
Abstract
Incretin-based therapies are highly successful in combatting obesity and type 2 diabetes1. Yet both activation and inhibition of the glucose-dependent insulinotropic polypeptide (GIP) receptor (GIPR) in combination with glucagon-like peptide-1 (GLP-1) receptor (GLP-1R) activation have resulted in similar clinical outcomes, as demonstrated by the GIPR–GLP-1R co-agonist tirzepatide2 and AMG-133 (ref. 3) combining GIPR antagonism with GLP-1R agonism. This underlines the importance of a better understanding of the GIP system. Here we show the necessity of β-arrestin recruitment for GIPR function, by combining in vitro pharmacological characterization of 47 GIPR variants with burden testing of clinical phenotypes and in vivo studies. Burden testing of variants with distinct ligand-binding capacity, Gs activation (cyclic adenosine monophosphate production) and β-arrestin 2 recruitment and internalization shows that unlike variants solely impaired in Gs signalling, variants impaired in both Gs and β-arrestin 2 recruitment contribute to lower adiposity-related traits. Endosomal Gs-mediated signalling of the variants shows a β-arrestin dependency and genetic ablation of β-arrestin 2 impairs cyclic adenosine monophosphate production and decreases GIP efficacy on glucose control in male mice. This study highlights a crucial impact of β-arrestins in regulating GIPR signalling and overall preservation of biological activity that may facilitate new developments in therapeutic targeting of the GIPR system.
Similar content being viewed by others


Main
Several naturally occurring G protein-coupled receptor (GPCR) genetic variants have been shown to affect receptor function and to result in undesired phenotypic traits and unintended responses to clinically used pharmaceuticals, a third of which target GPCRs4,5. Studying receptor genetic variability can provide a deeper understanding of the physiological roles of a given system. Optimally, such studies cover allelic series comprising both loss-of-function (LoF) variants and gain-of-function (GoF) variants, thereby strengthening the mechanistic interpretation of changed gene function. Genetic variants of glucose-dependent insulinotropic polypeptide receptor (GIPR) gene, including the common variant (E354Q) and two rare LoF variants (R190Q and E288G), have been associated with changes in cardiometabolic traits6,7,8,9,10,11. Yet there is a lack of comprehensive characterization of genetic variations in the GIPR to understand the role of GIPR signalling in human (patho)physiology.
In this Letter, we sequenced GIPR in four Danish study cohorts (N = 10,523), including patients newly diagnosed with type 2 diabetes (T2D) (DD2 cohort), individuals from a population-based study cohort (Inter99 cohort) screened for T2D, a cohort of obese children and adolescents (Holbaek Study) and a cohort of women with gestational diabetes mellitus (GDM cohort) in search for genetic variants in the GIPR. Of the identified 61 GIPR variants, we selected all 47 non-synonymous variants for further functional characterization (Supplementary Table 1).
First, we measured GIP-mediated Gs activity via cyclic adenosine monophosphate (cAMP) accumulation against the reference human wild-type (WT) GIPR and determined their maximal signal (Emax) and GIP potency (EC50) (Extended Data Fig. 1a,b). We used a range of GIP concentrations corresponding to those observed in response to ingestion of low, moderate and high amounts of glucose as measured in earlier clinical studies12,13 (Fig. 1a,b). Thus, ‘low’ corresponded to ~10 pM and ‘high’ to ~100 pM (Fig. 1c). Next, we ranked the 47 variants by their signalling activity at 100 pM GIP and identified 29 variants with an activity <50% of WT. These were classified as partial and complete LoF variants (referred to as LoF variants hereafter) (Fig. 1d), with a cumulative allele frequency of 1% in the combined Danish cohorts. The binding capacity of GIP (Bmax) was impaired for all 29 LoF variants (Fig. 1e). Five showed undetectable GIP binding (S64P, C84Pfs*70, R101H, Y258Sfs*50 and Q400Gfs*56; Fig. 1f). For the remaining 24 variants, high affinity (Kd) for GIP was retained (Fig. 1f), suggesting a loss of signal due to low receptor expression. Four variants (T116R, R183Q, R190Q and R300W) with maintained Bmax displayed no cAMP signalling at 100 pM GIP, unlike variants with comparable Bmax, for example, G144S, A185V and L304R (Fig. 1d,e). At higher GIP doses, cAMP production was restored in R183Q, R190Q and R300W, while T116R remained unable to signal up to 100 nM GIP (Extended Data Fig. 1a), suggesting these residues are critical for G protein interaction. Conversely, the T25R variant maintained over 50% signalling capacity at a 25% Bmax, indicating a possible structural modification that enhances signalling despite lower GIP binding. Gene dose titration experiments were performed to investigate the impact of low receptor expression on GIPR signalling. WT GIPR ranging from doses of 10 ng to 10 µg (1,000-fold increase) receptor DNA revealed an association between GIPR signalling and cell surface expression (Extended Data Fig. 1c). As expected, equal binding affinity of GIP was observed independently of receptor expression levels. In contrast, a right shift towards lower potency was observed with decreasing GIPR levels (Extended Data Fig. 1d,e). Combining the data for receptor expression and signalling at 100 pM GIP of the 47 GIPR variants as well as the WT GIPR transfected with increasing DNA doses, a linear correlation (r2 = 0.8) appeared (Extended Data Fig. 1f). This suggests that receptor surface expression is the main driver of activity of the GIPR variants, similar to what has been observed in the glucagon-like peptide-1 receptor (GLP-1R)14.

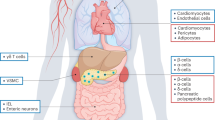
a, Localization of GIP-secreting cells in the gastrointestinal tract. b, GIP secretion in response to ingestion of low (25 g), moderate (50–75 g) or high (100–125 g) amounts of glucose according to previous human studies (N = 8)12,13. The blue box highlights the range from low (10 pM) to high (100 pM) levels of GIP. c, The corresponding area of the physiological range of GIP on a WT GIPR dose-response curve for cAMP production. d, cAMP production at 100 pM GIP, shown as percentage of activity compared with WT GIPR. Grey, WT-like activity and dark red, below 50% activity of WT. The data represent the mean ± s.e.m. WT GIPR: N = 18; R12Q, S64P, Y73H, R164Q, P195R–A207V, S382T–S382Y, S415I–G449A: N = 6; L13F–R38L, V99I–R101H, R164W, T168A, L261H, E461*–E463Q: N = 4; R43G, M67R, C84P–W90L, I221M, R336P: N = 5; T116R–A150T, H166Y, R183Q–A185V, W233*–Y258S, E291K–R300W, E354Q–I378M, K397N–V399M: N = 3; R190Q: N = 9; E288G: N = 8; L304R: N = 7. Independent experiments were performed in duplicate. e,f, Homologous competition binding of GIP, showing maximum binding capacity (Bmax) and affinity (Kd) compared with WT GIPR. The variants are arranged in the same order as in b. NB, no binding. The data represent the mean ± s.e.m. WT GIPR: N = 18; R12Q: N = 5; L13F–R101H, G144S–A207V, W233*, E288G, E354Q, S382T–G449A, E463Q: N = 3; T116R, I221M, Y258S–L261H, E291K–R336P, I378M, E461*: N = 4. Independent experiments were performed in duplicate. g, Illustration of the GIPR with variants located at functional sites (orange) and non-functional sites (blue). Green area: ligand binding site; yellow area: (micro) switches; red area: G protein interface (left). Mean difference in cAMP efficacy between variants at functional (N = 13) and non-functional (N = 34) sites (right). No significant difference was observed between the efficacy of 100 pM, P = 0.30. Statistical significance was assessed using the Wilcoxon rank-sum test (two sided).
The 29 LoF mutations were evenly distributed across the GIPR structure (Fig. 1g), which led us to speculate whether variants located at functional sites were more deleterious than those at non-functional sites. Thus, we mapped the location of variants to the ligand binding interface, the G protein interface and the activity-regulating (micro-)switches, which revealed a comparable cAMP production between variants located at functional sites (N = 13) and those at non-functional sites (Wilcoxon rank-sum test P = 0.3) (Fig. 1g). Variant effect predictors (VEPs) did not reveal any correlations between measured efficacy and VEP scores (mean r2 across 15 predictors: 0.09) (Extended Data Fig. 2a). For previously described VEP masks15, we observed an under-representation of variants predicted to cause impaired signalling compared with the in vitro LoF variants (Extended Data Fig. 2b). Hence, variant location and VEPs are insufficient for estimating nuanced GIPR variant outcomes in vitro.
Given that GIP binding leads to receptor phosphorylation by GPCR kinases, resulting in sustained recruitment of β-arrestins16, we determined β-arrestin recruitment for the 47 non-synonymous GIPR variants. Since the WT GIPR predominantly recruits β-arrestin 2 over β-arrestin 1 (ref. 17), we focused on β-arrestin 2 in this study (henceforth, β-arrestin 2 will be referred to as β-arrestin). We observed no major differences in potency for most variants, apart from a few lacking the ability to recruit β-arrestin (Fig. 2a). The variants with maintained β-arrestin recruitment displayed varying Emax (Fig. 2b), falling into four main groups. Of the 29 variants with impaired cAMP production, 21 also had impaired β-arrestin recruitment, hereafter classified as LoFcAMP/LoFarr (defined as <50% activity in either pathway). Eight variants (M67R, Y73H, V99I, T116R, A150T, A185V, P195R and A207V) with cAMP production LoF showed WT-like β-arrestin recruitment (LoFcAMP/WT-likearr). Six variants (T25R, R43G, R164Q, H166Y, I221M and K397N) showed maintained cAMP production but low β-arrestin recruitment (WT-likecAMP/LoFarr). A fourth group of ten variants (R12Q, L13F, R38L, A294V, I299V, E354Q, S415I, E418K, G449A and E463Q) were WT-like (WT-likecAMP/WT-likearr). The dose-response relationship in cAMP production versus β-arrestin recruitment of four variants (R12Q, S64P, M67R and I221M), typical of each of the four characterized groups, illustrated the distinct signalling profiles of the variants (Fig. 2c). Beyond these four groups, one variant (I378M) showed more β-arrestin recruitment (GoF) with WT-like cAMP production (WT-likecAMP/GoFarr) and another (E461*) had more cAMP production (GoF) and preserved β-arrestin recruitment (GoFcAMP/WT-likearr). Structural mapping revealed that variants with similar signalling preferences did not cluster in specific regions or domains (Supplementary Fig. 1), except for the LoFcAMP/WT-likearr. In this group, all variants except A185V were located in the extracellular domain (M67R, Y73H, V99I and T116R) and extracellular loop 1 (P195R and A207V), indicating that mutations in these areas specifically affected cAMP production.

a, β-arrestin 2 recruitment potency (logEC50) of GIP. Variants are arranged according to cAMP efficacy of 100 pM GIP. b, β-arrestin 2 recruitment maximal signalling (Emax), shown as a percentage of WT GIPR activity. NA, no activation. c, Dose-response curves of cAMP production (left) and β-arrestin 2 recruitment (right) of GIPR variants R12Q, I221M, M67R and S64P, respectively. Data represents the mean ± s.e.m. WT GIPR: N = 30; R12Q–S64P, Y73H–C84P, V99I–T168A, A185V, P195R: N = 5; M67R: N = 6; W90L, R183Q, A207V, R336P–E354Q: N = 4; R190Q, E288G: N = 6; I221M–L261H, E291K–L304R, I378M–E463Q: N = 3. Independent experiments were performed in duplicate.
Given the necessity of β-arrestin recruitment for GIPR internalization16, we probed the internalization profile of the eight LoFcAMP/WT-likearr variants together with the two LoFcAMP/LoFarr variants, R190Q and E288G (previously published to be impaired in internalization11). All LoFcAMP/WT-likearr variants demonstrated preserved internalization capabilities (Extended Data Fig. 3a,b). Of these, Y73H, T116R, A150T, A185V and A207V had nearly identical levels of internalization as the WT GIPR, while M67R, V99I and P195R displayed a slightly diminished internalization (Extended Data Fig. 3c,d). Conversely, the two LoFcAMP/LoFarr variants, R190Q and E288G, with maintained GIP binding but impaired cAMP signalling and β-arrestin recruitment, did not internalize, highlighting the importance of β-arrestin recruitment but not Gs activation for GIPR internalization.
Next, we sought to link the molecular pharmacology of the GIPR variants to cardiometabolic phenotypes using Danish cohorts (T2D: DD2 and Inter99, N = 7,173 and quantitative traits: Inter99, N = 5,711) and the UK Biobank18 (465,506 whole-exome-sequenced participants). We detected 365 non-synonymous GIPR variants in the UK Biobank (Supplementary Table 2), of which 32 of the 47 Danish non-synonymous GIPR variants were represented (Fig. 3a). We grouped the characterized variants according to their molecular characteristics (Fig. 3b) to perform burden testing. Additionally, we performed burden testing of all synonymous and predicted LoF (pLoF) GIPR sequenced variants in the Danish population and the UK Biobank, respectively (Supplementary Tables 1 and 2), which were not functionally characterized. We did not detect significant associations with any phenotype for any of the GIPR variant groups in the Danish cohorts (Extended Data Figs. 4 and 5, Supplementary Table 3 and Supplementary Fig. 2), probably due to low power given the few carriers in all variant groups. In the UK Biobank, ~3,800 carriers of LoFcAMP/LoFarr variants displayed lower body mass index (BMI) (effect size (β) = −0.10 s.d., 95% confidence interval (CI) −0.13 to −0.08), body fat percentage (β = −0.06 s.d., 95% CI −0.09 to −0.04), hip circumference (β = −0.12 s.d., 95% CI −0.15 to −0.09) and waist circumference (β = −0.07 s.d., 95% CI −0.09 to −0.04) than non-carriers (Fig. 3c). The LoFcAMP/LoFarr carriers also had a nominally lower odds ratio (OR) for obesity compared to non-carriers (OR 0.85, CI 0.75 to 0.90; Extended Data Fig. 6), agreeing with the adiposity protective effect of impaired GIPR signalling9,10,11. Similarly, carriers of the 105 pLoF variants found in the UK Biobank (488 carriers) displayed significantly lower hip circumference (β = −0.19 s.d., 95% CI −0.27 to −0.11) and slightly lower BMI (β = −0.13 s.d., 95% CI −0.21 to −0.04; Fig. 3c). In contrast to the LoFcAMP/LoFarr variants, carriers of the pLoF variants also showed slightly lower diastolic blood pressure (β = −0.16 s.d., 95% CI −0.25 to −0.08; Fig. 3c) with no increase in hypertension (Extended Data Fig. 6). Intriguingly, the lower adiposity phenotypes in LoFcAMP/LoFarr carriers were not found in the ~6,600 LoFcAMP/WT-likearr carriers (Fig. 3c). Likewise, no associations were observed in the other GIPR variant groups (Fig. 3c and Extended Data Fig. 6). Comparable results were found when restricting to 427,931 individuals of British ancestry (Extended Data Fig. 7). Finally, we conducted a meta-analysis regression using single-variant estimates of GIPR variants regressed on their molecular phenotype, revealing an impact of impaired β-arrestin recruitment (below 50% of WT) on BMI and hip circumference but not on blood pressure and no impact of impaired cAMP production (Extended Data Fig. 8).

a, The overlap between non-synonymous GIPR variants found in the Danish population and the UK Biobank. N, total cohort sample size. b, The pharmacological profile for all GIPR variants in terms of cAMP accumulation (efficacy (Eff.) at 100 pM GIP), binding ability and β-arrestin recruitment compared with WT receptor clustered into six in vitro phenotype groups. c, A forest plot showing burden test statistics in the UK Biobank cohort (N = ~440,000). Carriers represent the number of individuals for each phenotype carrying any of the GIPR variants within that GIPR variant group. q, FDR-adjusted P values of the burden tests (performed using ACAT-O). pLoF denotes variants predicted to cause loss of function with VEP47 and/or CADD score > 30, that is, pLoF variants were not functionally characterized. aThe WT-likecAMP/WT-likearr variant, E354Q, was excluded from burden testing due to high MAF (~20%). Beta represents the effect size as standard deviation of the phenotype with error bars representing the 95% CI.
These findings illustrate that differential GIPR signalling/coupling may contribute to altered phenotypic outcomes and underscore the importance of β-arrestin and GIPR internalization for proper function. This prompted us to further assess the role of β-arrestin for GIPR signalling by investigating the endosomal signalling in representative GIPR variants from each of the six in vitro-based groups. We coupled mini Gs (mGs, an engineered G protein that detects the active receptor component for engaging and activating heterotrimeric Gs) with enhanced bystander bioluminescence resonance energy transfer (BRET) at early endosomes following exposure to GIP19,20,21. Variants with diminished β-arrestin recruitment displayed minimal to no signalling from early endosomes. In contrast, those that retained β-arrestin recruitment effectively recruited mGs to early endosomes and continued their intracellular signalling to varying degrees, compared with the WT GIPR (Fig. 4a). These data demonstrate the crucial role of β-arrestin in sustaining Gs-mediated signalling of the GIPR from endosomes, supported by the positive correlation between β-arrestin recruitment and GIP-induced endosomal signalling (Fig. 4b). Additionally, we investigated the basal subcellular localization of the variants in relation to their distribution at the plasma membrane, endosomes, Golgi apparatus and endoplasmic reticulum (ER). Most variants showed distribution patterns similar to the WT GIPR, predominantly at the plasma membrane (Supplementary Fig. 3a). However, only the two LoFcAMP/LoFarr variants, R190Q and E288G, deviated slightly from WT GIPR as they appeared to be trapped intracellularly in the ER with reduced expression on the plasma membrane (Supplementary Fig. 3b), which is in line with their overall impaired receptor function.

a, Gs pathway engagement at early endosomes of representative GIPR variants from each of the six in vitro-based groups. The delta (Δ) BRET values are calculated by subtracting the vehicle value from the top values (1 µM) and evaluated by a one sample t-test (two-sided) performed on the mean of individual experiments with hypothetical value set to 0. The data represent mean ± s.e.m. of WT GIPR, N = 8 and GIPR variants, N = 4. Independent experiments were performed in duplicates. Exact P values are shown above the data points for variants significantly different from 0. b, Pearson correlation analysis of endosomal signalling (ΔBRET) with β-arrestin 2 recruitment (Emax % of WT) of the variants. LoF/LoF (LoFcAMP/LoFarr), LoF/WT (LoFcAMP/WT-likearr), WT/LoF (WT-likecAMP/LoFarr), WT/GoF, (WT-likecAMP/GoFarr), GoF/WT (GoFcAMP/WT-likearr) and WT/WT (WT-likecAMP/WT-likearr). The data represent the mean ± s.e.m. Independent experiments were performed in duplicates. The number of replicates is the same as Figs. 4a and 2. c, Workflow of the pharmacologic studies in Arrb2 knockout (KO) mice. t0, t15, t30, t60 and t120 represent timepoints (in minutes) after initiating an intraperitoneal glucose tolerance test (IPGTT). d,e, Glucose excursion during an IPGTT in WT male mice (N = 10–14 per group) (d) and male Arrb2 KO mice (N = 12–13 per group) (e) 15 min following vehicle (black circles), d-Ala GIP (orange triangles) or GLP-1 (teal squares). Mouse data are represented as mean ± s.e.m. Statistical significance was determined by two-way analysis of variance, and Holm-Šidák testing was used to correct for multiple testing. The P value for either GLP-1 or GIP compared with vehicle is shown. Figure 4c was created with BioRender.com.
GIP and GLP-1 are mostly known for enhancing glucose-stimulated insulin secretion from the pancreatic β cells22,23. We therefore explored the functional relationship between β-arrestin recruitment and GIP-stimulated glucose regulation by conducting pharmacological interventions in a murine genetic global germline β-arrestin 2 (Arrb2) knockout (KO) model. Male mice were injected with native GLP-1 and d-Ala GIP—a highly potent GIPR agonist24—before an intraperitoneal glucose tolerance test (IPGTT; Fig. 4c). As expected, both GLP-1 and d-Ala GIP significantly lowered blood glucose levels in WT male mice (Fig. 4d). In contrast, only GLP-1 was able to lower blood glucose in male Arrb2 KO mice (Fig. 4e). Consistent with this, a significantly lower GIPR-mediated cAMP production was observed in β-arrestin1/2 KO HEK293 cells compared with parental HEK293 cells (Extended Data Fig. 9a–d). This was not the case for the GLP-1R signalling, which was unaffected by the absence of arrestins (Extended Data Fig. 9e–g). Thus, in agreement with the physiological effects in humans, β-arrestins were critical for maintained GIPR intracellular signalling and for GIP-mediated lowering of blood glucose levels in mice, indicating a functional role of β-arrestins in sustaining GIP, but not GLP-1, action.
In summary, we show that 29 GIPR variants with impaired cAMP production exhibited a proportionally lower agonist binding capacity, indicating that an impaired cAMP-mediated receptor activation was caused primarily by low receptor cell surface expression. A similar observation was found for GLP-1R14, emphasizing the importance of cell surface expression in canonical GPCR activation. Among the 29 GIPR variants, 21 had impaired β-arrestin recruitment (LoFcAMP/LoFarr), with the affected residues distributed at various locations indicating allosteric mechanisms, converging towards similar molecular phenotypes.
We and others have previously shown a lowering effect on adiposity-associated phenotypes for the two LoFcAMP/LoFarr variants, R190Q and E288G9,10,11. This effect was also recapitulated in the UK Biobank for the pooled group of LoFcAMP/LoFarr variants. This adiposity-lowering effect was not found with sustained β-arrestin recruitment (LoFcAMP/WT-likearr), implying a plausible contribution of β-arrestins to GIPR function in the context of metabolic health. Additionally, enhanced β-arrestin recruitment with maintained cAMP accumulation (WT-likecAMP/GoFarr) was nominally associated with higher adiposity in this cohort. Our meta-analysis regression supported the impact of β-arrestin recruitment on adiposity. However, since the effect sizes were estimated solely from the variants investigated in vitro, the results may be highly underpowered. In contrast to the β-arrestin contribution in current associations, GLP-1R variants leading to impaired β-arrestin recruitment do not contribute to the associations between impaired cAMP signalling variants and higher haemoglobin A1c levels and BMI14. We also noticed that the GIPR variant groups GoFcAMP/WT-likearr and WT-likecAMP/GoFarr affected the blood pressure and adiposity measures in the opposite effect directions. We did not observe a changed risk of T2D in LoFcAMP/LoFarr variant carriers in the UK Biobank, which is in contrast to the worsened insulin resistance and increased risk of T2D reported for the LoF-mimicking GIPR variant E354Q6,7. The lack of association with T2D risk in the UK Biobank may, in part, be due to known limitations regarding the register-based diabetes phenotypes in the UK Biobank, even though attempts have been made to establish a more solid T2D definition25,26. The lack of phenotype associations in the Danish cohorts may be explained by limited statistical power, and thus we cannot exclude an impact of impaired GIPR signalling on these phenotypes in the Danish population. Furthermore, the nuanced influence of lifestyle factors, which contribute to variation in cardiometabolic phenotypes and the efficacy of pharmacological interventions, may partially shape our findings. Nonetheless, our genetic data support the demonstrated lowering effect of diminished GIPR signalling on adiposity in humans and that the burden of rare protein-coding GIPR variants affects body adiposity9,10,11.
Our observation that maintained β-arrestin recruitment may be a strong determinant of the clinical phenotypes (lower adiposity in the LoFcAMP/LoFarr group but not in the LoFcAMP/WT-likearr group) suggests that a preserved GIPR response may be present despite diminished cAMP signalling if the β-arrestin system is intact. We also supported the pivotal role of β-arrestins in GIPR internalization16,27,28 by the in vitro findings, that is, the eight LoFcAMP/WT-likearr variants maintained their internalization properties in contrast to the two LoFcAMP/LoFarr variants (R190Q and E288G). This is in contrast to the GLP-1R, which can internalize independently of β-arrestins29. Additionally, the endosomal signalling experiments showed a clear positive correlation with β-arrestin recruitment, confirming the pivotal role of β-arrestins in regulating the post-activation trafficking30 and signalling31 of the GIPR and thereby potentially contributing to the unchanged biological actions of GIP in the carriers of LoFcAMP/WT-likearr. We did, however, not see any major changes in subcellular localization of the variants compared with WT GIPR, except for the two LoFcAMP/LoFarr variants, R190Q and E288G, which appeared to be trapped in the ER relative to localization at the plasma membrane, explaining their diminished GIP binding and activity. These findings suggest that the altered function of most of the GIPR variants might result from changes in receptor conformation or impaired receptor cell surface expression but not altered subcellular receptor distribution.
In agreement with our in vitro data, GIP treatment in vivo had no glucose-lowering effect in male mice lacking β-arrestins, whereas GLP-1 maintained its insulinotropic properties in the β-arrestin KO mice. Conversely, a recent study showed reduced acute GLP-1-mediated glycaemic control in β cell-specific β-arrestin 2 KO female mice, with a similar trend for d-Ala GIP treatment in the same KO mice32. However, our study, along with others33, demonstrated consistent insulinotropic actions of GLP-1 regardless of β-arrestin 2 in mice. In contrast, GIP-potentiated insulin secretion relied on β-arrestin 2, underscoring distinct signalling preferences of these closely related receptors. Of note, our mouse studies were conducted in global germline β-arrestin 2 KO mice, which aligns more closely with inherited human genetics. However, it should be noted that these findings are solely derived from studies in male mice, raising uncertainty about the translatability to female mice.
Recent studies have comprehensively profiled coding GLP1R variants. One study demonstrated no association between variants impairing cAMP signalling and cardiometabolic phenotypes34. However, upon inclusion of expression, a recent study revealed an association between reduced cell surface expression and metabolic phenotypes14. Likewise, GLP1R variants have been suggested to affect the outcome of GLP-1R agonist treatment35, highlighting the importance of genetic influences on drug responses.
The marked clinical effects of tirzepatide may be explained by its imbalanced nature as a dual agonist with activation of GIPR as native GIP, but with less recruitment of β-arrestin to the GLP-1R with full activation of the cAMP pathway2. Development of similar Gs biased analogues for the GIPR has also been proposed27. Although it is not possible to genetically instrument the dual agonistic effects, our results and those of others33 highlight that decreased recruitment of β-arrestins might benefit the GLP-1R system36,37 but not for the GIPR regarding its role in glycaemic control. Yet our findings in the UK Biobank show beneficial effects on adiposity-associated traits of an overall reduced GIPR activation, probably due to the lost lipogenic action of GIP. Hence, our data suggest that balanced β-arrestin recruitment may drive the differences in weight-associated effects of the variants and that the contribution of β-arrestins to modulate Gs-mediated signalling of the GIPR is imperative for the biological functions of GIP (Extended Data Fig. 10).
In conclusion, we expand our current understanding of the GIP–GIPR system by highlighting the importance of β-arrestin recruitment in GIPR function. Our study points towards improving future therapeutic targeting of the GIPR system by finetuning the balance between G protein activation and β-arrestin recruitment.
Methods
Study populations
The Inter99 cohort is a Danish population-based cardiovascular and T2D prevention cohort initiated in 1999. All participants (30–60 years of age) underwent a comprehensive health evaluation, including measurement of anthropometrics and blood pressure, and received a 2-h 75 g oral glucose tolerance test with blood drawn throughout the test, followed by biochemical measurements. We performed targeted sequencing on 6,089 individuals from Inter99, among which 4,243 (exclusively individuals with normoglycaemia) and 5,711 individuals (without known diabetes at baseline) were available for T2D and quantitative association analyses, respectively. According to the World Health Organization 1999 criteria, 4,333 of the 5,711 individuals without diabetes at baseline had normal glucose tolerance and 1,378 had pre-diabetes. The protocol of the Inter99 study followed the Declaration of Helsinki and was approved by the local ethical committee (KA 98 155). All participants provided written consent before the study examination38,39.
The Danish Centre for Strategic Research in Type 2 Diabetes (DD2) cohort, initiated in 2010, is an ongoing nationwide population-based project cohort with continuous enrolment of patients with newly diagnosed T2D. At enrolment, all patients were interviewed about, for example, habitual lifestyle factors, weight gain and family history of T2D, and provided biological samples, strengthened by further linking to medical register databases with individual patient data. The DD2 study was approved by the Danish National Committee on Biomedical Research Ethics and the Danish Data Protection Agency, and all participants gave signed consent before examination40. We performed targeted sequencing in 2,930 individuals from this cohort. The same individuals were included in T2D association analyses.
The Holbaek Study, previously known as The Danish Childhood Obesity Biobank, is a Danish children and adolescent obesity case–control cohort initiated to manage and study childhood obesity41. All participants included in the current study (0.5–24.7 years of age, median age 11.7 years, N (>19 years) = 16, N (<1 year) = 1) were recruited through the Children’s Obesity Clinic (Copenhagen University Hospital Holbæk, Denmark) from January 2008 to June 2014. We performed targeted sequencing on 1,146 children or adolescents from this study cohort. The study followed the Declaration of Helsinki and was approved by the Ethics Committee of Region Zealand, Denmark (SJ-104) and the Danish Data Protection Agency. All participants provided written consent; for participants younger than 18 years, oral assent was provided by the participants and the parents provided written consent. This cohort was only used for the identification of GIPR variants.
The GDM cohort consists of women with diet-treated gestational diabetes between 1978 and 1985, or 1987 and 1996. The study followed the Declaration of Helsinki and was approved by the Copenhagen ethical committee (KF 11-082/01). All participants provided signed consent before examination42. In total, we performed targeted sequencing on 358 women from this cohort. This cohort was only used for the identification of GIPR variants.
The UK Biobank is a large prospective study cohort consisting of ~500,000 individuals (of mostly European ancestry) with in-depth genetic and biochemistry as well as health and lifestyle data. All participants (40–69 years of age) were recruited from across the United Kingdom in 2006–2010. The UK Biobank study was approved by the North West Centre for Research Ethics Committee (11/NW/0382), and all participants provided signed consent for health-related research43.
Targeted sequencing of GIPR in the Danish cohorts and variant annotation
We sequenced the coding regions of 265 selected genes involved in obesity and diabetes development, including GIPR, by using solution-based target region capture followed by next-generation sequencing in 10,523 individuals from all four Danish study cohorts to increase the likelihood of identifying every Danish GIPR variant. Extensive details on DNA extraction, capture of the targeted region and next-generation sequencing were as previously described44. Genomic DNA extracted from peripheral blood lymphocytes was amplified by polymerase chain reaction and the final captured DNA libraries were sequenced with the Illumina HiSeq2000 platform. With the Burrows–Wheeler Alignment Tool, we aligned the final reads to the reference human genome, GRCh37/hg19 (University of California, Santa Cruz Genome Browser), then identified the genetic variants with the Genome Analysis Toolkit45. Individual genotypes with genotype quality <20, read depth <20, allelic depth ratio <0.2 or allelic depth ratio with a binomial test P value <5 × 10−7 were set to missing. Variants with call rate <80%, mean depth <20 and Hardy–Weinberg P value <5 × 10−7 in the subset of Inter99 individuals were removed. Individuals with mean depth <20, call rate <80%, outlying heterozygosity, non-European ancestry, more than one-degree relatives, genotype sex differing from registered sex and not matching previous genotype data were excluded.
For GIPR, we reached a minimum per-base mean depth of 27× and a median per-base mean coverage for the target region of 160×. We extracted variants for annotation according to the exon locations (https://genome.ucsc.edu/cgi-bin/hgTables) with 50 base pair overhangs of GIPR (NM_000164.3, build 37)46,47. Before annotation, we used LiftOver to lift the Danish dataset from GRCh37 to GRCh38, then filtered out the intronic regions and the 3′ and 5′ untranslated regions. These steps left 12 synonymous variants, two splice variants and 47 non-synonymous variants (two nonsense mutations (premature stop gain), four frameshift mutations and 41 missense mutations (Supplementary Table 1)) available for further analysis. All but the missense variant, E354Q, were rare variants (minor allele frequency (MAF) of <0.01).
Materials
The human GIPR was inserted into the pcDNA 3.1 vector (GenBank accession number: NM_000164) and was synthesized by and purchased from Genscript along with the 47 non-synonymous GIPR variants (Supplementary Table 4). Human GIP(1–42) was purchased from Wuxi. HEK293 cells were purchased from American Type Culture Collection. Cell medium for HEK293 was purchased from Thermo Fisher Scientific. Other chemicals were purchased from standard commercial sources.
Transfection and tissue culture
HEK293 (CRL-1573, American Type Culture Collection) and β-arrestin 1/2 knockout cells, generated from HEK293A48, were used for cAMP measurement, homologues competition binding and β-arrestin 2 recruitment. For the internalization assay, endosomal signalling and basal receptor localization experiments, HEK293A (R70507, Thermo Fisher) cells were used. Both cell lines were cultured at 5% CO2 at 37 °C in Roswell Park Memorial Institute GlutaMAX-I supplemented with 10% fetal bovine serum, 180 units ml−1 penicillin, and 45 g ml−1 streptomycin. For the cAMP and binding experiments, cells were transfected with the calcium phosphate precipitation method49. The polyethyleneimine (PEI) transfection method was used for the β-arrestin 2 recruitment assays50, endosomal signalling and basal receptor localization assays, while the lipofectamine transfection method was used for the internalization assay.
cAMP accumulation assay
cAMP production was measured with the DiscoverX Hithunter cAMP assay according to the manufacturer’s protocol (DiscoverX). One day before the assay, the transiently transfected HEK293 cells expressing either WT GIPR or the 47 GIPR variants were seeded into white 96-well plates at a density of 35,000 cells per well. On the day of the assay, the cells were washed twice with HEPES-buffered saline (HBS) and incubated with HBS and 1 mM 3-isobutyl-1-methylxanthine for 30 min at 37 °C. Subsequently, the cells were stimulated with increasing concentrations of GIP(1–42) from 1 pM to 100 nM and incubated for 30 min at 37 °C. After the incubation, the assay medium was removed from the plate and the cells were washed with 30 µl PBS and treated with 40 µl enzyme donor–lysis buffer–chemiluminescent reagents and 10 µl cAMP antibodies for 60 min before 40 µl enzyme acceptor solution added. After a 3-h incubation on a shaking table in the dark, the cAMP accumulation was measured based on luminescence with a Perkin Elmer EnVision 2104 Multilabel Microplate Reader.
Homologue competition binding assay
Binding experiments were performed simultaneously with the same transiently transfected HEK293 cells used for the cAMP experiments to enable the correlation of receptor expression with signalling between assays. In brief, 1 day before the assay, the transfected cells were seeded in a clear 96-well plate. The number of cells per well was adjusted to achieve 5–10% specific binding of 125I-GIP(1–42). On the assay day, the cells were washed twice with 4 °C binding buffer (50 mM HEPES buffer (pH 7.2), 1 mM CaCl2, 5 mM MgCl2 and 0.5% (wt/vol) bovine serum albumin) and incubated for 15 minutes at 4 °C. After the addition of increasing concentrations from 1 pM to 1 µM of unlabelled GIP(1–42), 15–40 pM 125I-GIP(1–42) was added and the plate was incubated for 3 h at 4 °C. After incubation, the cells were washed in ice-cold binding buffer and lysed with 200 mmol l−1 NaOH with 1% sodium dodecyl sulphate for 30 min. The gamma radiation was measured with a 2470 Wizard2 Automatic Gamma Counter.
β-arrestin 2 recruitment assay
Measurement of β-arrestin 2 recruitment was performed with a BRET assay. Two days before the assay, the HEK293 cells were transiently transfected with either WT GIPR or the GIPR variants together with the donor Rluc8-Arrestin-3-Sp1, the acceptor mem-linker-citrine-SH3 and the GPCR kinase 2 to facilitate β-arrestin 2 recruitment. On assay day, the cells were washed with PBS and resuspended in PBS supplemented with 5 mmol l−1 glucose. Subsequently, 85 µl of the cell suspension was transferred to a white 96-well plate and 10 µl of 50 µM coelenterazine-h was added. After 10 min of incubation, increasing concentrations of GIP(1–42), from 1 pM to 1 µM, were added, the cells were incubated for 30 min at room temperature and luminescence (ratio of 535 nm over 480 nm emission) was measured with a Perkin Elmer EnVision 2104 Multilabel Microplate Reader.
Internalization assay
The time-resolved fluorescence resonance energy transfer-based internalization assay was performed with HEK293A cells expressing the various GIPR variants with an N-terminal SNAP tag. The cells were seeded in white 384-well plates the day after transfection with 20,000 cells per well. The medium was removed the following day, and the variants were labelled with the Tag-lite snap-lumi4-tb (donor) (0.1 pmol µl−1) in Opti-MEM for 60 min at 37 °C. The cells were washed four times with internalization buffer (Hanks’ balanced salt solution (HBSS) supplemented with 1 mmol l−1 CaCl2, 1 mmol l−1 MgCl2, 20 mmol l−1 HEPES and 0.1% bovine serum albumin (pH 7.4)). Subsequently, 10 μl of 50 μmol l−1 fluorescein-O′-acetic acid (acceptor) was added to each well except for the wells used to record the donor signal. We then added 10 μl of GIPR agonist, in doses ranging from 1 nM to 1 µM, in the internalization buffer to the plates to monitor agonist-induced internalization. The internalization was measured every 4 min at 37 °C in a Perkin Elmer Envision 2105 multilabel reader.
Endosomal signalling assay
HEK293A cells (350,000 per ml) were transfected in suspension with plasmid DNA expressing WT or GIPR variants, Rluc8-mGs, rGFP-FYVE and salmon sperm DNA (SSD) at a ratio of 2:1:6:11 that had been complexed with PEI (3:1 PEI:DNA ratio) and seeded (35,000 cells per well) in white 96-well plates pre-coated with poly-d-lysine. At 48 h post-transfection, cells were washed with HBSS and subsequently maintained in 80 µl HBSS. Cells were stimulated with 10 µl GIP(1–42) (100 pM to 1 µM) for 115 min, followed by a 5 min incubation with 10 µl NanoGlo Luciferase Assay Substrate (1:1,000, Promega). Light emission at 515/20 nm (acceptor; rGFP) and 400/70 nm (donor; Rluc8) was measured using a Tecan Spark multimode microplate reader (Männedorf).
Basal receptor localization assay
HEK293A cells (350,000 per ml) were transfected in suspension with plasmid DNA expressing Rluc8-tagged WT or GIPR variants, compartment-specific acceptor (rGFP-CAAX, rGFP-FYVE, tdrGFP-Giantin or tdrGFP-PTP1B) and salmon sperm DNA at a ratio of 1:6:13 that had been complexed with PEI (3:1 PEI:DNA ratio) and seeded (35,000 cells per well) in white 96-well plates pre-coated with poly-d-lysine. At 48 h post-transfection, cells were washed with HBSS and subsequently maintained in 90 µl HBSS. Cells were incubated for 5 min with 10 µl coelenterazine 400a (2.5 µM). Light emission at 515/20 nm (acceptor; rGFP) and 400/70 nm (donor; Rluc8) was measured using a Tecan Spark multimode microplate reader (Männedorf).
In vitro data analysis
To obtain Emax, EC50, Bmax, Kd and IC50 values, we analysed the data with the non-linear regression curve fitting program GraphPad Prism Software 9 (GraphPad). For cAMP accumulation, the data were fitted as a sigmoidal curve with a Hill slope of 1; for homologous competition binding, the data were fitted as a sigmoidal curve with a Hill slope of −1.
The Bmax was calculated according to the following equation51:
in which B0 represents the total specific binding and [L] is the ligand concentration. The equilibrium dissociation constant (Kd) was obtained with the equation51:
The area under the curve for GIPR variant internalization was calculated based on the trapezoid rule with GraphPad Prism 9.
The statistical analyses for the in vitro experiments were performed in GraphPad Prism. The order of the molecular testing of the variants was assigned randomly. Data collection and analysis were not performed blind to the conditions of the experiments.
Variant mapping and effect predictions
All variants were subjected to Ensembl’s VEP v108 (ref. 47), including all dbNSFP pathogenicity predictions with rank-normalized scores (higher scores indicate higher deleteriousness), which were clustered with ‘seaborn’ clustermap52. Individual predictions for frameshift and stop-gained mutations including Q400Gfs*56, C84Pfs*70, E461* and W233* could not be retrieved. We aggregated individual VEPs into masking groups15 to categorize variants (transcript id: ENST00000590918) into putative loss-of-signalling variants, defined by passing one of the following masks: (1) LOFTEE = HC; (2) VEST4 >0.9, CADD >0.9, DANN >0.9, Eigen-raw >0.9 and Eigen-PC-raw >0.9; (3) FATHMM pred = deleterious (D), FATHMM-MKL pred = deleterious (D), PROVEAN pred = deleterious (D), MetaSVM pred = deleterious (D), MetaLR pred = deleterious (D) and MCAP >0.025; or (4) PolyPhen HDIV pred = probably damaging (D), PolyPhen HVAR pred = probably damaging (D), SIFT 4 G pred = deleterious (D), LRT pred = deleterious (D), MutTaster pred = disease_causing (D) and disease_causing_automatic (A). We excluded the PROVEAN VEP from mask 3 because of its inapplicability to evaluate variants in the GIPR transcript (ENST00000590918). We made the estimation plot with Dabestr53 and used the R software (v.4.0.2)54 to visualize the four masks and the putative loss-of-signalling variants.
We captured the GIPR variants in both the Danish population (combining DD2 and Inter99) and the UK Biobank that we were confident would lead to a disruption of the protein by classifying variants that had CADD scores55 above 30 or were protein-truncating variants (resulting in a stop gain, a start loss, a splice donor site, a splice acceptor site or a frameshift) as pLoF variants. The pLoF group in the Danish population and the UK Biobank thus consisted of nine (Supplementary Table 1) and 103 GIPR variants (Supplementary Table 2), respectively.
Power calculations
No statistical methods were used to pre-determine sample sizes for the association analyses as we used fixed populations, but our sample sizes are similar to or higher than those reported in previous publications14,34,56.
Statistical analyses in the Danish population
We focused on cardiometabolic phenotypes in adults, leaving 8,641 individuals for association studies of Danish adults from DD2 (ref. 40) and Inter99 (refs. 38,39). To test for T2D associations, we used DD2 as T2D cases (N = 2,930) and Inter99 as controls (individuals with normoglycaemia at baseline based on fasting and 2-h oral glucose tolerance test plasma glucose values (World Health Organization 1999 criteria) and without diabetes at baseline and at register-based follow-up (until 2017); N = 4,234), yielding a total sample size of 7,173 individuals (men/women ratio of 1.17 and average 51 years old). For quantitative trait analyses, we used those Inter99 individuals without known diabetes at baseline (N = 5,711, men/women ratio of 1.02 and average 46 years old). We calculated the following surrogate indices to analyse beta cell function and insulin sensitivity: the beta cell function insulin sensitivity glucose tolerance test (BIGTT)–acute insulin response (the BIGTT–AIR estimate of insulin secretion)57; BIGTT–sensitivity index (the BIGTT–SI estimate of insulin sensitivity); insulinogenic index (s-insulin30 min – s-insulin0 min) / p-glucose30 min (estimate of insulin secretion) and the insulin sensitivity index Matsuda (estimate of insulin sensitivity)58.
We used R (v.4.0.2)54 to perform logistic regression for T2D risk analyses, linear regression for analysis of quantitative traits and burden tests for all phenotypes. For the burden tests (using equal weights for all variants), we used the R package ‘SKAT’ (v.2.2.5, https://cran.r-project.org/web/packages/SKAT/SKAT.pdf)59 and applied the SKAT burden test (‘r.corr = 1’) for variants tested in vitro and SKAT-O (‘method = ‘optimal.adj’’) for synonymous and pLoF variants60. We classified carriers as individuals carrying any of the GIPR variants, according to their in vitro result grouping. We rank-normalized all quantitative traits before analysis for data to have normal distribution, and we adjusted for sex, age, age2 and four principal components (PCs), as well as BMI where mentioned. We imputed missing genotype data but excluded individuals with missing phenotypes or covariates. We only included variant groups with more than five carriers to have acceptable representatives of carriers, excluding the GoFcAMP/WT-likearr and WT-likecAMP/GoFarr groups for burden testing. We did not perform sex-stratified analyses because of the limited number of carriers in the variant groups, which may further limit the statistical power. However, we accounted for sex-related effects by using sex (reported at recruitment) as a covariate in our statistical analyses. We controlled the false discovery rate (FDR) with p.adjust in R (v.4.0.2)54 to calculate Benjamini–Hochberg q-values61.
Statistical analyses in the UK Biobank
The UK Biobank 470K whole-exome sequencing pVCF file18 was filtered to samples passing the following filters: not outliers for heterozygosity and missing rate (field #22027), no sex chromosome aneuploidy (#22019), not of mixed ethnic background (#21000), call rate >0.9 and no withdrawn consent. We further removed single-nucleotide variation genotypes with sequencing depth (DP) <7 and indels with DP <10, entries with a genotype quality <20, homozygous reference call with allele balance (AB) >0.1, heterozygous alternate with AB <0.9 and heterozygous with AB not in (0.2, 0.9) (ref. 62). Variants were restricted to those in whole-exome sequencing capture regions, with more than 90% of genotypes having DP >10. Variants with a call rate <0.99 and Hardy–Weinberg equilibrium P < 10 × 10−15 were removed. We annotated the variants with VEP47. All phenotypes, including BMI (#21001), body fat percentage (#23099), waist circumference (#48), systolic blood pressure (#93), diastolic blood pressure (#94), obesity/E66 (#130792), hypertension/I10 (#131286) and non-insulin-dependent (type II) diabetes mellitus/E11 (#130708) were pre-processed with PHESANT63 and inverse rank normal transformed for data to have normal distribution. We used the ICD-10 code for obesity (E66) to provide a more nuanced picture of the body composition as BMI represents a single snapshot of an individual; in particular, 14,380 samples had an E66 diagnosis and BMI <30; 38,427 samples had an E66 diagnosis and BMI >30; 79,013 samples had no E66 diagnosis, but BMI >30; and 282 samples had neither E66 diagnosis nor a BMI measure. We opted not to filter related individuals, since Regenie is robust against percentages of related individuals as high as 50%64 and this cohort exhibited a percentage of approximately 22%65. In total, 465,506 samples (54% were women, average 57 years old and 91% of British ancestry) were available for analysis after quality control filtering and were not filtered further for ancestry. The 427,931 United Kingdom individuals were defined by Euclidean distance on principal component analysis space66. Variant quality control metrics are provided in Supplementary Table 2. All quality control was performed with Hail (Hail Team. Hail 0.2.64 https://github.com/hail-is/hail).
Multiple variant combination association tests were performed with Regenie 3.2.5 (ref. 65) adjusting for sex, age, age2, age × sex, age2 × sex and the first 20 ancestry-informative PCs. All analyses were performed on the UK Biobank Research Analysis Platform Spark clusters (Hail) or single nodes (Regenie). The Cauchy P value combination test was performed with the omnibus aggregated Cauchy association test (ACAT-O)67. Regenie step 1 was run according to UK Biobank recommendations (https://rgcgithub.github.io/regenie/recommendations/), and exact step two parameters are available in the GitHub repo scripts folder. Per tested phenotype association, individuals with missing values in either the phenotype, covariates or genotypes were excluded from the analysis. Benjamini–Hochberg q-values61 were calculated with p.adjust in R (v.4.0.2)54 to adjust for multiple comparisons. We did not perform sex-stratified analyses because of the limited number of carriers in the variant groups, which may further limit the statistical power. However, we accounted for sex-related effects by using sex (derived from the National Health Service or self-reported) as a covariate in our statistical analyses.
To create the forest plots of the association analyses from the Danish population and the UK Biobank, we used the R package ‘metafor’ (v.4.4.0, https://cran.r-project.org/web/packages/metafor/metafor.pdf)68.
For the meta-analysis regression, phenotypic estimates of single variants with an allele count >20 were obtained, leaving 94 variants for analysis, of which 17 were in vitro-tested variants. These estimates derived from the association tests using Regenie65. Single-variant estimates of phenotypes and in vitro cAMP production and β-arrestin recruitment were modelled in Bayesian regression models using Stan (BRMS)69 in R (v.4.3.0)54 as ‘phenotype | mi(SE) ~ mi(cAMP) * mi(Barr)’ using the approach described in the vignette ‘Handle Missing Values with brms’. We used normal(0,1) priors for effect sizes and exponential(1) priors for sigma. We used four chains with 10,000 iterations with 4,000 warmups. All chains converged with a potential scale reduction factor onsplit chains (Rhat) 1.00 or 1.01 and a satisfactory effective sample size. Each phenotype was modelled separately, and fixed effects were plotted based on 1,000 samples. See ‘Code availability’ for exact model parameters.
Pharmacologic studies in Arrb2 KO mice
Studies were approved by and performed according to the guidelines of the Institutional Animal Care and Use Committee of Duke University. Male Arrb2 KO mice (Arrb2−/−) ~12–15 weeks of age were generously provided by Drs Howard Rockman and Robert Lefkowitz at Duke University70. The mice were kept in standard housing conditions with a temperature of 20–26 °C and a 12-h dark/light cycle to ensure the physiological and psychological wellbeing of the mice. Mice were fed research diets 5053. We restrictedly used male mice because of the potential impact of the sexual dimorphism, that is, the oestrus cycle in female mice on cardiometabolic traits, resulting in unwanted variability and to ensure comparability to existing research predominantly using male mice71. The mice were subjected to an IPGTT. Test compounds, including native GLP-1 and d-Ala GIP (GIP(1–42) with a substitution of Ala with d-Ala at position 2), were injected 15 min before glucose in these experiments. Data collection and analysis were not randomized or blind to the conditions of the experiments. The statistical analysis was performed in GraphPad Prism.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The reference human genome GRCh37/hg19 is available at the University of California, Santa Cruz Genome Browser (https://genome.ucsc.edu/). Data from the Danish cohorts can only become available from corresponding author N.G. upon reasonable request due to handling of personal information. For requesting the DD2 data, submission of an application (https://dd2.dk/forskning/ansoeg-om-data) must be directed to Research Manager Kurt Højlund (Kurt.Hoejlund.rsyd.dk) and Program Manager Jens S. Nielsen (jsn@rsyd.dk). The application form for accessing the UK Biobank data sets can be found at https://www.ukbiobank.ac.uk/enable-your-research. Source data are provided with this paper.
Code availability
Code used for the analyses of the Danish cohorts can become available from corresponding author Niels Grarup upon request due to handling of personal information. All code used for the analyses of the UK Biobank data is available at https://github.com/HauserGroup/gogoGPCR.
References
Rosenkilde, M. M. Advances in incretin-based therapeutics for obesity. Nat. Rev. Endocrinol. 20, 67–68 (2024).
Gasbjerg, L. S., Rosenkilde, M. M., Meier, J. J., Holst, J. J. & Knop, F. K. The importance of glucose-dependent insulinotropic polypeptide receptor activation for the effects of tirzepatide. Diabetes Obes. Metab. 25, 3079–3092 (2023).
Véniant, M. M. et al. A GIPR antagonist conjugated to GLP-1 analogues promotes weight loss with improved metabolic parameters in preclinical and phase 1 settings. Nat. Metab. https://doi.org/10.1038/s42255-023-00966-w (2024).
Hauser, A. S. et al. Pharmacogenomics of GPCR drug targets. Cell 172, 41–54.e19 (2018).
Schöneberg, T. & Liebscher, I. Mutations in G protein-coupled receptors: mechanisms, pathophysiology and potential therapeutic approaches. Pharmacol. Rev. 73, 89–119 (2021).
Almind, K. et al. Discovery of amino acid variants in the human glucose-dependent insulinotropic polypeptide (GIP) receptor: the impact on the pancreatic beta cell responses and functional expression studies in Chinese hamster fibroblast cells. Diabetologia 41, 1194–1198 (1998).
Sauber, J. et al. Association of variants in gastric inhibitory polypeptide receptor gene with impaired glucose homeostasis in obese children and adolescents from Berlin. Eur. J. Endocrinol. 163, 259–264 (2010).
Saxena, R. et al. Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat. Genet. 42, 142–148 (2010).
Turcot, V. et al. Protein-altering variants associated with body mass index implicate pathways that control energy intake and expenditure in obesity. Nat. Genet. 50, 26–41 (2018).
Akbari, P. et al. Sequencing of 640,000 exomes identifies GPR75 variants associated with protection from obesity. Science 373, eabf8683 (2021).
Kizilkaya, H. S. et al. Loss of function glucose-dependent insulinotropic polypeptide receptor variants are associated with alterations in BMI, bone strength and cardiovascular outcomes. Front. Cell Dev. Biol. 9, 749607 (2021).
Bagger, J. I. et al. Impaired regulation of the incretin effect in patients with type 2 diabetes. J. Clin. Endocrinol. Metab. 96, 737–745 (2011).
Nauck, M. A. et al. Incretin effects of increasing glucose loads in man calculated from venous insulin and C-peptide responses. J. Clin. Endocrinol. Metab. 63, 492–498 (1986).
Gao, W. et al. Human GLP1R variants affecting GLP1R cell surface expression are associated with impaired glucose control and increased adiposity. Nat. Metab. 5, 1673–1684 (2023).
Flannick, J. et al. Exome sequencing of 20,791 cases of type 2 diabetes and 24,440 controls. Nature 570, 71–76 (2019).
Gabe, M. B. N. et al. Human GIP(3-30)NH2 inhibits G protein-dependent as well as G protein-independent signaling and is selective for the GIP receptor with high-affinity binding to primate but not rodent GIP receptors. Biochem. Pharmacol. 150, 97–107 (2018).
Harris, M. et al. RAMPs regulate signalling bias and internalisation of the GIPR. Preprint at bioRxiv https://doi.org/10.1101/2021.04.08.436756 (2021).
Backman, J. D. et al. Exome sequencing and analysis of 454,787 UK Biobank participants. Nature 599, 628–634 (2021).
Wan, Q. et al. Mini G protein probes for active G protein-coupled receptors (GPCRs) in live cells. J. Biol. Chem. 293, 7466–7473 (2018).
Wright, S. C. et al. BRET-based effector membrane translocation assay monitors GPCR-promoted and endocytosis-mediated Gq activation at early endosomes. Proc. Natl Acad. Sci. USA 118, e2025846118 (2021).
Wright, S. C. et al. GLP-1R signaling neighborhoods associate with the susceptibility to adverse drug reactions of incretin mimetics. Nat. Commun. 14, 6243 (2023).
Kreymann, B., Ghatei, M. A., Williams, G. & Bloom, S. R. Glucagon-like peptide-1 7-36: a physiological incretin in man. Lancet 330, 1300–1304 (1987).
Dupre, J., Ross, S. A., Watson, D. & Brown, J. C. Stimulation of insulin secretion by gastric inhibitory polypeptide in man. J. Clin. Endocrinol. Metab. 37, 826–828 (1973).
Hinke, S. A. et al. Dipeptidyl peptidase IV-resistant [D-Ala(2)]glucose-dependent insulinotropic polypeptide (GIP) improves glucose tolerance in normal and obese diabetic rats. Diabetes 51, 652–661 (2002).
Thomas, N. J. et al. Identifying type 1 and 2 diabetes in research datasets where classification biomarkers are unavailable: assessing the accuracy of published approaches. J. Clin. Epidemiol. 153, 34–44 (2023).
Eastwood, S. V. et al. Algorithms for the capture and adjudication of prevalent and incident diabetes in UK Biobank. PLoS ONE 11, e0162388 (2016).
Jones, B. et al. Genetic and biased agonist-mediated reductions in β-arrestin recruitment prolong cAMP signaling at glucagon family receptors. J. Biol. Chem. 296, 100133 (2021).
Møller, T. C., Moo, E. V., Inoue, A., Pedersen, M. F. & Bräuner-Osborne, H. Characterization of the real-time internalization of nine GPCRs reveals distinct dependence on arrestins and G proteins. Biochim. Biophys. Acta Mol. Cell Res. https://doi.org/10.1016/j.bbamcr.2023.119584 (2024).
Roed, S. N. et al. Real-time trafficking and signaling of the glucagon-like peptide-1 receptor. Mol. Cell. Endocrinol. 382, 938–949 (2014).
Abdullah, N., Beg, M., Soares, D., Dittman, J. S. & McGraw, T. E. Downregulation of a GPCR by β-arrestin2-mediated switch from an endosomal to a TGN recycling pathway. Cell Rep. 17, 2966–2978 (2016).
Ismail, S. et al. Internalized receptor for glucose-dependent insulinotropic peptide stimulates adenylyl cyclase on early endosomes. Biochem. Pharmacol. 120, 33–45 (2016).
Bitsi, S. et al. Divergent acute versus prolonged pharmacological GLP-1R responses in adult β cell-specific β-arrestin 2 knockout mice. Sci. Adv. 9, eadf7737 (2023).
Zaïmia, N. et al. GLP-1 and GIP receptors signal through distinct β-arrestin 2-dependent pathways to regulate pancreatic β cell function. Cell Rep. 42, 113326 (2023).
Melchiorsen, J. U. et al. Rare heterozygous loss-of-function variants in the human GLP-1 receptor do not associate with cardiometabolic phenotypes. J. Clin. Endocrinol. Metab. https://doi.org/10.1210/clinem/dgad290 (2023).
Lagou, V. et al. GWAS of random glucose in 476,326 individuals provide insights into diabetes pathophysiology, complications and treatment stratification. Nat. Genet. 55, 1448–1461 (2023).
Willard, F. S. et al. Tirzepatide is an imbalanced and biased dual GIP and GLP-1 receptor agonist. JCI Insight 5, e140532 (2020).
Jones, B. et al. Targeting GLP-1 receptor trafficking to improve agonist efficacy. Nat. Commun. 9, 1–17 (2018).
Glümer, C., Jørgensen, T. & Borch-Johnsen, K. Prevalences of diabetes and impaired glucose regulation in a danish population. The Inter99 study. Diabetes Care 26, 2335–2340 (2003).
Jørgensen, T. et al. A randomized non-pharmacological intervention study for prevention of ischaemic heart disease: baseline results Inter99 (1). Eur. J. Cardiovasc. Prev. Rehab. 10, 377–386 (2003).
Nielsen, J. S., Thomsen, R. W., Steffensen, C. & Christiansen, J. S. The Danish Centre for Strategic Research in Type 2 Diabetes (DD2) study: implementation of a nationwide patient enrollment system. Clin. Epidemiol. 4, 27–36 (2012).
Holm, J. C. et al. Chronic care treatment of obese children and adolescents. Int. J. Pediatr. Obes. 6, 188–196 (2011).
Lauenborg, J. et al. Increasing incidence of diabetes after gestational diabetesa long-term follow-up in a Danish population. Diabetes Care 27, 1194–1199 (2004).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Gao, R. et al. Evaluation of a target region capture sequencing platform using monogenic diabetes as a study-model. BMC Genet. 15, 1–9 (2014).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Liu, X., Jian, X. & Boerwinkle, E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 32, 894–899 (2011).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17, 1–14 (2016).
O’Hayre, M. et al. Genetic evidence that β-arrestins are dispensable for the initiation of β2-adrenergic receptor signaling to ERK. Sci. Signal. 10, eaal3395 (2017).
Velden, W. J. C. V. D. et al. GLP-1 Val8: a biased GLP-1R agonist with altered binding kinetics and impaired release of pancreatic hormones in rats. ACS Pharmacol. Transl. Sci. 4, 296–313 (2021).
Velden et al. Molecular and in vivo phenotyping of missense variants of the human glucagon receptor. J. Biol. Chem. 298, 101413 (2022).
DeBlasi, A., O’Reilly, K. & Motulsky, H. J. Calculating receptor number from binding experiments using same compound as radioligand and competitor. Trends Pharmacol. Sci. 10, 227–229 (1989).
Waskom, M. seaborn: statistical data visualization. J. Open Source Softw. 6, 3021 (2021).
Ho, J., Tumkaya, T., Aryal, S., Choi, H. & Claridge-Chang, A. Moving beyond P values: data analysis with estimation graphics. Nat. Methods 16, 565–566 (2019).
R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing (R Foundation for Statistical Computing, 2023).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014).
Lotta, L. A. et al. Human gain-of-function MC4R variants show signaling bias and protect against obesity. Cell 177, 597–607.e9 (2019).
Hansen, T. et al. The BIGTT test. Diabetes Care 30, 257–262 (2007).
Matsuda, M. & DeFronzo, R. A. Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care 22, 1462–1470 (1999).
Seunggeun, L., Zhangchen, Z., Miropolsky, L. & Wu, M. SKAT: SNP—set (sequence) kernel association test. https://CRAN.R-project.org/package=SKAT (2023).
Lee, S. et al. Optimal unified approach for rare-variant association testing with application to small-sample case–control whole-exome sequencing studies. Am. J. Hum. Genet. 91, 224–237 (2012).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc.: Ser. B 57, 289–300 (1995).
Pedersen, B. S. et al. Effective variant filtering and expected candidate variant yield in studies of rare human disease. NPJ Genom. Med. 6, 1–8 (2021).
Millard, L. A. C., Davies, N. M., Gaunt, T. R., Smith, G. D. & Tilling, K. Software application profile: PHESANT: a tool for performing automated phenome scans in UK Biobank. Int. J. Epidemiol. 47, 29–35 (2018).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
Mbatchou, J. et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat. Genet. 53, 1097–1103 (2021).
Privé, F. et al. Portability of 245 polygenic scores when derived from the UK Biobank and applied to 9 ancestry groups from the same cohort. Am. J. Hum. Genet. 109, 12–23 (2022).
Liu, Y. et al. ACAT: a fast and powerful P value combination method for rare-variant analysis in sequencing studies. Am. J. Hum. Genet. 104, 410 (2019).
Viechtbauer, W. Conducting meta-analyses in R with the metafor package. J. Stat. Softw. 36, 1–48 (2010).
Bürkner, P.-C. brms: an R package for bayesian multilevel models using stan. J. Stat. Softw. 80, 1–28 (2017).
Wisler, J. W. et al. The role of β-arrestin2-dependent signaling in thoracic aortic aneurysm formation in a murine model of Marfan syndrome. Am. J. Physiol. Heart Circ. Physiol. 309, H1516–H1527 (2015).
Kleinert, M. et al. Animal models of obesity and diabetes mellitus. Nat. Rev. Endocrinol. 14, 140–162 (2018).
Acknowledgements
This work was supported by a scholarship to H.S.K. from the Danish Diabetes Academy funded by the Novo Nordisk Foundation (grant no. NNF17SA0031406) and a grant to M.M.R. from the EFSD/Lilly European Diabetes Research Programme. M.M.R was also supported by a grant from the Novo Nordisk Foundation (grant no. NNF21OC0070347). K.V.S., J.B.-J., O.P., B.H., J.J.H., T.H. and N.G. were supported by an unrestricted grant (NNF18CC0034900 and NNF23SA0084103) donated by the Novo Nordisk Foundation to partly fund the independent research facility of the Novo Nordisk Foundation Center for Basic Metabolic Research. A.S.H. and J.S.M. gratefully acknowledge funding from the Lundbeck Foundation (R278-2018-180) and the Independent Research Fund Denmark (3122-00044B). A.V. was supported by The Swedish Research Council, Strategic Research Area Exodiab, Dnr 2009-1039, and Swedish Foundation for Strategic Research, Dnr ICR15-0067. S.C.W. is supported by the Swedish Society for Medical Research (PD20-0153). V.M.L. is supported by the Swedish Research Council (grant agreement numbers 2019-01837 and 2021-02801), by the EU/EFPIA/OICR/McGill/KTH/Diamond Innovative Medicines Initiative 2 Joint Undertaking (EUbOPEN grant number 875510) and by the Robert Bosch Foundation, Stuttgart, Germany.
DD2 is supported by Danish Agency for Science (grant no. 09-067009 and 09-075724). DD2 is also supported by The Danish Health and Medicines Authority, The Danish Diabetes Association and an unrestricted donation from Novo Nordisk Foundation. The partners of the project are listed on the project website at www.DD2.nu. This research has been conducted using the UK Biobank Resource, a major biomedical database with genotype and phenotype data open to all approved health researchers (https://www.ukbiobank.ac.uk/), under application number 55955. We thank A. Inoue (Tohoku University) for providing β-arrestin knockout cell lines; M. Bouvier (Université de Montréal) for rGFP-CAAX, rGFP-FYVE, tdrGFP-Giantin and tdrGFP-PTP1B; N.A. Lambert (Augusta University) for Rluc8-mGs; and M. Sigvardt Baggesen, S. Petersen, R. Regmi and A. Dragan for excellent technical assistance for the in vitro studies. Graphic illustrations were created with BioRender.com.
Author information
Authors and Affiliations
Contributions
Conceptualization: T.H., J.J.H., B.H., N.G., A.S.H. and M.M.R. Methodology: H.S.K., K.V.S., J.S.M., J.D.D., P.L., S.C.W., J.B.-J., N.G., A.S.H. and M.M.R. Data acquisition or curation: O.P., J.D.D., J.M., S.A.M., P.J.K., B.F., J.E.C., D.A.D., D.P.-T., F.F., S.M., J.R., H.T.S., A.V., J.S.N., J.-C.H., J.L., P.D., J.L., A.L., S.C.W. and V.M.L. Formal analysis: H.S.K., K.V.S., J.S.M., P.L., A.S.H. and J.D.D. Writing—original draft: H.S.K., K.V.S., J.S.M., J.B.-J., N.G., A.S.H. and M.M.R. Writing—review and editing, all authors. Visualization: H.S.K., K.V.S., J.S.M., A.B., A.S.H., P.L. and P.A.G. Funding acquisition: H.S.K., N.G., A.S.H. and M.M.R. Supervision, L.S.G., N.G., A.S.H. and M.M.R.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that the study was conducted without any commercial or financial relationships that may raise concerns about possible conflicts of interest. L.S.G., J.J.H. and M.M.R. are co-founders and minority shareholders of Antag Therapeutics ApS. J.J.H., B.H. and M.M.R. are co-founders and minority shareholders of Bainan Biotech ApS. M.M.R. is chairman of the board of directors (Bainan Biotech ApS), B.H. is CEO and a member of the board of directors (Bainan Biotech ApS) and J.J.H. is a member of the board of directors of Antag Therapeutics ApS and of Bainan Biotech ApS. V.M.L. is a co-founder, CEO and shareholder of HepaPredict AB, as well as co-founder, CSO and shareholder of Shanghai Hepa Biotechnology Ltd. D.P.-T. maintains research collaborations with Novo Nordisk, MBX Biosciences and BlueWater Biosciences, and is a minority shareholder of BlueWater Biosciences. N.G. is currently employed at Novo Nordisk. J.M. is an employee of Novo Nordisk and J.D.D., S.A.M., P.J.K. and B.F. were formerly employed of Novo Nordisk at the time the work was performed. J.D.D., J.M., S.A.M., P.J.K. and B.F. are shareholders of Novo Nordisk. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Metabolism thanks Nigel Irwin, Maik Pietzner and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Yanina-Yasmin Pesch, in collaboration with the Nature Metabolism team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Maximal signalling and potency of the 47 GIPR variants in cAMP production, and the relation between WT GIPR gene expression and signalling.
a, cAMP production Emax, shown as percentage of WT GIPR activity. The variants are arranged according to the cAMP production (efficacy at 100 pM GIP). Grey, WT-like; dark red, below 50% activity. b, cAMP production potency (logEC50). c, Emax (cAMP production) vs Bmax (homologues competitive binding) of WT GIPR, expressed at DNA doses ranging from 10 ng to 10 µg. d, GIP binding affinity (Kd) of WT GIPR at DNA doses ranging from 10 ng to 10 µg. e, Potency (logEC50) of WT GIPR at DNA doses ranging from 10 ng to 10 µg in cAMP production. f, Linear correlation between WT GIPR expression and signalling of the 47 GIPR variants. Expression was measured as Bmax, and signalling was measured as cAMP production efficacy at 100 pM. The data represent the mean ± s.e.m. of a minimum of N = 3. Independent experiments were performed in duplicate.
Extended Data Fig. 2 Variant effect predictor (VEP) correlations to laboratory-tested GIPR variants.
a, Cluster heatmap of the VEP normalized predicted deleteriousness scores, ranging from benign (0) to deleterious (1). The x-axis shows the included VEPs, and the classification of the laboratory-tested variants according to cyclic adenosine monophosphate (cAMP) accumulation (left). b, Prediction of variants potentially leading to a loss-of-signalling (LoS) molecular phenotype with four VEP masks (M1–M4)15. The 47 GIPR variants and their allele frequencies in the Danish population (all four study cohorts combined) are shown in the left panel, sorted according to their cAMP production (from low (bottom) to high (top)) compared with that of WT (Fig. 1d). M1, the Loss-of-Function Transcript Effect Estimator (LOFTEE) high-confidence (HC) mask with predicted LoS variants (red). M2–M4, grouped VEPs. A variant was considered a LoS variant if all VEPs in one of the four masks predicted the variant as a LoS variant (red). ‘pLoS’ represents the variants predicted to have a LoS molecular phenotype (bright red). ‘in vitro LoF’ represents the tested GIPR LoF variants with < 50% cAMP production (bright red). NAs are white. In total, 17 variants were pLoS variants, of which 15 overlapped with the in vitro cAMP LoF variants, and two were in vitro cAMP WT-like variants.
Extended Data Fig. 3 Internalization abilities of the eight LoFcAMP/WT-likearr variants and two LoFcAMP/LoFarr.
a, The donor signal represents an estimate of receptor surface expression of the cells transfected with SNAP-WT GIPR or variants. N = 4. b, Internalization ratios of the SNAP-GIPR variants over time following stimulation with GIP in doses ranging from 1 nM to 1 µM. N = 4. c, Area under the curves of the SNAP-WT GIPR vs. SNAP-GIPR variants in response to GIP stimulation in doses ranging from 1 nM to 1 µM. N = 4. d, Dose-response curves of maximal internalization of the SNAP-GIPR variants, shown as percentage of maximal SNAP-WT GIPR internalization. N = 4. Data are shown as the mean ± s.e.m. Independent experiments were performed in triplicates.
Extended Data Fig. 4 Burden association of the GIPR variant groups on cardiometabolic phenotypes in the Danish cohorts.
Forest plot showing burden test statistics in the Danish population (Inter99). NC, number of individuals for each phenotype carrying any of the variants within that variant group. NN-C, number of individuals not carrying any of the variants within that variant group. q, false discovery rate (FDR) adjusted p-values of the burden tests (performed using SKAT/SKAT-O). pLoF denotes variants predicted to cause loss of function with VEP. pLoF variants were not functionally characterized but include some of the LoFcAMP/LoFarr variants. The E354Q variant was excluded from burden testing of the WT-likecAMP/WT-likearr group due to high minor allele frequency (MAF; ~20%). Beta and error bars represent effect size as standard deviation and the 95% confidence interval, respectively, derived from a linear model.
Extended Data Fig. 5 Forest plots of burden tests of cardiometabolic phenotypes in the Danish population (Inter99).
Association results of metabolic-associated biochemistry measures, estimates of beta cell function and insulin sensitivity, and cardiovascular-associated phenotypes (the lipid profile) for all variant groups are shown. Synonym, WT/WT, LoF/WT, LoF/LoF, and pLoF represent the groups of synonymous, WT-likecAMP/WT-likearr, LoFcAMP/WT-likearr, LoFcAMP/LoFarr, and predicted LoF variants, respectively. The E354Q variant was excluded from burden testing of the WT-likecAMP/WT-likearr group due to high minor allele frequency (MAF; ~20%). Abbreviations: FPG, fasting plasma glucose levels; PG, plasma glucose levels; FSI, fasting serum insulin levels; SI, serum insulin levels; FS C-pep, fasting serum C-peptide levels; HbA1c, hemoglobin A1c; AUC30ig, area under curve 0–30 min insulin/glucose; BIGTT-AIR, beta cell function insulin sensitivity glucose tolerance test (BIGTT)-acute insulin response (insulin secretion); BIGTT-SI, BIGTT-sensitivity index (insulin sensitivity); Insu index, insulinogenic index (insulin secretion); HDL, high-density lipoprotein; LDL, low-density lipoprotein; VLDL, very low-density lipoprotein. NC represents the number of individuals for each phenotype carrying any of the variants within that variant group. NN-C represents the number of individuals not carrying any of the variants within that variant group. q, FDR adjusted p-values of the burden tests (performed using SKAT/SKAT-O). Beta and error bars represent effect size as standard deviation and the 95% confidence interval, respectively, derived from a linear model.
Extended Data Fig. 6 Forest plot of binary phenotype burden tests in the UK Biobank population.
For each GIPR variant group, the odds ratios (OR) are shown for binary traits with corresponding confident intervals. ‘Carriers’ represent the number of individuals for each phenotype carrying any of the GIPR variants within that GIPR variant group. We excluded the E354Q variant from the burden testing of the WT-likecAMP/WT-likearr group due to high minor allele frequency (MAF; ~20%). ‘N’ represents the total number of individuals for each phenotype. q, the false discovery rate (FDR) adjusted p-values of the burden tests (performed using ACAT-O). Error bars represent the 95% confidence interval of the OR.
Extended Data Fig. 7 Comparison of p-values between with all UK Biobank samples or only individuals from the United Kingdom.
-log10(P) for each phenotype/grouping combination when testing either all samples passing quality control or limited to those with United Kingdom (UK) ancestry. The x-axis shows p-values (derived from burden testing using ACAT-O) for testing of all 465,000 individuals (Source Data of Fig. 3 and Extended Data Fig. 6). The y-axis shows p-values (derived from burden testing using ACAT-O) for testing of only 428,000 UK individuals. a, Binary traits. Circles represent type 2 diabetes (T2D), triangles represent obesity, and squares represent hypertension. Error bands represent the 95% confidence interval (CI) of the regression line. b, Quantitative traits. Circles, triangles, squares, pluses, crossed squares, and asterisks represent body fat percentage, body mass index, diastolic blood pressure, hip circumference, systolic blood pressure, and waist circumference, respectively. Error bands represent 95% CI of the regression line.
Extended Data Fig. 8 Meta-analysis regression of single-variant phenotypic association estimates on cAMP signalling and β-arrestin recruitment.
UK Biobank single-variant phenotypic association effect sizes (see Fig. 3) regressed on each variant’s in vitro effect on cAMP production (see Fig. 1) and β-arrestin recruitment (see Fig. 2) compared to WT GIPR. Analysis performed as a linear interaction model of phenotypic effect size estimates (y-axis) as response of single-variant (points) β-arrestin recruitment (B-arr, blue) and cAMP signalling (red) impact (x-axis). We used Bayesian Gaussian model which accounts for uncertainty in both individual phenotype estimates (vertical bars as standard error of the beta (β) from the linear regression model) and in vitro uncertainty (not shown). Error bands represent 95% credible interval and grey dashed lines represent no effect.
Extended Data Fig. 9 In vitro comparison of parental HEK293A cells and β-arrestin 1/2 KO cells transfected with GIPR or GLP-1R.
a1, cAMP accumulation measured in parental HEK293A cells transfected with 10 µg or 0.5 µg of receptor DNA upon cognate ligand stimulation. GLP-1R stimulated by GLP-1 mediates a more efficacious cAMP response at both receptor concentrations compared to GIPR stimulated by GIP. a2-a3, Receptor surface expression (Bmax) determined by homologous radio-ligand competition binding shows that GLP-1R a2, and GIPR a3, are expressed to the same extent on the cell surface. The cAMP formed by GLP-1R is approximately double the magnitude compared to GIPR, considering the number of receptors on the surface. b, Potency (pEC50) of GIP in cells transfected with GIPR. In the absence of β-arrestin, the potency of GIP is significantly decreased (p < 0.01). c-d, Corresponding GIPR cAMP formation upon 100 c, and 10 pM d, GIP stimulation. The absence of β-arrestin leads to a significant decrease in cAMP formation (p < 0.01). e, Potency (pEC50) of GLP-1 in cells transfected with GLP-1R. A slightly higher, yet nonsignificantly, potency of GLP-1 on the GLP-1R was observed in β-arrestin KO cells compared to parental cells. f-g, Corresponding cAMP formation upon 100 pM f, and 10 pM g, GLP-1 stimulation. A nonsignificant reduction in cAMP formation was observed between WT and β-arrestin KO cells. Statistical significance was assessed using a two-tailed paired t-test. The data represent the mean ± s.e.m. of N = 3 performed in duplicates.
Extended Data Fig. 10 Graphical illustration of the molecular and cellular pharmacological consequences of the GIPR variant groups.
GIP binds to the GIPR, leading to distinguishable patterns of Gαs signalling and β-arrestin recruitment, hereunder receptor internalization and endosomal signalling, depending on the GIPR variant groups. Only the LoFcAMP/LoFarr variant group is significantly associated with lower adiposity in humans. Intracellular red arrows, including red blunted arrows, indicate which pathways are affected in the variant receptor function. A red cross indicates diminished activation of the pathway. Adapted from ‘Cellular Environment (background)’, by BioRender.com (2023). Retrieved from https://app.biorender.com/biorender-templates.
Supplementary information
Supplementary Information
Supplementary Figs. 1–3.
Supplementary Table 1
Discovery of Danish protein-coding GIPR variants. A list of the 59 protein-coding GIPR variations identified through targeted sequencing in four Danish study cohorts: HOLBAEK Study, GDM, DD2 and Inter99. AA change, amino acid change; alt allele, alternative allele; ref allele, reference allele; SNV, single-nucleotide variation; N, total number of individuals within the cohort; hetero/homo, heterozygous carriers of the alternative allele/homozygous carriers of the alternative allele; MAF, minor allele frequency.
Supplementary Table 2
Tab ‘All UKBB samples’: Supplementary Table 2. Discovery of protein-coding GIPR variants in the UK Biobank and the overlap with the in vitro-tested GIPR variants. A list of the protein-coding GIPR variants identified in the UK Biobank for all samples. The overlapping non-synonymous GIPR variants are shown in ‘Grouping’, denoted by the GIPR variant groups, as are the synonymous and pLoF variants. Abbreviations and column explanation: locus, chromosome and position; ref, reference allele; alt, alternate allele; VEP, variant effect predictor; CADD, combined annotation dependent depletion score; N, total sample size; N not called, samples excluded; N_QC, samples excluded during QC; heterozygous alt, number of heterozygous carriers of the alternate allele; N homozygous (ref, alt), number of individuals being homozygous carriers of the reference allele and alternate allele, respectively; total alt, total number of carriers of the alternate allele; P value HWE, P value for Hardy–Weinberg equilibrium (HWE), derived from a two-sided exact test with mid-P value correction of HWE via the Levene–Haldane distribution; alleles, the reference and alternate allele; NA, not applicable. Tab ‘UK ancestry samples only’: Supplementary Table 2. Discovery of protein-coding GIPR variants in the UK Biobank and the overlap with the laboratory-tested GIPR variants. A list of the protein-coding GIPR variants identified in the UK Biobank of individuals with United Kingdom ancestry. The overlapping nonsynonymous GIPR variants are shown in ‘Grouping’, denoted by the GIPR variant groups, as are the synonymous and pLoF variants. Abbreviations and column explanation: locus, chromosome and position; ref, reference allele; alt, alternate allele; VEP, variant effect predictor; CADD, combined annotation dependent depletion score; N, total sample size; N not called, samples excluded; N_QC, samples excluded during QC; heterozygous alt, number of heterozygous carriers of the alternate allele; N homozygous (ref, alt), number of individuals being homozygous carriers of the reference allele and alternate allele, respectively; total alt, total number of carriers of the alternate allele; P value HWE, P value for Hardy–Weinberg equilibrium (HWE), derived from a two-sided exact test with mid-P value correction of HWE via the Levene–Haldane distribution; alleles, the reference and alternate allele; NA, not applicable.
Supplementary Table 3
Summary statistics of the T2D association studies in the Danish population. s.d., standard deviation; P-glm, P value of a general linear model (logistic regression); P, P value of the burden test (performed using SKAT/SKAT-O); q, false discovery rate (FDR) adjusted P.
Supplementary Table 4
Sequence of GIPR variants. The nucleotide changes were introduced into the human GIPR (GenBank accession number: NM_000164) in pcDNA 3.1 vector (sequence not provided here). The site-directed mutagenesis was performed by Genscript (Piscataway, NJ).
Source data
Source Data Fig. 1
cAMP and binding in vitro source data.
Source Data Fig. 2
β-arrestin recruitment in vitro source data.
Source Data Fig. 3
Statistical UK Biobank source data.
Source Data Fig. 4
Endosomal signalling and in vivo source data.
Source Data Extended Data Fig. 1/Table 1
cAMP and binding in vitro source data.
Source Data Extended Data Fig. 2/Table 2
Variant effect predictor source data.
Source Data Extended Data Fig. 3/Table 3
Internalization in vitro source data.
Source Data Extended Data Fig. 4/Table 4
Statistical Danish cohorts source data.
Source Data Extended Data Fig. 5/Table 5
Statistical Danish cohorts source data.
Source Data Extended Data Fig. 6/Table 6
Statistical UK Biobank source data.
Source Data Extended Data Fig. 7/Table 7
Statistical UK Biobank source data.
Source Data Extended Data Fig. 8/Table 8
Single-variant UK Biobank source data.
Source Data Extended Data Fig. 9/Table 9
In vitro source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kizilkaya, H.S., Sørensen, K.V., Madsen, J.S. et al. Characterization of genetic variants of GIPR reveals a contribution of β-arrestin to metabolic phenotypes. Nat Metab 6, 1268–1281 (2024). https://doi.org/10.1038/s42255-024-01061-4
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s42255-024-01061-4
This article is cited by
-
GLP-1-based therapies for diabetes, obesity and beyond
Nature Reviews Drug Discovery (2025)
-
GIPR-Ab/GLP-1 peptide–antibody conjugate requires brain GIPR and GLP-1R for additive weight loss in obese mice
Nature Metabolism (2025)
-
Adipose Tissue, at the Core of the Action of Incretin and Glucagon-Based Anti-Obesity Drugs
Current Obesity Reports (2025)
-
Rare MTNR1B variants causing diminished MT2 signalling associate with elevated HbA1c levels but not with type 2 diabetes
Diabetologia (2025)
-
New insights into the regulation of GIPR signalling
Nature Reviews Endocrinology (2024)
