
Abstract
Benzo[a]pyrene (B[a]P), an environmental neurotoxin, induces cognitive decline through ferroptosis-mediated mechanisms. Transcriptomic analysis (GSE75206) of B[a]P-exposed mouse hippocampus identified 1668 differentially expressed genes, with Plin4 emerging as a key regulator linked to ferroptosis and lipid droplet (LD) accumulation. Behavioral tests confirmed hippocampal-dependent cognitive impairment and Plin4 upregulation. Molecular analyses demonstrated ferroptosis activation, evidenced by altered expression of related genes (Gpx4, Slc7a11, Ptgs2) and biochemical markers of lipid peroxidation and iron imbalance. In HT22 cells, Benzopyrene-7,8-Diol-9,10-Epoxide (BPDE) dose-dependently elevated Plin4 expression, inducing mitochondrial damage and ferroptosis. Silencing Plin4 reversed BPDE-induced ferroptosis by restoring redox balance, reducing LD accumulation, and improving mitochondrial integrity. Mechanistically, Plin4 amplifies B[a]P neurotoxicity by exacerbating iron overload and LD accumulation, sensitizing neurons to ferroptosis. This study identifies Plin4 as a central mediator of environmental pollutant-induced neurodegeneration and proposes it as a therapeutic target for ferroptosis-related cognitive disorders.
Similar content being viewed by others


Introduction
Benzo[a]pyrene (B[a]P), a potent polycyclic aromatic hydrocarbon and pervasive environmental contaminant, is primarily introduced into the human body through inhalation of polluted air, cigarette smoke, and the consumption of charred foods [1, 2]. While B[a]P is well established as a carcinogen, accumulating evidence suggests it also exerts substantial neurotoxic effects. Both epidemiological and experimental studies have linked B[a]P exposure to cognitive decline and an increased risk of neurodegenerative disorders, including Alzheimer’s disease (AD), Parkinson’s disease, Huntington’s disease, and amyotrophic lateral sclerosis [3, 4]. These conditions are commonly associated with oxidative stress, dysregulated iron metabolism, and cellular dysfunction [5,6,7]. However, the molecular mechanisms by which B[a]P contributes to neurotoxicity remain incompletely understood.
Ferroptosis, a recently identified form of programmed cell death, has emerged as a critical mechanism in a range of pathological conditions, including neurodegeneration, cancer, and organ injury [8]. It is characterized by iron-dependent lipid peroxidation, leading to excessive generation of reactive oxygen species (ROS), oxidative membrane damage, and ultimately, cell death [9]. Unlike apoptosis or necrosis, ferroptosis is governed by a complex interplay between iron metabolism and oxidative stress [10, 11]. Given the essential role of iron in maintaining neuronal function, dysregulation of iron homeostasis has been increasingly recognized as a major contributor to neurodegenerative diseases [12, 13].
Lipid peroxidation is a central feature of ferroptosis, primarily resulting from the oxidation of polyunsaturated fatty acids, such as arachidonic acid and linoleic acid, which are highly vulnerable to reactive oxygen species (ROS) during oxidative stress [11]. Lipid droplets (LDs), which govern intracellular lipid storage, have been increasingly linked to the modulation of oxidative stress responses and cellular susceptibility to ferroptosis [14, 15]. Nevertheless, the precise role of LD-associated mechanisms in B[a]P-induced ferroptosis remains poorly understood.
Our prior investigations revealed that 7,8-dihydroxy-9,10-epoxybenzo[a]pyrene (BPDE), the biologically active metabolite of B[a]P, induces ferroptosis in both rat primary cortical neurons and human SH-SY5Y neuroblastoma cells in a dose- and time-dependent manner. These findings implicate ferroptosis as a central mechanism in B[a]P-induced neurotoxicity. Nevertheless, the precise molecular pathways by which B[a]P initiates ferroptosis—particularly those involving lipid droplet–associated processes—remain largely unexplored.
Therefore, this study aims to elucidate the molecular mechanisms underlying B[a]P-induced ferroptosis. By identifying key regulatory pathways and mediators, we seek to advance understanding of the neurotoxic effects of environmental pollutants and inform the development of therapeutic strategies to mitigate ferroptosis-related neurodegeneration.
Results
Identification of Key Gene Plin4 in the hippocampus of B[a]P-exposed mice based on GEO data and bioinformatics analysis
In a prior study by Chepelev et al., behavioral assessments indicated impairments in learning and memory, suggesting significant neurotoxicity due to B[a]P exposure [16]. In their 2016 work, an acute B[a]P exposure model was established using adult male Muta™ Mice. Animals received oral doses of B[a]P at 0, 1, 35, or 70 mg/kg body weight per day for three consecutive days. Transcriptomic analysis revealed alterations in synaptic function—such as upregulation of N-methyl-D-aspartate receptor subunits Grina and Grin2a—without notable activation of DNA damage response genes, despite the presence of B[a]P-DNA adducts. These findings suggest that B[a]P-induced neurotoxicity may arise primarily from disrupted synaptic signaling rather than direct genotoxic damage.
To further investigate the molecular underpinnings of B[a]P neurotoxicity, we conducted a comprehensive bioinformatics analysis using the hippocampal transcriptome dataset generated by Chepelev et al., available from the NCBI GEO database (accession number GSE75206). High-throughput RNA sequencing data from hippocampal tissues were analyzed to identify transcriptional changes associated with B[a]P exposure. Differential expression analysis (FDR-adjusted P-value < 0.05, |log₂FC| ≥ 3) revealed 1,668 significantly differentially expressed genes (DEGs). A heatmap illustrated the clustering patterns of gene expression profiles (Fig. 1a), and a volcano plot displayed the distribution of DEGs (Fig. 1b), highlighting the extent of transcriptional perturbation caused by B[a]P.
![Fig. 1: Bioinformatics Analysis Based on GEO Data Identifies Key Gene Plin4 in Hippocampal Tissue of B[a]P-Treated Mice.](http://media.springernature.com/lw685/springer-static/image/art%3A10.1038%2Fs41420-025-02747-8/MediaObjects/41420_2025_2747_Fig1_HTML.png?as=webp)
a Heatmap showing gene expression changes in the hippocampus of mice following acute B[a]P exposure. b Volcano plot showing the overall distribution of differentially expressed genes, with upregulated and downregulated genes marked based on FDR correction (FDR P-value < 0.05, |log2FC| ≥ 3). c The GO and KEGG pathway enrichment analysis results show biological functions and signaling pathways associated with the DEGs, including neurotransmitter receptor activity, synaptic plasticity, membrane potential regulation, and neurodegenerative diseases such as AD and ferroptosis. d PPI network illustrating interactions between DEGs, with BC analysis identifying genes key to lipid metabolism and neurobiology. e KMeans clustering analysis categorizing genes into three clusters based on expression levels (log2FC) and BC scores, with Cluster 1 showing a strong association with lipid metabolism. f Venn diagram showing overlap between KMeans clustering and ferroptosis-related genes, identifying Plin4 and Slc2a1 as key genes, with Plin4 significantly upregulated in hippocampal tissue of B[a]P-treated mice, suggesting its role in neurotoxicity. g Comparison of expression levels between Plin4 and Slc2a1, with Plin4 identified as a core gene in B[a]P-induced neurotoxicity. Data are presented as mean ± SEM(n = 4 mice per group) *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To elucidate the functional implications of these DEGs, we performed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses (Fig. 1c). At the molecular function level, DEGs were significantly enriched in passive transmembrane transporter activity, metal ion transmembrane transporter activity, and neurotransmitter receptor activity—particularly in processes regulating postsynaptic membrane potential—suggesting critical roles in maintaining neuronal signaling and membrane stability. At the biological process level, enriched terms included synaptic plasticity, cognitive function, and membrane potential regulation. At the cellular component level, DEGs were mainly associated with synaptic and postsynaptic membranes as well as glutamatergic synapses. KEGG analysis further revealed enrichment in pathways related to neurodegenerative diseases, glutamatergic synapse signaling, calcium signaling, and notably, ferroptosis—implying a possible role for ferroptosis in B[a]P-induced neurotoxicity.
To examine gene–gene interactions and identify biologically relevant regulators, we constructed a PPI network using the STRING database (https://string-db.org/) and visualized it in Cytoscape. After removing non-interacting genes, we calculated betweenness centrality (BC) scores using the CytoNCA plugin to pinpoint hub genes with regulatory potential (Fig. 1d). This network analysis highlighted genes involved in adipocyte differentiation, fatty acid degradation, and lipid metabolism—processes increasingly recognized as critical in neural homeostasis and pathology.
We then normalized both log₂FC and BC scores and applied KMeans clustering to classify DEGs based on both expression magnitude and network centrality. This approach grouped genes into three distinct clusters (Cluster 0, Cluster 1, Cluster 2), with Cluster 1 showing the strongest association with lipid metabolic processes based on literature evidence (Fig. 1e). To further narrow down potential mechanistic targets, we intersected Cluster 1 genes with ferroptosis-related genes curated in the FerrDb database (http://www.zhounan.org/ferrdb), identifying two candidates: Slc2a1 and Plin4 (Fig. 1f). Comparative expression analysis demonstrated a more pronounced upregulation of Plin4 in the hippocampus of B[a]P-exposed mice relative to Slc2a1 (Fig. 1g). Given Plin4’s known role in lipid droplet formation and its emerging link to oxidative stress regulation, we identified Plin4 as a potential key mediator of B[a]P neurotoxicity.
Taken together, our analysis suggests that B[a]P exposure leads to upregulation of Plin4, which may disrupt lipid metabolism homeostasis and promote ferroptosis—an iron-dependent form of programmed cell death characterized by lipid peroxidation. This mechanism could underlie the neuronal damage and cognitive deficits observed in B[a]P-exposed mice, as initially reported by Chepelev et al. We therefore propose that Plin4-mediated ferroptosis plays a critical role in B[a]P-induced neurotoxicity. This mechanistic insight not only advances our understanding of environmental pollutant-related neurodegeneration but may also offer a novel therapeutic target for mitigating B[a]P-associated cognitive impairment.
Subacute Exposure to B[a]P Induces Ferroptosis and High Expression of Plin4 in Mouse Hippocampal Tissue
To test our hypothesis, we established a subacute B[a]P exposure mouse model to validate the bioinformatics analysis results. Mice received daily intraperitoneal injections of 10 mg/kg B[a]P for 30 consecutive days, successfully replicating subacute exposure conditions. Figure 2a outlines the experimental procedures, including model establishment, behavioral testing, and gene expression analysis. The movement trajectories of mice during the Morris water maze and Y-maze tests are shown in Fig. 2b, c, respectively.
![Fig. 2: Subacute B[a]P exposure induces ferroptosis and upregulates Plin4 expression in mouse hippocampal tissue.](http://media.springernature.com/lw685/springer-static/image/art%3A10.1038%2Fs41420-025-02747-8/MediaObjects/41420_2025_2747_Fig2_HTML.png?as=webp)
a Experimental workflow illustrating the steps involved in the construction of the B[a]P subacute exposure mouse model, followed by behavioral experiments and gene expression analysis. b Motion trajectory of mice in the MWM experiment. c Motion trajectory of mice in the Y-maze experiment. d Comparison of escape latency in the MWM experiment. e Comparison of platform crossings in the MWM experiment. f Comparison of time spent in the target quadrant in the MWM experiment. g Comparison of the number of new arm entries in the Y-maze experiment. h Comparison of the time spent exploring new arms in the Y-maze experiment. i–l qPCR results showing the relative expression levels of Gpx4, Slc7a11, Ptgs2, and Plin4 in hippocampal tissue. m Total GSH levels in hippocampal tissue. n GSH-Px activity in hippocampal tissue. o MDA content in hippocampal tissue. p Fe²⁺ levels in hippocampal tissue. Data are presented as mean ± SEM(n = 6 mice per group, Ctrl and B[a]P groups) *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
In the water maze, B[a]P-treated mice exhibited significantly prolonged escape latencies (Fig. 2d), as well as fewer platform crossings and reduced time spent in the target quadrant (Fig. 2e, f). The Y-maze results further supported cognitive impairment, as treated mice showed decreased entries into and time spent in the novel arm (Fig. 2g, h), indicating impaired spatial memory and exploratory behavior.
To investigate the molecular mechanisms underlying these behavioral deficits, we conducted qPCR analysis of hippocampal tissues. Expression of Gpx4 and Slc7a11 was significantly downregulated (Fig. 2i, j), while Ptgs2 expression was markedly upregulated (Fig. 2k) in the B[a]P-treated group. Notably, Plin4 expression was significantly increased in the hippocampus, consistent with our bioinformatics predictions (Fig. 2l).
We further evaluated ferroptosis-related biochemical markers. GSH levels were decreased (Fig. 2m), whereas GSH-Px activity was significantly reduced (Fig. 2n). Additionally, MDA content and Fe²⁺ levels were both significantly increased (Fig. 2o, p), supporting the activation of ferroptosis.
Our bioinformatics analysis of the GSE75206 dataset identified Plin4 as a candidate gene potentially involved in B[a]P-induced neurotoxicity. This was corroborated in vivo by our subacute exposure model, which demonstrated cognitive impairment alongside increased hippocampal Plin4 expression. The observed molecular and biochemical changes—including reduced Gpx4 and Slc7a11, elevated Ptgs2, and disrupted ferroptosis indicators—strongly implicate ferroptosis as a key mechanism in B[a]P-induced cognitive dysfunction. Together, these findings suggest that Plin4 may contribute to neurotoxicity through modulation of ferroptosis, providing novel insights into the mechanisms of B[a]P-related brain injury.
Plin4 regulates ferroptosis in BPDE-induced hippocampal neurons
Upregulation of Plin4 in BPDE-induced hippocampal neurons
Building on our in vivo observations that Plin4 expression is upregulated in the hippocampus of B[a]P-exposed mice, we further explored the role of Plin4 in B[a]P-induced neuronal injury at the cellular level. To dissect the specific contribution of Plin4, we conducted in vitro studies using primary mouse hippocampal neurons and the mouse hippocampal neuronal cell line HT22. Given that in vitro systems do not fully recapitulate the complexity of in vivo metabolic processes, we employed BPDE—the biologically active metabolite of B[a]P—as a surrogate for B[a]P exposure. The experimental workflow is depicted in Fig. 3a.
![Fig. 3: Expression of Plin4 in B[a]P-induced hippocampal neuronal death.](http://media.springernature.com/lw685/springer-static/image/art%3A10.1038%2Fs41420-025-02747-8/MediaObjects/41420_2025_2747_Fig3_HTML.png?as=webp)
a Experimental workflow summarizing the steps of the current analysis. b Survival rate changes of primary mouse hippocampal neurons under different concentrations of BPDE treatment. The survival rate decreases as BPDE concentration increases. c Relative expression of Plin4 in primary mouse hippocampal neurons under BPDE treatment, with a significant upregulation of Plin4 expression. d Survival rate changes of HT22 cells under different concentrations of BPDE treatment, showing a dose-dependent decrease in survival rate as BPDE concentration increases. e Relative expression of Plin4 in HT22 cells under BPDE treatment, with a significant upregulation of Plin4 expression as BPDE concentration increases. Data are presented as mean ± SEM(n = 6 per group, BPDE concentrations in μM) *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Cells were treated with increasing concentrations of BPDE (0 μM, 0.25 μM, 0.5 μM, 0.75 μM). As the concentration rose, both primary neurons and HT22 cells exhibited a dose-dependent decline in viability (Fig. 3b, d). Meanwhile, Plin4 expression was significantly upregulated in both cell types (Fig. 3c, e).
Primary mouse hippocampal neurons offer a physiologically relevant model that more closely mirrors in vivo conditions, while HT22 cells provide greater experimental reproducibility and technical ease. Using both models enabled cross-validation of our results, enhancing the robustness and biological significance of our findings.
In summary, our results demonstrate that neuronal Plin4 expression increases in a dose-dependent manner following BPDE exposure, supporting the hypothesis that Plin4 plays a critical role in the neurotoxic effects associated with B[a]P.
Silencing Plin4 attenuates BPDE-induced ferroptosis in HT22 cells
Based on our previous research, which showed a dose-dependent increase in Plin4 expression in hippocampal neurons after BPDE exposure, we aimed to investigate the role of Plin4 in BPDE-induced ferroptosis. Although bioinformatics analysis suggests a potential link between Plin4 and ferroptosis, the precise mechanisms remain unclear. Our prior studies demonstrated that BPDE, a metabolite of B[a]P, induces ferroptosis in hippocampal neurons. However, how the lipid metabolism-related gene Plin4 contributes to this process is still unknown. To explore the role of Plin4 in BPDE-induced ferroptosis, we performed Plin4 knockdown experiments and successfully established a Plin4-silenced HT22 cell model (Fig. 4a). The results in Fig. 4b showed that silencing Plin4 did not significantly affect cell viability.

a Schematic of the construction of the Plin4-silenced HT22 cell model. b Effect of Plin4 silencing on HT22 cell survival rate. c Antagonistic effect of Plin4 silencing on BPDE-induced HT22 cell ferroptosis, compared with the ferroptosis inhibitor Fer-1. d TEM images showing mitochondrial damage in HT22 cells after BPDE treatment. Yellow arrows indicate normal mitochondria, while red arrows indicate damaged mitochondria. e Protein bands of Slc7a11 and Gpx4, showing the expression changes after BPDE treatment and Plin4 silencing. f Relative expression level of Slc7a11 protein. g Relative expression level of Gpx4 protein. h Relative expression level of Slc7a11 mRNA. i Relative expression level of Gpx4 mRNA. j Effect of BPDE treatment on total GSH content. k Effect of BPDE treatment on GSH-Px activity. l Effect of BPDE treatment on lipid ROS levels. m Effect of BPDE treatment on MDA content. n Confocal fluorescence images showing Fe²⁺ levels, illustrating the effect of BPDE treatment and Plin4 silencing on Fe²⁺ levels. o Fluorescence intensity calculated using ImageJ software, showing the changes in Fe²⁺ levels in different groups. p Fe²⁺ level detected using a reagent kit, comparing Fe²⁺ levels between Plin4 silencing and Fer-1 treatment groups. Data are presented as mean ± SEM (n = 6 per group). Group details: a-b: Ctrl, si-NC, si-Plin4. c, f–m, o–p: Two BPDE concentrations (0 μM and 0.75 μM), treated with si-NC, si-Plin4, or Fer-1. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
We then compared the effects of the ferroptosis inhibitor Ferrostatin-1 (Fer-1) and Plin4 silencing on BPDE-induced ferroptosis in HT22 cells. BPDE treatment significantly reduced cell viability, which was partially restored by Fer-1. Similarly, Plin4 silencing significantly improved cell viability after BPDE exposure, though the recovery was less than that observed with Fer-1. This suggests that Plin4 plays a critical role in BPDE-induced ferroptosis, likely by regulating lipid peroxidation and iron metabolism (Fig. 4c).
Transmission electron microscopy (TEM) revealed that BPDE treatment caused mitochondrial cristae fragmentation, dissolution, and increased membrane density in HT22 cells. Notably, Plin4 silencing partially alleviated these mitochondrial structural defects, suggesting a protective effect on mitochondrial integrity (Fig. 4d). Yellow arrows point to normal mitochondria, while red arrows highlight damaged ones. The restoration of mitochondrial morphology in the Plin4-silenced group suggests that Plin4 may contribute to mitochondrial damage during ferroptosis, potentially through its regulation of lipid metabolism and oxidative stress.
To further elucidate the molecular mechanisms underlying BPDE-induced ferroptosis, we examined the expression of two key ferroptosis-related genes, Slc7a11 and Gpx4, as well as their corresponding proteins, Slc7a11 and Gpx4. As shown in Fig. 4h, i, BPDE (0.75 μM) treatment led to a significant downregulation of Slc7a11 and Gpx4 mRNA expression levels, indicating transcriptional suppression of ferroptosis-inhibitory factors. At the protein level, Western blot analysis (Fig. 4e; full blot images are shown in Supplementary Material) and quantification (Fig. 4f, g) revealed a marked decrease in Slc7a11 and a moderate but statistically significant reduction in Gpx4 protein levels following BPDE exposure, supporting a role for BPDE in promoting ferroptosis.
Interestingly, knockdown of Plin4 significantly restored Slc7a11 and Gpx4 protein expression (Fig. 4f, g), suggesting that Plin4 contributes to BPDE-induced ferroptosis signaling. However, Plin4 silencing had no statistically significant effect on the mRNA levels of Slc7a11 and Gpx4 (Fig. 4h and i), indicating that Plin4 may regulate these proteins primarily at the post-transcriptional level, possibly through modulation of translation efficiency or protein stability. Notably, treatment with the ferroptosis inhibitor Fer-1 selectively reversed the BPDE-induced reduction in Slc7a11 protein levels, while Gpx4 protein expression remained largely unchanged, suggesting that BPDE-induced suppression of Gpx4 may occur through a distinct or less ferroptosis-sensitive mechanism. Together, these findings suggest that Plin4 may facilitate BPDE-induced ferroptosis by promoting the post-transcriptional downregulation of Slc7a11 and Gpx4. Among them, Slc7a11 appears to be more tightly coupled to ferroptosis regulation. Thus, Plin4 may represent a previously unrecognized modulator of ferroptosis under BPDE-induced stress conditions.
After BPDE exposure, GSH levels and GSH-Px activity were significantly reduced, while lipid ROS and MDA levels increased. However, silencing Plin4 restored GSH levels and GSH-Px activity, while lipid peroxidation and MDA levels were significantly decreased, although not as strongly as with Fer-1 (Fig. 4j–m). These results suggest that silencing Plin4 helps restore cellular antioxidant defenses, reducing lipid peroxidation and oxidative damage—key events in ferroptosis.
We also assessed Fe²⁺ levels using an iron ion assay kit and confocal microscopy (Fig. 4n). The results showed that BPDE treatment significantly increased Fe²⁺ levels, while both Fer-1 and Plin4 silencing reduced Fe²⁺ levels, with the Plin4-silenced group showing effects similar to those of Fer-1 (Fig. 4o). Fluorescence intensity analysis revealed slightly lower Fe²⁺ levels in the Plin4-silenced group compared to the Fer-1 group (Fig. 4p). These findings reinforce the role of Plin4 in regulating ferroptosis-related molecules and Fe²⁺ levels, suggesting that Plin4 plays a role in maintaining iron homeostasis and preventing ferroptosis.
In conclusion, our study demonstrates that silencing Plin4 protects HT22 cells from BPDE-induced ferroptosis. This protective effect likely involves the regulation of iron homeostasis, oxidative stress, antioxidant defenses, and mitochondrial function. By influencing lipid metabolism and mitochondrial integrity, Plin4 emerges as a key regulator of ferroptosis and a potential therapeutic target for ferroptosis-related diseases. These findings offer new insights into lipid metabolism’s role in ferroptosis and suggest that targeting Plin4 may provide novel strategies for treating neurodegenerative diseases linked to environmental toxicants.
Plin4 may enhance the sensitivity of BPDE-induced ferroptosis in HT22 cells by regulating LD accumulation
To delve deeper into the mechanisms by which Plin4 contributes to BPDE-induced ferroptosis in HT22 cells, we conducted a comprehensive literature review. Previous studies have shown that Plin4, a LD-associated protein, inhibits mitochondrial autophagy and promotes neuronal apoptosis, thereby facilitating the development and progression of neurodegenerative diseases. These findings imply a critical role for Plin4 in cognitive dysfunction. Given the close interplay between LD metabolism, oxidative stress, and neuronal injury, we hypothesized that Plin4 may regulate ferroptosis sensitivity by modulating aberrant LD accumulation under BPDE exposure.
LDs are essential organelles for neutral lipid storage and are involved in various physiological processes, including energy metabolism, steroid hormone synthesis, endoplasmic reticulum stress responses, mitochondrial function, and membrane maintenance. Recent studies suggest that beyond their metabolic functions, LDs may also contribute to the pathogenesis of neurodegenerative disorders. Under normal conditions, LDs are primarily distributed in glial cells, ependymal cells, and microglia within the nervous system [17, 18]. However, under pathological conditions such as neuronal aging or oxidative stress, LDs abnormally accumulate in neurons, a phenomenon closely linked to neurodegenerative disease progression.
In our study, TEM and Oil Red O staining revealed a significant increase in LDs in BPDE-treated HT22 cells, with LDs clustering around neurons and accompanied by pronounced morphological damage. Notably, Plin4 knockdown markedly reduced LD accumulation and ameliorated cellular structural damage. These consistent findings from both ultrastructural and histological assessments suggest that Plin4 plays a key role in BPDE-induced LD accumulation and is closely associated with neuronal damage (Fig. 5a, b).

a Oil Red O staining images showing LD accumulation (red arrows) in HT22 cells under different treatments: si-NC, BPDE, and si-Plin4 + BPDE. b TEM images revealing ultrastructural changes in LDs (red arrows) in the same treatment groups. c Quantification of LD area (% of cytoplasmic area) based on Oil Red O staining images shown in (a). d Quantification of LD area (% of cytoplasmic area) based on TEM images shown in (b). All quantitative data correspond to the same treatment groups: si-NC, BPDE, and si-Plin4 + BPDE. Data are presented as mean ± SEM (n = 6); *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To quantitatively evaluate the extent of LD accumulation, we performed image-based analysis using ImageJ software. For the Oil Red O staining (Fig. 5c), we measured the LD area as a percentage of the total cytoplasmic area in each cell. Similarly, for the TEM images (Fig. 5d), we quantified the proportion of LDs within the cytoplasm based on grayscale contrast and defined structural boundaries. In both analyses, at least six independent fields per group were randomly selected, and all images were processed under identical thresholding and scale parameters to ensure consistency and comparability. The quantification results showed a significant increase in LD content following BPDE treatment, whereas Plin4 silencing significantly decreased LD accumulation. These quantitative findings further validate our qualitative observations and underscore the regulatory role of Plin4 in BPDE-induced lipid metabolic alterations.
In conclusion, our findings indicate that BPDE promotes LD accumulation and may increase ferroptosis sensitivity through Plin4-regulated LD dynamics. Nevertheless, further investigation is needed to fully elucidate the mechanisms by which Plin4-mediated lipid metabolism contributes to ferroptosis and neuronal injury.
Discussion
In our previous studies, we found that BPDE, the active metabolite of B[a]P, induces ferroptosis in HT22 cells and mouse primary hippocampal neurons [19, 20]. However, the specific mechanisms by which B[a]P triggers ferroptosis in neurons remain unclear. Ferroptosis is primarily characterized by reduced intracellular GSH levels, decreased antioxidant enzyme activity, and dysregulated iron homeostasis, leading to iron accumulation. Our previous study revealed that BPDE inhibits the system Xc⁻ antiporter, which consists of xCT and 4F2hc (encoded by SLC7A11 and SLC3A2, respectively), thereby reducing cystine uptake, depleting intracellular GSH synthesis, inactivating GPX4, and promoting lipid peroxidation [21,22,23]. Furthermore, BPDE disrupts iron metabolism by elevating Fe²⁺ levels, likely through impaired iron storage or export, thereby amplifying Fenton reaction-driven oxidative damage [24, 25]. Notably, BPDE suppresses ASCL3, a transcription factor critical for neuronal antioxidant responses, exacerbating hippocampal neuronal vulnerability to ferroptosis [19, 26,27,28].
Recent studies suggest that BPDE-mediated system Xc⁻ inhibition may be driven by the promotion of p53 and the suppression of NRF2, both of which lead to downregulation of SLC7A11 [21, 29]. Our group’s findings indicate that BPDE upregulates p53, a known ferroptosis enhancer that transcriptionally represses SLC7A11, thus decreasing cystine import and depleting intracellular GSH. Simultaneously, BPDE downregulates NFE2L2, a key transcription factor that normally activates SLC7A11 expression to maintain redox homeostasis [30, 31]. This dual regulation creates a more oxidative intracellular environment, further exacerbating lipid peroxidation and ferroptosis [32].
To further dissect the role of Plin4 in ferroptosis, we examined its impact on the expression of ferroptosis-associated genes and proteins. Our results revealed that while BPDE significantly downregulated both Slc7a11 and Gpx4 at the mRNA and protein levels, Plin4 knockdown selectively restored the protein—but not mRNA—levels of these targets. This finding suggests that Plin4 may regulate ferroptosis through post-transcriptional mechanisms, potentially by modulating protein translation or stability. Moreover, treatment with the ferroptosis inhibitor Fer-1 selectively rescued Slc7a11 protein expression, but had no significant effect on Gpx4 levels, indicating that Slc7a11 is more directly involved in the canonical ferroptosis pathway in this context. These results support the possibility that Plin4 contributes to BPDE-induced ferroptosis by downregulating key ferroptosis-inhibitory proteins, especially Slc7a11, through a post-transcriptional mechanism. Thus, Plin4 may represent a previously unrecognized modulator of ferroptosis vulnerability in neurons exposed to genotoxic stress.
Research has shown that lipid metabolism regulates ferroptosis through mechanisms such as phospholipid peroxidation and the modulation of various signaling pathways [33, 34]. Our study also focused on lipid metabolism and performed bioinformatics analysis of transcriptomic data from adult male mice exposed to acute B[a]P. We identified 1668 DEGs, and machine learning analysis revealed a gene cluster highly associated with lipid metabolism. Among these genes, Plin4 stood out due to its significantly higher expression compared to others in the cluster. Cross-referencing with ferroptosis-related genes identified Plin4 as a key ferroptosis-associated gene. Given its involvement in lipid metabolism, we selected Plin4 for further investigation to better understand its role in ferroptosis.
Plin4, a member of the Perilipin family, is crucial for LD formation [18, 35, 36]. Increasing evidence suggests that all Perilipin proteins are involved in LD formation and regulation under various conditions [15]. Over the past two decades, lipid droplets have been characterized based on size, lipid content, and protein composition [35, 37]. LDs are highly dynamic organelles within cells, capable of storing neutral lipids such as cholesterol esters (CEs) and triacylglycerols (TGs). They have recently been recognized as essential components in stress responses across various cell types [35, 36]. In the nervous system, LDs are primarily observed in glial cells, ependymal cells, and microglia. However, under oxidative stress or lipotoxicity, LDs may also form in neurons [17, 38, 39]. The accumulation of LDs has been linked to several neurodegenerative diseases. Apart from energy homeostasis, LDs also perform various stress-related functions, including alleviating endoplasmic reticulum stress, lipotoxicity, and maintaining protein quality control. In glial cells, increased LDs have been associated with disease, and in AD models, elevated cerebral CEs and TGs correlate with LD accumulation in neural stem cell niches [17, 40]. Recent studies have highlighted the role of LDs in preventing lipotoxicity and redox imbalance (e.g., high ROS) during metabolic stress in glial cells and other cell types [14, 41].
Notably, recent studies have confirmed that Plin4 modulates ferroptosis by inhibiting the Hippo signaling pathway. The Hippo pathway plays a crucial role in cell proliferation, apoptosis, and stress responses, and its dysregulation has been linked to ferroptosis. Specifically, inhibition of the Hippo pathway leads to the activation of Yes-associated protein (YAP), which upregulates ACSL4, a critical enzyme that promotes lipid peroxidation and ferroptosis. This suggests that the Plin4-mediated ferroptosis response may be driven, at least in part, by a Hippo-off/YAP-on/ACSL4-up cascade, further sensitizing cells to ferroptosis [42, 43].
In this study, we observed Plin4 upregulation in the hippocampal tissues of B[a]P-exposed mice, mouse primary hippocampal neurons, and HT22 cells. Given previous findings that B[a]P induces ferroptosis in these models, we hypothesized that Plin4 may contribute to this process. To test this, we knocked out Plin4 in HT22 cells and assessed ferroptosis-related markers, including GSH content, GSH-Px activity, MDA levels, and Fe²⁺ accumulation. Plin4 silencing attenuated ferroptosis damage and partially mitigated Erastin-induced cytotoxicity, thereby restoring cell viability [21]. These results indicate that Plin4 is involved in mediating B[a]P-induced ferroptosis in HT22 cells.
However, one limitation of our study is the lack of assessment of Plin4 protein expression in hippocampal tissues and neuronal cells following B[a]P exposure. While Plin4 was initially identified through transcriptomic analysis and its mRNA expression was validated by qPCR in both in vivo and in vitro models, protein-level confirmation would have further substantiated its regulatory role. Although functional experiments using gene silencing provided supporting evidence for its involvement in B[a]P-induced ferroptosis, direct quantification of Plin4 protein remains necessary to reinforce our conclusions. We appreciate this important point and will address it in future studies.
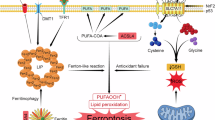
Taken together with our previous findings, our current results support a model in which BPDE triggers neuronal ferroptosis via a triad of mechanisms: inhibition of system Xc⁻ leading to GSH depletion, Fe²⁺ overload promoting lipid peroxidation, and Plin4-mediated lipid droplet accumulation exacerbating oxidative stress. These interactions are illustrated in Fig. 6.
![Fig. 6: Schematic diagram of B[a]P-induced neuronal ferroptosis mechanism.](http://media.springernature.com/lw685/springer-static/image/art%3A10.1038%2Fs41420-025-02747-8/MediaObjects/41420_2025_2747_Fig6_HTML.png?as=webp)
This diagram illustrates how B[a]P exposure leads to the inhibition of the system Xc⁻ antiporter, depletion of GSH, iron overload, lipid peroxidation, and LD accumulation, which collectively contribute to neuronal ferroptosis.
In summary, Plin4 is identified as a critical regulator of B[a]P/BPDE-induced ferroptosis via its role in lipid metabolism. Targeting Plin4 may disrupt this multi-hit ferroptosis pathway—addressing lipid peroxidation, iron overload, and system Xc⁻ dysfunction—and thus represents a promising therapeutic strategy for ferroptosis-associated neurodegeneration.
Despite these insights, several limitations should be acknowledged. First, the models employed—HT22 cells and acute B[a]P exposure in mice—may not fully replicate chronic or human-relevant conditions. Second, while Plin4 was validated, other genes within the lipid metabolism network remain unexplored. Third, the upstream regulatory mechanisms and in vivo functional relevance of Plin4 require further investigation. Future studies using chronic exposure models, conditional knockout mice, and integrated multi-omics approaches are needed to comprehensively elucidate Plin4’s role and therapeutic potential in neurodegenerative disease contexts.
Materials and methods
Transcriptome data collection and analysis
Transcriptome data were retrieved from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/) using the keywords “Benzo[a]pyrene” and “mouse”. The dataset GSE75206 was selected, which includes hippocampal tissues from male mice treated with Benzo[a]pyrene (B[a]P, 70 mg/kg·bw) for three consecutive days, with tissues harvested 24 h after the final exposure. The dataset consists of eight samples (n = 4 control, n = 4 B[a]P-treated) [16]. Each sample represents a biologically independent hippocampal tissue from a separate mouse. Gene expression data were normalized before analysis, and DEGs were identified using the “Limma” package (v3.56.2) in R (v4.3.0), with thresholds set at adjusted P-value < 0.05 (FDR) and |log2FC| ≥ 3. Visualization, including heatmaps and volcano plots, was performed using the “ggplot2” package (v3.4.3).
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis
GO and KEGG enrichment analyses were performed using the “ClusterProfiler” package (v3.18.1), “org.Hs.eg.db” (v3.11.0), and “AnnotationDbi” (v1.51.0) in R [44, 45]. Significance thresholds were set at P ≤ 0.05 and q ≤ 0.05. Results were visualized for interpretation.
Protein-protein interaction (PPI) network
The PPI network of DEGs was constructed using the STRING database (https://cn.string-db.org/) with a combined score >0.3 [46]. Network visualization and analysis were conducted using Cytoscape software (https://cytoscape.org/), and betweenness centrality (BC) scores were calculated to identify key genes [47].
Machine learning for candidate gene selection
The KMeans algorithm (K-means clustering) was applied to cluster genes based on expression levels and BC scores. The algorithm iteratively assigned genes to K clusters by minimizing the distance to cluster centroids until convergence [48].
Acquisition of ferroptosis-related genes
Ferroptosis-related genes were obtained from the FerrDb database (www.zhounan.org), developed by the Affiliated Brain Hospital of Guangzhou Medical University and Sichuan University [49]. FerrDb provides curated information on ferroptosis regulators (drivers, inhibitors, markers, and unclassified factors) and modulators (inducers and inhibitors), categorized by evidence level (validated, screened, predicted, deduced).
Venn diagram analysis
Venn diagrams were generated using the Venny 2.1 online tool (https://bioinfogp.cnb.csic.es/tools/venny/index.html) to identify overlapping genes between datasets and screen key genes for further analysis.
Ethics statement and animal housing conditions
All animal experiments were approved by the Ethics Committee of Shanxi Medical University (Approval No. 2021053). Healthy adult male C57BL/6J mice (clean grade) were obtained from the Laboratory Animal Center of Shanxi Medical University [Certificate No. SYSK (Jin) 2015-0001]. Animals were housed in a well-ventilated facility under natural light conditions, with 40–60% relative humidity, ambient noise below 60 dB, and free access to food and water. After a one-week acclimatization period, mice were randomly assigned to two groups (n = 6 biologically independent animals per group): a control group receiving intraperitoneal injections of corn oil (vehicle), and a treatment group receiving 10 mg/kg body weight B[a]P dissolved in corn oil. Injections were administered once daily for 30 consecutive days to establish a subacute B[a]P exposure model. The sample size (n = 6 per group) was chosen based on previous literature and preliminary studies to ensure sufficient statistical power while minimizing animal use in accordance with ethical standards. Each experiment was independently repeated 6 times (biological replicates), and each biological replicate included 3 technical replicates per assay to ensure reproducibility and accuracy. Data are presented as mean ± standard error of the mean (SEM). Statistical analyses were conducted using two-sided tests with corrections for multiple comparisons where appropriate. No blinding was performed during group allocation or outcome assessment. All procedures were performed following institutional guidelines and national regulations for the care and use of laboratory animals. No animals or samples were excluded from the analysis, and no pre-established exclusion criteria were applied.
Primary hippocampal neuron culture
Neonatal C57BL/6J mice (postnatal day 0–1) were obtained from the Experimental Animal Center of Shanxi Medical University [Certificate No. SCXK (Jin) 2019-0004; Ethics Approval No. 2021053]. After disinfection with 75% ethanol, brains were rapidly removed and placed in cold, calcium- and magnesium-free PBS. Under a stereomicroscope, meninges were carefully removed, and hippocampi were dissected out. The tissue was minced into ~1 mm³ pieces and digested with 0.25% trypsin and DNase I at 37 °C for 10 min. Digestion was terminated with DMEM containing 6% fetal bovine serum (FBS). The suspension was gently triturated, filtered, and centrifuged at 300 × g for 5 min. The cell pellet was resuspended in DMEM with 10% FBS and seeded onto poly-L-lysine-coated plates. After 4 h of incubation at 37 °C in 5% CO₂, the medium was replaced with serum-free Neurobasal®-A medium supplemented with 2% B-27. Half of the medium was changed every 3 days. Neurons were used for experiments on days in vitro (DIV) 6–7.
To simulate B[a]P exposure in vitro, cells were treated with benzo[a]pyrene diol epoxide (BPDE), the active metabolite of B[a]P, at concentrations of 0, 0.25, 0.5, or 0.75 μM for 24 h to assess its effects on cell viability and the expression of Plin4. All experiments were performed in 6 independent biological replicates, each with 3 technical replicates. Data are presented as mean ± SEM. Statistical analyses were performed using two-sided tests with adjustments for multiple comparisons where appropriate.
HT22 cell culture
HT22 mouse hippocampal neuronal cells (BNCC Biological Technology Co., Ltd, BNCC358041) were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Gibco, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, USA) and 1% penicillin-streptomycin (Gibco, USA), maintained at 37 °C in a humidified incubator with 5% CO₂. Cells were passaged when they reached 80–85% confluence using 0.25% trypsin (Gibco, USA). For recovery, cryopreserved cells were thawed in a 37 °C water bath, centrifuged at 1200 rpm for 5 min, resuspended in complete medium, and seeded into culture flasks. For cryopreservation, cells at 80–90% confluence were harvested, centrifuged at 1000 rpm for 5 min, resuspended in freezing medium containing DMSO, and gradually frozen in a programmed freezing container.
To evaluate the cytotoxic effects of BPDE in vitro, HT22 cells were exposed to increasing concentrations of BPDE (0, 0.25, 0.5, and 0.75 μM) for 24 h. Changes in cell viability and Plin4 expression were then assessed. For further investigation into the role of Plin4 in BPDE-induced ferroptosis, cells were divided into experimental groups with two BPDE concentrations (0 μM and 0.75 μM). Each group was treated with either si-NC (negative control), si-Plin4 (Plin4 silencing), or Fer-1 (ferroptosis inhibitor) to explore the effects of Plin4 knockdown and ferroptosis inhibition on BPDE-induced cell death and lipid peroxidation.
All in vitro experiments were performed in 6 independent biological replicates, and each biological replicate included 3 technical replicates. Data are expressed as mean ± SEM. Statistical analyses were conducted using two-sided tests, with corrections for multiple comparisons where applicable. Statistical significance was considered at P < 0.05. HT22 cells were authenticated by the supplier and tested for mycoplasma contamination prior to use.
Morris water maze (MWM) test
The MWM apparatus (purchased from the Department of Environmental Toxicology, Shanxi Medical University) consisted of a 150 cm diameter, 50 cm high pool with an overhead camera tracking system. The pool was divided into four quadrants (SW, NW, SE, NE), with water maintained at 22 ± 1 °C and a platform submerged 1 cm below the surface. After 1-h dark-room acclimation, mice underwent four daily training trials for 5 days, starting from different quadrants, with 60 seconds to locate the hidden platform. Escape latency was recorded, and mice remained on the platform for 15 s; failures were guided to the platform for a 20 s stay. On day 6, a spatial probe test was conducted by removing the platform and allowing mice to swim freely for 60 s. The swimming path and time spent in the target quadrant (SW) were recorded.
Y-Maze test
The Y-Maze apparatus (black acrylic, purchased from the Department of Environmental Toxicology, Shanxi Medical University) consisted of three arms at 120° angles: starting arm, other arm, and novel arm. In the first phase, mice explored the starting and other arms for 5 min, with the novel arm blocked. After a 1 h rest, they were reintroduced and allowed 5 min to explore all three arms. Between trials, the maze was cleaned with 70% ethanol. A video tracking system recorded behavior, and the number of entries and latency to enter the novel arm were analyzed.
Quantitative real-time PCR (qPCR)
Total RNA was extracted using RNA isolator Total RNA Extraction Reagent (Vazyme, R701-01, Nanjing), and cDNA was synthesized with HiScript II Q Select RT SuperMix for qPCR (+gDNA wiper) (Vazyme, R222-01, Nanjing) following the manufacturer’s instructions. qPCR was performed using PowerUP SYBR Green Master Mix (Thermo Fisher, USA) on an ABI StepOne Plus system (Applied Biosystems, USA). The thermal cycling conditions were as follows: initial denaturation at 95 °C for 5 min, followed by 40 cycles of 95 °C for 10 s and 60 °C for 30 s, with fluorescence signal acquisition during each extension phase. Melting curve analysis was conducted to verify specificity. The primer sequences were as follows:
-
Plin4
Forward: 5′-GTGTCCACCAACTCACAGATG-3′
Reverse: 5′-GGACCATTCCTTTTGCAGCAT-3′
-
Slc7a11
Forward: 5′-GGCACCGTCATCGGATCAG-3′
Reverse: 5′-GGACCAAAGACCTCCAGAATG-3′
-
Gpx4
Forward: 5′-TGTGCATCCCGCGATGATT-3′
Reverse: 5′-CCCTGTACTTATCCAGGCAGA-3′
-
Ptgs2
Forward: 5′-TTCCAATCCATGTCAAAACCGT-3′
Reverse: 5′-AGTCCGGGTACAGTCACACTT-3′
-
Gapdh (endogenous control)
Forward: 5′-AGGTCGGTGTGAACGGATTTG-3′
Reverse: 5′-TGTAGACCATGTAGTTGAGGTCA-3′
Relative gene expression levels were calculated using the 2−ΔΔCt method.
Cell viability assay
HT22 cells were seeded into 96-well plates at a density of 1 × 10⁵/mL to 1 × 10⁶/mL. Upon reaching appropriate confluence, cells were treated according to experimental groups. After 24 h, 10 μL of CCK-8 (Cell Counting Kit-8) solution was added to each well and incubated for 2 h at 37 °C in a 5% CO₂ incubator. Absorbance at 450 nm was measured using a microplate reader. Cell viability was calculated using the formula: Cell viability = [(Experimental group-Blank control)/(Control group-Blank control)] × 100%. Three replicate wells were used per group, with a blank control well.
Establishment of the cell gene silencing model for Plin4
Plin4 gene silencing was performed using siRNA and transfection reagents from GenePharma (Shanghai). Before transfection, 500 μL of growth medium was added to each well, and 1.5–3.5 × 10⁴ cells were seeded to reach 30–50% confluence 18–24 h prior. The siRNA-mate transfection reagent was mixed with 10 pmol (140 ng) of siRNA targeting Plin4 in 100 μL DMEM medium and vortexed. After a 10 min incubation at room temperature, 100 μL of the transfection mixture was added to each well, resulting in a final siRNA concentration of 16.7 nM. Cells were incubated at 37 °C, and Plin4 silencing efficiency was assessed by qPCR at 24–72 h post-transfection.
Cell morphology and ultrastructure
Ultrastructural analysis of cells was performed using TEM. After treatment, cells were centrifuged at 1200 rpm for 5 min, fixed in 2% glutaraldehyde for 2 h, and washed three times with PBS. The cells were dehydrated with graded acetone solutions, followed by infiltration and embedding. Thin sections were stained with 3% uranyl acetate and lead citrate, and the ultrastructural features were observed and imaged using a TEM.
Western blot
Proteins were separated using SDS-PAGE with gels corresponding to the target molecular weights, loading 10–20 μg protein per lane. The electrophoresis was carried out at 80 V for 30 min and then at 120 V until dye migration. Proteins were transferred to NC membranes using a wet transfer system (350 mA, 90 min) in Tris-glycine buffer. Membranes were blocked with 10% skim milk in TBST for 1 h and incubated overnight at 4 °C with the following primary antibodies: anti-SLC7A11 (ab307601, Abcam, 1:1000), anti-GPX4 (ab125066, Abcam, 1:2000), and anti-GAPDH (10494, Proteintech, 1:10 000). After TBST washes, membranes were incubated with HRP-conjugated secondary antibody (anti-rabbit IgG, AS014, Abclonal, 1:10,000) for 1 h at room temperature. Protein signals were visualized using ECL substrate (1:1 reagent A:B mixture) and quantified via ImageJ by normalizing target band intensities to loading controls. Full and uncropped western blots are shown in Supplementary Material.
Glutathione (GSH) content detection
The GSH content in HT22 cells and primary cells was measured using a colorimetric method. Reduced GSH reacts with 5,5′-dithiobis (2-nitrobenzoic acid) (DTNB) to form a yellow compound, quantified at 405 nm. After treatment, cells were lysed with RIPA buffer, and the supernatant was collected for analysis. Protein concentration and GSH content were measured according to the manufacturer’s protocol, and absorbance was read at 405 nm using a microplate reader.
Glutathione peroxidase (GSH-Px) activity detection
The GSH-Px activity in cells was measured using an enzyme-catalyzed colorimetric method. Reduced GSH reacts with DTNB to form a yellow compound, quantified at 412 nm. Cells were lysed as described, and GSH-Px activity was assessed using the manufacturer’s protocol. Absorbance was measured at 412 nm with a microplate reader.
Measurement of cellular Lipid ROS
HT22 cells and primary cells were seeded in 96-well plates and treated as per experimental groups. After 24 h, cells were washed with PBS and incubated with 8 μM C11-BODIPY (a fluorescent probe for lipid oxidation) at 37 °C for 30 min. Fluorescence signals for oxidized and non-oxidized lipids were measured using a multimode plate reader (non-oxidized: excitation/emission 540 nm/620 nm; oxidized: excitation/emission 428 nm/528 nm). The lipid ROS level was calculated by the ratio of oxidized to total fluorescence.
Malondialdehyde (MDA) content detection
MDA content in cells was measured using the thiobarbituric acid method. Cells were lysed with RIPA buffer, and the supernatant was collected for analysis. After adding the reagent mixture and sonication, the sample was incubated in a 95 °C water bath for 40 min, followed by cooling and centrifugation. Absorbance was measured at 532 nm using a microplate reader, and MDA content was calculated using the appropriate formula.
FerroOrange staining
HT22 cells were seeded at 100,000 cells/mL in confocal culture dishes and cultured overnight at 37 °C with 5% CO₂. After discarding the medium and washing with serum-free medium, cells were treated for 24 h as per the experimental groups. The cells were then stained with 1 μM FerroOrange (a fluorescent dye for detecting iron) for 30 min. Staining was observed and analyzed using a confocal microscope.
Ferrous Iron (Fe2+) content detection
HT22 cells were seeded in 96-well plates and treated as per the experimental design. After washing with serum-free medium, cells were incubated with 1 μM FerroOrange for 30 min at 37 °C. Fe²⁺ content was measured using a fluorescence microscope, with excitation at 542 nm and emission at 575 nm. Fluorescence intensity was quantified to assess intracellular Fe²⁺ levels.
Oil Red O staining
HT22 cells were cultured and treated as per the experimental design. After removing the culture medium and washing with PBS, cells were fixed with Oil Red O fixative for 20–30 min, followed by washing with distilled water. The cells were immersed in 60% isopropanol for 20–30 s and then stained with Oil Red O solution for 10–20 min. Mayer’s hematoxylin was applied for 1–2 min, and the cells were washed with Oil Red O buffer. Staining was observed under a microscope.
LD quantification
LD accumulation was quantitatively analyzed using ImageJ (National Institutes of Health, USA). For Oil Red O staining, images were captured under a bright-field microscope at the same magnification and exposure settings across all groups. The cytoplasmic region of each cell was manually outlined as the region of interest (ROI), and the total LD area was quantified by converting the image to 8-bit grayscale, followed by threshold adjustment to isolate positively stained regions. The LD area was then measured and expressed as a percentage of the total cytoplasmic area for each cell. At least six randomly selected fields per group were analyzed.
For TEM images, LDs were identified based on their electron-lucent appearance and spherical morphology. TEM images were converted to grayscale and analyzed using a similar thresholding approach in ImageJ. LD areas were measured and normalized to the total cytoplasmic area within each image. For each experimental group, six biological replicates (n = 6) were analyzed, and three independent regions were selected from each replicate for quantification. Quantitative values from all selected regions were averaged to calculate the LD content for each group. All image processing and analysis were performed using identical parameters across all groups to ensure consistency. Data are presented as mean ± SEM.
Quantification and statistical analysis
All data were analyzed using GraphPad Prism (Version 9.0, GraphPad Software, USA). Results are presented as mean ± SEM, unless otherwise specified. Each experiment was independently repeated six times (biological replicates), with three technical replicates per biological sample. For comparisons between two groups, two-sided unpaired Student’s t-tests were performed. For comparisons among multiple groups, one-way analysis of variance was used, followed by Tukey's post hoc test for multiple comparisons. Homogeneity of variances was confirmed prior to performing parametric tests. A P-value < 0.05 was considered statistically significant. Exact sample sizes (n), statistical tests used, and significance levels are detailed in the corresponding figure legends.
The sample sizes were chosen based on prior published studies and preliminary experiments conducted in our laboratory to ensure adequate statistical power to detect biologically relevant differences. Although formal power calculations were not performed, the selected group sizes (n = 6 biological replicates) are consistent with similar studies in the field. This, combined with three technical replicates per biological replicate and independent repetition of experiments, provides confidence in the reproducibility and robustness of the findings.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article
Data availability
The dataset analyzed in this study, GSE75206, was retrieved from the Gene Expression Omnibus (GEO) database. Additional data supporting the conclusions of this work are available from the corresponding author upon reasonable request.
Code availability
R codes used to analyze data and generate figures are available at Github (https://github.com/Sun0shy/BPDE_Ferroptosis).
References
Chepelev NL, Moffat ID, Bowers WJ, Yauk CL. Neurotoxicity may be an overlooked consequence of benzo[a]pyrene exposure that is relevant to human health risk assessment. Mutat Res Rev Mutat Res. 2015;764:64–89.
Knecht AL, Truong L, Marvel SW, Reif DM, Garcia A, Lu C, et al. Transgenerational inheritance of neurobehavioral and physiological deficits from developmental exposure to benzo[a]pyrene in zebrafish. Toxicol Appl Pharmacol. 2017;329:148–57.
Qiu C, Cheng S, Xia Y, Peng B, Tang Q, Tu B. Effects of subchronic benzo(a)pyrene exposure on neurotransmitter receptor gene expression in the rat hippocampus related with spatial learning and memory change. Toxicology. 2011;289:83–90.
Li F, Xie L, Xiao Q, Li J, Zhong H, Xu X, et al. Benzo[a]pyrene exposure induces anxiety-like behaviors in the mice through brain metabolic alterations. Sci Total Environ. 2024;954:176215.
Tartaglione AM, Racca A, Ricceri L. Developmental exposure to polycyclic aromatic hydrocarbons (PAHs): focus on benzo[a]pyrene neurotoxicity. Reprod Toxicol. 2023;119:108394.
Cheng S-Q, Xia Y-Y, He J-L, Liu X-Q, Chen X-M, Ding Y-B, et al. Neurotoxic effect of subacute benzo(a)pyrene exposure on gene and protein expression in Sprague-Dawley rats. Environ Toxicol Pharm. 2013;36:648–58.
Liu D, Zhao Y, Qi Y, Gao Y, Tu D, Wang Y, et al. Benzo(a)pyrene exposure induced neuronal loss, plaque deposition, and cognitive decline in APP/PS1 mice. J Neuroinflammation. 2020;17:258.
Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. 2021;18:280–96.
Cao Y, Huang M-Y, Mao C-H, Wang X, Xu Y-Y, Qian X-J, et al. Structural changes in cerebral microvasculature induced by ferroptosis contribute to blood-brain barrier disruption in Alzheimer’s disease: an autopsy study. Alzheimers Dement. 2025;21:e70103.
Sharma V, Sharma P, Singh TG. Ferroptosis and Alzheimer’s: unveiling new avenues for the treatment and prevention. Metab Brain Dis. 2025;40:167.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Stockwell BR. Ferroptosis turns 10: emerging mechanisms, physiological functions, and therapeutic applications. Cell. 2022;185:2401–21.
Zhang Y, Xie J. Ferroptosis implication in environmental-induced neurotoxicity. Sci Total Environ. 2024;934:172618.
You JH, Lee J, Roh J-L. PGRMC1-dependent lipophagy promotes ferroptosis in paclitaxel-tolerant persister cancer cells. J Exp Clin Cancer Res. 2021;40:350.
Bai Y, Meng L, Han L, Jia Y, Zhao Y, Gao H, et al. Lipid storage and lipophagy regulates ferroptosis. Biochem Biophys Res Commun. 2019;508:997–1003.
Chepelev NL, Long AS, Bowers WJ, Gagné R, Williams A, Kuo B, et al. Transcriptional profiling of the mouse hippocampus supports an NMDAR-mediated neurotoxic mode of action for benzo[a]pyrene. Environ Mol Mutagen. 2016;57:350–63.
Farmer BC, Walsh AE, Kluemper JC, Johnson LA. Lipid droplets in neurodegenerative disorders. Front Neurosci. 2020;14. https://doi.org/10.3389/fnins.2020.00742.
Islimye E, Girard V, Gould AP. Functions of stress-induced lipid droplets in the nervous system. Front Cell Dev Biol. 2022;10. https://doi.org/10.3389/fcell.2022.863907.
He H, Ji T, Lyu Y, Tian F, Mu E, Mu S, et al. BPDE induces ferroptosis in hippocampal neurons through ACSL3 suppression. Neurotoxicology. 2025;107:11–21.
Zhang Y, Yang Y, Chen W, Mi C, Xu X, Shen Y, et al. BaP/BPDE suppressed endothelial cell angiogenesis to induce miscarriage by promoting MARCHF1/GPX4-mediated ferroptosis. Environ Int. 2023;180:108237.
Lyu Y, Ren X-K, Guo C-C, Li Z-F, Zheng J-P. Benzo(a)pyrene-7,8-dihydrodiol-9,10-epoxide induces ferroptosis in neuroblastoma cells through redox imbalance. J Toxicol Sci. 2022;47:519–29.
Wen J, Li L, Ou D, Li J, Yang Y, Duan L, et al. Higenamine protects against doxorubicin-induced heart failure by attenuating ferroptosis via modulating the Nrf2/GPX4 signaling pathway. Phytomedicine. 2025;141:156670.
Xue X, Wang M, Cui J, Yang M, Ma L, Kang R, et al. Glutathione metabolism in ferroptosis and cancer therapy. Cancer Lett. 2025;621:217697.
Guo CC, Xu ZM, Lyu Y, Li XH, Li ZF, He H, et al. PM2.5 induces autophagy-dependent ferroptosis by endoplasmic reticulum stress in SH-SY5Y cells. J Appl Toxicol. 2023;43:1013–25.
Guo C, Lyu Y, Xia S, Ren X, Li Z, Tian F, et al. Organic extracts in PM2.5 are the major triggers to induce ferroptosis in SH-SY5Y cells. Ecotoxicol Environ Saf. 2023;249:114350.
Cao Y, Li J, Chen Y, Wang Y, Liu Z, Huang L, et al. Monounsaturated fatty acids promote cancer radioresistance by inhibiting ferroptosis through ACSL3. Cell Death Dis. 2025;16:184.
Sae-Fung A, Vinayavekhin N, Fadeel B, Jitkaew S. ACSL3 is an unfavorable prognostic marker in cholangiocarcinoma patients and confers ferroptosis resistance in cholangiocarcinoma cells. NPJ Precis Oncol. 2024;8:284.
Sheng W, Liao S, Cao S, Wei Y, Zhao J, Yue Y, et al. Reactive oxygen species-sensitive fenofibrate-loaded dextran nanoparticles in alleviation of osteoarthritis. Carbohydr Polym. 2025;347:122768.
Liu W, Zhu Y, Ye W, Xiong J, Wang H, Gao Y, et al. Redox regulation of TRIM28 facilitates neuronal ferroptosis by promoting SUMOylation and inhibiting OPTN-selective autophagic degradation of ACSL4. Cell Death Differ. 2025;32:1041–57.
Gao N, Huang Z, Xie J, Gao S, Wang B, Feng H, et al. Cryptotanshinone alleviates cerebral ischemia reperfusion injury by regulating ferroptosis through the PI3K/AKT/Nrf2 and SLC7A11/GPX4 signaling pathway. J Ethnopharmacol. 2025;348:119800.
Luo L, Zhao Q, Cui X, Dong S, Wang Y, Jiang N, et al. Male-specific lethal 1 (MSL1) promotes Erastin-induced ferroptosis in colon cancer cells by regulating the KCTD12-SLC7A11 axis. Cell Death Dis. 2025;16:281.
Huang C, Zeng Q, Chen J, Wen Q, Jin W, Dai X, et al. TMEM160 inhibits KEAP1 to suppress ferroptosis and induce chemoresistance in gastric cancer. Cell Death Dis. 2025;16:287.
Chen Y, Frost S, Khushi M, Cantrill LC, Yu H, Arthur JW, et al. Delayed recruiting of TPD52 to lipid droplets – evidence for a “second wave” of lipid droplet-associated proteins that respond to altered lipid storage induced by Brefeldin A treatment. Sci Rep. 2019;9:9790.
Itabe H, Yamaguchi T, Nimura S, Sasabe N. Perilipins: a diversity of intracellular lipid droplet proteins. Lipids Health Dis. 2017;16:83.
Han X, Liu Y, Dai Y, Xu T, Hu Q, Yi X, et al. Neuronal SH2B1 attenuates apoptosis in an MPTP mouse model of Parkinson’s disease via promoting PLIN4 degradation. Redox Biol. 2022;52:102308.
Zhang X-D, Sun J, Zheng X-M, Zhang J, Tan L-L, Fan L-L, et al. Plin4 exacerbates cadmium-decreased testosterone level via inducing ferroptosis in testicular Leydig cells. Redox Biol. 2024;76:103312.
Xiao L, Yang X, Sharma VK, Abebe D, Loh YP. Hippocampal delivery of neurotrophic factor-α1/carboxypeptidase E gene prevents neurodegeneration, amyloidosis, memory loss in Alzheimer’s Disease male mice. Mol Psychiatry. 2023;28:3332–42.
Arrese EL, Saudale FZ, Soulages JL. Lipid droplets as signaling platforms linking metabolic and cellular functions. Lipid Insights. 2014;7:7–16.
Pennetta G, Welte MA. Emerging links between lipid droplets and motor neuron diseases. Dev Cell. 2018;45:427–32.
Li Y, Chen Z, Han J, Ma X, Zheng X, Chen J Functional and Therapeutic Significance of Tumor-Associated Macrophages in Colorectal Cancer. Front Oncol. 2022;12. https://doi.org/10.3389/fonc.2022.781233.
Schott MB, Rozeveld CN, Weller SG, McNiven MA. Lipophagy at a glance. J Cell Sci. 2022;135:jcs259402.
Zeng F, Zhou M, Li Q, Hu H, Chen C. Sevoflurane promotes neuronal ferroptosis via upregulation of PLIN4 to modulate the hippo signaling pathway. Neurotoxicology. 2024;105:1–9.
She G, Hai X-X, Jia L-Y, Zhang Y-J, Ren Y-J, Pang Z-D, et al. Hippo pathway activation mediates cardiomyocyte ferroptosis to promote dilated cardiomyopathy through downregulating NFS1. Redox Biol. 2025;82:103597.
Gene Ontology Consortium. Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015;43:D1049–1056.
Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999;27:29–34.
Szklarczyk D, Kirsch R, Koutrouli M, Nastou K, Mehryary F, Hachilif R, et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023;51:D638–D646.
Sun H, Yang J, Li X, Lyu Y, Xu Z, He H, et al. Identification of feature genes and pathways for Alzheimer’s disease via WGCNA and LASSO regression. Front Comput Neurosci. 2022;16. https://doi.org/10.3389/fncom.2022.1001546.
Huang W, Peng Y, Ge Y, Kong W. A new Kmeans clustering model and its generalization achieved by joint spectral embedding and rotation. PeerJ Comput Sci. 2021;7:e450.
Zhou N, Bao J. FerrDb: a manually curated resource for regulators and markers of ferroptosis and ferroptosis-disease associations. Database. 2020;2020:baaa021.
Funding
This study was supported by the National Natural Science Foundation of China (Grant No. 30872137), the Shanxi 1331 Project (2021-5-2-2-B1), the Natural Science Foundation of Shanxi Province (Grant Nos. 202203021211106 and 20210302124301), the Innovation Team of Changzhi Medical College (CX202001), and the Key Laboratory of Shanxi Province for Aging Mechanism Research and Transformation. We thank scidraw.io for the mouse schematic.
Author information
Authors and Affiliations
Contributions
Hongyu Sun and Zhirui Ma performed the experiments, conducted bioinformatics analyses, prepared the figures, and drafted the manuscript. They are co-first authors of this work. Xingdi Guo, Jie Chen, Hui He, Xiaomin Tong, Tingyu Ji, Shihan Ding, and Chaoli Zhou assisted with data collection and experimental procedures. Yi Lyu and Fengjie Tian contributed to data analysis and interpretation. Jinping Zheng conceived and supervised the project, and critically revised the manuscript. All authors reviewed and approved the final version of the manuscript. All authors also commented on and approved the draft manuscript prior to submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
All animal procedures were approved by the Ethics Committee of Shanxi Medical University (Approval No. 2021053) and conducted in accordance with institutional guidelines and national regulations for the care and use of laboratory animals. Adult male C57BL/6J mice and neonatal C57BL/6J mice were obtained from the Experimental Animal Center of Shanxi Medical University [Certificate No. SCXK (Jin) 2019-0004]. No human subjects or data were involved in this study, and no additional ethics approval was required for the use of commercially available cell lines.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sun, H., Ma, Z., Guo, X. et al. Plin4 modulates lipid droplet accumulation and ferroptosis in neurons exposed to benzo[a]pyrene. Cell Death Discov. 11, 442 (2025). https://doi.org/10.1038/s41420-025-02747-8
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41420-025-02747-8
