Expression of A72_BBP59 changes the phosphoproteome of T. brucei. In SILAC-based phosphoproteomics (three independent experiments) A72_BBP59 expression was induced with tetracycline for 4 h in trypanosomes cultured in heavy SILAC medium. Un-induced A72_BBP59 cells were grown in light SILAC medium as a control. Phosphopeptides were enriched and identified using LC-FAIMS-MS/MS and a 1% False Discovery Rate. Phosphopeptides observed in at least two independent biological samples replicates with a two-fold change (at least) in abundance between light and heavy samples were accepted as positive results. Representative phosphopeptides are presented by plotting relative abundance against Qvality posterior error probability (PEP) score. Highlighted phosphopeptides (red) belong to proteins essential for cytokinesis.
For 183 proteins phosphopeptide abundance increased, whereas amounts of phosphopeptides from 117 proteins decreased (Table S1). Eleven proteins essential for cytokinesis were affected by A72_BBP59 (Table 1), providing a link from BBP59 to cytokinesis polypeptides (reviewed in78). T. brucei encodes 9068 polypeptides1,79 of which 2550 are phosphorylated6. Therefore, our finding that 11 proteins are associated with BBP59 regulation of cytokinesis (Table 1) documents significant selectivity of our approach.
Discussion
Effectors for CK1.2 regulation of cytokinesis
Cytokinesis and kDNA segregation are executed by groups of proteins, not all of which have been identified. Here we have used a bottom-up approach directed by a protein kinase to determine involvement of a hypothetical protein in both cytokinesis and kDNA division.
First, we confirmed a role for CK1.2 in cytokinesis using genetic and small molecule tools. Knockdown of CK1.2 (Fig. 1A) as well as chemical inhibition of the enzyme activity with SB-431542 (Fig. 1B) increased abundance of 2K2N (post-mitotic) trypanosomes (Fig. 1C), confirming a block of cytokinesis3. Participation of CK1.2 in cytokinesis could occur by direct interaction with proteins in that pathway (reviewed in80). However, its dispersal in the cytoplasm8 did not support a hypothesis that the kinase was concentrated at sites routinely associated with cytokinesis. For that reason, an alternative hypothesis that CK1.2 influenced cytokinesis indirectly, through substrates that it phosphorylated (i.e., effector proteins), was considered.
A “cytokinesis substrate effector protein” for CK1.2 must meet several criteria8. First, the candidate must be dephosphorylated after knockdown or chemical inhibition of CK1.2. Second, the candidate must be phosphorylated directly by CK1.2. Third, perturbation of the candidate’s expression (by knockdown or overexpression) must inhibit cytokinesis. BBP59 met these criteria. It is a phosphoprotein (Fig. 2A) that is dephosphorylated after CK1.2 knockdown8 (Fig. 4), suggesting interaction with CK1.2 in vivo. A peptide from BBP59 is phosphorylated directly by CK1.2 in vitro (Figs. 2 and 5). Ectopic expression of BBP59 inhibits cytokinesis (Fig. 6A).
A72_BBP59 changes the protein’s effects on cytokinesis and kDNA division
S72 is required for CK1.2 phosphorylation of BBP59 (Fig. 5). To assess importance of S72 in biological activities of BBP59, we studied effects of two S72 mutants, A72_BBP59 and D72_BBP59. A72_BBP59 cannot be phosphorylated because it lacks a hydroxyl on the side chain of Ala. Strikingly, that mutant selectively blocked cytokinesis while facilitating kDNA division (Fig. 6B and C), indicating that unphosphorylated S72 promotes kDNA division while preventing cytokinesis. In contrast, the phospho-mimetic D72_BBP59 facilitated cytokinesis while inhibiting kDNA division at the same level as S72_BBP59 (Fig. 6B and C), implying that phospho-Ser72 promotes cytokinesis (Fig. 6D). These data reveal a cycle of phosphorylation centered on Ser72 is a “molecular switch” for BBP59 promoting either cytokinesis or kDNA division (Fig. 6D).
Ser72 is outside both the kinase and the “activation loop” domains of BBP59 (Fig. S8). So, we propose that Ser72 is part of a linear motif that facilitates BBP59 interactions with partner proteins to promote cytokinesis or kDNA division (Fig. 6D). In support of this suggestion, four Eukaryotic Linear Motifs (ELMs) that might facilitate BBP59 interactions with other proteins, namely (i) 14-3-3_CanoR_1, (ii) FHA, (iii) PDZ_Class_2, and (iv) WD40_WDR5_VDV_2 are found proximal to Ser72 (Table S2).
BBP59 is conserved in trypanosomatids
BBP59 is a hypothetical protein that is predicted to be a protein kinase3 (Fig. S8A) with orthologs in other kinetoplastids, including Trypanosoma cruzi and Leishmania donovani (Fig. S8B), suggesting roles for the protein in trypanosomatids. In bloodstream T. brucei the functions of BBP59 are not clear. Evidence was presented for a role in differentiation of bloodstream to insect-stage T. brucei2, although a genome-wide screen for proteins necessary for that differentiation event did not identify BBP59 as a key factor81.
Our attempt to understand contributions of BBP59 phosphorylation to trypanosome biology relied on expression of ectopic copies of the gene for short periods (less than 6 h) in a wild-type background. This approach was necessary because we obtained conflicting data after RNAi knockdown of gene (Fig. S7) or deletion of both alleles of BBP59; heterozygous clones did not display obvious phenotypes. Further, we know now that trypanosomes adapt to deletion of the BBP59 gene by altering their proteome (work in progress). This observation, like that reported for non-essential yeast genes66, implies that knockout lines of BBP59 are not simply wild-type trypanosomes lacking the protein. That is, “genuine” knockout lines may not exist; they may be lost during “proteome adaptation” while selecting for stable transfectants. From this perspective it is more appealing to study mutant BBP59 genes in wild-type trypanosomes using a “dominant-negative” protocol than in mutant cells whose proteome has been altered due to loss of BBP59.
Establishing biochemically with purified PTM enzymes that a protein is a true substrate validates our in vivo strategy for selecting candidates after proteome-wide analysis (see Fig. 2). We concede, however, that this substrate confirmation step in vitro may not be possible in some biological systems. In that case in vivo confirmation of the action of a PTM enzyme (see Fig. 4) may be sufficient for the task at hand.
A protein kinase can guide functional annotation of hypothetical polypeptides
More than 3000 protein-coding genes (30% of the genome) in T. brucei are hypothetical1,63, presenting an opportunity to identify functions for uncharacterized polypeptides. Here we use CK1.2 to illustrate how a protein kinase may be deployed as an “in vivo guide” to assign functions to hypothetical proteins. We present a workflow (Fig. 8) that could be adapted to other posttranslational modification (PTM) enzymes (e.g., acetylases/deacetylases, phosphatases, or proteases) to guide the assignment of biological functions to hypothetical proteins. Results from such studies can contribute to functional annotation of genomes3,8,63,76.

Identification of effectors for CK1.2 signaling and discovery of functions for hypothetical proteins in the African trypanosome. Here we summarize a six-step workflow used to discover effectors of CK1.2. We first catalogued molecular/physiologic effects after knock down of the gene. Second, we assembled a list of 53 “candidate protein substrates” by determining polypeptides that were dephosphorylated after loss of the enzyme 8 (T. brucei has 9068 polypeptides1,79). Third, we verified substrates in vitro by assaying their phosphorylation by purified CK1.2 (Fig. 2). Fourth, we ascertained phosphorylation of the substrate BBP59 in vivo after addition of a small molecule inhibitor SB-431542 (of CK1.2) or after knockdown of CK1.2 (Fig. 4). Fifth, we confirmed the phosphosite of BBP59 by alanine scanning mutations of serines and threonines therein (Fig. 5). Sixth, we demonstrated that perturbation of BBP59 expression phenocopied knockdown of CK1.2 control of cytokinesis and kinetoplast division (Fig. 6) leading to its classification as an effector for CK1.2 signaling.
In phosphoproteomic experiments, we identified 53 polypeptides that were dephosphorylated after knockdown of CK1.28. Reasoning that some of these so-called “CK1.2-Pathway Proteins” could be phosphorylated by the enzyme we tested peptides from predicted phosphosites as substrates and found four that were modified by CK1.2; hypothetical proteins BBP59, LRRP1, BBP268, and CAP64 (Fig. 2). These polypeptides may be involved in biological pathways regulated by CK1.2 (e.g., cytokinesis, basal body biogenesis, kDNA division)3,8. Focusing on BBP59 as an example we demonstrate a role for that hypothetical protein in kDNA division and cytokinesis.
We outline steps used in our CK1.2-directed work in the African trypanosome (Fig. 8). The strategy employed can be adapted for protein kinases or other enzymes that modify proteins (e.g., acetyltransferases, phosphatases). Our objective was to deploy a protein kinase to direct placement of polypeptides in signaling pathways, when sequence alignments fail to predict possible functions. From 9068 polypeptides in the proteome1,79 we identified 53 candidate substrates of CK1.2 (i.e., dephosphorylated after knockdown of the gene)8. T. brucei has 40 essential protein kinases, so the study performed here could be expanded to 39 other kinases for 1491 hypothetical phosphoproteins6. In this scenario, assuming an equal number of unique substrates per kinase, each enzyme might phosphorylate 38 hypothetical phosphoproteins (or 162 phosphosites per kinase)6. Hence, the concepts presented here could be a roadmap for assigning function to over 1400 hypothetical phosphoproteins in T. brucei. Indeed, these principles can be extended to any cell in which knockdown of genes encoding PTM enzymes is feasible.
Materials and methods
Trypanosome culture
“Single Marker” (SM) bloodstream T. brucei74 was maintained in log phase growth (< 1.5 × 106 cells/mL) in HMI-9 medium83 or in tetracycline (Tet)-free HMI-9 (TF-HMI-9) at 37 °C with 5% CO2. TF-HMI9 contained 10% Tet-free fetal bovine serum (FBS) (Catalog number 631367, Takara Bio USA, Mountain View, CA) instead of normal FBS (Catalog number S11150H, R&D Systems, Minneapolis, MN.)
Fluorescence microscopy
Staining of trypanosomes with mCLING and DAPI Trypanosomes (106) were pelleted (3000 × g for 3 min) and rinsed in 500 μL ice-cold phosphate buffered saline containing 10 mM glucose (PBS-G), pelleted again (3000xg for 3 min), and resuspended in ice cold PBS-G (24 μL). mCLING labeling73 was modified as follows. One μL of 50 μM mCLING-488 (Catalog number 710-006AT3; Synaptic Systems GmbH, Goettingen, Germany) was added and the cells were mixed by repeated gentle pipetting. One hundred microliters of ice-cold 4% paraformaldehyde (PFA) in PBS with 0.05% glutaraldehyde was added and cells were gently mixed. Twenty microliter portions of cell suspension were spread on poly-L-lysine-coated glass cover slips (12 mm round) and dried, cell-side up, in an open Petri dish on a 50 °C bead bath until most of the liquid had evaporated. Coverslips were transferred to 24-well plates, briefly rinsed twice with 500 μL PBS, immersed in de-ionized water, dried and mounted on a microscope slide with Vectashield containing 4’,6-diamidino-2-phenylindole (DAPI) (Catalog number H-1200, Vector Laboratories, Burlingame, CA). Edges of slides were sealed with “Wet n Wild” clear nail polish (Electron Microscopy Services, Hatfield, PA). Z-stack images were captured with a Keyence BZ-X800 microscope (Keyence Corporation of America, Itasca, IL) equipped with a 60X (NA 1.4) objective, and files were processed using Fiji83,84.
Quantitation of kinetoplasts and nuclei: A grid of Z-stacks and maximum intensity projections were acquired, and the images stitched together using Keyence BZ-X800 Analyzer software (version 1.1.1.8) (“Correct shading” and “uncompressed” options were applied). Trypanosomes were grouped according to the number of kinetoplasts and nuclei per cell, and the data tabulated using the CellCounter plugin in Fiji83,84. Three independent experiments were conducted with at least 150 trypanosomes counted in each sample. The average percent of the population with a kinetoplast and nucleus combination per cell (e.g. 1K1N, 2K1N, 2K2N, 1K2N) from three (or more) experiments was determined for control and experimental populations. A χ2 test was used to test for statistical significance of differences in distributions of cell types between groups as specified in appropriate sections.
Concurrent detection of BBP59 and CK1.2 in trypanosomes BBP59 was tagged with 3X Myc (described above) in a trypanosome line containing V5-CK1.28. Trypanosomes were processed for fluorescence microscopy as described earlier and in previous work85. Images were acquired and analyzed with a BZ-X810 fluorescence microscope (Keyence Inc.) as described previously. Total number of cells used for overlap analysis was ~ 260 from three sets of independent experiments. Mander’s overlap coefficient was calculated using a colocalization plugin tool JACoP86 in Fiji.
Quantitative western blotting
Trypanosome pellets (2 × 106 cells) were resuspended in Laemmli buffer (50 mM Tris, pH 6.8, 2% SDS, 10% glycerol, 3% β-mercapto-ethanol, 0.05% bromophenol blue) and separated by SDS-PAGE (4–20% polyacrylamide Mini-PROTEAN TGX pre-cast gel; Bio-Rad; Hercules, CA, USA) before transfer to a polyvinylidene fluoride (PVDF) membrane (Bio-Rad) using a Trans-blot Turbo Transfer System (Bio-Rad; Hercules, CA). The membrane was incubated in Revert 700 Total Protein Stain (Li-COR, catalog number 926-11011), and a Li-COR Odyssey CLx system was used to capture an image for measuring total protein content per lane, following protocols described previously87,88.
Knockdown of CK1.2
CK1.2 RNAi cells8 (5 × 104 cells/mL) growing in TF-HMI-9 were incubated with or without tetracycline (1 μg/mL) for 12 h. Trypanosomes were harvested, stained with mCLING and DAPI, and kinetoplasts and nuclei were enumerated as described above.
Protein kinase assay in vitro
Reactions (final volume 20 µL) contained 50 nM purified CK1.2, 20 uM pS7 peptide (Anaspec; Fremont, CA, USA) in buffer (50 mM HEPES pH 7.6, 5 mM MgCl2, 2 mM dithiothreitol, 150 mM NaCl) with 20 µM ATP and 1 μCi γ[33P]ATP. Reactions were incubated for 20 min at 30 °C, and halted with two volumes of ice cold 10% trichloroacetic acid (TCA). Terminated reactions were spotted onto discs of Whatman P-81 filter paper that was washed three times with 75 mM H3PO4, once with 100% acetone, and then dried using a vacuum manifold. Radioactivity on the Whatman P-81 filter paper discs were analyzed in a scintillation counter (TriCarb 4810 TR; Perkin Elmer, Waltham, MA). The averages of two technical replicates from each sample were recorded. The experiment was performed three times with different biological samples.
For candidate substrate tests, peptides that were dephosphorylated after CK1.2 RNAi8 were synthesized with at least 6 amino acids flanking predicted phosphosite(s). If the peptide had a net charge less than + 2, lysines were added to the termini until the charge was + 2, to facilitate recovery on Whatman P-81 filter paper for scintillation counting (described above). Peptides (500 µM final concentration) were added to reaction mixtures and phosphorylation was quantified as described above. For enzyme inhibition assays, 1 µL of SB-431542 (Catalog number 13031-10, Caymen Chemical, Ann Arbor, MI) or PF-670462 (Catalog number 14588-10, Caymen Chemical) from 20X stocks in 100% dimethylsulfoxide (DMSO) were added to kinase reaction mixtures before incubation at 30 °C. Data obtained was analyzed using four parameter non-linear regression with Prism 7 (Graph Pad, San Diego, CA), to determine IC50 for each compound.
Treatment of trypanosomes with SB-431542
Single Marker (SM) trypanosomes (5 × 105 cells/mL) were resuspended in 5 mL of pre-warmed HMI-9 and incubated with SB-431542 or DMSO to final concentrations of 8 μM and 0.04%, respectively, for 4 h at 37 °C, after which they were stained with mCLING and DAPI (described above). Microscopy analyses were performed as described earlier.
Epitope tagging of BBP59
3X Myc tag- pMOTag23M vector54 (a gift from Roberto Docampo, University of Georgia) was used as a template in a PCR reaction with a forward primer 3X Myc tag- pMOTag23M vector54 (a gift from Roberto Docampo, University of Georgia) was used as a template in a PCR reaction with a forward primer aaaa a reverse primer bbbb to create a C-terminal 3X-Myc BBP59 tagging fragment. PCR product (~ 13 μg DNA) was resuspended in 100 µL human T Cell transfection buffer (Amaxa) and was transfected into SM trypanosomes (4 × 107) with an Amaxa Nucleofector (Protocol X-001). Stable clones were obtained by limiting dilution and were selected by culturing trypanosomes in medium containing 0.1 μg/mL puromycin and 6.5 μg/mL G418. Using the same protocol, BBP59 was tagged with 3X-Myc in a CK1.2 RNAi background create a C-terminal 3X-Myc BBP59 tagging fragment. PCR product (~ 13 μg DNA) was resuspended in 100 µL human T Cell transfection buffer (Amaxa) and was transfected into SM trypanosomes (4 × 107) with an Amaxa Nucleofector (Protocol X-001). Stable clones were obtained by limiting dilution and were selected by culturing trypanosomes in medium containing 0.1 μg/mL puromycin and 6.5 μg/mL G418. Using the same protocol, BBP59 was tagged with 3X-Myc in a CK1.2 RNAi background.
3X HA tag- A pMOTag4H vector54 was used as a template in a PCR reaction with the primers listed above. The HA-tagging product was transfected into SM trypanosomes as described above, and stable clones were selected in medium containing 5 μg/mL hygromycin and 6.5 μg/mL G418.
Spaghetti Monster-Myc tagging- A pMOT vector containing a spaghetti monster fluorescent protein with Myc-tags was used as a template for PCR reaction with primers described above. The BBP59-Myc-tagging fragment was transfected into SM trypanosomes as described earlier. Stable clones were selected in medium containing puromycin (0.1 μg/mL) and 6.5 μg/mL G418.
Electrophoretic mobility shift assays of BBP59
SB-431542 treatment: BBP59-Myc trypanosomes (5 × 105 cells/mL in HMI-9) were treated with DMSO (0.08%) or SB-431542 (8 μM) for 4 h at 37 °C. Trypanosomes (3 × 106) were pelleted (3000xg for 3 min), washed once with cold PBS-G, resuspended in 20 μL 1 × Laemmli buffer and heated at 95 °C for 5 min.
CK1.2 RNA interference: CK1.2 RNAi cells harboring an endogenous BBP59-Myc tag (2 × 105 cells/mL) were incubated with or without tetracycline (1 μg/mL) at 37 °C for 12 h. Cells were processed for SDS-PAGE as described earlier.
Phosphatase treatment: BBP59-Myc trypanosomes (5 × 106) were pelleted (3000xg for 3 min), washed once with PBS-G, and resuspended in 30 μL Tris-buffered saline (TBS) (pH 7.5) in a 1.5 mL microcentrifuge tube. The tube was immersed in an ice/ethanol bath at − 80 °C to rapidly freeze trypanosomes, then placed in a water bath at room-temperature to thaw cells. Three rounds of freeze/thaw lysis were performed, and trypanosome lysis was confirmed by light microscopy. Samples were mixed, divided into two equal aliquots, and MgCl2 added (1 mM final concentration). Lambda protein phosphatase (20 units) (Catalog number P0753, New England Biolabs) or water (0.5 µL) was added to each sample which was incubated at 30 °C for 25 min. Laemmli buffer (5X) was added to a final concentration of 1X followed by heating at 95 °C for 5 min, and SDS-PAGE.
Quantitation of BBP59-Myc band intensities: Lysates (2 × 106 cell equivalents) were loaded onto a 4–20% gradient gel and subjected to SDS-PAGE and western blotting as described above. Densitometry was performed with Empiria Studio 2.1 (Li-COR). Using the software, boxes were drawn around the upper and lower bands of the BBP59-Myc doublet in control cells (e.g. DMSO-treated, “no tetracycline”, or “no phosphatase” samples) and the anti-Myc antibody signal in the 800 nm channel was measured in each box for both control and treated lysates. Anti-Myc antibody signal was normalized to total protein (determined with Revert 700 Total Protein Stain (Li-COR) in each lane. The normalized signal intensity for the upper band was divided by the normalized signal intensity in the lower band to obtain the Upper:Lower BBP59-Myc doublet band ratio. Three biological replicates were analyzed. A Student’s T-test was used to test for statistical significance of differences in the means of Upper:Lower ratios between control and treatment groups.
Establishment of expression lines for BBP59 and its Ser72 variant
A BBP59 coding sequence (from TriTrypDB1) was synthesized as part of a pUC57 expression construct with a C-terminal 3X Myc tag inserted before the stop codon (GeneWiz). BamHI and HindIII restriction sites were appended to the 5′ end and 3′ ends (after the Myc tag). A HindIII site (AAGCTT) beginning at position 1219 was removed with a silent G to A substitution at position 1,221. BBP59 was cleaved from the pUC-57 expression vector with BamHI and HindIII, and ligated into BamHI- and HindIII-digested pDEX-77789 to create BBP59-pDEX777 which was sequenced to confirm identity of DNA fragments.
To generate S72A-BBP59-pDEX-777, site-directed mutagenesis was performed on BBP59-pDEX-777 using a Q5 mutagenesis kit (Catalog number E0554, New England Biolabs) with primers generated using the NEBaseChanger web tool (Forward, CTCTACGCCAgCCGTAACAGC; reverse, CCTGCACTTGAAACCCTCTG). The forward primer introduced a T > G mutation (indicated with lower case) at base 4671 in the plasmid sequence. The resulting plasmid was sequenced to confirm the S72A change.
S72D BBP59-pDEX-777 was produced using Q5 site-directed mutagenesis with the forward primer CTCTACGCCAGACGTAACAGCCG and the reverse primer CCTGCACTTGAAACCCTC.
Overexpression lines were established by transfecting NotI-linearized plasmids in SM T. brucei (described earlier). Stable clones were obtained by selection in medium containing phleomycin (2 μg/mL) and G418 (3.25 μg/mL).
Induction of BBP59 protein production
Trypanosomes were suspended (5 × 105 cells/mL) in pre-warmed TF-HMI-9 and were divided equally into two flasks. To one flask, tetracycline was added to a final concentration of 1 μg/mL and the cultures were incubated at 37 °C for 4 h after which 2 × 106 cells from each sample were processed for a quantitative western blotting as described earlier. The difference in average intensity of BBP59-Myc bands between cells with an endogenous BBP59-Myc tag (WT) and the induced overexpression trypanosomes was assessed for statistical significance using a paired Student’s T-test (Prism 8 software).
Kinetoplast and nucleus distribution was quantified as described earlier. Possible statistical significance of differences in distribution of trypanosome division stages (i.e., 1K1N, 2K1N, 1K2N and 2K2N) caused by expression of BBP59 proteins was determined with a χ2 test (Prism 8 software).
Proteome analysis
Media and trypanosome culture: Trypanosomes were cultured in Tet-free SILAC medium75 for seven days (at least) prior to harvesting cells for mass spectrometry. “Heavy” SILAC medium contained 13C6-L-Lys (Catalog number 88431, Thermo Scientific, Rockford, IL) and 13C6-L-Arg (Catalog number 88433, Thermo Scientific, Rockford, IL). “Light” SILAC medium was prepared with unlabeled lysine and arginine.
BBP59-Ser72Ala trypanosomes cultured in heavy SILAC medium were diluted to 5 × 105 cells/mL in pre-warmed SILAC medium and treated with tetracycline (1 μg/mL final concentration). An un-induced control sample (5 × 105 cells/mL) was prepared in pre-warmed light SILAC medium. Cultures were incubated for 4 h at 37 °C after which 108 trypanosomes from un-induced and induced trypanosomes were pelleted (3000xg for 3 min), washed with ice-cold PBS-G, resuspended in 500 μL of ice-cold PBS-G and combined in a 1.5 mL centrifuge tube. Cells were pelleted and resuspended in 350 μL lysis buffer (8 M urea, 50 mM HEPES, pH 8.0) with 1X Halt phosphatase and protease inhibitor cocktail (Thermo Fisher, Waltham, MA). The trypanosome suspension was sonicated (four pulses 3–5 s each, amplitude 60) using a sonicator (Q55 QSonica, Newton, CT), and resting 30 s on ice between pulses.
Approximately two milligram of protein lysate was buffered in 50 mM HEPES (pH 8.0), 8 M urea, and protease and phosphatase inhibitor cocktails. The samples were reduced by adding TCEP (final concentration of 5 mM) and mixing at room temperature for 15 min. Alkylation was carried out by adding chloroacetamide to a final concentration of 10 mM and mixing at room temperature for 30 min. The urea concentration was reduced to 1 M by adding 100 ammonium bicarbonate and proteolytic digestion was carried out with a LysC/trypsin mixture (Promega) at 1:25 enzyme to substrate ratio. Digestion was carried out overnight at 37 °C and the enzyme reaction was stopped with trifluoroacetic acid (added to a final concentration of 0.1%). The samples were split into 300 µg proteomic and 1.7 mg phosphoproteomic samples.
Basic reverse phase separation of proteomic samples: The proteomic samples were desalted on Oasis 10 mg/1 cc HLB cartridges (Waters), taken to dryness by vacuum centrifugation, and fractionated by basic reverse phase (bRP) chromatography. Fractionation was carried out on a Vanquish Horizon HPLC (ThermoScientific) connected to a 2.1 mm × 150 mm Zorbax Extend C18, 5-µm particle size column (Agilent) and using mobile phases of 10 mM ammonium bicarbonate, pH 10 (A) and 100% acetonitrile (B). The method was constructed with a flowrate of 0.25 mL/min and operating at 5% B for 5 min, ramping to 40% B over 50 min, increasing to 90% B over 5 min, and holding at 90% for 5 min. Fractions were collected in 250 µL well plates (ThermoFisher) and were concatenated down into eight pools (Pool 1 contained fractions 1, 9, 17, 25, 33, etc., Pool 2 contained fractions 2, 10, 18, 26, 34, etc., and so on). The pools were dried by vacuum centrifugation and stored at 4 °C until LC–MS/MS analysis was performed.
Phosphopeptide enrichment: Phosphoproteomic samples were desalted on SepPak 200 mg/3 cc cartridges (Waters) and taken to dryness by vacuum centrifugation. Phosphopeptide enrichment was carried out using the High-Select Fe-NTA phosphopeptide enrichment kit (ThermoScientific) following the manufacturer’s instructions. Enriched eluates were immediately dried by vacuum centrifugation, and resuspended in 0.1% TFA, desalted on Oasis 10 mg/1 cc cartridges (Waters), and dried. Eluates were stored at 4 °C until LC–MS/MS analysis.
Global Proteomic and Phosphoproteomic Analyses: NanoLC/ESI MS/MS was carried out on proteomic bRP pools or phosphopeptide enriched eluates using an Easy1200 nLC (ThermoScientific) coupled in-line with an Orbitrap Eclipse with a Field Asymmetric Ion Mobility Spectrometry (FAIMS) mass spectrometry system (ThermoScientific). Automated protein database searches were performed using Proteome Discoverer 2.4 (Thermo Scientific). The data were searched against a Trypanosoma brucei database (Uniprot Tb927 and BBP59 S72A) that included common contaminants. Searches were performed with settings for the proteolytic enzyme trypsin. Maximum missed cleavages were set to 2. The precursor ion tolerance was set to 10 ppm and the fragment ion tolerance was set to 0.6 Da. Dynamic peptide modifications included SILAC 13C(6) (+ 6.020 Da on K,R), oxidation (+ 15.995 Da on M). Dynamic modifications on the protein terminus included acetyl (+ 42.-11 Da on N-terminus), Met-loss (− 131.040 Da on M) and Met-loss + Acetyl (− 89.030 Da on M) and static modification carbamidomethyl (+ 57.021 on C). For phosphopeptide enriched samples, phosphorylation was set as a dynamic peptide modification (+ 79.966 Da on S, T and Y). Sequest HT was used for database searching. All search results were run through Percolator for scoring and phosphosite localization probability was calculated. Minora Feature Detector was used for quantification.
Linear motif analysis
Sequences were analyzed via PRATT (https://www.ebi.ac.uk/jdispatcher/pfa/pratt)64. To search for a CK1 target motif, twenty human sequences annotated under “MOD_CK1_1” were taken from Eukaryotic Linear Motif65. The search parameters used were as follows: C% (minimum percentage of considered sequences to match = 75), PL (maximum pattern length) = 20, PN (maximum number of pattern symbols = 20), PX (maximum number of consecutive wildcards) = 5, FN (maximum number of flexible spacers) = 3, FL (maximum flexibility) = 3, E (search greediness) = 3.
To search for a shared motif among in vitro substrates of CK1.2, the parameters used were as follows: C% (minimum percentage of considered sequences to match = 100), PL (maximum pattern length) = 20, PN (maximum number of pattern symbols = 20), PX (maximum number of consecutive wildcards) = 5, FN (maximum number of flexible spacers) = 3, FL (maximum flexibility) = 3, E (search greediness) = 3. Detection of a predicted CK1.2 phosphorylation site within ZC3H11 was done by scanning motif against sequence in ScanProsite90,91.
Data availability
All data are available in the main text or the supplementary materials.
References
Aslett, M. et al. TriTrypDB: A functional genomic resource for the Trypanosomatidae. Nucleic Acids Res. 38, D457-462. https://doi.org/10.1093/nar/gkp851 (2010).
Alsford, S. et al. High-throughput phenotyping using parallel sequencing of RNA interference targets in the African trypanosome. Genome Res. 21, 915–924. https://doi.org/10.1101/gr.115089.110 (2011).
Jones, N. G. et al. Regulators of Trypanosoma brucei cell cycle progression and differentiation identified using a kinome-wide RNAi Screen. PLoS Pathog. 10, e1003886. https://doi.org/10.1371/journal.ppat.1003886 (2014).
Urbaniak, M. D. et al. Chemical proteomic analysis reveals the drugability of the kinome of Trypanosoma brucei. ACS Chem. Biol. 7, 1858–1865. https://doi.org/10.1021/cb300326z (2012).
Nett, I. R. et al. The phosphoproteome of bloodstream form Trypanonosoma brucei, causative agent of African sleeping sickness. Mol Cell Proteomics https://doi.org/10.1074/mcp.M800556-MCP200[pii] (2009).
Urbaniak, M. D., Martin, D. M. & Ferguson, M. A. Global quantitative SILAC phosphoproteomics reveals differential phosphorylation is widespread between the procyclic and bloodstream form lifecycle stages of Trypanosoma brucei. J. Proteome Res. 12, 2233–2244. https://doi.org/10.1021/pr400086y (2013).
Guyett, P. J., Xia, S., Swinney, D. C., Pollastri, M. P. & Mensa-Wilmot, K. Glycogen synthase kinase 3beta promotes the endocytosis of transferrin in the African Trypanosome. ACS Infect. Dis. 2, 518–528. https://doi.org/10.1021/acsinfecdis.6b00077 (2016).
Sullenberger, C., Hoffman, B., Wiedeman, J., Kumar, G. & Mensa-Wilmot, K. Casein kinase TbCK1.2 regulates division of kinetoplast DNA, and movement of basal bodies in the African trypanosome. PLoS ONE 16, e0249908. https://doi.org/10.1371/journal.pone.0249908 (2021).
Pradel, L. C., Bonhivers, M., Landrein, N. & Robinson, D. R. NIMA-related kinase TbNRKC is involved in basal body separation in Trypanosoma brucei. J. Cell Sci. 119, 1852–1863. https://doi.org/10.1242/jcs.02900 (2006).
Hu, H., Zhou, Q., Han, X. & Li, Z. CRL4WDR1 controls polo-like kinase protein abundance to promote bilobe duplication, basal body segregation and flagellum attachment in Trypanosoma brucei. PLoS Pathog. 13, e1006146. https://doi.org/10.1371/journal.ppat.1006146 (2017).
Urbaniak, M. D. Casein kinase 1 isoform 2 is essential for bloodstream form Trypanosoma brucei. Mol. Biochem. Parasitol. 166, 183–185. https://doi.org/10.1016/j.molbiopara.2009.03.001 (2009).
Minia, I. & Clayton, C. Regulating a post-transcriptional regulator: Protein phosphorylation, degradation and translational blockage in control of the Trypanosome stress-response RNA-binding protein ZC3H11. PLoS Pathog. 12, e1005514. https://doi.org/10.1371/journal.ppat.1005514 (2016).
Lundgren, D. H., Hwang, S. I., Wu, L. & Han, D. K. Role of spectral counting in quantitative proteomics. Expert Rev. Proteomics 7, 39–53. https://doi.org/10.1586/epr.09.69 (2010).
Zhang, B. et al. Detecting differential and correlated protein expression in label-free shotgun proteomics. J. Proteome Res. 5, 2909–2918. https://doi.org/10.1021/pr0600273 (2006).
Unwin, R. D. et al. Multiple reaction monitoring to identify sites of protein phosphorylation with high sensitivity. Mol. Cell Proteomics 4, 1134–1144. https://doi.org/10.1074/mcp.M500113-MCP200 (2005).
Zhang, P. et al. Multiple reaction monitoring to identify site-specific troponin I phosphorylated residues in the failing human heart. Circulation 126, 1828–1837. https://doi.org/10.1161/CIRCULATIONAHA.112.096388 (2012).
Dang, H. Q. et al. Proximity interactions among basal body components in Trypanosoma brucei identify novel regulators of basal body biogenesis and inheritance. MBio https://doi.org/10.1128/mBio.02120-16 (2017).
Niemann, M. et al. Mitochondrial outer membrane proteome of Trypanosoma brucei reveals novel factors required to maintain mitochondrial morphology. Mol. Cell Proteomics 12, 515–528. https://doi.org/10.1074/mcp.M112.023093 (2013).
Sivashankari, S. & Shanmughavel, P. Functional annotation of hypothetical proteins—A review. Bioinformation 1, 335–338. https://doi.org/10.6026/97320630001335 (2006).
Salavati, R. & Najafabadi, H. S. Sequence-based functional annotation: What if most of the genes are unique to a genome?. Trends Parasitol. 26, 225–229. https://doi.org/10.1016/j.pt.2010.02.001 (2010).
Cherry, J. M. The saccharomyces genome database: Gene product annotation of function, process, and component. Cold Spring Harb. Protoc. 2015, pdb-prot088914. https://doi.org/10.1101/pdb.prot088914 (2015).
Lobb, B., Tremblay, B. J., Moreno-Hagelsieb, G. & Doxey, A. C. An assessment of genome annotation coverage across the bacterial tree of life. Microb. Genom. https://doi.org/10.1099/mgen.0.000341 (2020).
Carnielli, C. M., Winck, F. V. & Paes Leme, A. F. Functional annotation and biological interpretation of proteomics data. Biochim Biophys. Acta 1854, 46–54. https://doi.org/10.1016/j.bbapap.2014.10.019 (2015).
Giuffra, E., Tuggle, C. K., Faang Consortium. Functional annotation of animal genomes (FAANG): Current achievements and roadmap. Annu. Rev. Anim. Biosci. 7, 65–88. https://doi.org/10.1146/annurev-animal-020518-114913 (2019).
Ribeiro, A. J. M., Tyzack, J. D., Borkakoti, N. & Thornton, J. M. Identifying pseudoenzymes using functional annotation: Pitfalls of common practice. FEBS J. 287, 4128–4140. https://doi.org/10.1111/febs.15142 (2020).
Kim, Y. & Chang, S. Ever-expanding network of dynamin-interacting proteins. Mol. Neurobiol. 34, 129–136. https://doi.org/10.1385/MN:34:2:129 (2006).
Gallo, S., Folco, C. B. & Crepaldi, T. The MET oncogene network of interacting cell surface proteins. Int. J. Mol. Sci. 25, 13692. https://doi.org/10.3390/ijms252413692 (2024).
Alberts, B. The cell as a collection of protein machines: Preparing the next generation of molecular biologists. Cell 92, 291–294. https://doi.org/10.1016/s0092-8674(00)80922-8 (1998).
De Las Rivas, J. & Fontanillo, C. Protein-protein interaction networks: Unraveling the wiring of molecular machines within the cell. Brief Funct. Genomics 11, 489–496. https://doi.org/10.1093/bfgp/els036 (2012).
Suderman, R. & Deeds, E. J. Machines vs. ensembles: Effective MAPK signaling through heterogeneous sets of protein complexes. PLoS Comput. Biol. 9, e1003278. https://doi.org/10.1371/journal.pcbi.1003278 (2013).
Sanbonmatsu, K. Y. Computational studies of molecular machines: The ribosome. Curr. Opin. Struct. Biol. 22, 168–174. https://doi.org/10.1016/j.sbi.2012.01.008 (2012).
Naktinis, V., Turner, J. & O’Donnell, M. A molecular switch in a replication machine defined by an internal competition for protein rings. Cell 84, 137–145. https://doi.org/10.1016/s0092-8674(00)81000-4 (1996).
Beenken, A. et al. Structures of LRP2 reveal a molecular machine for endocytosis. Cell 186, 821-836.e813. https://doi.org/10.1016/j.cell.2023.01.016 (2023).
Uchida, N. et al. Endocytosis-like vesicle fission mediated by a membrane-expanding molecular machine enables virus encapsulation for in vivo delivery. J. Am. Chem. Soc. 145, 6210–6220. https://doi.org/10.1021/jacs.2c12348 (2023).
Kerr, C. H. et al. Dynamic rewiring of the human interactome by interferon signaling. Genome Biol. 21, 140. https://doi.org/10.1186/s13059-020-02050-y (2020).
Denis, G. V. et al. Identification of transcription complexes that contain the double bromodomain protein Brd2 and chromatin remodeling machines. J. Proteome Res. 5, 502–511. https://doi.org/10.1021/pr050430u (2006).
Wheeler, R. J., Gull, K. & Sunter, J. D. Coordination of the cell cycle in Trypanosomes. Annu. Rev. Microbiol. 73, 133–154. https://doi.org/10.1146/annurev-micro-020518-115617 (2019).
Sullenberger, C., Pique, D., Ogata, Y. & Mensa-Wilmot, K. AEE788 inhibits basal body assembly and blocks DNA replication in the African Trypanosome. Mol. Pharmacol. 91, 482–498. https://doi.org/10.1124/mol.116.106906 (2017).
Callahan, J. F. et al. Identification of novel inhibitors of the transforming growth factor β1 (TGF-β1) type 1 receptor (ALK5). J. Med. Chem. 45, 999–1001. https://doi.org/10.1021/jm010493y (2002).
Vogt, J., Traynor, R. & Sapkota, G. P. The specificities of small molecule inhibitors of the TGFss and BMP pathways. Cell Signal 23, 1831–1842. https://doi.org/10.1016/j.cellsig.2011.06.019 (2011).
Walton, K. M. et al. Selective inhibition of casein kinase 1 epsilon minimally alters circadian clock period. J. Pharmacol. Exp. Ther. 330, 430–439. https://doi.org/10.1124/jpet.109.151415 (2009).
Adler, P. Therapeutic targeting of casein kinase 1delta/epsilon in an Alzheimer’s disease mouse Model. J. Proteome Res. 18, 3383–3393 (2019).
Matsuyama, S. SB-431542 and Gleevec inhibit transforming growth factor-beta-induced proliferation of human osteosarcoma cells. Cancer Res. 63, 7791–7798 (2003).
Hjelmeland, M. D. SB-431542, a small molecule transforming growth factor-beta-receptor antagonist, inhibits human glioma cell line proliferation and motility. Mol. Cancer Ther. 3, 737–745 (2004).
Kim, D. K., Kim, J. & Park, H. J. Synthesis and biological evaluation of novel 2-pyridinyl-[1,2,3]triazoles as inhibitors of transforming growth factor beta 1 type 1 receptor. Bioorg. Med. Chem. Lett. 14, 2401–2405. https://doi.org/10.1016/j.bmcl.2004.03.024 (2004).
Beauchamp, R. L. et al. A high-throughput kinome screen reveals serum/glucocorticoid-regulated kinase 1 as a therapeutic target for NF2-deficient meningiomas. Oncotarget 6, 16981–16997. https://doi.org/10.18632/oncotarget.4858 (2015).
Bain, J. et al. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 408, 297–315. https://doi.org/10.1042/BJ20070797[pii] (2007).
Qiao, Y. et al. Small molecule modulators targeting protein kinase CK1 and CK2. Eur. J. Med. Chem. 181, 111581. https://doi.org/10.1016/j.ejmech.2019.111581 (2019).
Bienen, E. J., Maturi, R. K., Pollakis, G. & Clarkson, A. B. Jr. Non-cytochrome mediated mitochondrial ATP production in bloodstream form Trypanosoma brucei brucei. Eur. J. Biochem. 216, 75–80 (1993).
McAllaster, M. R. et al. Proteomic identification of novel cytoskeletal proteins associated with TbPLK, an essential regulator of cell morphogenesis in Trypanosoma brucei. Mol. Biol. Cell 26, 3013–3029. https://doi.org/10.1091/mbc.E15-04-0219 (2015).
Affolter, M., Hemphill, A., Roditi, I., Muller, N. & Seebeck, T. The repetitive microtubule-associated proteins MARP-1 and MARP-2 of Trypanosoma brucei. J. Struct. Biol. 112, 241–251. https://doi.org/10.1006/jsbi.1994.1024 (1994).
Zhou, Q., Hu, H. & Li, Z. KLIF-associated cytoskeletal proteins in Trypanosoma brucei regulate cytokinesis by promoting cleavage furrow positioning and ingression. J. Biol. Chem. 298, 101943. https://doi.org/10.1016/j.jbc.2022.101943 (2022).
Wheeler, R. J., Scheumann, N., Wickstead, B., Gull, K. & Vaughan, S. Cytokinesis in Trypanosoma brucei differs between bloodstream and tsetse trypomastigote forms: Implications for microtubule-based morphogenesis and mutant analysis. Mol. Microbiol. 90, 1339–1355. https://doi.org/10.1111/mmi.12436 (2013).
Oberholzer, M., Morand, S., Kunz, S. & Seebeck, T. A vector series for rapid PCR-mediated C-terminal in situ tagging of Trypanosoma brucei genes. Mol. Biochem. Parasitol. 145, 117–120. https://doi.org/10.1016/j.molbiopara.2005.09.002 (2006).
Manders, E. M. M., Verbeek, F. J. & Aten, J. A. Measurement of co-localization of objects in dual-colour confocal images. J. Microsc. 169, 375–382. https://doi.org/10.1111/j.1365-2818.1993.tb03313.x (1993).
Kumar, G., Thomas, B. & Mensa-Wilmot, K. Pseudokinase NRP1 facilitates endocytosis of transferrin in the African trypanosome. Sci. Rep. 12, 18572. https://doi.org/10.1038/s41598-022-22054-x (2022).
Torres Robles, J., Lou, H. J., Shi, G., Pan, P. L. & Turk, B. E. Linear motif specificity in signaling through p38alpha and ERK2 mitogen-activated protein kinases. Proc. Natl. Acad. Sci. U S A 120, e2316599120. https://doi.org/10.1073/pnas.2316599120 (2023).
Gogl, G. et al. Rewiring of RSK-PDZ interactome by linear motif phosphorylation. J. Mol. Biol. 431, 1234–1249. https://doi.org/10.1016/j.jmb.2019.01.038 (2019).
Kumar, M. et al. ELM-the eukaryotic linear motif resource-2024 update. Nucleic Acids Res. 52, D442–D455. https://doi.org/10.1093/nar/gkad1058 (2024).
Yang, H. et al. pSite: Amino acid confidence evaluation for quality control of De Novo peptide sequencing and modification site localization. J. Proteome Res. 17, 119–128. https://doi.org/10.1021/acs.jproteome.7b00428 (2018).
Suni, V., Imanishi, S. Y., Maiolica, A., Aebersold, R. & Corthals, G. L. Confident site localization using a simulated phosphopeptide spectral library. J. Proteome Res. 14, 2348–2359. https://doi.org/10.1021/acs.jproteome.5b00050 (2015).
Taus, T. et al. Universal and confident phosphorylation site localization using phosphoRS. J. Proteome Res. 10, 5354–5362. https://doi.org/10.1021/pr200611n (2011).
Cayla, M., Nievas, Y. R., Matthews, K. R. & Mottram, J. C. Distinguishing functions of trypanosomatid protein kinases. Trends Parasitol. 38, 950–961. https://doi.org/10.1016/j.pt.2022.08.009 (2022).
Jonassen, I. Efficient discovery of conserved patterns using a pattern graph. Comput. Appl. Biosci. 13, 509–522 (1997).
Kumar, M. et al. The eukaryotic linear motif resource: 2022 release. Nucleic Acids Res. 50, D497–D508. https://doi.org/10.1093/nar/gkab975 (2022).
Messner, C. B. et al. The proteomic landscape of genome-wide genetic perturbations. Cell 186, 2018-2034.e2021. https://doi.org/10.1016/j.cell.2023.03.026 (2023).
Cao, B. et al. Mutant dominant-negative rhodopsin(∆I256) causes protein aggregates degraded via ERAD and prevents normal rhodopsin from proper membrane trafficking. Front. Mol. Biosci. 11, 1369000. https://doi.org/10.3389/fmolb.2024.1369000 (2024).
Kuo, M. E., Parish, M., Jonatzke, K. E. & Antonellis, A. Comprehensive assessment of recessive, pathogenic AARS1 alleles in a humanized yeast model reveals loss-of-function and dominant-negative effects. bioRxiv https://doi.org/10.1101/2024.06.20.599900 (2024).
Veitia, R. A. Exploring the molecular etiology of dominant-negative mutations. Plant Cell 19, 3843–3851. https://doi.org/10.1105/tpc.107.055053 (2007).
Hu, H. et al. The Aurora B kinase in Trypanosoma brucei undergoes post-translational modifications and is targeted to various subcellular locations through binding to TbCPC1. Mol. Microbiol. 91, 256–274. https://doi.org/10.1111/mmi.12458 (2014).
Kumar, P. & Wang, C. C. Dissociation of cytokinesis initiation from mitotic control in a eukaryote. Eukaryot. Cell 5, 92–102. https://doi.org/10.1128/EC.5.1.92-102.2006 (2006).
Pradel, L. C., Bonhivers, M., Landrein, N. & Robinson, D. R. NIMA-related kinase TbNRKC is involved in basal body separation in Trypanosoma brucei. J. Cell Sci. 119, 1852–1863. https://doi.org/10.1242/jcs.02900[pii] (2006).
Wiedeman, J. & Mensa-Wilmot, K. A fixable probe for visualizing flagella and plasma membranes of the African trypanosome. PLoS ONE 13, e0197541. https://doi.org/10.1371/journal.pone.0197541 (2018).
Wirtz, E., Leal, S., Ochatt, C. & Cross, G. A. M. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 99, 89 (1999).
Butter, F. et al. Comparative proteomics of two life cycle stages of stable isotope-labeled Trypanosoma brucei reveals novel components of the parasite’s host adaptation machinery. Mol. Cell Proteomics 12, 172–179. https://doi.org/10.1074/mcp.M112.019224 (2013).
Guyett, P. J., Xia, S., Swinney, D. C., Pollastri, M. P. & Mensa-Wilmot, K. Glycogen synthase kinase 3β promotes the endocytosis of transferrin in the African Trypanosome. ACS Infect. Dis. 2, 518–528. https://doi.org/10.1021/acsinfecdis.6b00077 (2016).
Kall, L., Storey, J. D. & Noble, W. S. Non-parametric estimation of posterior error probabilities associated with peptides identified by tandem mass spectrometry. Bioinformatics 24, i42-48. https://doi.org/10.1093/bioinformatics/btn294 (2008).
Zhou, Q., Hu, H. & Li, Z. New insights into the molecular mechanisms of mitosis and cytokinesis in trypanosomes. Int. Rev. Cell Mol. Biol. 308, 127–166. https://doi.org/10.1016/B978-0-12-800097-7.00004-X (2014).
Berriman, M. et al. The genome of the African Trypanosome Trypanosoma brucei. Science 309, 416–422 (2005).
Hammarton, T. C. Cell cycle regulation in Trypanosoma brucei. Mol. Biochem. Parasitol. 153, 1–8. https://doi.org/10.1016/j.molbiopara.2007.01.017[pii] (2007).
Mony, B. M. et al. Genome-wide dissection of the quorum sensing signalling pathway in Trypanosoma brucei. Nature 505, 681–685. https://doi.org/10.1038/nature12864 (2014).
Hirumi, H. & Hirumi, K. Axenic culture of African trypanosome bloodstream forms. Parasitol. Today 10, 80–84 (1994).
Rueden, C. T. et al. Image J2: ImageJ for the next generation of scientific image data. BMC Bioinform. 18, 529. https://doi.org/10.1186/s12859-017-1934-z (2017).
Schindelin, J. et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682. https://doi.org/10.1038/nmeth.2019 (2012).
Luo, G. et al. Discovery and optimization of biaryl alkyl ethers as a novel class of highly selective, CNS-penetrable, and orally active adaptor protein-2-associated kinase 1 (AAK1) inhibitors for the potential treatment of neuropathic pain. J. Med. Chem. 65, 4534–4564. https://doi.org/10.1021/acs.jmedchem.1c02132 (2022).
Bolte, S. & Cordelieres, F. P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232. https://doi.org/10.1111/j.1365-2818.2006.01706.x (2006).
Sanz-Rodriguez, C. E. et al. Physiologic targets and modes of action for CBL0137, a lead for human African Trypanosomiasis drug development. Mol. Pharmacol. https://doi.org/10.1124/molpharm.121.000430 (2022).
Sharma, A., Cipriano, M., Ferrins, L., Hajduk, S. L. & Mensa-Wilmot, K. Hypothesis-generating proteome perturbation to identify NEU-4438 and acoziborole modes of action in the African Trypanosome. iScience 25, 105302. https://doi.org/10.1016/j.isci.2022.105302 (2022).
Poon, S. K., Peacock, L., Gibson, W., Gull, K. & Kelly, S. A modular and optimized single marker system for generating Trypanosoma brucei cell lines expressing T7 RNA polymerase and the tetracycline repressor. Open Biol. 2, 110037. https://doi.org/10.1098/rsob.110037 (2012).
de Castro, E. et al. ScanProsite: Detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 34, W362-365. https://doi.org/10.1093/nar/gkl124[pii] (2006).
Blum, M. et al. InterPro: The protein sequence classification resource in 2025. Nucleic Acids Res. https://doi.org/10.1093/nar/gkae1082 (2024).
Acknowledgements
We thank Drs. David Swinney and Shuangluo Xia (Institute for Rare and Neglected Diseases Drug Discovery, Mountain View, California) who purified recombinant CK1.2 and identified small molecule inhibitors of the enzyme. We thank Dr. Phil Gafken (Fred Hutchinson Cancer Center) for mass spectrometry studies.
Funding
This study was supported by the National Institutes of Health R01AI126311 and National Institutes of Health R01AI124046.
Author information
Authors and Affiliations
Contributions
Conceptualization: KM-W, JW, SGL Methodology: KM-W, JW,SGL Investigation: KM-W, JW, GK, BH Visualization: JW, SGL, GK, BH Funding acquisition: KM-W Project administration: KM-W Supervision: KM-W Writing – original draft: JW, KM-W Writing – review & editing: KM-W, JW, SGL, BH, GK.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wiedeman, J., Kumar, G., Hoffman, B. et al. Discovery of effectors for casein kinase signaling in the African trypanosome. Sci Rep 15, 23284 (2025). https://doi.org/10.1038/s41598-025-05668-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-05668-9
