
Abstract
Signaling pathways regulating cell proliferation and survival have become attractive targets for anticancer strategies. In the present study, we analyzed by immunohistochemistry, a panel of benign nevi, superficial spreading and nodular primary melanomas and metastases for expression of activated p38/mitogen-activated protein kinase (p-p38) and c-jun N-terminal kinase (JNK) (p-JNK) and correlated the findings with known prognostic variables. Twenty-five and 35% of the primaries and 9 and 25% of the metastases expressed variable levels of p-p38 and p-JNK, respectively. In benign nevi, 73.5% expressed p-JNK and 7% expressed p-p38. For patients with superficial spreading melanomas, high level of cytoplasmic p-JNK was associated with thicker tumors (P=0.017) and shorter disease-free survival (P=0.003) as well as with markers of cell proliferation (cyclin A (P=0.017) and p21 (P=0.021)). In nodular melanomas, nuclear p-p38 was associated with Ki-67 (P=0.012), but neither cytoplasmic nor nuclear localized p-p38 was associated with disease outcome. Of note, in superficial spreading melanomas, a positive correlation between cytoplasmic p-JNK and cytoplasmic p-extracellular signal-regulated kinase ERK½ (P=0.005) and p-p38 (P=0.003) was observed. Likewise, p-p38 in cytoplasm was positively associated with cytoplasmic p-ERK1/2 (P<0.0005) and p-Akt (P=0.047). In contrast, except for a positive correlation between nuclear p-p38 and membranous p-TrkA (P=0.02), no correlation between the activation status of the different signaling pathways was observed in nodular melanomas. In conclusion, our results suggest that in benign nevi activated JNK may have a role in restricting uncontrolled cell proliferation or survival. However, during tumor progression, activation of JNK is associated with cell proliferation and shorter relapse-free period for patients with superficial spreading melanomas, suggesting that the JNK activation status could be a marker for clinical outcome in at least a subgroup of malignant melanoma. In contrast, activation of p38 seems to play a less important role in development and progression of malignant melanomas.
Similar content being viewed by others

Main
Malignant melanoma is the most rapidly increasing cancer form in the western world today, a phenomenon that is most likely caused by increased exposure for UV-radiation. At an early stage, melanomas can be cured by surgery, but once the tumor has spread beyond the skin, it is almost incurable by available chemotherapy. Novel therapies directed against melanoma-selective targets are therefore urgently needed. Dysregulated signaling pathways that regulate cell proliferation and survival are commonly observed in cancer and in recent years these pathways have become attractive targets for anticancer strategies (reviewed in Bode and Dong1). Thus, it is of major importance to elucidate the activation status as well as the mechanisms leading to abrogated signaling in different forms of cancer.
The mitogen-activated protein kinases (MAPKs) represent a family of serine-threonine kinases involved in a wide range of cellular responses. Depending on the cellular context, activation of MAPKs has been correlated with proliferation, differentiation, cell survival and apoptosis (reviewed in Kyosseva2). In mammalian cells, three major groups of MAPKs have been identified; the extracellular signal-regulated kinases (ERK1/2), c-jun N-terminal kinase (JNK) and p38/MAPK. ERK1/2 is a component of the RAS-RAF-MEK-ERK1/2 pathway, and is mainly stimulated by growth factors and cytokines. JNK and p38, also called stressactivated protein kinases, are activated by cellular stress (eg, UV irradiation, osmotic stress, heat shock, protein synthesis inhibitors), inflammatory cytokines (eg, tumor necrosis factor-α and interleukin-1) and G-protein coupled agonists (eg, thrombin). Like ERK1/2, JNK and p38 are activated by dual phosphorylation on tyrosine and threonine residues by MAP kinase kinases (MKKs). Whereas MKK3 and MKK6 selectively phosphorylate and activate p38, JNK is activated by MKK4 and MKK7 (reviewed in Hagemann and Blank3).
p38 and JNK regulate the expression of a number of cytokines, transcription factors and cell surface receptors. Activated p38 has been shown to phosphorylate cellular targets like phospholipase A2, the microtubule-associated protein Tau, and the transcription factors ATF-1 and -2, MEF-2A, Sap-1, Elk-1, NF-κB, Ets-1 and p53 (reviewed in Roux and Blenis4). One of the main substrates of JNK is the transcription factor c-jun, which upon transactivation leads to increased expression of genes with AP-1 sites in their promoters, including the c-jun gene itself. Other JNK substrates include ATF-2, Elk-1, p53, DPC-4, Sap-1a and NFAT-4, all positive regulators of the transcription factor c-fos, further increasing AP-1 level. Activated JNK also phosphorylates JunB, JunD and the Ets-related transcription factor PEA3 (reviewed in Zhang and Liu5).
Constitutive activation of the MAPK cascades has been observed in various in vitro tumor cell lines,6, 7 and has been associated with carcinogenesis and metastatic potential of human cancers.8, 9 Until recently it has been anticipated that the MAPK/ERK1/2 signaling pathway is involved in cell proliferation and survival, whereas activated JNK and p38 have been suggested to act as proapoptotic tumor suppressors. Recent discoveries have, however, revealed that several MAPKs can phosphorylate the same substrates and also affect each other through cross-talk reactions and feedback mechanisms (reviewed in Engelberg10). In this regard, Aguirre-Gisho et al11 recently showed that ERK1/2 activation is regulated by p38.
Although the MAPK pathways have been thoroughly studied in in vitro models, few studies on the activation of JNK and p38, in particular, have been carried out in vivo. Activation of both JNK and p38 has been reported in breast carcinoma,12 metastatic serous ovarian carcinoma 13 and in gastric cancer 14 whereas activation of JNK and p38 alone has been reported in poorly differentiated liver tumors 15 and in prostate cancer,16 respectively. A correlation between activated JNK and early stages of non-small-cell lung cancers has been described.17 Moreover, deactivation of p38 and JNK has been found in human colon cancers compared to adjacent normal mucosa18 as well as in advanced rat prostate carcinoma.19
In this study, we have investigated the expression of activated p38 and JNK in human malignant melanoma biopsies using immunohistochemistry, and assessed the relationship between activated p38 and JNK and known prognostic variables, cell cycle factors and disease progression.
Materials and methods
Specimens
Formalin-fixed, paraffin-embedded tissue from 152 primary (94 superficial spreading melanomas and 58 nodular melanomas) and 68 metastatic melanomas, as well as 27 benign nevi, was examined for the expression of p-p38. Sections from 154 primary (93 superficial melanomas and 61 nodular melanomas) and 73 metastatic melanomas, together with 34 benign nevi, were examined for expression of p-JNK.
Immunohistochemical Analysis
Sections of formalin-fixed, paraffin-embedded tissue were immunostained using the two-step EnVision system (DAKO EnVisionTM DAKO A/S, Glostrup, Denmark). Deparaffinized sections were microwaved in low pH buffer (pH 6.0) (DAKO A/S, Glostrup, Denmark) at 750 W for 5 min and then at 500 W for 15 min to unmask the epitopes. After treatment with 1% hydrogen peroxide for 5 min to block endogenous peroxidase, the sections were incubated with polyclonal rabbit anti-phospho-JNK1/2 (Biosource, Camarillo, CA, USA) and polyclonal anti-phospho-p38 (Biosource) for 30 min at room temperature. The sections were then incubated with HRP-labeled secondary antibody for 30 min followed by 20 min incubation at 37°C with AEC substrate (DAKO A/S). The sections were counterstained with hematoxylin, and mounted in glycerine. All series included positive controls. Negative controls included substitution of the primary antibody with mouse myeloma protein of the same isotype and concentration as the monoclonal antibody. All controls gave satisfactory results. Immunostains for cyclins A, D1 and D3, Ki-67, p21WAF1/CIP1, p27kip1, pERK1/2, pAkt and TrkA are described previously.20, 21, 22, 23, 24, 25, 26 Five semiquantitative classes were used to describe the number of stained cells: negative, ≤5%, 6–25%, 26–75%, 76–100%. Both nuclear and cytoplasmic staining was scored. The staining was evaluated by an experienced surgical pathologist (BD).
Statistical Analysis
The relationship between activated JNK and p38 expression and mean tumor thickness was evaluated nonparametrically using the Mann–Whitney two-sample test. Owing to the relatively low numbers of specimens examined, the comparison between the expression of p-JNK and p-p38 and other markers of cell cycle progression and signaling was performed using the Fisher exact test. The Student's t-test was used to compare the activation of JNK and p38 in benign nevi and primary melanomas as well as for evaluating the relationship between activation of JNK and p38 and melanoma subtypes. Kaplan–Meier estimates and the log-rank test were used to evaluate the survival data statistically. P<0.05 was considered statistically significant.
Results
Expression of Activated p38 and JNK in Primary and Metastatic Melanoma Lesions
p-p38 and p-JNK staining results are summarized in Tables 1a, b and 2. For both proteins, a heterogeneous cytoplasmic and/or nuclear staining pattern was observed (Figure 1). Thirty-eight of 152 (25%) primary melanomas expressed detectable levels of activated p38 in the cytoplasm, nucleus or both (Table 1a). Notably, there was a clear decline in the number of tumors expressing activated p38 in the metastases compared to the primary tumors (P=0.005) as only six of 68 (9%) showed positive immunostaining in the cytoplasm and/or nucleus. In 31 cases, both primary and metastatic tumors from the same patient were available for p38 analysis. Seven (23%) showed positive immunostaining in the primary tumor only, three (10%) in the metastatic tumor only, whereas one case was positive in both the primary and the metastatic lesion. Activated p38 was detected in only two of 27 (7%) benign nevi.

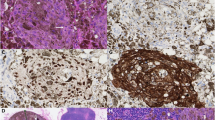
Immunohistochemical staining of activated p38 and JNK, demonstrating variable levels of nuclear and cytoplasmic expression of p-p38 in primary melanomas (a and b), negative expression of p-p38 in benign nevus (c), variable levels of cytoplasmic and nuclear expression of p-JNK in primary melanomas (d and e) and nuclear expression of p-JNK in benign nevus (f).
Activated JNK was observed in 54 of 154 (35%) primary melanomas, with p-JNK more frequently observed in the nucleus in nodular than in superficial spreading melanomas (P=0.042) (Table 1b). A moderate decline in JNK activation in the metastases as compared to the primary melanomas was observed, as only 18 of 73 metastases (25%) showed positive immunostaining. In 38 cases, both primary and metastatic tumor from the same patient were available for p-JNK analysis. Ten (27%) were positive in the primary tumor only, six (16%) in the metastatic tumor only and four (11%) cases showed positive immunoreactivity in both primary and metastatic lesion. Interestingly, we noted that activated JNK was significantly more frequently expressed in benign nevi than in primary (P<0.0005) and metastatic (P<0.0005) melanomas with positive staining in 25 of the 34 (74%) cases. In the nevi, p-JNK staining was exclusively localized to the nucleus.
Expression of Activated p38 and JNK in Relation to Clinical Parameters
As very few of the tumors expressed p-p38 or p-JNK in more than 5% of the cells (Table 2), we decided to categorize cases as negative vs positive rather than according to the percentage of positive cells. We did not find any correlations between p-p38 and p-JNK and clinical parameters such as tumor thickness, the presence of ulceration, relapse-free and overall survival when evaluating the total number of primary melanomas. However, when analyzing superficial spreading and nodular melanomas separately, we observed an association between cytoplasmic p-JNK and tumor thickness in superficial spreading melanomas (P=0.017), with higher activation level in thicker tumors (Table 3). Moreover, a significant correlation between lack of cytoplasmic p-JNK and longer relapse-free period was observed for patients with superficial spreading melanoma (P=0.003) (Figure 2).

Kaplan–Meier curve demonstrating the relationship between protein expression of activated cytoplasmic JNK and relapse-free survival (P=0.003) for patients with superficial spreading melanoma.
Relationship between Activated p38 and JNK Expression and Cell Cycle Markers
Several studies have demonstrated a correlation between cell proliferation and activation of p38 and/or JNK (reviewed in Engelberg10). The panel of malignant melanomas has been examined previously for expression of factors involved in cell cycle regulation and proliferation (cyclin A, D1, D3, Ki-67, p21WAF1/CIP1 and p27Kip1).20, 21, 22, 26 It was of interest, therefore, to study the possible association between these markers and the activation status of p38 and JNK in this cohort of tumors. A significant positive association between activated p38 in the nucleus and Ki-67 (P=0.012) was observed in nodular melanomas (Table 4a). We also observed a significant positive association between the presence of p-JNK in the cytoplasm and cyclin A (P=0.017) and p21WAF1/CIP1 (P=0.021) in superficial spreading melanomas (Table 4b). Moreover, we found an association, although not significant, between cytoplasmic p-JNK and cyclin D3 (P=0.075) and Ki-67 (P=0.088) in superficial spreading melanomas as well as between cytoplasmic p-JNK and cyclin D3 (P=0.082) in nodular melanomas. No correlation was seen between nuclear p-JNK and cell cycle markers.
Activated p38 and JNK are Associated with Each Other and with Activation of ERK1/2 and Akt
Our panel of melanomas has previously also been examined for the expression of activated ERK1/2 and Akt and the protein level of PTEN.23, 24 Because a number of studies have documented cross-talks between the different signaling pathways,11, 27, 28, 29, 30, 31, 32 it was of interest to examine whether such associations also exist in melanoma. A significant positive correlation between cytoplasmic p-p38 and p-ERK1/2 (P<0.0005) as well as between cytoplasmic p-JNK and p-ERK1/2 (P=0.005) in superficial spreading melanomas was found (Table 4a and b). Moreover, we observed a significant positive association between cytoplasmic localized p-p38 and p-Akt (P=0.047) in superficial spreading melanomas and a marginally positive association between nuclear expression of p-JNK and p-Akt (P=0.059) in nodular melanomas. We could not find any associations with PTEN. Finally, the presence of p-p38 and p-JNK in the cytoplasm was positively correlated to each other (P=0.003) in superficial spreading melanomas.
Activated p38 is Associated with Activated TrkA
Upon activation, tyrosine receptor kinases have been shown to activate several signaling cascades of which the MAPK/ERK1/2 and the phosphatidylinositol 3′-kinase (PI3K) are the most common ones.33 As we have previously found that these two pathways are not activated by TrkA in our melanoma specimens25 (unpublished results), we wanted to examine whether p-TrkA rather could activate the JNK and/or p38 pathways. As shown in Table 4a, a significant positive association between p-p38 in the nucleus and p-TrkA in the membrane (P=0.02) as well as a close to significant correlation between nuclear p-p38 and cytoplasmic p-TrkA (P=0.078) was observed in nodular melanomas. No correlation between JNK activation and TrkA activation was seen.
Discussion
In the present study we used immunohistochemistry to examine the activation status of p38 and JNK in a panel of melanocytic tissue. The obtained data were used to evaluate to what extent p38 and JNK activation had an impact on patient clinical outcome.
In agreement with other studies,12, 15, 16, 17 we observed a heterogeneous cytoplasmic and/or nuclear pattern of activated p38 and JNK in both primary and metastatic malignant melanomas. Owing to the fact that only 1/10 000 cells in the primary tumor is able to metastasize, it is important to detect tumor subpopulations that have markers of increased aggressiveness. We therefore believe that the 5% cutoff set in our study is of biological relevance. Of particular interest is our finding demonstrating the presence of exclusively nuclear JNK activity in a very high percentage of benign nevi. This finding is in contrast to Kunz et al34 who showed no p-JNK immunoreactivity in benign nevi. It is, however, well accepted that UV exposure may result in JNK activation followed by induction of c-jun, activation of the AP-1 transcription factor complex and apoptosis (reviewed in Shaulian and Karin35 and Silvers et al36). Benign nevi are located in the skin surface and are thoroughly exposed to UV-radiation and it may be speculated, therefore, that the profound JNK activation has a protective role restricting uncontrolled growth or survival of the nevus cells. Although p38 has been shown to also be activated by UV-radiation,37 the low degree of activation in benign nevi suggests a less obvious role for this kinase in early stages of melanogenesis. In support of the differential activation of JNK and p38 in benign nevi, Vicent et al17 recently observed activation of JNK but not p38 in normal cells of the lung. Similarly, JNK activation was seen predominantly in normal mammary ducts and less in breast cancer cells whereas the opposite activation pattern was observed for p38.38 In contrast, Vintman et al39 did not observe any differences in activation status of JNK and p38 in benign vs malignant mesotheliomas.
In accordance with the presence of activated JNK and p38 in the cytoplasm of malignant melanomas, observations are emerging suggesting the existence of cytoplasmic JNK and p38 targets as well. In this regard, activated JNK has been shown to phosphorylate and inactivate proapoptotic Bad protein40 as well as phosphorylate and inactive antiapoptotic Bcl-2 and Bcl-xL proteins (reviewed in Engelberg10). Likewise, p38 has been demonstrated to phosphorylate and inactivate Bcl-2 and Bcl-xL as well as to alter mitochondrial permeability leading to the release of cytochrome c.41, 42
Deactivation of JNK and p38 concomitant with increased tumor aggressiveness has been previously described in rat prostate and human colon cancer.18, 19 Furthermore, JNK activation was associated with early-stage non-small-cell lung cancer17 whereas deactivation of p38 has been associated with progression of hepatocellular carcinomas.18 In accordance with these studies, we observed a marked deactivation of p38 and a marginal deactivation of JNK in melanoma metastases as compared to primary tumors and it has been speculated that deactivation of p38 and JNK in advanced tumors provide cells with an antiapoptotic mechanism and growth advantage (reviewed in Engelberg10). In contrast to what was expected, our results showed that lack of cytoplasmic JNK activation in superficial spreading melanomas was associated with thinner primary tumors and prolonged disease-free survival. This observation is, however, in accordance with a similar observation made in breast cancer38 and supports the hypothesis put forward by Engelberg,10 suggesting that the stress-activated kinases play different roles in early stage vs advanced tumors. The effect on disease progression is, furthermore, supported by the observed association between p-JNK and the levels of proteins involved in regulating cell cycle progression. Thus, activated cytoplasmic JNK was significantly associated with cyclin A and p21CIP1/WAF1 and although not significant, with Ki-67 and cyclin D3 protein levels in superficial spreading melanomas. In this regard, we have previously reported an association between expression of cyclin A and cyclin D3, cell proliferation and disease outcome in superficial spreading melanomas.20, 21 Furthermore, in a previous study we have suggested that p21CIP1/WAF1 may play a role in facilitating cell cycle progression in early-stage superficial spreading melanomas.22 In agreement with our results, Margheri et al43 observed a decline in JNK phosphorylation concomitant with inhibition of cyclin D3 and cyclin A in response to blocking the urokinase receptor. Moreover, Nuntharatanapong et al44 and Kim et al45 reported a positive correlation between the cdk-inhibitor p21WAF1/CIP1 and cytoplasmic p-JNK.
Although our results suggest that JNK activation does not affect cell proliferation and disease outcome in nodular melanomas, we cannot exclude a role in invasion and/or motility. In wound healing, cytoplasmic p-JNK has been shown to be involved in control of cell motility by regulating protrusion and migration in a gene-expression independent manner.46 Moreover, a number of studies have demonstrated the induction of matrix metalloproteinase-2 (MMP-2) by JNK in several tumor forms, including melanomas.47 In fact, it has been suggested that the multiple MAPK pathways cooperate in the regulation of MMP transcription in response to different signals (reviewed in Westermarck and Kahari48).
In contrast to p-JNK, p38 activation was not associated with markers of cell cycle progression or disease outcome in superficial spreading melanomas. In nodular melanomas, on the other hand, the presence of activated p38 in the nucleus was associated with Ki-67 expression. However, we have previously shown that the level of Ki-67 has little impact on disease progression of this subtype.20 Together these findings suggest that p38 does not play a major role in the development or progression of melanocytic tumors and that the observed activation is rather an effector of other signaling pathways. In support of this, Aguirre-Ghiso et al11 suggested that the p38 pathway may be altered or dysfunctional in melanomas.
We have previously shown that p-TrkA is expressed by primary and metastatic melanomas and is associated with poor clinical outcome.25 However, we were not able to demonstrate any association between p-TrkA and the major downstream signaling pathways ERK1/2/MAPK or PI3K suggesting that other signaling pathways downstream of TrkA are activated. In the present study, we found that membranous p-TrkA was significantly associated with nuclear activated p38 in nodular melanomas and a close to significant association between cytoplasmic p-TrkA and nuclear p38 was seen as well. However, the lack of association between p38 activation and clinical parameters suggest that activation of TrkA may also lead to activation of other downstream targets.
Numerous studies have demonstrated both positive and negative cross-talk between different signaling pathways.11, 27, 28, 29, 30, 31, 32 We observed a strong significant positive association between the activation status of cytoplasmic JNK, p38, and ERK1/2 in superficial spreading melanomas as well as an association between activation of Akt and p38 in the cytoplasm. In a study by Pedram et al32 expression of vascular endothelial growth factor was shown to induce a positive ERK1/2-JNK cross-talk in endothelial cells. However, this is in contrast to the findings of Shen et al30 who concluded that sustained JNK activation uncouples ERK1/2 activation from MEK, indicating a negative cross-talk between these two pathways in Jurkat cells. Moreover, our data are not in agreement with studies showing a negative cross-talk mechanism between p38 and ERK1/2 in several tissues.11, 27, 28, 29 Common for these studies is the observation that inhibiting one pathway induces a robust increase in activation of the other and vice versa. This again demonstrates the complexity of signaling in different environments. Thus, in melanoma we might suggest a positive cross-talk mechanism between the different MAPKs.
We observed few correlations between pJNK and p38 in nodular melanomas compared to the superficial spreading melanomas. The different biology of these two tumor types may partly explain these variations, but we cannot exclude that this could be due to the relative small number of tumors.
We report that activated JNK and p38 are expressed by a subset of melanocytic tumors. In benign nevi, activated JNK suggest a role in restricting uncontrolled cell proliferation or survival. In contrast, expression of p-JNK might suggest an aggressive phenotype and poor clinical outcome for patients suffering from superficial spreading melanomas. Furthermore, our results suggest that activation of p38 does not play a major role in the development and/or progression of neither superficial spreading melanomas nor nodular melanomas.
References
Bode AM, Dong Z . Signal transduction pathways in cancer development and as targets for cancer prevention. Prog Nucleic Acid Res Mol Biol 2005;79:237–297.
Kyosseva SV . Mitogen-activated protein kinase signaling. Int Rev Neurobiol 2004;59:201–220.
Hagemann C, Blank JL . The ups and downs of MEK kinase interactions. Cell Signal 2001;13:863–875.
Roux PP, Blenis J . ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev 2004;68:320–344.
Zhang W, Liu HT . MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res 2002;12:9–18.
Manni A, Wechter R, Gilmour S, et al. Ornithine decarboxylase over-expression stimulates mitogen-activated protein kinase and anchorage-independent growth of human breast epithelial cells. Int J Cancer 1997;70:175–182.
Mansour SJ, Matten WT, Hermann AS, et al. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science 1994;265:966–970.
Ito Y, Sasaki Y, Horimoto M, et al. Activation of mitogen-activated protein kinases/extracellular signal- regulated kinases in human hepatocellular carcinoma. Hepatology 1998;27:951–958.
Salh B, Marotta A, Matthewson C, et al. Investigation of the Mek-MAP kinase-Rsk pathway in human breast cancer. Anticancer Res 1999;19:731–740.
Engelberg D . Stress-activated protein kinases-tumor suppressors or tumor initiators? Semin Cancer Biol 2004;14:271–282.
Aguirre-Ghiso JA, Estrada Y, Liu D, et al. ERK(MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38(SAPK). Cancer Res 2003;63:1684–1695.
Esteva FJ, Sahin AA, Smith TL, et al. Prognostic significance of phosphorylated P38 mitogen-activated protein kinase and HER-2 expression in lymph node-positive breast carcinoma. Cancer 2004;100:499–506.
Davidson B, Givant-Horwitz V, Lazarovici P, et al. Matrix metalloproteinases (MMP), EMMPRIN (extracellular matrix metalloproteinase inducer) and mitogen-activated protein kinases (MAPK): co-expression in metastatic serous ovarian carcinoma. Clin Exp Metastasis 2003;20:621–631.
Liang B, Wang S, Zhu XG, et al. Increased expression of mitogen-activated protein kinase and its upstream regulating signal in human gastric cancer. World J Gastroenterol 2005;11:623–628.
Guo L, Guo Y, Xiao S, et al. Protein kinase p-JNK is correlated with the activation of AP-1 and its associated Jun family proteins in hepatocellular carcinoma. Life Sci 2005;77:1869–1878.
Royuela M, Arenas MI, Bethencourt FR, et al. Regulation of proliferation/apoptosis equilibrium by mitogen-activated protein kinases in normal, hyperplastic, and carcinomatous human prostate. Hum Pathol 2002;33:299–306.
Vicent S, Garayoa M, Lopez-Picazo JM, et al. Mitogen-activated protein kinase phosphatase-1 is overexpressed in non-small cell lung cancer and is an independent predictor of outcome in patients. Clin Cancer Res 2004;10:3639–3649.
Wang Q, Ding Q, Dong Z, et al. Downregulation of mitogen-activated protein kinases in human colon cancers. Anticancer Res 2000;20:75–83.
Uzgare AR, Kaplan PJ, Greenberg NM . Differential expression and/or activation of P38MAPK, erk1/2, and jnk during the initiation and progression of prostate cancer. Prostate 2003;55:128–139.
Florenes VA, Maelandsmo GM, Faye R, et al. Cyclin A expression in superficial spreading malignant melanomas correlates with clinical outcome. J Pathol 2001;195:530–536.
Florenes VA, Faye RS, Maelandsmo GM, et al. Levels of cyclin D1 and D3 in malignant melanoma: deregulated cyclin D3 expression is associated with poor clinical outcome in superficial melanoma. Clin Cancer Res 2000;6:3614–3620.
Maelandsmo GM, Holm R, Fodstad O, et al. Cyclin kinase inhibitor p21WAF1/CIP1 in malignant melanoma: reduced expression in metastatic lesions. Am J Pathol 1996;149:1813–1822.
Jorgensen K, Holm R, Maelandsmo GM, et al. Expression of activated extracellular signal-regulated kinases 1/2 in malignant melanomas: relationship with clinical outcome. Clin Cancer Res 2003;9:5325–5331.
Slipicevic A, Holm R, Nguyen MT, et al. Expression of activated Akt and PTEN in malignant melanomas: relationship with clinical outcome. Am J Clin Pathol 2005;124:528–536.
Florenes VA, Maelandsmo GM, Holm R, et al. Expression of activated TrkA protein in melanocytic tumors: relationship to cell proliferation and clinical outcome. Am J Clin Pathol 2004;122:412–420.
Florenes VA, Maelandsmo GM, Kerbel RS, et al. Protein expression of the cell-cycle inhibitor p27Kip1 in malignant melanoma: inverse correlation with disease-free survival. Am J Pathol 1998;153:305–312.
Lee KH, Hyun MS, Kim JR . Growth factor-dependent activation of the MAPK pathway in human pancreatic cancer: MEK/ERK and p38 MAP kinase interaction in uPA synthesis. Clin Exp Metastasis 2003;20:499–505.
Liu Q, Hofmann PA . Protein phosphatase 2A-mediated cross-talk between p38 MAPK and ERK in apoptosis of cardiac myocytes. Am J Physiol Heart Circ Physiol 2004;286:H2204–H2212.
Sharma GD, He J, Bazan HE . p38 and ERK1/2 coordinate cellular migration and proliferation in epithelial wound healing: evidence of cross-talk activation between MAP kinase cascades. J Biol Chem 2003;278:21989–21997.
Shen YH, Godlewski J, Zhu J, et al. Cross-talk between JNK/SAPK and ERK/MAPK pathways: sustained activation of JNK blocks ERK activation by mitogenic factors. J Biol Chem 2003;278:26715–26721.
Liu SL, Lin X, Shi DY, et al. Reactive oxygen species stimulated human hepatoma cell proliferation via cross-talk between PI3-K/PKB and JNK signaling pathways. Arch Biochem Biophys 2002;406:173–182.
Pedram A, Razandi M, Levin ER . Extracellular signal-regulated protein kinase/Jun kinase cross-talk underlies vascular endothelial cell growth factor-induced endothelial cell proliferation. J Biol Chem 1998;273:26722–26728.
Kaplan DR, Miller FD . Signal transduction by the neurotrophin receptors. Curr Opin Cell Biol 1997;9:213–221.
Kunz M, Ibrahim S, Koczan D, et al. Activation of c-Jun NH2-terminal kinase/stress-activated protein kinase (JNK/SAPK) is critical for hypoxia-induced apoptosis of human malignant melanoma. Cell Growth Differ 2001;12:137–145.
Shaulian E, Karin M . AP-1 in cell proliferation and survival. Oncogene 2001;20:2390–2400.
Silvers AL, Bachelor MA, Bowden GT . The role of JNK and p38 MAPK activities in UVA-induced signaling pathways leading to AP-1 activation and c-Fos expression. Neoplasia 2003;5:319–329.
Tada A, Pereira E, Beitner-Johnson D, et al. Mitogen- and ultraviolet-B-induced signaling pathways in normal human melanocytes. J Invest Dermatol 2002;118:316–322.
Yeh YT, Hou MF, Chung YF, et al. Decreased expression of phosphorylated JNK in breast infiltrating ductal carcinoma is associated with a better overall survival. Int J Cancer 2005;118:2678–2684.
Vintman L, Nielsen S, Berner A, et al. Mitogen-activated protein kinase expression and activation does not differentiate benign from malignant mesothelial cells. Cancer 2005;103:2427–2433.
Yu C, Minemoto Y, Zhang J, et al. JNK suppresses apoptosis via phosphorylation of the proapoptotic Bcl-2 family protein BAD. Mol Cell 2004;13:329–340.
Grethe S, Ares MP, Andersson T, et al. p38 MAPK mediates TNF-induced apoptosis in endothelial cells via phosphorylation and downregulation of Bcl-x(L). Exp Cell Res 2004;298:632–642.
Kong JY, Klassen SS, Rabkin SW . Ceramide activates a mitochondrial p38 mitogen-activated protein kinase: a potential mechanism for loss of mitochondrial transmembrane potential and apoptosis. Mol Cell Biochem 2005;278:39–51.
Margheri F, D'Alessio S, Serrati S, et al. Effects of blocking urokinase receptor signaling by antisense oligonucleotides in a mouse model of experimental prostate cancer bone metastases. Gene Therapy 2005;12:702–714.
Nuntharatanapong N, Chen K, Sinhaseni P, et al. EGF receptor-dependent JNK activation is involved in arsenite-induced p21Cip1/Waf1 upregulation and endothelial apoptosis. Am J Physiol Heart Circ Physiol 2005;289:H99–H107.
Kim GY, Mercer SE, Ewton DZ, et al. The stress-activated protein kinases p38 alpha and JNK1 stabilize p21(Cip1) by phosphorylation. J Biol Chem 2002;277:29792–29802.
Altan ZM, Fenteany G . c-Jun N-terminal kinase regulates lamellipodial protrusion and cell sheet migration during epithelial wound closure by a gene expression-independent mechanism. Biochem Biophys Res Commun 2004;322:56–67.
Hong IK, Kim YM, Jeoung DI, et al. Tetraspanin CD9 induces MMP-2 expression by activating p38 MAPK, JNK and c-Jun pathways in human melanoma cells. Exp Mol Med 2005;37:230–239.
Westermarck J, Kahari VM . Regulation of matrix metalloproteinase expression in tumor invasion. FASEB J 1999;13:781–792.
Acknowledgements
This work was supported by The Norwegian Cancer Society. We thank Ellen Hellesylt, Liv Inger Håseth, Inger Liv Nordli, Mai Nguyen and Ann Larsen for excellent technical support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jørgensen, K., Davidson, B. & Flørenes, V. Activation of c-jun N-terminal kinase is associated with cell proliferation and shorter relapse-free period in superficial spreading malignant melanoma. Mod Pathol 19, 1446–1455 (2006). https://doi.org/10.1038/modpathol.3800662
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/modpathol.3800662
