
Abstract
The pro-inflammatory programmed cell death pathway, necroptosis, relies on phosphorylation of the terminal effector, MLKL, by RIPK3. RIPK3-deficient mice or those harboring the kinase-inactivating mutation, RIPK3K51A, are ostensibly normal in the absence of challenge, indicating that RIPK3 and its kinase activity are dispensable for development. However, another kinase-inactivating mutation, RIPK3D161N, results in embryonic lethality in mice due to widespread apoptosis. As a result, the RIPK3D161N mutation is thought to confer a toxic gain-of-function. Here, to further explore the impacts of RIPK3 inactivation, we compared the stability and cellular interactions of RIPK3D161N and RIPK3K51A to a third previously-uncharacterized kinase-dead variant, RIPK3D143N. We show that RIPK3K51A was unstable and did not associate with RIPK1, RIPK3D161N was unstable but interacted with RIPK1, whereas RIPK3D143N was stable and bound RIPK1 in a manner comparable to wild-type RIPK3. Thus, all three variants scaffold differently, suggesting that the assembly of cell death machinery by RIPK3 is finely tuned, not just by its kinase activity, but also by the conformation of its kinase domain. Physiologically, Ripk3D143N/D143N mice exhibited a partially penetrant lethality in utero. However, once born, Ripk3D143N/D143N mice were fertile and phenotypically indistinguishable from wild-type mice in the absence of challenge. Full blockade of necroptotic signaling was shown in cells from Ripk3D143N/D143N mice, with the RIPK3D143N mutation also protecting Casp8−/− mice from lethal necroptosis during embryogenesis and preventing necroptotic ileitis in mice that lacked intestinal epithelial caspase-8 expression. Our studies support the idea that RIPK3 is a nexus between apoptotic and necroptotic signaling, and highlight the importance of considering kinase domain conformation in RIPK3 inhibitor development.
Similar content being viewed by others


Introduction
Necroptosis is a caspase-independent, pro-inflammatory programmed cell death modality that has been implicated in the pathology of a wide range of diseases. Lysis caused by necroptotic cell death releases damage-associated molecular patterns (DAMPs) into the extracellular milieu [1]. DAMPs release, in turn, provokes an immune response, which reflects the likely origins of the pathway as an altruistic cell death mode that combats pathogens [2,3,4,5,6,7,8,9]. Over the past decade, intense interest has centered on the role of dysregulated necroptosis in disease pathologies, particularly in ischemic injury and inflammatory diseases in the kidney, lung, gut and skin [10,11,12,13,14,15,16,17,18,19,20,21,22].
Necroptotic signaling is initiated by ligation of death receptors, such as TNF Receptor 1 by TNF, or pathogen receptors, such as Toll-like receptors and the intracellular sensor protein, ZBP1, by microbial ligands. Under specific contexts, for example when the cellular inhibitors of apoptosis proteins (cIAP) E3 Ubiquitin ligase family and pro-apoptotic caspases are depleted, necroptosis can ensue by prompting assembly of an intracellular signaling platform known as the necrosome. This megadalton complex is nucleated by the Receptor-interacting serine/threonine protein kinases (RIPKs), RIPK1 and RIPK3 [23, 24]. RIPK1 and RIPK3 assemble into hetero-oligomers through the functional amyloid-forming RIP homotypic interaction motif (RHIM) within the sequence C-terminal to each kinase domain [25,26,27]. Autophosphorylation of both RIPK1 and RIPK3 is essential for necroptotic signaling, with autophosphorylation within the RIPK3 kinase domain C-lobe (T224/S227 in human RIPK3 [28,29,30]; T231/S232 in mouse RIPK3 [31]) essential for facilitating binding to, and recruitment of, the terminal pathway effector, the MLKL (mixed lineage kinase domain-like) pseudokinase, to the necrosome [30, 32]. Subsequent phosphorylation of MLKL’s pseudokinase domain by RIPK3 is the critical trigger in promoting MLKL disengagement from the necrosome, oligomerization, trafficking to the plasma membrane, and assembly into lytic hotspots where membrane permeabilization occurs [28, 33,34,35,36,37,38].
Because RIPK3-mediated phosphorylation of MLKL is a pivotal step in necroptotic signaling [30, 32], several mouse models have been established to probe the functions of RIPK3 [39,40,41,42,43,44]. Deletion of RIPK3 does not compromise mouse viability and, in the absence of challenge, these mice are largely indistinguishable from wild-type counterparts [40, 41]. In comparison, many pathological processes are influenced by RIPK3 deficiency [20, 45], although distinguishing whether these effects stem from RIPK3’s catalytic or non-enzymatic functions remains challenging. With the aim of deconvoluting this issue, three kinase-dead RIPK3 mouse strains have been generated: (1) The Ripk3K51A/K51A mouse strain harbors a mutation in the N-lobe of the kinase domain that eliminates canonical ATP-binding. Like Ripk3–/– mice, this strain is phenotypically normal under basal conditions [39]; (2) The Ripk3D161N/D161N mouse strain carries a substitution in an activation loop residue that is required for Mg2+ binding. This strain has a fully penetrant embryonic lethal phenotype driven by errant Caspase-8-mediated apoptosis [42]; (3) The Ripk3S165D/T166E mouse strain harbors two mutations in RIPK3’s activation loop that impair kinase activity and produces a phenotype similar to, albeit less severe than, Ripk3D161N/D161N mice [43]. Why these kinase-inactivating mutations in RIPK3 yield different phenotypes remains unclear. It may indicate that the RIPK3K51A mutation prevents both RIPK3’s necroptotic and apoptotic actions, whereas the RIPK3D161N and RIPK3S165D/T166E variants solely perturb necroptotic signaling. Understanding how RIPK3’s kinase domain regulates cell death remains a topic of great interest given that selective small inhibitors of RIPK3, while preventing necroptosis, can also trigger on-target apoptosis [39].
To further examine the role of RIPK3’s kinase domain in the necroptosis-to-apoptosis switch, we generated another knock-in mouse strain harboring a conservative Asp-to-Asn substitution at residue 143 within the catalytic loop of RIPK3’s kinase domain, herein referred to as Ripk3D143N/D143N mice. In contrast to other kinase-dead RIPK3 variants, Ripk3D143N/D143N mice were born at a sub-Mendelian ratio due to the partial loss of homozygotes during embryogenesis. However, once born, Ripk3D143N/D143N mice were viable, fertile and developmentally normal into adulthood. The distinct phenotypes of the various kinase-inactivating mutations could be attributed to differences in their protein stability and binding repertoires. We observed that RIPK3D143N was more stable than RIPK3K51A and RIPK3D161N, and only RIPK3D143N bound both RIPK1 and Caspase-8 in the presence of a caspase inhibitor. Overall, our data support the concept that the conformation of RIPK3’s kinase domain tightly regulates the higher-order assembly of a RIPK1- and Caspase-8-containing complex.
Results
The kinase domain of RIPK3 controls apoptosis
RIPK3 catalytic activity is essential for necroptotic signaling because of its ability to phosphorylate and activate MLKL [30, 32]. RIPK3’s kinase activity relies on three active site residues: the ATP-positioning lysine within the β3 strand of the N-lobe (K51 in mouse RIPK3); the catalytic Asp of the catalytic loop HRD motif (D143); and the Mg2+ cofactor binding Asp of the DFG motif in the activation loop (D161) (Fig. 1A). To confirm this, we showed the recombinant kinase domain of mouse RIPK3 lacked phosphotransferase activity (Fig. 1B) and was unable to phosphorylate serine-345 of mouse MLKL (Fig. 1C) when any of the K51A, D143N or the D161N mutations were introduced. To compare the functional impact of these kinase-inactivating mutations, we stably transduced Ripk3−/− mouse dermal fibroblasts (MDF) with lentiviral vectors encoding doxycycline-inducible wild-type RIPK3 (RIPK3WT), RIPK3K51A, RIPK3D143N and RIPK3D161N. A concentration of 10 ng/mL doxycycline (dox) was chosen to reconstitute RIPK3 expression to endogenous levels (Supplementary Fig. 1A). Unlike with RIPK3WT, reconstitution with RIPK3K51A, RIPK3D143N or RIPK3D161N did not restore sensitivity to the necroptotic stimulus, TNF/Smac mimetic/IDN-6556 (TSI; Fig. 1Di), consistent with the catalytic inactivity of these mutants observed in vitro using recombinant proteins (Fig.1B-C).

A Richardson (Ribbons) diagram of the mouse RIPK3 kinase domain structure (PDB, 4M66) [72] in which the three catalytic residues targeted for kinase dead mutations are shown as red sticks. B Relative ATP consumption (relative fluorescence units; RFU) by the recombinant kinase domain of mouse RIPK3WT, RIPK3K51A, RIPK3D143N or RIPK3D161N mutants when co-incubated with recombinant full-length mouse MLKL as detected by an ADP-Glo assay. Bar graph shows mean + SEM of n = 3 independent replicates. ***p < 0.001 by one-way ANOVA. C Immunoblot of reactions from Panel (B). D Quantitation of the death of Ripk3–/– mouse dermal fibroblasts (MDF) without or with doxycycline (dox) to express RIPK3WT, RIPK3K51A, RIPK3D143N or RIPK3D161N in the presence of the stipulated stimuli. Treatments: (i) TNF, Smac mimetic and IDN-6556 (TSI); (ii) vehicle, DMSO; (iii) IDN-6556; (iv) TNF and Smac mimetic (TS); (v) GSK′843; (vi) GSK′843, IDN-6556. Mean ± SEM for one experiment and representative of data from 2 independent experiments. Cell death calculated as the percentage of propidium iodide+ cells relative to the number of SPY700+ cells. E Immunoblot of MDFs treated for 8 h as in Panel D. Data representative of two independent experiments. Closed arrowheads indicate full-length proteins of interest. Open arrowheads indicate relevant cleavage products. F Immunofluorescence of Ripk3–/– MDFs in the absence (-dox; without doxycycline) or presence ( + dox+IDN; with doxycycline and pan-Caspase inhibitor, IDN-6556) of RIPK3WT, RIPK3K51A, RIPK3D143N or RIPK3D161N inducible expression. Data representative of two independent experiments. Hoechst nuclear signal is depicted in red. RIPK3 immunosignal is depicted in white. Yellow arrowhead exemplifies stimulus-independent clustering of RIPK3D161N.
In the absence of an external stimulus, the reconstitution of Ripk3–/– fibroblasts with RIPK3WT or the RIPK3K51A, RIPK3D143N and RIPK3D161N mutants yielded different cell fates. RIPK3K51A did not induce death, RIPK3WT and RIPK3D143N produced mild death, whereas RIPK3D161N caused extensive death (Fig. 1Dii). These constitutive death responses were due to RIPK3-mediated apoptosis as they were prevented by the pan-Caspase inhibitor, IDN-6556 (Fig. 1Diii). Thus, consistent with prior reports [39, 42], we found that RIPK3D161N is a stimulus-independent trigger for apoptosis, whereas the other kinase-dead variants of RIPK3 were comparatively less toxic. We next studied the role of RIPK3 mutation on TNF-induced apoptosis. Interestingly, RIPK3WT and RIPK3D143N, but not RIPK3K51A, increased the rate of apoptosis caused by TNF/Smac mimetic compared to cells in which exogene expression was not induced by dox (TS; Fig. 1Div). Due to constitutive killing activity of RIPK3D161N, the impact of RIPK3D161N on TNF-induced apoptosis could not be assessed. Thus, RIPK3’s role in TNF-induced apoptosis is blocked by the RIPK3K51A mutation, but largely unaffected by the RIPK3D143N mutation.
To further explore RIPK3’s apoptotic scaffolding function, we used GSK′843 – a RIPK3 inhibitor that targets the RIPK3 active site and induces caspase-8-dependent apoptosis [39]. GSK′843 induced high levels of apoptosis in cells expressing RIPK3WT and RIPK3K51A, but was well-tolerated by cells reconstituted with RIPK3D143N (Fig. 1Dv-vi). Again, the influence of RIPK3D161N on GSK′843-induced apoptosis could not be distinguished from its inherent toxicity. Thus, in contrast to TNF-induced apoptosis, RIPK3’s role in GSK′843-induced apoptosis is unaffected by the RIPK3K51A mutation, but attenuated by the RIPK3D143N mutation. Immunoblotting confirmed that, unlike the other RIPK3 mutants, only RIPK3D161N constitutively triggered caspase-3 activation, only cleavage of RIPK3K51A was not increased during TNF-induced apoptosis, and only RIPK3D143N failed to increase GSK′843-induced caspase-3 activation (Fig. 1E). Notably, the ability of RIPK3D161N and GSK′843 to trigger apoptosis was reflected by their shared propensity to drive RIPK3 into large cytoplasmic puncta (Fig. 1F and Supplementary Fig. 1B). Together, our data suggest that RIPK3’s kinase domain conformation finely tunes its apoptotic scaffolding function, with RIPK3D161N constitutively inducing apoptosis, RIPK3K51A obviating RIPK3’s involvement in TNF-induced apoptosis, and RIPK3D143N attenuating GSK′843-induced apoptosis.
Catalytically-dead RIPK3D143N retains stability and RIPK1-Caspase-8-binding
We hypothesized that conformational differences among the three kinase-dead versions of RIPK3 underlies their variable stability and scaffolding properties. To address this possibility, we expressed each exogene in Ripk3–/– MDFs, subjected each to a thermal gradient and examined RIPK3’s solubility by immunoblot. This assay was chosen on the basis that pro-apoptotic signaling by GSK′843 involves enhanced RIPK3 thermal stability (Supplementary Fig. 1C). Interestingly, RIPK3D143N was the only mutant that exhibited thermal stability comparable to wild-type RIPK3 (Fig. 2A-B), with the destabilization of RIPK3K51A and RIPK3D161N implying each is conformationally labile. These differences in the stability of RIPK3 had no bearing on the stability of endogenous MLKL (Fig. 2A-B), prompting us to examine which other interactions are modulated by RIPK3 mutation. Accordingly, we reconstituted Ripk3–/– fibroblasts with RIPK3WT or the kinase-dead mutants in the presence of IDN-6556 to prevent caspase-mediated cleavage of RIPK3 [44, 46], followed by a brief treatment with TSI to allow TNF-induced death complexes to form (Fig. 2C and Supplementary Fig. 2). Critically, the interactions of RIPK3D143N resembled that of wild-type RIPK3, where it bound RIPK1 and Caspase-8 following necroptotic stimulation with TSI. On the other hand, RIPK3K51A bound neither RIPK1 nor Caspase-8, which aligns with its inability to mediate TNF-driven cell death. While RIPK3D161N was able to retain RIPK1 binding, it no longer precipitated with Caspase-8, which may underlie its constitutive killing activity. More broadly, these data imply that the stability and conformation of RIPK3’s kinase domain, in addition to its catalytic activity, is a key determinant of apoptotic and necroptotic signaling.

Immunoblots (IB, Panel A) and quantitation (Panel B) of cellular thermal stability assay of RIPK3WT, RIPK3K51A, RIPK3D143N and RIPK3D161N. Representative of n = 4 for RIPK3WT, RIPK3K51A, RIPK3D143N, and n = 1 for RIPK3D161N. Mean ± SEM. C Immunoblot of input and co-immunoprecipitations (FLAG-IP) of reconstituted RIPK3WT, RIPK3K51A, RIPK3D143N and RIPK3D161N in Ripk3−/− mouse dermal fibroblasts (MDF) in the presence of IDN-6556 ± overnight doxycycline (dox), then 2 h of necroptotic stimuli (TSI). Data representative of n = 2. See Supplementary Fig. 2 for quantitation of RIPK3 (co)-immunoprecitation experiments. Uncropped immunoblot data are included as a supplementary data file.
Adult Ripk3 D143N/D143N mice are viable, fertile and ostensibly normal
In contrast to RIPK3K51A and RIPK3D161N mutants, RIPK3D143N retains RIPK3WT-like stability and cellular interactions, making it an ideal kinase-dead mutant to explore RIPK3’s non-necroptotic actions in vivo. Accordingly, we generated a mouse harboring a RIPK3D143N-encoding mutation within exon 3 of the mouse Ripk3 gene using CRISPR/Cas9 editing (Fig. 3A). Ripk3D143N/D143N mice were viable, fertile and developed normally into adulthood (Fig. 3B and Supplementary Fig. 3A–G). Intriguingly, while heterozygous matings produced a Mendelian distribution of genotypes at embryonic day 10 (E10.5), Ripk3D143N/D143N mice were slightly but significantly under-represented at weaning (Fig. 3C and Supplementary Fig. 3H; χ2 test for goodness of fit showed p = 0.88 at E10.5 and p = 0.02 at weaning). This observation suggests that Ripk3D143N/D143N mice are partially sensitized to the same peripartum checkpoint that causes lethality in Ripk3D161N/D161N mice and Ripk3S165D/T166E mice [42, 43]. In unchallenged adult Ripk3D143N/D143N mice, RIPK3D143N was expressed and distributed comparably to wild-type RIPK3 in all examined tissues (Fig. 3D and Supplementary Fig. 4), consistent with the stability of exogenous RIPK3D143N observed in our cellular studies. Importantly, fibroblasts and bone marrow-derived macrophages from Ripk3D143N/D143N mice confirmed that the endogenously-expressed RIPK3D143N was catalytically inactive because these cells were completely protected from TNF-induced necroptotic death yet could still undergo TNF-induced apoptosis (Fig. 3E-F). Collectively, these data show that endogenous RIPK3D143N, despite conferring a transient and partial toxicity in utero, is stable and well-tolerated in mouse cells and tissues.

A Clustered regularly interspaced short palindromic repeats (CRISPR) gene editing strategy for the generation of Ripk3D143N/D143N mice. B Ripk3WT/WT and Ripk3D143N/D143N littermates at 4 months-of-age. C Genotype distribution at E10.5 or at weaning from heterozygous Ripk3D143N/WT matings. Statistical test to compare observed versus expected genotype ratios: χ2 test for goodness of fit showed p = 0.88 at E10.5 and p = 0.02 at weaning. See Supplementary Fig. 3H for animal numbers per genotype and age. D Immunoblot of organ homogenates of littermates, n = 2 per genotype. E Quantitation of the death of mouse dermal fibroblasts (MDF) and bone marrow derived macrophages (BMDM) from Ripk3WT/WT and Ripk3D143N/D143N mice. Cells were untreated (UT) or stimulated with TNF+Smac mimetic (TS) or TS+IDN-6556 (TSI). Cell death was measured by the uptake of SYTOX Green. BMDM death was calculated as the percentage of SYTOX Green+ cells relative to the number of SPY620+ cells. Mean ± SEM from one experiment is shown. Representative of two independent experiments across three independent MDF lines, and one BMDM experiment. F Micrographs of Ripk3WT/WT and Ripk3D143N/D143N MDF after 19 h of treatment. Scale bars represents 200 µm.
RIPK3D143N protects mice from the embryonic lethality of caspase-8 deficiency
Caspase-8 knockout mice die mid-gestation from unrestrained RIPK3-MLKL-dependent necroptosis [47,48,49,50]. This lethal phenotype is fully penetrant with no Casp8–/– mice observed at birth unless they carry compound mutations that preclude necroptosis. For example, Casp8–/– mice that carry homozygous RIPK3K51A or RIPK3D161N substitutions survive into adulthood [39, 42]. Consistent with this notion, we find that Casp8–/–Mlkl+/– mice are viable when crossed onto a Ripk3D143N/D143N background, whereas no Casp8–/–Mlkl+/–Ripk3D143N/WT or Casp8–/–Mlkl+/–Ripk3WT/WT mice were observed in these litters at birth (Fig. 4A-B). These data definitively show that the RIPK3D143N mutation prevents necroptotic cell death in vivo.

A The number of neonates (genotyped at P1-P5) from seven heterozygous Casp8+/–Mlkl+/–Ripk3WT/D143N matings on C57BL/6 J background. Alleles annotated by a question mark (?) were either the wild-type (+) or the knock-out (–) allele. B Photo of Casp8+/+Mlkl+/–Ripk3WT/WT and Casp8–/–Mlkl+/–Ripk3D143N/D143N littermates at 3 days of age.
RIPK3D143N protects mice from MLKL-dependent intestinal inflammation
Germline loss of caspase-8 can cause very early onset inflammatory bowel disease in humans [51]. Similarly, conditional deletion of caspase-8 from intestinal epithelial cells causes ileitis in mice characterised by unrestrained RIPK1-RIPK3-MLKL-dependent necroptosis and Paneth cell loss [20, 21, 52,53,54]. To more finely address the role of necroptotic signaling in this disease model, we established a colony of Villin1000-Cre Casp8fl/fl mice (herein referred to as Casp8IEC mice) where the Villin promoter drives Cre expression to delete Caspase-8 from intestinal epithelial cells. We confirmed that Villin1000-driven Cre activity was on-target (Supplementary Fig. 5A; mild off-target Cre activity was found in the liver) and induced selective and complete deletion of caspase-8 from the intestinal epithelia in Casp8IEC mice (Supplementary Fig. 5B). Consistent with prior reports [20, 21, 52,53,54], Casp8IEC mice developed endoscopically-apparent colitis (Fig. 5A-B), with a significant proportion of the Lysozyme+ and MPTX2+ Paneth cells lost by 6 months-of-age (Fig. 5C-D and Supplementary Fig. 6A-B). Beyond these manifestations, Casp8IEC mice in our facility exhibited a mild phenotype without impacts on survival (Supplementary Fig. 6C), weight (Supplementary Fig. 6D), or Goblet cell numbers (Fig. 5E-F). Importantly, crossing Casp8IEC mice onto the Ripk3D143N/D143N background completely prevented Paneth cell loss (Fig. 5C-D). Similarly, Casp8IECMlklIEC mice with conditional deletion of both caspase-8 and MLKL were fully protected from Paneth cell loss (Fig. 5C-D). Interestingly, epithelial RIPK3 expression was increased in the Casp8IEC ileum, but remained at basal levels in Casp8IECRipk3D143N/D143N and Casp8IECMlklIEC mice (Fig. 5G). Collectively, these data show that cell-intrinsic necroptosis is solely responsible for Paneth cell loss, which in turn sparks ileitis and increased epithelial RIPK3 expression in the Casp8IEC model.

A Endoscopy images of a healthy wild-type (Casp8WT) mouse and a Casp8IEC mouse with colitis. Red arrow points to the loss of vasculature and black arrow points to a loose stool. B Murine endoscopic index of colitis severity (MEICS) scores for Casp8WT and Casp8IEC mice. Each dot represents the score from a mouse during their monthly endoscopy session between 6-24 weeks-of-age. Line indicates mean. ***p < 0.001 from unpaired non-parametric t-test with Mann-Whitney correction. Casp8WT n = 13 mice. Casp8IEC n = 12 mice. C Immunohistochemistry (brown) signals for lysozyme. Scale bars are 20 µm. Images representative of Casp8WT n = 3, Casp8IEC n = 4, Casp8IECMlklIEC n = 3, and Casp8IECRipk3D143N/D143N n = 4 mice. D Lysozyme+ cells as a percentage of total cells. Same cohort as in Panel C. An average of 35,000 cells were measured per mouse with each dot representing the mean value from one mouse. Bars indicate group mean ± SD. *p < 0.05 and **p < 0.01 by ordinary one-way ANOVA with Tukey’s correction. E Periodic Acid-Schiff (PAS; magenta) staining. Scale bars are 20 µm. Images representative of Casp8WT n = 5, Casp8IEC n = 6, Casp8IECMlklIEC n = 4, and Casp8IECRipk3D143N/D143N n = 3 mice. F PAS+ cells as a percentage of total cells. Same cohort as in Panel E. An average of 70,000 cells were measured per mouse with each dot representing the mean value from one mouse. Bars indicate group mean ± SD. n.s. indicates p > 0.05 by ordinary one-way ANOVA with Tukey’s correction. G, H Immunosignals for RIPK3 (Panel G) and pRIPK3 (Panel H) in the ileum of Casp8WT, Casp8IEC, Casp8IECMlklIEC, and Casp8IECRipk3D143N/D143N mice; representative of n = 3, 5, 5, and 4 mice. Scale bars are 100 µm. See Supplementary Fig. 7C, D for further details.
It was striking that Paneth cells are the only intestinal epithelial population that succumbs to caspase-8-deficiency in mice. To further investigate this observation, we developed an automated immunohistochemistry protocol that selectively detects phospho-activated RIPK3 (pRIPK3; Supplementary Fig. 7A-B). This protocol produced robust immunosignals in TSI-treated Ripk3–/– fibroblasts that were reconstituted with RIPK3WT, but not in TSI-treated Ripk3–/– fibroblasts reconstituted with RIPK3K51A, RIPK3D143N or RIPK3D161N (Supplementary Fig. 7A-B). Using this protocol, we find that pRIPK3 levels are elevated in the ileum of Casp8IEC mice relative to Casp8WT mice (Fig. 5H and Supplementary Fig. 7C-D). As expected, no pRIPK3+ve epithelial cells were found in the Casp8IEC Ripk3D143N ileum (Fig. 5H). Surprisingly, only a small number of pRIPK3+ve epithelial cells were found in the Casp8IEC ileum (Fig. 5H); with pRIPK3+ve epithelial cells also rare in the Casp8IECMlklIEC ileum where activated RIPK3 can in theory accumulate without triggering Paneth cell death. The finding that most Paneth cells in the Casp8IEC and Casp8IECMlklIEC ileum were pRIPK3-ve implies that another insult, in addition to caspase-8-deficiency, is responsible for triggering RIPK3 activation and Paneth cell necroptosis. The nature of this secondary insult remains of outstanding interest. More broadly, our study shows that the Ripk3D143N mouse model is an elegant way of preventing necroptosis in vivo and that the epithelial necroptotic pathway is a critical and complex regulator of gut inflammation.
Discussion
Until recently, observations that RIPK3 inhibition can promote apoptosis have raised concerns about RIPK3’s suitability as a therapeutic target [39, 42]. However, subsequent findings indicate that RIPK3 inhibitors can be well-tolerated in animals [55,56,57], suggesting RIPK3 kinase domain conformation, rather than loss of catalytic activity, is likely to underpin the previously reported caspase-8-driven toxicity. Here, we tested this idea by generating a knock-in mouse strain harboring the RIPK3D143N kinase-dead mutant. The resulting Ripk3D143N homozygote mice were viable, fertile, with no detectable pathology or hematopoietic defects in the absence of challenge, unlike the embryonic lethality found in the Ripk3D161N strain [42]. Collectively, our data support the idea that RIPK3D143N is akin to RIPK3WT in interactions and stability, leading us to propose that Ripk3D143N/D143N mice should be adopted as the preferred model for studying the non-catalytic roles of RIPK3 in vivo. We demonstrate the utility of the Ripk3D143N/D143N strain as a model for studying necroptosis in vivo by showing protection from ileitis caused by intestinal epithelial cell knockout of Casp8. The protection conferred by the RIPK3D143N mutation was phenocopied by deletion of Mlkl specifically within intestinal epithelial cells, indicating that blockade of necroptosis within intestinal epithelial cells, rather than infiltrating cells, confers protection.
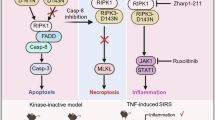
While adult Ripk3D143N/D143N mice grossly resembled wild-type counterparts, we observed a partially-penetrant embryonic lethality manifesting in sub-Mendelian ratio of homozygotes at weaning. Because embryonic lethality was previously observed for the Ripk3D161N strain [42] and Ripk3S165D/T166E strain [43], but not Ripk3K51A mice [39], it was known that loss of RIPK3 catalytic activity itself is not deleterious to development. Here, our findings with the Ripk3D143N knock-in mouse support this assertion, and instead indicate that the conformation of the RIPK3 kinase domain is a crucial determinant of cell, and organism, viability. Much emphasis has been placed on the role of the RIPK1 and RIPK3 RHIMs in assembling the necrosome. Whilst RIPK1 autophosphorylation of S161 and S166 within its kinase domain are crucial to propagation of necroptotic signals [18, 58,59,60], our data support a model in which the conformation of the RIPK3 kinase domain allosterically influences RIPK3’s interaction with RIPK1, which in turn influences downstream cell death signaling (Fig. 6). How this allosteric regulation extends to caspase-8 sequestration and activation, and impacts the stoichiometry and connectivity of RIPK3-containing complexes, remains a major focus for future investigation.

Star indicates mutant form of RIPK3. RIP homotypic interaction motif (RHIM), Death Effector Domain (DED), Death Domain (DD). Figure generated using BioRender.
Our study raises several questions about RIPK3 biology and the field’s renewed interest in targeting it therapeutically. For example, it will be interesting to see if small molecule inhibitors that, like the RIPK3D143N mutation, can preserve RIPK3’s kinase activity-independent scaffolding ability also have improved safety profiles. It is also interesting to note that mice harboring four different inactivating mutations in RIPK3 all have distinct phenotypes [39, 42, 43]. This degree of conformational lability and functional non-redundancy is remarkable and suggests that more sophisticated approaches, such as deep mutational scanning, may be needed to fully understand the kinase-independent functions of RIPK3.
Methods
Mice
Mice were housed at the Walter and Eliza Hall Institute of Medical Research (WEHI), Australia. Animal experiments were approved by the WEHI Animal Ethics Committee (2022.085) in accordance with the Prevention of Cruelty to Animals Act (1986) and the Australian National Health and Medical Research Council Code of Practice for the Care and Use of Animals for Scientific Purposes (1997). Mice were housed in a temperature and humidity controlled specific pathogen free facility with a 12 h:12 h day night cycle. Villin.cre1000 mice were purchased from Jackson Laboratories (Jackson Laboratories, Maine, United States; Strain #021504). The Casp8–/– mouse strain [47], the floxed Casp8 mouse strain [61], the floxed Mlkl mouse strain [32], the Casp8+/–Mlkl+/– mouse strain [50] and the Rosa26 Ai9 Cre reporter mouse strain [62] have been reported previously. All mice in this study were maintained on a C57BL/6 J background. No predefined inclusion/exclusion criteria and no prior estimates of sample sizes were used in this study. Co-housed littermate mice, rather than randomized cohorts, were preferentially used for the Casp8IEC model. Unless stipulated, mice in the Casp8IEC model were harvested at 5–6 months of age. This study used mice of both sexes without preference.
Generation and genotyping of the Ripk3 D143N/D143N mouse strain
Ripk3D143N/D143N mice on C57BL/6 J background were generated with CRISPR/Cas9 by the Melbourne Advanced Genome Editing Center laboratory. The Ripk3 gene on mouse chromosome 14 was targeted to induce the D143N mutation. One sgRNA of the sequence TGTTAGAGGGCTTGAGGTCC was used to create double stranded breaks within the Ripk3 locus to stimulate homologous recombination. An oligonucleotide donor with the sequence CCTGCTGCAGGAAGTGGTGCTGGGGATGTGCTACCTACACAGCTTGAACCCTCCGCTCCTGCACCGGAATCTCAAGCCCTCTAACATTCTGCTGGATCCAGAGCTCCACGCCAAGGTTAGTCCATCTACA was used to introduce the D143N mutation. To detect the integration of the donor, mice were genotyped using the forward primer GGGACAGGTACTCAACATGGTC and the reverse primer TTTCGGCGTGTAGGAAGAAG, with sequencing overhangs attached. The PCR conditions for genotyping were: 95 °C 2 min, (95°C 30 s, 60°C 30 s, 72°C 30 s) x 35, 72°C 5 min. F0 mice were backcrossed with wildtype C57BL/6 J mice for at least 2 generations to breed out any potential off-target mutations.
Immunoblotting
Cells/tissues were lysed in ice-cold RIPA buffer (10 mM Tris-HCl pH 8.0, 1 mM EGTA, 2 mM MgCl2, 0.5% v/v Triton X-100, 0.1% w/v sodium deoxycholate, 0.5% w/v sodium dodecyl sulfate (SDS), and 90 mM NaCl) supplemented with 1× Protease and Phosphatase Inhibitor Cocktail (Cell Signaling Technology Cat#5872) and 100 U/mL Benzonase (Sigma-Aldrich Cat#E1014) to a concentration of 50 mg/mL (w/v) with a stainless-steel bead using a Qiagen TissueLyser II (1 min at 30 Hz). Homogenates were boiled for 10 min in 1 × SDS sample buffer (126 mM Tris-HCl, pH 8, 20% v/v glycerol, 4% w/v SDS, 0.02% w/v bromophenol blue, 5% v/v 2-mercaptoethanol) and ran on 1.5 mm NuPAGE 4–12% Bis-Tris gels (ThermoFisher Scientific NP0335BOX) in MES Running Buffer (ThermoFisher Scientific NP000202) at 150 V for 60 min. Gels were transferred to a polyvinylidene difluoride membrane (Merck Cat# IPVH00010) at 100 V for 60 min and then blocked in 5% w/v skim milk powder in TBS-T (50 mM TrisHCl pH 7.4, 0.15 M NaCl, 0.1 v/v Tween-20). After transfer, gels were stained with SimplyBlue SafeStain (ThermoFisher Scientific LC6065) and membranes were probed with the following antibodies: RIPK3 (clone 8G7; ref. [8]; produced in-house; available from Millipore as MABC1595), RIPK3 (clone 1H12; ref. [63]; produced in-house), MLKLpS345 (clone D6E3G; Cell Signaling Technology), MLKL (clone 3H1; ref. [32]; produced in-house; available from Millipore as MABC604), Caspase-8 (clone F5K9P; Cell Signaling Technology), RIPK1pS166 (Cat#31122; Cell Signaling Technology), RIPK1 (clone D94C12; Cell Signaling Technology), RIPK3pT231pS232 (clone E7S1R; Cell Signaling Technology), RIPK3 (clone D4G2A; Cell Signaling Technology), caspase-3 (Cat#9662; Cell Signaling Technology), horseradish peroxidase (HRP)-conjugated Actin (Santa Cruz Biotechnology, Cat# sc-47778), and GAPDH (Millipore MAB374). Primary antibodies produced in-house were used at 1 μg/mL for immunoblot, and all other primary antibodies were used at a 1:1000-2000 dilution in Tris-Balanced Salt Solution containing 0.1% Tween 20 (TBS + T) supplemented with 5% w/v cow’s skim milk powder and 0.01% w/v sodium azide. Primary antibody probes were performed overnight at 4 °C on a rocker before washing twice in TBS + T, then membranes were probed with a 1:10000 dilution of HRP-conjugated secondary antibody: goat anti-rat immunoglobulin (Ig; Southern BioTech Cat#3010-05), goat anti-rabbit Ig (Southern BioTech Cat#4010-05), and goat anti-mouse Ig (Southern BioTech Cat#1010-05). Membranes were washed four times in TBS + T and signals revealed by enhanced chemiluminescence (Merck Cat#WBLUF0100) on a ChemiDoc Touch Imaging System (Bio-Rad). Between probing with primary antibodies from the same species, membranes were incubated in stripping buffer (200 mM glycine pH 2.9, 1% w/v SDS, 0.5 mM TCEP) for 30 min at room temperature and then re-blocked. Uncropped immunoblots are included as a Supplemental Data file.
Immunohistochemistry (IHC)
Mice were euthanized via gradual carbon dioxide asphyxiation, with bipedal and orbital reflexes checked to ensure death. Tissues were harvested and immediately placed in 10% Neutral-Buffered Formalin at a 1:10 tissue:formalin ratio at room temperature. Formalin fixed tissues were paraffin embedded using the standard 8-h auto-processing protocol of Tissue-Tek VIP® 6 AI Tissue Processor (Sakura Finetek USA) within 1–2 days of harvest. Four-micron-thick sections of paraffin-embedded tissues were cut onto adhesive slides (Menzel Gläser Superfrost PLUS). IHC and image acquisition of sections stained for Caspase-8, RIPK1, RIPK3 and cleaved Caspase-3 followed protocols described previously [14]; staining for lysozyme (Abcam, Cat#108508), MPTX2 (Abcam, Cat#EPR20920-19; marker of Paneth cells [64]), and pRIPK3 (CST, Cat#91702) followed the protocols detailed in Supplemental Table 1.
Cellular thermal stability assay
Ripk3–/– mouse dermal fibroblasts with doxycycline-inducible expression constructs for RIPK3WT, RIPK3K51A, RIPK3D143N or RIPK3D161N were seeded to ~50% confluency. After 2 h, cells were treated with 10 ng/mL doxycycline and 5 μM IDN-6556. The next day, cells were trypsinized, pelleted (700 × g, 5 min, room temperature), then resuspended in dPBS supplemented with cOmplete EDTA-free protease inhibitor (Merck, Cat#04693132001) and PhosSTOP phosphatase inhibitor (Merck, Cat#4906837001). 100 μL of 2 × 106 cells/mL were aliquoted into polymerase chain reaction (PCR) tubes and incubated across a gradient of 8 temperatures (45–57 °C) for 3 min, then at 21 °C for 3 min, then at 12 °C for 1 min. Samples were immediately frozen at –80 °C, thawed on ice, pelleted (20,000 × g, 20 min, 4 °C) and 35 μL of supernatant analyzed via immunoblot. Immunoblot signals were quantified with Image Lab v6.1 (Bio-Rad) using the raw full-resolution.scn Chemidoc files.
Immunoprecipitation
Ripk3–/– mouse dermal fibroblasts with doxycycline-inducible expression constructs for RIPK3WT, RIPK3K51A, RIPK3D143N or RIPK3D161N were seeded into 6-well plates at 2×105 cells/well. 2 h later, cells were treated with 5 μM IDN-6556 and ±10 ng/mL doxycycline. The next day, cells were treated with 100 ng/mL tumor necrosis factor, 500 nM Smac mimetic Compound A and 5 µM IDN-6556. 90 min later, cells were washing with PBS, then lysed with 1 mL/well of lysis buffer 50 mM Tris-HCl, pH 7.4, 1% v/v Triton X-100, 150 mM NaCl, cOmplete EDTA-free protease inhibitor (Merck, Cat#04693132001), PhosSTOP phosphatase inhibitor (Merck, Cat#4906837001) and 100 U/mL Benzonase (Sigma #E1014). Lysates were freeze-thawed and a 70 \({{{\rm{\mu }}}}\)L aliquot kept aside as the “input”. The remainder of each lysate was supplemented with 20 \({{{\rm{\mu }}}}\)L/sample of anti-HA magnetic beads (ThermoFisher Scientific Cat#88837), samples rotated (30 min, 4°C), beads pelleted and discarded, 120 \({{{\rm{\mu }}}}\)L/tube of anti-Flag magnetic beads (Merck, Cat#M8823) was added to the supernatant, samples rotated (90 min, 4 °C), beads pelleted and washed three times with 0.5 mL of lysis buffer, beads pelleted, resuspended in 75 \({{{\rm{\mu }}}}\)L of 2xSDS loading buffer, incubated (10 min, 100 °C), beads pelleted and the supernatant analyzed via immunoblot.
Cell pellet preparation
Trypsinized cells were centrifuged at 671 × g for 3 min at room temperature. The supernatant was discarded, cell pellets resuspended in 10% v/v Neutral Buffered Formalin, incubated for 20 min at room temperature, and centrifuged at 671 × g for 3 min at room temperature. Cell pellets were resuspended in 50–70 μL of HistoGel (Epredia Cat#HG-4000-012) pre-warmed to 70 °C and then pipetted onto ice-cold glass coverslips to set. Set pellets were stored in 70% (v/v) ethanol until paraffin-embedding.
Expression constructs and cell line generation
For expression in mammalian cells, wild-type full-length RIPK3 was amplified by PCR from a mouse RIPK3 template (synthesized by ATUM, CA) and subcloned into the doxycycline-inducible, puromycin-selectable mammalian expression vector, pF TRE3G PGK puro (Ampr) using BamHI and EcoRI restriction sites, as before [41]. Mutations (K51A, D143N, D161N) cDNAs were synthesized and subcloned into pF TRE3G PGK puro as BamHI-EcoRI fragments (ATUM, CA). Vector DNA was co-transfected into HEK293T cells with pVSVg and pCMVδR8.2 helper plasmids to generate lentiviral particles, which were transduced into MDF cell lines (Ripk3−/−, derived as previously described [41]) and selected for genomic integration using puromycin (2.5 μg/mL; StemCell Technologies) using established procedures [65, 66]. For recombinant protein expression, wild-type and mutant RIPK3 (amino acids 2-353) were amplified by PCR from mouse RIPK3 templates (synthesized by ATUM, CA) and subcloned into the insect expression vector pFastBac Htb SBP (Ampr), using ligation-independent cloning. Wild-type full-length mouse MLKL was subcloned into the insect expression vector pFastBac Htb, as described previously [32]. All insert sequences were verified by Sanger sequencing.
Recombinant expression and purification
Mouse RIPK3 (amino acids 2-353) was expressed and purified from ExpiSf9 insect cells using established procedures [67]. Briefly, the bacmid was prepared in DH10MultiBac E. coli (ATG Biosynthetics) from a pFastBac Htb-SBP vector encoding a TEV protease-cleavable His6-SBP tag N-terminal to RIPK3. ExpiSf9 insect cells were cultured in ExpiSf CD media (Thermo Fisher Scientific). Each bacmid (1 μg) was introduced into 0.9 × 106 ExpiSf9 cells by Cellfectin II (Thermo Fisher Scientific) mediated transfection in six-well plates using the Bac-to-Bac protocol (Thermo Fisher Scientific). After 4 days of static incubation at 27 °C in a humidified incubator, the resulting P1 baculovirus was harvested and added at 2% v/v to 50 mL ExpiSf9 cells at 1.0 × 106 cells/mL density, which were shaken at 27 °C, 130 RPM. The cell density was monitored daily using a haemocytometer slide and maintained at 0.5–3.0 × 106 cells/mL by diluting with fresh ExpiSf CD media when necessary, until growth arrest (defined as a cell density less than the twice the cell count 1 day prior). Approximately 24 h after growth arrest was recorded, the P2 baculovirus was harvested by collecting the supernatant after pelleting the cells at 500 × g for 5 min. P2 baculovirus was added to 0.5 L ExpiSf9 cells cultured at 5–6 × 106 cells/mL in 2.8 L Fernbach flasks at 0.6% v/v, and cultured at 27 °C, 90 RPM for 72 h. Cells were harvested at 500 × g and pellets snap frozen in liquid N2 and either thawed immediately for lysis or stored at −80 °C.
RIPK3 cell pellets were resuspended in Ni-NTA buffer (20 mM HEPES pH 7.5, 200 mM NaCl, 5% v/v glycerol), supplemented with 5 mM imidazole (pH 7.5), EDTA-free cOmplete Protease Inhibitor (Roche) and 0.5 mM Bond-Breaker TCEP and were lysed by sonication, before the lysate was clarified by centrifugation (40,000 ×g, 45 min, 4 °C). The clarified lysate was incubated with Ni-NTA resin (Roche) pre-equilibrated in Ni-NTA buffer with 5 mM imidazole for >1 h on rollers at 4 °C, before the beads were pelleted at 500 ×g and washed extensively with Ni-NTA buffer containing 35 mM imidazole. The proteins were eluted in Ni-NTA buffer containing 500 mM imidazole. The His6-SBP tag was cleaved using recombinant His6-TEV protease and dialysis in SEC buffer (20 mM HEPES pH 7.5, 200 mM NaCl, 5% v/v glycerol). The eluate was further purified by the addition of Ni-NTA resin (cOmplete His-Tag; Roche) to eliminate uncleaved material and the TEV protease. The sample was then spin concentrated (30 kDa MWCO; Millipore), before being loaded onto a Superdex 200 10/300 GL (Cytiva) pre-equilibrated with SEC buffer (20 mM HEPES pH 7.5, 200 mM NaCl, 5% v/v glycerol). Purified fractions, as assessed following resolution by reducing StainFree SDS-PAGE gel electrophoresis (Bio-Rad), were pooled, spin concentrated, aliquoted, and snap frozen in liquid N2 for storage at −80 °C. Mouse MLKL was expressed in ExpiSf9 insect cells and purified by Ni-NTA pulldown, using an equivalent method to mouse RIPK3, as detailed above.
Kinase activity assay
RIPK3 wild-type and mutant kinase activity was measured using an ADP-Glo kinase assay kit (Promega), following standard procedures. Briefly, a 10 μL kinase reaction was assembled, containing 50 ng recombinant mouse RIPK3 (amino acids 2-353), 50 ng recombinant full-length mouse MLKL, and 200 μM ATP in kinase buffer (50 mM HEPES pH 7.5, 20 mM MgCl2, 1 mM TCEP). Each reaction was performed at 30°C for 30 min and terminated by the addition of the ADP-Glo reagent. After 40 min, the Kinase Detection reagent was added and left for 30 min before luminescence was detected with a microplate reader (CLARIOstar, BMG LabTech). Assays were performed in triplicate, with wild-type RIPK3 assayed alongside RIPK3K51A, RIPK3D143N, and RIPK3D161N mutants.
Assessments of colitis severity
Colonoscopies on mice were performed every four weeks from 6 to 24 weeks of age using established method [68, 69]. Briefly, mice were anaesthetized via the gradual increase of isoflurane (0.5–5% v/v), pedal reflex was checked to ensure deep anesthesia, the endoscope probe inserted into the rectum (Coloview miniendoscopic system including Endovision Tricam (Karl Storz Cat#20212001-020); Xenon 175 light source with anti-fog pump (Karl Storz Cat#20134001); HOPKINS straight Forward Telescope (Karl Storz Cat#64301 A); Endoscopic Sheath (total diameter 3 mm; Kalr Stroz Cat#61029 C); Fiber Optic Light Cable (Kalr Stroz Cat#69495ND)) and then a video recorded with computer and media player software (Apple Inc., iMovie). Colonoscopy videos were scored using the murine endoscopic index of colitis severity (MEICS) rubric [68] in a blinded fashion.
Mouse dermal fibroblasts and bone marrow derived macrophages culturing
Mouse dermal fibroblasts (MDF) were prepared from the skin of mouse tails then immortalized via stable lentiviral transduction with SV40 large T antigen as described previously [32] and maintained in DMEM + 8% fetal calf serum. Bone marrow-derived macrophages (BMDM) were generated from the marrow harvested of mice femurs. Harvested cells were maintained in DMEM supplemented with 20% L929 conditioned media and 8% fetal calf serum for at least 7 days before experimentation. All cells were maintained at 37°C under humidified conditions and 10% v/v carbon dioxide.
Induction and quantification of cell death
MDF and BMDM were seeded at 20,000 cells/well and 120,000 cells/well in 48-well plates, respectively. The next day, cells were treated with stimuli in DMEM supplemented with 1:20000 SYTOX Green (Invitrogen, Cat#S7020) ± 1:1000 SPY620-DNA (Spirochrome, Cat#SC401) or in DMEM supplemented with 1:2000 propidium iodide (ThermoFisher Scientific; Cat#P3566) ± 1:2000 SPY700-DNA (Spirochrome, Cat#SC601). The stimuli used were: 10 ng/mL doxycycline, 5 μM IDN-6556, 10 μM GSK′843, 100 ng/mL tumor necrosis factor, 500 nM Smac mimetic compound A. Directly after stimulation, cells were moved into an IncuCyte SX5 System (Essen Bioscience) and imaged using the 10x objective over time using the default bright-field, green, red and far-red channel settings. SYTOX Green+ or propidium iodide+ dead cells were quantified using IncuCyte SX5 v2022B software (Essen Bioscience).
Immunofluorescence and image acquisition
Ripk3–/– MDFs with doxycycline-inducible expression constructs for RIPK3WT, RIPK3K51A, RIPK3D143N or RIPK3D161N were seeded at 15,000 cells/well into 8-well chamber slides (Ibidi Cat#80826). 2 h later cells were treated with 10 ng/mL doxycycline and 5 μm IDN-6556, then 9 h later cells were treated ±10 μM GSK′843. 90 min later, slide was ice-chilled for 3 min, then washed in ice-cold dPBS, then fixed for 15 min in ice-cold methanol. Cells were washed once in ice-cold dPBS, then blocked in ice-cold Tris-balanced salt solution with 0.05% v/v Triton-X100 (TBS + T) supplemented with 10% v/v donkey serum (Sigma #D9663) for >1 h. Cells were incubated in 2 μg/mL anti-RIPK3 antibody (clone 1H12; ref. [63]; produced in-house) overnight at 4 °C in TBS + T with 10% v/v donkey serum. Cells were washed twice in TBS + T then incubated in 1:10000 AlexaFluor488-conjugated donkey anti-rat IgG supplemented with 0.1 μg/mL Hoechst 33342 (ThermoFisher Scientific Cat#H3570) for 3 h at room temperature with gentle rocking. Cells were washed four times in ice-cold TBS + T then stored at 4 °C until being imaged on an Inverted Axio Observer.Z1 microscope (Zeiss) with the following specifications: Plan-Apochromat 100x/1.4 Oil Ph3 M27 lens, HXP 120 V excitation source, AlexaFluor488 imaged with a λExcitation = 450–490 nm; λbeamsplitter = 495 nm; λEmission = 500–550 nm, Hoechst 33342 imaged with a λExcitation = 359–371 nm; λbeamsplitter = 395 nm; λEmission = 397-∞ nm, a sCMOS PCO.edge 4.2 camera, ZEN blue v3.10.103 capture software and ImageJ 1.54p post-acquisition processing software [70]. Typically, for each independent experiment, 5–10 randomly selected fields were captured per treatment group, where only the Hoechst 33342 signal was visualised prior to multi-channel acquisition. To ensure consistent signal intensities across independent experiments, the same excitation, emission and camera settings were used throughout this study.
Histopathology of mice
Comprehensive full body necropsy, Haematoxylin and eosin (H&E) images, blood analysis reports of wildtype n = 3 and Ripk3D143N/D143N n = 3 littermates between 7-10 months of age were prepared by veterinary and medical pathologists from Phenomics Australia, Melbourne Victoria.
Hematological analysis
Submandibular bloods of mice from 2 to 13 months of age were collected into EDTA-coated tubes (Sarstedt Microvette® 500 EDTA K3E, 500 µL, Order number: 20.1341). Blood samples were diluted by a factor of 1-13-fold in dPBS and blood cells quantified with ADVIA 2120 hematological analyzer on the same day as blood collection.
Periodic acid–Schiff stain
The manual staining protocol for Periodic acid–Schiff stain (PAS, Sigma-Aldrich, Cat#1090330500) is detailed in Supplementary Table 1.
Acquisition of (immuno)histochemistry images
Stained slides were scanned on: Olympus VS200 (objective: 20x, numerical aperture 0.8, media dry; software: Olympus VS200 ASW 3.41). Representative full-resolution 8-bit RGB micrographs of tissues were imported into ImageJ 1.53t [70]. Capture settings and post-acquisition image transformations were held constant between any micrographs that were being compared.
Assessment of Villin.Cre specificity
Fresh frozen tissue sections from ROSA26 Ai9 Villin.Cre1000 mice and wild-type control mice were mounted in DAPI-containing media and imaged on a Vectra Polaris Imaging System (Akoya Biosciences), as described previously [63]. Acquisition software: Vectra Polaris v.1.0. Resolution: 0.5 μm/pixel (×20 objective). LED light source. Default filters for DAPI MSI (4 s exposure) and Texas Red (150 s exposure) were used.
Quantification of cells by IHC
Full resolution scans of IHC stained tissue sections were opened in QuPATH software v.0.5.1. In brief, a selection tool is used to highlight an area of an average of 50% of Swiss rolls. IHC signal was unmixed from Hematoxylin using the “Color Deconvolution 2” plugin [71]. IHC-stain positive cells were segmented and quantified. Percentage of PAS+ cells as total cells were calculated with two scripts, “PAS+ cell detection” and “Total cell count”. Percentage of Lysozyme+ or MPTX2+ cells were calculated with “Lysozyme+ cell detection and total cell count”. “Threshold” for each script was adjusted with each staining batch due to natural variations, and tissue sections from mouse models of inflammation were accompanied by wildtype littermate controls on the same slide. The scripts are provided in Supplementary Table 2.
Statistical analysis
The number of independent experiments for each dataset is stipulated in the respective figure legend. The employed statistical tests are stipulated in the respective figure legend and performed using Prism v.10 (GraphPad). Statistical tests were only applied to quantitative data collated from 3 or more independent replicas.
Data availability
The uncropped blots from this study are included as Supplementary Data. Other raw data from this study are available from the corresponding authors upon request.
Materials availability
Any materials from this study that are not commercially available will be provided under Materials Transfer Agreement upon request to the corresponding authors.
References
Nakano H, Murai S, Moriwaki K. Regulation of the release of damage-associated molecular patterns from necroptotic cells. Biochem J. 2022;479:677–85.
Fletcher-Etherington A, Nobre L, Nightingale K, Antrobus R, Nichols J, Davison AJ, et al. Human cytomegalovirus protein pUL36: a dual cell death pathway inhibitor. Proc Natl Acad Sci USA. 2020;117:18771–9.
Jiao H, Wachsmuth L, Kumari S, Schwarzer R, Lin J, Eren RO, et al. Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature. 2020;580:391–5.
Kitur K, Wachtel S, Brown A, Wickersham M, Paulino F, Penaloza HF, et al. Necroptosis promotes staphylococcus aureus clearance by inhibiting excessive inflammatory signaling. Cell Rep. 2016;16:2219–30.
Liu Z, Nailwal H, Rector J, Rahman MM, Sam R, McFadden G, et al. A class of viral inducer of degradation of the necroptosis adaptor RIPK3 regulates virus-induced inflammation. Immunity. 2021;54:247–58 e247.
Newton K, Wickliffe KE, Maltzman A, Dugger DL, Strasser A, Pham VC, et al. RIPK1 inhibits ZBP1-driven necroptosis during development. Nature. 2016;540:129–33.
Pearson JS, Giogha C, Muhlen S, Nachbur U, Pham CL, Zhang Y, et al. EspL is a bacterial cysteine protease effector that cleaves RHIM proteins to block necroptosis and inflammation. Nat Microbiol. 2017;2:16258.
Petrie EJ, Sandow JJ, Lehmann WIL, Liang LY, Coursier D, Young SN, et al. Viral MLKL Homologs Subvert Necroptotic Cell Death by Sequestering Cellular RIPK3. Cell Rep. 2019;28:3309–19 e3305.
Zhang T, Yin C, Boyd DF, Quarato G, Ingram JP, Shubina M, et al. Influenza virus Z-RNAs induce ZBP1-mediated necroptosis. Cell. 2020;180:1115–29 e1113.
Kolbrink B, von Samson-Himmelstjerna FA, Murphy JM, Krautwald S. Role of necroptosis in kidney health and disease. Nat Rev Nephrol. 2023;19:300–14.
Pefanis A, Ierino FL, Murphy JM, Cowan PJ. Regulated necrosis in kidney ischemia-reperfusion injury. Kidney Int. 2019;96:291–301.
Muller T, Dewitz C, Schmitz J, Schroder AS, Brasen JH, Stockwell BR, et al. Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure. Cell Mol Life Sci. 2017;74:3631–45.
Lu Z, Van Eeckhoutte HP, Liu G, Nair PM, Jones B, Gillis CM, et al. Necroptosis signaling promotes inflammation, airway remodeling, and emphysema in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2021;204:667–81.
Chiou S, Al-Ani AH, Pan Y, Patel KM, Kong IY, Whitehead LW, et al. An immunohistochemical atlas of necroptotic pathway expression. EMBO Mol Med. 2024;16:1717–49.
Pierdomenico M, Negroni A, Stronati L, Vitali R, Prete E, Bertin J, et al. Necroptosis is active in children with inflammatory bowel disease and contributes to heighten intestinal inflammation. Am J Gastroenterol. 2014;109:279–87.
Pang J, Al-Ani AH, Patel KM, Young SN, Kong I, Chen J-j, et al. A necroptotic-to-apoptotic signaling axis underlies inflammatory bowel disease. bioRxiv 2024: 2024.2011.2013.623307.
Anderton H, Alqudah S. Cell death in skin function, inflammation, and disease. Biochem J. 2022;479:1621–51.
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–9.
Devos M, Tanghe G, Gilbert B, Dierick E, Verheirstraeten M, Nemegeer J, et al. Sensing of endogenous nucleic acids by ZBP1 induces keratinocyte necroptosis and skin inflammation. J Exp Med. 2020;217:e20191913.
Newton K, Dugger DL, Maltzman A, Greve JM, Hedehus M, Martin-McNulty B, et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 2016;23:1565–76.
Schwarzer R, Jiao H, Wachsmuth L, Tresch A, Pasparakis M. FADD and Caspase-8 Regulate Gut Homeostasis and Inflammation by Controlling MLKL- and GSDMD-Mediated Death of Intestinal Epithelial Cells. Immunity. 2020;52:978–93 e976.
Wu MCL, Italiano E, Jarvis-Child R, Alwis I, Smythe R, Albornoz EA, et al. Ischaemic endothelial necroptosis induces haemolysis and COVID-19 angiopathy. Nature. 2025;643:182–91.
Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–23.
He S, Wang L, Miao L, Wang T, Du F, Zhao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–11.
Pham CLL, Titaux-Delgado GA, Varghese NR, Polonio P, Wilde KL, Sunde M, et al. NMR characterization of an assembling RHIM (RIP homotypic interaction motif) amyloid reveals a cryptic region for self-recognition. J Biol Chem. 2023;299:104568.
Wu X, Ma Y, Zhao K, Zhang J, Sun Y, Li Y, et al. The structure of a minimum amyloid fibril core formed by necroptosis-mediating RHIM of human RIPK3. Proc Natl Acad Sci USA. 2021;118:e2022933118.
Mompean M, Li W, Li J, Laage S, Siemer AB, Bozkurt G, et al. The Structure of the Necrosome RIPK1-RIPK3 Core, a Human Hetero-Amyloid Signaling Complex. Cell. 2018;173:1244–53 e1210.
Meng Y, Davies KA, Fitzgibbon C, Young SN, Garnish SE, Horne CR, et al. Human RIPK3 maintains MLKL in an inactive conformation prior to cell death by necroptosis. Nat Commun. 2021;12:6783.
Meng Y, Horne CR, Samson AL, Dagley LF, Young SN, Sandow JJ, et al. Human RIPK3 C-lobe phosphorylation is essential for necroptotic signaling. Cell Death Dis. 2022;13:565.
Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–27.
Chen W, Zhou Z, Li L, Zhong CQ, Zheng X, Wu X, et al. Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling. J Biol Chem. 2013;288:16247–61.
Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39:443–53.
Garnish SE, Meng Y, Koide A, Sandow JJ, Denbaum E, Jacobsen AV, et al. Conformational interconversion of MLKL and disengagement from RIPK3 precede cell death by necroptosis. Nat Commun. 2021;12:2211.
Meng Y, Garnish SE, Davies KA, Black KA, Leis AP, Horne CR, et al. Phosphorylation-dependent pseudokinase domain dimerization drives full-length MLKL oligomerization. Nat Commun. 2023;14:6804.
Petrie EJ, Birkinshaw RW, Koide A, Denbaum E, Hildebrand JM, Garnish SE, et al. Identification of MLKL membrane translocation as a checkpoint in necroptotic cell death using Monobodies. Proc Natl Acad Sci USA. 2020;117:8468–75.
Petrie EJ, Sandow JJ, Jacobsen AV, Smith BJ, Griffin MDW, Lucet IS, et al. Conformational switching of the pseudokinase domain promotes human MLKL tetramerization and cell death by necroptosis. Nat Commun. 2018;9:2422.
Samson AL, Garnish SE, Hildebrand JM, Murphy JM. Location, location, location: A compartmentalized view of TNF-induced necroptotic signaling. Sci Signal. 2021;14:eabc6178.
Samson AL, Zhang Y, Geoghegan ND, Gavin XJ, Davies KA, Mlodzianoski MJ, et al. MLKL trafficking and accumulation at the plasma membrane control the kinetics and threshold for necroptosis. Nat Commun. 2020;11:3151.
Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell. 2014;56:481–95.
Newton K, Sun X, Dixit VM. Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol. 2004;24:1464–9.
Tovey Crutchfield EC, Garnish SE, Day J, Anderton H, Chiou S, Hempel A, et al. MLKL deficiency protects against low-grade, sterile inflammation in aged mice. Cell Death Differ. 2023;30:1059–71.
Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343:1357–60.
Li D, Chen J, Guo J, Li L, Cai G, Chen S, et al. A phosphorylation of RIPK3 kinase initiates an intracellular apoptotic pathway that promotes prostaglandin(2alpha)-induced corpus luteum regression. Elife. 2021;10:e67409.
Newton K, Wickliffe KE, Maltzman A, Dugger DL, Webster JD, Guo H, et al. Caspase cleavage of RIPK3 after Asp(333) is dispensable for mouse embryogenesis. Cell Death Differ. 2024;31:254–62.
Tovey Crutchfield EC, Garnish SE, Hildebrand JM. The role of the key effector of necroptotic cell death, MLKL, in mouse models of disease. Biomolecules. 2021;11:biom11060803.
Tran HT, Kratina T, Coutansais A, Michalek D, Hogan BM, Lawlor KE, et al. RIPK3 cleavage is dispensable for necroptosis inhibition but restricts NLRP3 inflammasome activation. Cell Death Differ. 2024;31:662–71.
Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, et al. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 1998;9:267–76.
Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471:368–72.
Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363–7.
Alvarez-Diaz S, Dillon CP, Lalaoui N, Tanzer MC, Rodriguez DA, Lin A, et al. The pseudokinase MLKL and the kinase RIPK3 have distinct roles in autoimmune disease caused by loss of death-receptor-induced apoptosis. Immunity. 2016;45:513–26.
Lehle AS, Farin HF, Marquardt B, Michels BE, Magg T, Li Y, et al. Intestinal inflammation and dysregulated immunity in patients with inherited caspase-8 deficiency. Gastroenterology. 2019;156:275–8.
Gunther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, et al. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature. 2011;477:335–9.
Kaya GG, Schwarzer R, Dannappel M, Vlantis K, Gobel U, Kondylis V, et al. Unfolded protein response transcription factor XBP1 suppresses necroptosis-induced colitis by reinforcing the mucus barrier. Immunity. 2025;58:2208–25 e2206.
Patankar JV, Muller TM, Kantham S, Acera MG, Mascia F, Scheibe K, et al. E-type prostanoid receptor 4 drives resolution of intestinal inflammation by blocking epithelial necroptosis. Nat Cell Biol. 2021;23:796–807.
Gardner CR, Davies KA, Zhang Y, Brzozowski M, Czabotar PE, Murphy JM, et al. From (tool)bench to bedside: the potential of necroptosis inhibitors. J Med Chem. 2023;66:2361–85.
Gautam A, Boyd DF, Nikhar S, Zhang T, Siokas I, Van de Velde LA, et al. Necroptosis blockade prevents lung injury in severe influenza. Nature. 2024;628:835–43.
Xia K, Zhu F, Yang C, Wu S, Lin Y, Ma H, et al. Discovery of a potent RIPK3 inhibitor for the amelioration of necroptosis-associated inflammatory injury. Front Cell Dev Biol. 2020;8:606119.
Berger SB, Kasparcova V, Hoffman S, Swift B, Dare L, Schaeffer M, et al. Cutting Edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J Immunol. 2014;192:5476–80.
Laurien L, Nagata M, Schunke H, Delanghe T, Wiederstein JL, Kumari S, et al. Autophosphorylation at serine 166 regulates RIP kinase 1-mediated cell death and inflammation. Nat Commun. 2020;11:1747.
Koerner L, Li X, Silnov E, Laurien L, Pasparakis M. RIPK1 autophosphorylation at S161 mediates cell death and inflammation. J Exp Med. 2025;222:e20250279.
Salmena L, Lemmers B, Hakem A, Matysiak-Zablocki E, Murakami K, Au PY, et al. Essential role for caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev. 2003;17:883–95.
Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–40.
Samson AL, Fitzgibbon C, Patel KM, Hildebrand JM, Whitehead LW, Rimes JS, et al. A toolbox for imaging RIPK1, RIPK3, and MLKL in mouse and human cells. Cell Death Differ. 2021;28:2126–44.
Haber AL, Biton M, Rogel N, Herbst RH, Shekhar K, Smillie C, et al. A single-cell survey of the small intestinal epithelium. Nature. 2017;551:333–9.
Sethi A, Horne CR, Fitzgibbon C, Wilde K, Davies KA, Garnish SE, et al. Membrane permeabilization is mediated by distinct epitopes in mouse and human orthologs of the necroptosis effector, MLKL. Cell Death Differ. 2022;29:1804–15.
Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, Sharma P, et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci USA. 2014;111:15072–7.
Fitzgibbon C, Meng Y, Murphy JM. Co-expression of recombinant RIPK3:MLKL complexes using the baculovirus-insect cell system. Methods Enzymol. 2022;667:183–227.
Mielke L, Preaudet A, Belz G, Putoczki T. Confocal laser endomicroscopy to monitor the colonic mucosa of mice. J Immunol Methods. 2015;421:81–88.
Ernst M, Preaudet A, Putoczki T. Non-invasive assessment of the efficacy of new therapeutics for intestinal pathologies using serial endoscopic imaging of live mice. J Vis Exp. 2015;97:e52383.
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–82.
Landini G, Martinelli G, Piccinini F. Colour deconvolution: stain unmixing in histological imaging. Bioinformatics. 2021;37:1485–7.
Xie T, Peng W, Yan C, Wu J, Gong X, Shi Y. Structural insights into RIP3-mediated necroptotic signaling. Cell Rep. 2013;5:70–78.
Acknowledgements
We thank Bruce Rosengarten and Wayne Cawthorne for helpful discussions; WEHI Bioservices, WEHI Histology, the WEHI Monoclonal Antibody Facility for technical assistance; Gary Kasof and Cell Signaling Technology for generously sharing antibodies; Professor Marc Pellegrini for the Casp8fl/fl strain; and Tina Cardamone and Dr John Finnie from Phenomics Australia Histopathology and Slide Scanning Service for their assistance with phenotyping the Ripk3D143N/D143N mouse strain. The generation of Ripk3D143N/D143N mice used in this study was supported by Phenomics Australia and the Australian Government through the National Collaborative Research Infrastructure Strategy (NCRIS) program.
Funding
This work was supported by National Health and Medical Research Council of Australia grants (1172929 and 2034104 to JMM, 2008652 to EDH, 2002965 to ALS, 1195038 to JS, 2034044 to CRH; IRIISS), and by the Victorian State Government Operational Infrastructure Support scheme. TLP is supported by a Viertel Senior Medical Research Fellowship. JMM and JS received research funding from Anaxis Pharma Pty Ltd and AH was funded by Anaxis Pharma Pty Ltd. We acknowledge scholarship support for SC (Australian Government Research Training Program Stipend Scholarship, WEHI Handman PhD Scholarship). Open Access funding enabled and organized by CAUL and its Member Institutions.
Author information
Authors and Affiliations
Contributions
Conceptualization: SC, EDH, ALS, JMM. Investigation: SC, CRH, KMP, AP, JAR, SNY, AJ, SEG, AH, CH, JMH, AJK, ALS; Methodology: SC, KMP, AJK, TLP, ALS. Resources: JS, TLP, EDH, ALS, JMM. Supervision: EDH, ALS, JMM. Funding acquisition: EDH, ALS, JMM. Writing: SC, ALS and JMM co-wrote the paper with input from authors.
Corresponding authors
Ethics declarations
Competing interests
KMP, CRH, SNY, AH, JMH, JS, ALS and JMM contribute to or have contributed to a project developing necroptosis inhibitors in collaboration with Anaxis Pharma. TLP is cofounder and shareholder in Nelcanen Therapeutics. The other authors declare no competing interests.
Ethics
All experiments were approved by the WEHI Animal Ethics Committee following the Prevention of Cruelty to Animals Act (1996) and the Australian National Health and Medical Research Council Code of Practice for the Care and Use of Animals for Scientific Purposes (1997).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chiou, S., Horne, C.R., Patel, K.M. et al. The kinase domain of RIPK3 tunes its scaffolding functions. Cell Death Differ (2026). https://doi.org/10.1038/s41418-026-01677-x
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41418-026-01677-x
This article is cited by
-
RIPK3’s shape-shifting scaffold: how kinase domain conformation fine-tunes necroptosis
Cell Death & Differentiation (2026)
