
Abstract
In vitro cardiac model systems have rapidly advanced as complementary platforms to conventional two-dimensional (2D) cultures and animal models, which, despite their long-standing contributions, exhibit inherent limitations in predicting human cardiac responses. This review highlights recent progress in biomimetic platforms that more faithfully recapitulate the structure and function of the human myocardium, including engineered three-dimensional (3D) tissues, chambered ventricular constructs, self-organizing cardiac organoids, and microphysiological systems. These models are increasingly being applied as Drug Development Tools (DDTs) for safety pharmacology, efficacy testing, and cardiotoxicity assessment, offering improved predictive performance compared to traditional assays. By incorporating key features, such as three-dimensional tissue architecture, multicellular composition, electromechanical coupling, and physiological loading, these platforms enhance the translational relevance of preclinical studies. Recent innovations include maturation-enhanced organoids, vascularized engineered heart tissues, chamber models with physiological pressure–volume dynamics, and chip-based platforms that enable the real-time assessment of contractility and electrophysiology. Importantly, the integration of immune and vascular components, as well as multi-organ connectivity, further extends their applicability to systemic drug evaluations and disease modeling. Collectively, these advances bridge the gap between reductionist in vitro assays and clinical studies and align with emerging regulatory paradigms that emphasize human-relevant and non-animal testing methods. By enabling mechanistic insights into human cardiogenesis, cardiomyocyte maturation, and patient-specific disease modeling, advanced in vitro cardiac platforms hold great promise for precision pharmacology and regenerative medicine. Overall, in vitro cardiac models represent a transformative paradigm for advancing drug discovery, improving safety predictions, and reducing the reliance on animal testing in cardiovascular research.

Similar content being viewed by others

Introduction
Cardiovascular disease (CVD) is the leading cause of mortality worldwide, accounting for approximately one-third of all global deaths1. Despite this substantial disease burden, the development of new cardiovascular therapeutics continues to face high attrition rates during Phase II and III clinical trials, largely due to insufficient efficacy and unexpected toxicities2,3. A fundamental limitation underlying these failures is the poor predictive accuracy of conventional preclinical models, which fail to faithfully recapitulate key aspects of human cardiac physiology and pharmacodynamics4. Although animal models have long served as the backbone of preclinical cardiovascular research, they differ markedly from humans in heart size, ion channel expression profiles, metabolic pathways, and drug responses. These differences result in significant interspecies variability and limit translational predictability4,5. Similarly, widely used in vitro assays, such as hERG ion channel testing in transfected cell lines, capture only isolated electrophysiological effects. As a result, these assays often generate false-positive or false-negative predictions of arrhythmia risk6,7. Consequently, clinically relevant cardiac safety liabilities, including proarrhythmic and cardiodepressive effects, may remain undetected until late-stage development or even after market approval8. Therefore, drug-induced cardiotoxicity remains a major cause of late-stage clinical trial failure and post-marketing withdrawal, underscoring the urgent need for more predictive, human-relevant cardiac screening platforms. Human induced pluripotent stem cell–derived cardiomyocytes (hiPSC-CMs) have emerged as a promising solution to bridge this translational gap9. Reprogrammed from adult somatic cells, hiPSC-CMs retain the human genetic background and recapitulate the key electrophysiological properties of native cardiomyocytes10,11. Importantly, they can be generated at scale, maintained in long-term cultures, and integrated into reproducible, high-throughput drug screening workflows12. Their intrinsic human relevance enables the detection of cardiotoxic and arrhythmogenic effects that are frequently overlooked in animal models13, representing a critical advantage over conventional approaches. Furthermore, patient-specific and gene-edited hiPSC-CMs enable the modeling of a broad spectrum of cardiovascular diseases, ranging from inherited channelopathies to complex cardiomyopathies, thereby advancing precision pharmacology9. Contemporary in vitro cardiac models now span a wide continuum of structural and functional complexity. These range from simple two-dimensional (2D) monolayers to three-dimensional (3D) engineered heart tissues (EHTs), self-organizing cardiac organoids (COs), and microphysiological systems (MPSs), commonly referred to as heart-on-a-chip (HoC) platforms10,14. Each model class offers a distinct balance between experimental throughput and physiological fidelity, providing complementary tools for efficacy testing, safety, and mechanistic interrogation. In this review, we summarize the current landscape of hiPSC-derived cardiac model systems, critically evaluate their applications in preclinical drug development, and discuss persistent challenges, including tissue maturation, scalability, reproducibility, and regulatory validation of these models. Particular emphasis is placed on recent micro- and nanoscale engineering innovations that are driving these platforms toward enhanced translational relevance and clinical impacts.
In vitro cardiac model platforms for drug evaluation
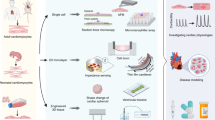
To overcome the inherent limitations of conventional in vivo animal models and overly reductionist in vitro assays, a wide range of in vitro cardiac platforms has been developed to improve the predictive accuracy of preclinical cardiovascular drug evaluation (Fig. 1). Whereas early cardiac models relied predominantly on animal-derived cells, such as neonatal rat ventricular myocytes (NRVMs), the field has rapidly transitioned toward human-based systems derived from human induced pluripotent stem cells (hiPSCs) to recapitulate human-specific cardiac physiology more closely. Collectively, these platforms span multiple levels of cardiac organization, ranging from single cardiomyocytes to multicellular engineered tissues and organ-level structures. Each model class entails a distinct trade-off between physiological fidelity and experimental throughput, enabling investigators to select the most appropriate platform based on their specific pharmacological objectives.

The top panel depicts cardiac organization across length scales, from subcellular structures and cardiomyocytes to myocardium, whole heart, and the circulation system. The middle panel summarizes major classes of in vitro cardiac models, including 2D monolayers, 3D engineered tissues, cardiac organoids, and microphysiological systems, highlighting their representative architectures, advantages, and limitations. The bottom timeline highlights key milestones in the evolution of in vitro cardiac models and is reproduced with permission from the indicated references (1991–2025); scale bars are shown for each representative image: (scale bar = 200 µm) 191(1991), (scale bar = 100 µm) 192(1997), 193(2000), (scale bar = 1 cm) 194(2002), (scale bar = 2 µm) 195(2004), [scale bars = 100 µm (left), 20 µm (right)] 196(2009), 20(2017), [scale bars = 100 µm (right-top), 20 µm (right-bottom)]29 (2018), (scale bar = 0.5 mm)30 (2019), (2021), [scale bars = 2 mm (left), 50 µm (right)]42 (2022), (2024), [scale bars = 2 mm (left), 1 cm (middle), 2 mm (right-top), 200 µm (right-bottom)]33 (2025). Red arrows indicate contraction direction; blue arrows, relaxation direction. SR sarcoplasmic reticulum, ECM extracellular matrix, AP action potential, EHT engineered heart tissue, hiPSC human induced pluripotent stem cells, CM cardiomyocyte, MPS microphysiological system, MΦ macrophage, EC endothelial cell
In this section, we systematically categorize and evaluate the major classes of in vitro cardiac models, including single-cell assays, geometrically patterned monolayers, three-dimensional engineered heart tissues, cardiac organoids, and microphysiological systems (MPSs). Particular emphasis is placed on their applications in drug efficacy testing, toxicity screening, and cardiac safety pharmacology.
Single-cell, cell-pair, and aligned monolayer in vitro models
The increasing availability of human-induced pluripotent stem cell–derived cardiomyocytes (hiPSC-CMs) has substantially accelerated the development of reproducible and human-relevant in vitro cardiac models4. Among the foundational enabling technologies, microcontact patterning provides precise control over cell–extracellular matrix (ECM) interactions and cellular geometries. Early studies demonstrated that single cardiomyocytes cultured on rectangular micropatterns with a 7:1 aspect ratio preferentially formed focal adhesions at the corners, thereby promoting anisotropic actin polymerization and aligned myofibrillar assembly15 (Fig. 2a). Subsequent comparisons between circular and rectangular geometries further confirmed that anisotropic constraints enhanced sarcomere organization16 (Fig. 2b). Extending these approaches to cell-pair configurations enabled a detailed investigation of electromechanical coupling and junctional protein dynamics under defined substrate stiffness conditions17 (Fig. 2c). Building on this concept, anisotropic monolayer systems, often referred to as “2.5D tissues,” have been developed. In these platforms, cardiomyocytes align on linearly patterned substrates, enabling coordinated contraction, junctional maturation, and directionally constrained action potential propagation. The mechanical performance of these aligned monolayers was quantitatively evaluated using muscular thin-film (MTF) platforms, initially fabricated on polydimethylsiloxane (PDMS) substrates to measure contractile bending forces, and later adapted with gelatin-based materials to improve physiological stiffness and long-term culture stability18 (Fig. 2d). Subsequent innovations, including optogenetic pacing–enabled MTFs (Opto-MTFs), provided spatiotemporal control of contraction while enabling simultaneous calcium imaging19. In parallel, multimaterial three-dimensional printing strategies have facilitated the integration of embedded strain sensors for high-content pharmacological analysis20. Additional refinements, such as fibronectin-based micropatterns21 and geometric bilayer constructs incorporating insulated pacemaker nodes22 (Fig. 2e), further advanced studies on wavefront initiation and propagation dynamics. Narrowed MTF geometries subsequently improved the precision of conduction velocity measurements, enhancing their utility for arrhythmia modeling and drug screening23 (Fig. 2f).

a Actin alignment is regulated by myocyte shape but not matrix elasticity, sarcomere content is dependent on myocyte shape and matrix elasticity (scale bars = 10 µm). Reproduced with permission16. Copyright 2014, The American Physiological Society. b CMs on circular and rectangular microcontact printed ECM island (scale bars = 10 µm). Reproduced with permission15. Copyright 2008, John Wiley and Sons. c (Top) cell–cell junctions had sigmoid-like contours. (Bottom) regulation of displacement, traction stress, and focal adhesion formation by substrate stiffness (scale bars = 10 µm). Reproduced with permission17. Copyright 2012, The National Academy of Sciences. d (Left) Engineering anisotropic cardiac tissues with micro-molded gelatin hydrogels. (Right) Extended culture of human iPSC-derived cardiac myocytes on micro-molded gelatin MTFs [scale bars = 10 µm (top-left), 1 mm (top-right), 10 µm (bottom)]. Reproduced with permission18. Copyright 2014, Elsevier. e Muscular bilayer construct showing representative mesoarchitecture and microarchitecture. Body kinematics and hydrodynamics of the biohybrid fish during one and a half tail-beat cycles [scale bars = 50 µm (top), 5 mm (bottom)]. Reproduced with permission22. Copyright 2022, The American Association for the Advancement of Science. f Calcium wavefront isochrone maps (scale bars = 50 µm). Reproduced with permission23. Copyright 2023, Elsevier. ACM arrhythmogenic cardiomyopathy, AR aspect ratio, ChR2 channelrhodopsin-2, CM cardiomyocyte, E elastic modulus, ECM extracellular matrix, MTF muscular thin film, Veh vehicle, WT wild type
Beyond geometric and structural control, hiPSC-CM monolayers constitute a robust and scalable platform for early-stage in vitro drug testing of human cardiac tissue. When cultured as confluent, electrically coupled monolayers in multiwell plate formats, these systems are highly amenable to high-throughput screening using automated imaging, electrophysiological, and impedance-based analyses. Within the Comprehensive in vitro Proarrhythmia Assay (CiPA) framework, 2D hiPSC-CM monolayers serve as the primary human cell–based model. These systems enable the evaluation of integrated electrophysiological responses and improve the prediction of proarrhythmic risk beyond conventional hERG assays24. Although these two-dimensional systems cannot fully recapitulate the biomechanical complexity or three-dimensional architecture of the native myocardium, their high reproducibility, scalability, and compatibility with industrial screening workflows render them indispensable for initial cardiotoxicity assessment25. Accordingly, hiPSC-CM monolayers provide a critical foundation for preclinical cardiac pharmacology and serve as a practical entry point for more physiologically sophisticated three-dimensional engineered heart tissues and chambered cardiac constructs.
Advanced 3D models of engineered heart tissue and ventricular architecture
Three-dimensional (3D) in vitro cardiac models provide advanced platforms that more closely recapitulate human cardiac development and physiology than conventional two-dimensional (2D) cultures do. Although 2D monolayers are widely employed for high-throughput applications, three-dimensional engineered tissues offer substantially enhanced structural and functional fidelity, enabling the detection of pathophysiological phenotypes that are often undetectable in planar systems. For example, titin (TTN) mutation–associated contractile dysfunction in hiPSC-CMs has been revealed exclusively in three-dimensional microtissue models26. Based on their architectural and functional design principles, advanced 3D cardiac models are broadly classified into two major categories: engineered heart tissue (EHTs) and ventricle-like chamber constructs. Each class offers distinct advantages for modeling cardiac mechanics, electrophysiological behavior, and disease-specific phenotypes in humans.
Engineered heart tissues (EHTs)
EHTs aim to recapitulate the myocardial microenvironment by integrating hiPSC-CMs with ECM components and applying defined mechanical and/or electrical stimulation27. Early ring-shaped EHT constructs enabled the quantitative measurement of contractile force within looped geometries while promoting sarcomeric alignment28 (Fig. 3a). Subsequent studies have demonstrated that electromechanical stimulation initiated at early developmental stages robustly enhances EHT maturation, leading to adult-like ultrastructural features, organized sarcomeres, T-tubule formation, and more physiological calcium handling29 (Fig. 3b). The Biowire II platform further advances EHT technology by generating atrial- and ventricular-specific tissues through spatially confined electrical conditioning, thereby enabling chamber-specific drug response testing30 (Fig. 3c). Recent efforts have focused on improving tissue maturity, multicellular complexity, and physiological relevance. In this context, EHT patches and multilayered human cardiac muscle patches (hCMPs) have demonstrated enhanced contractile performance and remuscularization in vivo, achieving functional repair and electromechanical integration in both primate and human hearts. These results represent a major translational milestone for EHT-based cardiac therapies31,32 (Fig. 3d). In parallel, large-animal studies have reported that epicardial EHT allografts support long-term engraftment, vascular integration, and recovery of ventricular function in rhesus macaques without inducing arrhythmias or tumorigenesis, further reinforcing clinical feasibility and highlighting critical translational considerations, including graft maturity, immune modulation, and Good Manufacturing Practice (GMP)-scale production33. Beyond cardiomyocyte-only constructs, the incorporation of non-myocyte populations, such as cardiac macrophages, has been shown to enhance hiPSC-CM alignment and force generation34 (Fig. 3e). In addition, prevascularization strategies employing endothelial cells (ECs) and fibroblasts (FBs) have further improved tissue integration, perfusion, and functional stability after transplantation35 (Fig. 3f).

a (Top) EHT condensation around the central Teflon cylinder in casting molds between culture days 1 to 4. EHTs after transfer in a stretch apparatus to continue culture under unidirectional and cyclic stretch (10%, 2 Hz) [scale bars = 10 mm (top), 20 µm (bottom-1), 100 µm (bottom-2), 1 µm (bottom-3, 4)]. Reproduced with permission28. Copyright 2002, Wolters Kluwer Health, Inc. (Bottom) EHTs consist of a dense network of mainly longitudinally oriented cell bundles. TEM of sarcomeric structures, cell–cell junctions, and basal membrane of cardiac myocytes in EHT. b Experimental design: early-stage or late-stage hiPSC-CMs and supporting fibroblasts were encapsulated in fibrin hydrogel to form tissues stretched between two elastic pillars and made to contract by electrical stimulation. Cross-sections taken to evaluate T-tubules [scale bars = 20 µm (left-1, 2, 3, 4), 500 µm (middle-top), 10 µm (middle-bottom), 10 µm (right)]. Reproduced with permission29. Copyright 2018, Springer Nature. c Biowire II platform generating atrial and ventricular-specific, and heteropolar cardiac tissues [scale bars = 30 µm (left-top-1, 2), 0.5 mm (left-bottom-1), 200 µm (left-bottom-2), 2 mm (right)]. Reproduced with permission30. Copyright 2019, Elsevier. d Large-scale EHT patches restoring heart function in a dose-dependent manner [scale bars = 2 mm (top-left), 20 µm (top-middle), 20 µm (top-right), 500 µm (bottom-1), 50 µm (bottom-2), 250 µm (bottom-3, 4)]. Reproduced with permission31. Copyright 2021, Wolters Kluwer Health, Inc. e Human engineered cardiac tissue (hECT) integrated with cardiac macrophages to enhance contractility (scale bars = 100 µm). Reproduced with permission34. Copyright 2024, Elsevier. f Pre-vascularized cardiac patches improving in vivo perfusion and function [scale bars = 1 mm (top), 100 µm (bottom-1), 25 µm (bottom-2, 3, 4)]. Adapted with permission35. Copyright 2024, Elsevier. A A-band, CD45 Leukocyte common antigen, CM Cardiomyocyte, cTnT Cardiac troponin T, Cx43 Connexin 43, DAPI 4′,6-diamidino-2-phenylindole, EC endothelial cell, H H-zone, hECT human engineered cardiac tissue, I I-band, iCF induced cardiac fibroblast from hiPSC, iCM induced cardiomyocyte from hiPSC, iMΦ induced macrophage from hiPSC, M M-line, mCherry fluorescent protein (red marker), MLC2a myosin light chain 2a, MLC2v myosin light chain 2v, SAA serum amyloid A, SR sarcoplasmic reticulum, TT T-tubule, WGA wheat germ agglutinin, YAP yes-associated protein, Z Z-line, α-actinin alpha-actinin
Ventricle chamber models
Beyond linear tissue constructs, ventricle chamber models are designed to recapitulate the macroscopic geometry and biomechanical pumping functions of the human heart. These systems enable the quantitative evaluation of pressure–volume (P–V) relationships, ejection fraction (EF), and directionally organized electrical conduction, which are essential parameters for translational and functional assessments. Early examples include engineered cardiac organoid chambers and miniature balloon catheter–based constructs that reproduce preload sensitivity and electrical responsiveness36,37 (Fig. 4a). Subsequent tissue-engineered ventricular models fabricated using rotational molding, three-dimensional bioprinting, or hydrogel casting have successfully replicated the anatomically relevant chamber curvature and wall architecture38. Recent advances in three-dimensional bioprinting have further expanded the structural and functional control of ventricular models. Notable examples include freeform reversible embedding of suspended hydrogels (FRESH)-printed collagen ventricles39, ventricles fabricated using fiber-reinforced gelatin-based bioinks40 (Fig. 4b), and the human chambered muscle pump (hChaMP) constructed using gelatin methacryloyl (GelMA) and collagen methacrylate (ColMA) bioinks41 (Fig. 4c). In parallel, rotary jet-spun scaffolds that reproduce helical myocardial fiber alignment have enabled ventricular constructs that exhibit apical twist and base-to-apex contraction patterns, closely resembling native cardiac mechanics42 (Fig. 4d). More recent studies have demonstrated that multi-lineage ventricular constructs incorporating cardiomyocytes (CMs), hiPSC-CMs, ECs, and FBs can sustain long-term functional stability and pharmacological responsiveness, supporting their application as advanced platforms for drug testing and disease modeling43. Collectively, these three-dimensional ventricular systems bridge the gap between conventional in vitro assays and native human myocardium, providing physiologically relevant tools for drug screening, mechanistic studies, and regenerative applications.

a Engineering human ventricular cardiac organoid chambers (hvCOC) [scale bars = 5 mm (1), 50 µm (2), 20 µm (3), 500 nm (4)]. Reproduced with permission37. Copyright 2018, Elsevier. b Fiber alignment that occurs under shear stress (τ) during 3D printing leads to native ECM anisotropic structural features in 3D scaffolds, promoting tissue alignment and organization to recapitulate in vivo heart muscle. Structural, electrophysiological, and contractile properties of 3D ventricle models (scale bar = 50 µm). Reproduced with permission40. Copyright 2020, Wolters Kluwer Health, Inc. c The optimized bioink formulation was combined with human iPSCs and bioprinted to form a hChaMP [scale bars = 2 mm (top-left), 5 mm (top-right), 5 mm (bottom-left), 5 µm (bottom-right)]. Reproduced with permission41. Copyright 2022, The American Association for the Advancement of Science. d Focused rotary jet spinning for producing helical structures [scale bars = 20 µm (top), 2 mm (bottom)]. Reproduced with permission42. Copyright 2023, The American Association for the Advancement of Science. COXIV cytochrome c oxidase subunit IV, CMs cardiomyocytes, cTnI cardiac troponin I, Cx43 Connexin 43, DAPI 4′,6-diamidino-2-phenylindole, ECM extracellular matrix, FN fibronectin, PIV particle image velocimetry, GelMA gelatin methacryloyl, ColMA collagen methacryloyl, LN laminin, Kir2.1 inward rectifier potassium channel, FRJS focused rotary jet spinning
While engineered heart tissues and chambered ventricular models offer robust macroscopic mimicry of myocardial structure and pump function, recent advances in CO technology have enabled the bottom-up generation of miniaturized, self-organizing cardiac units that closely emulate early human cardiogenesis and cellular diversity. Therefore, the following section focuses on the emerging landscape of cardiac organoids and their potential utility in preclinical drug evaluation.
Organoids
Organoids are self-organizing 3D cell culture systems that recapitulate the essential structural and functional features of native tissues through spontaneous spatial patterning and intrinsic cell–cell and cell–ECM interactions44. Cardiac organoids, primarily derived from hiPSCs, emulate the early stages of human heart development by incorporating multiple cardiac lineages, including CMs, ECs, epicardial cells, and mesenchymal stromal cells, within architecturally organized domains.
These systems exhibit hallmark morphogenetic processes, such as lumen formation, chamber-like compartmentalization, and coordinated lineage specification, providing powerful platforms for investigating cardiogenesis, congenital heart defects, and interlineage crosstalk under physiologically relevant conditions45. Heart-forming organoids (HFOs) represent some of the earliest cardiac organoid models that reproduce the spatiotemporal coordination between the cardiac mesoderm and foregut endoderm, a critical interaction during primitive heart tube formation46 (Fig. 5a). Through biphasic WNT signaling modulation and Matrigel embedding, HFOs generate vascular and septum transversum–like tissues that mimic early cardiogenic niches.

a Cardiac organoid with co-development of cardiac mesoderm and foregut endoderm, mimicking heart tube formation [scale bars = 200 µm (top-left), 100 µm (top-right), 200 µm (bottom)]. Reproduced with permission46. Copyright 2021, Springer Nature. b Self-organizing cavity-forming cardioid modeling early heart field and lumenogenesis [scale bars = 1 µm (top), 200 µm (bottom-left), 50 µm (bottom-right)]. Reproduced with permission47. Copyright 2021, Elsevier. c Epicardioid capturing epicardium formation and EMT processes [scale bars = 200 µm (top-left), 100 µm (top-middle), 50 µm (top-right), 50 µm (bottom)]. Reproduced with permission50. Copyright 2023, Springer Nature. d Multi-chamber cardiac organoid with spatially patterned atrial and ventricular domains [scale bars = 200 µm]. Reproduced with permission51. Copyright 2023, Elsevier. e Blood-generating organoid co-developing cardiac and hematopoietic lineages [scale bars = 500 µm]. Reproduced with permission52. Copyright 2024, Springer Nature. f Vascularized cardiac organoid with integrated hepatic and endothelial precursors [scale bars = 1 mm (top-left), 0.5 mm (top-right), 0.5 mm (bottom-left), 100 µm (bottom-middle), 20 µm (bottom-right)]. Reproduced with permission55. Copyright 2025, The American Association for the Advancement of Science. IC inner core, ML middle layer, OL outer layer, WT1 Wilms tumor 1, EPDCs epicardium-derived cells, EMT epithelial-mesenchymal transition, ID intercalated disk, RA right atrium, LV left ventricle, RV right ventricle, BG-HFO blood-generating heart-forming organoid, MES mesenchyme, CMs cardiomyocytes, ECs endothelial cells, SMCs smooth muscle cells, cVO cardiac vascularized organoid
Cardioids constitute another major milestone, as they reproduce lumenogenesis, coelomic cavity formation, and early chamber morphogenesis through intrinsic self-patterning mechanisms47 (Fig. 5b). The spatially segregated expression of transcription factors, such as HAND1 and NFATC1, further underscores their utility in dissecting the genetic programs governing cardiac morphogenesis. Chamber-forming heart organoids further extend this complexity by generating cavitated structures with atrial–ventricular compartmentalization and stratified myocardial and endocardial layers in the absence of externally imposed mechanical cues48. Elongating multi-lineage organized (EMLOC) neurocardiac gastruloids advance this paradigm by enabling the coordinated development of neuroectodermal and cardiac mesodermal tissues, facilitating investigations into early neurocardiac communication49. Epicardioids represent a particularly important innovation, as they recapitulate epicardial sheet formation, epithelial-to-mesenchymal transition (EMT), and dynamic interactions with the underlying myocardium and coronary progenitors, which are central to vascular development and fibrotic remodeling50 (Fig. 5c). In parallel, multi-chamber cardioid systems have demonstrated spatially organized atrial and ventricular domains, enabling high-resolution modeling of chamber-specific developmental abnormalities and disease phenotypes51 (Fig. 5d). To further enhance physiological relevance, blood-forming heart organoids have been developed that concurrently generate endocardial, myocardial, and hemogenic endothelial lineages, thereby recapitulating the integrated development of the cardiovascular and hematopoietic systems52 (Fig. 5e). Recently, gastruloid-based cardiac models have expanded the developmental scope of cardiac organoid technologies53,54,55. Notably, human gastruloids have been shown to self-organize into early cardiac and hepatic vascular domains, recapitulating the coordinated emergence of mesodermal and endodermal tissues during early cardiogenesis55 (Fig. 5f). This approach provides a powerful framework for interrogating early vascularization, inter-organ patterning, and cross-germ layer interactions in vitro, thereby bridging developmental biology and regenerative cardiac modeling.
Despite these strengths, cardiac organoids are primarily suited for modeling early cardiogenesis, congenital cardiac disorders, and multicellular developmental interactions. However, organoid morphogenesis remains inherently stochastic. Limited control over biophysical cues, therefore, constrains their utility for standardized pharmacological testing and arrhythmia risk assessment. Building on the developmental insights provided by organoid systems, MPSs have emerged as complementary platforms that offer defined microarchitectures, controlled perfusion, and programmable electrical stimulation. These features enable precise regulation of mechanical loading and real-time electrophysiological measurements, making MPSs particularly well-suited for high-throughput drug screening and cardiac safety pharmacology (Table 1).
Microphysiological systems (MPSs)
MPSs, often referred to as organs-on-a-chip, represent a transformative advance over traditional preclinical animal models. By reducing translational discrepancies, these systems improve clinical predictability56. These platforms integrate living tissues into microfluidic devices engineered to recapitulate the architecture, microenvironment, and dynamic functionality of native organs. In the cardiovascular field, cardiac-specific MPSs, particularly HoC systems, are designed to faithfully reproduce key physiological features, including electrical conduction, mechanical contraction, and pharmacological responsiveness57. In contrast to self-organizing COs, which are well-suited for modeling early cardiac morphogenesis and developmental disorders, HoC platforms offer precise experimental control. This includes spatiotemporal regulation of perfusion, electrical pacing, mechanical loading, and pharmacokinetic microenvironments (Table 1). HoC systems are especially advantageous for standardized drug screening, proarrhythmia risk assessment, and real-time evaluation of contractile and electrophysiological functions under well-defined conditions.
To further capture systemic physiology, multi-organ-on-a-chip (Multi-OoC) technologies have been developed to enable controlled inter-organ communication between cardiac, hepatic, and vascular tissues58. These integrated platforms are particularly valuable for assessing cardiotoxicity arising from both direct drug effects and indirect influences of hepatic metabolism or immune-mediated responses, thereby enhancing the physiological relevance of in vitro safety and efficacy testing59,60.
Early MPS designs combined MTFs with microfluidic architectures to enable the real-time measurement of contractile responses under dynamic flow conditions, facilitating higher-throughput drug screening61. The subsequent incorporation of three-dimensional cardiac microtissues subjected to mechanical stimulation further improved cellular alignment, tissue compaction, and contractile force generation62 (Fig. 6a). In parallel, metabolically driven maturation strategies have enhanced hiPSC-CM function by improving calcium handling and sarcomeric organization63. The advent of direct laser writing (DLW) has enabled the fabrication of precision-engineered HoC devices incorporating embedded transducers for real-time electrophysiological sensing and controlled perfusion64. Disease modeling capabilities have also substantially advanced. For example, infarct border zone-on-a-chip systems recreate spatial oxygen gradients to simulate ischemia–reperfusion injury, revealing region-specific physiological and electrophysiological responses65 (Fig. 6b). The incorporation of immune components into vascularized HoC platforms has enabled mechanistic studies on inflammation-induced cardiac dysfunction. In this context, extracellular mitochondrial DNA has been identified as a trigger of pathological inflammatory signaling, which can be attenuated by exosome-based therapeutic approaches66 (Fig. 6c). In addition, platforms incorporating primitive macrophages have demonstrated sustained vascularization and preserved tissue function during extended culture periods67 (Fig. 6d). Recently, tri-culture systems comprising ECs, FBs, and hiPSC-CMs have achieved synchronized contraction and long-term functional stability under continuous perfusion, enabling robust and reproducible functional assessments68. Concurrently, multi-OoC platforms have progressed toward greater modularity and physiological integration in recent years. Early systems employed plug-and-play designs that allowed the customizable assembly of individual tissue modules69. These architectures have since evolved into integrated platforms for modeling liver–heart interactions, thereby enabling the investigation of metabolism-mediated cardiotoxicity70,71. More advanced designs now incorporate multiple organoids within a single perfusable circuit, permitting simultaneous drug testing across interconnected tissues and yielding comprehensive and human-relevant safety profiles72,73 (Fig. 6e). hiPSC-derived cardiac–hepatic chips have proven to be effective in predicting drug-induced arrhythmias and hepatotoxicity. The most advanced iterations feature vascularized and maturation-enhanced organoids interconnected by functional flow networks that simulate nutrient delivery, hormonal signaling, and systemic pathophysiology74 (Fig. 6f).

a Integration of mechanical stimulation with microfluidics in micro-engineered cardiac tissues to promote tissue functionality in a beating heart-on-a-chip format [scale bars = 100 µm (left), 100 µm (right)]. Reproduced with permission62. Copyright 2016, Royal Society of Chemistry. b Creation of a myocardial infarct border-zone-on-a-chip system to simulate oxygen gradients and spatial tissue remodeling after ischemia [scale bars = 10 mm (top), 20 µm (bottom-left), 1 mm (bottom-right)]. Reproduced with permission65. Copyright 2022, The American Association for the Advancement of Science. c Immune-enhanced vascularized HoC model incorporating macrophages to recapitulate myocardial inflammation and evaluate immunomodulatory therapies [scale bar = 200 µm]. Reproduced with permission66. Copyright 2024, The American Association for the Advancement of Science. d Long-term vascularization of heart-on-a-chip achieved by incorporating primitive macrophages, promoting stable perfusion in engineered cardiac tissues [scale bars = 100 µm (top-left), 10 µm (top-right), 250 µm (bottom-left), 100 µm (bottom-right)]. Reproduced with permission67. Copyright 2024, Elsevier. e Multi-organoid chip derived from hiPSCs for assessing antidepressant-induced systemic toxicity [scale bars = 100 µm (i, ii), 200 µm (iii, iv)]. Reproduced with permission73. Copyright 2020, IOP Publishing. f A vascularly linked multi-tissue platform that connects matured organ compartments via perfusable vasculature [scale bars = 50 µm (top), 10 µm (bottom-1), 50 µm (bottom-2, 3, 4)]. Adapted with permission74. Copyright 2022, Springer Nature. µECT micro-engineered cardiac tissue, PDMS polydimethylsiloxane, MI myocardial infarction, EVs extracellular vesicles, DPSC dental pulp stem cell, MAC/MΦ macrophage, ALB albumin, CYP450 Cytochrome P450, OoC organ-on-a-chip, HoC heart-on-a-chip, hiPSCs human induced pluripotent stem cells, HUVECs human umbilical vein endothelial cells, PBMC peripheral blood mononuclear cell
Advances in MPS and multi-OoC technologies are reshaping preclinical cardiac research by providing physiologically relevant, scalable human-based platforms. In parallel with these technological developments, regulatory interest has increased, with agencies such as the U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMA) recognize MPS-based approaches as key New Approach Methodologies (NAMs) for cardiac safety assessment. Supported by initiatives, including the CiPA and the revised ICH S7B/E14 Q&A framework, these efforts emphasize rigorous electrophysiological validation, metabolic competence, and clinically aligned benchmark datasets. Ensuring the translational reliability of MPS platforms requires standardized multiparametric evaluation frameworks that integrate structural, functional, molecular, and metabolic analyses.
Comprehensive assessment of cardiac models: structural, functional, molecular, and metabolic evaluations
Evaluating the translational relevance of in vitro cardiac models requires a multidimensional framework encompassing structural integrity, physiological function, molecular identity, and metabolic competence (Fig. 7, Table 2). Together, these complementary dimensions ensure that engineered cardiac tissues faithfully reflect the key properties of the native human myocardium.

Schematic overview of structural, gene expression, and functional parameters used to evaluate engineered cardiac tissues and organoids. (Top left) Structural readouts span macrostructure (e.g., muscle alignment, chamber formation) and microstructure, including extracellular matrix composition, sarcomeric proteins, junctional complexes, and subcellular organelles. (Top right) Gene expression analysis integrates spatial and single-cell transcriptomics to map tissue architecture, spatiotemporal isoform switching, and predict intercellular communication pathways. (Bottom) Functional readouts encompass electrophysiology (e.g., action potential duration, arrhythmia patterns), mechanics (excitation–contraction coupling, calcium transients, sarcomere shortening), and metabolism (mitochondrial respiration, glycolytic activity), enabling holistic assessment of cardiac performance and disease phenotypes. SCN5A sodium voltage-gated channel alpha subunit 5 (NaV1.5), SER sarcoplasmic endoplasmic reticulum, PLN phospholamban, RYR2 ryanodine receptor 2, ACTA1 actin alpha 1, MYBPC3 myosin binding protein C, cardiac, TPM1 tropomyosin 1, MYH6 myosin heavy chain 6 (α-MHC), MYH7 myosin heavy chain 7 (β-MHC), TNNT2 troponin T type 2, TNNI3 Troponin I type 3, TTN titin, N2BA/N2B titin isoforms, LMNA lamin A/C, RBM20 RNA binding motif protein 20, TAZ tafazzin, DNAJC19 DnaJ heat shock protein family member C19, BIN1 bridging integrator 1, JPH2 Junctophilin 2, DSP desmoplakin, DSG desmoglein, DSC desmocollin, PKG plakoglobin, FPD field potential duration, APD30/APD50/APD90 action potential duration at 30%, 50%, 90% repolarization, EAD early afterdepolarization, DAD delayed afterdepolarization, SERCA2 sarcoplasmic reticulum Ca2+-ATPase 2, SL sarcomere length, CaT (F/F0) calcium transient, Tau decay time constant of Ca2+ transient, LTCC L-type calcium channel, NCX Na+/Ca2+ exchanger, NKA Na+/K+-ATPase, ATP adenosine triphosphate, PGC1A peroxisome proliferator-activated receptor gamma coactivator 1-alpha, TFAM transcription factor A, mitochondrial, CPT1B carnitine palmitoyl transferase 1B, FCCP carbonyl cyanide-p-trifluoromethoxy phenylhydrazone, 2-DG 2-deoxyglucose, ECAR extracellular acidification rate, OCR oxygen consumption rate, ECM extracellular matrix
Structural assessment provides foundational insights into cellular architecture, focusing on sarcomere organization, intercalated disc formation, and cytoskeletal integrity in cardiomyocytes. Structural readouts span multiple length scales, ranging from macroscopic features, such as tissue alignment and chamber formation, to microscopic and subcellular characteristics, including extracellular matrix composition, sarcomeric proteins, junctional complexes, and organelle organization. Standard approaches include immunostaining for α-actinin, connexin 43 (Cx43), and desmosomal proteins such as desmoplakin (DSP) and desmoglein (DSG), combined with confocal or super-resolution microscopy to visualize myofibrillar alignment and junctional morphology75.
Functional evaluation centers on excitation–contraction coupling (ECC)76. Upon membrane depolarization, voltage-gated calcium channels are activated, triggering calcium release from the sarcoplasmic reticulum (SR) via ryanodine receptors. Cytosolic Ca²⁺ binding to troponin C initiates contraction, followed by Ca²⁺ reuptake into the SR via sarco/endoplasmic reticulum Ca²⁺-ATPase (SERCA) and extrusion through the sodium–Ca²⁺ exchanger (NCX) and plasma membrane Ca2+-ATPase (PMCA) to enable relaxation. These processes can be quantitatively interrogated using patch-clamp electrophysiology and multielectrode arrays, calcium imaging with indicators such as Fluo-4 or genetically encoded sensors (e.g., GCaMP), and mechanical assays, including muscular thin films, cantilever deflection, or traction force microscopy. The integration of these functional readouts provides a composite measure of cardiomyocyte maturity and electromechanical competence.
Molecular profiling adds further depth to phenotypic characterization by resolving lineage specification, maturation states, and disease-associated signatures77. Bulk RNA sequencing enables pathway-level analysis, whereas single-cell RNA sequencing (scRNA-seq) reveals cellular heterogeneity, rare subpopulations, and developmental trajectories78,79. Spatial transcriptomic approaches, such as Slide-seq and Visium, preserve tissue architecture while enabling region-specific gene expression mapping, providing insights into morphogenesis and disease progression80. Complementary proteomic and epigenomic analyses further elucidate regulatory networks and structural protein landscapes that are not accessible at the transcript level alone.
Metabolic assessment is equally critical because cardiomyocyte function is tightly coupled to cellular bioenergetics81. During maturation, cardiomyocytes undergo a metabolic transition from glycolysis to oxidative phosphorylation. This shift can be evaluated using extracellular flux analysis to quantify oxygen consumption and glycolytic activity (Seahorse OCR/ECAR), mitochondrial imaging to assess morphology, membrane potential, and reactive oxygen species (ROS) production, and metabolomic and lipidomic profiling to characterize substrate utilization and mitochondrial efficiency. Integrating multimodal datasets that encompass structural, functional, molecular, and metabolic information enables rigorous validation of in vitro cardiac platforms. These datasets include imaging, electrophysiology, transcriptomics, and bioenergetic measurements, and lay the foundation for AI-assisted models optimization and more predictive in vitro-to-in vivo translation. Advanced deep learning architectures, including multimodal transformer models and graph neural networks (GNNs), are increasingly applied to in vitro cardiac datasets. These approaches uncover latent relationships between cellular form, function, and metabolism, enabling automated phenotype classification and early detection of cardiotoxic liabilities. For example, deep learning–based imaging pipelines have enhanced high-throughput screening sensitivity by identifying subtle cardiotoxic phenotypes across libraries of more than 1200 compounds82. In parallel, predictive simulations and in silico modeling approaches are being used to forecast complex tissue-level dynamics and drug responses, exemplified by machine learning–optimized bioinspired cardiac constructs83 and computational multielectrode array models that integrate heterogeneous hiPSC-CM populations to improve the predictive accuracy of cardiac safety testing84. These integrative and data-driven strategies represent a critical step toward establishing robust human-relevant cardiac platforms that support next-generation precision pharmacology and cardiovascular therapies.
Applications
Fundamental research: human cardiogenesis and cardiomyocyte maturation
The establishment of physiologically relevant in vitro cardiac models using human induced pluripotent stem cells (hiPSCs) requires a fundamental understanding of two key biological processes: human cardiogenesis85 and cardiomyocyte (CM) maturation86. Elucidating these processes provides the developmental and functional framework necessary for generating advanced cardiac organoids, engineered heart tissues (EHTs), and disease-specific cardiac models. Moreover, such knowledge is essential for investigating the mechanisms underlying congenital and acquired heart diseases, enabling the development of targeted therapeutic strategies and advancing precision medicine in the field of cardiology. Collectively, insights into human cardiogenesis and approaches to promote hiPSC-CM maturation form the biological foundation upon which next-generation in vitro cardiac platforms are built.
Advances in human cardiogenesis using cardiac organoids
Cardiac organoid platforms have emerged as powerful tools for modeling the early stages of human heart development. These systems recapitulate key processes of cardiogenesis, including cardiac mesoderm induction, first and second heart field specification, chamber morphogenesis, and coordinated development of vascular lineages87. Their self-organizing capabilities enable controlled and reproducible investigations of complex spatial and temporal developmental processes that are otherwise difficult to study in vivo. Recent innovations have improved the resolution and mechanistic fidelity of organoid-based studies on early cardiogenesis46,88. These platforms have facilitated mechanistic insights into lineage specification, spatial patterning, and signaling dynamics during the development of the heart. Beyond cardiac-specific models, advanced multi-lineage organoids have been developed to emulate integrated human embryogenesis in vitro. For instance, the modulation of Wnt signaling in hiPSC aggregates has yielded organoids comprising cardiac, endodermal, and ectodermal tissues, recapitulating early foregut–heart axis interactions49. Gastruloid platforms support the co-development of cardiac and hepatic vasculature55 and simulate the simultaneous emergence of hematopoietic and cardiac lineages52. Additionally, epicardioid systems enable modeling of epicardial sheet formation and myocardial interactions during vascular development50. These advanced organoid technologies, including cardioids, gastruloids, and epicardioids, provide high-resolution in vitro platforms for studying human cardiogenesis across germ layers, offering novel insights into tissue patterning and lineage crosstalk.
Strategies for promoting cardiomyocyte maturation
A major limitation of hiPSC-CMs is their intrinsic structural and functional immaturity, which constrains their translational applicability89. Therefore, promoting cardiomyocyte maturation is essential for enhancing the physiological relevance of in vitro cardiac models. Mature hiPSC-CMs closely recapitulate adult myocardial responses, thereby enabling more accurate drug testing, improved modeling of adult-onset cardiac diseases, and enhanced integration into regenerative therapeutic strategies.
A diverse array of maturation strategies has been developed to overcome these limitations. Biomaterial-based scaffolds provide biomechanical cues that promote sarcomere alignment and contractile development28. In parallel, mechanical and electrical stimulation, designed to mimic physiological loading and pacing, enhances cytoskeletal organization, mitochondrial biogenesis, and calcium-handling machinery, collectively driving a more adult-like electrophysiological phenotype29,32. Co-culture with supportive non-myocyte populations, including fibroblasts and endothelial cells, further facilitates paracrine and juxtacrine signaling pathways that improve hiPSC-CM functional maturation and tissue-level organization90.
Metabolic and hormonal conditioning also play pivotal roles in the maturation of cardiomyocytes. Induction of fatty acid oxidation, coupled with hormonal modulation using triiodothyronine (T3) and glucocorticoids, has been shown to advance both electrophysiological and metabolic maturation of hiPSC-CMs63. These integrated maturation approaches significantly enhance key phenotypic features, including sarcomere organization, force generation, action potential duration, calcium transient kinetics, and oxidative metabolism in cardiomyocytes. Consequently, mature hiPSC-CMs exhibit substantially improved utility for disease modeling, drug screening, and translational cardiac research75. Ongoing investigations into cardiomyocyte maturation pathways continue to drive the development of increasingly predictive and clinically relevant in vitro cardiac platforms. Together, advances in understanding human cardiogenesis and strategies for promoting cardiomyocyte maturation establish the essential biological foundation for next-generation cardiac model systems, whose ultimate translational value lies in their ability to model human cardiovascular diseases with both physiological and pathological properties.
Cardiac disease modeling
hiPSC-CMs have transformed cardiac disease modeling by providing patient-specific, ethically acceptable, and genetically tractable platforms for mechanistic investigations and drug screening. Unlike embryonic stem cells (ESCs), hiPSCs are reprogrammed from somatic cell sources, such as dermal fibroblasts or peripheral blood mononuclear cells (PBMCs), thereby enabling the generation of patient-derived cardiac models while avoiding ethical concerns associated with embryonic tissues91. This approach allows cardiac disease mechanisms to be studied in a human cellular context while preserving the unique genetic background92.
The utility of hiPSC-CM-based disease models is further enhanced by CRISPR/Cas9-mediated genome editing, which enables the precise insertion, correction, or deletion of pathogenic variants. When combined with isogenic control lines, this strategy allows for the direct attribution of observed phenotypes to specific genetic alterations and supports high-resolution mechanistic analyses93. Such genetic precision is particularly valuable for modeling monogenic cardiac disorders and evaluating mutation-targeted therapeutic strategies.
Broadly, hiPSC-based cardiac disease models can be classified into inherited and acquired models (Tables 3–7). Inherited cardiac diseases are typically modeled by generating hiPSCs from affected patients or by introducing disease-associated mutations into control lines using targeted genome editing, which recapitulates intrinsic genetic pathologies. In contrast, acquired cardiac disease models rely on the application of extrinsic stressors, such as hypoxia, inflammatory cytokines, electrical pacing, or metabolic challenge, to otherwise healthy hiPSC-CMs, mimicking environmental, metabolic, or systemic insults encountered in vivo.
hiPSC-derived cardiac platforms enable detailed mechanistic dissection of both inherited and acquired cardiac disorders, providing genetically defined and patient-specific models for elucidating arrhythmogenic mechanisms, disease progression, and therapeutic responses. These capabilities position hiPSC-based systems as powerful tools for precision disease modeling and the development of targeted therapies for cardiovascular diseases.
Modeling inherited cardiac diseases
Inherited cardiac diseases arise from pathogenic mutations affecting ion channels, sarcomeric proteins, developmental regulators, and metabolic pathways, ultimately leading to electrical instability, contractile dysfunction, and abnormal morphogenesis in the heart. Long QT syndrome (LQTS) (Table 3) is a prototypical ion channelopathy. KCNQ1 mutations (LQTS1) reduce the slow delayed rectifier potassium current (IKs), prolong action potential duration and field potential duration (APD/FPD), and induce arrhythmias that can be rescued by β-blockers or E-403194,95. KCNH2 mutations (LQTS2) similarly impair IKr, prolong repolarization, and predispose patients to early afterdepolarization (EAD), partially reversed by isoproterenol96,97,98, whereas SCN5A mutations (LQTS3) increase INaL, prolong APD, and trigger arrhythmias that are shortened by mexiletine99,100,101. Additional variants include KCNJ2 mutations in Andersen–Tawil syndrome (LQTS7), characterized by reduced IK1, prolonged APD, and delayed afterdepolarization (DAD) rescued by flecainide102, and CACNA1C mutations in Timothy syndrome (LQTS8), which impair Ca²⁺ channel inactivation, leading to prolonged APD that can be improved by roscovitine103. Calmodulin mutations (CALM1–3, LQTS14–16) impair calcium-dependent inactivation (CDI) of the L-type Ca²⁺ current, prolonging APD and producing severe arrhythmogenicity, which can be reversed by Ca²+ buffering or CRISPR-based gene correction104,105,106. Triadin knockout syndrome (TKOS) caused by TRDN mutations exhibits prolonged APD, Ca²⁺ mishandling, and arrhythmic alternans that are normalized by protein replacement107, whereas short QT syndrome (SQTS1, KCNH2) demonstrates increased IKr, abbreviated APD/FPD, and quinidine-sensitive EADs97,108. Beyond QT syndromes, catecholaminergic polymorphic ventricular tachycardia (CPVT) models carrying RYR2 or CASQ2 mutations show spontaneous Ca²⁺ waves and DADs under adrenergic stimulation, ameliorated by dantrolene or gene therapy19,109, and Brugada syndrome (SCN5A) models display reduced sodium currents, impaired conduction, and phase 2 reentry-like abnormalities110.
Inherited cardiomyopathies (Table 4) have also been extensively explored in vitro. Hypertrophic cardiomyopathy (HCM) caused by mutations in MYH7, MYBPC3, PRKAG2, ACTN2, or BRAF is associated with sarcomeric disarray, hypercontractility, increased Ca²⁺ sensitivity, and prolonged APD111,112,113. Dilated cardiomyopathy (DCM), modeled with mutations in TTN, TPM1, VCL, PLN, and RBM20, recapitulates sarcomere disorganization, reduced contractile force, impaired responses to mechanical and adrenergic stress, unfolded protein response activation, and defective calcium handling114,115,116,117,118,119, whereas correction of ABD-1 mutations restores both contractility and Ca²⁺ regulation. Duchenne muscular dystrophy (DMD) hiPSC-CMs carrying DMD mutations exhibit impaired contractile function, loss of dystrophin, Ca²⁺ mishandling, and stress hypersensitivity, with partial rescue by gene-editing strategies120,121,122,123. Arrhythmogenic cardiomyopathy (ACM) arising from PKP2 or DSP mutations demonstrates reduced desmosomal integrity, lipogenesis, apoptosis, impaired conduction, and spontaneous arrhythmias, particularly under mechanical loading124,125,126. Restrictive cardiomyopathy (RCM) models with FLNC mutations display impaired relaxation, increased passive tension, and diastolic dysfunction127, whereas mitochondrial cardiomyopathy, such as Barth syndrome (TAZ), produces sarcomere disorganization, diminished force, oxidative stress, and contractile instability93.
Congenital heart defects (Table 5) have been modeled by introducing mutations into developmental regulators, such as HAND1, TBX5, ISL1, NKX2.5, and FOXF1. COs carrying these mutations reveal impaired cavity formation, atrial and outflow tract malformations, delayed contraction onset, and abnormal chamber morphogenesis, thereby highlighting the developmental origins of arrhythmogenic substrates47,51,128.
Inherited metabolic disorders (Table 6) also contribute to the structural and electrical dysfunction. Fabry disease (GLA) models show Gb3 accumulation, hypertrophy, Ca²⁺ mishandling, and heightened excitability129. Danon disease (LAMP2) recapitulates impaired autophagy, oxidative stress, sarcomere disarray, and hypertrophy130,131 and Pompe disease (GAA) reproduces glycogen overload, prolonged APD, impaired calcium kinetics, and contractile dysfunction132. These inherited cardiac disease models elucidate direct mechanistic links between genetic perturbations and arrhythmogenic remodeling and provide powerful in vitro platforms for testing mutation-specific therapies and advancing precision cardiology research.
Modeling acquired cardiac diseases
Acquired cardiac disorders (Table 7) arise from environmental stressors, systemic diseases, or physiological overload and are modeled in vitro by exposing hiPSC-derived cardiac tissues to specific extrinsic stimuli. Although these conditions are not genetically inherited, they involve maladaptive remodeling of electrophysiological and contractile properties, providing clinically relevant systems for studying arrhythmogenesis.
Myocardial infarction (MI) has been modeled using cryoinjury, which induces fibronectin and collagen deposition, cardiomyocyte necrosis, and limited regenerative responses47, as well as oxygen-gradient systems combined with noradrenaline exposure, which triggers metabolic reprogramming, fibrosis, and Ca²⁺ dysregulation133. Chemical hypoxia induced by cobalt chloride (CoCl₂) further reproduces cardiomyocyte death and arrhythmogenic electrophysiological phenotypes134. Myocardial ischemia has also been simulated in microfluidic platforms that generate spatial oxygen gradients under continuous perfusion, resulting in impaired calcium handling, reduced contractile force, and transcriptional activation of inflammatory programs65,135. Similarly, global restriction of oxygen delivery evokes contractile irregularities, metabolic acidosis, and lactate normalization upon reperfusion, closely mimicking ischemia–reperfusion dynamics136.
Heart failure and pathological remodeling can be induced by chronic catecholamine overstimulation in EHTs. These models exhibit progressive contractile decline, cardiomyocyte hypertrophy, and elevated natriuretic peptide secretion, consistent with maladaptive ventricular remodeling observed in vivo137. Tachycardia-induced cardiomyopathy has been modeled through chronic optical tachypacing of EHTs, which accelerates contraction kinetics, shortens action potential duration and refractory periods, and increases susceptibility to proarrhythmic activity138. Diabetic cardiomyopathy is reproduced by culturing two-dimensional monolayers under hyperglycemic and lipotoxic conditions supplemented with endothelin-1 and cortisol, leading to sarcomere disorganization, oxidative stress, lipid accumulation, cellular hypertrophy, and Ca²⁺ handling abnormalities139. Finally, inflammatory cardiomyopathy (myocarditis) has been modeled using proinflammatory cytokine cocktails, including IL-1β, TNF-α, and IFN-γ, or viral mimetics that induce sarcomeric loss, electrophysiological instability, and cytokine release. Notably, several pathological phenotypes can be partially rescued by endothelial cell–derived exosomes66. These acquired disease models capture the multifactorial nature of non-genetic cardiac pathologies and provide mechanistically relevant in vitro platforms for investigating arrhythmogenic mechanisms and evaluating therapeutic interventions.
Cardiotoxicity evaluation and drug screening using multi-organ platforms
Cardiotoxicity remains a major concern in drug development, particularly for compounds that do not primarily target the cardiovascular system140. Accurate assessment of cardiotoxicity risk requires consideration of inter-organ interactions, such as the influence of hepatic metabolism on cardiac function, which are often overlooked in conventional monoculture-based in vitro systems58. To address these limitations, multi-organ-on-a-chip (Multi-OoC) platforms have been developed to integrate multiple tissue types, thereby enabling the physiologically relevant evaluation of systemic drug responses in human-based models.
A representative example is a liver–heart microphysiological system (MPS) incorporating hiPSC-derived cardiac and hepatic tissues, which has been used to assess the metabolism-mediated cardiotoxicity of terfenadine. In this platform, terfenadine impaired cardiac electrophysiology and contractility only in the presence of functional hepatic tissue, underscoring the critical role of metabolic coupling in accurate toxicity prediction70. Recently, vascularized cardiac MPS platforms have been used to evaluate the cardiotoxic effects of anticancer therapeutics on both myocardial and endothelial compartments. For instance, vandetanib, a vascular endothelial growth factor receptor (VEGFR) inhibitor, was tested using a vascularized cardiac spheroid-on-a-chip model, where it induced impaired contractile performance and rhythm disturbances141. Similarly, a multi-lineage heart-on-a-chip integrating cardiomyocytes, endothelial cells, and fibroblasts enabled the assessment of VEGFR/platelet-derived growth factor receptor–targeting tyrosine kinase inhibitors (VPTKIs), capturing their combined toxicity on vascular integrity and myocardial function142.
While these studies have substantially advanced our understanding of acute and short-term cardiotoxic effects, it is increasingly recognized that clinically relevant cardiotoxicity is predominantly chronic and cumulative in nature143. Most existing in vitro assays evaluate drug responses over timescales of hours to days, whereas clinical cardiotoxic manifestations often emerge only after weeks or months of continuous exposure. This temporal mismatch represents a critical gap between preclinical testing and clinical observations in cardiotoxicity assessment. Accordingly, the establishment and systematic classification of long-term cardiotoxicity models are essential for improving the predictive accuracy and translational relevance. Extended culture of hiPSC-CM–based tissues within chip-based platforms enables continuous monitoring of progressive functional deterioration, including declining contractile force, impaired calcium handling, and increasing electrophysiological instability, which are hallmarks of chronic cardiomyopathy. Maintaining tissue stability and maturation over prolonged culture periods provides further insight into metabolic adaptation, mitochondrial stress, and fibrosis-like remodeling under sustained pharmacological exposure. Consequently, future cardiotoxicity assessment strategies should incorporate long-term dynamic culture conditions and repeated dosing paradigms to more faithfully reproduce chronic exposure scenarios and bridge the gap between experimental models and clinical outcomes.
Regenerative medicine and therapeutic potential of engineered cardiac tissues
Engineered cardiac tissues developed in vitro are increasingly being explored for regenerative medicine applications, particularly for the repair of the damaged myocardium following myocardial144,145,146. Among the most extensively investigated platforms, cardiac patches and cardiac organoids (COs) have demonstrated promising potential for transplantation-based therapies147,148. Cardiac patches are typically fabricated by combining hiPSC-CMs with biomaterial scaffolds such as fibrin, collagen, or synthetic polymers. Following transplantation into infarcted hearts, these constructs have shown partial restoration of contractile function and evidence of electrical coupling with the host myocardium149. COs, which self-organize into three-dimensional structures that recapitulate early cardiac development, have also been investigated as reparative grafts. For example, transplantation of hiPSC-derived COs into murine models of myocardial infarction has been shown to promote neovascularization and partial recovery of myocardial structure and function150. To further enhance post-transplantation outcomes, recent studies have emphasized prevascularization strategies, including co-culture with endothelial cells and the incorporation of proangiogenic factors, such as vascular endothelial growth factor (VEGF). These approaches accelerate anastomosis with the host vasculature, improve graft perfusion, and reduce ischemic necrosis after implantation151,152.
In parallel, advances in immune engineering have led to the development of hypoimmunogenic iPSC lines through the targeted deletion of human leukocyte antigen (HLA) class I and II genes and the overexpression of immunomodulatory molecules, such as programmed death-ligand 1 (PD-L1). These genetic modifications substantially reduce host immune rejection and prolong graft survival in allogeneic transplantation settings153,154. These developments highlight the significant translational potential of engineered cardiac tissues for myocardial repair. Nonetheless, critical challenges remain, including achieving durable graft integration, long-term electrical synchrony with the host myocardium, clinical-scale manufacturing, and regulatory feasibility, which are addressed in the following section.
Challenges
Technical limitations of current hiPSC-derived cardiac models
Despite substantial advances, current hiPSC-derived cardiac models remain insufficient for fully recapitulating the biological complexities of the adult human heart. A central limitation is the immaturity of hiPSC-derived cardiomyocytes (hiPSC-CMs), which retain fetal-like structural, electrophysiological, and metabolic characteristics rather than adult phenotypes75. Specifically, hiPSC-CMs lack mature features, including dense sarcomeric organization and transverse tubules. They also exhibit immature calcium handling and metabolic profiles that favor glycolysis over fatty acid oxidation, which are hallmarks of an early developmental state. This immaturity constrains contractile force generation, conduction velocity, and responsiveness to physiological stimuli, thereby limiting the model’s overall fidelity. Cellular heterogeneity and variability pose additional technical challenges. Even isogenic hiPSC lines derived from a single donor can display substantial line-to-line variability, resulting in inconsistent differentiation efficiency and functional readouts155. Moreover, experiments performed across different laboratories frequently yield divergent results because of variations in cell lines, differentiation protocols, and culture conditions. This interlaboratory and inter-donor variability undermines reproducibility and complicates cross-study comparisons156, posing a major obstacle to the standardization of hiPSC-CM–based platforms. Another key limitation is the incomplete recreation of the native cardiac microenvironment. Engineered cardiac constructs often rely on simplified or artificial matrices that fail to fully replicate the biochemical composition, structural hierarchy, and biomechanical properties of native extracellular matrix (ECM). Consequently, critical tissue-level features, including anisotropic fiber alignment, spatial stiffness gradients, and physiologically relevant cell–matrix signaling, are inadequately reproduced.
A particularly critical deficiency is the lack of functional vascularization in most human iPSC-derived cardiac tissues. In the absence of a perfusable capillary network, three-dimensional constructs are subject to diffusion limitations, with oxygen and nutrient transport restricted to a few hundred micrometers in the tissue. This limitation leads to hypoxic cores, metabolic stress, and cell death in thicker tissues157 and is widely recognized as the primary cause of necrotic core formation in cardiac organoids and engineered heart tissues. Although substantial efforts are underway to incorporate endothelial networks and angiogenic cues into cardiac organoids and heart-on-a-chip platforms, the generation of stable perfusable microvasculature remains a significant engineering challenge. Multicellular integration complicates model fidelity. Incorporating multiple cardiac cell types, including cardiomyocytes, fibroblasts, vascular cells, and immune cells, into a single construct is technically challenging, as these populations have distinct maturation timelines, metabolic requirements, and signaling dependencies. Consequently, one cell type may dominate the culture or fail to mature appropriately within the engineered niche. These microenvironmental and multicellular integration challenges often result in an electromechanical mismatch between engineered tissues and the adult myocardium.
Such mismatches have important functional consequences, particularly in translational and transplantation contexts. Tissue-engineered grafts frequently struggle to achieve robust electrical and mechanical coupling with the host myocardium, in part due to immature ion channel expression in hiPSC-CMs and mechanical incompatibility between engineered constructs and native cardiac tissue. Inadequate electrical integration can lead to conduction blocks or arrhythmogenesis, whereas mechanical mismatch arising from disparities in stiffness or force generation can impair effective force transmission. Several studies have demonstrated that immature hiPSC-CMs with limited coupling capacity can provoke arrhythmogenic events when introduced into adult cardiac tissue158. These technical limitations related to cellular maturity, tissue architecture, vascularization, and host integration underscore the need for continued bioengineering innovation to enhance the physiological fidelity, robustness, and translational utility of hiPSC-derived cardiac models.
Challenges in translational research and clinical applicability
Translating hiPSC-based cardiac models from the bench to bedside introduces a distinct set of translational challenges. One of the primary hurdles is scalability, the ability to produce cardiac cells or tissues at the volume, consistency, and quality required for industrial drug screening or therapeutic applications. The large-scale production of hiPSC-CMs with uniform quality remains difficult, as current differentiation protocols are labor-intensive and subject to substantial batch-to-batch variability159. Consequently, the routine generation of millions of mature, functionally consistent cardiomyocytes or large, clinically relevant tissue constructs for high-throughput screening or allogeneic cell therapy remains unachieved.
The scaling of engineered cardiac tissues presents additional challenges. Although miniature cardiac organoids or microtissues are relatively straightforward to culture and manipulate, they often lack adult-level structural and functional complexity. In contrast, larger engineered tissues more closely approximate the myocardial architecture but frequently suffer from limited viability, including core necrosis and insufficient integration of supporting cell populations. These trade-offs complicate efforts to balance physiological relevance and manufacturability.
Another major translational challenge is the inter-donor and inter-line variability inherent to hiPSC-based models. Because hiPSC lines retain the genetic background of their donors, phenotypic differences, such as baseline electrophysiological properties, maturation states, and drug responsiveness, can arise even among lines derived from healthy individuals. Such variability complicates the interpretation of disease phenotypes and pharmacological responses, as it can be difficult to distinguish genuine biological effects from line-specific idiosyncrasies. Although standardization of differentiation protocols and comprehensive phenotypic characterization of each hiPSC line can mitigate some of these issues, a degree of heterogeneity remains intrinsic to the system155.
Regulatory acceptance is an additional bottleneck for clinical translation. Although regulatory agencies have begun to recognize hiPSC-derived cardiac models as emerging New Approach Methodologies (NAMs), particularly in the context of cardiac safety pharmacology, clear regulatory standards and validation frameworks are still evolving. Recent analyses by U.S. The Food and Drug Administration (FDA) and the Center for Drug Evaluation and Research (CDER) indicate that although submissions incorporating hiPSC-CM–based cardiotoxicity data—often derived from multielectrode array electrophysiology assays—are increasing, their influence on regulatory decision-making has thus far been limited. In many cases, submitted studies lacked a clearly defined context of use or failed to convincingly link in vitro phenotypes with clinically meaningful risks, limiting their regulatory utility. These observations underscore the need for standardized performance metrics and systematic validation of hiPSC-based cardiac assays using well-characterized human clinical data.
Finally, bridging the gap between in vitro findings and human pathophysiology remains a challenge. Reductionist cardiac models, even when engineered with high fidelity, cannot fully capture the multi-organ and systemic complexities of cardiovascular disease. Neurohumoral regulation, immune system interactions, and circulatory feedback mechanisms are largely absent in isolated cardiac tissues, making it difficult to reproduce disease manifestations driven by systemic inflammation or neuroendocrine remodeling. In addition, the immature phenotype of hiPSC-CMs limits their ability to faithfully replicate adult disease states and long-term pharmacological response. Addressing these translational barriers will require continued advances in tissue maturation, multicellular complexity, and long-term culture stability, as well as the accumulation of large-scale datasets linking hiPSC-based readouts to the patient outcomes. Until such correlations are firmly established, caution is warranted when extrapolating in vitro results to clinical settings, and hiPSC-derived cardiac platforms are likely to serve as complementary—rather than replacement—tools alongside existing preclinical models in the near term.
Limitations in preclinical drug evaluation and safety testing
hiPSC-derived cardiac platforms play an increasingly important role in preclinical drug evaluation; however, clear limitations remain in the predictive power and scope of these assays. On the positive side, hiPSC-CMs have substantially improved the detection of certain cardiotoxic effects in vivo. For example, in the context of proarrhythmia risk assessment, multielectrode array (MEA) recordings from hiPSC-CM monolayers can reveal drug-induced alterations in repolarization and arrhythmic events, such as early afterdepolarizations, which may be overlooked in animal models. The incorporation of hiPSC-CM assays into safety pharmacology pipelines, most notably through the Comprehensive in vitro Proarrhythmia Assay (CiPA) initiative, has demonstrated more human-relevant predictions of QT prolongation and arrhythmia risk for selected compounds160. However, the reliability of these platforms is not uniform across all forms of cardiotoxicity. Their predictive performance is well established for compounds that directly perturb cardiac ion channels, such as hERG blockers that induce arrhythmias, but remains less validated for other mechanisms of cardiac injury. In particular, chronic structural cardiotoxicities, including anthracycline-induced cardiomyopathy, or subtle, long-term functional impairments are difficult to detect using current acute hiPSC-CM–based assays.
Most hiPSC-CM studies continue to focus on short-term endpoints, including acute changes in electrophysiology, contractility, and cell viability following drug exposure over hours to days after exposure. In contrast, clinically relevant cardiotoxicity often emerges only after weeks or months of treatment with these agents. Although modeling chronic drug responses in vitro is an active area of investigation, maintaining stable adult-like cardiomyocyte function during extended culture and repeated dosing remains technically challenging.
Several advanced three-dimensional (3D) models have begun to address this limitation by applying sustained pathological stress to induce chronic injuries. For instance, engineered heart tissues exposed to prolonged afterload or continuous neurohormonal stimulation, such as norepinephrine or transforming growth factor-β (TGF-β), exhibit progressive contractile decline, prolonged calcium transients, and arrhythmic bursts, which are characteristic of heart failure and maladaptive remodeling. While these studies demonstrate that chronic disease phenotypes can be recapitulated in vitro, such approaches are not yet routinely adopted in drug testing because of their experimental complexity and limited throughput. Consequently, reliable prediction of long-term drug effects, including fibrosis, hypertrophy, and cumulative cardiotoxicity, remains a significant challenge in hiPSC-derived systems.
Another important limitation of preclinical applications is the incomplete integration of pharmacokinetic (PK) and multi-organ factors. In vivo, cardiac drug responses are shaped by metabolism, systemic distribution, and off-target effects in the organs. To address this, multi-organ microphysiological systems (MPSs) have been developed, such as heart–liver co-culture platforms that combine hiPSC-derived cardiac tissues with liver organoids or liver-on-a-chip modules, to account for drug bioactivation and clearance. These systems can uncover cardiotoxic effects mediated by metabolites that would be missed in heart-only models. Nevertheless, constructing robust multi-organ platforms presents practical challenges, including the need to optimize tissue-specific media, match physiologically relevant flow rates and scale across organs, and enable inter-organ communication that accurately reflects human physiology. At present, no universally standardized “body-on-a-chip” platform exists, and many multi-organ systems remain bespoke, limiting their reproducibility and widespread adoption. Consequently, the routine integration of PK/pharmacodynamic considerations into in vitro cardiotoxicity testing remains uncommon.
Beyond the biological and engineering limitations, computational integration has emerged as an important complementary dimension in preclinical cardiac safety assessment. Recent advances in artificial intelligence (AI) and machine learning (ML) enable multimodal fusion of imaging, electrophysiology, calcium dynamics, and transcriptomic data to train predictive models for cardiotoxicity risk and phenotype classification82,158,161. In particular, self-supervised and transformer-based architectures have demonstrated superior performance over traditional ML approaches in detecting subtle morphological, electrophysiological, and conduction abnormalities in hiPSC-CM systems. These computational strategies complement organoid and microphysiological platforms by supporting automated screening, early detection of drug-induced risk signatures, and patient-specific risk stratification in drug-induced cardiotoxicity. Although widespread adoption will require standardized data pipelines and benchmarking frameworks, AI-enhanced cardiac platforms represent a promising avenue for improving predictive accuracy and reducing the reliance on animal models.
In parallel with advances in biological fidelity and computational sophistication, regulatory alignment remains essential for broader clinical and industrial applications. Recent guidance from regulatory agencies, including the U.S. FDA and the European Medicines Agency (EMA) emphasize the qualification of human-relevant NAMs and the integration of hiPSC-based assays and MPSs into cardiac safety workflows. Initiatives such as CiPA and the revised ICH S7B/E14 Q&A framework highlight the importance of standardized electrophysiological validation, reproducible functional benchmarks, and demonstrated clinical concordance for regulatory acceptance in investigational new drug (IND) submissions. Accordingly, future progress will depend not only on continued bioengineering and computational innovation but also on the development of harmonized validation pipelines, reference compound sets, and cross-laboratory reproducibility standards to support regulatory qualification and scalable deployment of these technologies.
Conclusions and future perspectives
Human induced pluripotent stem cell (hiPSC)-derived in vitro cardiac systems represent a major advance in nonclinical drug evaluation and regenerative medicine. Given the intrinsic structure–function dependency of the heart, physiologically accurate modeling is essential for developing predictive and reliable platforms. The ability of hiPSC-based models to recapitulate human cardiac electrophysiology, contractility, and pharmacological responsiveness underscores their value as next-generation tools that address the key limitations of animal studies and immortalized cell lines. Recent advances in three-dimensional cardiac tissue engineering, including engineered heart tissues, chamber-like organoids, and heart-on-a-chip systems, have substantially enhanced the biological fidelity of these models. These constructs support long-term culture, multicellular integration, and biomechanical conditioning, enabling more accurate modeling of chronic drug responses and patient-specific phenotypes compared to conventional approaches. In safety pharmacology, hiPSC-based assays have been incorporated into the Comprehensive in vitro Proarrhythmia Assay (CiPA) framework and are increasingly recognized as Drug Development Tools by regulatory agencies.
Beyond preclinical testing, hiPSC-derived cardiac tissues are progressing toward clinical applications. Preclinical studies using engineered cardiac patches and myocardial grafts have demonstrated partial remuscularization and functional recovery in animal models of myocardial infarction, and early clinical trials of allogeneic hiPSC-based cardiac products have reported encouraging safety profiles. Together, these findings highlight the therapeutic potential of engineered cardiac tissues in patients with advanced cardiovascular diseases. Nevertheless, important challenges remain, including incomplete cardiomyocyte maturation, limited vascularization, inter-line variability, and a lack of standardized manufacturing and validation pipelines. Ongoing interdisciplinary efforts spanning biofabrication, bioreactor engineering, immune modulation, and AI-driven data integration actively address these limitations.
Looking forward, the continued integration of advanced computational and systems-level approaches with biologically engineered cardiac platforms will be essential to accelerate clinical translation. The convergence of AI-enabled multimodal analysis, large-scale biobanks, and patient-specific genomic information is expected to enhance predictive cardiotoxicity assessments and precision drug screening. In parallel, multi-organ and vascularized tissue-chip circuits will enable the modeling of systemic interactions, such as cardio-hepatic drug metabolism, immune activation, and neurohumoral regulation, thereby improving translational robustness beyond single-organ systems.
Emerging directions include: (i) multimodal deep-learning frameworks that integrate imaging, electrophysiology, calcium dynamics, and transcriptomics to detect subtle structural and functional phenotypes; (ii) generative and self-supervised AI platforms for phenotype discovery and drug-response prediction; (iii) standardized data pipelines and cloud-native cardiac data repositories to support reproducible AI benchmarking; (iv) patient-matched hiPSC models combined with gene editing and disease-specific stratification for personalized medicine; and (v) scalable Good Manufacturing Practice (GMP) workflows for clinical-grade cardiac tissues and future autologous and allogeneic cell therapies. The integration of bioengineering, stem cell biology, computational modeling, and regulatory science is establishing a next-generation ecosystem for cardiac drug development and regenerative therapies. As these technologies continue to mature, hiPSC-derived cardiac platforms are poised to complement and ultimately transform conventional nonclinical testing paradigms, ushering in an era of human-relevant, precision-engineered, and clinically actionable cardiovascular innovation.
References
Roth, G. A. et al. Global burden of cardiovascular diseases and risk factors, 1990–2019: update from the GBD 2019 study. J. Am. Coll. Cardiol. 76, 2982–3021 (2020).
Waring, M. J. et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 14, 475–486 (2015).
Hay, M., Thomas, D. W., Craighead, J. L., Economides, C. & Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 32, 40–51 (2014).
Denning, C. et al. Cardiomyocytes from human pluripotent stem cells: from laboratory curiosity to industrial biomedical platform. Biochim. Biophys. Acta Mol. Cell Res. 1863, 1728–1748 (2016).
Olson, H. et al. Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul. Toxicol. Pharmacol. 32, 56–67 (2000).
Fermini, B. et al. A new perspective in the field of cardiac safety testing through the comprehensive in vitro proarrhythmia assay paradigm. J. Biomol. Screen. 21, 1–11 (2016).
Gintant, G., Sager, P. T. & Stockbridge, N. Evolution of strategies to improve preclinical cardiac safety testing. Nat. Rev. Drug Discov. 15, 457–471 (2016).
Laverty, H. et al. How can we improve our understanding of cardiovascular safety liabilities to develop safer medicines? Br. J. Pharmacol. 163, 675–693 (2011).
Sharma, A., Wu, J. C. & Wu, S. M. Induced pluripotent stem cell-derived cardiomyocytes for cardiovascular disease modeling and drug screening. Stem Cell Res. Ther. 4, 150 (2013).
Karakikes, I., Ameen, M., Termglinchan, V. & Wu, J. C. Human induced pluripotent stem cell–derived cardiomyocytes: insights into molecular, cellular, and functional phenotypes. Circ. Res. 117, 80–88 (2015).
Lian, X. et al. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat. Protoc. 8, 162–175 (2013).
Burridge, P. W., Holmström, A. & Wu, J. C. Chemically defined culture and cardiomyocyte differentiation of human pluripotent stem cells. Curr. Protoc. Hum. Genet. 87, 21.3.1-21.3.15 (2015).
Sala, L., Bellin, M. & Mummery, C. L. Integrating cardiomyocytes from human pluripotent stem cells in safety pharmacology: Has the time come? Br. J. Pharmacol. 174, 3749–3765 (2017).
Mathur, A. et al. Human iPSC-based cardiac microphysiological system for drug screening applications. Sci. Rep. 5, 8883 (2015).
McCain, M. L., Yuan, H., Pasqualini, F. S., Campbell, P. H. & Parker, K. K. Matrix elasticity regulates the optimal cardiac myocyte shape for contractility. Am. J. Physiol. Heart Circ. Physiol. 306, H1525–H1539 (2014).
Bray, M. A., Sheehy, S. P. & Parker, K. K. Sarcomere alignment is regulated by myocyte shape. Cell Motil. Cytoskelet. 65, 641–651 (2008).
McCain, M. L., Lee, H., Aratyn-Schaus, Y., Kléber, A. G. & Parker, K. K. Cooperative coupling of cell-matrix and cell–cell adhesions in cardiac muscle. Proc. Natl. Acad. Sci. USA 109, 9881–9886 (2012).
McCain, M. L., Agarwal, A., Nesmith, H. W., Nesmith, A. P. & Parker, K. K. Micromolded gelatin hydrogels for extended culture of engineered cardiac tissues. Biomaterials 35, 5462–5471 (2014).
Park, S.-J. et al. Insights into the pathogenesis of catecholaminergic polymorphic ventricular tachycardia from engineered human heart tissue. Circulation 140, 390–404 (2019).
Lind, J. U. et al. Instrumented cardiac microphysiological devices via multimaterial three-dimensional printing. Nat. Mater. 16, 303–308 (2017).
Batalov, I., Jallerat, Q., Kim, S., Bliley, J. & Feinberg, A. W. Engineering aligned human cardiac muscle using developmentally inspired fibronectin micropatterns. Sci. Rep. 11, 11502 (2021).
Lee, K. Y. et al. An autonomously swimming biohybrid fish designed with human cardiac biophysics. Science 375, 639–647 (2022).
Kim, S. L. et al. Spatiotemporal cell junction assembly in human iPSC-CM models of arrhythmogenic cardiomyopathy. Stem Cell Rep. 18, 1811–1826 (2023).
Colatsky, T. et al. The comprehensive in vitro proarrhythmia assay (CiPA) initiative—update on progress. J. Pharmacol. Toxicol. Methods 81, 15–20 (2016).
Burridge, P. W. et al. Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat. Med. 22, 547–556 (2016).
Huang, G. et al. Titin-truncating variants in hiPSC cardiomyocytes induce pathogenic proteinopathy and sarcomere defects with preserved core contractile machinery. Stem Cell Rep. 18, 220–236 (2023).
Ketabat, F., Alcorn, J., Kelly, M. E., Badea, I. & Chen, X. Cardiac tissue engineering: a journey from scaffold fabrication to in vitro characterization. Small Sci. 4, 2400079 (2024).
Zimmermann, W.-H. et al. Tissue engineering of a differentiated cardiac muscle construct. Circ. Res. 90, 223–230 (2002).
Ronaldson-Bouchard, K. et al. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 556, 239–243 (2018).
Zhao, Y. et al. A platform for generation of chamber-specific cardiac tissues and disease modeling. Cell 176, 913–927.e918 (2019).
Querdel, E. et al. Human engineered heart tissue patches remuscularize the injured heart in a dose-dependent manner. Circulation 143, 1991–2006 (2021).
Pretorius, D., Kahn-Krell, A. M., LaBarge, W. C., Lou, X. & Zhang, J. Engineering of thick human functional myocardium via static stretching and electrical stimulation. Iscience 25, 103824 (2022).
Jebran, A.-F. et al. Engineered heart muscle allografts for heart repair in primates and humans. Nature 639, 503–511 (2025).
Lock, R. I. et al. Macrophages enhance contractile force in iPSC-derived human engineered cardiac tissue. Cell Rep. 43, 114302 (2024).
Li, H. et al. In vitro vascularization improves in vivo functionality of human engineered cardiac tissues. Acta Biomater. 211, 61–73 (2026).
Lee, E. J., Kim, D. E., Azeloglu, E. U. & Costa, K. D. Engineered cardiac organoid chambers: toward a functional biological model ventricle. Tissue Eng. Part A 14, 215–225 (2008).
Li, R. A. et al. Bioengineering an electro-mechanically functional miniature ventricular heart chamber from human pluripotent stem cells. Biomaterials 163, 116–127 (2018).
MacQueen, L. A. et al. A tissue-engineered scale model of the heart ventricle. Nat. Biomed. Eng. 2, 930–941 (2018).
Lee, A. et al. 3D bioprinting of collagen to rebuild components of the human heart. Science 365, 482–487 (2019).
Choi, S. et al. Fibre-infused gel scaffolds guide cardiomyocyte alignment in 3D-printed ventricles. Nat. Mater. 22, 1039–1046 (2023).
Kupfer, M. E. et al. In situ expansion, differentiation, and electromechanical coupling of human cardiac muscle in a 3D bioprinted, chambered organoid. Circ. Res. 127, 207–224 (2020).
Chang, H. et al. Recreating the heart’s helical structure-function relationship with focused rotary jet spinning. Science 377, 180–185 (2022).
Kuckelkorn, C. et al. Engineered in vitro multi-cell type ventricle model generates long-term pulsatile flow and modulates cardiac output in response to cardioactive drugs. Adv. Healthc. Mater. 14, 2403897 (2025).
Lancaster, M. A. & Knoblich, J. A. Organogenesis in a dish: modeling development and disease using organoid technologies. Science 345, 1247125 (2014).
Kim, H., Kamm, R. D., Vunjak-Novakovic, G. & Wu, J. C. Progress in multicellular human cardiac organoids for clinical applications. Cell Stem Cell 29, 503–514 (2022).
Drakhlis, L. et al. Human heart-forming organoids recapitulate early heart and foregut development. Nat. Biotechnol. 39, 737–746 (2021).
Hofbauer, P. et al. Cardioids reveal self-organizing principles of human cardiogenesis. Cell 184, 3299–3317.e3222 (2021).
Lee, S.-G. et al. Generation of human iPSCs derived heart organoids structurally and functionally similar to heart. Biomaterials 290, 121860 (2022).
Olmsted, Z. T. & Paluh, J. L. A combined human gastruloid model of cardiogenesis and neurogenesis. Iscience 25, 104486 (2022).
Meier, A. B. et al. Epicardioid single-cell genomics uncovers principles of human epicardium biology in heart development and disease. Nat. Biotechnol. 41, 1787–1800 (2023).
Schmidt, C. et al. Multi-chamber cardioids unravel human heart development and cardiac defects. Cell 186, 5587–5605.e5527 (2023).
Dardano, M. et al. Blood-generating heart-forming organoids recapitulate co-development of the human haematopoietic system and the embryonic heart. Nat. Cell Biol. 26, 1984–1996 (2024).
Rossi, G. et al. Capturing cardiogenesis in gastruloids. Cell Stem Cell 28, 230–240.e236 (2021).
Silva, A. C. et al. Co-emergence of cardiac and gut tissues promotes cardiomyocyte maturation within human iPSC-derived organoids. Cell Stem Cell 28, 2137–2152.e2136 (2021).
Abilez, O. J. et al. Gastruloids enable modeling of the earliest stages of human cardiac and hepatic vascularization. Science 388, eadu9375 (2025).
Wang, K. et al. Microphysiological systems: design, fabrication, and applications. ACS Biomater. Sci. Eng. 6, 3231–3257 (2020).
Butler, D. & Reyes, D. R. Heart-on-a-chip systems: disease modeling and drug screening applications. Lab Chip 24, 1494–1528 (2024).
Skardal, A., Shupe, T. & Atala, A. Organoid-on-a-chip and body-on-a-chip systems for drug screening and disease modeling. Drug Discov. Today 21, 1399–1411 (2016).
Ewart, L. et al. Navigating tissue chips from development to dissemination: a pharmaceutical industry perspective. Exp. Biol. Med. 242, 1579–1585 (2017).
Sung, J. H. et al. Recent advances in body-on-a-chip systems. Anal. Chem. 91, 330–351 (2018).
Agarwal, A., Goss, J. A., Cho, A., McCain, M. L. & Parker, K. K. Microfluidic heart on a chip for higher throughput pharmacological studies. Lab Chip 13, 3599–3608 (2013).
Marsano, A. et al. Beating heart on a chip: a novel microfluidic platform to generate functional 3D cardiac microtissues. Lab Chip 16, 599–610 (2016).
Huebsch, N. et al. Metabolically driven maturation of human-induced-pluripotent-stem-cell-derived cardiac microtissues on microfluidic chips. Nat. Biomed. Eng. 6, 372–388 (2022).
Jayne, R. K. et al. Direct laser writing for cardiac tissue engineering: a microfluidic heart on a chip with integrated transducers. Lab Chip 21, 1724–1737 (2021).
Rexius-Hall, M. L. et al. A myocardial infarct border-zone-on-a-chip demonstrates distinct regulation of cardiac tissue function by an oxygen gradient. Sci. Adv. 8, eabn7097 (2022).
Lu, R. X. Z. et al. Cardiac tissue model of immune-induced dysfunction reveals the role of free mitochondrial DNA and the therapeutic effects of exosomes. Sci. Adv. 10, eadk0164 (2024).
Landau, S. et al. Primitive macrophages enable long-term vascularization of human heart-on-a-chip platforms. Cell Stem Cell 31, 1222–1238.e1210 (2024).
Liu, Y. et al. Human heart-on-a-chip microphysiological system comprising endothelial cells, fibroblasts, and iPSC-derived cardiomyocytes. Sci. Rep. 14, 18063 (2024).
Loskill, P., Marcus, S. G., Mathur, A., Reese, W. M. & Healy, K. E. μOrgano: a Lego®-like plug & play system for modular multi-organ-chips. PLoS ONE 10, e0139587 (2015).
Oleaga, C. et al. Investigation of the effect of hepatic metabolism on off-target cardiotoxicity in a multi-organ human-on-a-chip system. Biomaterials 182, 176–190 (2018).
Rajan, S. A. P. et al. Probing prodrug metabolism and reciprocal toxicity with an integrated and humanized multi-tissue organ-on-a-chip platform. Acta Biomater. 106, 124–135 (2020).
Skardal, A. et al. Drug compound screening in single and integrated multi-organoid body-on-a-chip systems. Biofabrication 12, 025017 (2020).
Yin, F. et al. HiPSC-derived multi-organoids-on-chip system for safety assessment of antidepressant drugs. Lab Chip 21, 571–581 (2021).
Ronaldson-Bouchard, K. et al. A multi-organ chip with matured tissue niches linked by vascular flow. Nat. Biomed. Eng. 6, 351–371 (2022).
Karbassi, E. et al. Cardiomyocyte maturation: advances in knowledge and implications for regenerative medicine. Nat. Rev. Cardiol. 17, 341–359 (2020).
Bers, D. M. Cardiac excitation–contraction coupling. Nature 415, 198–205 (2002).
Litviňuková, M. et al. Cells of the adult human heart. Nature 588, 466–472 (2020).
Tucker, N. R. et al. Transcriptional and cellular diversity of the human heart. Circulation 142, 466–482 (2020).
Miranda, A. M. et al. Single-cell transcriptomics for the assessment of cardiac disease. Nat. Rev. Cardiol. 20, 289–308 (2023).
Farah, E. N. et al. Spatially organized cellular communities form the developing human heart. Nature 627, 854–864 (2024).
Bornstein, M. R., Tian, R. & Arany, Z. Human cardiac metabolism. Cell Metab. 36, 1456–1481 (2024).
Grafton, F. et al. Deep learning detects cardiotoxicity in a high-content screen with induced pluripotent stem cell-derived cardiomyocytes. eLife 10, e68714 (2021).
Zimmerman, J. F. et al. Bioinspired design of a tissue-engineered ray with machine learning. Sci. Robot. 10, eadr6472 (2025).
Botti, S., Krause, R. & Pavarino, L. F. In silico modelling of multi-electrode arrays for enhancing cardiac drug testing on hiPSC-CM heterogeneous tissues. J. Physiol. https://doi.org/10.1113/JP287276 (2025).
Li, Y. et al. The molecular mechanisms of cardiac development and related diseases. Signal Transduct. Target. Ther. 9, 368 (2024).
Singh, B. N. et al. Proliferation and maturation: janus and the art of cardiac tissue engineering. Circ. Res. 132, 519–540 (2023).
Ni, B., Ye, L., Zhang, Y., Hu, S. & Lei, W. Advances in humanoid organoid-based research on inter-organ communications during cardiac organogenesis and cardiovascular diseases. J. Transl. Med. 23, 380 (2025).
Lewis-Israeli, Y. R. et al. Self-assembling human heart organoids for the modeling of cardiac development and congenital heart disease. Nat. Commun. 12, 5142 (2021).
Li, W. et al. Comprehensive promotion of iPSC-CM maturation by integrating metabolic medium with nanopatterning and electrostimulation. Nat. Commun. 16, 2785 (2025).
Giacomelli, E. et al. Human-iPSC-derived cardiac stromal cells enhance maturation in 3D cardiac microtissues and reveal non-cardiomyocyte contributions to heart disease. Cell Stem Cell 26, 862–879.e811 (2020).
Takahashi, K. et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007).
Musunuru, K. et al. Induced pluripotent stem cells for cardiovascular disease modeling and precision medicine: a scientific statement from the American Heart Association. Circ. Genom. Precis. Med. 11, e000043 (2018).
Wang, G. et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med. 20, 616–623 (2014).
Moretti, A. et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N. Engl. J. Med. 363, 1397–1409 (2010).
Giacomelli, E., Sala, L., Ward-van Oostwaard, D. & Bellin, M. Cardiac microtissues from human pluripotent stem cells recapitulate the phenotype of long-QT syndrome. Biochem. Biophys. Res. Commun. 572, 118–124 (2021).
Chang, Y. et al. hERG-deficient human embryonic stem cell-derived cardiomyocytes for modelling QT prolongation. Stem Cell Res. Ther. 12, 278 (2021).
Goldfracht, I. et al. Engineered heart tissue models from hiPSC-derived cardiomyocytes and cardiac ECM for disease modeling and drug testing applications. Acta Biomater. 92, 145–159 (2019).
Veldhuizen, J. et al. Modeling long QT syndrome type 2 on-a-chip via in-depth assessment of isogenic gene-edited 3D cardiac tissues. Sci. Adv. 8, eabq6720 (2022).
McKeithan, W. L. et al. Reengineering an antiarrhythmic drug using patient hiPSC cardiomyocytes to improve therapeutic potential and reduce toxicity. Cell Stem Cell 27, 813–821.e816 (2020).
Malan, D. et al. Human iPS cell model of type 3 long QT syndrome recapitulates drug-based phenotype correction. Basic Res. Cardiol. 111, 14 (2016).
Ma, D. et al. Modeling type 3 long QT syndrome with cardiomyocytes derived from patient-specific induced pluripotent stem cells. Int. J. Cardiol. 168, 5277–5286 (2013).
Kuroda, Y. et al. Flecainide ameliorates arrhythmogenicity through NCX flux in Andersen-Tawil syndrome-iPS cell-derived cardiomyocytes. Biochem. Biophys. Rep. 9, 245–256 (2017).
Yazawa, M. et al. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature 471, 230–234 (2011).
Rocchetti, M. et al. Elucidating arrhythmogenic mechanisms of long-QT syndrome CALM1-F142L mutation in patient-specific induced pluripotent stem cell-derived cardiomyocytes. Cardiovasc. Res. 113, 531–541 (2017).
Limpitikul, W. B. et al. A precision medicine approach to the rescue of function on malignant calmodulinopathic long-QT syndrome. Circ. Res. 120, 39–48 (2017).
Wren, L. M. et al. Genetic mosaicism in calmodulinopathy. Circ. Genom. Precis. Med. 12, e002581 (2019).
Clemens, D. J. et al. Cellular and electrophysiological characterization of triadin knockout syndrome using induced pluripotent stem cell-derived cardiomyocytes. Stem Cell Rep. 18, 1075–1089 (2023).
El-Battrawy, I. et al. Modeling short QT syndrome using human-induced pluripotent stem cell–derived cardiomyocytes. J. Am. Heart Assoc. 7, e007394 (2018).
Lodola, F. et al. Adeno-associated virus-mediated CASQ2 delivery rescues phenotypic alterations in a patient-specific model of recessive catecholaminergic polymorphic ventricular tachycardia. Cell Death Dis. 7, e2393–e2393 (2016).
Liang, P. et al. Patient-specific and genome-edited induced pluripotent stem cell–derived cardiomyocytes elucidate single-cell phenotype of Brugada syndrome. J. Am. Coll. Cardiol. 68, 2086–2096 (2016).
Cohn, R. et al. A contraction stress model of hypertrophic cardiomyopathy due to sarcomere mutations. Stem Cell Rep. 12, 71–83 (2019).
Flenner, F. et al. Translational investigation of electrophysiology in hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 157, 77–89 (2021).
Cashman, T. J., Josowitz, R., Johnson, B. V., Gelb, B. D. & Costa, K. D. Human engineered cardiac tissues created using induced pluripotent stem cells reveal functional characteristics of BRAF-mediated hypertrophic cardiomyopathy. PLoS ONE 11, e0146697 (2016).
Dai, Y. et al. Troponin destabilization impairs sarcomere-cytoskeleton interactions in iPSC-derived cardiomyocytes from dilated cardiomyopathy patients. Sci. Rep. 10, 209 (2020).
Pettinato, A. M. et al. Development of a cardiac sarcomere functional genomics platform to enable scalable interrogation of human TNNT2 variants. Circulation 142, 2262–2275 (2020).
Hinson, J. T. et al. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 349, 982–986 (2015).
Deacon, D. C. et al. Combinatorial interactions of genetic variants in human cardiomyopathy. Nat. Biomed. Eng. 3, 147–157 (2019).
Feyen, D. A. et al. Unfolded protein response as a compensatory mechanism and potential therapeutic target in PLN R14del cardiomyopathy. Circulation 144, 382–392 (2021).
Streckfuss-Bömeke, K. et al. Severe DCM phenotype of patient harboring RBM20 mutation S635A can be modeled by patient-specific induced pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell. Cardiol. 113, 9–21 (2017).
Kyrychenko, V. et al. Functional correction of dystrophin actin binding domain mutations by genome editing. JCI insight 2, e95918 (2017).
Guan, X. et al. Dystrophin-deficient cardiomyocytes derived from human urine: new biologic reagents for drug discovery. Stem Cell Res. 12, 467–480 (2014).
Long, C. et al. Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing. Sci. Adv. 4, eaap9004 (2018).
Marini, V. et al. Long-term culture of patient-derived cardiac organoids recapitulated Duchenne muscular dystrophy cardiomyopathy and disease progression. Front. Cell Dev. Biol. 10, 878311 (2022).
Kim, C. et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature 494, 105–110 (2013).
Blazeski, A. et al. Engineered heart slice model of arrhythmogenic cardiomyopathy using plakophilin-2 mutant myocytes. Tissue Eng. Part A 25, 725–735 (2019).
Bliley, J. M. et al. Dynamic loading of human engineered heart tissue enhances contractile function and drives a desmosome-linked disease phenotype. Sci. Transl. Med. 13, eabd1817 (2021).
Wang, B. Z. et al. Engineered cardiac tissue model of restrictive cardiomyopathy for drug discovery. Cell Rep. Med. 4, 100976 (2023).
Feng, W. et al. Computational profiling of hiPSC-derived heart organoids reveals chamber defects associated with NKX2-5 deficiency. Commun. Biol. 5, 399 (2022).
Birket, M. J. et al. A human stem cell model of Fabry disease implicates LIMP-2 accumulation in cardiomyocyte pathology. Stem Cell Rep. 13, 380–393 (2019).
Hashem, S. I. et al. Brief report: Oxidative stress mediates cardiomyocyte apoptosis in a human model of Danon disease and heart failure. Stem Cells 33, 2343–2350 (2015).
Barndt, R. J. et al. Metabolic maturation exaggerates abnormal calcium handling in a Lamp2 knockout human pluripotent stem cell-derived cardiomyocyte model of Danon Disease. Biomolecules 13, 69 (2022).
Raval, K. K. et al. Pompe disease results in a Golgi-based glycosylation deficit in human induced pluripotent stem cell-derived cardiomyocytes. J. Biol. Chem. 290, 3121–3136 (2015).
Richards, D. J. et al. Human cardiac organoids for the modelling of myocardial infarction and drug cardiotoxicity. Nat. Biomed. Eng. 4, 446–462 (2020).
Song, M. et al. Modeling acute myocardial infarction and cardiac fibrosis using human induced pluripotent stem cell-derived multi-cellular heart organoids. Cell Death Dis. 15, 308 (2024).
Sebastião, M. J. et al. Bioreactor-based 3D human myocardial ischemia/reperfusion in vitro model: a novel tool to unveil key paracrine factors upon acute myocardial infarction. Transl. Res. 215, 57–74 (2020).
Veldhuizen, J. et al. Cardiac ischemia on-a-chip to investigate cellular and molecular response of myocardial tissue under hypoxia. Biomaterials 281, 121336 (2022).
Tiburcy, M. et al. Defined engineered human myocardium with advanced maturation for applications in heart failure modeling and repair. Circulation 135, 1832–1847 (2017).
Lemme, M. et al. Chronic intermittent tachypacing by an optogenetic approach induces arrhythmia vulnerability in human engineered heart tissue. Cardiovasc. Res. 116, 1487–1499 (2020).
Drawnel, F. M. et al. Disease modeling and phenotypic drug screening for diabetic cardiomyopathy using human induced pluripotent stem cells. Cell Rep. 9, 810–820 (2014).
Patel, C. N. et al. Strategies for redesigning withdrawn drugs to enhance therapeutic efficacy and safety: a review. Wiley Interdiscip. Rev. Comput. Mol. Sci. 15, e70004 (2025).
Di Cio, S., Marhuenda, E., Haddrick, M. & Gautrot, J. E. Vascularised cardiac spheroids-on-a-chip for testing the toxicity of therapeutics. Sci. Rep. 14, 3370 (2024).
Mozneb, M. et al. Multi-lineage heart-chip models drug cardiotoxicity and enhances maturation of human stem cell-derived cardiovascular cells. Lab Chip 24, 869–881 (2024).
Pabel, S. et al. Long-term effects of empagliflozin on excitation-contraction-coupling in human induced pluripotent stem cell cardiomyocytes. J. Mol. Med. 98, 1689–1700 (2020).
Hashimoto, H., Olson, E. N. & Bassel-Duby, R. Therapeutic approaches for cardiac regeneration and repair. Nat. Rev. Cardiol. 15, 585–600 (2018).
Coulombe, K. L., Bajpai, V. K., Andreadis, S. T. & Murry, C. E. Heart regeneration with engineered myocardial tissue. Annu. Rev. Biomed. Eng. 16, 1–28 (2014).
Chien, K. R. et al. Regenerating the field of cardiovascular cell therapy. Nat. Biotechnol. 37, 232–237 (2019).
Riegler, J., Gillich, A., Shen, Q., Gold, J. D. & Wu, J. C. Cardiac tissue slice transplantation as a model to assess tissue-engineered graft thickness, survival, and function. Circulation 130, S77–S86 (2014).
Riegler, J. et al. Human engineered heart muscles engraft and survive long term in a rodent myocardial infarction model. Circ. Res. 117, 720–730 (2015).
Huethorst, E. et al. Evidence for intermittent coupling of intramyocardial small, engineered heart tissues acutely implanted into rabbit myocardium. Cardiovasc. Res. 121, 1697–1711 (2025).
Silver, S. E. et al. Hypoimmunogenic hPSC-derived cardiac organoids for immune evasion and heart repair. Preprint at bioRxiv https://doi.org/10.1101/2025.04.09.648007 (2025).
Gao, L. et al. Myocardial tissue engineering with cells derived from human-induced pluripotent stem cells and a native-like, high-resolution, 3-dimensionally printed scaffold. Circ. Res. 120, 1318–1325 (2017).
Zhang, D. et al. Tissue-engineered cardiac patch for advanced functional maturation of human ESC-derived cardiomyocytes. Biomaterials 34, 5813–5820 (2013).
Han, X. et al. Generation of hypoimmunogenic human pluripotent stem cells. Proc. Natl. Acad. Sci. USA 116, 10441–10446 (2019).
Tsuneyoshi, N. et al. Hypoimmunogenic human iPSCs expressing HLA-G, PD-L1, and PD-L2 evade innate and adaptive immunity. Stem Cell Res. Ther. 15, 193 (2024).
Tereshchenko, Y., Petkov, S. G. & Behr, R. The efficiency of in vitro differentiation of primate iPSCs into cardiomyocytes depending on their cell seeding density and cell line specificity. Int. J. Mol. Sci. 25, 8449 (2024).
Ismaili, D. et al. Human induced pluripotent stem cell-derived cardiomyocytes as an electrophysiological model: Opportunities and challenges—the Hamburg perspective. Front. Physiol. 14, 1132165 (2023).
Grebenyuk, S. et al. Large-scale perfused tissues via synthetic 3D soft microfluidics. Nat. Commun. 14, 193 (2023).
Juhola, M., Penttinen, K., Joutsijoki, H. & Aalto-Setälä, K. Analysis of drug effects on iPSC cardiomyocytes with machine learning. Ann. Biomed. Eng. 49, 129–138 (2021).
Yang, H., Yang, Y., Kiskin, F. N., Shen, M. & Zhang, J. Z. Recent advances in regulating the proliferation or maturation of human-induced pluripotent stem cell-derived cardiomyocytes. Stem Cell Res. Ther. 14, 228 (2023).
Konala, V. B. R. et al. CiPA-qualified human iPSC-derived cardiomyocytes: A new frontier in toxicity testing by evaluating drug-induced arrhythmias. Toxicol. In Vitro 108, 106100 (2025).
Kowalczewski, A. et al. Integrating nonlinear analysis and machine learning for human induced pluripotent stem cell-based drug cardiotoxicity testing. J. Tissue Eng. Regen. Med. 16, 732–743 (2022).
Gerdes, A. M. et al. Structural remodeling of cardiac myocytes in patients with ischemic cardiomyopathy. Circulation 86, 426–430 (1992).
Ball, A. J. & Levine, F. Telomere-independent cellular senescence in human fetal cardiomyocytes. Aging Cell 4, 21–30 (2005).
Shih, H.-T. Anatomy of the action potential in the heart. Tex. Heart Inst. J. 21, 30 (1994).
Vigmond, E. J., Massé, S., Roney, C. H., Bayer, J. D. & Nanthakumar, K. The accuracy of cardiac surface conduction velocity measurements. Clin. Electrophysiol. 11, 694–705 (2025).
Lu, X. et al. Measuring local gradients of intramitochondrial [Ca2+] in cardiac myocytes during sarcoplasmic reticulum Ca2+ release. Circ. Res. 112, 424–431 (2013).
Pioner, J. M. et al. Optical investigation of action potential and calcium handling maturation of hiPSC-cardiomyocytes on biomimetic substrates. Int. J. Mol. Sci. 20, 3799 (2019).
Hwang, H. S. et al. Comparable calcium handling of human iPSC-derived cardiomyocytes generated by multiple laboratories. J. Mol. Cell. Cardiol. 85, 79–88 (2015).
Alvarez, J. A. E., Jafri, M. S. & Ullah, A. Local control model of a human ventricular myocyte: an exploration of frequency-dependent changes and calcium sparks. Biomolecules 13, 1259 (2023).
Hasenfuss, G. et al. Energetics of isometric force development in control and volume-overload human myocardium. Comparison with animal species. Circ. Res. 68, 836–846 (1991).
King, J. & Lowery, D. R. Physiology, Cardiac Output (StatPearls, 2017).
Lopaschuk, G. D., Spafford, M. A. & Marsh, D. R. Glycolysis is predominant source of myocardial ATP production immediately after birth. Am. J. Physiol. Heart Circ. Physiol. 261, H1698–H1705 (1991).
Scuderi, G. J. & Butcher, J. Naturally engineered maturation of cardiomyocytes. Front. Cell Dev. Biol. 5, 50 (2017).
Jacot, J. G., McCulloch, A. D. & Omens, J. H. Substrate stiffness affects the functional maturation of neonatal rat ventricular myocytes. Biophys. J. 95, 3479–3487 (2008).
Bhana, B. et al. Influence of substrate stiffness on the phenotype of heart cells. Biotechnol. Bioeng. 105, 1148–1160 (2010).
Kijlstra, J. D. et al. Integrated analysis of contractile kinetics, force generation, and electrical activity in single human stem cell-derived cardiomyocytes. Stem Cell Rep. 5, 1226–1238 (2015).
Yang, X. et al. Tri-iodo-l-thyronine promotes the maturation of human cardiomyocytes-derived from induced pluripotent stem cells. J. Mol. Cell. Cardiol. 72, 296–304 (2014).
Feyen, D. A. et al. Metabolic maturation media improve physiological function of human iPSC-derived cardiomyocytes. Cell Rep. 32, 107925 (2020).
Yadid, M. et al. Endothelial extracellular vesicles contain protective proteins and rescue ischemia-reperfusion injury in a human heart-on-chip. Sci. Transl. Med. 12, eaax8005 (2020).
Kim, D.-S. et al. Highly durable crack sensor integrated with silicone rubber cantilever for measuring cardiac contractility. Nat. Commun. 11, 535 (2020).
Li, J. et al. Circulating re-entrant waves promote maturation of hiPSC-derived cardiomyocytes in self-organized tissue ring. Commun. Biol. 3, 122 (2020).
Goldfracht, I. et al. Generating ring-shaped engineered heart tissues from ventricular and atrial human pluripotent stem cell-derived cardiomyocytes. Nat. Commun. 11, 75 (2020).
Jackman, C. P., Carlson, A. L. & Bursac, N. Dynamic culture yields engineered myocardium with near-adult functional output. Biomaterials 111, 66–79 (2016).
Wang, E. Y. et al. Biowire model of interstitial and focal cardiac fibrosis. ACS Cent. Sci. 5, 1146–1158 (2019).
Ma, Z. et al. Contractile deficits in engineered cardiac microtissues as a result of MYBPC3 deficiency and mechanical overload. Nat. Biomed. Eng. 2, 955–967 (2018).
Shadrin, I. Y. et al. Cardiopatch platform enables maturation and scale-up of human pluripotent stem cell-derived engineered heart tissues. Nat. Commun. 8, 1825 (2017).
Visone, R. et al. A microscale biomimetic platform for generation and electro-mechanical stimulation of 3D cardiac microtissues. APL Bioeng. 2, 046102 (2018).
Schneider, O. et al. Fusing spheroids to aligned μ-tissues in a heart-on-chip featuring oxygen sensing and electrical pacing capabilities. Mater. Today Bio 15, 100280 (2022).
Hinson, J. T. et al. Integrative analysis of PRKAG2 cardiomyopathy iPS and microtissue models identifies AMPK as a regulator of metabolism, survival, and fibrosis. Cell Rep. 17, 3292–3304 (2016).
Prondzynski, M. et al. Disease modeling of a mutation in α-actinin 2 guides clinical therapy in hypertrophic cardiomyopathy. EMBO Mol. Med. 11, e11115 (2019).
Rohr, S., Schölly, D. & Kleber, A. G. Patterned growth of neonatal rat heart cells in culture. Morphological and electrophysiological characterization. Circ. Res. 68, 114–130 (1991).
Rohr, S., Kucera, J. P., Fast, V. G. & Kléber, A. G. Paradoxical improvement of impulse conduction in cardiac tissue by partial cellular uncoupling. Science 275, 841–844 (1997).
Fink, C. et al. Chronic stretch of engineered heart tissue induces hypertrophy and functional improvement. FASEB J. 14, 669–679 (2000).
Zimmermann, W.-H. et al. Cardiac grafting of engineered heart tissue in syngenic rats. Circulation 106, I151-7 (2002).
Radisic, M. et al. Functional assembly of engineered myocardium by electrical stimulation of cardiac myocytes cultured on scaffolds. Proc. Natl. Acad. Sci. USA 101, 18129–18134 (2004).
Zhang, J. et al. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ. Res. 104, e30–e41 (2009).
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (RS-2025-23323900) and by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (RS-2025-24536077).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, Y.H., Son, Y.H., Choi, Y. et al. Advanced in vitro cardiac models for drug evaluation: integration of organoids, engineered tissues, and microphysiological systems. Microsyst Nanoeng 12, 162 (2026). https://doi.org/10.1038/s41378-026-01249-6
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41378-026-01249-6
