
Abstract
Previous reports showed that long non-coding RNA (lncRNA) participates in the development and progression of tumors. Nevertheless, the effect of LINC02139 and its mechanism on gastric cancer (GC) is still unknown. We revealed that LINC02139 is upregulated in GC cell lines and tissues and high LINC02139 expression was correlated with the advancement of GC in patients. Functionally, overexpression of LINC02139 promoted, while knockdown of LINC02139 impaired GC cell proliferation, migration, and invasion in vitro and impeded tumorigenesis in a tumor xenograft model in vivo. Mechanistically, LINC02139 directly bound to XIAP and increased the protein level by maintaining its protein stability through inhibition of the ubiquitination and proteasome-dependent degradation pathway. Importantly, the regulatory function of XIAP in LINC02139-mediated oncogenic effects was demonstrated. Both in vitro and in vivo experiments showed that LINC02139 and XIAP collaboratively modulate GC cell growth and apoptosis. Analysis of clinical GC tissues further confirmed the upregulation of XIAP and the positive association between LINC02139 and XIAP expression. These findings established LINC02139 as a driver of tumorigenesis and highlighted the crucial involvement of the LINC02139-XIAP axis in GC progression, suggesting its potential as a promising therapeutic target for combating GC advancement.
Similar content being viewed by others


Introduction
In terms of incidence, gastric cancer (GC) is the third most common type of cancer worldwide and the fourth most common cause of cancer-related mortality1. Despite the enormous achievements that have been approached for improving clinical prognosis, such as endoscopic resection, surgery, perioperative or adjuvant chemotherapy, and targeted therapies, patients with advanced GC are still suffering from a quite dismal prognosis2. As is reported GC is a heterogeneous disease in molecular and phenotype with extremely complex intrinsic mechanisms of malignancy and metastasis3,4. Hence, it is of utmost importance for us to get a better understanding of the underlying mechanism of GC development and progression, which could contribute to the early diagnosis, treatment, and prognosis of GC.
Long non-coding RNA (lncRNA) has been defined as RNA that does not code functional proteins and is longer than 200 nucleotides in length. To date, a growing body of research has shown that lncRNA functions as a crucial mediator in malignancies through multiple mechanisms, including cis and trans regulation of genes, nucleus-based epigenetic modification, and cytoplasmic post-transcriptional modulation5,6,7. For instance, PAXIP1-AS1 was downregulated in GC and regulated tumor progression through mediating epithelial-to-mesenchymal transition8. LINC00501 was elevated in GC and was correlated with poor clinicopathological features, which advanced GC progression9. The overexpression of LINC00346 in GC was correlated with advanced pathological stage, large tumor size, and unfavorable prognosis, suggesting its crucial role as a regulator in GC tumorigenesis and progression10. However, LINC02139 is a long intergenic non-protein-coding RNA that is located on the 16q24.1 chromosome, whose biological function and the specific mechanism involved in GC remain to be fully understood.
The X-linked inhibitor of apoptosis protein (XIAP), a leading member of the family of inhibitors of apoptosis proteins, functions as an apoptosis-suppressing protein by obstructing the specific caspase activities, particularly caspase-3, caspase-7, and caspase-911,12,13. Accumulating evidence indicates that other than inhibiting apoptosis, XIAP is implicated in tumorigenesis, progression, chemoresistance, and poor prognosis, and is a heavily compelling target for anti-cancer therapy14,15. For example, XIAP inhibited miR-200a transcription and upregulated EGFR, thereby promoting bladder cancer cell growth16. Drug-cannabidiol suppressed XIAP protein expression by increasing XIAP ubiquitination and induced cell apoptosis, thereby inhibiting GC progression17. In endometrial cancer, the inhibition of XIAP transcription and translation led to the occurrence of cancer cell apoptosis and the cessation of proliferation18. Nevertheless, the precise role of XIAP in LINC02139-modulated GC progression is largely unknown.
In this investigation, we identified that LINC02139 was aberrantly upregulated and was associated with adverse clinicopathologic features. Overexpression of LINC02139 promoted, while the silence of LINC02139 impaired GC cell proliferation, migration, and invasion in vitro and impeded tumorigenesis in a tumor xenograft model in vivo. Furthermore, a molecular mechanism by which LINC02139 bound and stabilized XIAP and accelerated GC cell proliferation, migration, invasion, and suppressed cell apoptosis was confirmed. Together, these findings shed light on LINC02139’s role and underlying mechanism in driving GC progression, suggesting a potential biomarker and therapeutic target.
Results
LINC02139 is upregulated in GC and the high LINC02139 level is associated with poor clinicopathologic features
To identify differentially expressed lncRNAs that expressed above the threshold level (1.2-fold change and P < 0.05) in GC tissues, we searched the gene-expression datasets GSE53137 and GSE95667. The result showed that 19 lncRNAs were highly expressed and 30 lncRNAs were down-expressed in the GSE53137 dataset, whereas 150 lncRNAs were elevated and 49 lncRNAs were decreased in the GSE95667 dataset. The overlap of the two datasets displayed that there were seven annotated lncRNAs were upregulated, namely CPEB1-AS1, LINC02632, LINC01713, LINC01680, LINC01594, LINC01997, and LINC02139. Additionally, three lncRNAs were found to be downregulated, specifically LINC02256, LINC01399, and TBX18-AS1 (Fig. 1A). The primary focus of this research was on the lncRNAs that exhibited high expression levels.

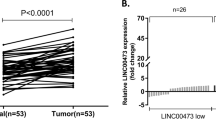
A Differentially expressed lncRNAs are displayed as heatmaps. Red represents upregulated lncRNAs and blue represents downregulated lncRNAs. Venn diagrams show the overlaps of GSE53137 and GSE95667. B LINC02139 expression levels in GSE53137 and GSE95667 are shown as scatter plots. ***P < 0.01, ****P < 0.001. C LINC02139 expression levels in GC and normal tissues measured by qRT-PCR are displayed as waterfall plots and scatter plots. ****P < 0.001. D qRT-PCR was conducted to test LINC02139 expression in GC cell lines and immortalized gastric mucosal cell line GES-1. One-way ANOVA and Dunnett’s T3 multiple comparison tests. **P < 0.05. E In situ hybridization (ISH) analysis was adopted to ascertain the LINC02139 expression in normal gastric mucosa and GC tissues. F LINC02139 expression in the cytoplasmic and nuclear fractions in GES-1 and AGS cells was determined by qRT-PCR. GAPDH represents cytoplasmic control and nuclear-enriched abundant transcript 1 (NEAT1) represents nuclear control. G The coding potential of LINC02139 was explored through the Coding Potential Assessment Tool (CPAT) and Coding Potential Calculator (CPC). HOX transcript antisense RNA (HOTAIR) and GAPDH were set as non-coding and coding RNA control, respectively. Scale bars, 100 μm in (E).
Then, we detected the expression of CPEB1-AS1, LINC02632, LINC01713, LINC01680, LINC01594, LINC01997, and LINC02139 in 32 paired human GC tissues. qRT-PCR analyses showed that the high expression rates of CPEB1-AS1, LINC02632, LINC01713, LINC01680, LINC01594, LINC01997, and LINC02139 were 50.0% (16/32), 47.0% (15/32), 60.0% (19/32), 34.4% (11/32), 50.0% (16/32), 41.0% (13/32), and 72.0% (23/32) respectively. Among those, LINC02139 was the most remarkably upregulated (Supplementary Fig. 1) and was selected as the research object, which was further validated by analyzing the expression data from GSE53137 and GSE95667 (Fig. 1B). Subsequently, we examined LINC02139 expression in 72 paired human GC tissues. The findings indicated that LINC02139 (52/72, 72.2%) was dramatically upregulated in GC tissues (Fig. 1C). Moreover, the qRT-PCR assay unveiled a significant rise in LINC02139 expression in almost all GC cell lines, including SNU-719, SNU-5, SNU-1, HGC-27, and AGS, except MKN-45, as compared with human gastric epithelial mucosa cell line GES-1 (Fig. 1D). These findings consistently confirmed that LINC02139 was upregulated in GC. Following that, ISH assays were utilized to identify LINC02139 subcellular distribution, revealing that LINC02139 was present in both cytoplasm and nucleus of gastric cancer tissues (Fig. 1E). The subcellular fractionation analysis showed similar results (Fig. 1F). In addition, we assessed if LINC02139 can encode proteins using the Coding Potential Assessment Tool (CPAT) and Coding Potential Calculator (CPC). The findings from both studies indicated that LINC02139 does not possess the ability to code for proteins (Fig. 1G).
To estimate correlations between LINC02139 expression and GC clinicopathological subgroups that were stratified based on the patient’s gender, age, tumor size, differentiation, depth of invasion, lymph node metastasis, distant metastasis, and AJCC TNM stage, the median expression level of LINC02139 was used as the cut-off value and the 72 pairs of GC patients were stratified into low- and high-LINC02139 expression groups. Greater tumor size, deeper depth of invasion (T1-2 vs. T3-4), increased lymph node metastasis, distant metastasis, and clinical stage (I-II vs. III-IV stage) were observed in the high-LINC02139 expression group compared with the low-LINC02139 expression group. However, no correlations were observed between high LINC02139 expression and gender, age, or tumor differentiation (Supplementary Table 4).
Altogether, these results demonstrate that LINC01239 may exert a crucial role in GC tumorigenesis and progression, and serve as a valuable indicator in GC.
LINC02139 facilitates the malignant phenotypes of GC cells in vitro and in vivo
We next investigated the effects of LINC02139 on GC cell proliferation, migration, and invasion. To this end, AGS and HGC-27 cells were initially transfected with Vector or LINC02139 overexpression plasmids, Scr-siRNA, or three independent siRNAs for 48 h to overexpress or knockdown LINC02139. qRT-PCR assays were utilized to measure the transfection efficiencies (Supplementary Fig. 2). We then performed gain of and loss of function experiments to identify the potential role of LINC02139 in GC malignant phenotypes. EdU and colony formation assays were adopted to assess the proliferative ability of GC cells. The EdU results revealed a significant increase in the EdU-positive GC cells after overexpressing LINC02139. Conversely, there was a reduction in the EdU-positive GC cells after silencing LINC02139 (Fig. 2A, Supplementary Fig. 3A). Congruously, overexpression of LINC02139 facilitated GC cell colony formation capacity, whereas silence of LINC02139 exhibited the reverse results (Fig. 2B, Supplementary Fig. 3B). We also conducted Transwell assays to estimate GC cell migratory and invasive abilities and validated that ectopic expression of LINC02139 markedly promoted GC cell migration and invasion, whereas silence of LINC02139 impaired the migratory and invasive abilities (Fig. 2C, D, Supplementary Fig. 3C, D). In addition, in the wound healing assays, the motility of LINC02139-overexpressing GC cells was dramatically enhanced. Inversely, the motility of LINC02139-silenced GC cells was weakened (Fig. 2E, Supplementary Fig. 3E).

A EdU assays were executed to detect the effects of LINC02139 on the DNA synthesis of GC cells. B Colony formation assays were exploited to determine the effects of LINC02139 on the proliferative abilities of GC cells. C, D Transwell assays were employed to examine the migratory (C) and invasive (D) capacities of GC cells after being transfected with vector plasmids, LINC02139 overexpression plasmids, Scr-siRNA, or LINC02139-siRNAp. Scr-siRNA: Scramble-siRNA; siRNAp: siRNA pool, siRNA1, siRNA2, siRNA3. E Wound healing assays were utilized to observe the motility of the transfected GC cells. F The images of the whole body and subcutaneous tumor of the nude mice. G The growth curves of the subcutaneous tumors. ****P < 0.001. H, I Ki-67 and CD105 expression levels in the subcutaneous tumors were tested using IHC assays. Scale bars, 50 μm in (A), 100 μm in (H) and (I).
To explore whether LINC02139 affects GC cell proliferation in vivo, a subcutaneous tumor model was constructed in nude mice with AGS cells. As shown in Fig. 2F, G, Supplementary Fig. 3F, mice subcutaneously injected with LINC02139-silenced cells exhibited significantly smaller tumor volumes, lower growth, and less weight compared with those in the counterpart control group. Subcutaneous tumor tissues of these two groups were confirmed by HE staining (Supplementary Fig. 3G). Subsequently, IHC assays were conducted to detect tumor proliferation ability, indicating that LINC02139 depletion led to a significant reduction of the Ki-67 positive rates compared with the control group (Fig. 2H, Supplementary Fig. 3H). Consistently, the tumors in the LINC02139-silencing group also showed a lower microvascular density compared with those in the control group (Fig. 2I, Supplementary Fig. 3I).
In summary, the obtained results suggest that LINC02139 may significantly promote GC cell growth both in vitro and in vivo.
LINC02139 binds and stabilizes XIAP protein in human GC cells
Accumulating evidence reveals that lncRNAs are prone to be involved in modulating cancers by directly interacting with various proteins19,20,21. Thus, we endeavored to identify LINC02139-associated proteins using RNA pull-down and protein mass spectrometry (MS) in the wild-type AGS cells. The obtained protein samples were separated by SDS-PAGE, followed by silver staining, and subsequently subjected to protein MS. A differential band between the LINC02139 sense and the antisense near 55 kDa was observed in silver staining (Fig. 3A). MS identified XIAP as a LINC02139-interacting protein (Supplementary Table 5), which was further confirmed through RNA pull-down assays in AGS and HGC-27 cells (Fig. 3B). Meanwhile, RIP assays also validated a remarkable enrichment of LINC02139 in AGS and HGC-27 cells with an anti-XIAP antibody (Fig. 3C). To further determine the specific region that XIAP binds to LINC02139, a series of LINC02139 deletion mutants fragments, including full-length (FL:1–2015), F1 (1–700), F2 (701–1400), and F3 (1401–2015) were constructed accordingly. Through RNA pull-down and western blotting assays, we found that all three truncated fragments of LINC02139 owned binding regions for XIAP (Fig. 3D). Besides, His-tagged full-length (FL:1–497aa) and truncated XIAP mutants, BIR 1 (baculoviral IAP repeat domain 1) (F1:1–130aa), BIR 2 (baculoviral IAP repeat domain 2) (F2:131–250aa), BIR 3 (baculoviral IAP repeat domain 3) (F3:251–370aa), and RING (Zinc finger domain) (F4:371–497aa) were designed to investigate which domain of XIAP was responsible for binding to LINC02139. As shown, the results of RIP indicated that the BIR 1 domain of XIAP was essential for the binding of XIAP to LINC02139 (Fig. 3E). Taken together, these results demonstrate that LINC02139 could bind with XIAP in GC cells.

A Proteins retrieved from the LINC02139 pull-down assay in AGS cells were investigated by SDS-PAGE, followed by silver staining. The specific band is denoted with an arrow. B The in vitro transcribed biotinylated full-length of LINC02139 and the antisense control were employed in RNA pull-down assays. The combination of LINC02139 and XIAP was evaluated through western blotting. C RIP assays were conducted with an anti-XIAP antibody in AGS and HGC-27 cells. The enrichment of LINC02139 was measured via qRT-PCR. Anti-IgG was used as a negative control. ****P < 0.001. D Immunoblot detection of the XIAP protein in AGS cells as retrieved by in vitro transcribed biotinylated different constructions of LINC02139. E 6×His-tagged XIAP full-length and truncations were constructed. RIP-qRT-PCR assays were performed to verify the enrichment of LINC02139 in AGS cells by using an anti-His antibody. Anti-IgG was used as a negative control. *P > 0.05, ***P < 0.01.
To dissect the underlying molecular mechanism of LINC02139 in regulating XIAP, XIAP mRNA and protein expression in LINC02139 overexpressed or silenced AGS and HGC-27 were initially validated. It was shown that XIAP protein expression was significantly upregulated in LINC02139-overexpressing GC cells and vice versa (Fig. 4A), but XIAP mRNA expression remained unchanged in both AGS and HGC-27 cells (Fig. 4B). However, the gain of and loss of function of XIAP did not affect LINC02139 expression (Supplementary Fig. 4A–D), suggesting that LINC02139 modulates XIAP at the post-transcriptional level. Subsequently, a protein half-life assay was employed to further investigate the specific mechanism, the results of which revealed that the silence of LINC02139 in AGS cells could distinctly reduce the stability of the XIAP protein (Fig. 4C), accompanied by particularly increased XIAP polyubiquitination (Fig. 4D). Moreover, LINC02139 silencing-modulated down-regulation of XIAP could be restored by treatment with proteasome inhibitor MG 132 (Fig. 4E). Above all, these findings illustrate that LINC02139 could interact with and stabilize XIAP protein by protecting XIAP from the ubiquitin-mediated degradation.

A Western blotting analyses of XIAP expression in LINC02139-overexpressed and LINC02139-silenced GC cells. B qRT-PCR analyses of XIAP expression in LINC02139-overexpressed and LINC02139-silenced GC cells. *P > 0.05, ***P < 0.01, ****P < 0.001. C The half-life of XIAP protein was tested after the transfected AGS cells were treated with 100 μM CHX for the indicated time by using western blotting. These pictures were the representatives of three independent experiments with identical results. D The ubiquitin level of XIAP in GC cells was measured using immunoprecipitation and western blotting. IP immunoprecipitation, IB immunoblotting. E The AGS cells were co-incubated with CHX (100 μM) and MG 132 (10 nM) for the indicated periods, followed by western blotting.
LINC02139 augments GC cell proliferation, migration, and invasion by targeting XIAP
To point out whether XIAP functions in LINC02139-mediated GC cell proliferation, migration, and invasion, XIAP was knocked down in LINC02139-overexpressing AGS and HGC-27 cells. As exhibited in Fig. 5A, B, EdU and colony formation assays indicated that XIAP silence strikingly abrogated LINC02139 overexpressing-induced facilitation of proliferative ability as well as the colony formation ability of GC cells. Furthermore, Transwell migration and invasion assays were conducted and showed that XIAP depletion attenuated the ascent effects of LINC02139 overexpression on GC cell migratory and invasive abilities (Fig. 5C, D). Additionally, wound healing experiments also validated that the knockdown of XIAP reduced the promotion function of LINC02139 overexpression on GC cell motility (Fig. 5E, Supplementary Fig. 5).

A, B The proliferative ability was assessed using EdU (A) and colony formation assays (B) in the transfected GC cells. The EdU-positive cells and colony numbers were counted. ****P < 0.001. C, D Transwell assays unveiled migratory (C) and invasive (D) abilities of the transfected GC cells. The numbers were estimated. ***P < 0.01, ****P < 0.001. E Wound healing assays were employed to detect the motility of the transfected GC cells. Scale bars, 50 μm in (A).
Accordingly, these results uncover that XIAP is an essential modulator of LINC02139-mediated GC cell malignant phenotypes.
LINC02139 is positively correlated with XIAP in the human GC samples and participates in mediating GC cell apoptosis
The data obtained from the starbase (https://rnasysu.com/encori/) showed that XIAP is more highly expressed in GC tissues than in normal tissues (Supplementary Fig. 6A). Moreover, Kaplan–Meier survival analysis (https://kmplot.com/) revealed that patients with high XIAP expression had poorer overall survival (Supplementary Fig. 6B). In addition, we detected XIAP expression in 12 pairs of fresh human GC samples by using IHC assays. It was shown that XIAP was dramatically upregulated in most cancer tissues compared with the adjacent cancerous tissues (Supplementary Fig. 6C). To explore the correlations between LINC02139 and XIAP expression levels, we detected XIAP protein expression in 12 pairs of GC tissues via western blotting, and LINC02139 expression was tested by using qRT-PCR assays. The results exhibited that XIAP was overexpressed in nine pairs of GC tissues, while LINC02139 expression was elevated in ten pairs of GC tissues (Fig. 6A, B). Pearson correlation analysis demonstrated a positive correlation between LINC02139 and XIAP (Fig. 6C). As previous research reported, XIAP is one of the members of the inhibitor of apoptosis proteins (IAP), exhibiting the most promising anti-apoptotic effect via various pathways22,23. To validate the functions of XIAP in LINC02139-mediated GC cell apoptosis, we performed Hoechst 33258 and flow cytometry assays. Overexpression of LINC02139 significantly reduced GC cell apoptosis in Hoechst 33258 assays, whereas XIAP depletion impaired the inhibitory effects of LINC02139 overexpression (Fig. 6D). Congruously, flow cytometry assays also exhibited that XIAP silencing attenuated the suppressive effects of LINC02139 overexpression on GC cell apoptosis (Fig. 6E).

A XIAP protein expression in 12 pairs of human GC samples was detected using western blotting, normalized to GAPDH. The relative protein expression level was quantified by comparing the gray level of each band. N, normal; T, tumor. *P > 0.05; ****P < 0.001. B LINC02139 expression in the 12 pairs of human GC samples was measured using qRT-PCR. Normalized to GAPDH. N, normal; T, tumor. *P > 0.05; **P < 0.05, ***P < 0.01, ****P < 0.001. C A Pearson correlation analysis was performed to determine the correlations between LINC02139 and XIAP expression. D A fluorescent microscopy technique was applied to visualize the Hoechst 33258 staining in the nucleus of the indicated transfected GC cells. Arrows in the images indicate the cells with condensed chromatin and fragmented nuclei. The apoptotic ratios were displayed. ***P < 0.01, ****P < 0.001. E Flow cytometry assays were used to inspect the apoptosis in the transfected GC cells. The apoptotic ratios were shown as histograms. ****P < 0.001. Scale bars, 50 μm in (D).
These findings together show that LINC02139 and XIAP have a favorable correlation in human GC samples. Apoptosis was decreased in LINC02139 overexpression GC cells, yet XIAP knockdown partially reversed the inhibitory effects of LINC02139.
LINC02139 reinforces GC cell proliferation and restrains apoptosis by targeting XIAP in vivo
To investigate the functions of XIAP in LINC02139-induced GC cell proliferation and apoptosis in vivo, a subcutaneous tumor model was employed with AGS cells. As shown in Fig. 7A–C, mice subcutaneously injected with LINC02139 overexpression cells yielded bigger tumor volumes, faster growth, and heavier tumor weight compared with those in the control groups, whereas XIAP silence reverted the promotion effects of LINC02139 overexpression on GC cell proliferation. We then performed HE staining to validate subcutaneous tumor tissues in each group (Fig. 7D). Subsequently, IHC assays were employed to detect the indicated protein expression levels, including Ki-67, CD105, and Cleaved Caspase-3. It was obvious that Ki-67 (Fig. 7E, F) and CD105 (Fig. 7G, H) were all elevated in the LINC02139 overexpression group compared with the control group, whereas a restoration was observed in the XIAP silence group, indicating that exogenous LINC02139 increased GC cell proliferation and tumor vessel density, but the silence of XIAP reversed the promotion effects induced by LINC02139. On the contrary, compared with the control group, Cleaved Caspase-3 expression was decreased in the LINC02139 overexpression group, but partially rescued in the XIAP silence group, indicating that ectopic LINC02139 restrained GC cell apoptosis, whereas XIAP depletion rescued the inhibition effects of LINC02139 overexpression on GC cell apoptosis (Fig. 7I, J).

A The whole body and subcutaneous tumors of the nude mice. B The growth curves of the subcutaneous tumor. ****P < 0.001. C The tumor weight of different groups. ****P < 0.001. D HE staining was performed to verify the subcutaneous tumors. E–J Ki-67, CD105, and Cleaved Caspase 3 expression levels in the subcutaneous tumors were detected using IHC (E, G, I). The proliferation index, microvascular density, and positive percentage of Cleaved Caspase 3 were calculated (F, H, J). ****P < 0.001. Scale bars, 100 μm in (D) and (E, G, I).
According to these findings, LINC02139-induced GC cell proliferation and apoptosis are crucially regulated by XIAP.
Discussion
Although the biological functions of lncRNAs in GC have been extensively elaborated, the potential molecular mechanisms of LINC02139 involved in tumorigenesis and aggressiveness in GC are required to be fully investigated. The results from this present study indicated that LINC02139 was upregulated in human GC cell lines and tissues. Reduction of LINC02139 restrained GC cell proliferation, migration, and invasion both in vitro and in vivo. Moreover, LINC02139 directly interacted with and stabilized XIAP by inhibiting its ubiquitination degradation, resulting in cell proliferation and an arrest in apoptosis, thus expediting GC progression. Additionally, the results emphasized that the LINC02139-XIAP aix was confirmed to play a critical role in GC progression, implying that it may function as a promising biomarker and therapeutic target for GC management.
Mountain evidence has indicated that lncRNAs widely participate in regulating the cellular biological processes of various malignancies, including GC. In epithelial ovarian cancer, RNF157-AS1 interacted with the EZH2 and HMGA1 proteins, suppressing DIRAS3 and ULK1-modulated autophagy, thus impeding cancer cell proliferation, invasion, and migration24. LncRNA ROR was proved to be overexpressed in breast cancer and acted as a cancer driver via recruiting histone transmethylase MLL1 to the TIMP3 promoter region25. LncRNA NEAT1 enhanced the stability of RPRD1B, accelerating the implantation of GC cells in lymph nodes through the c-Jun/c-Fos/SREBP1 axis26. LncRNA DIAPH2-AS1 functioned as an oncogenic gene and reflected a worse prognosis for GC patients, which promoted GC cell migration, invasion, and neural invasion by regulating the stabilization of NSUN2 and modulating the m5C modification of NTN127. Nevertheless, little is known about the biological function of LINC02139 underlying GC onset and progression. Here, we revealed the dramatically increased expression of LINC02139 in GC. Meanwhile, The cancer specimens from GC patients with ≥5 cm tumor sizes, T3-T4 invasion, lymph node metastasis, distant metastasis, and stage III-IV were more likely to exhibit high expression of LINC02139. Furthermore, in vivo and in vitro assays confirmed that overexpression of LINC02139 promoted, whereas the silence of LINC02139 suppressed GC cell proliferation, migration, and invasion, especially cell growth. These results consistently suggest that LINC02139 acts as a pivotal driver of cancer phenotypes in GC.
Distinctive functions of lncRNAs for regulating malignant biological processes were predominately dependent on lncRNA cellular localization. Generally speaking, nuclear lncRNAs are implicated in chromatin interactions, transcriptional modulation, and RNA processing, while cytoplasmic lncRNAs act as modulators for mRNA stability or translation and mediate cellular signaling pathways28,29,30. In this study, the results of subcellular fractionation analysis together with ISH assays showed that LINC02139 was distributed in the cell cytoplasm and nucleus of GC. LncRNA is inclined to exert biological effects by interacting with the targeted proteins31. For example, in triple-negative breast cancer, lncRNA GATA3-AS1 destabilized the GATA3 protein by advancing GATA3 ubiquitination, improving tumor progression and immune escape32. LncRNA TP53TG1 bound to CIP2A and triggered the ubiquitination-mediated degradation of the protein, resulting in the restraint of the PI3K/AKT pathway in GC33. Herein, we identified that XIAP was the specific binding protein of LINC02139. LINC02139 interacted with and stabilized XIAP by inhibiting the ubiquitin-proteasome degradation pathway, thereby upregulating XIAP protein expression. Through the aforementioned experiments, it was observed that overexpression of LINC02139 could promote GC cell proliferation, migration, and invasion, but hinder cell apoptosis in vivo and in vitro. Intriguingly, such effects could be rescued by XIAP depletion, hinting that the regulatory function of LINC02139 in the malignant biological processes is dependent on XIAP. The deubiquitination-regulated overexpression of XIAP is a critical mediator of LINC02139 to promote GC progression. Furthermore, the obtained results provided insight into the potential of the LINC02139-XIAP signaling axis for GC therapeutic targets.
XIAP is a potent and versatile suppressor of apoptosis that directly inhibits specific members of the caspase family of cysteine proteases11,34,35. In addition to caspase inhibition, XIAP is also found to be aberrantly elevated in numerous human cancer types, such as bladder cancer16, renal cell carcinoma36, Colorectal cancer37, glioma38, lung adenocarcinoma39, and melanoma40. However, the role that XIAP played in GC progression remains unclear and still needs to be further explored. In the present study, through bioinformatics methods and molecular biology experiments, an increase of the XIAP protein and mRNA expression was observed in human clinical GC tissues, predicting a poor prognosis for patients with GC. Moreover, there exists a positive correlation between LINC02139 and XIAP expression, implying these genes synergistically advanced GC. XIAP may be a promising therapy target for GC.
Taken together, we assessed the regulatory function of the LINC02139-XIAP axis in GC in this research. However, the detailed signaling pathway underlying XIAP in regulating GC needs to be explored in future studies.
In conclusion, this study reveals a mechanism that accounted for the tumor-driver role of LINC02139 in GC progression (Fig. 8). LINC02139 promotes GC cell growth, migration, and invasion and reduces apoptosis by binding to and deubiquitinating XIAP.

A hypothetical model portrays the roles that LINC02139 and XIAP exerted in GC.
These findings provide a new perspective for targeting the LINC02139-XIAP axis as a molecular biologic marker and therapeutic strategy for GC.
Methods
Cell culture and antibodies
GES-1, along with the MKN-45, SNU-719, SNU-5, SNU-1, HGC-27, and AGS were acquired from the American Type Culture Collection (ATCC; Manassas, VA, USA). All the cell lines were cultured in either a Dulbecco’s modified Eagle medium (DMEM; Gibco, Gaithersburg, MD, USA) or an RPMI-1640 medium (Sigma, St. Louis, MO, USA). These mediums were supplemented with 10% fetal bovine serum (FBS; Sigma, St. Louis, MO, USA). The culture took place in a humidified atmosphere at 37 °C with 5% CO2. The antibodies applied in the according experiments are listed in Supplementary Table 1.
Data source
RNA-Seq datasets GSE53137 and GSE95667 were downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo). The GSE53137 dataset consists of six pairs of GC tissues, while the GSE95667 dataset includes four pairs of GC tissues.
Tissue specimens
Seventy-two fresh human GC tissues and the corresponding normal samples were collected from patients who visited Nanfang Hospital, Southern Medical University (Guangzhou, China). The diagnosis was confirmed and staged by two experienced pathologists according to the 8th edition of the cancer staging manual from the American Joint Committee on Cancer (AJCC). All patients had not received any preoperative treatments, such as radiation therapy, chemo, or targeted therapy. The Ethics Committee of the Nanfang Hospital, Southern Medical University, gave its approval to this scheme.
In situ hybridization (ISH)
To investigate the expression of LINC02139 in GC tissues that were embedded in paraffin, an ISH assay was utilized. Boster (Wuhan, China) provided the oligonucleotide probes of LINC02139, which were labeled with digoxigenin. The specific investigation sequences were displayed as follows: 5’-CTAAATGCTCCCTGGGGCATCTCGTGTAACCTTTGCAGGA-3’; 5’-CTTGGCCTCCCAAAGTGCTGGGATTACAGGCATGAGCCAC-3’;5’-CCAGCAGTTCCACTTTTCGGTATTTATCTAGGAGAAATGA-3’. According to the previous study, this experiment was conducted following the guidelines provided by the relevant manufacturer41.
RNA extraction and qRT-PCR
The extraction of RNA was performed by utilizing TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the guidelines provided by the manufacturer. Using a Prime Script RT reagent kit (Takara Bio, Inc., Shiga, Japan), 500 ng of RNA was transcribed in reverse to generate first-strand cDNA. For qPCR, a SYBR Green Pro Taq HS Premixed kit from Takara Bio, Inc. in Shiga, Japan, was used. The cycling conditions included 30 s at 95 °C, followed by 40 cycles of 5 s at 95 °C and 30 s at 60 °C. The expression of gene mRNA was assessed using the 2−△△Ct method, with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) selected as the control. Supplementary Table 2 contains the listed primer sequences.
Protein isolation and western blotting
Cells or tissues were isolated and the total protein was determined using bicinchoninic acid to measure the concentration. After obtaining the cell lysate, it was analyzed using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred onto polyvinylidene fluoride membranes (PVDF, Merck Millipore, Billerica, MA, USA). Following the blocking step with 5% skimmed milk, the membranes were then exposed to the primary antibodies and incubated overnight at 4 °C. The following day, the membranes were incubated with the secondary antibodies at room temperature for 1 h, followed by thorough washing with TBST. Finally, chemiluminescence was applied to visualize the potential protein bands.
Oligonucleotide and plasmid transfections
The siRNAs for LINC02139 and XIAP and the controls (Scr-siRNA, scrambled-siRNA) were purchased from GenePharma (Shanghai, China). The full-length sequence and the truncations of human LINC02139 were cloned into a pcDNA3.1 plasmid vector. XIAP sequence and the truncations were constructed into a pcDNA3.1-3×Flag-6×His (tag) plasmid vector. Set empty vector as a control (Kidan Biosciences Co., Ltd, Guangzhou, China). Lipofectamine 3000 reagents (Invitrogen, Carlsbad, CA, USA) were applied for the transfection of siRNAs and plasmids followed by the manufacturer’s protocol. The specific sequences of siRNAs are shown in Supplementary Table 3.
Coding potential analysis
LINC02139 coding capacity was estimated using the Coding Potential Calculator (CPC) and the Coding-Potential Assessment Tool (CPAT). GAPDH and HOX transcript antisense RNA (HOTAIR) were set as the represents of protein-coding gene and non-coding gene, respectively.
Subcellular fractionation analysis
A Cytoplasmic and Nuclear RNA Purification Kit (Norgenbiotek Corporation, Thorold, ON, Canada) was utilized for the RNA extraction from GES-1 and AGS cells under the manufacturer’s instructions. qRT-PCR assays were performed to analyze the cytoplasmic and nuclear fractions distribution. We chose nuclear-enriched abundant transcript 1 (NEAT1) as the endogenous control for the nucleus, and GAPDH as the endogenous control for the cytoplasm. Related primers are displayed in Supplementary Table 2.
Colony formation assays
Cells were plated in 12-well plates with 400 cells/well. After being cultured for two weeks, the cells underwent two rounds of PBS washing for 5 min, followed by fixation with 4% paraformaldehyde for 15 min, and stained with 0.1% crystal violet for 10 min at ambient temperature. The number of colonies was counted by using a microscope.
5-ethynyl-2′-deoxyuridine (EdU) incorporation assays
The cells were placed in 96-well dishes in groups of three, with a density of 1 × 104 cells per well, and left to incubate for one night. Cell proliferation was evaluated using the Cell-Light EdU Apollo567 In Vitro Kit (RiboBio, Guangzhou, China) according to the instructions provided by the manufacturer. For the visualization of cells that were positive for EdU, an Olympus IX73 inverted fluorescence microscope (Olympus Corporation, Tokyo, Japan) was utilized.
Detection of apoptotic cells by flow cytometry
Cells after being transfected for 48 h with vector plus Scr-siRNA, or LINC02139 plus Scr- siRNA, or LINC02139 plus XIAP-siRNAp (siRNA 1; siRNA 2; siRNA 3; siRNAp, siRNA pool; 33.3%) were trypsinized and fixed by ice‐cold 70% ethanol for 30 min. After that, the transfected cells were incubated with 20 mg/mL RNase (Sigma‐Aldrich) at 37 °C for 1 h. For cell apoptosis analysis, cells were stained with Annexin V-FITC kit (YeaSen, Shanghai, China), and then apoptotic cells were detected by flow cytometry.
Hoechst 33258 staining for cell apoptosis analysis
Cells were seeded in 24-well plates at a density of 40–50% and cultured overnight. After being washed with PBS twice, the cells were fixed with 4% paraformaldehyde for 10 min, stained with 1 mg/mL Hoechst 33258 for 10 min, and washed extensively using PBS for 5 min. The apoptosis cells with fragmented and condensed nuclei were observed in three randomly selected fields under an inverted fluorescent microscopy and the apoptosis rate in each group was counted which was identified as the count of apoptotic cells as a percentage of the total number of observed cells.
Transwell migration and invasion assays
2 × 104 cells were resuspended with serum-free medium and incubated in the 8-μm pores Transwell chambers (Corning, New York, USA) for the examination of cell migration. For cell invasion analysis, 4 × 104 cells were plated in the Matrigel-coated chambers (BD Biosciences, New Jersey, USA). Medium containing 10% FBS was added to the 24-well plate that inserted chambers and continued the culture for 48 h in the incubator. Cells stuck to the lower side of the chambers were fixed with the 4% paraformaldehyde for 15 min and stained with 0.1% crystal violet for 10 min. After being washed with PBS extensively, cells were counted under an upright fluorescence microscope (Olympus CS43, Japan) in three randomly selected fields.
Wound healing assays
In cell wound healing analysis, transfected cells were seeded in 6-well plates and cultured to confluence. A 10 μL pipette tip was used to scratch a vertical wound per well, and the cell debris was eliminated with PBS. The cells were continuously incubated in a medium with 1% FBS. Throughout the process, pictures were taken to assess the dimensions of the existing injuries. The assessment of the migration index involved calculating the migration index using the formula [(initial wound width − width of the wound at the pictured time point) / initial wound width] × 100%.
RNA pull-down and mass spectrometry
An RNA pull-down kit (BersinBio, Guangzhou, China) was used to identify LINC02139-interacting proteins. To summarize, a biotin-labeled LINC02139 sense chain and an antisense control were synthesized and purified using T7 RNA polymerase and the RNAmax-T7 kit (RiboBio, Guangzhou, China). At room temperature, 50 μL of streptavidin magnetic beads and 50 pM RNA probes labeled with biotin were mixed and stirred for 30 min, followed by a 2-h incubation with AGS cell lysates. Proteins associated with RNA were gathered and separated using SDS-PAGE. The separation was followed by silver staining, as per the instructions provided by the manufacturer (Bio-Rad Laboratories, Hercules, CA). After undergoing mass spectrometry, the proteins that were obtained are listed in Supplementary Table 5.
RNA immunoprecipitation (RIP)
A Magna RIP Kit (Merck Millipore, US) was employed to perform the RIP experiment. In short, cells were lysed using RNA immunoprecipitation lysis buffer and then incubated overnight at 4 °C with 5 μg of either anti-XIAP, anti-6×His, or IgG. Next, RNA-protein immunocomplexes were gathered using protein A/G magnetic beads, followed by isolation, purification, and detection of RNA using qRT-PCR. The primers utilized in qRT-PCR can be found in Supplementary Table 2.
Protein stability assays
AGS cells were transfected with LINC02139-siRNAp or Scr-siRNA and were consequently incubated with 100 μM CHX (Sigma–Aldrich, USA) for the periods of 0, 3, 6, 9, and 12 h. At the indicated time points, cells were harvested and the proteins were resolved using western blotting to identify the abundance of endogenous XIAP protein.
Protein degradation assays
Ubiquitination assays were performed as reported previously42. Briefly, to examine the effects of LINC02139 on the ubiquitination of XIAP, cells were transfected with LINC02139-siRNAp or Scr-siRNA for 48 h. Cell protein samples were harvested after being treated with the proteasome inhibitor MG 132 (10 nM; Sigma–Aldrich, USA) for 8 h, and immunoprecipitated with anti-XIAP antibodies using protein A/G beads (Santa Cruz Biotechnology, Texas, USA) at 4 °C overnight. Then washed and boiled the magnetic beads in a loading buffer, and the immunoprecipitated protein complexes were separated by western blotting with anti-ubiquitination antibodies.
Haematoxylin and eosin (HE) staining
GC tissues embedded in paraffin with a size of 4 μm were subjected to baking at a temperature of 65 °C for 2 h. Subsequently, deparaffinization was carried out using xylene, followed by rehydration using a sequence of alcohol solutions. Next, the sections were treated with hematoxylin for 3 min, followed by differentiation using a 1% hydrochloric alcohol solution for 2 s, and finally stained with eosin solution for 1 min. Following that, the slides underwent dehydration and were subsequently sealed using neutral balsam.
Immunohistochemistry (IHC)
The GC tissues, which were embedded in paraffin, were sliced into 4 μm sections and then subjected to baking at 65 °C for 2 h. After that, they were deparaffinized in xylene for 20 min and rehydrated using a sequence of alcohol solutions. Following a 10-min immersion in 3% hydrogen peroxide to inhibit the peroxidase, the slides underwent microwave treatment in citrate buffer (pH 6.0) for antigen retrieval. Following the cooling to ambient temperature, the sections were obstructed using 5% BSA for 1 h and subsequently subjected to incubation with the specified primary antibodies overnight at 4 °C. On the next day, the sections were rewarmed for 1 h and washed with PBS. Afterward, they were exposed to a secondary antibody conjugated with HRP and incubated at room temperature for 1 h. To visualize the results, the chromogen tetrahydrochloride of 3,3’-diaminobenzidine was employed. Following that, the slides were stained with hematoxylin, dried out, and then protected with neutral resins. The protein expression was observed under a microscope.
Lentivirus and mouse models
The lentiviruses expressing LINC02139 were constructed and synthesized by Genechem (Shanghai, China) using the Ubi-MCS-SV40-Cherry vector. The empty vector was selected as a control. qRT-PCR assays were utilized to verify the infection efficiency. The shRNA of LINC02139 and XIAP was constructed and synthesized according to the corresponding siRNA2 by Genechem (Shanghai, China). The infection efficiency was verified using qRT-PCR and western blotting assays, respectively. To establish the LINC02139 overexpression and XIAP knockdown stable cell lines, lentivirus expressing LINC02139 and XIAP shRNA were co-transfected into the AGS cells. After 48 h transfection, puromycin or G418 (Sigma–Aldrich, USA) was employed to screen the stable clones.
BALB/c-nu/nu female mice (15–25 g, 4–6 weeks old) used in this study were all purchased from the Guangdong Medical Laboratory Animal Center. The mice were raised in a specific pathogen-free environment at the Laboratory of Medical Animal Center of NanFang Hospital. About 1 × 107 LINC02139-silencing cells or the control cells, Vector + Scr shRNA, LINC02139 + Scr shRNA, or LINC02139 + XIAP shRNA transduced AGS cells suspended in 100 μL PBS were subcutaneously injected into the right flanks of the mice. The tumor growth progression was monitored every three days with a vernier caliper and the tumor volumes were calculated by the measurements of tumor length (L), and width (W), V = L × W2/2. On day 28 after injection, all mice were anesthetized using ether and killed by cervical dislocation, and the tumors were isolated for the weight and image. HE and IHC staining were performed to confirm the tumor tissues. The animal experiments were accomplished according to the Guide for the Institutional Animal Ethical Committee with the approval of the Experimental Animal Ethics Committee of Southern Medical University.
Statistical analysis
All in vitro experiments were performed with three independent biological replicates and triple technical replicates. Quantitative data are exhibited as mean ± SD. Two-tailed Student’s t-tests were applied to analyze the quantitative data. Statistical analyses of the associations between LINC02139 expression and clinicopathological data were conducted by using the Chi-square test or Fisher’s exact test appropriately. We used the Kaplan–Meier method to plot survival curves and analyzed them using log-rank tests. Linear regression and Pearson correlation coefficients were employed to evaluate the correlation between gene expressions. For in vivo experiments, the nude mice were randomly grouped into five per group using a blinding method. Statistical analyses were conducted using the SPSS software (version 20.0, Chicago, IL, USA). A two-tailed P-value less than 0.05 was thought to be statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data generated or analyzed during this study are included in the article and supplemental files, or available from the corresponding author on reasonable request. All source data underlying the graphs and charts shown in the figures are presented in Supplementary Data File 1. Uncropped and unedited blots/gels are presented in Supplementary Fig. 7.
References
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021).
Smyth, E. C., Nilsson, M., Grabsch, H. I., van Grieken, N. C. & Lordick, F. Gastric cancer. Lancet 396, 635–648 (2020).
Zeng, Y. & Jin, R. U. Molecular pathogenesis, targeted therapies, and future perspectives for gastric cancer. Semin. Cancer Biol. 86, 566–582 (2022).
Onoyama, T., Ishikawa, S. & Isomoto, H. Gastric cancer and genomics: review of literature. J. Gastroenterol. 57, 505–516 (2022).
Mercer, T., Dinger, M. & Mattick, J. Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 10, 155–159 (2009).
Ulitsky, I. & Bartel, D. P. lincRNAs: genomics, evolution, and mechanisms. Cell 154, 26–46 (2013).
Sun, M. & Kraus, W. L. From discovery to function: the expanding roles of long noncoding RNAs in physiology and disease. Endocr. Rev. 36, 25–64 (2015).
Li, J. et al. The HOXD9-mediated PAXIP1-AS1 regulates gastric cancer progression through PABPC1/PAK1 modulation. Cell Death Dis. 14, 341 (2023).
Pei, M. et al. The LINC00501-HSP90B1-STAT3 positive feedback loop promotes malignant behavior in gastric cancer cells. Cell Signal. 108, 110711 (2023).
Xu, T. P. et al. KLF5 and MYC modulated LINC00346 contributes to gastric cancer progression through acting as a competing endogeous RNA and indicates poor outcome. Cell Death Differ. 26, 2179–2193 (2019).
Deveraux, Q. L., Takahashi, R., Salvesen, G. S. & Reed, J. C. X-linked IAP is a direct inhibitor of cell-death proteases. Nature 388, 300–304 (1997).
Eckelman, B. P., Salvesen, G. S. & Scott, F. L. Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep. 7, 988–994 (2006).
Edison, N. et al. Degradation of Bcl-2 by XIAP and ARTS promotes apoptosis. Cell Rep. 21, 442–454 (2017).
Lu, M. et al. XIAP induces NF-κB activation via the BIR1/TAB1 interaction and BIR1 dimerization. Mol. Cell. 26, 689–702 (2007).
Schimmer, A. D., Dalili, S., Batey, R. A. & Riedl, S. J. Targeting XIAP for the treatment of malignancy. Cell Death Differ. 13, 179–188 (2006).
Huang, C. et al. XIAP BIR domain suppresses miR-200a expression and subsequently promotes EGFR protein translation and anchorage-independent growth of bladder cancer cell. J. Hematol. Oncol. 10, 6 (2017).
Jeong, S. et al. Cannabidiol promotes apoptosis via regulation of XIAP/Smac in gastric cancer. Cell Death Dis. 10, 846 (2019).
Jiang, R. et al. Esculetin inhibits endometrial cancer proliferation and promotes apoptosis via hnRNPA1 to downregulate BCLXL and XIAP. Cancer Lett. 521, 308–321 (2021).
Hang, M. X. et al. Positive feedback regulation of lncRNA PVT1 and HIF2α contributes to clear cell renal cell carcinoma tumorigenesis and metastasis. Oncogene 40, 5639–5650 (2021).
Zhang, F. et al. LncRNA CRNDE attenuates chemoresistance in gastric cancer via SRSF6-regulated alternative splicing of PICALM. Mol. Cancer 20, 6 (2021).
Zhu, Y. et al. LncRNA LINC00942 promotes chemoresistance in gastric cancer by suppressing MSI2 degradation to enhance c-Myc mRNA stability. Clin. Transl. Med. 12, e703 (2022).
Silke, J. & Meier, P. Inhibitor of apoptosis (IAP) proteins-modulators of cell death and inflammation. Cold Spring Harb. Perspect. Biol. 5, a008730 (2013).
Suzuki, Y., Nakabayashi, Y., Nakata, K., Reed, J. C. & Takahashi, R. X-linked inhibitor of apoptosis protein (XIAP) inhibits caspase-3 and -7 in distinct modes. J. Biol. Chem. 276, 27058–27063 (2001).
Xu, P. et al. Differential effects of the LncRNA RNF157-AS1 on epithelial ovarian cancer cells through suppression of DIRAS3- and ULK1-mediated autophagy. Cell Death Dis. 14, 140 (2023).
Hu, A. et al. Long non-coding RNA ROR recruits histone transmethylase MLL1 to up-regulate TIMP3 expression and promote breast cancer progression. J. Transl. Med. 19, 95 (2021).
Jia, Y. et al. Long non-coding RNA NEAT1 mediated RPRD1B stability facilitates fatty acid metabolism and lymph node metastasis via c-Jun/c-Fos/SREBP1 axis in gastric cancer. J. Exp. Clin. Cancer Res. 41, 287 (2022).
Li, Y. et al. Long noncoding RNA DIAPH2-AS1 promotes neural invasion of gastric cancer via stabilizing NSUN2 to enhance the m5C modification of NTN1. Cell Death Dis. 14, 260 (2023).
Guttman, M. & Rinn, J. L. Modular regulatory principles of large non-coding RNAs. Nature 482, 339–346 (2012).
Batista, P. J. & Chang, H. Y. Long noncoding RNAs: cellular address codes in development and disease. Cell 152, 1298–1307 (2013).
Schmitt, A. M. & Chang, H. Y. Long noncoding RNAs in cancer pathways. Cancer Cell. 29, 452–463 (2016).
McDonel, P. & Guttman, M. Approaches for understanding the mechanisms of long noncoding RNA regulation of gene expression. Cold Spring Harb. Perspect. Biol. 11, a032151 (2019).
Zhang, M. et al. LncRNA GATA3-AS1 facilitates tumour progression and immune escape in triple-negative breast cancer through destabilization of GATA3 but stabilization of PD-L1. Cell Prolif. 53, e12855 (2020).
Fang, D. et al. m6A modification-mediated lncRNA TP53TG1 inhibits gastric cancer progression by regulating CIP2A stability. Cancer Sci. 113, 4135–4150 (2022).
Liston, P. et al. Suppression of apoptosis in mammalian cells by NAIP and a related family of IAP genes. Nature 379, 349–353 (1996).
Shi, Y. Caspase activation, inhibition, and reactivation: a mechanistic view. Protein Sci. 13, 1979–1987 (2004).
Li, F. et al. Kidney cancer biomarkers and targets for therapeutics: survivin (BIRC5), XIAP, MCL-1, HIF1α, HIF2α, NRF2, MDM2, MDM4, p53, KRAS and AKT in renal cell carcinoma. J. Exp. Clin. Cancer Res. 40, 254 (2021).
Liao, Y. et al. Inflammation mobilizes copper metabolism to promote colon tumorigenesis via an IL-17-STEAP4-XIAP axis. Nat. Commun. 11, 900 (2020).
Saha, G. et al. USP7 targets XIAP for cancer progression: establishment of a p53-independent therapeutic avenue for glioma. Oncogene 41, 5061–5075 (2022).
Wang, Y., Shen, L., Li, G., Chen, J. & Ge, R. Upregulation of XIAP promotes lung adenocarcinoma brain metastasis by modulating ceRNA network. Front. Oncol. 12, 946253 (2022).
Daoud, M. et al. XIAP promotes melanoma growth by inducing tumour neutrophil infiltration. EMBO Rep. 23, e53608 (2022).
Tang, W. et al. The p300/YY1/miR-500a-5p/HDAC2 signalling axis regulates cell proliferation in human colorectal cancer. Nat. Commun. 10, 663 (2019).
Wu, X. et al. USP3 promotes gastric cancer progression and metastasis by deubiquitination-dependent COL9A3/COL6A5 stabilisation. Cell Death Dis. 13, 10 (2021).
Acknowledgements
We sincerely acknowledge the generous support of the Guangdong Provincial Key Laboratory of Gastroenterology, Department of Gastroenterology, Nanfang Hospital, Southern Medical University and the Department of Gastroenterology, The Second Affiliated Hospital, School of Medicine, The Chinese University of Hong Kong, Shenzhen & Longgang District People’s Hospital of Shenzhen. This work was supported by the National Natural Science Foundation of China (Grant/Award numbers: 82273354 and 82372955), Guangdong Basic and Applied Basic Research Foundation (Grant/Award number: 2020A1515110059 and 2023A1515111049), Science and Technology Planning Project of Guangdong Province (Grant/Award number: 2017B020209003), Natural Science Foundation of Guangdong Province (Grant/Award number: 2024A1515012891), Shenzhen Science and Technology Innovation Commission Fund (Grant/Award number: JCYJ20210324135005013).
Author information
Authors and Affiliations
Contributions
J.Y.L., L.X., and J.D.W. designed and conceived this study. M.M.P., J.M.Z., and Z.Y. performed the experiments in vitro. Experiments in vivo were performed by M.M.P., J.K.W., and Y.P. Tissue samples were collected by Y.D.C., S.Y.P., X.Y.W., and P.Y. X.D.H., Y.C.X., and L.J.H. contributed to data analysis and interpretation. X.T.H., X.S.W., and W.M.T. performed the statistical analysis. S.D.L. and Y.C. helped with technical support. S.D.L. and J.D.W. coordinated the project. J.D.W. and J.J.L. supervised the project. M.M.P. wrote the manuscript and J.D.W. revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This study was approved by the Medical Ethics Committee of Nanfang Hospital, Southern Medical University, and Southern Medical University Experimental Animal Ethics Committee.
Peer review
Peer review information
Communications Biology thanks Han Tian and the other anonymous reviewer(s) for their contribution to the peer review of this work. Primary handling editors: Tuan Anh Nguyen and Mengtan Xing.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pei, M., Zhang, J., Yu, Z. et al. LINC02139 interacts with and stabilizes XIAP to regulate cell proliferation and apoptosis in gastric cancer. Commun Biol 7, 1497 (2024). https://doi.org/10.1038/s42003-024-07202-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-024-07202-5
