
Abstract
Expression profiling in spatially defined regions is crucial for systematically understanding tissue complexity. Here, we report a method of photo-irradiation for in-situ barcoding hybridization and ligation sequencing, named PBHL-seq, which allows targeted expression profiling from the photo-irradiated region of interest in intact fresh frozen and formalin fixation and paraffin embedding (FFPE) tissue samples. PBHL-seq uses photo-caged oligodeoxynucleotides for in situ reverse transcription followed by spatially targeted barcoding of cDNAs to create spatially indexed transcriptomes of photo-illuminated regions. We recover thousands of differentially enriched transcripts from different regions by applying PBHL-seq to OCT-embedded tissue (E14.5 mouse embryo and mouse brain) and FFPE mouse embryo (E15.5). We also apply PBHL-seq to the subcellular microstructures (cytoplasm and nucleus, respectively) and detect thousands of differential expression genes. Thus, PBHL-seq provides an accessible workflow for expression profiles from the region of interest in frozen and FFPE tissue at subcellular resolution with areas expandable to centimeter scale, while preserving the sample intact for downstream analysis to promote the development of transcriptomics.
Similar content being viewed by others


Introduction
Single-cell RNA sequencing (scRNA-seq) technologies reveal tissue heterogeneity and expand the understanding of cell populations and states among complex tissues1,2,3,4,5,6,7,8,9. However, cells do not function in isolation as their spatial location and neighboring cells are essential for cellular function within complex tissue10,11,12,13. Multiple spatial transcriptomic profiling methodologies have been developed over the last decade12,14,15,16,17,18,19,20,21,22,23,24,25, which broadly fall into three major categories in terms of how positional information of RNA is acquired: (1) imaging-based in-situ RNA hybridization16,17,19,22,26,27,28,29,30,31 and/or sequencing14,32,33,34, (2) next-generation sequencing (NGS) with spatial barcoding18,24,35, (3) region of interest (ROI) study36,37,38,39,40,41,42,43.
Imaging-based strategies have evolved rapidly from detecting a handful of genes to hundreds or thousands and the whole transcriptome, for example, seqFISH44 and MERFISH17 for in-situ RNA hybridization and ISS32 and cPAL33 for in-situ RNA sequencing. However, the general caveats of these methods are that they rely on high-sensitive fluorescence imaging systems and sophisticated image analysis processes. Moreover, they are usually based on probes that hybridize with known mRNA sequences, which can limit the potential to discover new sequences and variants.
For NGS-based methods, the most prominent form is to utilize in-situ barcoded DNA arrays to capture the spatial location of mRNA transcripts. Such an array can be manufactured by commercial microarray slide12, beads45, Illumina flow cells18, or polymerase colonies46. Two commercial platforms, the Visium system (based on ST12) and Stereo-seq47, are available for general use. However, the high cost of these products restricts its access to general laboratories. Several laboratory stage techniques such as slide-seq V245, HDST24, and seq-scope18 have comparable resolutions to the commercial products48, but DNA barcode location needs to be determined by pre-sequencing steps, which is inaccessible to most research laboratories.
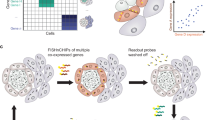
As for the third group, the region of interest of the known spatial locations and tissue shapes can be obtained by physical isolation or optical making. While microdissection technologies such as LCM-seq49 and GEO-seq50,51 are flexible for selecting interested tissue regions, they require complex instrumentation. Nanostring’s commercial optical ROI technology, Digital Spatial Profiler (DSP)43, is based on probes that hybridize with known mRNA sequences, which limits its application. To overcome this, non-probe-based approaches such as PIC52 and Light-seq53 are used to obtain gene expression in spatially defined areas. However, they can only analyze a region of interest or need to synthesize complex light-sensitive compounds for reaction, which is poor in applicability. Here, we present a unique approach of photo-irradiation for in-situ barcoding hybridization and ligation sequencing, named PBHL-seq, which can perform whole transcriptome analysis from the region of interest in frozen and FFPE tissue without sample destruction. This is achieved by two key steps illustrated in Fig. 1. Firstly, we take advantage of a photo-caged reverse transcription primer which enables control delivery of barcoded adapters to different ROI via ligation. Secondly, we collect the double cDNA in a non-destructive way and employ in vitro transcription (IVT) to convert barcoded cDNAs to sequencing libraries. We combine photo-controlled barcoding and collection double-strand cDNA strategy to make it possible to analyze larger areas of tissue such as mouse embryos at E15.5 ( ~ 70.8 mm2). As the double cDNA is collected from tissue in a non-destructive way, the section can be revisited for further assays, e.g., morphology or protein labeling. As the proof of concept, we utilize PBHL-seq to identify cell types from mixed cell cultures. We also demonstrate the application on frozen and FFPE mouse embryo sections. Finally, we show that PBHL-seq can identify genes uniquely expressed in the nuclear compartment.

PBHL-seq enables selective barcoding of user-selected ROIs from frozen and FFPE tissue sections for transcriptome sequencing. Step (1): In situ RT with NPOM-caged primer is performed. Before photo irradiation, target ROIs can be selected based on phenotypic factors including spatial location, morphology, and protein biomarkers. The user selection allows large or small regions and contiguous or disjointed cell groups to be flexibly labeled by photo-irradiation and in-situ hybridization and ligation of DNA barcodes, which are then converted into sequenceable indices. The process can be re-iterated using different barcode sets for multiplexed targeting of different regions. Step (2): After photo-irradiation labeling, double-strand cDNAs are synthesized and released from the section, and sequencing libraries are prepared. Optionally, the same sample can be revisited after sequencing to perform follow-up assays, such as high-resolution imaging, morphology, or protein labeling. Different shapes: different ROIs. ROIs regions of interest, RT reverse transcription, IVT in vitro transcription, NPOM eight 6-nitropiperonyloxylmethyl, RT-PCR reverse transcription-polymerase chain reaction, NGS next-generation sequencing, IF immunofluorescence, HE hematoxylin-eosin staining.
Results
PBHL-seq overview
To understand the spatial organization and cell-specific gene expression, we attempt to develop a profiling method to obtain spatial resolution transcriptomic profiles from optically selected areas. The method, termed PBHL-seq, employs photo-caged oligodeoxynucleotides (ODNs) in response to photo-irradiation to attach DNA barcodes to cDNAs from multiple regions of interest in frozen and FFPE samples. Experimentally, in situ reverse transcription (RT) is performed on tissue sections with caged RT primer containing photo caged-ODNs. We employ a conventional fluorescence microscope equipped with a special Mosaic 3 system to selectively illuminate ROI based on phenotypic parameters such as spatial location and morphology, and protein biomarkers. Upon photo-irradiation, the caged moieties on RT primer are cleaved from the 1st strand cDNA. Only the uncaged ODNs can be hybridized and ligated to a barcode adapter, which contains an upstream T7 promoter for linear amplification of cDNA (termed in vitro transcription or IVT54). In this design, only the gene expression of photo-irradiation ROIs can be detected.
The most critical step in this experimental design is the suppression of cDNA amplification from nonirradiated regions. A caged RT primer with eight 6-nitropiperonyloxylmethyl (NPOM) conjugated thymidine upstream of dT (for mRNA capture) and UMI (unique molecular identifier) sequence is employed, which is unable to base-pair42,55,56 with barcode adaptor. Upon local illumination with 365 nm UV light, the photo-caged ODNs are released. A T7 promoter primer containing a partially double-stranded DNA adaptor, which is complementary to the NPOM-modified ODNs, is applied to the section and T4 ligase is used to attach the barcode adaptor to cDNA within the area of photo illumination. In areas not illuminated by UV, the NPOM moiety fails to base pair and ligate to the adaptor52. Thus, these minimize cDNA amplification from non-irradiated areas.
To set up the PBHL-seq platform, we use a digital micromirror device (DMD)57 attached to a standard widefield imaging setup to custom-select ROIs. Using a ×20 objective, the system can yield a theoretical resolution of about 2 µm. We optimize the mosaic system exposure time and light intensity and find that 10 s exposure time and 100% illumination intensity produce the most efficient uncaging of ODNs (Supplement Fig. 1). We first validate the photo-controlled barcoding strategy in vitro on a Codelink glass surface immobilized with NPOM-caged DNA strands (Fig. 2A). We select a geometry for photo-irradiation on the glass surface and hybridize a fluorescently labeled barcode adapter that can base-pair with NPOM-uncaged DNA strands. Indeed, we can pattern multiple regions on the slide, such as the two-color geometry (Fig. 2B).

A Schematic for PBHL-directed barcode attachment on glass slides. Amino-modified single-stranded poly A oligos are immobilized onto Codelink glass surfaces and NPOM-caged oligodeoxynucleotides are hybridized. Target pixels corresponding to ROIs in the field of view are photo-illuminated using the mosaic system by the selected pattern. Fluorescent barcode strands are hybridized to the uncaged barcode oligos. Unhybridized strands are removed by washes, which reveals the illuminated area with fluorescence. B Iterative photo-irradiation using two geometry that defines two ROIs to attach two pattern barcode strands onto a DNA-coated glass slide. Scale bar, 50 μm.
Establishment of PBHL-seq for ROI expression analysis
To use PBHL-seq for spatial transcriptomics, we first perform an in-situ RT reaction on fixed culture cell or tissue sections (Fig. 3A, step 1) with NPOM-caged RT primer to synthesize 1st strand cDNA (Fig. 3A, step 2). The RT primer contains the NPOM-caged hybridization arm, UMI for transcript quantification, and poly dT for mRNA capture. We then photo-irradiate the ROI to uncaged the NPOM-caged ODNs (Fig. 3A, step 3), which can hybridize and ligate to the barcode adapter (Fig. 3A, step 4 and Supplement Table 1). The barcode adapter consists T7 promoter, PCR handle, barcode, and ODNs complementary with the NPOM-caged ODNs on RT primer. This process can be repeated to label multiple ROIs with different barcode adapters (Fig. 3A, steps 5, 6). After all barcoding rounds are completed, second-strand cDNA synthesis is performed, and double-stranded cDNA is collected from the section (Supplement Fig. 2A). Among these, only NPOM-uncaged DNA strands can generate double-strand cDNA containing T7 promoter from the ligated barcode adaptor, which is used for cDNA linear amplification with in vitro transcription. Besides, this process leaves the tissue section intact for further analysis (Fig. 3A, steps 7, 8).

A The PBHL-seq workflow for in situ transcriptomic sequencing: (1) The sample is fixed and permeabilized. (2) In-situ RT is performed on fixed samples with primers containing NPOM-caged hybridization arm, unique molecular identifier (UMI), and dT oligo. (3–6) Within each ROI, a unique DNA barcode adaptor with T7 promoter, PCR handle, barcode N, and hybridization arm is hybridized and ligated to uncage-RT primers after UV illumination. (7) Double-strand is synthesized on the sample section, afterward, the double-strand cDNA is collected in an EP tube. (8) IVT, RT, and PCR amplification are performed. The sequencing library is prepared. NPOM eight 6-nitropiperonyloxylmethyl, ODNs oligodeoxynucleotides, RT reverse transcription, ROI region of interest, T7 T7 promoter, T7 Pol T7 polymerase, UMI unique molecular identifier, IVT in vitro transcription, RT-PCR reverse transcription-polymerase chain reaction. B Efficiency of hybridization by different hybridization buffers with multiplex analysis for ROIs. The ligation buffer contains 2×SSC, 10% formamide, 10% dextran sulfate sodium salt, Recombinant RNase Inhibitor, and primer. The tris buffer contains 0.3 M NaCl, 20 mM Tris-HCl pH 7.4, and 0.2% Tween 20. SSC buffer: Saline Sodium Citrate buffer. C, D Background from nonirradiated cells is performed with human-mouse mixed cultures. C DAPI staining marks the nuclei position of 293T and 3T3 cells (blue). Scale bar, 1000 μm. E Sequentially fluorescent labeling of multi-ROIs. Scale bar, 50 μm. F, G, H Cell mixing experiments with human 293T and mouse 3T3 cells show little species mixing. G After the second strand synthesis, the sample is stained by DRAQ5 (blue camouflage color). A subset of ~60 3T3 cells and ~80 293T cells are barcoded in the whole well (n = 3 biological replicate, representative image shown). Scale bar, 250 μm. I Illustration of the experiments for DMD-assisted UV irradiation of 1, 10, ~100, and ~1000 cells. Scale bar, 250 μm. J, K Boxplot of numbers of gene-assigned UMIs and detected genes. Midline marks the median and edges indicate the 25th and 75th percentiles, and the meter shape represents the average value.
Firstly, for multiple ROIs analysis, it is essential to avoid the barcode cross-over from later rounds of barcoding. To demonstrate the specificity of PBHL-seq, we perform a barcode mixing experiment with two barcode adaptors, A1 and A2, on cultured cells. We first illuminate an ROI and ligate the A1 adaptor. Then, A1 is washed away and the A2 adaptor is applied to the section. In the resulting cDNA library, A2 barcode-derived genic reads consist of 1.75% ± 0.96% (mean ± s.d., n = 4 biological replicates, Fig. 3B), suggesting that a ROI area will be nearly completely ligated to a barcode adaptor.
Secondly, the utility of PBHL-seq requires suppression of cDNA amplification from nonirradiated regions. To evaluate the ability of PBHL-seq to detect ROI-specific expression from a larger background area, we separately culture 293T cells (human) and 3T3 cells (mouse), then adhere them onto the same coverslip (Fig. 3C). Only 3T3 cells are photo-irradiated under a fluorescence microscope by a mosaic system. At this point, we ensure that the proportion of no photo-irradiated 293T cells and photo-irradiated 3T3 cells on different coverslips are 100:1, 1000:1, and 10,000:1, and the total number of 293T cells and 3T3 cells is similar. After irradiation, we hybridize and ligate the barcode adapter on the cell coverslip. In the cDNA library, the human genic reads rate is 0.12% ± 0.26%, when the proportion of no photo-irradiated 293T cells to photo-irradiated 3T3 cells is 100:1 (mean ± s.d., n = 3 biological replicates, Fig. 3D). Even when the proportion increases to 10,000:1, the human genic reads rate is still negligible, 0.43% ± 0.30% (mean ± s.d., n = 3 biological replicates, Fig. 3D). Thus, we conclude that PBHL-seq is highly specific for detecting gene expression in illuminated areas.
Thirdly, we set to test the multiplexing ability of PBHL-seq to label multi-ROIs. We first confirm the efficiency of photo-irradiation on cells with fluorescently labeled barcode adaptors. 3T3 cells are fixed, permeabilized, and then performed in situ RT with NPOM-caged ODNs. We then illuminate a small patch of cells with a mosaic system and add fluorescently labeled barcode strands that can base-pair with NPOM-uncaged ODNs. Indeed, we can label two areas on the cell coverslip with two fluorescent barcode strands (Fig. 3E). Next, we perform a cell mixing experiment with 3T3 and 293T cells co-cultured on the coverslip (Fig. 3F). Based on the distinct morphology of the two cell types, we photo-illuminate ~60 mouse cells and ~80 human cells for sequential ligation barcoding (Fig. 3G and Supplement Table 2). We observe the majority of species-specific reads correspond to their labeled barcodes (97.6% ± 0.43% of mouse reads, 95.1% ± 3.39% of human reads, n = 3 biological replicates, mean ± s.d., Fig. 3H), which is higher than Light-seq (89.1% ± 0.7% of mouse reads, 87.3% ± 0.7% of human reads, n = 3 biological replicates, mean ± s.d.)53. In addition, Normalized gene expression levels expressed as log2-transformed transcripts per kilobase per million reads (TPM) display good correlation across biological replicates (Pearson correlation coefficient >0.7) and the top 200 expressed genes correlate highly across biological replicates (Pearson correlation coefficient >0.8, Supplement Fig. 2B, C), which illustrates that the expression profile across biological replicate has good correlation with PBHL-seq. Lastly, after dcDNA is collected, the coverslip cell is then used for immunofluorescence (IF) with anti-tubulin antibody and nuclear staining to demonstrate the integrity of the sample. (Supplement Fig. 2D).
Lastly, we provide quantitative validation for wider acceptance and application. Four thousand 3T3 cells are seeded onto the coverslip and subject to in situ RT with NPOM-caged ODNs. Then 1, 10, ~100, and ~1000 cells are photo-irradiated and ligated different barcodes on the same coverslip (Fig. 3I). We find the average UMI counts are 4.6 × 103, 2.0 × 105, 3.0 × 105, and 4.0 × 105 for 1, 10, ~100, or ~1000 cells, respectively (n = 7 biological replicate, Fig. 3J), which demonstrates linearity between the detected UMIs and the number of cells (p = 3.633e−16, R2 = 0.9852, student’s t-test). The numbers of genes detected in 1, 10, ~100, or ~1000 cells are 2529 ± 171, 11,060 ± 201, 11,742 ± 190, and 12,412 ± 150 genes in replicate averages (n = 7 biological replicate, mean ± s.d., Fig. 3K), which is still a linear correlation (p = 9.209e−32, R2 = 0.9983, student’s t-test). Therefore, the number of cells labeled with different barcodes shows a good correlation with the sequencing results, demonstrating that this method truly reflects the transcriptome information in the irradiated region samples.
Spatial sequencing with PBHL-seq in frozen sections
RNA sequencing of specific cell populations within tissue samples remains challenging, especially when tissue samples are large. Therefore, we test PBHL-seq on frozen sections from mouse embryos at E14.5 to select ROIs based on morphological identification. First, in-situ RT is performed in a fixed 10 µm sagittal section of the mouse embryo. We then arbitrarily select two different regions around the eye to label different fluorescent barcodes base-pairing with NPOM-uncaged ODNs after photo-irradiation, which can label different regions with two fluorescent barcode strands on the same view, leaving the intact sample for DAPI dye (Supplement Fig. 4A). Another, We select two different regions of the eye and viscus to perform photo-irradiation and ligation barcoding (Supplement Fig. 3A). For the sequencing results, we first calculate the sensitivity of PBHL-seq. At a sequencing depth of 9 million reads per biological replicate, we observe an average of 14,781 ± 156 genes and 9576 ± 432 genes for ROIs of the eye (150 µm x 150 µm) and viscus (80 µm x 80 µm) (n = 3 biological replicate, mean ± s.d., Supplement Fig. 3B) and an average of 1506 ± 77 UMIs and 594 ± 60 UMIs per 100 µm2 area for eye and viscus, respectively (n = 3 biological replicate, mean±s.d., Supplement Fig. 3C), whose sensitivity is correlated with that of DBiT-seq58 (~5000 UMIs, mouse embryo), Slide-SeqV245 (500–1000 UMIs, mouse embryo). Replicates show consistent read filtering throughout the sequence-processing pipeline (Supplement Fig. 4B) and are well correlated based on principal component analysis (PCA) for each region (Supplement Fig. 3D). What’s more, the correlation of the same tissue region is high and the correlation between the eye and viscus is low (Supplement Fig. 4C). We discover 3,691 genes with significant differential expression between eye and viscus by volcano plots (Supplement Fig. 3E) and heatmap (Supplement Fig. 4D). Three known genes are identified within the optic vesicle with spatially correlated expression patterns only at the eye region. Pax6 is expressed in region-specific neural progenitors in the neural tube, which lines the optic vesicle and optic stalk, and Pmel is observed around the optic vesicle which is a pigment cell-specific gene involved in developing fibrillar sheets, and Six6 is a gene known for specification and proliferation of retinal cells in vertebrate embryos which is mainly lined within the optical vesicle59. Similarly, different expression genes (DEGs) are identified for the viscus region. Apoa2, which encodes apolipoprotein E, is a liver-specific gene60. Alb, is a marker gene of hepatoblasts cells, and Mt1 enables copper ion binding activity and acts upstream of or within several processes, which is broadly expressed in liver E14.561. What’s more, twenty-four differential expression genes with known spatial locations including the above six genes are shown with heatmap (Supplement Fig. 4E) and ISH experiments from EMAP home web (EMAGE home (emouseatlas.org)) demonstrate that the expression levels of these genes are indeed different in the eye and viscus part of the mouse embryo. Thus, PBHL-seq can detect region-specific gene expression in mouse embryo tissue sections.
To demonstrate the ability of PBHL-seq to perform transcriptome analysis on more regions of interest and closer proximity, we analyze the CA1, CA3, DG (dentate gyrus), and RSP (retrosplenial area) regions of frozen mouse brain tissue, which is the most complex organ in the body and have different cell types and functions in different regions. A 10 µm coronal section of the mouse brain (containing hippocampus) is performed PBHL-seq and four biological biological replicates are performed for each region. Before selecting the region of interest, the tissue is nuclear stained with DRAQ5 to give it a more pronounced histological appearance. Afterward, the CA1, CA3, DG, and RSP regions are manually selected based on mouse brain histomorphology (Interactive.Atlas.Viewer:Atlas.Viewer.(brain-map.org)) and illuminated (Fig. 4A, blue pseudo-color). At the sequencing depth of 20 million reads, we detect the average number of genes in the CA1 (80 µm × 80 µm), CA3 (60 µm × 60 µm), DG (60 µm × 60 µm), and RSP (80 µm × 80 µm) regions is 7509 ± 221, 4978 ± 347, 6701 ± 366 and 7003 ± 330, respectively (mean ± s.d., n = 4 biological replicate, Fig. 4B). Relative to the total irradiated area, the number of UMIs detected in the CA1, CA3, DG, and RSP regions is 766 ± 144, 406 ± 67,771 ± 143 and 785 ± 166 UMIs/100 μm2, respectively (mean ± s.d., n = 4 biological replicate, Fig. 4C), whose sensitivity is correlated with that of PIC52 (2500–5000 UMIs, mouse brain tissue) and Light-seq53 (~1500 UMIs, mouse retinal tissue). PCA indicates that expression profiles are clearly distinct according to the photo-irradiated brain regions (Fig. 4D). We discover 1349 DEGs between CA1 and CA3, 1535 DEGs between CA1 and DG, 1275 DEGs between DG and CA3, 1283 DEGs between CA1 and RSP, 1679 DEGs between DG and RSP, and 1524 DEGs between RSP and CA3 by Volcano plots (Fig. 4E), which include the genes verified by ISH in the mouse brain gene database -Allen Brain Atlas (Brain.Map-brain-map.org) for their specific expression in CA1 (Ociad2, Fibcd1), CA3 (Parp8, Chst1, Dgkb), DG (Lct, Dock10, Trpc6, Ryr1) and RSP (Etv1, Lamp5, Stmn1). Thus, PBHL-seq can detect region-specific gene expression in freshly frozen mouse brain tissue slices with closer spatial proximity and more regions of interest.

A Four regions of the mouse brain are uniquely barcoded for sequencing. Scale bar, 1000 µm. B, C Boxplot of numbers of detected genes (B, n = 4 biological replicate) and Unit-area UMIs for each ROI (C, n = 4 biological replicate). Midline marks the median and edges indicate the 25th and 75th percentiles, and the x mark represents the average value. D Two-dimensional principal component analysis (PCA) for the expression profiling is shown. E Volcano plots of differentially expressed genes, with selecting markers labeled. The x and y axes show the log2(fold-change) and the −log10(P-value), respectively.
Spatial sequencing with PBHL-seq in FFPE tissue sections
Many clinical samples are formalin-fixed and paraffin-embedded (FFPE) for a high degree of preservation of morphological detail and long-term storage62. However, due to formalin-mediated strand cleavage and the formation of crosslinks between RNA and other biomolecules, formalin fixation negatively affects nucleic acid integrity and accessibility63, which hampers the development of sequencing-based spatial transcriptomic method64. Currently, spatial mRNA profile strategies have been developed for application on FFPE samples, such as DBiT-seq65 and Visium for FFPE tissue66. However, the DSP43 platform remains the only method for ROIs unbiased spatial mRNA profiling of FFPE tissue but it relies on gene probe hybridization. We seek to test the applicability of PBHL-seq to recover spatial mRNA profiles from FFPE tissue sections.
Recovery of fixed mRNA for spatial FFPE tissue analysis is achieved by removing paraffin and cross-links in situ. Firstly, the FFPE tissue section on a slide is deparaffinized by successive immersions in xylene and ethanol, which is a deparaffinization strategy commonly used in immunohistochemistry (IHC). Secondly, the tissue sections are pre-permeabilized with collagenase followed by cross-link reversal. Collagenase is used in early spatial transcriptomics protocols to aid in the disruption of the extracellular matrix structure67. Cross-link reversal is performed at 70 °C with Tris-EDTA buffer at pH 8.0, by which the sequencing data do not show any major differences66. Finally, the tissue sections are enzymatically permeabilized, at which point the section is ready for PBHL-seq.
We apply PBHL-seq to profile sagittal FFPE sections from embryonic day 15.5 (E15.5) mouse embryos (C57BL/6N). Two domains, the eye field and the viscus, of the same mouse embryo section are independently photo-irradiated (eye and viscus sites; n = 3 biological replicate, Fig. 5A). The double-strand cDNAs are collected and libraries are prepared and sequenced to about 7.0 × 106 reads per sample. We detect an average of 12,843 ± 47 genes and 4151 ± 114 genes from the eye (250 µm × 250 µm) and viscus (80 µm × 80 µm) ROI (n = 3 biological replicate, mean±s.d., Fig. 5B), with the numbers of UMIs relative to the total irradiated areas being 203 ± 2 and 129 ± 3 UMIs/100 μm2 for eye and viscus, respectively (n = 3 biological replicate, mean ± s.d., Fig. 5C). We next validate whether spatially specific gene expression profiles are consistent with their embryonic origin. Two-dimensional PCA indicates that the expression profiles are distinct between two photo-irradiated embryo regions (Fig. 5D). Additionally, the correlation of the same region is high and the correlation of different regions is low (Supplement Fig. 5A) for three biological replicates. We discover 2241 genes with significant differential expression between the eye and viscus (Fig. 5E and Supplement Fig. 5C), including known genes such as Pax6 and Pmel for the eye region and Mt1, Apoa2, and Alb for the visceral region. What’s more, twenty-six differential expression genes with known spatial locations (EMAGE home (emouseatlas.org)) including the above five genes are shown with a heatmap (Supplement Fig. 5B). Thus, PBHL-seq provides an accessible workflow for genome-wide gene expression with multiple ROI in mouse embryo FFPE tissue.

A Two ROIs on the mouse embryo section, the eye, and viscus, are uniquely barcoded with PBHL-seq. Scale bar, 500 μm. B, C Boxplot of numbers of detected genes (B, n = 3 biological replicate) and Unit-area UMIs (C, n = 3 biological replicate). The midline marks the median and the edges indicate the 25th and 75th percentiles, and the x mark represents the average value. D Two-dimensional PCA for ROI expression profiling is shown. E Heatmap of twenty-two genes with known differential expression between eye and viscus (P-value < 0.05 and |log2(fold-change)| > 1 are shown, n = 3 biological replicate). E Volcano plots of differentially expressed genes between the eye and viscus, with only a few better characteristic marker genes labeled. The x and y axes show the log2(fold-change) and the log10(P-value), respectively.
PBHL-seq for subcellular microstructures
Many RNAs display subcellular localization that correlates with their function68,69,70. In addition, gene expression analysis of subcellular structures is essential for understanding a wide range of biological processes17,19, e.g., synaptic RNA localization in dendritic spines. Imaging-based techniques such as seqFISH, MERFISH, FISSEQ, and ExSeq can determine the subcellular location of transcripts14,17,19,71. We want to explore the feasibility of PBHL-seq for such an assay.
We use the Hela cell line and attempt to detect RNAs in the nucleus and cytoplasm. Firstly, to verify that subcellular structures can be specifically labeled, we distinguish the two regions by subsequent irradiation and ligation of different fluorescence barcodes (Fig. 6A). Then, we perform PBHL-seq for nuclear and cytoplasmic areas to profile transcripts. After in-situ RT, cells are stained for anti-tubulin antibody for cytosol and DRAQ5 for nuclei to distinguish the two areas. We then photo-irradiate the two areas for about 1000 nuclei areas (labeled with DRAQ5, ~10 μm with diameter) and about 2000 cytosol areas (stained with anti-tubulin antibody, ~5 μm with diameter) (only part is shown in Fig. 6B). The double-strand cDNAs are collected and libraries are prepared and sequenced to about 6.0 × 106 reads per sample. We detect over 10,000 genes with the numbers of UMIs relative to the total irradiated areas being 450 ± 47 UMIs/100 μm2 for cytoplasm and 402 ± 100 UMIs/100 μm2 for nucleus (n = 4 biological replicate, mean ± s.d., Fig. 6C, D). Two-dimensional PCA indicates that the expression profiles are distinct between the nucleus and cytoplasm (Fig. 6E). Additionally, a total of 7336 differentially expressed genes are detected (Fig. 6F) for their specific expression in the cytoplasm and nucleus, which include the genes verified in the human gene database (GeneCards-Human.Genes.Gene.Database.Gen.Search) for their specific expression in the nucleus (MLAT1 and NEAT1) and the cytoplasm (GAPDH) (Fig. 6F). Another, to further validate the differential genes identified in the cytoplasm (3809 DEGs) and nucleus (3527 DEGs), Hela cells are partitioned into standard nuclei and cytoplasm using the Cytoplasmic & Nuclear RNA Purification Kit72. The efficacy of the separation is confirmed to be superior (Supplement Fig. 6A). Subsequently, RNA-seq and differential expression analysis are conducted on the standard nuclei and cytoplasm. The Venn diagram (Supplement Fig. 6B) and upset plot (Supplement Fig. 6C) analysis reveals that 80% of the DEGs in the nucleus are present in the standard nucleus, while 82% of the DEGs in the cytoplasm are found in the standard cytoplasm. These results demonstrate the applicability of PBHL-seq for subcellular localization.

A Two regions of Hela cells are uniquely fluorescent barcoded with ROX and FAM dye primers. Scale bar, 25 μm. B Images of green immunofluorescence (IF) for cytoplasm and red DRAQ5 for nucleus. The UV irradiation part region is shown with cycles. Scale bar, 10 μm. C, D Boxplot of numbers of detected genes (C, n = 4 biological replicate) and Unit-area UMIs (D, n = 4 biological replicate). Midline marks the median and edges indicate the 25th and 75th percentiles, and the x mark represents the average value. E Two-dimensional PCA for the expression profiling is shown. F Volcano plots of differentially expressed genes between the nucleus and cytoplasm, with selecting markers labeled. The x and y axes show the log (fold-change) and the -log10 (P-value), respectively.
The small number of cells transcriptomics in tissue with PBHL-seq
Only a small number of cells of interest in complex spatial contexts are captured for transcriptomes remains a major challenge. PBHL-seq is used to analyze mouse embryonic retinal pigment epithelial (RPE) cells, which consist of a mono-cell layer in the eye cup (Fig. 7A). We selectively photo-illuminated ~60 RPE cells identified with melanin deposits in the eye region (Fig. 7A, HE staining, arrow). To test the ability of PBHL-seq to distinguish RPE gene expression from neighboring neural retina (NR), we also illuminate 120 µm × 120 µm square region in the neural retina region. We detect over 10,000 genes with the numbers of UMIs being 774 ± 99 UMIs/100 μm2 for the neural retina region and 817 ± 109 UMIs/cell for RPE cells (n = 4 biological replicate, mean ± s.d., Fig. 7B, C) with 1.2 × 107 reads per sample. The correlation of the same tissue region is high whereas the correlation between the RPE cells and neural retina region is low (Fig. 7D). What’s more, the PCA result shows that expression profiles are clearly distinct according to the photo-irradiated eye regions (Fig. 7E). We discover 3511 genes with significant differential expression between RPE cells and neural retina by volcano plots (Fig. 7F). These DEGs include RPE marker genes, such as Tyr, Mitf and involved in pigment synthesis genes, such as Tyrp1, Rdh5, Mertk, Igf2r, Serpinf1, Bace2, Nid2, Dct, Lypd1, Cfh, Ihh, Adgre5, Axl, H1973,74. Thus, we conclude that PBHL-seq can detect a small number of gene expressions in complex tissue.

A Two ROIs on the mouse embryo section, retinal pigment epithelial (RPE) cells (show part photo-illuminate cells) and neural retina (NR), are uniquely barcoded with PBHL-seq. The scale bar of the mouse embryo is 500 μm. In particular, the scale bar of zoomed-in images is 25 μm. B, C Boxplot of numbers of detected genes (B, n = 4 biological replicate) and Unit-area UMIs (C, n = 4 biological replicate). The midline marks the median and edges indicate the 25th and 75th percentiles, and the x mark represents the average value. D The correction of different replications (n = 4 biological replicate). E Two-dimensional PCA for ROI expression profiling is shown. F Volcano plots of differentially expressed genes between the eye and viscus, with only a few better characteristic marker genes labeled. The x and y axes show the log2(fold-change) and the log10(P-value), respectively.
Discussion
In recent years, array-based spatial transcriptome technologies such as the 10× Genomics Visium and Stereo-seq47 greatly expanded our knowledge of positional gene expression information in intact tissue. In contrast, these methods indiscriminately obtain complete spatial expression information of the whole tissue. Targeted transcriptome profiling of regions of interest, traditionally done by laser microdissection, allows precise interrogation of targeted cell populations only. To overcome the limitation of LCM, which requires physical isolation and destruction of the sample, we developed PBHL-seq to detect spatial gene expression in specific areas. In terms of cost, PBHL-seq requires only the commercially available NPOM-caged oligos, which costs only a few dollars per section labeling reaction. Whereas other methods like commercial DSP43 costs several thousand dollars per reaction. In terms of robustness, PBHL-seq uses in situ reverse transcription followed by spatially targeted barcoding of cDNAs to create spatially indexed transcriptomes of photo-illuminated regions, which routinely recovers ~1000 per 10 × 10 µm2 unit area in fresh frozen tissues, comparable to Slide-SeqV245 (500–1000 UMIs, mouse embryo), PIC52 (2500–5000 UMIs, mouse brain tissue) and Light-seq53 (~1500 UMIs, mouse retinal tissue). In terms of throughput, we demonstrate sequential labeling of four ROI on a single slide, and could easily expand to tens of areas. In contrast, PIC52 can only detect single regions. In addition, PBHL-seq has additional advantages following (1) PBHL-seq is not based on probe contrast to DSP43; (2) PBHL-seq can work on both OCT-embedded tissue and FFPE tissue; (3) PBHL-seq maintains tissue morphology and allows for multiple follow-up analyses; (4) The PBHL-seq method can compatible with multi-omics spatial profiling of ROI, such as ATAC, CUT&Tag, protein, etc.
PBHL-seq can detect spatially heterogeneous cells in frozen tissues and FFPE tissue with areas expandable to centimeter scale, such as mouse embryo E15.5 (70.8 mm2), far greater than the typical capture area of array-based methods. At the same time, it can resolve subcellular areas in μm scale, as it is allowed by the Mosaic illumination system. Meanwhile, the use of barcodes allows for labeling multiple regions of interest on the same sample and in a single sequencing library preparation. For the further development of spatial transcriptomics, it will be useful in the future to benchmark and compare with various techniques at the single-cell level.
Another PBHL-directed barcoding permits precise labeling of individual cells within the dense tissue environment, but some light-scattering can induce out-of-ROI crosslinking, which effect is mitigated by drawing photo-illuminated region slightly smaller than the intended ROIs inside the cellular boundary. Further, a laser-based point-scanning microscope offers higher barcoding resolution and may be used in place of a DMD in the future. We expect that PBHL-seq’s sensitivity can be further improved with optimization of the in-situ RT, barcoding hybridization and ligation, design of barcode, use of targeted ISH probes, and targeted ribosomal RNA depletion. Although the spatial resolution of PBHL-seq is up to single cells and subcellular levels, we think it is possible to ultimately enable the profiling of organelles by improvement of photo-irradiation.
The PBHL-seq method is also compatible with genome-wide level or protein-level spatial profiling of ROI. We envision that by using in situ ATAC or CUT&Tag with an NPOM caged Tn5 transposome. Our method would allow one to achieve epigenetic profiling of multiple ROI on tissue slides. Similarly, various protein levels can be measured with labeled antibodies, such as CITE-seq75, Abseq76, and LIBRA-seq77, which commonly use ODNs as an index material, and caged ODNs provide the advantage of being able to amplify sequence libraries only from photo-irradiated ROIs. What’s more, the PBHL-seq technology can be used to analyze spatial multip-omics. Simultaneous analysis of such ROI will be enabled by the combination of ODNs caged with NPOM or/and other caging groups, which have different wavelength-selectivity78,79,80. Different caging groups have unique barcodes for labeling. After the in-situ RT with such mixed ODNs, multi-color irradiation, and barcode sequencing will separate the sample information. Thus, PBHL-seq will be able to determine epigenetic landscapes and protein landscapes as well as expression profiles in an ROI-specific manner.
Methods
Cell culture
NIH/3T3 (ATCC, CRL-1658) and HEK/293T cells (ATCC, CRL-3216) were cultured at 37 °C in a humidified 5% CO2 atmosphere, in high-glucose DMEM (Gibco, 11965092) supplemented with 10% calf serum (Gibco, 10270-106), 1% penicillin/streptomycin (Nacalai Tesque, 0503), and 1% l-glutamine (Nacalai Tesque, 16948-04). HeLa (ATCC, CCL-2; Cell lines were tested for mycoplasma by the manufacturer before shipment and none of the cell lines used were authenticated) cells for verifying subcellular applications with PBHL-seq were cultured in the same conditions. For cell preparation, Poly-l-lysine-coated chambers were coated with sterile PDL (Sigma Aldrich, P6407) at 0.3 mg/ml overnight at 4 °C. Afterwards, the chamber was dried for 1 h, washed with UltraPure water (Invitrogen, 10977023). Chambers were then seeded with cells per well and placed in an incubator (37 °C with 5% CO2) overnight for sequencing or fluorescent analysis.
Tissue handling
The study was approved by the Animal Care and Use Committee of Ocean University of China. These C57BL/6 mice were purchased from Shandong First Medical University & Shandong Academy of Medical Sciences. We have complied with all relevant ethical regulations for animal use. We used wild-type adult C57BL/6 mice in this study and this study did not involve wild animals. Adult pregnant C57BL/6J mice (6–8 weeks) were sacrificed on the day of arrival and collected the embryo (E 14.5 days) and adult C57BL/6J mice (6–8 weeks, sex was not a relevant variable in our analysis, but the randomly selected mice were female) were sacrificed and collected the brain. The freshly dissected embryos and brain were immersed into OCT (SAKURA; 4583) snapped frozen with dry ice and directly kept at −80 °C for long-time storage. The frozen tissue block was warmed to −20 °C in a cryostat (Leica, CM3050S) for 60 min before sectioning. The tissue block was then sliced at a 5° cutting angle and sectioned at a thickness of 10 µm on a cryostat. For FFPE tissue, embryos (E 15.5 day) were collected, and immediately fixed by 3.7% PFA, then made to standard paraffin embed and sectioned at a thickness of 7 µm onto a poly-Llysine coated glass slide.
Caged ODNs
NPOM-caged ODNs (RT primer caged NPOM) were purchased from Bio-Synthesis (Supplement Table 1). The synthesized caged ODNs were shielded from the light during transport. After receipt, a solution of caged ODNs was immediately aliquoted into single-use volumes and freeze-stored in a light-shielded box.
Deparaffinization of FFPE tissue section
Deparaffinization was performed by two times washing with Xylene (VWR, 28975.291) for 15 min each. To remove the remaining xylene, the section was washed for 2 min with 99% ethanol (VWR, 84835.290) two times. Tissue was then rehydrated by immersing in 96% ethanol (VWR, 20823.290) and 70% ethanol (VWR, 97064-768) for 2 min each by two times, and finally placed in RNase-free water for 5 min.
Permeabilization of FFPE tissue section
For FFPE samples, a collagenase mix (98.6 μl HBSS buffer (Life Technologies, 14025-050), 1 μl BSA (Bionordika, B9000S), 0.4 μl collagenase I (50 U/μl, Life Technologies, 17018-029)) was equilibrated to 37 °C and then were added to a tissue section. The slides were sealed and incubated for 20 min at 37 °C. Once the incubation was complete, the collagenase mix was pipetted off and the slides were washed with 100 μl of 0.1× SSC buffer (Sigma-Aldrich, 93017), which is diluted in RNase DNase-free water. Subsequently, 100 μl TE buffer pH 8.0 (ThermoFisher, AM9849) was added, and the slides were sealed and incubated for 1 h at 70 °C. After the incubation, the slides were left to equilibrate at room temperature for 5 min. Meanwhile, 0.1% pepsin solution (Sigma-Aldrich, P7000-25G) is dissolved in 0.1 M HCl (Sigma-Aldrich, 318965-1000M) and was equilibrated to 37 °C, then 75 μl of 0.1% pepsin solution were added to the tissue section for 30 min at 37 °C. After this step, 0.1× SSC buffer was added to wash the pepsin solution. The permeabilization of the FFPE tissue section was performed PBHL-seq protocol, except Fixation and permeabilization step.
PBHL-seq protocol for sequencing library
Fixation and permeabilization of the tissue section
Cells on the coverslip or frozen tissue sections were washed twice with PBS-diethylpyrocarbonate (DEPC) (Sangon-biotech, B540627-0500) and fixed with 3.7% formaldehyde solution (Sigma-Aldrich, F8775-25ML) in PBS-DEPC for 10 min at room temperature. Specimens were permeabilized with 0.1% TritonX-100 (Sigma Aldrich, T8787-50ML) in PBS-DEPC for 20 min and then with 0.1 N HCl (Sigma-Aldrich, 318965-1000ML) for 5 min, followed by neutralization with 1 M Tris-HCl, pH 8.0 (Invitrogen, 15568025) for 10 min at room temperature.
In situ RT
Permeabilized specimens were incubated in PBS-DEPC for 5 min at 65 °C and quickly cooled in ice-cold PBS-DEPC. Primer mix [0.5 μl of 100 μM RT primer caged NPOM (Supplement table 1), 0.5 μl of Recombinant RNase Inhibitor (Takara, 2313 A), 5 μl of 20× SSC (Sigma-Aldrich, 93017), 5 μl of formamide (Invitrogen, AM9342), 25 μl 20% dextran sulfate sodium salt (Sigma-Aldrich, D8906) and 14 μl of RNase free water] was added for 30 min. The RT reaction mix [20 μl of 5× RT buffer (Invitrogen, EP0751), 10 μl of 10 mM dNTPs (Sigma-Aldrich, 71004-M), 1 μl of Recombinant RNase Inhibitor (Takara, 2313 A), 1 μl of Maxima H Minus Reverse Transcriptase (Invitrogen, EP0751), and 68 μl of RNase free water) was applied to the specimens, and incubated at 42 °C for 90 min.
Block reaction
To block stripped NPOM of NPOM-caged ODNs, the block hybridization buffer was prepared as follows: 1 μl of 100 μM block adapter (Supplement Table 1), 1 μl of 100 μM linker NPOM (Supplement Table 1), 1 μl of Recombinant RNase Inhibitor (Takara, 2313 A), 10 μl of 20× SSC (Sigma-Aldrich, 93017), 10 μl formamide (Invitrogen, AM9342), 50 μl 20% dextran sulfate sodium salt (Sigma-Aldrich, D8906) and 28 μl of RNase free water, for 30 min. Then the ligation mix [10 μl of T4 DNA ligase buffer (New England Biolabs, B0202SVIAL), 5 μl of T4 DNA ligase (New England Biolabs, M0202LVIAL), and 85 μl of RNase-free water] was performed at 37 °C for 30 min. After this step, the sections were washed with 65% formamide (Invitrogen, AM9342) and PBS two times.
Photo-irradiation
Photo-irradiation of cell cultures and tissue sections for uncaging was performed under a Nikon ECLIPSE Ti2 fluorescence microscope irradiated with an ANDOR Mosaic3 (100% power) through a Nikon objective lens [20×/0.75 Plan-APO] and a Nikon A filter cube at a wavelength of 465–495 nm and a wavelength of 352 nm–402 nm for 10 s, unless otherwise indicated. The target ROIs can be selected based on phenotypic factors including spatial location, morphology, and protein biomarkers.
Hybridization and ligation barcode oligo 1
After photo-irradiation, the first hybridization buffer was prepared as follows: 1 μl of 100 μM T7 read1 bar1 adapter (Supplement table 1), 1 μl of 100 μM linker NPOM (Supplement table 1), 1 μl of Recombinant RNase Inhibitor (Takara, 2313 A), 20 μl of 20× SSC (Sigma-Aldrich, 93017), and 77 μl of RNase free water, for 30 min. Then the ligation mix [10 μl of T4 DNA ligase buffer (New England Biolabs, B0202SVIAL), 5 μl of T4 DNA ligase (New England Biolabs, M0202LVIAL), and 85 μl of RNase-free water] was performed at 37 °C for 30 min.
Iterative barcoding rounds
Different ROIs were photo-irradiated for unique barcodes, following as above Photo-irradiation and Hybridization and ligation barcode oligo.
Second-strand DNA synthesis and collection
A second-strand mix [10 μl of NEBNext® Second Strand Synthesis Reaction Buffer (New England Biolabs, E6113AAVIAL), 2.5 μl of NEBNext® Second Strand Synthesis Enzyme Mix (New England Biolabs, E6112AAVIAL), and 42.5 μl of RNase-free water] was applied to the specimens and incubated for 2 h at 16 °C, then was collected to EP tube. The collected double-stranded cDNA solution was purified with 1.8 × Speedbead magnetic carboxylate (Cytiva, 17357672) and eluted with 6 μl of 10 mM Tris-HCl, pH 8.0.
IVT
The eluted double-stranded cDNAs (6 μl) were combined with IVT mix [2 μl each of A/G/C/UTP solution, 4 μl of 10× T7 reaction buffer, and 2 μl of T7 enzyme from the TranscriptAid T7 High Yield Transcription Kit (Invitrogen, K0441)] and incubated for 13 h at 37 °C. 3 μl of Exonuclease I (New England Biolabs, M0293L) and 3 μl of Shrimp Alkaline Phosphatase (rSAP) (New England Biolabs, M0371L) were added and incubated for 15 min at 37 °C, afterward, 2.6 μl of fragmentation buffer (Invitrogen, AM8740) was added and incubated for 15 min at 70 °C. After adding 2.86 μl of stop buffer, the aRNAs were purified using 3× Speedbead magnetic carboxylate (Cytiva, 17357672) and eluted with 7 μl of 10 mM Tris-HCl, pH 8.0.
Library preparation and sequencing
The eluted aRNAs (7 μl) were combined with RT primer mix [1 μl of 10 μM random RT primer (Supplement table 1) and of 10 mM dNTPs (Sigma-Aldrich, 71004-M)], and incubated for 5 min at 65 °C and then quickly put in ice. The RT reaction mix was then added [3 μl of 5× RT Buffer (Invitrogen, EP0751), 0.1 μl of Recombinant RNase Inhibitor (Takara, 2313 A), 0.2 μl of Maxima H Minus Reverse Transcriptase (Invitrogen, EP0751), and 2.7 μl of Rnase free water], incubated for 10 min at 25 °C and further incubated for 90 min at 42 °C. The RT products (15 μl) were combined with PCR mix [1 μl each of 10 μM PCR primers P701, P501, and 10 μl 5× KAPA HiFi Fidelity buffer (KAPA Biosystems, KB2500), 1 μl of 10 mM dNTPs (Sigma-Aldrich, 71004-M), 1 μl of 25 mM MgCI2 (KAPA Biosystems, KB1001), 1 μl of KAPA HiFi HotStart enzyme (KAPA Biosystems, KE2502) and 19.5 μl of RNA-free water], and amplified by PCR (98 °C for 3 min, 98 °C for 20 s, followed by 16 cycles of 98 °C for 20 s, 67 °C for 15 s and 72 °C for 6 min, with a final extension at 72 °C for 5 min). Fragments of 250–1000 bp were then purified and size-selected with Speedbead magnetic carboxylate (Cytiva, 17357672). The quality of the resulting cDNA library was quantified using a Qubit dsDNA HS assay kit (Invitrogen, Q32854) with Qubit 3.0 Fluorometer (Invitrogen), before sequencing on the Illumina HiSeq 1500 platform.
Data analysis
Drop-seq core computational protocol (v.2.0.0) was used to process data to generate gene expression matrices. Noteworthy, the Drop-seq core computational protocol (v.2.0.0) allowed the program to extract both intronic data as well as the typical coding+utr data. Before STAR alignment, data was collected on tag cell barcodes UMIs and RNA quality control. Specifically, the following operations were performed: example cell barcode, example molecular barcode, filterBAM, TrimStartingSequence, PolyATrimmer, SamToFastq. Then STAR alignment, genome quantification (coding+intron+utr) for mapped Reads, correcting barcode, and generating gene expression matrices were performed. Specifically, STAR alignment, SortSam, MergeBamAlignment, TagReadWithGeneExon, DetectBeadSynthesisErrors Detecting and repairing barcode synthesis errors, Digital Gene Expression and Cell The selection was performed. The variance analysis was performed with the DESeq2 package of the R project (version 4.1.2). The heatmap analysis was performed with the heatmap of the R project (version 4.1.2). The volcano map analysis was performed with the ggplot2 of the R project (version 4.1.2). The correlation map analysis was performed with the corrplot of the R project (version 4.1.2). The PCA map analysis was performed with the ggplot2 of the R project (version 4.1.2).
Further analysis
After next-generation sequencing, the sample was intact for further analysis. For FFPE tissue, HE staining was performed according to standard protocol. For frozen tissue, they were incubated for 5 min at room temperature with DAPI.
Fluorescence-labeled barcode in tissue and cultured cells
For the fluorescent label, samples were fixed in 4% formaldehyde (Sigma-Aldrich, F8775-25ML) for 10 min at room temperature. Then, samples were washed twice with 1× PBS and permeabilized with 0.1% Triton X-100 (vol/vol) (Sigma Aldrich, T8787-50ML) for 20 min. Samples were then washed twice with 1× PBS, and performed in-suit RT and photo-irradiation as described above. The fluorescent barcode hybridization buffer was prepared as follows: 1 μl of 100 μM barcode-FAM (Supplement Table 1), 1 μl of 100 μM linker NPOM (Supplement table 1), 1 μl of Recombinant RNase Inhibitor (Takara, 2313 A), 10 μl of 20× SSC (Sigma-Aldrich, 93017), 10 μl formamide (Invitrogen, AM9342), 50 μl 20% dextran sulfate sodium salt (Sigma-Aldrich, D8906) and 28 μl of RNase free water, for 30 min. Then the ligation mix [10 μl of T4 DNA ligase buffer (New England Biolabs, B0202SVIAL), 5 μl of T4 DNA ligase (New England Biolabs, M0202LVIAL), and 85 μl of RNase-free water] was performed at 37 °C for 30 min. After this step, the sections were washed with 65% formamide (Invitrogen, AM9342) and PBS two times. For block uncaged-ODNs, the fluorescent block buffer was prepared as follows: 1 μl of 100 μM block adapter (Supplement Table 2), 1 μl of 100 μM linker NPOM (Supplement Table 1), 1 μl of Recombinant RNase Inhibitor (Takara, 2313 A), 20 μl of 20× SSC (Sigma-Aldrich, 93017), and 77 μl of RNase free water, for 30 min. Then the ligation mix [10 μl of T4 DNA ligase buffer (New England Biolabs, B0202SVIAL), 5 μl of T4 DNA ligase (New England Biolabs, M0202LVIAL), and 85 μl of RNase-free water] was performed at 37 °C for 30 min. For the second fluorescent label, the buffer was prepared as follows: 1 μl of 100 μM barcode-ROX (Supplement Table 1), 1 μl of 100 μM linker NPOM (Supplement Table 1), 1 μl of Recombinant RNase Inhibitor (Takara, 2313 A), 10 μl of 20× SSC (Sigma-Aldrich, 93017), 10 μl formamide (Invitrogen, AM9342), 50 μl 20% dextran sulfate sodium salt (Sigma-Aldrich, D8906) and 28 μl of RNase free water, for 30 min. Then the ligation mix [10 μl of T4 DNA ligase buffer (New England Biolabs, B0202SVIAL), 5 μl of T4 DNA ligase (New England Biolabs, M0202LVIAL), and 85 μl of RNase-free water] was performed at 37 °C for 30 min. After this step, the sections were washed with 65% formamide (Invitrogen, AM9342) and PBS two times for imaging.
PBHL-seq analysis on cultured NIH/3T3 and HEK/293 T
Poly-L-lysine-coated chambers were coated with sterile PDL (Sigma Aldrich, P6407) at 0.3 mg/ml overnight at 4 °C. Afterwards, the chamber was dried for 1 h, washed with UltraPure water (Invitrogen, 10977) and dried again before cell seeding. Chambers were then seeded with ~4000 293T and ~5,000 NIH/3T3 cells and placed in an incubator (37 °C with 5% CO2) overnight. Samples were gently washed with PBS-DEPC (pre-warmed to 37 °C, Sangon-biotech, B540627-0500) and fixed in 4% formaldehyde (wt/vol, Sigma-Aldrich, F8775-25ML) in 1× PBS for 10 min at room temperature. Then, samples were washed twice with 1 × PBS and permeabilized with 0.1% Triton X-100 (vol/vol, Sigma Aldrich, T8787-50ML)) for 20 min. Samples were then washed twice with 1× PBS, and the following protocol was performed as described for tissue samples above. After extraction of double-strand cDNAs to prepare the library and sequence, cells were washed twice in PBST and then kept in fresh PBST at 4 °C until further analysis. For the cell mixing experiment, multiplexed IF was performed. All antibodies were spun down at 10,000 g for 10 min at 4 °C before use. Cells were incubated in 1% BSA to block for 1 h, then with Alexa Fluor® 488 Anti-Tubulin antibody [YOL1/34] (Abcam, ab195883) diluted 1:100 in 1× PBS and 1% BSA overnight. Cells were washed in 1× PBS 2× 1 min and then incubated with 20 µM DRAQ5 (Invitrogen, 62254) in 1× PBS for 5 min, and washed 2× 1 min with 1× PBS, followed by imaging in fresh 1× PBS.
Cells quantitative analysis
The 3T3 cells section was performed PBHL-seq protocol, except for Photo-irradiation and Hybridization and ligation barcode oligo step. In this step, 1 cell was photo-irradiated and hybridization and ligation barcode 4 oligo; 10 cells were photo-irradiated and hybridization and ligation barcode 2 oligo; 100 cells were photo-irradiated and hybridization and ligation barcode 3 oligo; 1000 cells were photo-irradiated and hybridization and ligation barcode 1 oligo.
PBHL-seq analyzes mouse brain ROIs
The mouse brain tissue is performed PBHL-seq protocol. All were the same except for the in situ RT reaction followed by incubation at room temperature with 20 µM DRAQ5 solution for 30 min and four rounds of barcode hybridization and ligation.
PBHL-seq for subcellular microstructures
The subcellular microstructures are performed by PBHL-seq protocol. However, this needs to include an immunostaining step with an in situ RT step and block reaction step. The immunostaining step was performed for subcellar microstructure profiling. Briefly, HeLa cells were blocked with 1% BSA solution (Bionordika, B9000S) for 1 h at room temperature. To visualize cytoplasm, cells were incubated for 2 h at room temperature with the Anti-Tubulin antibody Alexa Fluor® 488 (Abcam, ab195883). All antibodies were spun down at 10,000 g for 10 min at 4 °C before use. Alexa Fluor® 488 Anti-Tubulin antibody [YOL1/34] (Abcam, ab195883) diluted 1:100 in 1× PBS and 1% BSA were added to cells for two hours. To visualize cell nuclear, cells were washed in 1× PBS 2× 1 min and then incubated with 20 µM DRAQ5 (Invitrogen, 62254) in 1 × PBS for 5 min, and washed 2× 1 min with 1× PBS.
PBHL-seq analyzes small number cell transcriptomics in tissue
Small number (RPE) cell transcriptomics in tissue is performed in PBHL-seq protocol. All were the same except for the in situ RT reaction followed by H&E staining and two rounds of barcode hybridization and ligation.
Standard cytoplasmic and nuclear RNA purification and sequencing
Hela cells were separated into standard cytoplasm RNA and nucleus RNA with cytoplasmic & Nuclear RNA Purification Kits (Norgen Biotek, 21000). The purification RNA of standard cytoplasm and nucleus was performed with qPCR with a cytoplasmic marker (S14) and nuclear marker (Human U2sn), which sequence information as follows:
S14 Forward (GGCAGACCGAGATGAATCCTC),
S14 Reverse (CAGGTCCAGGGGTCTTGGTCC)
Human U2sn-Forward 5’-CATCGCTTCTCGGCCTTTTG-3’
Human U2sn-Reverse 5’-TGGAGGTACTGCAATACCAGG-3’
Standard cytoplasmic and nuclear RNA were performed in RNA-seq, and sequencing data and the subcellular microstructures’ sequencing data of PBHL-seq have performed the variance analysis with the DESeq2 package of the R project (version 4.1.2). The upset analysis with the upset UpSetR (1.4.0).
Statistics and reproducibility
All PBHL-seq data with brain tissue, mouse embryo tissue, FFPE tissue, sub-cell, and small number cell transcriptomics in tissue made at least three biological replicates, and difference analysis was performed with P-value < 0.05 and |Log2(Fold-Change)| >1. Cells quantitative analysis made seven biological replicates with student’s t-test for correction analysis.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw sequencing data are available online in NCBI’s Gene Expression Omnibus and are accessible through GEO series accession number GSE268148. All other data are available from the corresponding author (or other sources, as applicable) on reasonable request. Source data are provided in the supplement data file with this paper.
References
Tanay, A. & Regev, A. Scaling single-cell genomics from phenomenology to mechanism. Nature 541, 331–338 (2017).
Park, J.-E. et al. A cell atlas of human thymic development defines T cell repertoire formation. Science 367, eaay3224 (2020).
Harris, J. A. et al. Hierarchical organization of cortical and thalamic connectivity. Nature 575, 195–202 (2019).
Hagemann-Jensen, M. et al. Single-cell RNA counting at allele and isoform resolution using Smart-seq3. Nat. Biotechnol. 38, 708–714 (2020).
Zhu, L. et al. Single-cell sequencing of peripheral mononuclear cells reveals distinct immune response landscapes of COVID-19 and influenza patients. Immunity 53, 685–696. e683 (2020).
Macosko, E. Z. et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015).
Klein, A. M. et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201 (2015).
Han, X. et al. Mapping the mouse cell atlas by microwell-seq. Cell 172, 1091–1107. e1017 (2018).
Rosenberg, A. B. et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 360, 176–182 (2018).
Knipple, D. C., Seifert, E., Rosenberg, U. B., Preiss, A. & Jäckle, H. Spatial and temporal patterns of Krüppel gene expression in early Drosophila embryos. Nature 317, 40–44 (1985).
Scadden, D. T. Nice neighborhood: emerging concepts of the stem cell niche. Cell 157, 41–50 (2014).
Stahl, P. L. et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82 (2016).
Van Vliet, S. et al. Spatially correlated gene expression in bacterial groups: the role of lineage history, spatial gradients, and cell-cell interactions. Cell Syst. 6, 496–507. e496 (2018).
Alon, S. et al. Expansion sequencing: spatially precise in situ transcriptomics in intact biological systems. Science 371, 481–48 (2021).
Andersson, A. et al. Single-cell and spatial transcriptomics enables probabilistic inference of cell type topography. Commun. Biol. 3, 8 (2020).
Battich, N., Stoeger, T. & Pelkmans, L. Image-based transcriptomics in thousands of single human cells at single-molecule resolution. Nat. Methods 10, 1127–1133 (2013).
Chen, K. H., Boettiger, A. N., Moffitt, J. R., Wang, S. & Zhuang, X. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, https://doi.org/10.1126/science.aaa6090 (2015).
Cho, C.-S. et al. Microscopic examination of spatial transcriptome using Seq-Scope. Cell 184, 3559 (2021).
Eng, C. H. L. et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 568, 235 (2019).
Fazal, F. M. et al. Atlas of subcellular RNA localization revealed by APEX-Seq. Cell 178, 473 (2019).
Gyllborg, D. et al. Hybridization-based in situ sequencing (HybISS) for spatially resolved transcriptomics in human and mouse brain tissue. Nucleic Acids Res. 48, 11 (2020).
Goh, J. J. L. et al. Highly specific multiplexed RNA imaging in tissues with split-FISH. Nat. Methods 17, 689 (2020).
Vickovic, S. et al. SM-Omics is an automated platform for high-throughput spatial multi-omics. Nat. Commun. 13, 13 (2022).
Vickovic, S. et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat. Methods 16, 987 (2019).
Srivatsan, S. R. et al. Embryo-scale, single-cell spatial transcriptomics. Science 373, 111 (2021).
Lubeck, E. & Cai, L. Single-cell systems biology by super-resolution imaging and combinatorial labeling. Nat. Methods 9, 743–U159 (2012).
Shah, S., Lubeck, E., Zhou, W. & Cai, L. In situ transcription profiling of single cells reveals spatial organization of cells in the mouse hippocampus. Neuron 92, 342–357 (2016).
Eng, C. H. L., Shah, S., Thomassie, J. & Cai, L. Profiling the transcriptome with RNA SPOTs. Nat. Methods 14, 1153 (2017).
Kishi, J. Y. et al. SABER amplifies FISH: enhanced multiplexed imaging of RNA and DNA in cells and tissues. Nat. Methods 16, 533 (2019).
Wang, X. et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 361, 9 (2018).
Coskun, A. F. & Cai, L. Dense transcript profiling in single cells by image correlation decoding. Nat. Methods 13, 657 (2016).
Ke, R. Q. et al. In situ sequencing for RNA analysis in preserved tissue and cells. Nat. Methods 10, 857 (2013).
Shendure, J. et al. Accurate multiplex polony sequencing of an evolved bacterial genome. Science 309, 1728–1732 (2005).
Lee, J. H. et al. Highly multiplexed subcellular RNA sequencing in situ. Science 343, 1360–1363 (2014).
Rodriques, S. G. et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463 (2019).
Brown, V. M. et al. Multiplex three-dimensional brain gene expression mapping in a mouse model of Parkinson’s disease. Genome Res. 12, 868–884 (2002).
Junker, J. P. et al. Genome-wide RNA tomography in the Zebrafish Embryo. Cell 159, 662–675 (2014).
Peng, G. D. et al. Spatial transcriptome for the molecular annotation of lineage fates and cell identity in mid-gastrula mouse embryo (vol 36, 681, 2016). Dev. Cell 55, 802–804 (2020).
Schede, H. H. et al. Spatial tissue profiling by imaging-free molecular tomography. Nat. Biotechnol. 39, 968 (2021).
Hufnagel, B. et al. High-quality genome sequence of white lupin provides insight into soil exploration and seed quality. Nat. Commun. 11, 12 (2020).
Medaglia, C. et al. Spatial reconstruction of immune niches by combining photoactivatable reporters and scRNA-seq. Science 358, 1622–1626 (2017).
Hu, K. H. et al. ZipSeq: barcoding for real-time mapping of single cell transcriptomes. Nat. Methods 17, 833 (2020).
Merritt, C. R. et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nature Biotechnol. 38, 586 (2020).
Lubeck, E., Coskun, A. F., Zhiyentayev, T., Ahmad, M. & Cai, L. Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods 11, 360–361 (2014).
Stickels, R. R. et al. Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat. Biotechnol. 39, 313–319 (2021).
Fu, X. A. et al. Polony gels enable amplifiable DNA stamping and spatial transcriptomics of chronic pain. Cell 185, 4621 (2022).
Chen, A. et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell 185, 1777 (2022).
Moses, L. & Pachter, L. Museum of spatial transcriptomics. Nat. Methods 19, 534–546 (2022).
Nichterwitz, S. et al. Laser capture microscopy coupled with Smart-seq2 for precise spatial transcriptomic profiling. Nat. Commun. 7, 1–11 (2016).
Peng, G. D. et al. Spatial transcriptome for the molecular annotation of lineage fates and cell identity in mid-gastrula mouse embryo. Dev. Cell 36, 681–697 (2016).
Chen, J. et al. Spatial transcriptomic analysis of cryosectioned tissue samples with Geo-seq. Nat. Protocols 12, 566–580 (2017).
Honda, M. et al. High-depth spatial transcriptome analysis by photo-isolation chemistry. Nat. Commun. 12, https://doi.org/10.1038/s41467-021-24691-8 (2021).
Kishi, J. Y. et al. Light-Seq: light-directed in situ barcoding of biomolecules in fixed cells and tissues for spatially indexed sequencing. Nat. Methods 19, 1393 (2022).
Vangelder, R. N. et al. Amplified RNA Synthesized from limited quantities of heterogeneous cDNA. Proc. Natl Acad. Sci. USA 87, 1663–1667 (1990).
Govan, J. M. et al. Optochemical control of RNA interference in mammalian cells. Nucleic Acids Res. 41, 10518–10528 (2013).
Deiters, A. Light activation as a method of regulating and studying gene expression. Curr. Opin. Chem. Biol. 13, 678–686 (2009).
Singh-Gasson, S. et al. Maskless fabrication of light-directed oligonucleotide microarrays using a digital micromirror array. Nat. Biotechnol. 17, 974–978 (1999).
Liu, Y. et al. High-spatial-resolution multi-omics sequencing via deterministic barcoding in tissue. Cell 183, 1665 (2020).
Heavner, W. & Pevny, L. Eye Development and retinogenesis. Cold Spring Harb. Perspect. Biol. 4, https://doi.org/10.1101/cshperspect.a008391 (2012).
Pijuan-Sala, B. et al. Single-cell chromatin accessibility maps reveal regulatory programs driving early mouse organogenesis. Nat. Cell Biol. 22, https://doi.org/10.1038/s41556-020-0489-9 (2020).
Lotto, J. et al. Single-cell transcriptomics reveals early emergence of liver parenchymal and non-parenchymal cell lineages. Cell 183, 702 (2020).
Mathieson, W. & Thomas, G. Using FFPE tissue in genomic analyses: advantages, disadvantages and the role of biospecimen science. Curr. Pathobiol. Rep. 7, 35–40 (2019).
Hoffman, E. A., Frey, B. L., Smith, L. M. & Auble, D. T. Formaldehyde crosslinking: a tool for the study of chromatin complexes. J. Biol. Chem. 290, 26404–26411 (2015).
Civita, P. et al. Laser capture microdissection and RNA-Seq analysis: high sensitivity approaches to explain histopathological heterogeneity in human glioblastoma FFPE archived tissues. Front. Oncol. 9, https://doi.org/10.3389/fonc.2019.00482 (2019).
Bai, Z. et al. Spatially exploring RNA biology in archival formalin-fixed paraffin-embedded tissues. Preprint at bioRxiv https://doi.org/10.1101/2024.02.06.579143 (2024).
Gracia Villacampa, E. et al. Genome-wide spatial expression profiling in formalin-fixed tissues. Cell Genom. 1, 100065 (2021).
Salmen, F. et al. Barcoded solid-phase RNA capture for spatial transcriptomics profiling in mammalian tissue sections. Nat. Protocol. 13, 2501–2534 (2018).
Crosetto, N., Bienko, M. & van Oudenaarden, A. Spatially resolved transcriptomics and beyond. Nat. Rev. Genet. 16, 57–66 (2015).
Moor, A. E. et al. Global mRNA polarization regulates translation efficiency in the intestinal epithelium. Science 357, 1299 (2017).
Halpern, K. B. et al. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 542, 352 (2017).
Nguyen, H. Q. et al. 3D mapping and accelerated super-resolution imaging of the human genome using in situ sequencing. Nat. Methods 17, 822 (2020).
Hwang, H. W., Wentzel, E. A. & Mendell, J. T. A hexanucleotide element directs microRNA nuclear import. Science 315, 97–100 (2007).
Bumsted, K. M. & Barnstable, C. J. Dorsal retinal pigment epithelium differentiates as neural retina in the microphthalmia (mi/mi) mouse. Investig. Ophthalmol. Vis. Sci. 41, 903–908 (2000).
Haeseleer, F. et al. Dual-substrate specificity short chain retinol dehydrogenases from the vertebrate retina. J. Biol. Chem. 277, 45537–45546 (2002).
Stoeckius, M. et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865 (2017).
Shahi, P., Kim, S. C., Haliburton, J. R., Gartner, Z. J. & Abate, A. R. Abseq: Ultrahigh-throughput single cell protein profiling with droplet microfluidic barcoding. Sci. Rep. 7, https://doi.org/10.1038/srep44447 (2017).
Setliff, I. et al. High-throughput mapping of B cell receptor sequences to antigen specificity. Cell 179, 1636 (2019).
Menge, C. & Heckel, A. Coumarin-caged dG for improved wavelength-selective uncaging of DNA. Org. Lett. 13, 4620–4623 (2011).
Rodrigues-Correia, A., Weyel, X. M. M. & Heckel, A. Four levels of wavelength-selective uncaging for oligonucleotides. Org. Lett. 15, 5500–5503 (2013).
Tang, X. et al. Caged nucleotides/nucleosides and their photochemical biology. Org. Biomol. Chem. 11, 7814–7824, (2013).
Acknowledgements
This work is supported by the Funding Project of the National Key Research and Development Program of China (2018YFD0900604) and the Natural Science Foundation of China (41676119 and 41476120).
Author information
Authors and Affiliations
Contributions
Xiaonan Cui was the main author behind the development of PBHL-seq, who provided intellectual input to the project, designed, and performed the experiments. Xiaonan Cui, Xue Dong, and Mengzhu Hu contributed to the formulation of the project, performed the statistical analysis, and generated the figures. Xue Dong performed initial experiments. Mengzhu Hu provided tissue sections. Wenjian Zhou supported cell cultivation. Weiyang Shi played a supervisory role throughout the project, conceiving and overseeing the study. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare the following competing interests: Weiyang Shi has filed a patent (PCTxxx) based on the technique described in this paper. All other authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Jia-Ren Lin, and Shih-Yu Chen for their contribution to the peer review of this work. Primary handling editors: Ruby Huang and Johannes Stortz. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cui, X., Dong, X., Hu, M. et al. Large field of view and spatial region of interest transcriptomics in fixed tissue. Commun Biol 7, 1020 (2024). https://doi.org/10.1038/s42003-024-06694-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-024-06694-5
