
Abstract
Plasmodium species replicate via schizogony, which involves asynchronous nuclear divisions followed by semi-synchronous segmentation and cytokinesis. Successful segmentation requires a double-membranous structure known as the inner membrane complex (IMC). Here we demonstrate that PfFBXO1 (PF3D7_0619700) is critical for both asexual segmentation and gametocyte maturation. In Toxoplasma gondii, the FBXO1 homolog, TgFBXO1, is essential for the development of the daughter cell scaffold and a component of the daughter cell IMC. We demonstrate PfFBXO1 forming a similar IMC initiation scaffold near the apical region of developing merozoites and unilaterally positioned in gametocytes of P. falciparum. While PfFBXO1 initially localizes to the apical region of dividing parasites, it displays an IMC-like localization as segmentation progresses. Similarly, PfFBXO1 localizes to the IMC region in gametocytes. Following inducible knockout of PfFBXO1, parasites undergo abnormal segmentation and karyokinesis, generating inviable daughters. PfFBXO1-deficient gametocytes are abnormally shaped and fail to fully mature. Proteomic analysis identified PfSKP1 as one of PfBXO1’s stable interacting partners, while other major proteins included multiple IMC pellicle and membrane proteins. We hypothesize that PfFBXO1 is necessary for IMC biogenesis, chromosomal maintenance, vesicular transport, and ubiquitin-mediated translational regulation of proteins in both sexual and asexual stages of P. falciparum.
Similar content being viewed by others

Introduction
Malaria remains a major healthcare challenge in many parts of the world. Plasmodium falciparum, the causative agent of the deadliest form of malaria in humans, continues to pose a significant global health threat, with >200 million cases and >500,000 fatalities reported each year1. Its lifecycle is a remarkable tale of adaptation and survival due to precisely orchestrated events within the mosquito and human hosts. The sporozoites, injected by the Anopheline mosquito, travel to the liver and invade hepatocytes, where they multiply and are released into the blood stream to invade red blood cells (RBCs) and proliferate. The intra-erythrocytic stages consists of two cycles, (i) the asexual cell division or schizogony and (ii) the sexual or gametocytogenesis for transmission2,3,4. During schizogony, the parasite within the RBC undergoes multiple asynchronous rounds of nuclear replications prior to a final semi-synchronous nuclear and cytoplasmic division by the developing cytoskeleton to form 16-36 individual daughter cells, known as merozoites5. These daughter merozoites egress and re-invade uninfected RBCs, resulting in the clinical symptoms observed in patients.
The parasite cytoskeleton plays a central role in maintaining its structural integrity and is essential for cell division and merozoite formation6. It is comprised of three membrane bilayers, the outermost parasite plasma membrane followed by the double membranous inner membrane complex (IMC)7. The IMC serves as a scaffold for organelle organization and segregation during schizogony8,9. Upon RBC invasion, the merozoite disassembles its cytoskeleton and establishes itself in a parasitophorous vacuole forming a ring stage. The ring stage develops into a metabolically active trophozoite stage, and undergo nuclear replication where parasite chromosomes are tethered to the centrosome by formation of microtubule organizing centers (MTOCs)3,10,11. Later in schizogony, a scaffold of proteins assembles to initiate IMC biogenesis, likely mediated by the vesicles at the apical end prior to nuclear segregation9,12. As the scaffold grows, associated with one end of the MTOC, the nuclear material at the other end replicates. Following centrosomal duplication, nuclear material within an intact nuclear membrane is separated by the mitotic spindles resulting in two nuclei, each with separate MTOCs13. This atypical mitotic division continues asynchronously without cytokinesis to form a multinucleated schizont, simultaneously replicating the cytoskeletal scaffold10,14. During the last few hours of schizogony, the replicating scaffolds on the periphery develop further, segregating the pool of cytoplasmic contents and the nuclei to form individual merozoites in a process known as segmentation5. The IMC anchors the development of an actomyosin complex known as glideosome on its outer surface, while associating with the alveolins forming the pellicle and the sub-pellicular microtubules (SPMTs) on the interior15. A robust glideosome and apical complex is crucial for the motility and invasion of the individual merozoites, while the alveolins and SPMTs provide the structural support and stability, enabling efficient invasion7,16,17. The IMC is also critical in establishing the polarity of the cell with the apical ring and other apical organelles towards one end and the basal complex at the other end6,18.
Beyond asexual replication, the parasite cytoskeleton is equally indispensable during the sexual stages of gametocytogenesis. The precise regulation of microtubules and IMC development promote sequestration of these stages until full maturation of male and female gametocytes19,20. Mature gametocytes then re-enter circulation and are available for further transmission by a mosquito. Within the mosquito midgut, the gametocytes transform into male and female gametes and this process of gametogenesis is heavily dependent on swift disintegration of the IMC and other cytoskeletal components, assisting zygote formation. Post gametogenesis, the IMC and other cytoskeletal components are synthesized de novo and are important for both ookinete and sporozoite development and motility16,21. Absence of the IMC or IMC-associated proteins such as alveolins resulted in fragile and inviable ookinetes. Successful traversing of midgut epithelium to reach the basal lamina and oocyst formation is dependent on a robust cytoskeleton, a critical step in the transmission of the parasite21. Our knowledge of proteins involved in IMC and its biogenesis have primarily been derived from the model apicomplexan Toxoplasma gondii. Vesicular RabGTPases such as Rab11a and Rab11b are known to be important for IMC biogenesis9,22. The IMC is dynamic with a variety of protein components that are specific for each stage of the parasite. Proteins such as GAP40, GAP50, and GAPMs are targeted directly to the IMC by their membrane domains, while other components such as MyoA and MTIP form a complex with GAP45 and are then transported to the IMC through the interaction with GAP45 and GAP5023,24,25. The alveolins are a group of 13 proteins that form the sub-pellicular network in association with the SPMTs. The alveolin composition varies with the stage with some proteins predominantly or exclusively present in the asexual, sexual, or mosquito stages16,26. Several other proteins such as PhIL1, Coronin, and DHHCs (palmitoyl transferases) have been identified to be part of IMC and their absence is reported to affect the growth and development of the parasite16,27,28. Post-translational modifications such as phosphorylation, N-myristoylation, and palmitoylation are important for the localization and function of several of these IMC proteins23. Tight regulation of the assembly and disassembly of these components is crucial for growth and development of the parasite. Yet, how this regulation is brought about, and the factors involved remain largely unknown.
F-box proteins are an evolutionarily diverse family of proteins that share a domain of about 50 amino acid residues known as the F-box domain. First identified in the human F-box protein/cyclin F, the F-box domain is a protein interaction domain that binds SKP1, an adaptor that links F-box proteins to cullin-1 to form the SCF (SKP1/Cullin-1/F-box protein) complex. The SCF-complex is one of several Cullin-RING-ligase families that dock with ubiquitin-charged E2 proteins to assemble polyubiquitin chains on target proteins as a signal for their recognition by factors that deliver them to the 26S-proteasome for degradation29,30. Most F-box proteins possess C-terminal protein-protein interaction domains such as WD40-repeat domains and leucine-rich repeat domains, that determine the nomenclature of these proteins and mediate substrate selection. Some F-box proteins appear to lack substrate interaction domains, suggesting that their F-box domain mediates conditional self-destruction as has been proposed for the Dictyostelium discoideum FBXO protein, JcdI31. Besides degradation, ubiquitination of substrates is a versatile post-translational modification that regulates a plethora of functions, including subcellular localization and cell cycle regulation within the cell32,33. Recently 18 putative F-box domain-containing proteins were identified in Toxoplasma gondii, with four having homologs in Plasmodium falciparum34. PfFBXO1 (PF3D7_0619700) is a homolog of TgFBXO1 (TGGT1_310930). In T. gondii, TgFBXO1 localizes to the apical region of the developing daughter cells, in the region where the developing IMC and SPMTs nucleate during its cell division, known as the Daughter Cell Scaffold (DCS). This protein is important for the growth and development of T. gondii34. The role of this F-box containing protein in P. falciparum remains largely unknown, though a recent study in P. berghei showed that PbFBXO1 is important for schizont development and crucial for gametogenesis and ookinete development35. In the current study, we study the localization and importance of PfFBXO1 in the intra-erythrocytic stages of P. falciparum using epitope-tagged and inducible knockout transgenic strains. In addition, we performed affinity-based and proximity-based interactome analysis using TurboID36 to identify the interacting partners of the PfFBXO1 in schizogony and gametocytogenesis. Understanding the role of FBXO1 in P. falciparum and its connection to ubiquitination pathways will not only help unravel the regulatory process that is fundamental for the maintenance of protein homeostasis and cell cycle progression in the parasite, but also holds promise for the possibility of novel therapeutic target identification.
Results
Expression and Localization of PfFBXO1 in intra-erythrocytic stages
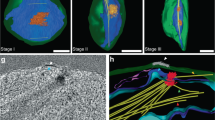
PfFBXO1 is a 633 amino acid protein with a single F-box domain, a predicted N-terminal myristoylation site, and no accompanying C-terminal domains. To investigate the localization of PfFBXO1, we attached the high-avidity epitope tag, spaghetti monster-V5 (smV5)37 to the C-terminus of PfFBXO1, and integration of the tag was confirmed by PCR (Supplementary Fig. 1A). By immunofluorescence assay (IFA), PfFBXO1 initially appears as a pair of dots near the outer edges of each dividing nucleus in the early schizont stage (5–8 nuclei). As the schizont develops, it forms a semicircular structure around the centrosome that extends to form a ring (Fig. 1A). PfFBXO1 initially colocalizes with PfGAP4538, an IMC-associated protein, but becomes more diffuse as segmentation progresses and the IMC envelopes the nascent merozoites (Fig. 1B). By comparison, PfFBXO1 is close but does not colocalize with the epitope-tagged basal complex protein, PfBLEBmyc (Fig. 1C). Given the challenges of resolving cellular structures in schizonts, we utilized ultrastructure expansion microscopy (U-ExM)39,40 to better characterize the localization of PfFBXO1.

PfFBXO1 localization by immunofluorescence stained with anti-V5 (PfFBXO1) and A anti-CrCen (Centrin, centrosome marker), (B) anti-PfGAP45 (IMC marker), or (C) anti-myc (PfBLEB, basal complex marker). Scale bar = 2 µm for (A–C). Ultrastructure Expansion Microscopy (U-ExM) of early stage schizont showing PfFBXO1 localizing to the developing daughter cell scaffold and co-stained with (D) anti-CrCen (Centrin) or (E) anti-PfGAP45 (IMC). Scale bar = 5 µm for (D, E). Inset panel shows 5X zoomed image. Shown here are representative images from at least 3 biological replicates. F Western blot of smV5-tagged PfFBXO1 showing the expression throughout schizont development, indicated by hours post-infection above each lane. G Graph showing the relative expression of smV5-tagged PfFBXO1 protein at respective time points. Values represented are mean with SEM from two biological replicates, each in done in technical triplicate.
By U-ExM, PfFBXO1 is associated with the cytoplasmic extension of the nuclear envelope-embedded microtubule-organizing center or centriolar plaque41,42 (Fig. 1D). The centriolar plaque, often referred to as the centrosome, resembles a tree-like structure in a cross-section, with microtubules forming the roots embedded in the dividing nucleus. In centriolar plaques of actively dividing nuclei, the cytoplasmic extension of the centriolar plaque has two branches with centrins at the junction of the branches (Fig. 1D), and the peripheral region of these branches exhibit staining for PfFBXO1 with PfGAP45 nearby (Fig. 1D, E). These branches resemble two “C” shaped/hook-shaped structures facing each other. As the nuclear division progresses, the branches are pulled apart, and rhoptries begin to develop beneath each of the branches. These PfFBXO1-stained regions mature to take on a disc-like shape, which might be a precursor to the apical rings. As segmentation concludes and daughter merozoites are released, PfFBXO1 becomes undetectable by IFA. Immunoblot analysis of tightly synchronized parasites during the different stages of schizont development shows that PfFBXO1 is first detectable at 34–38 h post-invasion (h.p.i.) with a peak expression at 44–48 h.p.i. (Fig. 1F). PfFBXO1 protein expression in late stages was seen by western blot and not by IFA, likely reflecting the bulk nature of the lysate from many parasites and the increased sensitivity of the immunoblot technique.
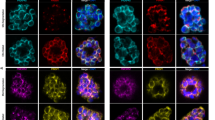
In the sexual transmission stage of the parasite, known as gametocytes, PfFBXO1 localizes to the IMC. In stage I/II gametocytes, PfFBXO1 is visible and localizes along the developing IMC-plates (Fig. 2A), identified by co-staining with antisera for PfGAP45. Additionally, PfFBXO1 is peripheral to the SPMT network (Fig. 2B), providing additional evidence for the IMC-associated localization of PfFBXO1. PfBLEB is another gametocyte marker that localizes to the regions of the gametocyte plasma membrane that do not have underlying IMC43. As expected, PfBLEB localizes to the opposite side from PfFBXO1 (Fig. 2C). U-ExM analysis provides a more detailed localization of PfFBXO1, which appears speckled and in close proximity to the IMC (Fig. 2D). It is worth noting that, though PfFBXO1 localization is close to PfGAP45, it does not exactly co-localize with it by U-ExM (Fig. 2D), it is exterior to the SPMTs (Fig. 2E, F and Supplementary Fig. 1B) and is distinct from the PfBLEB compartment (Fig. 2G). Considering the IMC-like localization of PfFBXO1 in both asexual and sexual stages of development, we sought to uncover its role using an inducible knockout parasite line.

Localization of PfFBXO1 by immunofluorescence in gametocyte stages stained with anti-V5 (PfFBXO1) and (A) anti-PfGAP45 (IMC marker), (B) anti-alpha-Tubulin (sub-pellicular microtubule, SPMT, marker), or (C). anti-myc (PfBLEB). Shown here are max-projections with Scale bar = 2 µm for (A–C). U-ExM (max-projections) showing PfFBXO1 localization at (D) the IMC plates similar to PfGAP45 in stage II gametocyte, (E) exterior to that of SPMTs visualized by anti-alpha-Tubulin in a stage IV gametocyte, and (F) single z-stack of an unexpanded stage IV gametocyte measuring the distance between two peaks of SPMT staining by anti-alpha-tubulin and anti-V5 (PfFBXO1) staining along the gray line. G Max-projection of U-ExM showing PfFBXO1 distinct from PfBLEB compartment in a stage III gametocyte. Scale bar for U-ExM = 10 µm (D, E and G). Shown here are representative images of results from at least 3 biological replicates.
PfFBXO1 is essential for schizogony and proper segmentation
To evaluate if PfFBXO1 was required for asexual replication, we generated an inducible knockout (iKO) strain by flanking its coding region with loxP sites in a 3D7-DiCre parental strain, that expresses the rapamycin-dimerizable split-Cre recombinase44,45 (Fig. 3A). In this strain, we also included a mNeonGreen (mNG) fluorescent tag fused to the codon altered PfFBXO1 to follow PfFBXO1 localization in live parasites. The strain was designed such that mRuby2 would be expressed upon excision of PfFBXO1mNG following rapamycin treatment (Fig. 3A). The integration of the construct at the endogenous locus and successful excision upon rapamycin treatment was confirmed by locus PCR (Supplementary Fig. 2A, B) and whole-genome sequencing (sequence reads deposited in NCBI Sequence Read Archive, #PRJNA1151667). Despite our clear visualization of PfFBXO1 when it was fused to the smV5 tag by immunofluorescence, we were unable to detect PfFBXO1-mNG expression in live schizonts, likely due to its low expression level. Despite this, we noticed a clear growth phenotype in the parasites upon rapamycin treatment.

A Schematic representation of DiCre system used for creating the inducible knockout parasite strain. B Flow cytometry-based growth curve analysis shows inability of PfFBXO1-iKO parasites to replicate. C Graph depicting reduced parasitemia at the end of first replication cycle in rapamycin treated parasites. D Graph representing SYBR green-based DNA content assay within a single asexual replication cycle showing the ratio of 1 N, 2 N, 3 N, 4 N and ≥5 N stages to the total parasitemia at specific time-points post-infection, in control and PfFBXO1-iKO parasites. * (Asterisk) denotes parasites that are likely inviable (see E), but SYBR green staining cannot distinguish dead 1 N parasites from viable rings. Data represented in the graphs (B, C and D) are means with error bars representing the standard deviation from technical triplicates from one representative biological replicate experiment (out of three). Individual data points are available in the Supplementary Data 2 file. E Hemacolor-stained smears of parasites treated with DMSO (control) and rapamycin (PfFBXO1 knockout) sampled at different time-points post-infection showing an inability of PfFBXO1-deficient parasites to form new rings. Scale bar = 5 µm.
Following rapamycin-induced knockout of PfFBXO1, parasites arrest within a single asexual cycle. This result was confirmed quantitatively in a flow cytometry-based replication analysis of these parasites with and without induced PfFBXO1 knockout (Fig. 3B). To better characterize the timing of the replication defect, we monitored parasitemia by flow cytometry at 4-h intervals and additionally confirmed that the defect was noticed at the end of the first asexual cycle (Fig. 3C). These same data also allowed quantification of DNA content per parasite as well (Fig. 3D). No significant difference in the amount of nuclear material per parasite was present until 46 h.p.i. At 46 and 50 h.p.i. the control line, as expected, showed a significant decrease in ≥5 N stages of schizonts and marked increase in 1 N ring stages, while the rapamycin-treated iKO parasite line showed a majority of ≥5 N stage schizonts (Fig. 3D). Data from additional replicates are provided in the Supplementary Fig. 3A, B. Field staining of the knockout parasites at corresponding time-points during the cycle showed aberrant schizonts starting from about 38 h.p.i with significantly fewer number of newly formed rings (Fig. 3E). It also showed that though the rapamycin-treated parasites were able to egress, fewer merozoites were released, with a considerable number of them stuck to the residual bodies. Most of the seemingly free merozoites were unable to re-invade to form rings, while those that formed ring-like structures within the RBCs did not survive to become trophozoites. We quantified and confirmed these results by manually counting the parasites using hemacolor-stained thin smears at 4-hour intervals (summarized in Supplementary Fig. 3C). It is worth noting that in rapamycin treated parasites, infected RBCs were difficult to find in the smear at 60 h.p.i., and the counts represent the overall number of ring-like structures identified and not necessarily live parasites. We next set out to identify the reason behind the observed growth phenotype using IFA and U-ExM ultrastructure analysis.
PfFBXO1 is important for IMC biogenesis and organization of apical organelles
PfFBXO1-deficient parasites failed to form new viable rings. No significant defects were observed at the outset in DNA replication or in the development of early stage schizonts by hemacolor-staining (Fig. 3E). To further evaluate the defect, we performed IFAs of the DMSO-treated and rapamycin-treated knockout parasites. Monitoring the IMC development by IFA using anti-PfGAP45 and anti-PfIMC1g antibodies, we noticed impaired IMC in the PfFBXO1-deficient parasites (Fig. 4A) during later stages of schizogony and segmentation. Plasma membrane staining of parasites with anti-PfMSP1 antibody showed dysmorphic plasma membrane staining with aggregates of incompletely segmented merozoites, similar to the IMC abnormalities (Fig. 4B). Alveolin network formation was also affected and showed abnormalities with PfIMC1g staining (Fig. 4C). Careful evaluation of the IFAs revealed that though the nuclear material replicated normally (by flow-based assay as shown in Fig. 3D), the process of karyokinesis remained incomplete resulting in connected nuclei within the aggregates of the knockout parasites (Fig. 4A–C). In addition, staining of SPMTs by alpha-tubulin and rhoptries using PfRON4 showed a disorganized pattern compared to the controls (Fig. 4A, C). The observed phenotype in PfFBXO1-deficient parasites could be due to either (i) a delay in the progression of karyokinesis as evidenced by the presence of still dividing nuclei within FBXO1-deficient schizonts or (ii) a result of impaired IMC development, leading to improper segmentation of the daughter merozoites. To ensure that this was not a result of improper staging but a phenotype in similarly staged parasites, we quantified the extent of nuclear packaging within the PfGAP45/PfIMC1g-stained membranes in E64-arrested “post-egress” parasites, identified by PfAMA1 translocation46,47. This quantification of proper daughter cell packaging performed by counting the number of IMC enclosed nuclei into uninucleate, multi-nucleate, and anucleate daughter cells/unit showed a significant difference between the DMSO-treated controls and rapamycin-treated PfFBXO1 knockout parasites (Fig. 4D). Supplementary Video 1 shows the complete z-stacks of the control and rapamycin treated parasites, depicting the extent of the abnormal development in the PfFBXO1-deficient parasites.

A Representative IFAs of E-64 treated egress-ready DMSO or rapamycin treated PfFBXO1-iKO parasites showing defects in SPMTs (visualized by anti-alpha-Tubulin), IMC (anti-PfGAP45), and nuclear staining (Hoechst). B Similar parasites co-stained with anti-PfMSP1 (parasite plasma membrane marker) and anti-PfGAP45 (IMC). C Similar parasites co-stained with anti-PfIMC1g (alveolin marker) and PfRON4 (rhoptry marker). For (A–C), PfAMA1 translocation was used as a marker to identify E-64 stalled “post-egress” schizonts. Scale bar = 2 µm. D Violin-plot representation quantifying the number of normally segmented merozoites compared to abnormal (multi or a-nucleated zoites) in PfAMA1 translocated schizonts in DMSO treated controls (N = 36) and rapamycin treated PfFBXO1-iKO parasites (N = 36) from biological triplicate experiments. Significance calculated by two-way ANOVA (**** represents p < 0.0001). Representative z-projected U-ExM images of E Early-stage control and PfFBXO1-knockout parasites showing no significant difference in centrosome replication or nuclear microtubules. F U-ExM of late-stage parasites showing segmentation defects, and mislocalization of SPMTs (marked by red arrow heads), long and convoluted rhoptries (indicated by white arrows) in rapamycin treated parasites. Basal complex (BC) is used as an indicator of similar stages for comparison. 5X zoomed panel highlights an anucleated zoite containing only the apical organelles. Scale bar = 10 µm. G Graph showing significant increase of rhoptry-length in rapamycin treated PfFBXO1 knockouts (N = 9) compared to DMSO treated ones (N = 7) measured from 3 biological replicate preparations of U-ExM samples (**** represents a p < 0.0001, calculated by unpaired two-tailed parametric t-test with Welch’s correction). All IFA and U-ExM images are representative images of experiments performed with at least three biological replicates.
U-ExM of control and rapamycin-treated parasites further clarified the severity of segmentation defects in PfFBXO1-deficient parasites. The NHS ester general protein staining of early schizont stages were largely similar in both control and knockout parasites, with similar MTOC, centrin, and tubulin within the mitotic spindles (Fig. 4E), despite a few additional ill-defined regions of staining in NHS ester of the knockout parasites. However, in the late stage schizonts, the phenotype was more pronounced with the presence of anucleate units and central aggregates with multiple nuclei and apical organelles within a large “megazoite” surrounded by a connected membrane structure (Fig. 4F). This finding is similar to what was shown by PfGAP45, PfIMC1g, and PfMSP1 staining in standard IFAs. Sub-pellicular microtubules in the PfFBXO1-deficient parasites were mislocalized. For example, the SPMTs were occasionally displaced from each other or went in opposite directions, and in some cases, we noticed short extensions beyond the apical polar ring (highlighted by red arrowheads in Fig. 4F). In addition, we noticed that the rhoptries in PfFBXO1 knockout parasites had elongated necks and were convoluted, unlike a shorter and straighter rhoptry necks of the controls (indicated by white arrows). Quantification of the rhoptry lengths from the apical polar ring to the base of the bulb showed significantly longer rhoptries in the PfFBXO1-deficient parasites with a median expanded length of 2.53 microns compared to a median of 2.26 µm in the PfFBXO1-sufficient expanded parasites (Fig. 4G). The basal complex rings were noticeable and contracted in the few normally appearing or anucleate zoites/units, while they could not be distinguished in the aggregates (Fig. 4C). The stage of the parasite was gauged based on the condensed shape of the nuclei and the contraction of the basal membrane ring as seen in the NHS ester staining (arrows). BODIPY-TR ceramide staining to monitor the overall membrane structure further confirmed the improper segmentation phenotype (Supplementary Fig. 4). Knockout parasites showed less-defined membrane staining compared to the uniformity of staining of the membrane structures in controls (Supplementary Fig. 4 and Supplementary Video 2). Building on these findings, our next step was to characterize the role of PfFBXO1 in gametocyte development.
PfFBXO1 is essential for maintaining structural integrity and maturation of gametocytes
We further used the iKO line to study the effect of PfFBXO1 knockout during gametocytogenesis. PfFBXO1mNG knockout was induced in parasites by addition of rapamycin at the stage of committed rings post-gametocyte induction. This stage was deliberately chosen because the PfFBXO1-knockout parasites failed to re-invade, resulting in the absence of committed rings if treated with rapamycin earlier. However, this prevented us from studying the effect of PfFBXO1 on gametocyte induction itself. The PfFBXO1 knockout gametocytes showed significant defects in gametocyte maturation. Standard IFAs with antibodies against the IMC and tubulin of the gametocytes showed that the knockout parasites had abnormal shapes, while the PfFBXO1-sufficient control gametocytes preserved their morphological structure (Fig. 5A). U-ExM showed defects in IMC biogenesis and assembly of SPMTs in knockouts (Fig. 5B). Staining with IMC-markers, PfGAP45 or PfPhIL1, showed decreased levels of staining along the putative IMC in PfFBXO1-deficient parasites compared to the control parasites (Fig. 5A, B and Supplementary Fig. 5). In addition, the SPMTs also showed significant assembly defects with reduced staining and occasional breaks. These results demonstrate a defective IMC in PfFBXO1-deficient gametocytes. BODIPY-TR ceramide staining showed that there was indeed a defective membrane structure in the PfFBXO1-deficient parasites as compared to the controls. While the control parasites showed a clear IMC plate structure and plasma membrane staining along the region of expected IMC, in the PfFBXO1-deficient parasites, we were unable to visualize these features (Fig. 5C). We also noted that PfFBXO1-deficient parasites in U-ExM appeared to be flatter in three-dimensional reconstructions compared to their control counterparts (Supplementary Video 3). We further set out to determine if PfFBXO1-deficient gametocytes were able to progress through gametocytogenesis and survive until maturation. Monitoring of the gametocyte cultures by microscopy of hemacolor-stained cultures showed an increase in abnormal looking parasites post-stage III (corresponding to day 6) in PfFBXO1-knockout line (Fig. 5D). Furthermore, staging of the parasites on days 4, 6, 8, 10 and 12 using hemacolor-staining showed that the rapamycin-treated line accumulated increased numbers of abnormal/dead parasites throughout their maturation. We noticed similar number of stage II and stage III gametocytes as that in control, however, in the absence of PfFBXO1, the majority of the gametocytes could not progress to normal stage IV and stage V (Fig. 5D). Upon measuring gametocytes in ~100 fields of view, we found that the roundness (calculated by 4*area*π-1*major_axis-2) of the rapamycin-treated gametocytes was significantly higher than that of the DMSO-treated controls with the progression of gametocytogenesis. While there was no significant difference in the shape of the gametocytes on Day 6, the roundness of PfFBXO1-deficient gametocytes increased significantly on days 8 and 10 (Fig. 5E) in contrast to expected elongation (thereby decrease in roundness) as seen in controls. Both the control and knockout gametocytes had a mean roundness score of 0.57 ± 0.08 and 0.59 ± 0.12, respectively on day 6 (mostly stage III). While the roundness score of control gametocytes decreased to a median of 0.38 ± 0.12 on Day 8 (mostly stage IV) and 0.49 ± 0.19 on day 10 (mostly stage V), the roundness score of PfFBXO1-knockout gametocytes had a median of 0.56 ± 0.12 on day 8 and increased to 0.69 ± 0.19 on day 10, respectively.

A Representative IFA of a stage III gametocyte showing the effect of PfFBXO1 depletion on SPMTs (anti-alpha-Tubulin) and IMC (anti-PfGAP45). Scale bar = 2 µm. B U-ExM of PfFBXO1-deficient parasites showing dysmorphic IMC and SPMTs compared to controls. Insets highlight the reduced PfGAP45 and tubulin staining at one of the ends of a stage III gametocyte. C U-ExM image of a stage III gametocyte showing absence of IMC sutures and broken membranes visualized by NHS ester and BODIPY-TR Ceramide staining in rapamycin treated parasites compared to the uniform membrane staining and distinct IMC-sutures in controls. Arrowheads show the presence of IMC plates. Scale bar = 10 µm for (B, C). D Area-plot showing the progression of gametocytogenesis and gametocyte maturation. The values plotted are number of gametocytes at a given stage on the specified days after induction, determined by manual counting of 50–100 high power fields of Hemacolor-stained smears from two biological replicates. Mean values are plotted with error bars representing the standard deviation. Representative image of the staging is provided as an index. Scale bar = 2 µm. E Scatterplot quantifying the morphological abnormality observed in the rapamycin treated PfFBXO1-deficient gametocytes based on their shape. A roundness score of 1 indicates a perfectly round or spherical structure. Bars represent the median value with the 95% CI. The quantification was performed on 3 technical replicates each, in two separate induction experiments (biological replicates). Shown here is the result from one biological replicate. Significance is calculated using unpaired t-test with Welch’s correction.
To decrease any potential artifacts induced during the process of fixation for IFA or U-ExM, we corroborated our results by live-cell microscopy. Published transcriptomic data showed PfFBXO1 expression to be significantly higher in gametocytes compared to that of schizonts48, which prompted us to try to detect PfFBXO1mNG in gametocytes by live-cell microscopy. Confocal imaging of live gametocytes showed IMC-like expression of PfFBXO1mNG starting from stage II gametocytes until stage V gametocytes (Fig. 6A). Live cell microscopy of progression of gametocytogenesis quantified by staging about 25 fields of view indicated similar results to that of the fixed hemacolor-staining. While the DMSO treated PfFBXO1-sufficient parasites showed normal progression of gametocytes, forming stage IV and stage IV gametocytes on day10 and day12, the rapamycin-treated PfFBXO1-deficient parasites showed increasingly shrunken and abnormally shaped parasites expressing mRuby2 (Fig. 6A) beyond stage III (or since day 8). SiR-tubulin staining corroborated the SPMT mislocalization, observed in IFAs and U-ExM (Fig. 6A and Supplementary Video 4). Quantification of the gametocyte shape in live cells showed a significant increase in the roundness of the rapamycin-treated gametocytes on day 8 compared to the DMSO-treated controls or rapamycin treated parental control (Fig. 6B). Live cell microscopy also corroborated the subsequent increase in the degree of roundness in PfFBXO1-deficient gametocytes past stage III (Supplementary Fig. 6). All measurements were performed in at least two independent biological replicates of gametocyte inductions.

A Representative images of control and knockout stage II gametocyte on Day 4, stage III on Day 6, and stage IV on Day 8 post-induction showing increased abnormalities in shape and SPMTs (visualized by SirTubulin) upon rapamycin treatment as gametocytogenesis progresses. Scale bar = 2 µm. B Scatter plot quantifying the shape of the gametocyte depicts increased roundness upon rapamycin treatment in PfFBXOmNG gametocytes compared to controls, as measured in live gametocytes by confocal microscopy. The results plotted in the graph are values from three technical triplicates showing median (line) with 95%CI (bars). Significance is calculated using unpaired two-tailed t-test with Welch’s correction.
Protein Interactors of PfFBXO1
FBXO1 is known to be part of the SCF (SKP1, cullin and F-box) E3 ubiquitin ligase protein complex in yeast and human cells49,50. To evaluate the presence of the complex in P. falciparum, we performed affinity pull-down assays using the smV5-tag. We successfully identified only PfSKP1 in the immunoprecipitations, while cullin and ring ligases were absent (Supplementary File 1). Considering the fragile nature of interaction of this complex, we hypothesized that the stringent RIPA buffer might have been too harsh to preserve the interactions, and so we repeated the affinity-mass spectrometry in milder buffer conditions using TritonX-100 instead of RIPA. However, even under these milder conditions, PfSKP1 was the only reliable interacting partner identified (Supplementary File 1). This corroborated with previously established data that FBXO1 primarily binds to SKP1 which then binds to the other components of the complex. This could be a result of several factors including the strength of the interaction and solubility of interacting proteins. To overcome this limitation, we used an enzyme-catalyzed proximity labeling method, TurboID36,51.
A promiscuous biotin ligase (TurboID) was genetically fused to PfFBXO1 (Supplementary Fig. 7A). This biotin ligase covalently adds a biotin to the free amine-group of lysine residues of proximal proteins, including direct, indirect, and merely proximal proteins, upon incubating with exogenous biotin under native conditions36. Owing to the strength of the covalent bond between the biotin and streptavidin anchors, the labeled proteins can be purified under harsh conditions. Despite the caveat of also capturing non-interactors that are merely within the proximity of the tagged protein, this enables capture of putative weak-interactors and transiently interacting proteins. After checking the expression of PfFBXO1 during schizogony and the biotinylation pattern of parasites at different stages of schizogony by western blot, we decided on 42–46 h.p.i schizonts for our analysis. Efficacy of biotinylation was confirmed by multiple methods including U-ExM in both schizonts and gametocytes (Supplementary Fig. 7B, C). Streptavidin pull-down of 42–46 h.p.i. synchronized PfFBXO1-turboID tagged schizonts identified a total of 890 biotinylated proteins present in 3 biological replicates, after excluding contaminants and streptavidin. Of these, 866 proteins were found to be significantly highly biotinylated with a fold-change of ≥ 2 and identified with 2 or more unique peptides and an FDR of <0.001 (Fig. 7A). PfSKP1 was identified as one of the significant proteins with a 5-fold difference. Several IMC pellicle and glideosome proteins, including PfIMC1c, PfIMC1g, PfIMC32, PfGAP40, PfGAP50, PfGAPM1, PfGAPM2, PfPhIL1, and PhIL 1-interacting proteins along with basal complex proteins such as PfBLEB, PfCINCH, and PfMORN1 were among the top proteins identified. This corroborates the fact that PfFBXO1 is localized to the IMC. In addition to these, several cell cycle regulators, DNA replication, and centrosome associated proteins including CRKs, PfCDPK1, PfCentrin 2, spindle and kinetochore-associated protein 2 (PfSKA2), chromatin assembly factor (PfCAF1), PfSMC3 and MCM proteins were also identified. Gene Ontology (GO)-based enrichment analysis of the significant proteins revealed the enrichment of proteins localized to IMC pellicle, nucleus and vesicles (Fig. 7B). Although we identified two ubiquitin-like proteins, four E3 ligases, including two HECT-type ubiquitin ligases (Ubiquitin activating enzyme 1 and deubiquitinating enzyme MINDY), we did not identify cullin proteins or ring ligases in our list of significantly enriched proteins.

A Volcano-plot depicting the highly biotinylated proteins (in red) against the proteins identified in ~42 h.p.i. schizonts (compiled from 3 biological replicates). B Bubble plot representing the top 10 enriched Gene Ontology-based cellular component terms of the significantly biotinylated protein list in schizont stages. The enrichment analysis was performed using PlasmoDB. C Significantly biotinylated proteins (from 3 biological replicates) in stage III gametocytes represented by red in a volcano plot. D Top ten enriched categories within the GO-based cellular component analysis of the proteins significantly biotinylated in stage III gametocytes. The bait protein, PfFBXO1 and its known interactor PfSKP1 are highlighted in green. E Venn-diagram showing the overlap of the proteins identified by RIPA-based affinity MS/MS, triton X100-based affinity MS/MS in schizonts, and TurboID experiments in schizonts and stage III gametocytes.
We also performed proximity-based TurboID analysis in stage III gametocytes to study the interacting partners during gametocytogenesis. For this, we employed the NF54iGP252 strain of parasites transfected with TurboID-tagged PfFBXO1. We analyzed 3 biological replicates of biotin-treated and untreated control gametocytes harvested as stage III gametocytes. Data-independent acquisition mass spectrometry analysis of the samples resulted in the identification of a total of 1579 proteins. Of these, 581 proteins were found to be significantly upregulated (≥2-fold and strict FDR of <0.001) in biotin-treated samples compared to that of the untreated controls (Fig. 7C). The list included numerous expected proteins including PfSKP1, IMC and glideosome proteins like PfGAP40/45/50, PfGAPM1-3, PfMTIP, PfPhil1, PfPIP1/2/3, and PfPhIL1 interacting candidates (PIC) 3 and 6 among other IMC proteins like PfISP1/3, PfIMC32 and PfIMC1i. In addition to these, we also identified PfBLEB, PfCDPK1 and 7, along with several CCR4-NOT complex associated proteins. In gametocytes, we identified both cullin 1 and 2 proteins, but they failed to cross the significant threshold. GO-based enrichment of the significantly identified proteins showed an enrichment of IMC, plasma membrane, and vesicular proteins (Fig. 7D). Components of translational initiation complex were also found to be enriched in the gametocyte TurboID.
Twelve proteins were consistently identified to be significant in at least 3 of the four IP-MS experiments. These included the bait protein PfFBXO1 (PF3D7_0619700), PfSKP1 (PF3D7_1367000), Glideosome proteins such as PfGAP40/50 and PfGAPM2, along with a putative FACT complex subunit, FACT-L (Fig. 7E). Among the commonly identified proteins were two conserved Plasmodium proteins of unknown function PF3D7_1409400 with four transmembrane domains and PF3D7_0821000, a P-loop NTPase containing Dynamin superfamily protein with two transmembrane domains. Both proteins are predicted to be essential by piggyBac transposon insertion mutagenesis study53. The T. gondii ortholog of PF3D7_1409400 is predicted to be in the IMC by HyperLOPIT analysis54. PF3D7_0821000 is conserved in Plasmodium with an expression pattern comparable to PfFBXO1 at the transcriptome level; in that they are both expressed mainly in schizonts, stage V gametocytes, and ookinetes.
F-box domain of PfFBXO1 successfully complemented the F-box domain region in TgFBXO1
PfFBXO1 was initially identified as a homolog of Toxoplasma gondii F-box protein 1 (TgFBXO1), and it shares the similar localization to an analogous daughter cell scaffold (DCS) in P. falciparum. Also, TgFBXO1 is important for parasite growth due to its function in the development of DCS34. Therefore, we sought to determine whether PfFBXO1 was able to complement the growth defect of a parasite strain in which the TgFBXO1 native promoter was replaced with an anhydrotetracycline (ATc)-repressible promoter55 to generate a N-terminal HA-tagged HATgFBXO1 parasite strain (Fig. 8A). In these lines, addition of ATc to the media depletes HATgFBXO1 levels (Fig. 8B), allowing us to test the function of a C-terminal myc-tagged PfFBXO1 in complementation strains. Since TgFBXO1 and PfFBXO1 are not well conserved in the N-terminal portion of these proteins, we also generated a chimeric form of FBXO1 in which the N-terminal of TgFBXO1 was cloned in frame with the C-terminal domain containing the F-box region of PfFBXO1 (FBXO1-Chimera complement). After confirming the expression of both myc-tagged PfFBXO1 constructs by immunoblot (Fig. 8C), we performed plaque growth assays using HATgFBXO1 complemented with a myc-tagged TgFBXO1 (TgFBXO1 complement) as positive control. Parasites complemented with TgFBXO1myc and FBXO1-Chimeramyc, but not PfFBXO1myc, were able to form plaques after 5 days when grown in the presence of ATc (Fig. 8D, E), indicating that the function of PfFBXO1 is conserved in the C-terminal but is insufficient by itself to complement TgFBXO1.

A List of complement parasite strains generated with details of FBXO1 protein expressed. Endogenous HATgFBXO1 expression is controlled by ATc. Constitutive expression of myc-tagged FBXO1 complements is controlled by SAG4 promoter. B Western blot depicting the efficacy of HA-tagged endogenous TgFBXO1 knockdown. C Western blot showing the expression of different myc-tagged complements. The top band in each lane is the full-length protein, and lower bands likely represent breakdown products. D Plaque assays showing the effect of complementation with exogenous myc-tagged-FBXO1 in the absence of endogenous HATgFBXO1. Scale bar = 2 mm. E Scatter plot quantifying the plaque areas measured upon complementation shows successful complementation with a chimeric-FBXO1 but not with full-length PfFBXO1 alone. The plaque assays were performed at the same time in biological triplicates and the combined data is plotted (N = 30 for each condition). The significance was calculated using ordinary one-way ANOVA (**** represents p < 0.0001).
Discussion
The first characterization of an apicomplexan FBXO1 was in T. gondii where it was found to be important for its growth as a component of the DCS, aiding development and proper organization of the IMC34. We present evidence for a similar role for PfFBXO1 in the development of the IMC and the formation of an analogous daughter cell scaffold-like structure during Plasmodium schizogony. We show that PfFBXO1 is one of the first proteins localizing to this DCS-like region of the cell. PfFBXO1 is essential for IMC biogenesis during both asexual and sexual stages, and its absence results in improper segmentation of schizonts and loss of gametocyte progression to stages IV and V. A recent study of PbFXBO1 in the rodent parasite, P. berghei, demonstrated its localization to the apical region of early schizonts which became faint, dotted and peripheral, concentrating towards the basal end of merozoites as segmentation progressed35. Though we observed a similar localization of PfFBXO1 during segmentation, we did not observe the basal end concentration in late schizonts. We find that the basal complex, visualized by PfBLEB, remained distinct to that of the IMC-like staining during early and mid-segmentation of PfFBXO1 (Fig. 1B, C). In addition, we found a more severe growth defect of PfFBXO1 knockout parasites than the partial defect witnessed in schizonts of P. berghei35. We also show that the nuclear segregation and organization of the apical organelles such as rhoptries and SPMTs are affected by the absence of PfFBXO1.
The most well-studied and accepted function of F-box proteins is their role as the substrate recruiting partner of SKP1 in the SCF complex. The SCF complex serves as an adaptor protein for the E3-ubiquitin ligase system, which transfers either a single ubiquitin or a polyubiquitin chain to a substrate. RING-type ligases are known to be associated with the SCF complex forming the cullin ring ligase (CRL) systems. In T. gondii, TgFBXO1 and TgCul1 were identified in SKP1 pull-down assays, using both polyclonal anti-TgSKP1 and anti-HA antibody pull-downs, for tagged version of TgSKP134. These data suggest that, in Toxoplasma, the role of FBXO1 as the recruiting component of the SCF E3 ubiquitin ligase complex is likely conserved34. However, a recent study characterizing the P. falciparum cullin ring ligases revealed that PfFBXO1 binds only to PfSKP1 using PfSKP1-IP and dot-blot assays but was not pulled down by cullins or RBX1 proteins56. Our results also demonstrate the interaction between PfFBXO1 and PfSKP1 but do not identify other known components of the complex such as cullins or RBX1 proteins. Interestingly, a recent study in Toxoplasma also reports the successful identification TgSKP1 in co-IPs of HA-tagged TgFBXO1 but not the other SCF components57.
PbFBXO1 plays a critical role in gamete development following activation of gametocytes35. Nuclear division is critical at this stage with swift replication and division of the single male gametocyte nucleus into 8 male gametes via meiosis. We compared the ubiquitinome dataset of wild type gametocyte pre-activation with our dataset of significantly biotinylated proteins in gametocytes to identify putative substrates. In all, 66 proteins were common between the two datasets, and they were all highly ubiquitinated in the wild type compared to the PbFBXO1 knockouts, post-activation. This list includes the HECT-like E3 ligase (PF3D7_0826100), other E3 ligases, cdc48 and CDPK1 in addition to the protein of unknown function, PF3D7_1409400. The P. berghei study also highlighted a potential role for CDPK1 as a negative regulator of PbFBXO1 though no direct evidence of this interaction or mechanism was identified35,58. Thus, while it seems reasonable to hypothesize that PfFBXO1 has an essential role in the process of cell division, whether these are mediated solely by ubiquitination is hard to determine. Previous studies in yeast and other organisms have highlighted an SCF-independent, but SKP1-dependent, role of some F-box proteins. For example, Ctf13 is an F-box protein crucial for kinetochore assembly in S. cerevisiae that is dependent on SKP1 binding but independent of SCF function59. Human FBXO1 (Cyclin F) is also known to regulate the G2-M-phase transition by promoting nuclear localization of cyclin-B1 and forming a complex with cyclin-B1 and cdc2, both of which are independent of its SCF activity. Other studies have shown additional examples of SCF-independent roles of F-box proteins, either by themselves or as dimers with SKP1, in processes including centromere assembly and endosomal component recycling59,60,61,62.
The Apicomplexan FBXO proteins were named sequentially and not by their direct homology to the human FBXO proteins34. PfFBXO1 shows good conservation within the Apicomplexan clade34, suggesting a specialized role that has evolved within this parasite group. Though the mechanism of cell division (endodyogeny) in T. gondii differs from that of schizogony in Plasmodium species63, our results suggest that the mechanisms of IMC biogenesis, and FBXO1’s role in apicomplexan cell division remains conserved and may have emerged evolutionarily before the divergence in the mode of cell division. In T. gondii, centrin duplication precedes TgFBXO1 localization, and TgFBXO1 depletion resulted in aberrant IMC as shown by the ISP1 and IMC3 staining34. Consistent with this data, our results indicate no visible defect in centrosome duplication (Fig. 4E) but show aberrant nuclear division and IMC biogenesis in the absence of PfFBXO1 (Fig. 4A–D). Approximately 275 proteins identified by our schizont TurboID experiment overlapped with that of the chromatin-bound proteome identified in a recent study64. This could be either due to mere proximity of the site of IMC nucleation to the centromere or a potential role of PfFBXO1 in chromatin assembly and replication. The identification of two proteins, PfSKP1 and PF3D7_0517400 (FACT-L), in the V5 pull-down and TurboID assays suggests a putative interaction with chromatin proteins. Several other proteins including SMC1, SMC3, components of the MCM complex, and the FACT complex were significantly biotinylated in our study. In addition, the identification of HECT-type ubiquitin ligases suggests that these interactions, and the role of PfFBXO1 in chromatin assembly and replication, might be mediated either through the SCF complex or a SKP1-dependent but cullin-independent mechanism.
In our co-immunoprecipitation and TurboID assays, we consistently identified multiple IMC proteins, including PfGAP40, PfGAP50 and PfGAPM2 proteins. PfGAP40 and PfGAP50 are trafficked to the IMC from the endoplasmic reticulum and are essential for asexual replication23,24,25. PfGAP50 likely localizes to the IMC lumen and interacts with the other components of the glideosome through its C-terminal helix extension24,65. Their absence results in morphological abnormality, impaired IMC integrity, and failure of glideosome assembly23,24. Similarly, GAPMs are known to be an integral part of the IMC via their transmembrane domains and important for maintaining IMC integrity8,25. During the early stages of IMC development, PfGAP45 is hypothesized to form a soluble complex with the PfMyoA and PfMTIP and then be recruited and anchored to the IMC via the interaction between PfGAP45 and PfGAP40/50 proteins66,67. Although the mechanism of this interaction is not completely understood, PfGAP45 was shown to be associated only with the plasma membrane in the absence of GAP40/50 in T. gondii25. Considering that the PfGAP45 and basal complex protein localization to the apical region is initially unaffected, we hypothesize that PfFBXO1 is likely needed for maintenance of the IMC. Given that PfFBXO1-deficient parasites are unable to establish viable rings upon invasion, we hypothesize that PfFBXO1 may interact with PfGAP40/50 and PfGAPMs and facilitate the anchoring of PfGAP45 and other glideosome components to the IMC, failing which, the IMC integrity is compromised. It is worth noting that the conservation of IMC-related processes across Apicomplexa, despite the differences in cell division methods, highlights importance of these structures and mechanisms in Apicomplexan biology. The ability of the P. falciparum F-box domain to complement its T. gondii counterpart further supports this hypothesis.
Similarly, loss of PfFBXO1 in gametocytes does not inhibit the initial IMC formation but rather affects the stability of the IMC and thereby gametocyte maturation. Gametocyte maturation involves a morphological change in the shape aided by the subpellicular microtubules and the IMC19. Studies have indicated the site of IMC biogenesis to be crucial for SPMT assembly and removal of IMC proteins like PfGAP40 or PfPhIL1 contributed to loss of maturation beyond stage III23,27. A recent study identified an additional protein PfSPM3 to be crucial for gametocytogenesis and its absence resulted in round and non-falciform gametocytes that is similar to that of PfFBXO1-deficient gametocytes68. PfSPM3 along with other known IMC proteins like PfISP1, PfGAPMs, PfPhIL1 and other PfPhIL-interacting (PIC) proteins were identified to be significantly biotinylated our gametocyte TurboID assay. This further corroborates the interaction of these proteins and role of PfFBXO1 in maintaining the IMC integrity. Reduced staining for the IMC proteins and sub-pellicular network, along with overall NHS-ester and BODIPY-TR ceramide staining in PfFBXO1 knockouts indicate a potential lack of protein stability. We hypothesize that absence of a robust IMC results in collapse of the cells to reduce their surface-tension, contributing to a more rounded shape as observed in our live-cell experiments.
Though the molecular mechanism remains unclear, PfFBXO1 likely stabilizes these membrane proteins within the membrane either directly as an integral component of the IMC or mediated by post-translational modifications of other component proteins. Absence of PfFBXO1 resulted in reduced clarity of the membrane staining by BODIPY-TR ceramide in late schizonts (Supplementary Fig. 4). To assess the possibility, we compared our dataset of significant interactome gene IDs with the ubiquitinome data from previous study in schizonts69. We identified 250 common proteins in all, of which 57 proteins were ubiquitinated in schizont stage, consisting mainly of 60S ribosomal proteins, HECT-like E3 ligase (PF3D7_0826100). The majority of the pellicle proteins, except PfGAPM2, PfIMC1c and PfCDPK1, were ubiquitinated in the merozoite stage when PfFBXO1 protein is reduced or absent. While these proteins are ubiquitinated in schizonts, the extent of ubiquitination at the site increases in merozoites. Western blot analysis of total ubiquitination and K63- or K48-specific ubiquitination showed no significant differences in the presence or absence of PfFBXO1 in schizonts (Supplementary Fig. 8). In addition, we performed drug assays to identify the effect of PfFBXO1 knockdown on susceptibility to MLN7243, an inhibitor of ubiquitin activating enzyme, and an SCF inhibitor, SMER3. Data from both assays revealed no significant effect of the PfFBXO1 knockdown, suggesting either a different mechanism of ubiquitination or irrelevance of the protein in these pathways (Supplementary Fig. 9). These data suggest that PfFBXO1 may not be involved in the conventional ubiquitination pathway, at least in schizogony, or that the level of knockdown was insufficient to bring about a significant change.
Finally, we conclude that the cytoskeleton is a dynamic network of structural proteins, and finely orchestrated post-translational modifications of key proteins play pivotal roles in mediating essential processes throughout the life cycle of P. falciparum. We hypothesize that PfFBXO1 plays a crucial role in these processes in both asexual and sexual stages of P. falciparum development. Although the exact mechanism of action could not be elucidated in this study, we suggest a few (not mutually exclusive) hypotheses for PfFBXO1 function. (i) PfFBXO1 mediates the process of chromosome and nuclear division via SKP1 and FACT complex. (ii) PfFBXO1 is an integral component of IMC and maintains the stability of IMC by forming a complex with PfGAP40/50 and PfGAPMs. This complex mediates the anchoring of glideosome components to the IMC via PfGAP45. (iii) Given that ubiquitination increases at the end of segmentation when PfFBXO1 levels decrease, there is a possibility that PfFBXO1 stabilizes the pellicle proteins and maintains the IMC by preventing their ubiquitination until late segmentation. At this stage, the absence of PfFBXO1 from IMC allows the ubiquitination of these proteins which presumably helps in IMC disintegration upon reinvasion. To gain a comprehensive understanding of PfFBXO1’s precise function, further investigations from various angles are imperative.
Materials and Methods
Plasmid construction and PCRs
All PCRs were performed using PrimeSTAR GXL enzyme (Clonetech) and plasmid sequences were confirmed by sanger sequencing from Genewiz.
PfFBXO1-smV510x,Tet (pJPM55). The 3’ and 5’ homology region (HR) was PCR amplified from genomic DNA (gDNA) of P. falciparum 3D7 strain (PF3D7) using oligos oJDD4726/4727 and oJDD4728/4729 respectively. By appending the codon-altered C-terminal region of PF3D7_0619700 (PfFBXO1) gene to the 5’HR using oJDD4754/4755 the two fragments were spliced by overlap-extension PCR (PCR SOE) with oJDD4716/4717 and cloned as NotI/NcoI fragments into pRR92, containing smV5 epitope tag and human dihydrofolate reductase (hDHFR).
PfFBXO1-mNeonGreen-LoxP plasmid (pSKS3) was constructed using Golden-Gate assembly of 8 fragment pieces consisting of (i) 3’HR (PCR amplified using oJDD5617/5618); (ii) 5’HR and loxP site (nested PCR amplification using oJDD5610/5612 and oJDD5619/5620) from PF3D7 gDNA; (iii) codon-altered PfFBXO1 (PF3D7_0619700) coding sequence using oJDD5621/5622; (iv) mNeonGreen with loxP site using oJDD5631/5632 from pRR208; (v) mRuby2 fragment using oJDD5633/5634; (vi) 2myc+UTR + CAM fragment using oJDD5635/5636; (vii) hDHFR expression cassette using oJDD5637/5628 and (viii) pGEM backbone using oJDD5615/5616. All pieces assembled through golden gate reaction using BsaI-HFv2 enzyme.
PfFBXO1-V5-turboID (pSKS10) was constructed by restriction digestion of pJPM55 using NotI and NcoI and inserting the 1.2 kb piece into pBNA0343.
CRISPR-Cas9 guide plasmids were all generated by annealing and ligating the oligos corresponding to the respective guides into BpiI-digested pRR21670 plasmid containing the SpCas9 and a U6 guide cassette. The guides targeting PF3D7_0619700 to obtain PfFBXO1smv5-10x-Tet and PfFBXO1v5-TurboID, guide oligos oJDD4722/4723 were used with pJPM55 and pSKS10, respectively. For guides with pSKS3, oJDD6129/6130 and oJDD6131/6132 in combination. For guides with pSKS07, oJDD5872/5873 and oJDD5870/5871 in combination.
Parasite strains used and Plasmodium culture
Majority of the experiments were performed in P. falciparum 3D7 laboratory strain, obtained from the Walter and Eliza Hall Institute (Melbourne, Australia). 3D7-DiCre44 parasites, obtained from Ellen Knuepfer, and NF54-DiCre71 parasites, obtained from Moritz Treeck, were used for all iKO systems. NF54-iGP252 parasites, obtained from Till Voss, were used for gametocyte TurboID experiments. Human RBCs and serum were purchased as anonymous samples from a commercial vendor (BioIVT). All ethical regulations relevant to human research participants were followed. This study was determined not to be human subjects research by the Boston Children’s Hospital Institutional Review Board.
PfFBXO1smV5-10xTet lines were obtained by transfecting 100 μg of linearized HDR plasmid pJPM55) with 50 μg of guide plasmid (pJPM57) in 3D7-Cas9 parasites (obtained previously by transfection of parental 3D7 line with pUF1-Cas9 plasmid and selected using PfDHODH inhibitor N-(3-chloro-4-methylphenyl)-5-methyl-2-(trifluoromethyl)[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (MMV665874 or AD1) at 150nM43. This parasite line was used for localization studies and drug assays.
PfFBXO1mNG (SKS3 and SKS3nf) were obtained by schizont transfection of pSKS3 HDR plasmid with guides pSKS5 and pSKS6 into 3D7-DiCre parasite line and NF54-DiCre parasite line, respectively. The parasites were selected using WR99210 (Jacobus Pharmaceuticals) at 2.5 nM. The 3D7-DiCre parasite line was used for all asexual assays and studies while the NF54-DiCre parasite line (SKS3nf) were used for the gametocyte progression analysis and gametocyte live-cell assays, IFAs and U-ExM studies.
PfFBXO1v5-TurboID (SKS10 and SKS10-iGP2) lines were obtained by schizont transfection of pSKS10 plasmids with guide pJPM57 into 3D7 and NF54-iGP252 parasite lines, respectively. The parasites were selected using WR99210 at 2.5 nM. 3D7-based line (SKS10) was used for all schizont stage IPs and TurboID, while NF54-iGP2-based line (SKS10-iGP2s) were used for the gametocyte TurboID analyses.
All parasites except the TurboID lines were cultured in RPMI-1640 (Sigma) supplemented with 25 mM HEPES (4-(2-hydroxyethel)-1-piperazineethanesulfonic acid) (EMD Biosciences), 50 mg/L hypoxanthine (Sigma), 0.216% sodium bicarbonate (Sigma) and 0.5% Albumax II (Invitrogen). The TurboID parasite lines, SKS10 and SKS10-iGP2s were cultured in Biotin-free and phenol red-free RPMI-1640 with all the other components same as for the 3D7 parasites. For SKS10-iGP2s the media was supplemented with 2.5 mM D-Glucosamine hydrochloride (Sigma) to prevent GDV1 expression. Parasites were cultured at 37 °C with a gas mixture of 90% N2, 5% O2 and 5% CO2 (Airgas) with addition of packed human RBCs at 4% hematocrit and O+ serum (for gametocytes) obtained from BioIVT.
P. falciparum transfections
Parasite lines PfFBXO1PfalKO and PfFBXO1smV5-Tet was constructed by co-transfecting 100 μg of linearized HDR plasmid (by digesting with StuI and treated with CIP to prevent re-ligation) and 50 μg of corresponding guide plasmids into P. falciparum 3D7 strain using electroporation. Electroporation was performed at ring stage using settings of 310 V, 950 μF and infinite Ω in a 0.2 cm cuvette on BioRad GenePulser.
Schizont transfections of PfFBXO1mNG-loxP (SKS3 and SKS3nf) and PfFBXO1v5-TurboID (SKS10 and SKS10-iGP2) lines were made by co-transfecting late-stage percoll/MACS-purified schizonts with 30 μg of linearized HDR plasmids and 10 μg each of corresponding guide plasmids into P. falciparum 3D7, 3D7 DiCre72, NF54 DiCre44 and NF54-iGP252 strains in a 100 μL cuvette using P3 Primary Cell 4D-Nucleofector X Kit and FP158 program on Amaxa 4D nucleofector unit from Lonza. Electroporated parasites were transferred to 10 cm culture dishes at a final hematocrit of 4% with stabilizing agents such as anhydrotetracycline (ATc) at 500 nM and D-Glucosamine hydrochloride at 2.5 mM. Media was changed every day for transfections and selection pressure was applied by addition of WR99210 (2.5 nM) (Jacobus Pharmaceuticals) from the next day of transfection.
Parasite synchronizations
Mature schizonts were prepared by centrifugation at 2000 rpm for 15 min after layering the parasites on 60% percoll solution. The interface containing schizonts were collected and transferred to culture with fresh media and RBCs after washes. For tight synchronization, the parasites were allowed to reinvade for about 2 h followed by sorbitol synchronization. The infected RBC (iRBC) pellet was treated with 5% sorbitol after removing the media and incubated at 37 °C for 10 min followed by replacing the sorbitol by media. Sorbitol treatment selectively preserves rings.
Limiting dilution cloning and cryopreservation
Single cell clones of parasite transfections were obtained by limiting dilution. For this, we prepare 3 dilutions: 2 cells/100 μL, 0.6 cells/100 μL and 0.2 cells/100 μL from 1:10,000 dilution of culture. Culture the plate for at 2% hematocrit with media replacements on day 4, day 7 and day 10 without drug selection. Wells are then screened on day 12 or 14 and clones are picked, ideally from the lower dilutions. These clones were then expanded in the presence of drug selection, followed by confirmation of plasmid integration into the genome through PCR amplification of 3’ and 5’HR regions and whole locus PCRs before proceeding with the other experiments. For PfFBXO1mNG-loxP clone used in the experiments, we further confirmed the integration by whole genome sequencing of the genomic DNA.
For cryopreservation, 400 μL of iRBC pellet was resuspended in 600 μL of serum. To this, 1 ml of freezing solution (28% glycerol, 3% sorbitol, 0.65% NaCl), mixed gently and frozen and stored in liquid nitrogen as 1 ml aliquots.
Flow cytometry-based parasite replication analysis
Following ring stage parasite synchronization with sorbitol, parasites were seeded at 0.25% in triplicates (three wells each) and treated either with DMSO as control or rapamycin (100 nM) and cultured at 2% hematocrit. At every time point, 100 μL of culture from each well was placed in a well of round bottomed 96 well plate. The iRBCs from culture was washed with 0.5% wt/vol BSA-PBS solution, followed by incubation with 1:1000 SYBR Green I solution (Life Technologies) for 20 min at room temperature in dark. Cells were washed again with 0.5% BSA-PBS solution and resuspended in PBS. Flow cytometry data was collected using a BD FACS Calibur machine (BD Biosciences) with an acquisition of 100,000 events per sample using CellQuest Pro software. Initial gating was performed with unstained, but SYBR treated, uninfected erythrocytes to account for erythrocyte autofluorescence as shown in Supplementary Fig. 10. The flow cytometry data was analyzed using FlowJo X software and further data analysis and graph plots were prepared using GraphPad Prism 9 software. All data represented are mean ± SD calculated from triplicate data. The growth curve experiments were performed on two parasite clones (as biological replicates). Time-based replication analyses were performed three separate times as biological replicates. Shown in the results is the data from one biological replicate containing technical triplicates. Gating strategy shown in Supplementary Fig. 10.
Gametocyte induction and culture
Gametocytes were induced using 3 methods depending on the parasite parental strain. Gametocyte induction for the 3D7 strains of PfFBXO1smV5-10xTet was performed by culturing percoll-synchronized parasites at 5–6% parasitemia in 4% hematocrit in choline-free media and stressed by replacing two-thirds of the spent media with fresh media, at trophozoite stage. The parasites were then cultured in this media for a day and maintained on static mode. Everyday media changes were performed after the initial stress by addition of 50% fresh media and 50% conditioned media (collected spent media from regular culture of 1% parasitemia at 4% hematocrit and centrifuged to remove any cells). The cells were treated with 0.25 mg/ml of Heparin (Sigma) and 50 mM N-acetyl glucosamine (Glc-NAC) (Sigma) from days 2 to 5 after induction to prevent asexual stages.
Gametocytes were induced by minimal fatty-acid media (MFA) method in PfFBXO1mNG-loxP in NF54-DiCre background (SKS3.1nf). Briefly, synchronized parasites were cultured in choline-free media and at 28–32 h.p.i (trophozoite stage), the culture media was replaced with MFA media (30 μM Oleic acid, 30 μM palmitic acid, 60 μM, RPMI and 0.216% Sodium bicarbonate) and cultured for 20–24 h until re-invasion to induce stress and gametocytogenesis. The MFA media was replaced with regular complete media just before re-invasion and cultured on static with daily media changes. The culture was supplemented with 0.25 mg/ml Heparin and 50 mM GlcNAC to prevent asexual stages and pure gametocytes were harvested at desired time-points.
Gametocyte induction in PfFBXO1v5-turboID in NF54-iGP2 background (SKS10.1-iGP2s) was induced simply by removal of D-Glucosamine hydrochloride from synchronized culture at 1–16-h rings for a full cycle to promote expression of GDV1. After re-invasion, the media was replaced with biotin-free complete media with 50:50 albumax:serum and supplemented with 2.5 mM D-Glucosamine hydrochloride. The culture was treated with 50 mM GlcNAC from day 2 until day 5 to prevent asexual stage parasites. The gametocytes were harvested as stage III parasites for mass spectrometry analysis of proximity-based biotinylation using TurboID to identify the interacting partners of PfFBXO1 in gametocytes.
Immunofluorescence assays (IFAs) and microscopy
iRBCs were smeared on slides or allowed to settle on poly-D lysine coated coverslips and fixed with 4% vol/vol paraformaldehyde in PBS (Electron Microscopy Sciences). Permeabilization was carried out with 0.1% vol/vol Triton X-100 in PBS for 10 min and blocking with 3% wt/vol BSA in PBS for 1 h at RT or overnight at 4 °C. Primary antibodies were incubated for 1 h at RT in BSA-PBS dilutions as mentioned in Supplementary Table 1. Subsequently, the cells were washed in PBS thrice and incubated with secondary antibodies, AlexaFluor 488, 555, 594 or 647 (Life Technologies) at 1:1000 dilutions in PBS for 45 min. Post incubation, the cells were incubated with Hoechst DNA dye (1:5000 in PBS; Life Technologies) for 15 min, followed by 3 PBS washes to remove unbound antibodies and dye. The slides and coverslips were mounted with Vecta-shield Vibrance antifade mounting media (Vector Laboratories Inc. H-1700) and kept at 4 °C until microscopic evaluation. All IFAs were performed at least three separate times and representative images are shown in the results.
Super-resolution microscopy z-stacks were captured using Airyscan2 with a 1.4 numerical aperture 63X oil-objective and excitation wavelengths of 405, 561 and 647 nm on LSM 980 or LSM 900 microscopes (Carl Zeiss Microscopy). Sixteen-bit SR images were captured by maintaining constant laser powers for respective channels and master gain of 650–750 with frame-switching and no averaging.
Ultrastructure Expansion Microscopy (U-ExM)
Percoll-purified schizonts or parasites in culture were allowed to settle on poly-D-lysine coated coverslips for 25–20 min and fixed at 37 °C with 4% vol/vol paraformaldehyde (Electron Microscopy Sciences) after removing the additional media. Following removal of PFA and 3 PBS washes, the cells were incubated in 1.4% formaldehyde (FA)/2% acrylamide (AA) solution in PBS, overnight. The treated coverslips were then used for gelation with monomer solution the next day by addition of 35 μL of monomer solution (19% sodium acrylate, 10% acrylamide, 0.1% bis-acrylamide in PBS) mixed with APS and TEMED and allowed to solidify for 1 h at 37 °C. The solidified gels were then denatured in denaturation buffer (200 mM SDS, 200 mM NaCl, 50 mM Tris at pH 9) for 15 min on a rocker at RT followed by 90 min at 95 °C. Post-denaturation, the gels were washed with deionized water and allowed to expand overnight at RT. The gels were initially shrunken in PBS for 15 min and then blocked in 3% wt/vol BSA-PBS for 3 h at RT and incubated with primary antibodies for 2.5 h at RT or overnight. The gels were subsequently washed 3 × 15 min with 0.5% PBST and incubated in secondary antibodies along with NHS-ester and Hoechst/Sytox dye in PBST for 2.5 h at RT on a rocker. Unbound secondary antibodies were removed by three 15 min PBST washes on a rocker and the final gels were allowed to expand overnight in deionized water and imaged. For BODIPY-TR staining, the expanded gels were incubated overnight in 0.2% wt/vol propyl gallate solution containing BODIPY-TR dye and imaged within a week.
For imaging, stained gels were placed on poly-D-lysine coated #1.5 glass-bottom imaging dishes (Cellvis) and imaged as super-resolution z-stacks using Airyscan on LSM 980 and LSM 900 (Carl Zeiss Microscopy) using 1.4 numerical aperture 63X oil-immersion objective. Eight-bit images without averaging were acquired using excitation lasers of 405, 488, 561 and 647 with suitable filters and master gain of 850. The laser power for each of the channels were maintained constant for imaging control and treated samples to enable comparison. The LUT range was inverted, and color range modified to 0–225 only for the NHS ester channel to enable clarity of the structures. Gamma was maintained at 1.
Live-cell microscopy
For monitoring the gametocyte progression by live cell microscopy, gametocytes were induced as mentioned before in PfFBXO1mNG-Dicre and NF54-dicre parental parasite lines using MFA media. Induced parasites were split into two 6-well plates (three wells for each condition as triplicates) and treated with either DMSO as control or rapamycin at committed ring stage, respectively. Cultured gametocytes were sampled on days 4, 6, 8, 10 and 12 to be imaged. For imaging, about 50 μL of culture was sampled from each sample and stained with SiR-tubulin (1:1000) staining kit and allowed to settle in a quadrant of concanavalin A coated #1.5 glass-bottom imaging dish (CellView) for an hour followed by 15-min staining with Hoechst 33342 at a dilution of 1:1000 in media. Unbound cells were then washed at least thrice using phenol-red free media with bicarbonate. Each quadrant was then filled with phenol-red free media and imaged. The gametocytes were always maintained at 37 °C and the microscopic chamber, and the insert were pre-warmed to 37 °C before transferring the gametocytes for imaging. At least 20–25 field of view were selected in each quadrant consisting of multiple gametocytes in each field of view under 63X oil immersion with numerical aperture of 1.4 on the Zeiss LSM 900 with AiryScan2. The fields were imaged using multiplex confocal imaging option using 405, 488, 561 and 647 channels at about 4% laser power with 2 × 2 binning. In addition, about 3-5 gametocytes were imaged using Airyscan settings after the confocal imaging. The experiments were repeated three different times each time in triplicates. In one biological replicate, the gametocytes were unhealthy, likely due to the donor RBCs, and had to be excluded. The data presented are from two biological replicates (two separate inductions in different blood) each with technical triplicates (three wells of a 6-well plate). Results presented for the NF54 DiCre parental parasites (Fig. 6B) are from a single biological replicate consisting of technical triplicates. Scatter plots depict the individual values with the median value and the 95% CI represented as lines and range. Significance was calculated using the un-paired two-tailed t-test with Welch’s correction.
Image analyses and measurements
For most of the displayed images, z-stack acquired on LSM 980 with AiryScan2 or LSM900 with AiryScan2 were processed in Fiji/ImageJ. The brightness or contrast were adjusted solely by the reset option for localization/qualitative purposes. For the images showing the control and knockout parasites, the minimum and maximum of the PfFBXO1 knockdown of knockout parasites were set to the range of control images to enable proper comparison. In addition, all control and knockout sample images were acquired using similar settings of exposure and laser power.
Hemacolor-staining of gametocyte smears collected on days 4, 6, 8, 10, and 12 were imaged on Olympus microscope using the 100X oil immersion lens (NA 1.4). Gametocytes were staged using cell counters. Roundness measurements were made in Fiji by manually tracing the shape of the gametocytes in z-projected images of the hemacolor-stained and confocal image fields. The ROI measurements were exported and plotted using GraphPad Prism software.
Western blot
iRBC pellet was treated with 0.2% wt/vol saponin solution on ice and the extracted pellets were resuspended in Laemmli buffer and boiled at 95 °C for 10 min to extract P. falciparum proteins. The protein samples were frozen at −20 °C until use. All protein samples were separated using mini-Protean TGX gels (4–20%) (Bio-Rad) and transferred to nitrocellulose membranes using wet transfer with low methanol at 90 V for 90 min. Membranes were probed with primary antibodies after blocking with LiCor blocking buffer in dilutions of 1:3000 for anti-V5, 1:2500 for anti-histone H3 in TBST; 1:1000 for anti-Ub (FK2 clone) and K63-specific antibodies and 1:10,000 for K48-linkage specific antibody in PBS. Detection of primary antibodies was carried out with near-infrared dye labeled secondary antibodies (LiCor) at a dilution of 1:10,000 and scanned using LiCor Odessey CLx imager. Quantification of western blot data was performed using Image Studio software from LiCor and ImageJ. All uncropped western blots are shown in Supplementary Fig. 11.
Drug assays
The effect of two drugs, MLN7243 (ubiquitin activating enzyme inhibitor) and SMER3 (SCF inhibitor) on the growth of PfFBXO1-deficient parasites (without ATc) and PfFBXO1-sufficient controls (with ATc) was tested using SYBR Green-based flow cytometry analysis. For this, synchronized PfFBXO1smV5 parasite clonal lines (JPM55.1) were treated with two-fold serial dilutions of MLN7243 [Cayman chemicals, cat# 30108] and SMER3 [Tocris Bioscience, cat# 4375] with a maximum concentration of 100 µM and cultured at 1% parasitemia and 1% hematocrit in triplicates in a 96-well plate. DMSO was used as negative control at a concentration of 0.1% and dihydroartemisinin (DHA) at 5 µM concentration was used as positive control. The parasitemia was monitored by SYBR Green-based flow cytometry assay, 72 h post treatment. Recorded parasitemia was normalized and plotted as a graph after log transformation and IC50 values were calculated using GraphPad Prism software. Two separate experiments were performed using parasites grown in different batches of RBCs (biological replicates). Each biological experiment consisted of 3 technical replicates.
Affinity-based immunoprecipitation Mass spectrometry
PfFBXO1smV5-10xTet parasite line (JPM55.1) cultures of 300 mL at 2% parasitemia were harvested on ice. iRBC pellets collected were treated with 0.2% wt/vol saponin in the presence of protease inhibitors (SigmaFast Protease inhibitor Cocktail, Sigma) and washed with PBS+protease inhibitors until the supernatant was clear. The parasite pellets were then lysed in chilled RIPA buffer (50 mM Tris-HCl, pH 8, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate and 0.1% sodium dodecyl sulfate) containing protease inhibitors followed by probe sonication at 30% amplitude for three cycles of three 10 s pulses with 10 s breaks, on ice. The cell suspension was allowed to chill on ice for 3 min in between each cycle. Following sonication, the cell lysates were cleared by centrifugation for 30 min at 4 °C at 14,000 rpm using Eppendorf 5425 centrifuge in cold room. The supernatant was collected and incubated with magnetic anti-V5 beads (MBL, Product #M167-11) for 1 h at RT. Beads were then washed 3X with RIPA+protease inhibitors followed by three washes with PBS+protease inhibitors to remove detergents. The beads were then resuspended in 50 μL 50 mM ammonium bicarbonate solution and submitted to Harvard’s Taplin Mass Spectrometry core facility for LC-MS/MS analysis.
For Triton-IP, the saponin lysed parasite pellet was resuspended in 0.5% vol/vol Triton X-100 in PBS with protease inhibitors and incubated on ice for 30 min with intermittent vortexing every 10 min. The lysate was then centrifuged, and supernatant collected was incubated with magnetic anti-V5 beads for 1 h at RT, followed by cold IP buffer washes 5 times and 3 PBS washes before resuspending in 50 μL 50 mM ammonium bicarbonate solution and submitting to the core facility for analysis. At the core facility, the samples were subjected to on-bead digestion with trypsin and analyzed as a single 2 h run per sample on Thermo Orbitrap mass spectrometer using the DDA acquisition mode. Mass spectrometry data was then searched using SEQUEST algorithm against Plasmodium falciparum protein database (2019 release) from UniProtKB for protein identifications. Sum intensities for each protein in 3D7 parental control and epitope-tagged sample were compared and proteins identified only in sample but not in control with at least 2 unique peptide evidence was considered for further analysis.
Proximity-based biotinylation using TurboID and mass spectrometry for identification of interacting partners
PfFBXO1V5-turboID parasites in 3D7 parental line (SKS10.1) were tightly synchronized by percoll followed by sorbitol treatment after 2 h. Synchronized parasites were cultured at about 3% parasitemia in biotin-free media at 4% hematocrit to obtain ~500 ml culture. At ~42 h.p.i. (hours post-invasion) 200 mM biotin was added to ~250 ml of culture at a final concentration of 150 μM final concentration and incubated at 37 °C for 1 h. Following the incubation, the culture was harvested by centrifugation at 4 °C for 15 min at 2000 rpm. The harvested untreated control samples and biotin treated sample RBC pellets were subjected to saponin lysis with addition of 0.05% wt/vol saponin in PBS containing protease inhibitors (SigmaFast Protease Inhibitor cocktail, Sigma) and allowed to incubate on ice for 10 min. Parasite pellets were collected after repeat washes in ice-cold PBS with protease inhibitors and stored in −80 °C until use. Parasite pellets were resuspended in 1 ml RIPA (50 mM Tris-HCl, pH 8, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate and 0.1% sodium dodecyl sulfate) containing protease inhibitors and sonicated and cleared as mentioned before for affinity-based immunoprecipitation. The cleared lysates were incubated with anti-streptavidin magnetic beads (Pierce™ Streptavidin Magnetic Beads, ThermoFisher Scientific) overnight on a rotor at 4 °C. The beads were then washed with ice-cold RIPA buffer + protease inhibitors thrice, followed by 3 × 2% wt/vol SDS washes, 3X with wash buffer 2 (50 mM HEPES pH7.5, 500 mM NaCl, 1 mM EDTA, 1% Triton-X 100 and 0.1% sodium deoxycholate) followed by 3 subsequent washes with wash buffer 3 (10 mM Tris-HCl, pH 8, 250 mM Lithium chloride, 1 mM EDTA, 0.5% NP-40 and 0.5% sodium deoxycholate) and 50 mM ammonium bicarbonate solution to remove all traces of salts and detergents before resuspending in 50 μL of 50 mM ammonium bicarbonate solution and submitted to the mass spectrometry core facility for solid-phase extraction and LC-MS/MS analysis.
Reduction, Alkylation, tryptic digestion, and solid-phase extraction
The proteins isolated via affinity purification were initially reduced and alkylated at 70 °C for 15 min in a digestion buffer composed of 100 mM TEAB (pH 8.5), 40 mM CAA, and 10 mM TCEP. Subsequently, a 1 μg aliquot of a trypsin/LysC mix was introduced in 1 μL of 100 mM TEAB, and the proteins were subjected to an overnight digestion at 37 °C in a shaker set to 115 RPM. The following morning, the digest was supplemented with an additional 1 μg of the trypsin/LysC mix, and digestion was extended for another 4 h at 37 °C. The resulting tryptic peptides were purified using stage-tips following the method outlined previously73. The purified peptides were then dried using a speed-vac concentrator and reconstituted in 12 μL of 0.2% (v/v) formic acid prepared in MS-grade water. Peptide concentrations were measured using the Pierce Quantitative Fluorometric Peptide Assay (Thermo Scientific, Waltham, MA, USA) as per the manufacturer’s instructions.
nanoLC-Orbitrap-MS Data Collection
LC-MS/MS data acquisition was performed on a Vanquish Neo nanoLC system coupled to an Orbitrap Eclipse mass spectrometer, together with an Easy Spray ESI ion source and a FAIMS Pro ion mobility separation unit (Thermo Fisher Scientific, Waltham, MA, USA). For high-resolution separation of peptides, an Acclaim PepMap trap column (75 μm × 2 cm) was used in tandem with a TS25 analytical column (75 µm × 25 cm, 100 Ȧ) from Ionopticks (Fitzroy, VIC, Australia). Peptide extracts were loaded with a 5 µL injection volume. Fractionation of peptides was achieved using a mobile phase consisting of 0.1% (v/v) formic acid in water (solution A) and 0.1% (v/v) formic acid in 80% (v/v) acetonitrile (solution B) at a flow rate of 300 nL/min, with the column temperature set to 40 °C. The column conditions were adjusted with 3% solution B before applying a linear gradient up to 40% solution B over 60 min, followed by a 6-minute wash at 95% solvent B to remove any peptides binding to the C18 resin.
In positive mode operation, the ion source temperature was adjusted to 305 °C with an applied voltage of 2000 V. Ionized peptides were directed through the FAIMS Pro unit set at −50 V. Mass spectra were acquired in MS1 mode at a resolution of 120,000, across a mass range of 350–2000 m/z, using standard automatic gain control settings and automatic injection time. For MS2 data, the mass spectrometer was operated in data-independent acquisition (DIA) mode at a resolution of 30,000. MS2 spectra were collected across a precursor mass range of m/z 375-1200, using 25 m/z isolation windows with 0.5 m/z overlaps, custom AGC target (1000 units), 30% normalized collision energy, and a resolution of 30,000.
Mass spectrometry searches and data analysis
For V5-based AP-MS, the searches were carried out at the core-facility using SEQUEST search engines and sum intensities for the proteins identified were provided in the form of excel files. Significant interactors were identified by filtering for the proteins that were identified only in the PfFBXO1smV5 tagged lysates and not in the 3D7 controls with at least 2 peptides.
For the asexual schizont TurboID dataset, raw files were searched using Andromeda search engine within the MaxQuant software (version 2.4.9.0) against latest release of Plasmodium falciparum 3D7 and human proteome databases (downloaded March 2024) from UniProtKB. The search parameters were set to identify tryptic peptides allowing maximum of 2 missed cleavages with N-terminal acetylation, methionine oxidation and biotinylation as variable modifications and Carbamidomethylation of Cysteine residues as permanent modification. Label free quantification was performed after enabling match between runs (MBR option) with minimum of 2 peptide requirement using the MaxLFQ algorithm74. FDR was set to 0.01 for peptides and proteins and other parameters were set at default values in the software. Filtering of contaminants, statistical analysis and significant protein identification was performed based on the LFQ values using Perseus software (version 2.4).
In-silico predicted spectral library was generated using DIA-NN advanced library free module from FASTA files of P. falciparum 3D7 reference proteome downloaded from UniprotkB along with MaxQuant contaminant database and list of human proteins identified in the asexual DDA searches. The libraries were created according to the developer instructions with trypsin as the enzyme and without the variable modifications using the DIA-NN (ver 1.8)75. The DIA-raw files were then searched against this spectral library for tryptic peptides allowing a maximum of 2 missed cleavages, methionine oxidation and N-terminal acetylation as variable modification and cysteine carbamydomethylation as constant modification. MBR option was enabled, and biotin as variable modification was avoided. MaxLFQ values were computed on the DIA-NN report file using the diann R package (https://github.com/vdemichev/diann-rpackage). The statistical analysis and identification of significant proteins (So = 1 and FDR cut-off of 0.001) were performed using Perseus software as before. The list of significant protein IDs was parsed through PlasmoDB to obtain Gene Ontology data and enrichment analyses.
T. gondii Complementation Studies
The HATgFBXO1 conditional expression mutant containing an N-terminal 3x-HA tag was generated as previously described34. For complementation of HATgFBXO1, the coding sequence of PfFBXO1myc and FBXO1-Chimeramyc (N-terminal amino acids 1–433 from TgFBXO1 fused with C-terminus amino acids 308-633 of PfFBXO1) were synthesized (GenScript) and cloned into pUPRT-DHFR-D plasmid with 50 bp of homology arms to the UPRT gene locus. The integration cassette was amplified by PCR using primer Comp_FWD and Comp_REV. For transfection, about 10 μg of pSAG1:CAS9-U6:sgUPRT plasmid76 and 10 μg of the integration cassette containing either PfFBXO1myc or PfFBXO1Chimeramyc were co-transfected into purified tachyzoites of HATgFBXO1 parasite. Following three rounds of drug selection with 10 µM 5-fluorodeoxyuracil, the single clones were isolated via limiting dilution in 96-well plates.
For the plaque assay, HFF monolayers in a 24-wells plate (Thermo Fisher Scientific) were infected with freshly egressed tachyzoites (approximately 100 tachyzoites per well). The plates were incubated at 37 °C, 5% CO2 for 5 days without any interference. Then, the culture medium was removed, and the infected monolayers were fixed with ice-cold methanol. For plaque visualization, the sample was stained with 0.2% crystal violet for 20 min. The size and number of plaques per well were analyzed using the ImageJ 1.46 software. Three biological replicates were performed for each sample and the combined results were plotted and analyzed using GraphPad prism software.
Statistics and reproducibility
All the experiments were performed with a minimum of three replicates to ensure reproducibility. In this manuscript, we refer to experiments conducted in separate (independent) culture vessels, treated individually as “technical replicates”. These experiments were conducted simultaneously and hence referred to as technical replicates, while experiments conducted at different time points or with different parasite clones are referred to as “biological replicates”. Replicate information for each experiment is provided in the figure legends and respective methods section. Statistical analyses for all experiments except mass spectrometry analysis were performed using GraphPad Prism software. Descriptive statistics and the significance calculation methods are provided for individual figures in their respective figure legends, along with the replicate information. Statistical significance, when calculated, is from technical triplicates included within the biological replicate when individual graphs are provided separately for each biological replicate. In Fig. 5D, E, and Supplementary Fig. 6, data from multiple biological replicates, each consisting of technical triplicates are combined into single graphs for analysis.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Whole genome sequencing data has been submitted to NCBI Sequence Read Archive (PRJNA1151667, https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA1151667). The raw mass spectrometry files have been submitted to the Proteome Xchange PRIDE repository (PXD059558). The authors declare that all other data supporting the findings of this study are available within the paper, its supplementary information files, or the public data archives noted above. Supplementary Data 1 contains the list of proteins identified in all the immunoprecipitation and Turbo-ID assays. Supplementary Data 2 consists of the source data for the graphs in the main figures.
References
World Health Organization, World Malaria Report 2023. (2023).
Striepen, B., Jordan, C. N., Reiff, S. & van Dooren, G. G. Building the perfect parasite: cell division in apicomplexa. PLoS Pathog. 3, e78 (2007).
White, M. W. & Suvorova, E. S. Apicomplexa cell cycles: something old, borrowed, lost, and new. Trends Parasitol. 34, 759–771 (2018).
McDonald, J. & Merrick, C. J. DNA replication dynamics during erythrocytic schizogony in the malaria parasites Plasmodium falciparum and Plasmodium knowlesi. PLoS Pathog. 18, e1010595 (2022).
Rudlaff, R. M., Kraemer, S., Marshman, J. & Dvorin, J. D. Three-dimensional ultrastructure of Plasmodium falciparum throughout cytokinesis. PLoS Pathog. 16, e1008587 (2020).
Kono, M. et al. Pellicle formation in the malaria parasite. J. Cell Sci. 129, 673–680 (2016).
Morrissette, N. S & Sibley, L. D Cytoskeleton of apicomplexan parasites. Microbiol. Mol. Biol. Rev. 66, 21–38 (2002).
Bullen, H. E. et al. A novel family of Apicomplexan glideosome-associated proteins with an inner membrane-anchoring role. J. Biol. Chem. 284, 25353–25363 (2009).
Agop-Nersesian, C. et al. Biogenesis of the inner membrane complex is dependent on vesicular transport by the alveolate specific GTPase Rab11B. PLoS Pathog. 6, e1001029 (2010).
Arnot, D. E., Ronander, E. & Bengtsson, D. C. The progression of the intra-erythrocytic cell cycle of Plasmodium falciparum and the role of the centriolar plaques in asynchronous mitotic division during schizogony. Int. J. Parasitol. 41, 71–80 (2011).
Tomasina, R., Gonzalez, F. C. & Francia, M. E. Structural and functional insights into the microtubule organizing centers of toxoplasma gondii and plasmodium spp. Microorganisms 9, https://doi.org/10.3390/microorganisms9122503 (2021).
Pieperhoff, M. S., Schmitt, M., Ferguson, D. J. & Meissner, M. The role of clathrin in post-Golgi trafficking in Toxoplasma gondii. PLoS One 8, e77620 (2013).
Voss, Y., Klaus, S., Guizetti, J. & Ganter, M. Plasmodium schizogony, a chronology of the parasite’s cell cycle in the blood stage. PLoS Pathog. 19, e1011157 (2023).
Gerald, N., Mahajan, B. & Kumar, S. Mitosis in the human malaria parasite Plasmodium falciparum. Eukaryot Cell 10, 474–482 (2011).
Kono, M. et al. Evolution and architecture of the inner membrane complex in asexual and sexual stages of the malaria parasite. Mol. Biol. Evol. 29, 2113–2132 (2012).
Ferreira, J. L. et al. The dynamic roles of the inner membrane complex in the multiple stages of the malaria parasite. Front. Cell Infect Microbiol. 10, 611801 (2020).
Hu, K. et al. Cytoskeletal components of an invasion machine-the apical complex of Toxoplasma gondii. PLoS Pathog. 2, e13 (2006).
Kono, M, Prusty, D, Parkinson, J & Gilberger, T. W The apicomplexan inner membrane complex. Front. Biosci. 18, 982–992 (2013).
Li, J. et al. Repurposing the mitotic machinery to drive cellular elongation and chromatin reorganisation in Plasmodium falciparum gametocytes. Nat. Commun. 13, 5054 (2022).
Dixon, M. W., Dearnley, M. K., Hanssen, E., Gilberger, T. & Tilley, L. Shape-shifting gametocytes: how and why does P. falciparum go banana-shaped? Trends Parasitol. 28, 471–478 (2012).
Bennink, S., Kiesow, M. J. & Pradel, G. The development of malaria parasites in the mosquito midgut. Cell Microbiol. 18, 905–918 (2016).
Agop-Nersesian, C. et al. Rab11A-controlled assembly of the inner membrane complex is required for completion of apicomplexan cytokinesis. PLoS Pathog. 5, e1000270 (2009).
He, L. et al. Plasmodium falciparum GAP40 plays an essential role in merozoite invasion and gametocytogenesis. Microbiol. Spectr. 11, e0143423 (2023).
Yeoman, J. A. et al. Tracking Glideosome-associated protein 50 reveals the development and organization of the inner membrane complex of Plasmodium falciparum. Eukaryot Cell 10, 556–564 (2011).
Harding, C. R. et al. Gliding associated proteins play essential roles during the formation of the inner membrane complex of toxoplasma gondii. PLoS Pathog. 12, e1005403 (2016).
Cepeda Diaz, A. K., Rudlaff, R. M., Farringer, M. & Dvorin, J. D. Essential function of alveolin PfIMC1g in the Plasmodium falciparum asexual blood stage. mBio 14, e0150723 (2023).
Parkyn Schneider, M. et al. Disrupting assembly of the inner membrane complex blocks Plasmodium falciparum sexual stage development. PLoS Pathog. 13, e1006659 (2017).
Saini, E. et al. Plasmodium falciparum PhIL1-associated complex plays an essential role in merozoite reorientation and invasion of host erythrocytes. PLoS Pathog. 17, e1009750 (2021).
Willems, A. R., Schwab, M. & Tyers, M. A hitchhiker’s guide to the cullin ubiquitin ligases: SCF and its kin. Biochim. Biophys. Acta 1695, 133–170 (2004).
Harper, J. W. & Schulman, B. A. Cullin-RING ubiquitin ligase regulatory circuits: a quarter century beyond the F-Box hypothesis. Annu. Rev. Biochem. 90, 403–429 (2021).
Boland, A. W. et al. Oxygen-dependent regulation of E3(SCF)ubiquitin ligases and a Skp1-associated JmjD6 homolog in development of the social amoeba Dictyostelium. J. Biol. Chem. 298, 102305 (2022).
Obara, K., Nishimura, K. & Kamura, T. E3 ligases regulate organelle inheritance in yeast. Cells 13, https://doi.org/10.3390/cells13040292 (2024).
Carroll, E. C. & Marqusee, S. Site-specific ubiquitination: deconstructing the degradation tag. Curr. Opin. Struct. Biol. 73, 102345 (2022).
Baptista, C. G. et al. Toxoplasma F-box protein 1 is required for daughter cell scaffold function during parasite replication. PLoS Pathog. 15, e1007946 (2019).
Rashpa, R., Klages, N., Schvartz, D., Pasquarello, C. & Brochet, M. The Skp1-Cullin1-FBXO1 complex is a pleiotropic regulator required for the formation of gametes and motile forms in Plasmodium berghei. Nat. Commun. 14, 1312 (2023).
Branon, T. C. et al. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 36, 880–887 (2018). PMID - 30125270.
Viswanathan, S. et al. High-performance probes for light and electron microscopy. Nat. Methods 12, 568–576 (2015).
Jones, M. L., Kitson, E. L. & Rayner, J. C. Plasmodium falciparum erythrocyte invasion: a conserved myosin associated complex. Mol. Biochem. Parasitol. 147, 74–84 (2006).
Bertiaux, E. et al. Expansion microscopy provides new insights into the cytoskeleton of malaria parasites including the conservation of a conoid. PLoS Biol. 19, e3001020 (2021).
Gambarotto, D., Hamel, V. & Guichard, P. Ultrastructure expansion microscopy (U-ExM). Methods Cell Biol. 161, 57–81 (2021).
Simon, C. S. et al. An extended DNA-free intranuclear compartment organizes centrosome microtubules in malaria parasites. Life Sci. Alliance 4, https://doi.org/10.26508/lsa.202101199 (2021).
Liffner, B. et al. Atlas of Plasmodium falciparum intraerythrocytic development using expansion microscopy. Elife 12, https://doi.org/10.7554/eLife.88088 (2023).
Clements, R. L. et al. Identification of basal complex protein that is essential for maturation of transmission-stage malaria parasites. Proc. Natl Acad. Sci. USA 119, e2204167119 (2022).
Knuepfer, E., Napiorkowska, M., van Ooij, C. & Holder, A. A. Generating conditional gene knockouts in Plasmodium - a toolkit to produce stable DiCre recombinase-expressing parasite lines using CRISPR/Cas9. Sci. Rep. 7, 3881 (2017).
Collins, C. R. et al. Robust inducible Cre recombinase activity in the human malaria parasite Plasmodium falciparum enables efficient gene deletion within a single asexual erythrocytic growth cycle. Mol. Microbiol. 88, 687–701 (2013).
Collins, C. R. et al. Malaria parasite cGMP-dependent protein kinase regulates blood stage merozoite secretory organelle discharge and egress. PLoS Pathog. 9, e1003344 (2013).
Absalon, S., Robbins, J. A. & Dvorin, J. D. An essential malaria protein defines the architecture of blood-stage and transmission-stage parasites. Nat. Commun. 7, 11449 (2016).
Aurrecoechea, C, et al. PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res. 37, D539 (2009).
Bai, C. et al. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell 86, 263–274 (1996).
D’Angiolella, V. et al. SCF(Cyclin F) controls centrosome homeostasis and mitotic fidelity through CP110 degradation. Nature 466, 138–142 (2010).
Roux, K. J., Kim, D. I., Raida, M. & Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 196, 801–810 (2012).
Boltryk, S. D. et al. CRISPR/Cas9-engineered inducible gametocyte producer lines as a valuable tool for Plasmodium falciparum malaria transmission research. Nat. Commun. 12, 4806 (2021).
Zhang, M. et al. Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science 360, https://doi.org/10.1126/science.aap7847 (2018).
Barylyuk, K. et al. A comprehensive subcellular atlas of the toxoplasma proteome via hyperLOPIT provides spatial context for protein functions. Cell Host Microbe 28, 752–766.e759 (2020).
Alvarez, C. A. & Suvorova, E. S. Checkpoints of apicomplexan cell division identified in Toxoplasma gondii. PLoS Pathog. 13, e1006483 (2017).
Rizvi, Z. et al. Plasmodium falciparum contains functional SCF and CRL4 ubiquitin E3 ligases, and CRL4 is critical for cell division and membrane integrity. PLoS Pathog. 20, e1012045 (2024).
Mandalasi, M. N. et al. Oxygen-dependent regulation of F-box proteins in Toxoplasma gondii is mediated by Skp1 glycosylation. J. Biol. Chem. 300, 107801 (2024).
Kumar, S. et al. PfCDPK1 mediated signaling in erythrocytic stages of Plasmodium falciparum. Nat. Commun. 8, 63 (2017).
Jonkers, W. & Rep, M. Lessons from fungal F-box proteins. Eukaryot Cell 8, 677–695 (2009).
Nelson, D. E., Randle, S. J. & Laman, H. Beyond ubiquitination: the atypical functions of Fbxo7 and other F-box proteins. Open Biol. 3, 130131 (2013).
Hermand, D. F-box proteins: more than baits for the SCF? Cell Div. 1, 30 (2006).
Kipreos, E. T. & Pagano, M. The F-box protein family. Genome Biol. 1, REVIEWS3002 (2000).
Gubbels, M. J. et al. Fussing about fission: defining variety among mainstream and exotic apicomplexan cell division modes. Front. Cell Infect Microbiol. 10, 269 (2020).
Batugedara, G. et al. The chromatin bound proteome of the human malaria parasite. Microb. Genom 6, https://doi.org/10.1099/mgen.0.000327 (2020).
Johnson, T. M., Rajfur, Z., Jacobson, K. & Beckers, C. J. Immobilization of the type XIV myosin complex in Toxoplasma gondii. Mol. Biol. Cell 18, 3039–3046 (2007).
Rees-Channer, R. R. et al. Dual acylation of the 45 kDa gliding-associated protein (GAP45) in Plasmodium falciparum merozoites. Mol. Biochem. Parasitol. 149, 113–116 (2006).
Rompikuntal, P. K. et al. Blocking Palmitoylation of Toxoplasma gondii Myosin Light Chain 1 Disrupts Glideosome Composition but Has Little Impact on Parasite Motility. mSphere 6, https://doi.org/10.1128/mSphere.00823-20 (2021).
Wichers-Misterek, J. S. et al. A microtubule-associated protein is essential for malaria parasite transmission. mBio 14, e0331822 (2023).
Green, J. L. et al. Ubiquitin activation is essential for schizont maturation in Plasmodium falciparum blood-stage development. PLoS Pathog. 16, e1008640 (2020).
Rudlaff, R. M., Kraemer, S., Streva, V. A. & Dvorin, J. D. An essential contractile ring protein controls cell division in Plasmodium falciparum. Nat. Commun. 10, 2181 (2019).
Jones, M. L. et al. A versatile strategy for rapid conditional genome engineering using loxP sites in a small synthetic intron in Plasmodium falciparum. Sci. Rep. 6, 21800 (2016).
Tiburcio, M. et al. A novel tool for the generation of conditional knockouts to study gene function across the plasmodium falciparum life cycle. mBio 10, https://doi.org/10.1128/mBio.01170-19 (2019).
Rappsilber, J., Mann, M. & Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 (2007).
Cox, J. et al. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell Proteom. 13, 2513–2526 (2014).
Demichev, V., Messner, C. B., Vernardis, S. I., Lilley, K. S. & Ralser, M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 17, 41–44 (2020).
Shen, B., Brown, K., Long, S. & Sibley, L. D. Development of CRISPR/Cas9 for efficient genome editing in toxoplasma gondii. Methods Mol. Biol. 1498, 79–103 (2017).
Acknowledgements
We thank Fabian Schulte at the Whitehead Institute Quantitative Proteomics Core and Ross Tomaino at the Taplin Mass Spectrometry Facility for their advice and core services. We thank Robin Anders, Julian Rayner, Alan Cowman, Matt Dixon, and Anthony Holder for providing antibodies; Ellen Knuepfer (3D7-DiCre), Moritz Treeck (NF54-DiCre), and Till Voss (NF54-iGP2) for providing parasites; and James McGee for cloning and transfecting the smV5-tagged parasite strain. The study was supported by the National Institutes of Health R01 AI150240 (I.J.B., C.M.W., and J.D.D.).
Author information
Authors and Affiliations
Contributions
S.K.S. - conceptualization, methodology, validation, formal analysis, investigation, writing – original draft. C.G.B. – methodology, investigation, formal analysis. C.M.W. – conceptualization, funding acquisition, writing – review and editing. I.J.B. – conceptualization, funding acquisition, writing – review and editing. J.D.D. – conceptualization, funding acquisition, methodology, formal analysis, project administration, writing – review and editing
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Nishith Gupta and Johannes Stortz. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sreenivasamurthy, S.K., Baptista, C.G., West, C.M. et al. PfFBXO1 is essential for inner membrane complex formation in Plasmodium falciparum during both asexual and transmission stages. Commun Biol 8, 190 (2025). https://doi.org/10.1038/s42003-025-07619-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-025-07619-6
