
Abstract
Transient Receptor Potential Vanilloid 1 (TRPV1) plays a central role in pain sensation and is thus an attractive pharmacological drug target. SAF312 is a potent, selective, and non-competitive antagonist of TRPV1 and shows promising potential in treating ocular surface pain. However, the precise mechanism by which SAF312 inhibits TRPV1 remains poorly understood. Here, we present the cryo-EM structure of human TRPV1 in complex with SAF312, elucidating the structural foundation of its antagonistic effects on TRPV1. SAF312 binds to the vanilloid binding pocket, preventing conformational changes in S4 and S5 helices, which are essential for channel gating. Unexpectedly, a putative cholesterol was found to contribute to SAF312’s inhibition. Complemented by mutagenesis experiments and molecular dynamics simulations, our research offers substantial mechanistic insights into the regulation of TRPV1 by SAF312, highlighting the interplay between the antagonist and cholesterol in modulating TRPV1 function. This work not only expands our understanding of TRPV1 inhibition by SAF312 but also lays the groundwork for further developments in the design and optimization of TRPV1-related therapies.
Similar content being viewed by others


Introduction
Transient receptor potential vanilloid 1 (TRPV1) is one of the most comprehensively studied members of the TRP family1. It can be activated by an array of physical and chemical stimuli, including low pH (below 6) and high temperature (over 43 °C)2, as well as natural compounds such as capsaicin, double-knot toxin (DkTx), and resiniferatoxin (RTX)3. Because TRPV1 is broadly expressed in the peripheral nervous system, it plays a pivotal role in pain and inflammation4,5. It is a potential target for treating various pain syndromes, including chronic, inflammatory, and neuropathic pain6. Consequently, various small-molecule compounds that can alter TRPV1 functions have emerged7. Despite their therapeutic potential, developing TRPV1-specific drugs is challenging because the sequence and structure of the members of the TRPV subfamily (TRPV1-6) are highly conserved in sequence and structure8. SAF312 (also known as Libvatrep), a selective and non-competitive inhibitor of TRPV1, is currently under investigation in phase II clinical trials for treating ocular surface pain (OSP)9,10. This drug candidate is expected to become the first approved TRPV1 antagonist for pain treatment. Although many structures of TRPV1 in complex with various ligands have greatly enriched the understanding of TRPV1 gating and modulation11,12,13,14,15, the structures of TRPV1 in complex with antagonists are relatively limited, with only one reported structure of TRPV1 bound to capsazepine13 and another one with SB-366791 at the time of writing this manuscript16. However, the molecular mechanism of TRPV1 inhibition by SAF312 remains unclear.
In this study, we present the cryo-electron microscopy (cryo-EM) structure of human TRPV1 (hTRPV1) in complex with SAF312 at a resolution of 2.75 Å, revealing the mechanism of action of SAF312 in antagonizing hTRPV1. We further investigate the relationship between the structure and function using whole-cell patch-clamp, mutagenesis, fluorescent imaging plate reader (FLIPR) assay, and molecular dynamics (MD) simulations. In addition, we unexpectedly observed an endogenous cholesterol (CHL) molecule that might contribute to the SAF312 binding and hTRPV1 inhibition. Our results provide significant insights into the molecular basis of the inhibitory effects of SAF312 on TRPV1, which could facilitate the development of TRPV1 antagonists for anti-pain therapies.
Results and discussion
SAF312 is a potent antagonist of hTRPV1
We evaluated the function of hTRPV1 using the FLIPR assay17 as well as whole-cell patch-clamp recordings. Capsaicin was used to evoke hTRPV1, yielding a half maximal effective concentration (EC50) of 0.39 µM (Supplementary Fig. 1a, b), consistent with the findings of a previous study17. We next assayed the concentration-dependent response of SAF312 (Fig. 1a, b) in inhibiting hTRPV1 elicited by 3.33 µM capsaicin, a concentration that activates hTRPV1 at 80–90% (Supplementary Fig. 1b). The resulting half maximal inhibitory concentration (IC50) value for SAF312 was 0.35 µM (Fig. 1c), in agreement with a previous report18. In addition, we examined the inhibitory function of SAF312 on pH 5-stimulated TRPV1 activation through whole-cell patch-clamp experiments. When exposed to a pH 5 solution three times, hTRPV1-expressing HEK293T cells exhibited robust and comparable currents (Fig. 1d, e). In line with the inhibition of capsaicin-activated TRPV1 by SAF312, a 30 s preincubation with 3 μM SAF312 greatly reduced the currents of TRPV1 elicited by pH 5 (Fig. 1f, g). With the preincubation of increased concentrations of SAF312, the currents evoked by pH 5 were gradually decreased, yielding an IC50 value of 0.69 µM for SAF312 (Fig. 1f, h).

a Chemical structure of the hTRPV1 antagonist SAF312. b The representative FLIPR capsaicin responses of hTRPV1 to preincubation with SAF312 at various concentrations (ranging from 66.67 μM to 0 nM) are shown, with an arrow indicating when capsaicin was added. c Curve fitting of dose-dependent SAF312 inhibition of hTRPV1WT calcium fluorescence signal evoked by 3.33 μM capsaicin (IC50 = 0.35 μM, n = 3 at each concentration point), data are shown as mean ± SD. d, e Representative acid (pH 5)-evoked hTRPV1-related current traces recorded at a holding potential of −60 mV with ramp pulses from −100 mV to +100 mV. hTRPV1-expressing HEK293T cell exposed to pH 5 solution three times (d), I-V curves were generated from the peak currents (peak1, peak2, and peak3) at each stimulation time (e). f, g hTRPV1-expressing HEKT293 cells exposed to pH 5 solution alone, pH 5 solution with 0.1 μM SAF312 following ~30 s preincubation with 0.1 μM SAF312, and pH 5 solution with 3 μM SAF312 following around 30 s preincubation with 3 μM SAF312 (f), I-V curves were generated from the peak currents (peak1, peak2, and peak3) at each stimulation time (g). h SAF312 dose-dependent curves in responding to pH 5 solution for HEK293T cells expressing hTRPV1 WT (n = 4 at each concentration point), data are shown as mean ± SD.
Structure of hTRPV1 complexed with SAF312 and CHL
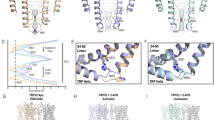
To reveal the precise mechanism of how SAF312 inhibits hTRPV1, we conducted cryo-EM single particle analysis of the hTRPV1 samples supplemented with SAF312 (Supplementary Fig. 1c). Subsequently, a high-resolution EM map of hTRPV1 was achieved at 2.75 Å with fourfold (C4) symmetry imposed (Supplementary Fig. 2). This high-quality EM map rich in side-chain densities enabled accurate model building of TRPV1 and assignments of SAF312 and lipids (Fig. 2a–c and Supplementary Fig. 2b, e). The overall structure of the hTRPV1-SAF312 (hTRPV1SAF312) complex assumes a closed conformation, which is essentially identical to that of the closed apo-form TRPV1 (PDB code: 7L2P), with a main-chain root mean squared deviation (RMSD) of 0.8 Å (Supplementary Fig. 3). We observed a piece of unambiguous EM density in the vanilloid binding pocket (VBP) in each of the voltage-sensing-like domains (VSLDs) (Fig. 2a), which fits a SAF312 molecule snugly (Fig. 2b, c). Unexpectedly, a piece of flat EM density close to SAF312 was observed, which may represent a bound CHL molecule (Fig. 2a–c), in line with the observation of an endogenous CHL molecule binding to a similar position of TRPV219.

a The cryo-EM map of hTRPV1SAF312 illustrates the binding of SAF312 and CHL to hTRPV1. The SAF312 map is represented in magenta, and the CHL map is depicted in cyan. b The overall structure model of hTRPV1SAF312 is viewed parallel to the membrane. SAF312 and CHL bind to the VBP pocket. c SAF312 and CHL bind to the VBP pocket. d The cryo-EM map of hTRPV1SAF312-2 illustrates the binding of SAF312 and CHL to hTRPV1. The SAF312 map is represented in magenta, and the CHL map is depicted in orange.
Considering that we added cholesteryl hemisuccinate tris salt (CHS) during protein purification, an analogy of CHL, we could not rule out the possibility of a CHS rather than a CHL binding to TRPV1. To ensure whether the endogenous CHL binds to this position, we purified the protein samples without adding CHS and determined the cryo-EM structure (hTRPV1SAF312-2) (Supplementary Figs. 1d, 4). The results showed that a similar flat EM density still existed, confirming endogenous CHL’s binding to the TRPV1-SAF312 complex (Fig. 2d and Supplementary Fig. 4).
SAF312 binding site in TRPV1
Our high-resolution structure defines the SAF312 binding pocket in hTRPV1 (Fig. 3a). SAF312 resides in the VBP formed by the S3, S4, S4-S5 linker helices and TRP-helix, as well as the CHL molecule (Fig. 2b, c). SAF312 is vertically placed in the pocket with its benzonitrile group positioned on top of the TRP-helix and its quinazolinone group pointing upwards (Fig. 3a). Specifically, SAF312 is surrounded by hydrophobic interactions from Y511, S512, L515, L553, A566, and CHL, additionally, N551, and R557 form hydrogen bonds and electrostatic interactions with SAF312. To assess the binding mode of SAF312 in hTRPV1, we first investigated the inhibitory effects of SAF312 on hTRPV1 with site-directed mutations in the pocket based on alanine scanning or the equivalent position residue of other TRPV family members (Fig. 3b) by FLIPR. Most of these mutations resulted in lost activity in response to capsaicin (Supplementary Fig. 5a), consistent with the previous reports20. The inhibitory effects of SAF312 on the single-site hTRPV1 mutants of A566S, E570Q, and R557K, which can be activated by capsaicin, were evaluated (Supplementary Fig. 5b–d). Among these, A566S and E570Q exhibited 7.1- and 4.5-fold lower potency of SAF312 than that of the wild-type hTRPV1 (hTRPV1WT) (Fig. 3c, d). Additionally, R557K strongly attenuated the inhibitory effect of SAF312 (Fig. 3c, d and Supplementary Fig. 5d), supporting the observed SAF312 binding pocket and the key interactions. We then tested the mutations via patch-clamp recording of hTRPV1 mutant-expressing HEKT293 cells exposed to pH 5 only or gradient SAF312 preincubation. Y511F shifted the inhibition curve toward the right, and its IC50 value increased by 5.8-fold compared to hTRPV1WT (Fig. 3e–g). Additionally, N551A almost eliminated the inhibitory effect of SAF312. Conversely, T550 and T556 do not form a close interaction with SAF312, the T550L and T556A mutations showed similar inhibition by SAF312 to that of WT (Fig. 3h, i, g). We also tested the effects of 1 µM SAF312 on other mutants, including L553A, Y554F, A566S, and V567I. Most of these mutants showed decreased inhibition (Supplementary Fig. 6a–e), although some of the mutations also showed small currents (Supplementary Fig. 6f). We further calculated the per-residue binding energy contributing to the binding of the SAF312 within hTRPV1 by using the molecular mechanics-generalized born surface area (MM/GBSA) method. The results showed that the binding energy contributions are consistent with the key residues identified by the complex structure (Fig. 3j).

a A closeup side view is shown for the detailed interactions between hTRPV1 and SAF312. The side chains of crucial residues involved in the interaction with SAF312 are shown in the sticks. b The sequence alignment of human TRPV channels is presented, with residues color-coded based on sequence similarity. Highly conserved residues are shown in blue, and less conserved residues are depicted in red. Residues that contribute to SAF312 binding and the corresponding domain are labeled. c, d Curve fitting (c) and IC50 (d) of SAF312 dose-dependent inhibition on different hTRPV1 mutants evoked by 3.33 μM capsaicin (n = 3 for E570Q; n = 4 for A566S and R557K respectively), data are shown as mean ± SD for c. e, f hTRPV1-Y511F expressing HEKT293 cells exposed to pH 5 solution alone, pH 5 solution with 0.1 μM SAF312 following approximately 30 s preincubation with 0.1 μM SAF312, and pH 5 solution with 3 μM SAF312 following around 30 s preincubation with 3 μM SAF312. I-V curves were generated from the peak currents (peak1, peak2, and peak3) at each stimulation time. g SAF312 dose-dependent curves in responding to pH 5 solution for HEK293T cells expressing hTRPV1WT (black), Y511F (blue), T550L (orange), T556A (green), or N551A (purple) (n = 4 at each concentration point). The currents at +80 mV induced by acid (pH 5) with different concentrations of SAF312 were normalized to the current induced by pH 5 alone (peak1). The series points were fitted with the Hill equation except for data from N551A exhibiting a connecting curve. The best-fitted IC50 values for WT, Y511F, T550L, and T556A were 0.69 μM, 4.02 μM, 0.60 μM, and 0.86 μM, respectively. Data were shown as mean ± SEM. h, i hTRPV1-T550L expressing HEKT293 cells exposed to pH 5 solution alone, pH 5 solution with 0.1 μM SAF312 following ~30 s preincubation with 0.1 μM SAF312, and pH 5 solution with 3 μM SAF312 following around 30 s preincubation with 3 μM SAF312. I-V curves were generated from the peak currents (peak1, peak2, and peak3) at each stimulation time. j Per residues contribution for SAF312 calculated by MM/GBSA (n = 3), data were shown as mean ± SD.
SAF312 was reported to be a TRPV1-specific antagonist18,21. To determine the potential molecular determinants of SAF312 specificity, we compared the sequences of the six TRPV channels around the pocket. The sequence alignments revealed that the majority of the residues involved in the interaction are relatively conserved, except that S512 of TRPV1 is significantly different from Phe at the equivalent position of all the other five TRPV channels (Fig. 3b). To assess the role of S512 in SAF312 specificity, we generated the substituent S512F, which is unresponsive to capsaicin or pH (Supplementary Fig. 5a), consistent with the findings of a previous report20. We next compared the SAF312 pocket in TRPV1 with the same regions in TRPV219 (PDB code: 7XEM), TRPV322 (PDB code: 7XJ0), TRPV423 (PDB code: 8JU6), TRPV524 (PDB code: 7T6M), and TRPV625 (PDB code: 7S8B). The superpositions revealed that all the Phe residues in the other TRPV channels clash with SAF312 (Supplementary Fig. 7). Furthermore, there is no space for this Phe to assume an alternative rotamer to avoid the clash. These observations strongly suggest that S512 could be the molecular determinant of the high selectivity of SAF312 for TRPV1.
CHL contributing to the SAF312 inhibition
TRPV1 has been reported to be inhibited by CHL26,27, but no structural information has been reported. Recently, a possible mechanism of TRPV2 regulation by CHL has been proposed; in addition, they found TRPV1 was not inhibited by CHL even at concentrations up to 250 μM for the currents evoked by 2-APB19. In our 2.75-Å hTRPV1SAF312 structure, strong EM density revealed that CHL bound to TRPV1 adjacent to SAF312 (Fig. 4a, b), forming hydrophobic interactions with SAF312, Y511, L515, and I573 (Fig. 4b). A superposition of hTRPV1SAF312 with the CHL-bound TRPV2 (PDB code: 7XEM) showed that the SAF312 partially occupies the CHL site in TRPV2, thereby shifting the CHL outwards in TRPV1 (Fig. 4c). L515A contributed significantly to the SAF312 inhibitory revealed by the whole-cell patch-clamp analysis. The patch-clamp results showed that compared to TRPV1WT, the inhibitory effect of 1 µM SAF312 on the L515A mutant was substantially decreased (Fig. 4d–f). L515 is situated between SAF312 and CHL within distances of 3.5–4.3 Å (Fig. 4b). The per residue contribution analysis from MD simulations indicates that L515 contributes more to CHL binding than to SAF312 (Figs. 3j, 4g). Thus, L515 might contribute to the SAF312 inhibitory by stabilizing CHL. We also tested the effects of SAF312 on I573A in response to pH 5, which showed decreased inhibition (Supplementary Fig. 8a–c), although this mutation also showed small currents (Supplementary Fig. 8d). These results are also consistent with the per-residue binding energy contributing to the binding of the CHL calculated by MM/GBSA (Fig. 4g). To assess better CHL’s influence on the binding of SAF312 to hTRPV1, we reduced the CHL content in the cell membrane by employing MβCD, a strong and commonly utilized CHL-depleting agent28. The results showed that with increased concentrations of MβCD (from 0.5 to 5 mM), the TPRV1 inhibition by SAF312 decreased gradually (Fig. 4h–j and Supplementary Fig. 8e–g), indicating that CHL is involved in the binding and inhibitory effect of SAF312.

a, b CHL binds to the VBP pocket, and the corresponding cryo-EM densities are shown as blue and orange meshes. b The stick models of CHL and the side chains of its binding residues are displayed. The side chains of crucial residues involved in the interaction with CHL are shown in sticks and their distances to SAF312 and CHL are labeled. c Superposition of the CHL from hTRPV1SAF312 (wheat) and TRPV2CHL (light blue, PDB 7XEM). The ligands partially overlap. d, e I-V curves of hTRPV1WT (d) and L515A (e) were generated from the 3 μM capsaicin-induced peak currents without (peak1, blue) or with 1 μM SAF312 (purple). f 3 μM capsaicin-induced current with 1 μM SAF312 at +80 mV for hTRPV1 WT and L515A was normalized to the first current of 3 μM capsaicin (peak1). Data were shown as mean ± SEM from five cells. g The residue contribution for CHL was calculated by MM/GBSA (n = 3), data are shown as mean ± SD. h, i Representative I-V curves were obtained from pH 5-induced peak currents without (peak1, blue) or with 1 μM SAF312 under the MβCD treatment with different concentrations (red) 0.5 mM (h) and 5 mM (i). j Dose-dependent recovery of MβCD on SAF312 inhibition to hTRPV1 current. The membrane CHL was depleted by different concentrations of MβCD for 15 min at 37 °C in the absence of serum before each whole-cell patch-clamp recording. The inhibition percentage of MβCD was calculated with I pH 5 + 1 μM SAF312/I pH 5 (peak1) %. Data were shown as mean ± SEM (n = 6, 5, 7, 6, and 5 for control, 0.5, 1, 3 and 5 mM MβCD). k The RMSD of CHL with and without SAF312 is plotted for three independent 400 ns MD simulations.
To gain insights into how SAF312 and CHL interact with hTRPV1, we conducted all-atom MD simulations using the hTRPV1SAF312 structure with or without CHL or SAF312, respectively. During the 400 ns MD simulation of the three independent repeats, both the SAF312 and CHL molecules reached a steady state within the first 10 ns (Supplementary Fig. 9a, b). During the parallel MD studies with/without SAF312, CHL exhibited stability binds to the pocket with RMSD within 2 Å when SAF312 was present. However, after SAF312 was removed, CHL showed higher flexibility (Fig. 4k), indicating it can no longer steadily stay in the binding pocket. During the parallel MD studies with/without CHL, the RMSD values of SAF312 in TRPV1 without CHL (1–1.5 Å) are slightly higher than those of SAF312 in TRPV1 with CHL (0.5–1 Å) (Supplementary Fig. 9c, d), suggesting an increased mobility of SAF312 in the absence of CHL. This result is consistent with our structural observation showing that SAF312 interacts majorly with the protein. Additionally, we observed that Y511 is very dynamic without CHL (Supplementary Fig. 9e, f), a key residue’s reorientation toward the vanilloid pocket correlates with ejection of the resident phosphatidylinositol (PI) lipid upon ligand binding13,16,29,30, suggesting that CHL contributes to SAF312 binding. These MD results further reinforce the tight binding of SAF312 to hTRPV1 (Supplementary Fig. 9c), which stabilizes hTRPV1 in the closed state and indicates that CHL can only bind to the VBP when proper ligand exist and function as a coregulator.
Inhibition mechanism of SAF312
In the absence of exogenous ligands, the vanilloid binding pocket of TRPV1 was found to be occupied by a PI lipid (PDB code: 7L2P). It is proposed that this PI lipid stabilizes the channel in its closed state in the apo-state. However, upon binding a vanilloid agonist, the PI lipid is displaced from the pocket, leading to channel opening and activation13,29. Previous studies have demonstrated that the agonist RTX binds to the vacant space left by the inositol head group. This binding event promoted the formation of a salt bridge between R557 of the S4 helix and E570 of the S4-S5 linker (PDB code:5IRX), resulting in the pulling of the S4-S5 linker away from the central axis. This conformational change facilitates the opening of the lower gate13. The validity of this mechanism is further supported by the analysis of a capsazepine-bound structure (PDB code: 5IS0), where the competitive vanilloid antagonist capsazepine occupies the same hydrophobic pocket as RTX. However, capsazepine does not facilitate the key interaction between R557 and E57013.
To reveal how SAF312 inhibits hTRPV1, we compared our SAF312-bound TRPV1 structure with the closed apo-form TRPV1 (PDB code: 7L2P) and the agonist-bound open TRPV1 (PDB code: 5IRX). The structural comparison revealed that hTRPV1SAF312 was captured in the closed state, with a selectivity filter and gate diameter of 4.7 and 5.2 Å, respectively (Fig. 5a), similar to the previously reported apo-form closed structure with diameters of 4.4 and 5.0 Å29. A superposition of hTRPV1SAF312 with the apo TRPV1 (PDB code: 7L2P) revealed that only subtle local conformational changes were observed in the S2-S4 of VSLD, the S4_S5 linker, and the TRP_helix between the two closed structures (Fig. 5b and Supplementary Fig 3). In addition, SAF312 partially overlaps with the ester group of PIP2; meanwhile, the CHL overlaps with the acyl chain of PIP2 (Supplementary Fig. 3). These findings suggest that lipids play dual roles in regulating channel gating and small-molecule binding processes. Overlay of hTRPV1SAF312 with the RTX-bound open TRPV1 (PDB code: 5IRX) revealed marked conformational changes in the S2-S4 of VSLD, the S4_S5 linker and the TRP_helix (Fig. 5c), resulting in shrinkage of the SAF312 structure at the inner gate (Supplementary Fig. 10). In contrast to RTX, SAF312 binds deeper in the pocket. The benzonitrile group is positioned right between R557 and E570 (Fig. 5a). As a result, these two charged residues may form a salt bridge which has been proposed to be important for activating TRPV113. SAF312 might have a greater affinity than PI, which could outcompete PI from binding to the VBP. The bound CHL may reinforce the closed state as it is placed between the S4 and the S4_S5 linker (Figs. 2b, c, 4b). By preventing the movement of the S4-S5 linker, the binding of SAF312 blocks the initial phase of the conformational transformation from a closed to an open state, thereby keeping the channel closed. Similarly, Neuberger et al. reported that SB-366791 binds to the vanilloid site and acts as an allosteric hTRPV1 inhibitor recently16 (Fig. 5d). Surprisingly, the superposition of the hTRPV1SAF312 with the hTRPV1SB-366791 showed striking differences in the binding modes of these two antagonists, as well as in the antagonist-bound induced conformational changes (Supplementary Fig. 11). These observations further underscore that different antagonists may employ different mechanisms when regulating the target proteins.

a From top to bottom, the VBP from hTRPV1SAF312 (wheat), the ion-conduction pore of TRPV1 calculated with HOLE is shown as the white sphere when SAF312 ligand bond, and a closeup view showing distances between R557 and E570 when SAF312 bond. b From top to bottom, the VBP from TRPV1PI PDB:7L2P (gray), the ion-conduction pore of TRPV1 calculated with HOLE is shown as the white sphere when PI bond, and a closeup view showing distances between R557 and E570 when PI bond. c From top to bottom, the VBP from TRPV1RTX/DkTX PDB:5IRX(pink), the ion-conduction pore of TRPV1 calculated with HOLE is shown as the white sphere when RTX/DkTX bond, and closeup view showing distances between R557 and E570 when RTX/DkTX bond. d From top to bottom, the VBP from TRPV1SB-3667791 PDB:8GFA (marine), the ion-conduction pore of TRPV1 calculated with HOLE is shown as the white sphere when SB-3667791 bond, and a closeup view showing distances between R557 and E570 when SB-3667791 bond.
Lipid regulation is a common feature of many TRP channel family members. Normally, the channel binding pocket is either occupied by endogenous lipid or displaced by agonist or antagonist31. Using the cryo-EM approach, this study captured the SAF312-bound hTRPV1 structure in the closed state and revealed that SAF312 binds to the VBP pocket. Combining the functional assays and MD simulations, we defined the key residues contributing to SAF312 binding and the molecular determinants of SAF312 specificity. For the TRP channels, most antagonists work independently and do not require interaction with endogenous lipids32. This SAF312_CHL interaction pattern in the ligand-binding site have to be paid attention to when doing optimization of SAF31233. The structural basis of SAF312-mediated inhibition of TRPV1 also offers valuable insights into further designing ligands to target TRPV1, facilitating the development of more effective drug candidates targeting TRPV1.
Methods
Plasmid constructs
The full-length cDNA of hTRPV1 (NM_018727.5) was amplified from a HEK293 cDNA library. The gene was subsequently cloned and inserted into two different pEG BacMam vectors. One vector was fused with a GFP-twin-Strep tag at the C-terminus for expression, while the other was fused with a mCherry-his tag at the C-terminus for the FLIPR assay. All the hTRPV1 mutants were generated using site-directed mutagenesis, and the primer information is provided in Supplementary Data. All the constructs used in this study were confirmed by DNA sequencing.
Calcium influx assay
HEK293T (ATCC, CRL-3216) cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (Thermo Scientific) and 1% penicillin/streptomycin (Thermo Scientific) and seeded onto 96-well plates. According to the manufacturer’s instructions, hTRPV1 and mutant plasmids were transfected using PEI at a 1:3 mass ratio. After 12 h of culture at 37 °C with 5% CO2, the cells were loaded with Fluo-8 AM dye (AAT Bioquest, cat. # 36316) for 40 min to 1 h. The cells were then treated with SAF312 with a 5 X gradient for 5–10 min. Next, agonist capsaicin was added at a final concentration of 3.33 μM to stimulate intracellular calcium influx, and Fluo-8 signals were measured 30 s before and after for 10 min using the FLIPR TETRA system (PerkinElmer). The calcium influx fluorescence signal was analyzed by calculating the area under the curve after adding capsaicin, from the lowest data points to 200 seconds with various concentrations of SAF312 (F), and normalized to the signal without any antagonist (F0).
Whole-cell patch-clamp
Human wild-type TRPV1 and mutant plasmids were transfected into HEK293T cells using Lipofectamine reagent (Invitrogen, 18324020) and PLUS reagent (Invitrogen, 11514015). All patch-clamp recordings were performed 26–32 h post-transfection. The pipette with a resistance of 3–5 MΩ was used. Currents were recorded at a −60 mV holding potential from −100 to +100 mV voltage ramp every 3 s, sampled at 10 kHz (pCLAMP), and filtered at 5 kHz (Clampfit). The standard extracellular bath solution contained 140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 10 mM glucose, and 10 mM HEPES, pH 7.4 adjusted with NaOH. The pH 5 bath solution contained 140 mM NaCl, 10 mM glucose, 10 mM MES, 5 mM KCl, 2 mM MgCl2, and 2 mM CaCl2, pH 5 was adjusted with NaOH. The pipette solution contains 140 mM KCl, 5 mM EGTA, and 10 mM HEPES, pH 7.4 adjusted with KOH. Capsaicin was diluted into the standard bath solution. Fresh SAF312 was prepared in standard or pH 5 bath solution with a 1000- to 10000-fold dilution. For membrane CHL depletion, Methyl-β-cyclodextrin (MβCD) (MedChemExpress, HY-101461) was pre-incubated with the transfected HEK293 T cells for 15 min at 37 °C in the absence of serum before each recording. For analysis, whole-cell currents obtained at +80 mV were used to calculate current densities and generate dose-dependent curves. All curves and statistics were generated with Prism 10.
Protein expression and purification
The recombinant baculovirus was generated in insect Sf9 cells following the manufacturer’s protocol. P2 viruses were used to infect HEK293F GnTI− (FreeStyle 293F cells, Gibco, USA) cells at 2 × 106/ml cells. To boost protein expression, 10 mM sodium butyrate was added to the culture after 8–10 h at 37 °C. After 48 h, cells were harvested by centrifugation at 1800×g and were subsequently frozen until further purification.
The entire purification process was carried out at 4 °C. Cells were resuspended in lysis buffer A (20 mM Tris, 150 mM NaCl, pH 8.0) supplemented with protease inhibitors including 250 μM phenylmethylsulfonyl fluoride (PMSF), 0.8 μM Aprotinin, 4.7 μM Leupeptin, and 2 μM Pepstatin A. Cells were disrupted by the Dounce homogenizer. Then membrane debris was collected through ultracentrifugation for 1 h (Beckman, Ti32 rotor, 100,000 × g). The membrane fraction was homogenized and resuspended in buffer A supplemented with 1% (w/v) dodecyl-β-d-maltopyranoside (DDM) (Jiejing Tech Inc., China) plus 0.2% CHS (Anatrace, USA) and was slowly agitated for 2 h. The supernatant was collected through another 40 min ultracentrifugation and passed through a 0.22-μm filter before loading onto the gravitate column packed with Streptactin Beads (Smart-Lifesciences, China). The resin was then washed with ten column volumes of buffer A supplemented with 0.06% (w/v) GDN (Anatrace, USA). Protein was eluted with buffer A containing 0.06% (w/v) GDN and 5 mM desthiobiotin (Sigma, USA). The eluted sample was concentrated and loaded onto a Superose 6 column (GE Healthcare, USA) equilibrated with buffer A plus 0.007% (w/v) GDN. Peak fractions corresponding to the tetrameric hTRPV1 were pooled and concentrated to ~10 mg/ml for cryo-EM sample preparation. The purification processes were supplemented with 10 μM SAF312 and an additional 100 μM SAF312 was added to the final concentrated protein sample. For the hTRPV1SAF312-2, the expression and purification methods are similar, except for the membrane extraction step, only DDM was used.
Cryo-EM grid preparation and data collection
A droplet of 3 µl of hTRPV1 samples was applied onto a glow-discharged gold grid (Quantifoil R1.2/1.3, Germany). The grid was then blotted for 2.0–4.0 s at 4 °C and 100% humidity using a Vitrobot Mark IV (Thermo Fisher Scientific, USA), and flash-frozen with liquid ethane cooled by liquid nitrogen. Cryo-EM data were collected on an FEI Titan Krios (Thermo Fisher Scientific, USA) electron microscope operating at an acceleration voltage of 300 kV using a K2 or K3 Summit direct electron detector positioned after a GIF quantum energy filter (Gatan, USA). Raw movies were automatically acquired in super-resolution mode with a physical pixel size of 1.04 Å for the K2 detector utilizing SerialEM34 and 0.85 Å for the K3 detector using EPU35 software. Each movie stack was recorded for 6.4 s fractionated into 32 frames with a total accumulated dose of ~60 e/Å2. A total of 2521 movie stacks were collected for hTRPV1SAF312 and 1322 movie stacks for hTRPV1SAF312-2.
Image processing
All the movie stacks were motion-corrected, binned twofold, and dose-weighted using MotionCorr236. Defocus values of the resulting summed micrographs were estimated with Gctf37. Template particle picking, 2D and 3D classification, particle polishing, and CTF refinement were carried out in Relion338. The final best class for hTRPV1SAF312 and hTRPV1SAF312-2 containing 239,934 and 183,350 particles was imported in cryoSPARC39 and was refined to 2.75 and 2.55 Å. Detailed data processing diagrams were presented in Supplementary Figs. 2,4.
Model building
The structure of TRPV1 (PDB code: 7L2P) was fitted in the cryo-EM density map of hTRPV1SAF312 using Chimera40. For the structure of hTRPV1SAF312-2, the hTRPV1SAF312 was used as the initial model. The fitted models were manually inspected and corrected in COOT41 and subsequently refined in real space using Phenix42. Statistics for cryo-EM data collection and model refinement are summarized in Supplementary Table 1. All figures were prepared with PyMOL (Schrödinger, LLC)43 and ChimeraX44.
MD simulation
The bilayer membrane system with protein complexs were prepared using the CHARMM-GUI45. The hTRPV1SAF312-CHL complex and the hTRPV1-CHL complex (SAF312 manually deleted) was merged into the POPC bilayer membrane containing 10% CHL, respectively. The hTRPV1SAF312 complex (CHL manually deleted) was merged into the POPC bilayer membrane. All systems were solvated in water with 150 mM NaCl. We used the Amber ff19SB force field for the protein, the TIP3P force field for the waters, the Lipid21 parameter for POPC and CHL in the bilayer membrane, and the gaff2 parameter for ligands and CHL in the complex. The simulation was done by Amber20 and AmberTools2146. Using the particle mesh Ewald method with periodic boundary conditions to model long-range electrostatic effects. A 9 Å cutoff was applied to Lennard-Jones and electrostatic interactions. SHAKE algorithm was used to constrain the bonds containing hydrogen. For each system, three independent simulations were performed in the following steps: (1) Minimization was performed without the SHAKE algorithm with a maximum cycle of 10,000 and with the steepest decent algorithm for the first 5000 cycles. Position restraints of 5 kcal mol−1 Å−2 were applied on the protein and the ligand. (2) Minimization was performed without the SHAKE algorithm with a maximum cycle of 10,000 and with the steepest decent algorithm for the first 5000 cycles. (3) The temperature was increased to 310 K in 1 ns with a periodic boundary for constant volume by Langevin dynamics with a collision frequency of 2 ps−1. (4) 1 ns density equilibrium with a periodic boundary for constant pressure was performed, and Berendsen’s coupling was used to maintain the system’s pressure at 1 bar with semi-isotropic pressure scaling. (5) A 400 ns production with a periodic boundary for constant pressure and with a constant temperature of 310 K was performed with. Binding affinities and the per-residue contributions were calculated by the MM/GBSA method. The MMPBSA.py in the AmberTools21 suite was used. For each trajectory, 100 frames from the 400 ns production run were used for MM/GBSA calculation. The final binding affinity was obtained by taking the average of the three independent trajectories. VMD1.9.347 was used to align the protein’s backbone and the cpptraj module in AmberTools21 was used to calculate the b-factor of each residue in the trajectory.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support this study are available from the corresponding authors upon request. The cryo-EM maps for hTRPV1SAF312 and hTRPV1SAF312-2 have been deposited in the Electron Microscopy Data Bank (EMDB) under accession codes EMD-36577 and EMD-38161. The atomic coordinates for hTRPV1SAF312 and hTRPV1SAF312-2 have been deposited in the protein data bank (PDB) under accession codes 8JQR and 8X94. The PDB accession codes for the published structures used in this study are 5IRX, 5IS0, 7L2P, 7S8B, 7T6M, 7XEM, 7XJ0, 8GFA, and 8JU6. Molecular Dynamics simulation parameters, initial and final trajectories as well as system set up are supplied as Supplementary Data 1 in a zip folder. Source data are provided with this paper.
References
Caterina, M. J. et al. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389, 816–824 (1997).
Kashio, M. & Tominaga, M. TRP channels in thermosensation. Curr. Opin. Neurobiol. 75, 102591 (2022).
Jordt, S. E. & Julius, D. Molecular basis for species-specific sensitivity to “hot” chili peppers. Cell 108, 421–430 (2002).
Tominaga, M. et al. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 21, 531–543 (1998).
Koivisto, A. P., Belvisi, M. G., Gaudet, R. & Szallasi, A. Advances in TRP channel drug discovery: from target validation to clinical studies. Nat. Rev. Drug Discov. 21, 41–59 (2021).
Szallasi, A., Cortright, D. N., Blum, C. A. & Eid, S. R. The vanilloid receptor TRPV1: 10 years from channel cloning to antagonist proof-of-concept. Nat. Rev. Drug Discov. 6, 357–372 (2007).
Patapoutian, A., Tate, S. & Woolf, C. J. Transient receptor potential channels: targeting pain at the source. Nat. Rev. Drug Discov. 8, 55–68 (2009).
Moran, M. M., McAlexander, M. A., Biro, T. & Szallasi, A. Transient receptor potential channels as therapeutic targets. Nat. Rev. Drug Discov. 10, 601–620 (2011).
Stasi, K., Alshare, Q., Jain, M., Wald, M. & Li, Y. Topical ocular TRPV1 antagonist SAF312 (Libvatrep) demonstrates safety, low systemic exposure, and no anesthetic effect in healthy participants. Transl. Vis. Sci. Technol. 11, 15 (2022).
Thompson, V. et al. Topical ocular TRPV1 antagonist SAF312 (Libvatrep) for postoperative pain after photorefractive keratectomy. Transl. Vis. Sci. Technol. 12, 7 (2023).
Liao, M., Cao, E., Julius, D. & Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 504, 107–112 (2013).
Cao, E., Liao, M., Cheng, Y. & Julius, D. TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 504, 113–118 (2013).
Gao, Y., Cao, E., Julius, D. & Cheng, Y. TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 534, 347–351 (2016).
Kwon, D. H. et al. Heat-dependent opening of TRPV1 in the presence of capsaicin. Nat. Struct. Mol. Biol. 28, 554–563 (2021).
Kwon, D. H., Zhang, F., Fedor, J. G., Suo, Y. & Lee, S. Y. Vanilloid-dependent TRPV1 opening trajectory from cryoEM ensemble analysis. Nat. Commun. 13, 2874 (2022).
Neuberger, A. et al. Human TRPV1 structure and inhibition by the analgesic SB-366791. Nat. Commun. 14, 2451 (2023).
Smart, D. et al. Characterisation using FLIPR of human vanilloid VR1 receptor pharmacology. Eur. J. Pharmacol. 417, 51–58 (2001).
Medley, Q. et al. In-vitro and in-vivo pharmacology of SAF312 as a TRPV1 inhibitor for ocular surface pain. Invest. Ophthalmol. Vis. Sci. 62, 722–722 (2021).
Su, N. et al. Structural mechanisms of TRPV2 modulation by endogenous and exogenous ligands. Nat. Chem. Biol. 19:72–80 (2022).
Yang, F. et al. Structural mechanism underlying capsaicin binding and activation of the TRPV1 ion channel. Nat. Chem. Biol. 11, 518–524 (2015).
Mogi, M. et al. Ocular pharmacology and toxicology of TRPV1 antagonist SAF312 (Libvatrep). Transl. Vis. Sci. Technol. 12, 5 (2023).
Fan, J. et al. Structural basis of TRPV3 inhibition by an antagonist. Nat. Chem. Biol. 19, 81–90 (2023).
Fan, J. et al. Structural pharmacology of TRPV4 antagonists. Adv. Sci. 11, e2401583 (2024).
Fluck, E. C., Yazici, A. T., Rohacs, T. & Moiseenkova-Bell, V. Y. Structural basis of TRPV5 regulation by physiological and pathophysiological modulators. Cell Rep. 39, 110737 (2022).
Neuberger, A., Nadezhdin, K. D. & Sobolevsky, A. I. Structural mechanisms of TRPV6 inhibition by ruthenium red and econazole. Nat. Commun. 12, 6284 (2021).
Picazo-Juarez, G. et al. Identification of a binding motif in the S5 helix that confers cholesterol sensitivity to the TRPV1 ion channel. J. Biol. Chem. 286, 24966–24976 (2011).
Liu, B., Hui, K. & Qin, F. Thermodynamics of heat activation of single capsaicin ion channels VR1. Biophys. J. 85, 2988–3006 (2003).
Mahammad, S. & Parmryd, I. Cholesterol depletion using methyl-beta-cyclodextrin. Methods Mol. Biol. 1232, 91–102 (2015).
Zhang, K., Julius, D. & Cheng, Y. Structural snapshots of TRPV1 reveal mechanism of polymodal functionality. Cell 184, 5138–5150 e5112 (2021).
Arnold, W. R. et al. Structural basis of TRPV1 modulation by endogenous bioactive lipids. Nat. Struct. Mol. Biol. (2024).
Rohacs, T. Phosphoinositide regulation of TRP channels. Handb. Exp. Pharmacol. 223, 1143–1176 (2014).
Yelshanskaya, M. V. & Sobolevsky, A. I. Ligand-binding sites in vanilloid-subtype TRP channels. Front. Pharmacol. 13, 900623 (2022).
Payandeh, J. & Volgraf, M. Ligand binding at the protein-lipid interface: strategic considerations for drug design. Nat. Rev. Drug Discov. 20, 710–722 (2021).
Schorb, M., Haberbosch, I., Hagen, W. J. H., Schwab, Y. & Mastronarde, D. N. Software tools for automated transmission electron microscopy. Nat. Methods 16, 471–477 (2019).
Thompson, R. F., Iadanza, M. G., Hesketh, E. L., Rawson, S. & Ranson, N. A. Collection, pre-processing and on-the-fly analysis of data for high-resolution, single-particle cryo-electron microscopy. Nat. Protoc. 14, 100–118 (2019).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Zhang, K. Gctf: real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, e42166 (2018).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Pettersen, E. F. et al. UCSF Chimera−a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
DeLano, W. L. PyMOL: an open-source molecular graphics tool. CCP4 Newsletter on Protein Crystallography 40, 82–92 (2002).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Jo, S., Kim, T., Iyer, V. G. & Im, W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 (2008).
Case, D. et al. Amber 2021 (University of California, 2021).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996).
Acknowledgements
We thank X. Huang, B. Zhu, X. Li, and L. Chen at the Center for Biological Imaging (CBI), Core Facilities for Protein Science at the Institute of Biophysics, Chinese Academy of Science (IBP, CAS), J. Zhao, B. Xu, and D. Wu at the Cryo-EM Center of School of Advanced Agricultural Sciences of Peking University, and Di Wu at the Institute of Physics, Chinese Academy of Sciences for support in cryo-EM data collection. We thank Junlei Du and Yihua Huang for their valuable input and discussions and Wenjun Xie’s initial assistance with some experiments. We also sincerely thank Dr. Yi Rao for generously sharing the FLIPR instruments. The MD simulation was carried out on the High-performance Computing Platform of Peking University. This work is supported by the National Key Research & Development Plan (2022YFC2502500 to X.L.), the National Natural Science Foundation of China (22193073 and 92253305 to X.L.; 22177006 to J.F.), and Beijing National Laboratory for Molecular Sciences (BNLMS- CXX-202106 to X.L.). X.L. is also supported by the New Cornerstone Science Foundation through the XPLORER PRIZE. A special research grant for biocatalyst development from Novartis Pharma AG for X.L. is acknowledged.
Author information
Authors and Affiliations
Contributions
X.L. initiated the project. X.L. and J.F. supervised the project. J.F. performed the cryo-EM study and wrote the draft of the manuscript. J.F. and H.K. performed the FLIPR assays. J.W. and H.K. synthesized the compounds. H.K. performed the MM/GBSA calculations and molecular dynamic simulation. X.L., J.F., and H.K. analyzed the data. J.L. performed the patch-clamp assays and analyzed the data with M.T. All the authors contributed to the manuscript preparation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fan, J., Ke, H., Lei, J. et al. Structural basis of TRPV1 inhibition by SAF312 and cholesterol. Nat Commun 15, 6689 (2024). https://doi.org/10.1038/s41467-024-51085-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-51085-3
