
Abstract
This study investigates the role of RNF4-mediated ubiquitination and degradation of PDHA1 in colorectal cancer (CRC) metabolism and metastasis. Integrating (The Cancer Genome Atlas) TCGA and Clinical Proteomic Tumor Analysis Consortium (CPTAC) databases, proteomic, clinical, and metabolomic analyses were performed, revealing PDHA1 as a prognostic marker in CRC. Immunohistochemical staining confirmed lower PDHA1 expression in metastatic CRC tissues. In vitro experiments demonstrated that PDHA1 overexpression inhibited CRC cell proliferation, migration, and invasion. RNF4 was identified as a key mediator in the ubiquitination degradation of PDHA1, influencing glycolytic pathways in CRC cells. Metabolomic analysis of serum samples from metastatic CRC patients further supported these findings. In vivo experiments, including xenograft and metastasis models, validated that RNF4 knockdown stabilized PDHA1, inhibiting tumor formation and metastasis. This study highlights the critical role of RNF4-mediated PDHA1 ubiquitination in promoting glycolytic metabolism, proliferation, and metastasis in CRC.
Similar content being viewed by others


Introduction
Colorectal cancer (CRC) is a prevalent malignancy globally, increasing incidence and mortality rates over time. Consequently, it has become a noteworthy public health concern1. Despite the substantial progress in early detection and treatment of colorectal cancer in recent years, the survival rate for patients with metastatic CRC remains low2,3,4. Research has indicated that the metabolic reprogramming of tumor cells plays a crucial role in their high invasiveness and metastatic potential. Thus, comprehending the metabolism of CRC cells is highly important for developing novel therapeutic approaches5.
Ubiquitination and SUMOylation are two prominent post-translational modification pathways in cells, which contribute to the regulation of protein stability, activity, and subcellular localization6,7. RNF4 is a specialized E3 ubiquitin ligase that targets SUMO-modified proteins for ubiquitination and degradation8. However, the mechanisms by which RNF4 regulates tumor cells’ metabolism and metastatic potential through the ubiquitination-dependent degradation of specific target proteins remain unclear9.
The PDHA1 protein is pivotal in the pyruvate dehydrogenase complex and is intricately linked to cellular metabolism, growth, and survival10. Recent studies have shown that the PDHA1 protein is dysregulated in various types of cancer and is closely linked to tumor cell proliferation, migration, and invasion10,11. However, the precise mechanism by which the PDHA1 protein is regulated by the SUMO-targeted ubiquitin ligase RNF4 and its impact on the glycolytic metabolism and metastatic capacity of CRC cells has yet to be fully elucidated10.
Metabolomics and proteomics are two branches of modern biology that offer powerful tools for studying cellular metabolic mechanisms12. Furthermore, metabolic dysregulation frequently occurs in various cancer cells and has been recognized as a significant hallmark of cancer13. PDHA1, a crucial component of the pyruvate dehydrogenase complex, plays a vital role in linking glycolysis and the mitochondrial tricarboxylic acid cycle. Previous studies have revealed a close association between the dysfunction of PDHA1 and metabolic reprogramming in cancer14. However, the exact role of PDHA1 in metabolism in colorectal cancer (CRC) remains incompletely understood. Therefore, by integrating metabolomics and proteomics techniques, a more systematic elucidation of the cellular metabolic network and its connection with protein regulation can be achieved15. Furthermore, in-depth analysis of these data can provide crucial clues for discovering novel therapeutic targets and strategies for CRC treatment.
This study aims to investigate how the SUMO-targeted ubiquitin ligase RNF4 mediates the ubiquitination degradation of PDHA1 protein and how this mechanism affects glycolysis metabolism and the metastatic capacity of CRC cells, considering the background provided.
Results
Development and validation of an 11-protein prognostic risk model for predicting survival in colorectal cancer patients
Colorectal cancer (CRC) is a global health concern, ranking second leading cause of cancer-related death in developed nations. By employing proteomics, valuable biomarkers could be identified for the diagnosis, prognosis, and precise management of CRC16. To identify prognostic-related proteins for colorectal cancer (CRC), proteomic data and clinical data for CRC patients were obtained from the TCGA database. The patients were randomly divided into train and test sets with a 1:1 ratio allocation. Subsequently, univariate Cox analysis was performed on the training dataset, revealing 29 prognostic-related proteins. These findings are visualized in the prognostic volcano plot (Fig. 1A). Moreover, implementing LASSO regression yielded a panel of 19 prognostic proteins along with their respective LASSO coefficients, as shown in Fig. 1B, C. Consequently, a prognostic risk model employing the expression levels of 11 proteins (Sox2, PDHA1, HSP27, PAI1, GATA6, TFRC, VHL, CYCLINE2, FOXM1, MYOSINIIA_pS1943, and IGFBP2) was developed. The Kaplan-Meier curve analysis demonstrated that patients with high-risk CRC exhibited overall survival (OS) rates that were lower in all the datasets, including the train and test datasets. Moreover, the high-risk group of CRC patients in all datasets also displayed lower progression-free survival (PFS) rates (Fig. 1D–F; Supplementary Fig. 1A).

A Volcano plot of prognostic-related proteins obtained from univariate COX analysis. Red dots represent high-risk proteins; blue dots represent low-risk proteins; B LASSO regression analysis curve, with different colored curves representing different proteins; C Cross-validation error curve of LASSO regression. The x-axis represents the log(λ) value and the y-axis represents the Partial Likelihood Deviance. The number of proteins retained for each log(λ) value used for calculation is shown above the graph with a dashed line indicating the log(λ) value and the number of genes retained when the Partial Likelihood Deviance is optimal; Kaplan–Meier curves analyzing the relationship between risk scores of the prognostic risk model and OS in the all (D), train (E), and test (F) datasets of CRC patients. n = 336 (excluding samples with unknown survival status and survival time of 0).
Further analysis of the ROC curve revealed that our prognostic risk model accurately predicts the survival of patients with colorectal cancer (CRC). The model accurately predicts the 1-year, 3-year, and 5-year survival rates of CRC patients, outperforming age, gender, and TMN stage (Supplementary Fig. 1B, C). A prognostic composite column plot was constructed to predict the overall survival probabilities of colorectal cancer (CRC) patients at 1, 3, and 5 years. The calibration curve results demonstrated the composite column plot’s high accuracy in predicting CRC patients’ survival. The column plot revealed survival rates of CRC patients to be 0.822 at 1 year, 0.616 at 3 years, and 0.429 at 5 years (Supplementary Fig. 1D, E).
Finally, we assessed the applicability of the developed prognostic risk model to colorectal cancer patients across various clinical groups. Kaplan-Meier curve analysis demonstrated the applicability of the prognostic risk model we developed in colorectal cancer (CRC) patients across various characteristics, including gender, age, M, N, and stage. However, the model only applied to CRC patients with T3-4 stage in T staging (Supplementary Fig. 2).
The results suggest that a prognostic risk model founded on 11 key proteins could effectively forecast the prognosis of patients with CRC.
Differential expression of PDHA1 in CRC and its prognostic significance: insights from proteomic analysis and immunohistochemistry
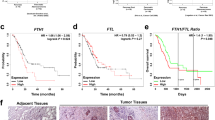
To further identify prognostically relevant differentially expressed proteins in CRC patients, we retrieved protein expression data from the CPTAC database and analyzed protein differential expression. Five thousand seven hundred thirty-six differentially expressed proteins were identified, consisting of 3059 upregulated proteins and 2677 downregulated proteins (Fig. 2A). The identified differentially expressed proteins were compared to the proteins in the prognostic risk model, identifying five prognostic-related differentially expressed proteins. These proteins include HSPB1/HSP27, IGFBP2, PDHA1, TFRC, and MYH9/MYOSINIIA_pS1943 (Fig. 2B). Among them, HSPB1, IGFBP2, and PDHA1 were found to be downregulated in tumor tissues of colorectal cancer (CRC) patients, while TFRC and MYH9 were upregulated in tumor tissues of CRC patients (Fig. 2C). According to the Kaplan-Meier curve analysis, patients with lower expression of PDHA1, TFRC, and MYOSINIIA_pS1943 proteins had a worse prognosis, while patients with higher expression of HSP27 and IGFBP2 proteins also had a worse prognosis. (Fig. 2D; Supplementary Fig. 3). Based on integrating proteomic differential expression and prognosis analysis, we discovered PDHA1 to be downregulated in CRC, and its downregulation is linked to a negative prognosis in patients.

A Volcano plot of differentially expressed proteins in CRC patients’ tumor tissue and normal tissue. Normal: n = 100, Tumor: n = 95; B Venn diagram of the intersection between differentially expressed proteins and prognosis-related proteins in the prognosis risk model; C Violin plots of the differential expression of the five prognosis-related proteins. Normal: n = 100, Tumor: n = 95; D Kaplan–Meier curves analyzing the relationship between PDHA1 protein expression and prognosis in CRC patients; E Immunohistochemical staining of PDHA1 protein expression in adjacent normal tissue, primary CRC tissue, and metastatic CRC tissue (scale bar=200 μm or 50 μm). Control: n = 26, p-CRC: n = 26, m-CRC: n = 28; * indicates P < 0.05; F Western blot to detect the protein expression levels of PDHA1 in adjacent normal tissues, primary colorectal cancer tissues, and metastatic colorectal cancer tissues. * indicates a statistical significance compared to the FHC group with a p < 0.05.
Previous studies have indicated a downregulation of PDHA1 in dextran sulfate sodium (DSS)-induced ulcerative colitis17; however, there is a lack of research on the expression and function of PDHA1 in colorectal cancer (CRC). Therefore, we further investigated the expression of PDHA1 in both metastatic and primary CRC tissues. Immunohistochemical and immunoblotting analyses revealed that compared to normal tissues, primary CRC tissues exhibited a significant decrease in PDHA1 protein expression. Moreover, the reduction in PDHA1 protein expression was even more pronounced in metastatic CRC tissues compared to primary CRC tissues. (Fig. 2E, F).
The results above indicate the downregulation of the PDHA1 protein in CRC tissue, suggesting that it may be a crucial protein differentially associated with the prognosis of patients with CRC.
Role of PDHA1 in modulating proliferation, migration, and invasion in CRC cell lines: insights from overexpression and knockdown studies
To investigate the impact of PDHA1 on the biological functions of CRC cells, we initially evaluated the expression of PDHA1 in several human CRC cell lines, namely DLD-1, SW480, SW620, HCT-116, and normal human colorectal mucosal cells (FHC). The Western blot analysis revealed a reduction in PDHA1 protein expression in all CRC cell lines compared to FHC cells, with SW620 cells demonstrating the lowest expression levels (Fig. 3A). Thus, we chose SW620 and DLD-1 cells, which exhibited relatively high protein expression of PDHA1, for further experimental validation. PDHA1 was overexpressed in SW620 cells, while the knockdown of PDHA1 was observed in DLD-1 cells. Western blot analysis demonstrated an increase in the protein expression of PDHA1 in the PDHA1 group compared to the pcDNA group. Moreover, the protein expression of PDHA1 was reduced in both the sh-PDHA1#1 and sh-PDHA1#2 groups compared to the sh-NC group, with a more pronounced knockdown effect in the sh-PDHA1#1 group (Fig. 3B).

A Western blot analysis of PDHA1 protein expression in various CRC cell lines and human normal colon mucosal cells (FHC). * indicates difference compared to the FHC group, P < 0.05; B Western blot analysis of PDHA1 protein expression in cells after overexpression or knockdown of PDHA1; C CCK-8 assay measuring changes in cell viability after overexpression or knockdown of PDHA1 at 24, 48, and 72 h; D Scratch assays measuring cell migration after overexpression or knockdown of PDHA1 (scale bar = 100 μm); E Transwell assay measuring cell migration and invasion after overexpression or knockdown of PDHA1 (scale bar = 50 μm); F RT-qPCR analysis of changes in mRNA levels of Snail, Slug, Twist, Vimentin, and E-cadherin after overexpression or knockdown of PDHA1. * indicates difference compared to the pcDNA group, P < 0.05; # indicates difference compared to the sh-NC group, P < 0.05. Cell experiments were repeated three times.
Moreover, we investigated the impact of modified PDHA1 expression or knockdown on CRC cells’ proliferation, migration, and invasion. CCK-8 assays demonstrated that SW620 cells in the PDHA1 group displayed reduced viability compared to the pcDNA group. In contrast, DLD-1 cells in the sh-PDHA1 group exhibited enhanced viability compared to the sh-NC group (Fig. 3C). The results of the Scratch and Transwell assays demonstrated that the PDHA1 group had reduced cell migration and invasion abilities compared to the pcDNA group. Conversely, the sh-PDHA1 group exhibited enhanced cell migration and invasion abilities compared to the sh-NC group (Fig. 3D, E). Lastly, the epithelial-mesenchymal transition (EMT)-related factors were examined. The findings indicated that compared to the pcDNA group, the mRNA levels of Snail, Slug, Twist, and Vimentin were considerably reduced in the PDHA1 group, while the mRNA level of E-cadherin was increased. Likewise, the sh-PDHA1 group demonstrated an increase in the mRNA levels of Snail, Slug, Twist, and Vimentin, accompanied by a decrease in the mRNA level of E-cadherin, in comparison to the sh-NC group (Fig. 3F).
The results above indicate that the overexpression of PDHA1 inhibits the proliferation, migration, and invasion of CRC cells, whereas the knockdown of PDHA1 promotes the proliferation, migration, and invasion of CRC cells.
RNF4 mediates the ubiquitination and degradation of SUMOylated PDHA1 in colorectal cancer cells: insights from SUMOylation and protein-protein interaction studies
Research reports suggest that the E3 ubiquitin ligase UBE3A/E6AP ubiquitinates and degrades PDHA1, thereby influencing the progression of non-alcoholic fatty liver disease18. However, it remains unclear whether the decreased expression of PDHA1 in CRC is also associated with the ubiquitin-mediated degradation of the PDHA1 protein. Protein ubiquitination and small ubiquitin-related modifiers (SUMOylation) are currently areas of intense research in the field of protein post-translational modification (PTM)19. Research has revealed a crosstalk between protein ubiquitination and SUMOylation mediated by SUMO-targeted ubiquitin ligases (STUbLs). Among these ligases, RNF4, a human SUMO-targeted ubiquitin E3 ligase, has been extensively investigated and demonstrated to operate in a SUMO2/3-dependent manner20. Our research has also demonstrated that the protein level of RNF4 exhibits high expression in both metastatic and primary colorectal cancer (CRC) tissues and in various CRC cell lines (Supplementary Fig. 4).
To investigate the potential regulatory role of RNF4 in the progression of CRC through the proteasomal pathway, we initially referenced established methods from previously published literature21,22. Additionally, we utilized the JASSA and SUMOPLOT websites for the prediction of SUMOylation sites on PDHA1 protein (Supplementary Fig. 5A). We also determined the SIM of the RNF4 protein (Supplementary Fig. 5B) and identified the specific ubiquitination site where the RNF4 protein interacts with PDHA1 using the biocuckoo website (Supplementary Table 1).
We then examined the impact of SUMOylation on the degradation of PDHA1 through ubiquitination. Previous research has indicated that MG132, as an effective inhibitor of proteasomal degradation23,24,. Thus, in SW620 cells co-expressing FLAG-PDHA1 and EGFP-tagged SUMO homolog, we examined the expression levels of PDHA1 and its SUMOylation status with or without the proteasome inhibitor MG132 treatment. Analysis from Ni-NTA revealed a significant increase in SUMOylated (SUMO2/3) PDHA1 abundance post MG132 treatment, while the levels of unmodified and SUMO1-conjugated PDHA1 remained largely unaffected (Fig. 4A). Subsequently, we examined the SUMOylation and ubiquitination of PDHA1 in SW620 cells. The results revealed that in untreated control cells, SUMOylation bands of PDHA1 were observed, with low levels of ubiquitination. Following MG132 treatment, the expression of PDHA1, SUMOylated bands, and ubiquitinated Ub were significantly upregulated, indicating a noticeable decrease in SUMOylation and ubiquitination levels of PDHA1 post-MG132 treatment.

A Co-transfection of FLAG-PDHA1 and EGFP-SUMO (SUMO1, SUMO2, or SUMO3) in SW620 cells, with or without treatment of 20 μM MG132 for 6 h. Ni-NTA analyzed SUMOylation of PDHA1; B Co-transfection of FLAG-PDHA1-His, HA-Ub, and T7-SUMO3 in SW620 cells, followed by treatment with CHX (20 μg/ml), MG132 (20 μM), or CHX + MG132 for 6 h. SUMOylation and ubiquitination of PDHA1 were examined by Ni2+ pull-down assay; C Co-transfection of SW620 cells with FLAG-PDHA1-His (WT, 2 KR (UMO-deleted double mutation), K77 (SUMO-deleted single mutation), K186 (another SUMO-deleted single mutation)), HA-Ub, and T7-SUMO3, followed by treatment with 20 μM MG132 for 6 h and Ni2+ pull-down experiments to detect the SUMOylation and ubiquitination of PDHA1; D Performance of Western blot analysis to evaluate the protein expression of PDHA1, SAE1, SAE2, Ubc9, SUMO-1, and SUMO-2 in different SW620 cell groups; E Transfection of SW620 cells with Flag-PDHA1 WT, Flag-PDHA1 2 KR, Flag-PDHA1 K77, and Flag-PDHA1 K186 for 48 h. The protein expression of PDHA1 was measured by Western blot after different incubation durations with 20 μg/ml CHX. * indicates difference compared to the Flag-PDHA1 WT group, P < 0.05. Cell experiments were repeated three times.
Conversely, when cells were treated with CHX alone or in combination with MG132, the levels of SUMOylation and ubiquitination bands of PDHA1 markedly decreased, suggesting that these treatments significantly increased the SUMOylation and ubiquitination levels of PDHA1 (Fig. 4B). Therefore, the above results preliminarily suggest that the downregulation of PDHA1 expression may be associated with ubiquitination degradation following SUMOylation of PDHA1. Furthermore, co-transfection of FLAG-PDHA1-His (WT, 2 KR: SUMOylation double-site mutants, K77: SUMOylation single-site mutant, K186: another SUMOylation single-site mutant), HA-Ub, and T7-SUMO3 in cells showed a significant decrease in SUMOylation and ubiquitination bands in the 2 KR, K77, and K186 groups compared to the control group (Fig. 4C). Moreover, we transfected wild-type PDHA1, as well as PDHA1 plasmids with mutations at the K77 and K186 sites, and measured the expression levels of SUMO E1 activating enzyme (SAE1/SAE2), SUMO-conjugating enzyme (Ubc9), and SUMO-1 or SUMO-2. Immunoblot results indicated that the aforementioned proteins showed no significant changes (Fig. 4D). Subsequently, analysis of the PDHA1 protein half-life revealed a significant increase in the half-life of Flag-PDHA1 2 KR, Flag-PDHA1 K77, and Flag-PDHA1 K186 compared to Flag-PDHA1 WT (Fig. 4E). These findings suggest that SUMOylation of PDHA1 promotes its ubiquitination degradation.
To further explore the protein interaction between RNF4 and PDHA1, the co-immunoprecipitation (co-IP) experiment results showed that both exogenous and endogenous RNF4 could co-immunoprecipitate with PDHA1 (Fig. 5A), indicating the presence of a protein-protein interaction between RNF4 and PDHA1. The PLA experiments also detected a PLA signal in the cytoplasm between endogenous RNF4 and PDHA1. To enhance protein quality and signal intensity, we treated the cells with MG-132 and transfected them with RNF4 overexpression plasmids, resulting in an increased PLA signal (Fig. 5B). After using shRNA to knock down RNF4, the expression of RNF4 was detected, and compared to the control group, RNF4 protein expression in the shRNA group was significantly reduced, suggesting that the decrease in RNF4 levels could reduce the interaction between RNF4 and PDHA1 (Fig. 5C). Moreover, knocking down endogenous RNF4 protein levels with specific shRNAs strongly reduced the PLA signal (Fig. 5D). The co-IP experiment results also revealed that the SUMOylation-defective mutations of PDHA1 (2KR, K77, K186) and the SIMs mutation of RNF4 (mSIM) significantly inhibited the protein-protein interaction between RNF4 and PDHA1 (Fig. 5E, F).

A Co-immunoprecipitation (co-IP) assay measuring the protein interaction between RNF4 and PDHA1 in SW620 cells after 24 h of transfection or co-transfection with Myc-RNF4 and Flag-PDHA1; B PLA assay measuring the proximity of RNF4 and PDHA1 in SW620 cells treated with 20 μM MG132 for 4 h (scale bar = 25 μm). Red fluorescence indicates the proximity of the two proteins. * indicates P < 0.05; C Western blot analysis of RNF4 protein expression in SW620 cells after knockdown of RNF4. * indicates difference compared to the sh-NC group, P < 0.05; D PLA assay measuring the proximity of RNF4 and PDHA1 in SW620 cells after knockdown of RNF4 (scale bar = 25 μm). Red fluorescence indicates the proximity of the two proteins. * indicates P < 0.05; E, F Co-IP assay measuring the protein interaction between RNF4 and FLAG-PDHA1 (WT, 2 KR, K77, K186) or Myc-RNF4 (WT or mSIM) in SW620 cells after co-transfection; G Ni2+ pull-down assay measuring the SUMOylation and ubiquitination of PDHA1 in SW620 cells co-transfected with FLAG-PDHA1-His and HA-Ub or T7-SUMO3 and Myc-RNF4 (WT or mSIM). Cell experiments were repeated three times.
In conclusion, our study revealed that RNF4 overexpression facilitates the ubiquitination of SUMOylated PDHA1, leading to a notable increase in the levels of SUMOylated PDHA1 bands. Conversely, the transfection of RNF4 SIMs mutant mSIM markedly suppresses the ubiquitination of PDHA1 (Fig. 5G).
The results above indicate that the RNF4 ubiquitin ligase, specifically targeting SUMO, can facilitate the ubiquitination and subsequent degradation of the PDHA1 protein.
Inhibition of RNF4 enhances PDHA1 stability and suppresses proliferation, migration, and invasion in CRC cells: insights from combined silencing experiments
Next, we further investigated the impact of RNF4 regulation on PDHA1 protein in CRC cell proliferation and migration. In SW620 cells, simultaneous knockdown of RNF4 and PDHA1 was performed. RT-qPCR analysis showed that compared to the sh-NC group, the sh-RNF4 group exhibited a significant decrease in RNF4 mRNA levels, with no significant change in PDHA1 mRNA levels. In contrast, compared to the sh-RNF4 group, the sh-RNF4+sh-PDHA1 group showed no significant change in RNF4 mRNA levels, while PDHA1 mRNA levels decreased significantly (Fig. 6A). Furthermore, Western blot analysis confirmed alterations in protein expression when RNF4 and PDHA1 were individually knocked down and when both were knocked down simultaneously. Compared to the sh-NC group, the sh-RNF4 group displayed a marked decrease in RNF4 protein levels and a significant increase in PDHA1 expression. In comparison, the sh-PDHA1 group showed no significant change in RNF4 protein levels but a notable decrease in PDHA1 expression compared to the sh-NC group. Additionally, compared to the sh-PDHA1 group, the sh-RNF4+sh-PDHA1 group exhibited a considerable decrease in RNF4 protein levels and a significant increase in PDHA1 expression, while there was no significant difference in RNF4 protein levels between the sh-RNF4 group and the sh-RNF4+sh-PDHA1 group, indicating a significant decrease in PDHA1 expression in the latter (Fig. 6B).

A RT-qPCR detection of RNF4 and PDHA1 mRNA expression in SW620 cells from each group; B Western blot detection of RNF4 and PDHA1 protein expression in SW620 cells from each group; C CCK-8 assay to measure the viability changes of SW620 cells from each group at 24, 48, and 72 h; D Transwell assay to measure the migration and invasion ability of SW620 cells from each group (scale bar = 50 μm); E Scratch assay to measure the migration of SW620 cells from each group (scale bar = 100 μm); F RT-qPCR detection of Snail, Slug, Twist, Vimentin, and E-cad mRNA levels in SW620 cells from each group. * indicates P < 0.05 compared to sh-NC group, # indicates P < 0.05 compared to sh-RNF4 group, & indicates P < 0.05 compared to the sh- PDHA1 group. The experiments were repeated three times.
CCK-8 detection revealed changes in cell viability. The sh-RNF4 group showed a decrease in cell viability compared to the sh-NC group. Conversely, the sh-RNF4+sh-PDHA1 group exhibited increased cell viability compared to the sh-RNF4 group (Fig. 6C). Scratch and Transwell experiments demonstrated that the cell migration and invasion abilities of the sh-RNF4 group decreased compared to the sh-NC group. In contrast, the cell migration and invasion abilities increased in the sh-RNF4+sh-PDHA1 group compared to the sh-RNF4 group (Fig. 6D, E). Analysis of EMT-related factors revealed that in the sh-RNF4 group, there was a significant decrease in the mRNA levels of Snail, Slug, Twist, and Vimentin, while the mRNA level of E-cadherin was notably increased compared to the sh-NC group. Compared to the sh-RNF4 group, the sh-RNF4+sh-PDHA1 group showed a significant increase in mRNA levels of Snail, Slug, Twist, and Vimentin, and a marked decrease in mRNA levels of E-cad (Fig. 6F).
The above findings suggest that suppressing RNF4 could enhance the stability of PDHA1 protein, leading to the inhibition of proliferation, migration, and invasion of CRC cells.
RNF4/PDHA1 axis modulates glycolytic metabolism in CRC: insights from non-targeted metabolomics and cellular glycolytic activity assessments
To further investigate the pathways by which RNF4/PDHA1 regulates CRC proliferation and metastasis, we collected serum samples from patients categorized into the m-CRC and p-CRC groups for non-targeted metabolomics analysis. The differential analysis revealed 30 metabolites that differed between the m-CRC and p-CRC groups (Fig. 7A). The differential metabolites were imported into the MetaboAnalyst website for pathway enrichment analysis. The results of the analysis revealed that these metabolites were predominantly enriched in the pathways associated with Glyoxylate and dicarboxylate metabolism, Alanine, aspartate, and glutamate metabolism, Purine metabolism, Glycolysis/Gluconeogenesis, as well as D-Glutamine and D-glutamate metabolism (Fig. 7B, C). According to research, aerobic glycolysis, commonly known as the Warburg effect, is a characteristic feature of cancer and has a vital role in the initiation and spread of tumors. PDHA1 is the E1 alpha subunit of the pyruvate dehydrogenase complex and is considered one of the crucial enzymes that aid in the transition from oxidative phosphorylation to aerobic glycolysis. A deficiency in PDHA1 could result in mitochondrial dysfunction and promote glycolysis25,26,27. Hence, we hypothesize that glycolysis is a crucial pathway controlled by PDHA1 in the growth and spread of CRC.

A Volcano plots showing the differential metabolites between the m-CRC and p-CRC groups, p-CRC: n = 26, m-CRC: n = 28; B Pathway enrichment analysis of differential metabolites using the MetaboAnalyst database; C Function enrichment analysis of differential metabolites using the MetaboAnalyst database; D, E Extracellular acid ratio (ECAR) and oxygen consumption rate (OCR) of cells overexpressing or knockdown of PDHA1 measured using Seahorse XF; 2-DG: 2-deoxyglucose, FCCP: Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone; F Lactate production in cells overexpressing or knockdown of PDHA1; G, H Extracellular acid ratio (ECAR) and oxygen consumption rate (OCR) of SW620 cells from each group measured using Seahorse XF; I Lactate production in SW620 cells from each group. In D–F, * indicates P < 0.05 compared to the pcDNA group, # indicates P < 0.05 compared to sh-NC group; in G–I, * indicates P < 0.05 compared to sh-NC group, # indicates P < 0.05 compared to sh-RNF4 group. The experiments were repeated three times.
In addition, we investigated the alterations in glycolytic metabolism levels in colorectal cancer (CRC) cells that either overexpressed or knocked down PDHA1. The findings demonstrated that PDHA1-overexpressing SW620 cells exhibited a noteworthy reduction in ECAR and lactate production levels, along with a substantial escalation in OCR levels, compared to the pcDNA group. Similarly, the sh-PDHA1 group of DLD-1 cells showcased an elevation in ECAR levels and lactate production and a considerable decrease in OCR levels relative to the sh-NC group (Fig. 7D–F).
Additionally, we assessed the glycolytic activity in cells where RNF4 and PDHA1 were simultaneously knocked down. The findings revealed a reduction in both the ECAR level and lactate production in SW620 cells of the sh-RNF4 group compared to the sh-NC group. Conversely, the OCR level was elevated. Moreover, in the sh-RNF4+sh-PDHA1 group of DLD-1 cells, there was an increase in both the ECAR level and lactate production compared to the sh-RNF4 group, while the OCR level showed a decrease (Fig. 7G–I).
The results above suggest that the RNF4/PDHA1 axis can enhance glycolytic metabolism, consequently facilitating the proliferation and metastasis of CRC cells.
RNF4/PDHA1 axis influences tumorigenesis and metastasis of CRC in vivo: insights from orthotopic tumor and metastasis models in nude mice
Finally, we developed orthotopic tumor models and metastasis models in nude mice to further confirm the influence of the RNF4/PDHA1 axis on the growth and metastasis of CRC cells in vivo. The tumor volume and weight measurement in each group showed that both tumor volume and weight were lower in the sh-RNF4 group than in the sh-NC group. Furthermore, the sh-RNF4+sh-PDHA1 group increased tumor volume and weight compared to the sh-RNF4 group (Fig. 8A–C). Immunohistochemical staining results demonstrated a decrease in the expression of ki67 and PCNA proteins in the sh-RNF4 group compared to the sh-NC group. Conversely, the sh-RNF4+sh-PDHA1 group exhibited increased expression of ki67 and PCNA proteins compared to the sh-RNF4 group (Fig. 8D).

A Subcutaneous tumor dissection images of mice from each group on day 35; B Line graph showing the tumor volume growth from day 5 to day 35 in each group of mice; C Tumor weight statistics on day 35 in each group of mice; D Immunohistochemical staining detecting the positive expression of proliferation markers Ki67 and PCNA in subcutaneous tumor tissues of mice from each group (scale bar = 50 μm or 25 μm); E IVIS Lumina II imaging system measuring lung metastatic nodules in mice from each group; F Representative images of lung metastatic nodules in mice from each group, indicated by black arrows; G Representative images of liver metastatic nodules in mice from each group. n = 8, * indicates P < 0.05 compared to sh-NC group, # indicates P < 0.05 compared to sh-RNF4 group.
Additionally, lung metastasis and liver metastasis models were established by injecting CRC cells into the tail vein or spleen. The metastasis status of each group was compared by monitoring lesions with a volume exceeding 2 mm3 using the IVIS Lumina II imaging system. The results indicated a reduction in lung metastasis in the sh-RNF4 group compared to the sh-NC group. Moreover, lung metastasis was increased in the sh-RNF4+sh-PDHA1 group compared to the sh-RNF4 group (Fig. 8E, F). In the liver metastasis model, we observed a reduction in liver metastasis in the sh-RNF4 group compared to the sh-NC group. Moreover, there was a notable increase in liver metastasis in the sh-RNF4+sh-PDHA1 group compared to the sh-RNF4 group, as shown in Fig. 8G.
The results above indicate that the destabilization of RNF4 leads to stabilizing the PDHA1 protein, inhibiting tumorigenesis and metastasis in CRC cells.
Discussion
This study identified PDHA1 as a crucial protein marker for CRC patient prognosis, aligning with the observed downregulation of PDHA1 in metastatic CRC tissues. This finding aligns with prior research, which suggests that PDHA1 is downregulated in specific cancer types28. Abnormal expression of PDHA1, being one of the key enzymes in the glycolytic pathway, could potentially result in metabolic disorders, consequently impacting tumor development and metastasis29. It gives us additional clues to investigate its role in CRC research.
This study demonstrates that the RNF4, a SUMO-targeted ubiquitin ligase, mediates the ubiquitination and degradation of PDHA1. Furthermore, downregulation of RNF4 expression leads to PDHA1 protein stabilization, thereby inhibiting the proliferation, migration, and invasion of CRC cells. This finding emphasizes the crucial role of RNF4 in governing the stability of PDHA1 and its involvement in CRC metastasis. Previous studies did not make clear the direct association between RNF4 and PDHA1. Consequently, this newly discovered mechanism offers a more precise therapeutic target30.
The metabolomic analysis indicated that the serum of metastatic CRC patients contained differentially expressed metabolites mainly enriched in glycolysis and related pathways31. Glycolysis plays a pivotal role in energizing cancer cells and serves as a vital fuel for the rapid proliferation of tumor cells32. Previous studies have reported a significant downregulation of PDHA1 expression in gastric cancer. The main mechanism involves miR-21-5p directly targeting PDHA1 to inhibit its expression. Subsequent downregulation of PDHA1 accelerates glycolytic function in gastric cancer cells and promotes cancer progression. Moreover, colorectal cancer (CRC), another crucial malignancy within the digestive system, was found to exhibit inhibited glycolytic metabolism upon knocking down RNF4 and overexpressing PDHA1. This inhibition subsequently impacts proliferation and metastasis, providing further evidence for the critical role of glycolysis in CRC development.
We tested our hypothesis by transplanting a tumor model subcutaneously in nude mice. This experiment confirmed that the suppression of RNF4 leads to the stabilization of PDHA1 protein. Consequently, tumor formation in vivo and liver and lung metastasis in CRC cells was inhibited. This study provides robust empirical evidence and demonstrates the role of this mechanism in actual organisms.
This study presents the first comprehensive analysis of the molecular mechanism through which the SUMO-targeted ubiquitin ligase RNF4 mediates the ubiquitination and degradation of PDHA1. Additionally, it explores the influence of this process on CRC metastasis, with particular emphasis on the relationship between RNF4 and PDHA1. Although previous studies have investigated the correlation between PDHA1 or RNF4 and CRC, the exact association and underlying mechanisms have not been completely elucidated10,33. Understanding the regulation of PDHA1 by RNF4 and its role in colorectal cancer (CRC) provides valuable insights for developing novel therapeutic strategies. In the future, drug or gene therapy targeting RNF4 or PDHA1 may be effective strategies for inhibiting colorectal cancer metastasis.
Based on the above findings, several preliminary conclusions could be drawn. Firstly, SUMO targets the ubiquitin ligase RNF4 to facilitate the ubiquitination and degradation of the PDHA1 protein. This process promotes the glycolytic metabolism of CRC cells, consequently leading to the proliferation and metastasis of CRC cells both intra- and extracellularly (Fig. 9). This study revealed that the SUMO-targeted ubiquitin ligase RNF4 is responsible for mediating the ubiquitin-dependent degradation of the PDHA1 protein, playing a crucial role in colorectal cancer (CRC). This discovery offers a fresh perspective and expands the knowledge base for molecular biology research on CRC.

Created with BioRender.com.
This study uncovers the pivotal roles of RNF4 and PDHA1 in the proliferation and metastasis of CRC cells. This finding suggests that targeting these two proteins or their interaction may serve as a promising therapeutic strategy, offering new treatment possibilities for patients with CRC. This study employed proteomic and integrated metabolomic technology to conduct a comprehensive analysis, offering a novel approach for future cancer research combining multiple techniques.
While multiple human colorectal cancer cell lines were utilized in this study, it is important to note that these cell lines may not completely replicate the complex physiological context of the human body. Hence, the research findings require additional Validation using animal models and clinical samples. This study identified metabolites in the serum of patients with metastatic CRC through metabolomics analysis. However, further investigation is required to understand these metabolites’ network relationships and detailed mechanisms. While the interaction between RNF4 and PDHA1 has been confirmed in CRC, whether other unidentified proteins are involved and how these interact remains unknown.
Building upon the findings of this study, future research should explore the potential of drugs or small molecule inhibitors that target RNF4 and PDHA1 as novel therapeutic options for patients with colorectal cancer (CRC). This study presents a model investigating the connection between SUMOylation, ubiquitination, and metabolic alterations in various cancer types. This strategy can potentially be applied in future research on different types of cancer. Metabolomics analysis has revealed that the identified metabolites are predominantly enriched in pathways, including glycolysis. It suggests that future research should delve into the specific roles of these metabolic pathways in CRC and potential alterations in other important metabolites or enzymes.
Methods
Proteomics data acquisition
The protein expression and clinical data of 358 colorectal cancer patients (TCGA-COAD) were acquired from The Cancer Genome Atlas (TCGA) database at https://portal.gdc.cancer.gov/. Retrieve the protein expression dataset of colon cancer patients from the Clinical Proteomic Tumor Analysis Consortium (CPTAC) database, available at https://pdc.cancer.gov/pdc/. The dataset comprises 95 colon cancer tumor tissue samples and 100 normal tissue samples34,35.
Prognostic risk model construction and prognosis-related analysis
This study randomly allocated 336 colorectal cancer (CRC) patient samples from the TCGA database. Samples with unknown patient survival status and zero survival time were excluded. The samples were allocated in a 1:1 ratio to the training and test sets, each containing 168 patient samples. We used the “survival” package in the R language to identify proteins associated with overall survival using hazard ratio (HR) and univariate Cox regression analysis. Proteins with a p-value less than 0.05 were selected as candidate biomarkers.
Based on the univariate Cox analysis results, we generated a prognostic volcano plot using the “ggplot2” package in the R language. Candidate proteins were included for further multivariable COX analysis. The LASSO Cox regression model is employed to ascertain the optimal coefficients and estimate the biased likelihood. A set of prognostic proteins and their corresponding LASSO coefficients were obtained based on the highest lambda value (lambda.min) selected through 1000-fold cross-validation in the LASSO method. These proteins and coefficients were then used to construct a prognostic risk model based on protein expression levels. The prognosis score is calculated as the sum of protein expression values multiplied by their corresponding coefficients. The prognostic score is calculated as the sum of the product between the expression level of each protein and its corresponding coefficient (β1, β2, etc.). (βn × expression level of protein n).
The train, test, and all sets (collectively) are divided into high-risk and low-risk groups based on the median of the prognostic scores in the training set. Using Kaplan–Meier survival curve analysis, the study compared the overall survival (OS) and progression-free survival (PFS) between high-risk and low-risk groups. Furthermore, it evaluated the variation in OS between patients with high protein expression and low expression in the prognostic risk model, using a cutoff value of P < 0.05.
The time-dependent ROC curve of a prognostic risk model for colorectal cancer (CRC) patients and the joint ROC curve of clinical features, including age, gender, T stage, M stage, and N stage, were generated using the “survivalROC” package in R. The accuracy of the prognostic risk model in predicting the prognosis of CRC patients was assessed based on the Area under the Curve (AUC) of the ROC curve.
We constructed a prognostic nomogram to assess the overall survival probabilities of colorectal cancer (CRC) patients at 1, 3, and 5 years. This analysis used the “rms” package in R (https://cran.r-project.org/web/packages/rms/). The calibration curve was plotted to compare the predicted overall survival (OS) and actual survival. The patients were categorized into two groups based on clinical characteristics: age, gender, stage, T, M, and N staging. Kaplan–Meier survival curve analysis was conducted to examine the applicability of the constructed prognostic risk model across different clinical groups36,37,38.
Differential protein expression analysis
Differential expression analysis was performed on the protein expression dataset of colon cancer patients obtained from the CPTAC database. The Wilcoxon test was employed to compare the protein expression differences between colon cancer tumor tissues and normal tissue samples. Proteins with a p-value < 0.05 were selected as differentially expressed. Please switch back to English: Generate volcano and violin plots to visualize differential protein expression. Construct a Venn diagram depicting the overlapping sets of differential proteins from the CPTAC database and prognostic risk model proteins from the TCGA database, utilizing the VennDiagram package39.
Ubiquitination and SUMOylation site prediction
To predict the SUMOylation sites of PDHA1, the JASSA (http://www.jassa.fr/) and SUMOPLOT (https://www.abcepta.com/sumoplot) websites were utilized. The JASSA website predicts the SUMO-binding motifs (SIM) of the RNF4 protein. Ubiquitination sites for the interaction between RNF4 protein and PDHA1 were predicted using the BioCuckoo website (http://gpsuber.biocuckoo.cn/online.php). Subsequent experimental validation was conducted on the two highest-ranking loci selected through a combination of scores, composite scores, and significance filtering40,41.
Clinical sample collection
The study recruited 28 patients diagnosed with metastatic colorectal cancer (m-CRC group) and 26 patients diagnosed with primary colorectal cancer (p-CRC group). The two groups were matched in terms of age and gender. The m-CRC group had a median age of 59.9 years, comprising 15 males and 13 females. The p-CRC group had a median age of 60.3 years, with 14 males and 12 females. Metastatic tumor tissues were collected from patients in the m-CRC group, in situ, tumor tissues were resected from patients in the p-CRC group, and adjacent normal tissues were collected as well (adjacent normal tissues refer to tissue samples located within a 3 cm range from the tumor edge; referred to as the Control-m-CRC group, with a total of 26 cases). These tissue samples were fixed using 4% paraformaldehyde for immunohistochemical staining.
To enhance the reproducibility of our research, we minimized variations in patient and sampling parameters, including fasting status and sampling time. Hence, during the time interval of 8 a.m. to 9 a.m., we collected fasting blood samples from patients in the m-CRC and p-CRC groups, after obtaining written informed consent and before commencing any treatment. These samples were collected specifically for subsequent metabolomic analysis. Clinical information of patients is shown in (Supplementary Table 2). The research involving human participants was conducted in accordance with the Declaration of Helsinki. The study protocol was approved by the Ethics Committee of Guangdong Provincial People’s Hospital (Guangdong Academy of Medical Sciences), Southern Medical University (Approval Number: KY-Z-2021-015-02). Informed consent was obtained from all participants prior to their inclusion in the study. For participants under the age of 16, consent was obtained from a parent or legal guardian.
Immunohistochemical staining
The expression of PDHA1 and RNF4 proteins in the tissues of patients with colorectal cancer (CRC) was detected using immunohistochemistry with streptavidin peroxidase (SP) labeling. The expression of proliferation-related markers Ki67 and PCNA was also examined in subcutaneous transplant tumor tissues of mice. The paraffin-embedded specimens of subcutaneous transplanted tumor tissue and tissue samples from CRC patients were harvested. These specimens were cut into continuous sections that are 5-μm thick. Immunohistochemical staining was performed after the routine dehydration process using the standard procedure.
To block endogenous peroxidase, the samples were treated with 3% hydrogen peroxide at room temperature for 10 min. Following this, normal goat serum was added and incubated for 10 min. The following antibodies were added subsequently: rabbit anti-Ki67 (1:2000, 28074-1-AP, Proteintech, Wuhan, China), rabbit anti-PCNA (1:200, ab92552, Abcam, UK), rabbit anti-PDHA1 (1:200, ab168379, Abcam, UK), or rabbit anti-RNF4 (1:200, HPA049356, Sigma-Aldrich, USA).
Overnight incubation at 4 °C is performed. Subsequently, the solution is exposed to the biotin-labeled secondary antibody (1:500, BA1003, BioDee, Wuhan, China) and maintained at 37 °C for 20 min. Following this, 50 μL of streptavidin-peroxidase solution is added and left to incubate at room temperature for 10 min. The experimental procedure included DAB staining, hematoxylin restaining, dehydration, clearing, mounting, and observation under a light microscope.
The criteria for identifying protein-positive cells entail that normal positive cells exhibited a brownish-yellow color and be evaluated according to the extent and intensity of staining. The percentage of positive stained Area determines the scoring for staining extent. A score of 0 indicates an extent less than 5%, 1 indicates an extent between 5% and 25%, 2 indicates an extent between 25% and 50%, 3 indicates an extent between 50% and 75%, and 4 indicates an extent greater than 75%. Staining intensity is classified into four levels: 0, 1, 2, and 3, representing negative (no staining), mild (weak), moderate (distinct), and severe (strong) staining, respectively. Next, calculate the weighted score by multiplying the intensity of staining by the percentage of Area42,43.
Metabolomics analysis
The serum samples were thawed at room temperature. Next, transfer 300 μL of each sample to 1.5 mL polypropylene tubes. Mix the samples with 900 μL of a solution containing 80% methanol and 0.1% formic acid, and vortex them for 2 min. After vortexing, centrifuge the tubes at 12,000 × g for 10 min. Transfer the supernatant into the small vial of the autosampler.
The Plasma metabolomics analysis combined the LC20 ultra-high performance liquid chromatography apparatus (Shimadzu, Japan) with the Triple TOF-6600 mass spectrometer (AB Sciex). Select the Waters ACQUITY UPLC HSS T3 C18 (100 × 2.1 mm, 1.8 μm) column for chromatographic analysis. The column is kept at a temperature of 40 °C, while elution is carried out with a flow rate of 0.4 mL/min. The mobile phase comprises an aqueous acetonitrile solution with a 0.1% formic acid concentration. The gradient elution program for mobile phase B is 5% from 0.0 to 11.0 min, 90% from 11.0 to 12.0 min, and 5% from 12.1 to 14 min. The rinse solution is introduced directly into the mass spectrometer without undergoing fragmentation44.
The conditions employed for mass spectrometry included the following settings: ionization voltage of 5500 V, capillary temperature of 550 °C, spray gas flow rate of 50 psi and auxiliary heating gas flow rate of 60 psi. The preprocessed data was analyzed using Orthogonal Partial Least Squares-Discriminant Analysis (OPLS-DA) and a permutation test with 100 permutations to prevent overfitting. Metabolites with VIP scores greater than 1 and p-values less than 0.05 in the OPLS-DA model were identified as differential metabolites (DMs). Furthermore, considering a fold change of at least 2 or a fold change of at most 0.5, along with univariate analysis, metabolites with a p-value less than 0.05 in the Student’s t-test were chosen as the final set of differential metabolites. MetaboAnalyst (Version 5.0) is utilized to identify relevant metabolic pathways45.
Cell culture
The colorectal cancer (CRC) cell lines employed in this study consist of DLD-1 (CC-Y1143), SW480 (CC-Y1500), SW620 (CC-Y1502), HCT-116 (CC-Y1194), and the FHC human normal colonic epithelial cell line (CC-Y1159)46. Human embryonic kidney cells 293T (CC-Y1010) were procured from Enzyme Research in Shanghai, China. 293T cells were cultured in DMEM high glucose medium (11965084, Thermo Fisher Scientific, USA), supplemented with 10% FBS (26140079, Thermo Fisher Scientific, USA) and 1% antibiotics (100 U/mL penicillin and 100 μg/mL streptomycin). SW480 and SW620 cells were cultured in Leibovitz’s L-15 medium (11415064, Thermo Fisher Scientific, USA), supplemented with 10% FBS and 1% antibiotics. HCT-116 cells were cultured in McCoy’s 5 A medium (16600082, Thermo Fisher Scientific, USA), supplemented with 10% FBS and 1% antibiotics. FHC and DLD-1 cells were cultured in RPMI-1640 medium (catalog number 11875101, Thermo Fisher Scientific, USA) supplemented with 10% fetal bovine serum (FBS) and 1% antibiotics. The cell cultures were maintained at 37 °C in a CO2 incubator (model BB15, Thermo Fisher Scientific, USA)47.
CCK-8 assay
The rapidly increasing CRC cells were seeded in 96-well plates at a density of 8 ×103 cells per well. The plates were then incubated in a cell culture incubator for specific periods (24 h, 48 h, and 72 h). Subsequently, 10 μL of CCK-8 solution (96992, Sigma-Aldrich, USA) was added to each well. Following a 1-h incubation in a humidified incubator at 37 °C, the absorbance of each sample at 450 nm was measured using the Bio-Tek Epoch microplate spectrophotometer (Bio-Tek, Winooski, VT, USA). Each group consists of six replicate holes, and the experiment is repeated three times48,49.
Transwell experiment
A Transwell invasion experiment was conducted on CRC cells following various treatments for 48 h. 50 μL of matrix gel (354234, BD Biosciences, USA) was added to the Transwell chamber and incubated at 37 °C for 30 min to solidify the gel. Following the rinse of the chamber with FBS-free medium, the small chamber is prepared. The cells are diluted in FBS-free medium to 2.5 ×104 cells/mL. Subsequently, 100 μL of the cell suspension is added to each well in the upper chamber, and 500 μL of medium containing 10% FBS is added to the lower chamber. After 24 h, the small chamber was removed, and a cotton swab was employed to eliminate the cells from the upper chamber. The cells were then fixed with 4% paraformaldehyde (PFA) at room temperature for 30 min. Subsequently, a 0.1% crystal violet staining was conducted for 30 min. Next, five random regions were selected for inverted microscope imaging (IXplore Pro, Olympus, Japan) and subsequent cell counting50,51. Cell migration experiments do not necessitate the incorporation of a matrix gel. The subsequent steps mirror those in the invasion experiment and are replicated three times.
Scratch test
CRC cells were inoculated into a 24-well plate for cultivation. Upon reaching confluence, they were carefully scraped using a 200 μL pipette tip. The cells were then washed with PBS and the medium was replaced with FBS-free RPMI 1640 for an additional 48-h incubation period. Photographs were taken using an inverted microscope at a magnification of ×5 at the beginning and end of the experiment. The scratch area was analyzed with ImageJ software, and the wound healing rate was calculated50,52.
Lentivirus and plasmid transfection
The lentiviral plasmids for PDHA1 shRNA and RNF4 shRNA (sh-PDHA1 and sh-RNF4), as well as their control lentiviral particles (sh-NC), were obtained from Sigma-Aldrich (USA). 293T cells were transfected with sh-PDHA1 or sh-RNF4 plasmids, together with pCMV-dR8.91 (ZT1466, Fenghui Biotech, Changsha, Hunan) and pMD2.G (#12259, Addgene, USA) packaging plasmids, in a 3:3:1 ratio of calcium phosphate. One day after transfection, the original culture medium was with a small serum-free medium, and the cells were cultured for 24–30 h. The conditioned medium was collected, passed through a 0.45 μm filter, and concentrated by centrifugation at 80,000 × g for 2 h at 4 °C. The lentivirus was resuspended in a serum-free culture medium and titrated using the p24 ELISA kit (L00938, Nanjing, Jiangsu) in accordance with the manufacturer’s instructions.
The supernatant containing the virus was incubated with SW620 or DLD-1 cells at 100 ng p24 per million cells. Following 72 h of viral infection, the culture medium was replaced with a medium containing 4 μg/mL puromycin, and the SW620 or DLD-1 cells were cultured for a minimum of 14 days. The puromycin-resistant cells were amplified in a medium containing 2 μg/mL of puromycin for 9 days. Following this, they were transferred to a puromycin-free medium to achieve stable knockdown of either PDHA1 or RNF4 in SW620 or DLD-1 cells. The information regarding the knockdown sequences is as follows: sh-PDHA1#1 (TRCN0000028582; target sequence: GCCAATCAGTGGATCAAGTTT), sh-PDHA1#2 (TRCN0000028602; target sequence: GTGAGAATAATCGCTATGGAA), sh-RNF4#1 (TRCN0000281894; target sequence: GACAAGCTCAGAAGCGAACTC), sh-RNF4#2 (TRCN0000272669; target sequence: ACGTATATGTGACTACCCATA), and sh-NC (CTCGCTTGGGCGAGAGTAA)53,54. To obtain the desired plasmid, the PDHA1 gene was connected to the eukaryotic expression vector pcDNA3.1 (V79520, Invitrogen, ThermoFisher Scientific, USA). SW620 cells were seeded at a density of 2 ×105 cells per well in a 6-well plate and incubated in a CO2 incubator for 24 h. Next, the target plasmid was transfected into the SW620 cells using Lipofectamine 3000 (L3000150, Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA).
The plasmid concentration was determined based on the instruction manual (approximately 50 nM) and then adjusted per specific circumstances. Following transfection, the cells were incubated at 37 °C with 5% CO2 for 6–8 h. Subsequently, the complete medium was replaced, and the cells were further incubated for an additional 48 h to facilitate the extraction of RNA and proteins for subsequent experiments55.
The groups were classified as follows: (1) DLD-1 cells were assigned to sh-NC, sh-PDHA1#1 (sh-PDHA1), and sh-PDHA1#2 groups, which were transfected with lentivirus for PDHA1 knockdown and control lentivirus; (2) SW620 cells were assigned to pcDNA and PDHA1 groups, which were transfected with plasmids for PDHA1 overexpression and control plasmid; (3) SW620 cells were assigned to sh-NC, sh-RNF4#1 (sh-RNF4), and sh-RNF4#2 groups, which were transfected with lentivirus for RNF4 knockdown and control lentivirus; (4) SW620 cells were assigned to sh-NC, sh-RNF4, and sh-RNF4+sh-PDHA1 groups56,57.
Construction of plasmids and their mutants
The human PDHA1 cDNA was inserted into the pBICEP-CMV™-3 vector containing a Flag tag (E7029, Sigma-Aldrich, USA) to construct the flag-PDHA1 plasmid. Alternatively, specific primers with a His6 tag sequence could be used to amplify the PDHA1 and achieve in-frame fusion at the C terminus, resulting in the construction of an expression plasmid that contains an N-terminal FLAG tag and a C-terminal His tag for PDHA1 (FLAG-PDHA1-His). To construct the T7-SUMO3 expression plasmid, the human SUMO3 (1e92) cDNA was inserted into the pcDNA3.1/T7 vector (138610, addgene, USA). HA-tagged ubiquitin (HA-Ub, 18712, Addgene, USA) was expressed using the pMT123 vector. The RNF4 cDNA was cloned into the pcDNA3.1 Myc vector (176045, addgene, USA) to generate the Myc-RNF4 plasmid. EGFP-tagged SUMO1, SUMO2, and SUMO3 were created by inserting processed forms of human cDNAs encoding SUMO1 (1-97), SUMO2 (1-92), and SUMO3 (1-93) into the pEGFP vector (165830, addgene, USA).
By substituting the lysine residues at positions K77 (LKAR) and K186 (GKDE)—which have been identified by the website as the SUMO sites of PDHA1—with arginine, a mutant variant of PDHA1 lacking the SUMO sites (Flag-PDHA1 2KR) was generated. Furthermore, we generated site-specific mutants for K77 (Flag-PDHA1 K77) and K186 (Flag-PDHA1 K186). Site-directed mutagenesis was performed on the SUMO-binding motifs (SIM) of RNF4 to generate RNF4 mSIM mutants (Myc-RNF4 mSIM) with the following mutations: aa36-39 IELV/AEAA; aa46-50 IVDL/AADA; aa58-62 VVDL/AADA; aa67-70 VVIV/AAAA. Flag-PDHA1 2KR and Myc-RNF4 mSIM constructs were produced using site-directed mutagenesis with the QuickChange® II XL kit from Agilent Technologies. This method used specific mutation primers and PCR58,59,60,61.
SUMOylation and ubiquitination analysis
The analysis of PDHA1 was conducted to examine SUMOylation and ubiquitination using the nickel-nitrilotriacetic acid (Ni-NTA) pull-down method. Initially, we co-transfected SW620 cells with FLAG-PDHA1-His, HA-Ub, T7-SUMO3, and other relevant expression plasmids. After 24 h, the cells were treated with 20 μM MG132 (474790, Sigma-Aldrich, USA), 20 μg/ml cycloheximide (CHX, 239765, Sigma-Aldrich, USA), or a combination of CHX + MG132 for 6 h. Subsequently, the cells were lysed using buffer A. His6-tagged PDHA1 was affinity purified using Ni-NTA agarose beads (78605, Thermo Fisher Scientific, Waltham, MA, USA).
Antibodies including anti-Flag (rabbit; 1:1000; F7425; Sigma-Aldrich, USA), anti-HA (rabbit; 1:1000; #3724; Cell Signaling, USA; https://www.cellsignal.cn/), and anti-T7 (mouse; 1:1000; T8823; Sigma-Aldrich, USA) were used for the detection of total PDHA1 and its ubiquitinated or sumoylated forms59.
Protein half-life determination
SW620 cells transfected with Flag-PDHA1 WT (Flag-PDHA1), Flag-PDHA1 2KR, Flag-PDHA1 K77, and Flag-PDHA1 K186 were treated with 20 μg/ml CHX for various durations (0, 2, 4, 6, 8, and 10 h) to inhibit protein synthesis. Subsequently, protein from each group was extracted and PDHA1 protein levels were assessed using Western blot analysis62.
Co-IP experiment detects protein-protein interactions
SW620 cells were utilized for Co-IP experiments to identify the interaction between endogenous and exogenous proteins. Co-immunoprecipitation (Co-IP) experiments were conducted in cells transfected with Myc-RNF4 and Flag-PDHA1 constructs. Subsequently, the cells were incubated for 24 h and treated with 20 μM MG-132 for 6 h. Initially, the cells are lysed using an IP lysis buffer (P0013, Beyotime, Shanghai, China) that includes protease and phosphatase inhibitors. Next, the cell lysate was centrifuged at 12,000 × g for 20 min at 4 °C. Subsequently, the cell lysates containing 200 μg of protein were incubated with the anti-Flag antibody (mouse; 1:50, F3165, Sigma-Aldrich, USA) or the anti-Myc antibody (mouse; 1:50, M4439, Sigma-Aldrich, USA) at 4 °C for 4 h. Alternatively, the cell lysis buffer could include either an anti-PDHA1 antibody (rabbit; 1:100, ab168379, Abcam, UK) or IgG (rabbit; 1:50, #3900, Cell Signaling, USA). The mixture was incubated overnight at 4 °C. Subsequently, the antibody-protein complex was captured using Protein A/G Sepharose beads (sc-2003, Santa Cruz Biotechnology, USA, https://www.scbt.com/zh/home). After thorough washing, boil the sample in 1× SDS sample buffer and analyze it using Western blot62,63.
In situ proximity ligation assay (PLA)
First, SW620 cells were seeded on glass coverslips. After 24 h, cells were either left untreated or transfected with designated plasmids (Myc-RNF4, sh-NC, and sh-RNF4), followed by treatment with 10 μM MG-132 for 4 h. The cells were fixed with 4% paraformaldehyde at room temperature for 20 min, and then washed with 0.1 M Glycine buffer (pH = 7). Permeabilization was done using a PBS solution containing 0.2% Triton X-100 for 10 min, followed by a PBS wash. The proximity between proteins was detected using the Duolink® In Situ kit (DUO92101, Sigma-Aldrich, USA) according to the manufacturer’s instructions. Cell analysis was conducted using an IX53 fluorescence microscope (CLSM; LSM 510 META, Carl Zeiss AG). Image processing was performed using Image J software. The number of puncta per transfected cell was estimated on a single slice, with approximately 100 cells per condition. Automatic estimation was done using plugins in the Image J software58.
Seahorse XF extracellular flux analysis
The Seahorse XF96 extracellular flux analyzer (Seahorse Bioscience, North Billerica, MA, USA) was employed, following the manufacturer’s instructions, to assess the extracellular acidification rate (EACR) and oxygen consumption rate (OCR) of cells in vitro. Inoculate 10,000 cells per well in an XF96 cell culture plate and incubate at 37 °C overnight. On the second day, the medium was substituted with L-15 medium that did not include sodium bicarbonate but contained 1 mM glutamine. Subsequently, the cells were incubated in a CO2 incubator at 37 °C for 60 min to attain pH and temperature equilibrium within the medium. The ECAR and OCR were monitored under baseline conditions and in the presence of 10 mM glucose, 1 µM oligomycin, 5 mM 2-deoxyglucose (2-DG), 1 µM FCCP, or 0.5 µM rotenone/antimycin A. Furthermore, the values for Glycolysis, Glycolytic Reserve, Basal Respiration, and Maximal Respiration must be calculated. Data is standardized according to protein quantification standards64,65.
Lactate production detection
The intracellular lactate levels were measured using the Lactic Acid Assay Kit II (MAK064, Sigma-Aldrich, USA). Cell samples were mixed with four times the volume of lactate assay buffer and dissolved, followed by centrifugation at 13,000 × g for 10 min. The supernatant (50 μL) was transferred to a 96-well plate, discarding the precipitate. Subsequently, LDH was removed using a 10 kDa ultrafiltration centrifuge tube. Lactate assay buffer (50 μL) was added to each well and mixed with a pipette. After an incubation of 30 min at room temperature in the dark, absorbance was measured at 450 nm66.
RT-qPCR
Total RNA was extracted from CRC cells using Trizol reagent (15596026, Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA). The concentration and purity of the total RNA were determined using a Nanodrop2000 micro-UV spectrophotometer. RNA was reverse transcribed into cDNA following the instructions provided by the PrimeScript RT reagent Kit (RR047A, Takara, Japan). The reverse transcription conditions are 42 °C for 30–50 min and 85 °C for 5 s. qRT-PCR detection was conducted utilizing the Fast SYBR Green PCR Kit (RR820A, Takara, Japan) and the ABI PRISM 7300 RT-PCR System (Applied Biosystems). The reaction conditions consisted of an initial denaturation at 95 °C for 5 min, followed by 40 cycles of denaturation at 95 °C for 30 s, annealing at 57 °C for 30 s, and extension at 72 °C for 30 s. Three repetitions are set for each hole. The relative expression level of a gene using the 2−ΔΔCt method is analyzed by calculating ΔΔCt. It is achieved by subtracting the average Ct value of the target gene in the experimental group from the average Ct value of the reference gene in the experimental group and then subtracting the difference between the average Ct value of the target gene in the control group and the average Ct value of the reference gene in the control group67. The experiment is repeated three times. The primer sequences are shown in Supplementary Table 3.
Western blot
The cell groups were collected and added individually to RIPA lysis buffer (P0013B, Beyotime, Shanghai, China) supplemented with 1% PMSF on ice for 30 min. The mixture was then centrifuged at 14,000 × g at 4 °C, and the supernatant was collected. Protein extraction was carried out, and the protein concentration of the samples was determined using the BCA method (P0012S, Beyotime, Shanghai, China). Add an appropriate amount of 5× loading buffer to denature the proteins and boil at 100 °C for 10 min. The protein loading amount was 50 μg. The gel was separated and concentrated before electrophoresis. Following electrophoresis, the target protein bands were transferred onto a PVDF membrane.
The PVDF membrane was immersed in 5% skim milk powder and sealed at room temperature for 1 h. Afterward, it was incubated overnight with primary antibodies, which included an anti-PDHA1 antibody (rabbit; 1:1000, ab110330, Abcam, UK), anti-RNF4 antibody (rabbit; 1:1000, 17810-1-AP, Proteintech, Wuhan Sanying, China), anti-Flag antibody (rabbit; 1:1000, F7425, Sigma-Aldrich, USA), anti-HA antibody (rabbit; 1:1000, #3724, Cell Signaling, USA), anti-Myc antibody (rabbit; 1:1000, SAB4501941, Sigma-Aldrich, USA), anti-T7 antibody (mouse; 1:1000, T8823, Sigma-Aldrich, USA), anti-EGFP antibody (mouse; 1:1000, MAB3580, Sigma-Aldrich, USA), anti-SAE1 antibody (rabbit; 1:1000, ab97523, Abcam, USA), anti-SAE2 antibody (rabbit; 1:1000, ab95835, Abcam, USA), anti-UBC9 antibody (goat; 1:1000, ab21193, Abcam, USA), anti-SUMO1 antibody (rabbit; 1:1000, S8070, Sigma-Aldrich, USA), anti-SUMO2/3 antibody (rabbit; 1:1000, 07-2167, Sigma-Aldrich, USA) and anti-α-Tubulin antibody (mouse; 1:5000, ab7291, Abcam, UK). Alpha-tubulin was used as an internal reference. After washing with PBST buffer at room temperature, membranes were incubated with Horseradish Peroxidase (HRP)-conjugated secondary antibodies, including goat anti-rabbit/goat anti-mouse/rabbit anti-goat IgG (1:10000, BA1054/BA1050/BA1040, Biod, Wuhan, China), for 1 h at room temperature. Subsequently, the membranes were washed six times with PBST for 5 min each. ECL detection reagent (AR1172, Biod, Wuhan, China) was evenly applied onto the membrane, followed by exposure in an imaging system (Amersham Imager 600, USA). Then use Image J for grayscale analysis68,69. The experiment is repeated three times. Please refer to Supplementary 6–52 for original WB images.
Subcutaneous tumor growth and metastasis experiments in nude mice
Seventy-two male BALB/c nude mice, aged 4–5 weeks and weighing 18–22 g, were procured from Vital River (401, Beijing, China). Animals in the SPF animal room were acclimated to the experimental environment by being raised for one week under constant humidity (45–50%) and temperature (25–27 °C), with a 12-h light-dark cycle. The mice were subjected to a 12-h fasting period before the experiment while having unrestricted access to food and water during all other times. All experiments involving live vertebrates were conducted in accordance with relevant guidelines and regulations and approved by the Ethics Committee of Guangdong Provincial People’s Hospital (Guangdong Academy of Medical Sciences), Southern Medical University, and conducted in strict accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health, ensuring all efforts were made to minimize animal suffering (Approval Number: KY-Z-2021-015-02). Additionally, the study adheres to the ARRIVE Essential 10 checklist, ensuring rigorous reporting standards.
To establish a subcutaneous xenograft model, 1 ×107 stably transfected SW620 cells were injected into the subcutaneous space on the back of nude mice. A total of 24 nude mice were randomly divided into three groups, namely the sh-NC group, the sh-RNF4 group, and the sh-RNF4+sh-PDHA1 group, each consisting of 8 mice. To track the progression of tumors, the width (W) and length (L) of tumors in each group of nude mice were measured every 5 days using a caliper. Tumor volume (V) was calculated using the formula V = (W^2 × L)/2. On the 35th day after injection, the nude mice were euthanized, and their tumors were dissected, photographed, and weighed48,70.
For constructing a metastasis model, the remaining 48 mice were divided into three groups: the sh-NC group, the sh-RNF4 group, and the sh-RNF4+sh-PDHA1 group, each consisting of 16 mice. Specifically, 8 mice were used for the construction of the lung metastasis model, while another 8 mice were used for the construction of the liver metastasis model. Stably transfected SW620 cells were suspended in 20 μL of phosphate buffer and injected into nude mice’s tail veins to establish a lung metastasis model. The development of lung metastases was monitored weekly using the IVIS® Spectrum small animal in vivo fluorescence imaging system (PerkinElmer). To establish a liver metastasis model, 1 ×106 SW620 cells were stably transfected and injected into the spleen. After 35 days, the mice were euthanized to evaluate their lung and liver metastatic capabilities71.
Statistical analysis
The research data was subjected to statistical analysis using SPSS software (version 21.0, IBM, USA). The data is presented as the mean ± standard deviation for each measurement. First, normality and homogeneity of variance were assessed. If both tests were passed, a non-paired t-test was used for comparing the two groups. If multiple groups were involved, a one-way analysis of variance (ANOVA) or repeated measures ANOVA was conducted, followed by a post-hoc Tukey’s test. Survival curves are compared using the log-rank test. The hazard ratio (HR) and the corresponding 95% confidence interval (CI) for risk were estimated using univariate Cox regression43,72,73. A significance level of P < 0.05 indicates that the observed difference is statistically significant.
Data availability
The data that supports the findings of this study are available in the manuscript and supplementary materials.
References
Hultcrantz, R. Aspects of colorectal cancer screening, methods, age and gender. J. Intern. Med. 289, 493–507 (2021).
Simó-Riudalbas, L. et al. Transposon-activated POU5F1B promotes colorectal cancer growth and metastasis. Nat. Commun. 13, 4913 (2022).
Biller, L. H. & Schrag, D. Diagnosis and Treatment of Metastatic Colorectal Cancer: A Review. JAMA 325, 669–685 (2021).
Mitsala, A., Tsalikidis, C., Pitiakoudis, M., Simopoulos, C. & Tsaroucha, A. K. Artificial Intelligence in Colorectal Cancer Screening, Diagnosis and Treatment. A New Era. Curr. Oncol. 28, 1581–1607 (2021).
Wang, Y., Wang, D., Yang, L. & Zhang, Y. Metabolic reprogramming in the immunosuppression of tumor-associated macrophages. Chin. Med. J. 135, 2405–2416 (2022).
Liu, X. et al. Protein modification: A critical modulator in the interaction between geminiviruses and host plants. Plant Cell Environ. 44, 1707–1715 (2021).
Fan, Y. et al. SUMOylation in Viral Replication and Antiviral Defense. Adv. Sci. 9, e2104126 (2022).
Chelbi-Alix, M. K. & Thibault, P. Crosstalk Between SUMO and Ubiquitin-Like Proteins: Implication for Antiviral Defense. Front. Cell Dev. Biol. 9, 671067 (2021).
Huang, J. et al. RNF4 promotes tumorigenesis, therapy resistance of cholangiocarcinoma and affects cell cycle by regulating the ubiquitination degradation of p27kip1 in the nucleus. Exp. Cell Res. 419, 113295 (2022).
Bian, Z., Fan, R. & Xie, L. A Novel Cuproptosis-Related Prognostic Gene Signature and Validation of Differential Expression in Clear Cell Renal Cell Carcinoma. Genes 13, 851 (2022).
Karissa, P. et al. Comparison Between Dichloroacetate and Phenylbutyrate Treatment for Pyruvate Dehydrogenase Deficiency. Br. J. Biomed. Sci. 79, 10382 (2022).
Joshi, S. K. et al. The AML microenvironment catalyzes a stepwise evolution to gilteritinib resistance. Cancer Cell. 39, 999–1014.e8 (2021).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Li, Y. et al. PDHA1 gene knockout in prostate cancer cells results in metabolic reprogramming towards greater glutamine dependence. Oncotarget 7, 53837–53852 (2016).
Wu, S. C. et al. Stomatin modulates adipogenesis through the ERK pathway and regulates fatty acid uptake and lipid droplet growth. Nat. Commun. 13, 4174 (2022).
Zhu, G., Jin, L., Sun, W., Wang, S. & Liu, N. Proteomics of post-translational modifications in colorectal cancer: Discovery of new biomarkers. Biochim. Biophys. Acta Rev. Cancer 1877, 188735 (2022).
Yang, C. et al. Identification of cuproptosis hub genes contributing to the immune microenvironment in ulcerative colitis using bioinformatic analysis and experimental verification. Front. Immunol. 14, 1113385 (2023).
Peng, K. et al. Effects of UBE3A on Cell and Liver Metabolism through the Ubiquitination of PDHA1 and ACAT1. Biochemistry 62, 1274–1286 (2023).
Jalal, D., Chalissery, J. & Hassan, A. H. Genome maintenance in Saccharomyces cerevisiae: the role of SUMO and SUMO-targeted ubiquitin ligases. Nucleic Acids Res. 45, 2242–2261 (2017).
Wang, M. et al. SENP3 regulates the global protein turnover and the Sp1 level via antagonizing SUMO2/3-targeted ubiquitination and degradation. Protein Cell. 7, 63–77 (2016).
Xu, Y. et al. Structural insight into SUMO chain recognition and manipulation by the ubiquitin ligase RNF4. Nat. Commun. 5, 4217 (2014).
Tatham, M. H. et al. RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat. Cell Biol. 10, 538–546 (2008).
Zhang, L. et al. MG132-mediated inhibition of the ubiquitin-proteasome pathway ameliorates cancer cachexia. J. Cancer Res. Clin. Oncol. 139, 1105–1115 (2013).
Kumar, R., González-Prieto, R., Xiao, Z., Verlaan-de Vries, M. & Vertegaal, A. C. O. The STUbL RNF4 regulates protein group SUMOylation by targeting the SUMO conjugation machinery. Nat. Commun. 8, 1809 (2017).
Zhao, G. et al. DDX39B drives colorectal cancer progression by promoting the stability and nuclear translocation of PKM2. Signal Transduct. Target Ther. 7, 275 (2022).
Liu, L. et al. PDHA1 Gene Knockout In Human Esophageal Squamous Cancer Cells Resulted In Greater Warburg Effect And Aggressive Features In Vitro And In Vivo. Onco Targets Ther. 12, 9899–9913 (2019).
Wei, T. et al. SIRT3 (Sirtuin-3) Prevents Ang II (Angiotensin II)-Induced Macrophage Metabolic Switch Improving Perivascular Adipose Tissue Function. Arterioscler Thromb. Vasc. Biol. 41, 714–730 (2021).
Li, G. et al. Appropriate level of cuproptosis may be involved in alleviating pulmonary fibrosis. Front. Immunol. 13, 1039510 (2022).
Gu, C. et al. Microglial MT1 activation inhibits LPS-induced neuroinflammation via regulation of metabolic reprogramming [published correction appears in Aging Cell. 2024 Feb;23:e14068]. Aging Cell. 20, e13375 (2021).
Young, M. J. et al. Estradiol-mediated inhibition of Sp1 decreases miR-3194-5p expression to enhance CD44 expression during lung cancer progression. J. Biomed. Sci. 29, 3 (2022).
Bauermeister, A., Mannochio-Russo, H., Costa-Lotufo, L. V., Jarmusch, A. K. & Dorrestein, P. C. Mass spectrometry-based metabolomics in microbiome investigations. Nat. Rev. Microbiol. 20, 143–160 (2022).
Tillie, R. J. H. A. et al. Partial Inhibition of the 6-Phosphofructo-2-Kinase/Fructose-2,6-Bisphosphatase-3 (PFKFB3) Enzyme in Myeloid Cells Does Not Affect Atherosclerosis. Front. Cell Dev. Biol. 9, 695684 (2021).
Quyun, C., Ye, Z., Lin, S. C. & Lin, B. Recent patents and advances in genomic biomarker discovery for colorectal cancers. Recent Pat. DNA Gene Seq. 4, 86–93 (2010).
Wu, P. et al. Integration and Analysis of CPTAC Proteomics Data in the Context of Cancer Genomics in the cBioPortal. Mol. Cell Proteom. 18, 1893–1898 (2019).
Vasaikar, S. V., Straub, P., Wang, J. & Zhang, B. LinkedOmics: analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 46, D956–D963 (2018).
Zheng, G. L., Zhang, G. J., Zhao, Y. & Zheng, Z. C. Screening Protein Prognostic Biomarkers for Stomach Adenocarcinoma Based on The Cancer Proteome Atlas. Front. Oncol. 12, 901182 (2022).
Wen, Y. D. et al. Proteomics-based prognostic signature and nomogram construction of hypoxia microenvironment on deteriorating glioblastoma (GBM) pathogenesis. Sci. Rep. 11, 17170 (2021).
Zheng, H., Liu, H., Li, H., Dou, W. & Wang, X. Weighted Gene Co-expression Network Analysis Identifies a Cancer-Associated Fibroblast Signature for Predicting Prognosis and Therapeutic Responses in Gastric Cancer. Front Mol. Biosci. 8, 744677 (2021).
Jiang, Y. et al. TANK-Binding Kinase 1 (TBK1) Serves as a Potential Target for Hepatocellular Carcinoma by Enhancing Tumor Immune Infiltration. Front. Immunol. 12, 612139 (2021).
Beauclair, G., Bridier-Nahmias, A., Zagury, J. F., Saïb, A. & Zamborlini, A. JASSA: a comprehensive tool for prediction of SUMOylation sites and SIMs. Bioinformatics 31, 3483–3491 (2015).
Wang, C. et al. GPS-Uber: a hybrid-learning framework for prediction of general and E3-specific lysine ubiquitination sites. Brief. Bioinform. 23, bbab574 (2022).
Li, L. et al. Phosphorylated Myosin Light Chain 2 (p-MLC2) as a Molecular Marker of Antemortem Coronary Artery Spasm. Med. Sci. Monit. 22, 3316–3327 (2016).
Tian, J. & Yuan, L. Sirtuin 6 inhibits colon cancer progression by modulating PTEN/AKT signaling. Biomed. Pharmacother. 106, 109–116 (2018).
Chi, N. F. et al. Untargeted metabolomics predicts the functional outcome of ischemic stroke. J. Formos. Med. Assoc. 120, 234–241 (2021).
Hou, J. Y. et al. Integrated transcriptomics and metabolomics analysis reveals that C3 and C5 are vital targets of DuZhi Wan in protecting against cerebral ischemic injury. Biomed. Pharmacother. 155, 113703 (2022).
Zeng, M., Zhu, L., Li, L. & Kang, C. miR-378 suppresses the proliferation, migration and invasion of colon cancer cells by inhibiting SDAD1. Cell Mol. Biol. Lett. 22, 12 (2017).
Li, M. et al. Lactobacillus-derived extracellular vesicles enhance host immune responses against vancomycin-resistant enterococci. BMC Microbiol. 17, 66 (2017).
Pan, X., Hong, X., Li, S., Meng, P. & Xiao, F. METTL3 promotes adriamycin resistance in MCF-7 breast cancer cells by accelerating pri-microRNA-221-3p maturation in a m6A-dependent manner. Exp. Mol. Med. 53, 91–102 (2021).
Wang, S. et al. Interactions Between lncRNA TUG1 and miR-9-5p Modulate the Resistance of Breast Cancer Cells to Doxorubicin by Regulating eIF5A2. Onco Targets Ther. 13, 13159–13170 (2020).
Zhu, G. et al. MyD88 mediates colorectal cancer cell proliferation, migration and invasion via NF‑κB/AP‑1 signaling pathway. Int J. Mol. Med. 45, 131–140 (2020).
Liu, P. et al. LncRNA-MALAT1 promotes neovascularization in diabetic retinopathy through regulating miR-125b/VE-cadherin axis. Biosci. Rep. 39, BSR20181469 (2019).
Niu, J. et al. DKK1 inhibits breast cancer cell migration and invasion through suppression of β-catenin/MMP7 signaling pathway. Cancer Cell Int. 19, 168 (2019).
Jin, W. et al. MUC1 induces acquired chemoresistance by upregulating ABCB1 in EGFR-dependent manner. Cell Death Dis. 8, e2980 (2017).
Lassot, I. et al. The E3 Ubiquitin Ligases TRIM17 and TRIM41 Modulate α-Synuclein Expression by Regulating ZSCAN21. Cell Rep. 25, 2484–2496.e9 (2018).
Sato, Y., Shinka, T., Sakamoto, K., Ewis, A. A. & Nakahori, Y. The male-determining gene SRY is a hybrid of DGCR8 and SOX3, and is regulated by the transcription factor CP2. Mol. Cell Biochem. 337, 267–275 (2010).
Yang, R. et al. IFN-γ promoted exosomes from mesenchymal stem cells to attenuate colitis via miR-125a and miR-125b. Cell Death Dis. 11, 603 (2020).
Ye, M., Dong, S., Hou, H., Zhang, T. & Shen, M. Oncogenic Role of Long Noncoding RNAMALAT1 in Thyroid Cancer Progression through Regulation of the miR-204/IGF2BP2/m6A-MYC Signaling. Mol. Ther. Nucleic Acids 23, 1–12 (2020).
Basu-Shrivastava, M. et al. Trim39 regulates neuronal apoptosis by acting as a SUMO-targeted E3 ubiquitin-ligase for the transcription factor NFATc3. Cell Death Differ. 29, 2107–2122 (2022).
Nishida, T. & Yamada, Y. RNF4-mediated SUMO-targeted ubiquitination relieves PARIS/ZNF746-mediated transcriptional repression. Biochem. Biophys. Res. Commun. 526, 110–116 (2020).
Nishida, T. & Yamada, Y. SUMOylation of the KRAB zinc-finger transcription factor PARIS/ZNF746 regulates its transcriptional activity. Biochem. Biophys. Res. Commun. 473, 1261–1267 (2016).
Nishida, T. & Yamada, Y. SMT3IP1, a nucleolar SUMO-specific protease, deconjugates SUMO-2 from nucleolar and cytoplasmic nucleophosmin. Biochem. Biophys. Res. Commun. 374, 382–387 (2008).
Zhang, L. et al. FBXO22 promotes the development of hepatocellular carcinoma by regulating the ubiquitination and degradation of p21. J. Exp. Clin. Cancer Res. 38, 101 (2019).
Zhang, H. et al. SET8 prevents excessive DNA methylation by methylation-mediated degradation of UHRF1 and DNMT1. Nucleic Acids Res. 47, 9053–9068 (2019).
Hu, T. et al. Tumor-intrinsic CD47 signal regulates glycolysis and promotes colorectal cancer cell growth and metastasis. Theranostics 10, 4056–4072 (2020).
Zhou, B. et al. Upregulation of mitochondrial ATPase inhibitory factor 1 (ATPIF1) mediates increased glycolysis in mouse hearts. J. Clin. Invest. 132, e155333 (2022).
Fan, Q. et al. Autophagy promotes metastasis and glycolysis by upregulating MCT1 expression and Wnt/β-catenin signaling pathway activation in hepatocellular carcinoma cells. J. Exp. Clin. Cancer Res. 37, 9 (2018).
Jin, D. et al. m6A mRNA methylation initiated by METTL3 directly promotes YAP translation and increases YAP activity by regulating the MALAT1-miR-1914-3p-YAP axis to induce NSCLC drug resistance and metastasis [retracted in: J Hematol Oncol. 2023 Feb 22;16:14]. J. Hematol. Oncol. 12, 135 (2019).
Salem, M., Shan, Y., Bernaudo, S. & Peng, C. miR-590-3p Targets Cyclin G2 and FOXO3 to Promote Ovarian Cancer Cell Proliferation, Invasion, and Spheroid Formation. Int. J. Mol. Sci. 20, 1810 (2019).
Shu, M. et al. Targeting oncogenic miR-335 inhibits growth and invasion of malignant astrocytoma cells. Mol. Cancer 10, 59 (2011).
Liu, R., Guo, H. & Lu, S. MiR-335-5p restores cisplatin sensitivity in ovarian cancer cells through targeting BCL2L2. Cancer Med. 7, 4598–4609 (2018).
Tang, J. et al. CircRNA circ_0124554 blocked the ubiquitination of AKT promoting the skip lymphovascular invasion on hepatic metastasis in colorectal cancer. Cell Death Dis. 12, 270 (2021).
Velazquez, R. et al. Lifelong choline supplementation ameliorates Alzheimer’s disease pathology and associated cognitive deficits by attenuating microglia activation. Aging Cell. 18, e13037 (2019).
Wang, W. et al. Ferroptosis inducers enhanced cuproptosis induced by copper ionophores in primary liver cancer. J. Exp. Clin. Cancer Res. 42, 142 (2023).
Acknowledgements
This study was supported by National Natural Science Foundation of China (82102497) and Natural Science Foundation of Guangdong (2023A1515030063) and Guangzhou Municipality Basic and Applied Basic Research Theme Outstanding Doctoral ‘Continuing Voyage’ Project 2024 (2024A04J10021).
Author information
Authors and Affiliations
Contributions
Jierong Chen, Zi-Yue Li and Guansheng Zheng wrote the paper and conceived and designed the experiments; Lixue Cao, Yun-Miao Guo and Qizhou Lian analyzed the data; Bing Gu and Cai-Feng Yue collected and provided the sample for this study. All authors have read and approved the final submitted manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, J., Li, ZY., Zheng, G. et al. RNF4 mediated degradation of PDHA1 promotes colorectal cancer metabolism and metastasis. npj Precis. Onc. 8, 258 (2024). https://doi.org/10.1038/s41698-024-00724-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41698-024-00724-5
This article is cited by
-
Modulation of copper homeostasis and cuproptosis by PDHA1 in acute myeloid leukemia
Discover Oncology (2025)
-
PDHA1 Orchestrates Hepatocellular Carcinoma Progression Through LINC00607-Mediated Regulation of Cuproptosis and Immune Evasion
Digestive Diseases and Sciences (2025)
