
Abstract
The link between the oral microbiome and neurodevelopmental disorders remains a compelling hypothesis, still requiring confirmation in large-scale datasets. Leveraging over 7000 whole-genome sequenced salivary samples from 2025 US families with children diagnosed with autism spectrum disorders (ASD), our cross-sectional study shows that the oral microbiome composition can discriminate ASD subjects from neurotypical siblings (NTs, AUC = 0.66), with 108 differentiating species (q < 0.005). The relative abundance of these species is highly correlated with cognitive impairment as measured by Full-Scale Intelligence Quotient (IQ). ASD children with IQ < 70 also exhibit lower microbiome strain sharing with parents (p < 10−6) with respect to NTs. A two-pronged functional enrichment analysis suggests the contribution of enzymes from the serotonin, GABA, and dopamine degradation pathways to the distinct microbial community compositions observed between ASD and NT samples. Although measures of restrictive eating diet and proxies of oral hygiene show relatively minor effects on the microbiome composition, the observed associations with ASD and IQ may still represent unaccounted-for underlying differences in lifestyle among groups. While causal relationships could not be established, our study provides substantial support to the investigation of oral microbiome biomarkers in ASD.
Similar content being viewed by others

Introduction
Autism spectrum disorders (ASD)1 are neurodevelopmental impairments involving the cognitive, emotional, relational, and behavioural spheres2,3,4. ASD children show repetitive behaviours and sensory hyper-sensibility5,6. Estimates of ASD prevalence per 1000 children in the US range from 18.5 to 27.67. ASD are thought to arise from the interaction between genetic and environmental factors3,8,9,10,11,12, with small-effect and de novo variants contributing to disease risk13,14,15,16. Although more than 180 ASD-implicated genes have been identified from joint analysis of protein-truncating variants, missense variants and copy number variants16, they represent only a fraction of the estimated up to 1000 genes possibly involved in ASD aetiology17,18. In addition to the genetic makeup inherited from the parents and de novo variants, the microbial community acquired at birth and shaped during childhood19 has been proposed as an element for a better understanding of ASD pathophysiology20,21,22.
The human microbiome is considered an element able to influence peripheral and central nervous system development and functioning. Although largely uncharacterized, a bidirectional communication system integrating the gut, the gut microbiome, and the brain has been proposed: the gut-brain axis23,24,25,26,27,28,29,30,31,32,33. There is mounting evidence for the link between the gut microbiome and ASD20, and a potential role for the gut-brain axis in the pathophysiology of ASD has been suggested. Three recent meta-analyses identified robust differences in the gut microbiome composition of children diagnosed with ASD34,35,36. In mice, transplantation of the human gut microbiota from ASD subjects induced autistic-like behaviours37. Dietary restrictiveness, finally, has been proposed as the main factor inducing the gut microbiome shift in ASD38. The human oral microbiome has also been shown to be altered in children with ASD; specifically, an increase in bacterial species from the genera Streptococcus and Haemophilus, and a decrease in the abundance of the genera Prevotella and Actinomyces have been described39. Another study focusing on the oral microbiome40 reported a depletion of the species Campylobacter concisus and Streptococcus vestibularis in ASD children. It was recently shown that an orally-administered gut microbiome neuroactive metabolite sequestrant alleviated anxiety symptoms in 30 adolescents41,42. Despite similar preliminary evidence, the putative role of the oral microbiome in neurodevelopment and specifically in ASD, and to which extent the oral microbiome is affected by dietary restrictiveness or by other ASD-related behaviours, remains largely not understood.
In this study, we explored the oral microbiome of 2025 US families of the SPARK-WGS cohort (N = 7812 participants, including 2154 ASD children, 1646 neurotypical siblings (NTs), 2012 mothers, and 2000 fathers). We performed metagenomic analysis of the non-human reads derived from whole genome sequencing data of saliva samples, which allowed the investigation of the oral microbiome in ASD at an unprecedented size and unprecedented depth (~2.2 times larger and 11 times deeper in terms of microbial read depth than the largest published salivary dataset based on the shotgun technology43). We characterized the oral microbiome composition and function in ASD children and NTs, then correlated metagenomic features to IQ, social communication, developmental coordination, and restrictive behaviours, including restrictive eating. Our metagenomic approach, coupled with the family cohort design, enabled high resolution of microbiome functional potential as well as the analysis of the microbiome strain-sharing between family members.
Results
The SPARK-WGS cohort
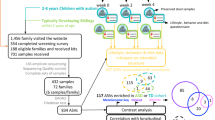
The SPARK-WGS cohort (N = 7812 individuals from 2025 US families, avg. family-size = 3.9), was established to study the oral microbiome in individuals with ASD (Fig. 1). Participants are members of families with an individual having a professional diagnosis of ASD, most of them recruited through clinical sites associated with the SPARK consortium44. Saliva samples were collected from all participants and sequenced at an average depth of 856 million reads per sample (42X) generating a total of 821 billions non-human reads (avg. per sample = 105 × 106, 95% CI [104 × 106, 107 × 106], Suppl. Data 1). The cohort comprises fathers (n = 2000, avg. age 41 years [40, 41]), mothers (n = 2012, avg. age 38 years [38, 39]), NTs (n = 1646, avg. age 8 years [7.8, 8.2]), and children diagnosed with ASD (n = 2154, avg. age 9 years [8.6, 9.1], Fig. 1). In total, 788 males and 658 females were NTs, while 1743 males and 411 females were children with a diagnosis of ASD. All children were biologically related to their parents as confirmed by the available genomic information (Methods). The male-to-female ratio in the ASD group was the expected 4:1 ratio45,46, and thus sex is moderately correlated with ASD diagnosis (Matthews correlation coefficient (MCC) = 0.35). Age also showed a small but significant correlation with ASD diagnosis (point-biserial ρ = 0.08, p = 9.4 × 10−7, n = 3800, Fig. 1). As sex and age are also associated with microbiome composition, they were also included in all analyses as potential confounders. Family relatedness, genetic ancestry of the individuals, and read-depth were also included when relevant.

The SPARK-WGS cohort was established for the study of the genetic basis of ASD through deep whole genome sequencing, collecting salivary DNA from families with ASD subjects across diverse locations across the US, thus allowing analysis of human and microbial sequences (with an initial coverage of 42X in total and 105 M microbial reads on average per sample). curatedMetagenomicData 3 was used to compare the SPARK-WGS cohort with other oral microbiome datasets. Participants included fathers, mothers, NTs and ASD-diagnosed children. The analysed cohort included 7,812 subjects from 2,025 families, the largest portion of which are quartets (n = 1541) or triads (n = 353). Dietary information was included via ARFID score and Picky Factor for diet restrictiveness. Medication history was included in the analysis. Diagnostic scores (social communication questionnaires (SCQ)), developmental coordination disorder questionnaire (DCDQ), repetitive behaviours scale (revised) (RBS-R), and full-scale Intelligence quotient (IQ) (estimated via a machine learning algorithm, Methods) were also collected. The human genomic information was used to compute ASD Polygenic Risk Score (PRS), genetic ancestry principal components, and to estimate the microbial load as an indicator of oral hygiene. Metagenomic sequences were used for taxonomic and strain-level profiling via MetaPhlAn 3 and StrainPhlAn 3, and metabolic repertoire profiling with HUMAnN 3.0 followed by imputation of GBM (gut-brain modules) to assess the microbiome neuroactive potential.
The SPARK-WGS cohort is a high resolution oral microbiome dataset
Non-human sequencing reads of the participants in the SPARK-WGS study (N = 7812) were profiled with MetaPhlAn 347. We detected in total 1320 bacterial species, 11 fungal, and 2 archaeal species (avg. species per sample = 161, 95% CI [104, 218], Supplementary Fig. 1). We investigated whether the SPARK-WGS cohort was similar to other salivary microbiome datasets. Beta-diversity analysis including 856 samples from curatedMetagenomicData (cMD) 348 revealed that the SPARK-WGS samples overlap with the salivary samples in cMD 3 but not with other oral cavity derived samples (Supplementary Fig. 2a–c). We wondered whether the increase in the number of species found in SPARK-WGS was due to multiple episodes of contamination. Potential contaminants were evaluated according to the list of 119 species determined by ref. 49, further collapsed to 59 that were present in the MetaPhlAn 3 database. We found hypothetical evidence of six contaminant species in total, four were found in < 10 samples and two were found in 37 and 38 samples, respectively. All were found at an abundance < 0.1% with the exception of Ralstonia pickettii, found at an abundance between 1% and 6% in seven samples (Methods, Supplementary Fig. 2d). Potential contaminants were thus at worst rare and we maintained a specific focus on them in the analysis by making sure such species were not included in any discriminating microbial signature.
The ASD and neurotypical oral microbiomes differ at the species level
Since a heatmap showing the 30 most abundant species in the cohort did not reveal obvious clusters driven by diagnosis, ethnicity, or sex (Methods), we next investigated the factors associated with the variation observed in the microbiome by two statistical decomposition methods. Family ID (a proxy for relatedness, co-housing, and common habits), followed by age, alpha-diversity, and sequencing depth, were found to be the variables that explain most of the microbiome interpersonal variation according to variance decomposition analysis and multivariate permanova (Suppl. Data 2, Supplementary Fig. 3a–c, Methods). ASD diagnosis was found significant by the permanova analysis (permanova p = 0.001, R2 = 2%, Supplementary Fig. 3a, b, Suppl. Data 2). In addition, a ten-times repeated permanova analysis on ASD diagnosis only sampling one child per family was also significant by both Aitchison and Bray-Curtis dissimilarities (p = 0.001, n = 2024, 999 iterations, R2 ranges 1.6-1.8% and 1-1.2%, respectively, Supplementary Fig. 3d, e), and pairwise beta diversities between ASD children were also on average higher than between neurotypical siblings (Mann-Whitney p < 10−7 in both, Supplementary Fig. 3f, g). Alpha-diversity of ASD oral microbiomes did not show significant differences compared to controls in crude nor in adjusted analysis (Mann-Whitney and Wald p > 0.05). These analyses revealed a modest, albeit significant, difference in beta-diversity between ASD and NT children.
We next applied two standard complementary methodologies to assess the differences in the microbiome composition of ASD children vs NTs. We first used a Random Forest (RF) algorithm, following a procedure concordant with the DOME guidelines on bioinformatics machine learning experiments50. We generated hundred datasets containing microbiome data from a random selection of 500 ASD children and 500 NTs from different families, having a female:male ratio 1:1 in each group, and ensuring that each set was also balanced by age and sequencing depth (Methods). Importantly, this approach allowed us to consider structural zeros in the natural behaviour of the algorithm. The average area under the curve (AUC), an unbiased measure of accuracy, out of a hundred ten-fold, ten-times iterated, cross-validations, was 0.66 (Fig. 2a). We evaluated RF feature importances by averaging the rate at which each species was used in the final tree-graph in the training set only51.

a 100 Receiver operating characteristic curves (ROCs) each from a 10-fold, 10 times evaluated Random Forest (RF) classifier discriminating ASD children from NTs using species-level relative abundances (ASD = 2,154, NTs = 1646). Each set was obtained by random sampling at most one child per family and balanced for ASD diagnosis, sex, age, and sequencing depth. b Scatterplot of the per-species RF feature importance (computed only from training folds to avoid overfitting) vs minus the log-10 of the ASD-related q value from a linear mixed model linking ASD diagnosis with centered log-ratio transformed MetaPhlAn 3 species abundances adjusted for sex, age, genetic ancestry, sequencing depth, and blocked by family ID. Orange line marks a lowess regression, and light-blue shaded area its 95% confidence interval. ρ refers to Spearman’s correlations. c ASD diagnosis-related betas from the aforementioned model considering the top-15 ASD-associated (with beta > 0) and the top-15 NTs-associated (beta < 0) at q < 0.005. Horizontal lines mark the 95% confidence intervals. Relative abundances (right) are presented in log-10 scale and coloured by enrichment in diagnostic groups. d Vertical colour bars indicate whether the species is positively, negatively (at q < 0.1), or not associated with use of medications, whether the species is aerotolerant, and whether the species is associated at q < 0.005 with the overall degree of microbial load. e Results for a model including microbial load estimation instead of microbial read depth. Yellow diamonds mark a positive association with microbial load, green diamonds mark a negative association with microbial load. Top-20 species per group are shown.
We then assessed differences in the species-level taxonomic composition of the microbiome between ASD-diagnosed children and the NTs using linear mixed models blocked by family ID and adjusted for sex, age, genetic ancestry, and sequencing depth. We found a total of 108 species associated with ASD or NTs at a stringent criterion for significance (q < 0.005, Suppl. Data 3). Notably, the two models were highly concordant, based on the correlation of ASD diagnosis q values with the RF feature importance of individual species in the classifier (ρ = 0.62, Fig. 2b). Fifty-two out of 108 identified species were more abundant in the saliva of ASD children (ASD beta > 0.0, q < 0.005). The five strongest associations were the species Actinomyces hongkongensis, Actinomyces johnsonii, Cutibacterium acnes, the Eikenella species NML 130454, and Rothia dentocariosa (betas 0.76 [0.6, 0.93], 0.76 [0.59, 0.93], 0.71 [0.53, 0.88], 0.70 [0.54, 0.86], 0.64 [0.53, 0.76]), corresponding to median increases of the relative abundances of 18.5, 2.6, 3.7, 2.8, and 2.2 times (Fig. 2c, Suppl. Data 3, 4). Fifty-six species were associated with neurotypicality (ASD beta < 0.0, q < 0.005), including Eubacterium sulci, Oribacterium parvum, Prevotella pallens, Prevotella shaii, and Prevotella jejunii (beta = −1.24 [−1.46, −1.03], −1.14 [−1.33, −0.95], −1.05 [−1.24, −0.87], −1.01 [−1.24, −0.78], −0.97 [−1.2, −0.75]), corresponding to an increase of the medians in the NTs group of 5.5, 7.3, 4.6, > 20, and 15.4 times. Repeating the analysis at the genus level, twenty-nine genera were associated (q < 0.005) with ASD diagnosis, including Cutibacterium and Eikenella (associated with NTs), and Oribacterium and Prevotella (associated with ASD, median fold change = 3.7, 1.6, 2, and 1.8, Supplementary Fig. 4a, Suppl. Data 5, 6). The Prevotella genus in particular accounted for twelve species-level associations with NTs in contrast to none with ASD.
Medications have a limited influence on the ASD oral microbiome
We then hypothesized that environmental factors shaping the oral microbiome might be the cause of the observed ASD-related species. First, we investigated the difference between ASD children who were reported to have been treated with medications (n = 802) and those who were not (n = 958). We identified a total 34 species associated with use of medications at q < 0.1, but no species was associated at q < 0.005. Among the species associated with medications at q < 0.1, four were among the top-30 associated with ASD, including R. dentocariosa (associated with ASD), Prevotella histicola, and Megasphaera micronuciformis (associated with NTs) which were all positively associated with medication use (Fig. 2d).
The ASD oral microbiome is characterized by higher load of aerobic species
Since medications was not the main driver of the observed associations, next we manually annotated the set of 108 species associated to ASD or NTs as aerobic, facultative aerobic, or anaerobic according to BacDive in DSMZ. Anaerobic species were mostly associated with NTs (only six aerotolerant associated species out of 56), while the ASD children microbiome was found to be enriched in aerobic or aerotolerant species (25 out of 52, Fisher exact test p < 0.0001). The enrichment of aero-tolerant species in ASD oral microbiome, possibly due to increased mouth-breathing52, is consistent with the increase in the species R. dentocariosa, S. gordonii, C. durum, and C. acnes (Fig. 2d). These findings suggest a potential contribution of altered mouth breathing in ASD to the observed ASD associations.
Salivary microbial load has a limited influence on the ASD oral microbiome
Since higher microbial load has been linked to poor oral hygiene and compromised oral health53,54, we estimated the microbial load in each subject (Methods) to investigate the potential impact of differences in oral hygiene between ASD and NTs on the observed associations. Such an indirect measure of oral hygiene allows us to draft some preliminary conclusions on the relationship between oral hygiene, microbial load, and oral microbiome in autism. A total of 123 species were associated with microbial load (as a surrogate for oral hygiene) while 110 species were associated with ASD after adjusting by the oral hygiene proxy. However, of the 52 species associated (q < 0.005) with both variables, twelve species were associated with ASD and high microbial load and eight with NTs and low microbial load (Fig. 2d, e, Suppl. Data 7), indicating a relatively limited influence of the inferred oral hygiene in the observed differences between ASD children and controls. A. hongkongensis, the Eikenella species NLM130154, the Streptococcus oral species 056, Actinomyces georgiae, and the skin commensal Propionibacterium propionicum, all found among the top associations with ASD, were also associated at q < 0.005 with higher microbial load (Fig. 2e). C. acnes, another skin commensal, was instead not associated with microbial load (poor hygiene). To ensure that the presence of C. acnes was not due to multiple episodes of contamination, we assessed the prevalence of C. acnes in 17 oral datasets from cMD 3, confirming its presence at a non-negligible prevalence (Supplementary Fig. 4b). The increased presence of C. acnes (aerotolerant) in ASD might be more likely motivated by mouth breathing than by oral hygiene. To ensure that the oral C. acnes is not a different species with respect to the typically found skin commensal, we performed a phylogenetic analysis of 508 C. acnes genomes including twenty reference genomes, 415 skin-derived assemblies, 24 airways assemblies, and 32 genomes from the SPARK-WGS cohort (Methods). No isolated cluster of any oral cavity or airwise-derived C. acnes was observed, suggesting that the same species is identified in the mouth, in the airways, and in the skin (Supplementary Fig. 4c). Conversely, commensal species such as R. dentocariosa and R. aerea were negatively associated (q < 0.005) with the estimated microbial load and positively with ASD, further suggesting that oral hygiene is not the sole reason driving the associations. In spite of the observed overlaps, covarying for the microbial load did not substantially modify the results of the ASD or NTs associated species in terms of q value and effect size (the ASD betas and q values of the two models showing a correlation of 0.99, Suppl. Data 8). Collectively, this suggests phenomena beyond oral hygiene at the basis of the identified associations between ASD and the oral microbiome.
Genetic predisposition to ASD does not correlate with any of the observed species
To investigate the role of the oral microbiome in the aetiology of ASD, we explored the association between microbiome composition and genetic predisposition to ASD, estimated by the inherited predisposition to a condition of a given individual due to common polymorphisms measured by the Polygenic Risk Score (PRS)55. No species were found associated with ASD-PRS, either adjusting or not adjusting for ASD diagnosis (data not shown). Since the PRS was computed from an ASD reference GWAS based on European ancestry subjects, we ran a model adjusted only for sex, age, and sequencing depth and including only caucasian individuals as defined by the ancestry principal component analysis (n = 2873). This analysis showed an association at q < 0.1 between PRS and five species (Haemophilus paraphrohaemolyticus, Anaerococcus vaginalis, Staphylococcus cohnii, Rothia sp, Corynebacterium testudinoris, Suppl. Data 8) but none of them reached statistical significance. These results suggest a limited contribution, if any, of the genetic risk component for ASD on the observed differences in microbiome compositions between children with ASD and NTs.
Cognitive functions are highly correlated with microbiome features
To explore phenotypic aspects capable of explaining the observed differences, we exploited ASD screening measures such as the Social Communication Questionnaire (SCQ) scale56, the Developmental Coordination Disorder Questionnaires (DCDQ)57, and the Repetitive Behaviours Scale (Revised) (RBS-R)58, which were available for the majority of ASD subjects. We also considered ASD cognitive impairment as a putative correlate of the microbiome composition. Given that intelligence quotients rated by licensed clinical psychologists were available only for a subset of subjects in our cohort, we took advantage of the full-scale intelligence quotients (IQ) estimated from parent-reported data by an algorithm developed to estimate with high accuracy cognitive levels in large cohorts of ASD children59. The predicted IQ determined for 1795 ASD subjects was highly correlated with the clinically-rated IQ In the subset of 116 ASD children (ρ = 0.77, p = 8.6 × 10−19, Supplementary Fig. 5a). The predicted IQ was correlated with the three above scales in the expected directions, i.e. positively correlated with the degree of motion coordination (DCDQ, ρ = 0.34, p = 1.3 × 10−31 as in60), negatively correlated with the social communication deficit (SCQ, ρ = −0.32, p = 4.9 × 10−32), and negatively correlated with the degree of repetitiveness (RBS-R, ρ = −0.12, p = 3 × 10−5). Consistently with previous findings61, the SCQ was positively correlated with the RBS-R (ρ = 0.44, p = 7.7 × 10−57) and negatively correlated with the DCDQ (ρ = −0.33, p = 4.9 × 10−28, Supplementary Fig. 5a–d, Suppl. Data 9). We then used linear models to assess the association of microbial species with each scale within the ASD group. Repetitive behaviours (RBS-R, n = 1728) did not show statistically significant associations with oral species (Suppl. Data 10); social communication questionnaire (SCQ, n = 1772) and developmental coordination disorder (DCDQ, n = 1491) showed 393 and 49 associations, respectively (q < 0.005, Suppl. Data 11, 12, Fig. 3a). However, when adjusting the above linear models for IQ, none of the identified associations for DCDQ and SCQ survived to FDR correction (no associations found at q < 0.005, Supplementary Fig. 6a–c). Furthermore, IQ showed 137 associations (54 positive and 83 negative), which were only minimally affected by adjusting for DCDQ or SCQ (Supplementary Fig. 6a, d). The above findings indicate that the oral microbiome signals in the study primarily reflect differences in intellectual functioning as measured by IQ, which likely mediates the observed signals for SCQ and DCDQ. We hypothesized that the microbial species - IQ association might be driven by poor oral hygiene and high microbial load in the ASD cohort. As expected, IQ displayed a significant negative correlation with the proportion of microbial reads (as cognitive impairment may lead to reduced oral hygiene habits, ρ = −0.24, p = 2.4 × 10−24, Supplementary Fig. 6e). After adjusting the association with IQ by the estimated microbial load, a total of 131 significant associations with IQ (q < 0.005) were found. While 44/131 significant associations were linked to both, only 14 species were positively associated with microbial load and negatively with IQ, suggesting IQ and oral hygiene as at least partially independent drivers of oral microbiome composition. Notably, the betas for the IQ species association were highly correlated with the ASD betas (ρ = −0.51, p = 2.7 × 10−88, Suppl. Data 13, Supplementary Fig. 7a), the predicted IQ effect sizes were highly correlated with the clinically-rated IQ model effect sizes (ρ = 0.44, p = 2 × 10−64), and the model on clinically-rated IQ showed 38 out of 39 significant associations (q < 0.2) overlapping with the full-dataset model on ASD diagnosis (Supplementary Fig. 5d). Among the four parameters considered, IQ was therefore the factor showing the highest correlation with the observed associations between ASD diagnosis and oral microbiome features.

a correlation plot of the betas from the model on the full cohort (ASD = 2154, controls = 1646) vs the betas from an identical model considering the subset of 291 ASD children with available dietary habits data and 241 NTs. b correlation plots of the betas from the differential abundance model considering the above subset and the betas from case-only models assessing the relationship between oral microbiome composition and ARFID score, Picky Factor, and full-scale IQ, within the 291 children with available dietary habits data. ρ refers to Spearman’s correlations. c The top-15 ASD associated and the top-15 NTs associated species from the full cohort differential abundance model are shown for the different models run within the subset. The fifth model refers to the association between IQ and oral microbiome composition adjusted for Picky Factor. Colours for the significant (q < 0.2) associations are reported in the legend. Grey refers to q > 0.2. d Variable importance assessed via permutation test in constraints ordination (Ordistep) on Aitchison pairwise distances among 291 ASD individuals reveals importance of Picky Factor and predominance of IQ over Picky Factor in determining oral microbiome dispersion. Variables are the same as Ext.DataFig. b With the exclusion of family ID and ASD diagnosis. (right) Picky Factor is excluded (non-significant) by the stepwise model selection when IQ is included.
Dietary behaviours show little impact on the observed microbiome differences
Dietary restrictiveness is common in ASD and has been recently proposed as a modulator of the gut microbiome38. To test to which extent our findings on the oral microbiome should be ascribed to differences in dietary habits, we took advantage of data collected using the SPARK Research Match platform on SPARK families that investigated eating behaviours62, by conducting an ad-hoc analysis on a subset in which both imputed IQ and avoidant/restrictive food intake disorders (ARFID) data were available. In total, we considered 291 ASD children with available data for ARFID based on the Nine-Items ARFID Screen (NIAS)63, focusing on picky eating (Picky Factor from the NIAS) and on the “ARFID score” (derived from a model including the three NIAS factors and survey questionnaires on measures of eating behaviours and GI symptoms)62. We compared the microbiome differential abundance results obtained from ASD children with avoidant/restrictive food intake disorders (ARFID) data and their siblings (total n = 532) to those obtained from the full cohort (n = 3800), using a less stringent threshold for false discovery rate (q < 0.2). We found a high correlation (ρ = 0.51, p = 9.2 × 10−88) between the two models, with 44 species with q < 0.2 in common, indicating that this subset is a fair representation of the entire cohort (Fig. 3a, Suppl. Data 14). We ran three separate case-only models, assessing the correlation between oral microbiome composition and ARFID Score, Picky Factor and imputed IQ. When comparing these models with those of the differential abundance model on the subset, the ARFID Score betas showed a low correlation with ASD (ρ = 0.11, p = 1.1 × 10−4), and the Picky Factor score showed a slightly higher correlation (ρ = 0.22, p = 10−16), but neither ARFID nor the Picky Factor scores showed significant associations at q < 0.2 (Suppl. Data 15, 16). Interestingly, IQ showed the highest number of statistical associations, either including or not including Picky Factor in the same model (n = 94 and 84, respectively, Suppl. Data 17, 18), as well as the highest correlation with the differential abundance model run in the subset (ρ = −0.33, p = 1.8 × 10−34, Fig. 3b), suggesting a major role in the observed ASD-related microbiome differences with respect to dietary habits. Considering the top-15 species either positively or negatively associated with ASD in the original cohort, 18 were still significantly associated (q < 0.2) in the differential abundance model run in the subset, but none was associated with ARFID score and Picky Factor, while 24 remained significantly associated with IQ (Fig. 3c, Supplementary Fig. 7b). To determine the importance of Picky Factor and IQ in explaining compositional samples’ dispersion (measured by Aitchison pairwise distances) in the oral microbiome profiles, we next conducted a multivariate beta-diversity analysis (with permutation tests in constrained ordination) with the available experimental variables. Picky Factor was associated with the overall microbiome sample dispersion when IQ was not included in the model (R2 = 0.6%, p = 0.028, Fig. 3d). IQ became predominant when included in the model (IQ R2 = 6%, p = 0.001), marginalizing the importance of Picky Factor (which was not significant when IQ was included in the stepwise model selection). Although the importance of IQ appeared predominant also in a relatively small dataset, future studies with greater power would be needed to confirm and further explore the specific role of picky eating in shaping the oral microbiome.
Reduced oral microbiome transmission in ASD and lower strain sharing rate in cases with severe-to-mild cognitive impairment
We hypothesised that there could be a difference between ASD and NTs children in the degree of microbiome strain sharing with their family members, given that person-to-person microbiome transmission has been found to be affected by social interactions64. A proxy for microbiome strain transmission has been recently obtained assessing genetic similarity of microbial strains64,65 as the presence of the same strain in two different individuals is very likely to be due to a strain sharing event64,66. Using strain-level metagenomic profiling (Methods) and considering 1525 families of four individuals (quartets) with two children discordant on ASD diagnosis, we computed strain sharing rates by dividing the number of shared strains between two individuals by the number of species detected in both (Suppl. Data 19, 20). Children with ASD displayed significantly lower strain sharing rates with both parents compared to NTs (post-hoc Dunn q = 3.9 × 10−21 and 2 × 10−20, with the father and with the mother: median 11% and 14% in ASD, respectively, vs. 12% and 15% in NTs), albeit with a low standardized mean difference (SMD, 0.07 and 0.06, respectively, Suppl. Data 21, Fig. 4a).

a person-to-person strain sharing (left: with father, right: with mother) coloured by diagnosis (pink = ASD, blue = NTs, n = 1,525 and 1,525). b between sibling strain sharing rates for 111 species evaluated at the strain level divided by IQ category (≤70, >70 & ≤85, >85) (each dot represents a sample average rate of strain sharing, n = 1525). c father-child strain sharing. ASD children (n = 1525) are divided by IQ category (≤70, >70 & ≤85, >85). NTs are reported in blue. Red line represents the median of the ≤70 IQ group. d mother-child strain sharing. ASD children (n = 1525) are divided by IQ category (≤70, >70 & ≤85, >85). NTs are reported in blue. Red line represents the median of the ≤70 IQ group. Numbers refer to Post-hoc Dunn-test q values; ns (non-significant) refers to q > 0.05. ‘d’ stands for Cohen’s d (standardized mean difference).
We then looked at the correlation between strain sharing rates and clinical data available for ASD children. The imputed IQ was significantly correlated with strain sharing between ASD children and their parents (n = 1525, ρ = 0.22, p = 3.2 ×10−16, ρ = 0.18, p = 2.1 ×10−11, with father and with mother, Supplementary Fig. 8a). Among the ASD-related scales, SCQ was negatively correlated with the ASD children sharing rates with the parents (p < 0.05, r = 0.05 in both mothers and fathers), suggesting reduced social interactions as a potential contributor to the strain transmission. However, the correlation was much weaker compared to the correlation with IQ, indicating that IQ is likely the primary factor involved in the observed decrease in microbiome strain sharing rate (Supplementary Fig. 8b). Notably, imputed IQ was correlated with the strain sharing rate average in the set of ASD children with available dietary information, while ARFID score and Picky Factor were not (n = 223, parent-to-child average strain-sharing IQ ρ = 0.2, p = 0.02, Supplementary Fig. 8c). We additionally investigated the influence of cognitive impairment on strain sharing rates by dividing the ASD children into commonly used IQ categories for severe or moderate (≤70) and mild or borderline ( > 70 and ≤ 85) cognitive impairment, or a normal IQ-range ( > 85)67. In total, 413 ASD participants had an IQ ≤ 70, 328 individuals had an IQ > 70 and ≤ 85, and 784 had an IQ > 85. First, the IQ of the ASD children did not seem to determine any difference in the average strain-sharing rate with their siblings (anova p = 0.65, Fig. 4b). ASD children with severe or moderate cognitive impairment (n = 328) showed the lowest strain sharing rate with both parents (median 9% and 12%, vs 12% and 15% of the NTs, post-hoc Dunn q = 2.4 × 10−11 and 10−6, SMD = 0.36 and 0.25), followed by ASD children with mild or borderline cognitive impairment as defined by IQ ranges (Fig. 4c, d, Suppl. Data 22, 23). ASD children in a normal IQ range ( > 85) showed higher strain sharing with both parents than the cognitively impaired ones (post-hoc Dunn q < 10−15 and 1.2 × 10−7 in sharing with the father and the mother, respectively, SMD 0.45 and 0.3), indicating cognitive impairment as a putative driver for the observed lower microbiome transmission. To then evaluate whether the oral hygiene proxy is responsible for this trend (i.e. if a higher degree of oral hygiene is positively correlated with microbial engraftment), we built linear models to assess the relationship between the average rate of transmission with the IQ-class, adjusting for the estimated microbial load. Cognitive impairment was the best predictor of the log-transformed strain-sharing rate (betas = 0.28 and 0.21, p < 1.6 × 10−11 and 5 × 10−8 with the fathers and the mothers, respectively). The estimated microbial load was also a significant predictor (betas = 0.01 and 0.01, p < 1.8 × 10−4 and 4.2 × 10−9), indicating both microbial load and cognitive impairment as factors contributing to the decreased microbial strain sharing rate. Finally, we exploited the three classes of IQ (severe or moderate cognitive impairment [≤ 75], mild or borderline [> 75 & ≤ 85], and normal IQ range [>85]) to conduct differential abundance analysis among the different classes. We observed a substantial correlation between species’ coefficients from differential abundance analysis in ASD vs NT and the differential abundance analyses contrasting severely or moderately vs mild or borderline impaired ASD children and vs normal IQ range ASD children (ρ = 0.47 and 0.51, respectively, p < 1 × 10−20 in both contrasts, for their correlation with the ASD vs NT coefficients, Supplementary Fig. 8d).
Functional potential differences in the genetic repertoire of ASD and NTs oral microbiomes
We compared the functional potential of the oral microbiome of ASD children and NTs inferred from the microbiome genetic repertoire with HUMAnN 3.047. We performed differential abundance analysis of MetaCyc pathways, adjusting by the same covariates used in the species abundance model, plus alpha diversity to account for the potential effect of species richness on the pathway contribution. At q < 10−7, mixed linear models on CLR-transformed pathway counts of read per kilobases identified 66 MetaCyc pathways associated with ASD (n = 41) or NTs (n = 25, Suppl. Data 24, 25). Aside, the strongest associations were also replicated with IQ but not with ARFID and Picky Factor (Fig. 5a, Suppl. Data 26–28). Many of the strongest associations with NTs were pathways related to the metabolism of glutamate (L-glutamate degradation VIII to propanoate, L-glutamate and L-glutamine biosynthesis, D-galacturonate degradation I, D-galactarate degradation I, Fig. 5a). On the opposite side, tryptophan and serotonin degradation, GABA degradation, and the pathway aromatic biogenic amine degradation, were all associated with the metabolic repertoire of ASD children (Fig. 5a). We examined the reference pathway graphs and identified three key-enzymes (EC 1.4.3.4, serotonin oxidase, 1.1.1.1, 5-hydroxyindole acetaldehyde reductase, and 1.2.1.3, 5-hydroxyindole acetaldehyde dehydrogenase) which showed higher mean coverage in ASD compared to NTs, and thus represent putative biomarkers for a potential selective advantage of the species associated with ASD in the salivary microbiome. In particular, the three-steps enzymatic chain leading from serotonin to 5-hydroxytryptophol consistently showed higher reads counts in ASD individuals compared to NTs (Fig. 5b).

a the top-15 ASD associated and the top-15 control associated (q < 10−7) MetaCyc pathways from a linear mixed model of ASD diagnosis adjusted for sex, age, genetic ancestry, sequencing depth, and alpha-diversity, and blocked by family ID. The beta from the same pathways is reported for an identical model on 291 ASD children with available diet information and 241 NTs, and linear models assessing the relationship between MetaCyc pathway and ARFID score, Picky Factor, and full-scale IQ on 291 ASD children. b (left) ASD-associated species contributing to EC numbers that are in the MetaCyc serotonin degradation pathway are reported, with the mean difference in enzyme coverage between ASD and NTs, and the number of ASD samples in which the contribution is observed. (right) the serotonin degradation MetaCyc pathway is reported. The three enzymes identified in microbial reads are highlighted.
Dopamine and GABA degradation potential in ASD oral microbiomes
We complemented these results with a separate differential abundance analysis based on the database of the gut-brain modules (GBM)68, a manually curated database of microbial genes potentially encoding neuroactive functions including non-gut and non-human ones68. Out of 56 modules, 19 were associated (q < 0.005) with ASD and 9 were associated with NTs (Suppl. Data 29, 30). The two functions that emerged mostly in ASD were dopamine degradation and GABA degradation (betas 0.55 and 0.5, respectively, Fig. 6a). The identification of dopamine degradation could be reconducted to the same enzymes previously identified for the serotonin degradation, specifically via dopamine oxidase and 3,4-dihydroxyphenylacetaldeyde dehydrogenase (EC 1.4.3.4 and 1.2.1.3, Fig. 6b). GABA degradation was identified also in the previous analysis as 4-aminobutanoate degradation V pathway (Fig. 5a), and, by exploring this pathway, we identified three enzymes (EC 2.6.1.19, 1.4.1.2, and 2.8.3.8) which are enriched in species associated with ASD, and are involved in the synthesis of ammonium via 4-aminobutyrate transaminase action followed by glutamate dehydrogenization, as well as in the synthesis of butyrate via ButCoA acetyl transferases (Fig. 6c). Notably, the product of this reaction is butyrate, which is also the product of the reaction representing the strongest association with NTs. Overall, these findings constitute potential directions for the investigation of the ASD oral microbiome at the enzymatic level.

a (left) 19 associated (q < 0.005) GB modules from a linear mixed model of ASD diagnosis in 3,785 salivary microbiome samples, adjusted for sex, age, genetic ancestry, sequencing depth, and alpha-diversity, and blocked by family ID. b reactions related to dopamine degradation in the MetaCyc biogenic amine degradation pathway pathway. c (top) reactions related to GABA degradation to ammonium and butyrate in the MetaCyc GABA degradation pathway; (bottom) ASD-associated species contributing to EC numbers that are in the MetaCyc GABA degradation pathway are reported, with the mean difference in enzyme coverage between ASD and NTs, and the number of ASD samples in which the contribution is observed.
Oral microbiome functional potential correlates with cognitive impairment
To help interpret the association between neuroactive pathways and ASD we assessed the association between ASD screening scales (SCQ, DCDQ, RBS-R) and IQ with the functional potential modules and compared the results with the differential abundance analysis. Considering the pathways enriched either in ASD or NTs at q < 10−7 in the full cohort, analogous trends were observed for SCQ and DCDQ, but with lower statistical significance (SCQ, opposite direction, no significant pathways; DCDQ, same direction, one significant pathway, Supplementary Fig. 9a, Suppl. Data 31–34). When replicating the same analysis using the GBM modules, IQ showed a high concordance with the differential abundance analysis between ASD children and NTs (ρ = −0.79, p = 8.9 × 10−11, Supplementary Fig. 9b), and similar results for other ASD diagnostic instruments, with no significant pattern for the RBS-R scale for repetitive behaviours (Supplementary Fig. 9c, Suppl. Data 35–38).
Discussion
We analysed a total of 7812 oral microbiomes from 2025 US families in the SPARK-WGS cohort sequenced at very high depth (avg. 105 million microbial reads per sample) to identify potential relationships between the salivary microbiome and ASD. After showing the relative abundances of multiple species differed between ASD children and NTs, we explored behavioural aspects capable of explaining the observed differences, including dietary habits and cognitive functions. Neither avoidant/restrictive food intake nor the use of medications or the estimated degree of oral hygiene showed an impact of the same scale of the one displayed by the imputed full-scale IQ in the ASD subjects. Repetitive behaviours also had a small influence on the oral microbial composition, whilst additional factors such as social communication and developmental coordination revealed significant associations, also mainly driven by IQ. The investigation of the person-to-person strain sharing of the oral microbiome using strain-level profiling and differential abundance analysis among different classes of cognitively impaired ASD children confirmed the influence of cognitive impairment in shaping the oral microbiome in ASD. ASD children exhibited lower degrees of microbial strain sharing with their parents compared to NTs, and this effect was found especially in subjects with severe to moderate cognitive impairment (IQ ≤ 70). Our analysis revealed significant differences in metabolic pathways related to serotonin, GABA, and dopamine degradation. This was particularly due to enzymes converting serotonin to 5-hydroxytryptophol and dopamine to (3-4-dihydroxyphenyl)acetate. Conversely, NT samples were enriched in glutamate degradation and direct butyrate biosynthesis pathways. Notably, as part of the GABA-degradation pathway we observed an enrichment in ButCoA acetyl transferases, whose product is butyrate. This suggests a potential distinction in butyrate production routes between ASD and NT individuals, albeit this warrants further investigations. The observed associations appear to be mainly linked to cognitive deficits (as measured by the imputed IQ values), although further studies may be needed in order to disentangle the contribution of different factors, including more accurate measures of dietary habits and oral hygiene, as possible mediators of the observed signal.
Key neurotransmitters such as gamma amino butyric acid (GABA), serotonin, glutamate, dopamine, and the SCFAs propionate, acetate, and butyrate are metabolised by the gut microbiome and might play a role in ASD69. Serotonin and its precursor tryptophan have been linked to behavioural impairments in ASD70,71,72,73, possible causal mechanisms have been suggested74,75,76. We also observed an enrichment of L-tryptophan degradation via tryptamine that induces the synthesis of serotonin by the enterochromaffin cells and helps regulate transit time in the gut77. The role of central and peripheral serotonin and its metabolites in the social and cognitive functions in ASD has been also the object on numerous investigations78, and the role of serotonin as a key mediator of the gut-brain-microbiome axis in ASD has been proposed79. Most notably, it is well known that elevated blood levels of serotonin are found in >25% of ASD subjects80, and further studies will prove able to verify the hypotheses here suggested, in particular, whether the molecular exchange between blood and saliva facilitated by the high vascularization of the salivary glands may result in a selective advantage for oral microbes carrying the key-reactions from serotonin to 5-hydroxytryptophol in their generic carriage. In addition to the serotonergic system, the balance between the glutamatergic (excitatory) and the GABA-ergic (inhibitory) systems has also been shown to be altered in ASD81,82,83,84. Here, we observed an enrichment in glutamate and butyrate synthesis in NTs, while the GABA degradation potential to butyrate was enriched in ASD. Several magnetic resonance spectroscopy studies have revealed decreased GABA levels in the brain of ASD children85,86, and lower GABA/Cre levels in the anterior cingulate cortex of ASD children were associated with lower IQ and greater impairments in social cognition87. Higher levels of glutamate as well as lower concentration of GABA were found in a metabolomic investigation of the urinary neurotransmitters in ASD, which were suggested to be driven by the gut microbiota88. Recently, the oral microbiome has raised considerable interest due to its link with host characteristics43,64 and recent findings have suggested that oral microbes can influence neurological processes and shape cognition and behaviour, suggesting the hypothesis of an oral microbiome-brain axis89. It has recently been shown that a mouse model of ASD with reduced dopamine signalling displays an altered oral microbiome with a significant decrease in Fusobacterium abundance, similarly to what we observed in our study90. Our study shows in a large cohort that, in ASD, impaired cognitive functions are associated with an oral microbiome harbouring altered taxonomic and neuroactive pathway composition. Notably, the largest study published so far on the ASD oral microbiome totalled 350 subjects39,40,91,92,93,94, while the largest meta-analysis considering the more-studied gut microbiome comprised 1740 subjects in total35. While most existing studies were performed with 16S rRNA gene sequencing, our metagenomic approach coupled with the family design enabled higher resolution and analysis of microbiome strain sharing patterns.
Still, this cross-sectional study has important limitations. First, we were not able to verify the true biological activity underlying the inferred evidence. Second, we could not control dietary patterns or retrieve food frequency data at high resolution, which might represent a key confounder. Recently, a gut metagenomics study in ASD children (N = 247) proposed that microbiome differences may reflect restricted dietary preferences in ASD, which results with reduced microbial taxonomic diversity in the stool38. Indeed, picky eating has been associated with alteration in the gut microbiome of ASD children95, however no data are available so far on its effect on the oral microbiome. In our investigation, we did not observe any significant association between picky eating and any microbial species. In contrast, we found significant associations between specific microbiome components and IQ. Additionally, we considered two key putative confounding aspects, namely the use of medication and oral hygiene (proxied by total microbial load), which turned out to have only a limited contribution to the observed results. Since the majority of ASD medications, including serotonin reuptake inhibitors and antipsychotics, are used to address common repetitive behaviours, anxiety and irritability symptoms, and are not restricted to cognitively impaired subjects, it is therefore conceivable that medications plays a minor role in the observed IQ-related alterations. We cannot fully exclude the presence of additional potentially confounding factors, possibly indirectly related to cognitive abilities, which might impact on the oral microbiome composition. Among them, factors linked to the salivary sampling (time of collection, time of last oral hygiene, and time of last meal) were not available, although instructions were given to not eat, drink, smoke or chew gum for 30 min before collecting the saliva sample. In our study, the sampling procedure was nevertheless highly homogenous across subjects (parents, ASD and NTs), with <0.5% of individuals requiring assisted sampling. Although we cannot rule out some of the identified species (i.e. Streptococcus sanguinis, Fusobacterium periodonticum) may be correlated to oral hygiene factors, their increase was not specific to the ASD group, and are unlikely to be driving the overall neuroactive potential related differences. Nevertheless, future studies will be needed to assess the generalizability of our findings to a more diverse population, unravel the role of specific medications, food frequency and oral hygiene practices on the oral microbiome, and explore the potential influence of gastrointestinal dysfunctions on the oral microbiome functional potential in ASD.
Our findings suggest cognitive impairment as the main factor underlying the differences observed between ASD and NT oral microbiomes, which may represent a mediator of effects on the microbiome due to group differences in lifestyle factors. The identified associations point to microbial pathways related to neurotransmitter metabolism, suggesting a potential link between cognitive function, lifestyle factors, the oral microbiome, and neurotransmitter-related pathways, though the nature of these relationships requires further investigation. Given the low permeability of the blood brain barrier, it is unlikely that salivary neurotransmitters entering into the bloodstream could reach the CNS and exert a direct effect at the central level. An alternative scenario worth considering is that certain neurotransmitter-degrading bacteria may translocate to the gut, or neurotransmitters in the oral cavity potentially reach and impact enteric neurons in the gut, where bidirectional communication with the central nervous system may lead to alterations in brain function96. However, the hypothesis of a selective pressure exerted by neurotransmitter levels on the salivary microbiome may also represent a compelling avenue for further exploration of ASD biomarkers. Empirical studies, especially focusing on the metabolomic profiles of the oral microbiome and their systemic effects, will be crucial for validating this hypothesis.
Methods
Subject details
The study is a cross-sectional analysis of a family cohort based on the SPARK collection coordinated by the Simons Foundation Autism Research Initiative (SFARI). The procedure collection for the SPARK-WGS cohort complied with all ethical regulations and with the Declaration of Helsinki (2013). Subjects being part of the SPARK initiative (Western IRB (https://www.wcgirb.com/), Protocol tracking number: WIRB20151664) are US children reported to have a professional diagnosis of ASD, and their family members. Once an ASD-diagnosed individual or their legally authorized representative starts the enrolment procedure, all the members of the family are invited to enrol. Parents or legal guardians are required to provide all the necessary information relative to their ASD dependent(s) and sibling(s), and independent adults report on themselves. The enrollment happens online and consists of informed consent to the study and to DNA collection/analysis, and submission of brief medical and psychiatric history and behavioural questionnaires on the ASD-diagnosed individual and other members of the family. All participants provided written informed consent to take part in the study. Written informed consent was obtained from all legal guardians or parents for all participants aged 18 and younger and all older participants who have a legal guardian. Assent was also obtained from dependent participants aged 10 and older. For a description of methods, see ref. 44.
Data sequencing, quality-preprocessing, taxonomic and functional profiling
Sample size was not pre-determined by statistical methods. A total of 7861 subjects providing saliva samples were enroled in the SPARK-WGS initiative between 2016 and 2021 and sequenced at the New York Genome Center between 2020 and 2021 (SPARK integrated WGS1-3; ID: SFARI_SPARK_iWGS_v1.1). Saliva was collected at home or on-site for site-affiliated participants, with the OGD-500 kit (DNA Genotek, Canada) which is based on induced drooling and avoids swabbing the mucosal surfaces, and sent to the New York Genome Center. Participants were instructed to not eat, drink, smoke or chew gum for 30 min before collecting the saliva sample. Sampling strategy was illustrated to the participant by written and video instruction. Genetic material was extracted using a Chemomagic MSM1/360 DNA extraction instrument and eluted into 110ul of TE buffer at PreventionGenetics (Marshfield, WI, USA). The biological sex and parental relationships of all samples were confirmed by a panel of microsatellite markers. Samples that failed these checks were not processed further. Libraries were prepared with the Illumina DNA PCR-Free Library Prep kit following manufacturer’s guidelines. Sequencing was performed using an Illumina NovaSeq 6000 platform using S2/S4 flow cells and following manufacturer protocols. An average of 856 million reads per sample (at 150 nt reads) was obtained (mean coverage 42X). Biological sex and parental relationships were confirmed again using PLINK software97. Any samples that failed these checks were not processed further. The final number of participants retained was further reduced to 7812 subjects from 2025 families due to metadata availability constraints. Samples were preprocessed using a modified version of the pipeline at https://github.com/SegataLab/preprocessing. Briefly, samples were mapped with BWA version 0.7.1598 against hg38 Human genome to remove human contamination. Non-mapping reads were extracted with samtools version 1.1099,100 and were then quality-screened with Trim Galore (version 0.6.4, parameters: --stringency 5 --length 75 --quality 20 --max_n 2 --trim-n)101 to remove short ( < 75 bp) and low quality (Phred quality score <20) reads as well as reads with more than two ambiguous nucleotides. The remaining high-quality reads were sorted into one forward, reverse and unpaired read file for each metagenome. Metagenomic sequences of the 7812 participants were profiled using MetaPhlAn 347 (ver. 3.0.13; default parameters) which estimates the taxonomic composition. HUMAnN 3.047 (ver. 3.0: default parameters) which estimates MetaCyc pathways and UniRef90 gene families’ abundances from metagenomic reads, was run on 3785 metagenomic samples. HUMAnN 3 gene families were grouped into KEGG Orthologs102 using the HUMAnN 3.0 utility script humann_renorm_tables, and gut-brain module (GBM, v1.0) 64 relative abundances, covering neuroactive potential pathways, were calculated with omixer-rpmR (v0.3.3)103.
SPARK-WGS as a salivary microbiome dataset
We retrieved 856 oral cavity microbiome profiles available in curatedMetagenomicData48. We computed pairwise distances between all samples and the SPARK-WGS cohort showed the lowest dissimilarity to the HMP_201219, BritoIL_2016104, and LassalleF_2017105 (avg. Bray-Curtis dissimilarity 0.61, 0.61, and 0.63, respectively) datasets, which are the three datasets available from saliva. Tongue dorsum, throat swabs, tonsils, and buccal mucosal samples (arguably closer to saliva samples with respect to teeth) from the HMP_2012 and the tongue dorsum datasets Castro-NallarE_2015106 and FerrettiP_2018107 were the next most similar to the SPARK-WGS cohort (avg. Bray-Curtis dissimilarity from our samples = 0.67, 0.69, 0.74, 0.75, 0.75, and 0.8, respectively). Plaque and keratinized gingival samples from the HMP_2012, GhensiP_2018108, ShiB_2015109, ChuDM_2017110 datasets showed the highest distances from SPARK-WGS (avg. Bray-Curtis dissimilarity = 0.81, 0.82, 0.83, 0.87, 0.93, 0.86). The sole exception was the dataset OlmMR_2017111 (tongue-dorsum) (avg. dissimilarity = 0.96, Supplementary Fig. 1d). These results are consistent with previous observations112. We used the same 856 samples to assess the prevalence of C. acnes in oral microbiome datasets. Metagenomic assembly of C. acnes was conducted on 1286 samples of the SPARK-WGS cohort as previously described113; 34 high and medium-quality genomes were integrated with other 474 high quality C. acnes genomes; phylogeny of C. acnes was built using PhyloPhlAn 3114 by aligning the C. acnes proteomes against the 400 universal marker genes. Tree was plotted using GraPhlAn115. Other potential contaminants were evaluated according to the list of 119 species determined by ref. 49, further collapsed to 59 that were present in the MetaPhlAn 3 database. Four potential contaminant species (Achromobacter xylosoxidans, Alcaligenes faecalis, Kocuria rosea, Variovorax paradoxus) were found at a non-null abundance in 13, five, five, and eight samples respectively. Two species (Ralstonia pickettii and Cutibacterium avidum) were found in 37 and 38 samples, respectively. All the potential contaminants identified were found at an abundance <0.1% with the exception of Ralstonia pickettii, which showed abundances between 1 and 2% in five samples, between 2 and 5% in one sample, and at 5.7% in one sample (this sample potentially affected by a contamination episode). Based on this analysis, we excluded the possibility of contamination affecting considerably the subsequent analyses.
Principal components analysis of human genetics
The genetic ethnicity of the individuals in SFARI-WGS cohort and their relatedness (ancestry PCAs) was quantified by the extraction of the principal components from the genetic relatedness matrix (GRM), computed with Plink 1.9 (option --make-grm-rel and --pca)97. The extracted features were used in the variance component estimation to account for relatedness between individuals. Moreover, we derived genetic components from the GRM matrix for each ethnic cluster found in 1000 Genomes phase 3116 dataset. Using the Genetic relatedness matrix of SPARK-WGS we projected each individual onto the major components of the ethnicity clusters derived from the 1000 Genome. Then, we applied cluster analysis (k-mean) on the principal components and assigned each individual to a cluster, resulting with the following distribution: 15% American, 3% African, 2% east Asian, 77% European, 3% south Asian. GRM computation was performed with Plink 1.9117, projection and cluster analysis with R (3.4.4) packages prcomp, kmeans and factoextra.
Compositionality issue
The issue of data compositionality was properly addressed by the centered log-ratio (CLR) transformation of the features used118. We used the scikit-bio python library (ver. 0.5.6). Zeroes in the MetaPhlAn profiles were imputed with a multiplicative replacement strategy. Zeroes in the MetaCyc and in the GBM profiles were imputed by pseudo-count of one.
Variable contribution analysis
Variable importance was assessed on CLR-transformed MetaPhlan 3 species profiles in 7812 metagenomes. We used the ordistep function from the R package vegan (ver. 2.5.7)119. Ordistep was run starting from the result from the capscale function120. We run a variance decomposition analysis121 fitting the model: “species ~ 1|proband or control + 1|male or female + 1|family-id + age (months) + Shannon entropy + log(sequencing depth) + ancestry principal component (aPC) n. 1 + aPC n. 2 + aPC n. 3”43.
Alpha and beta diversity assessments
Alpha diversity (Shannon entropy) of MetaPhlAn 3 species was evaluated by Kruskall Wallis test over the diversity distributions of the 4 individual types in our cohort (father, mother, sibling, proband) and between the two main diagnostic groups. We then fit a linear mixed model (statsmodels python library, ver. 0.11.1) with Shannon-diversity as response and diagnosis, age, gender, sequencing depth as predictors, blocked by family ID. Significance was assessed by Wald test. Beta diversity was used to contrast siblings and probands, and significance was assessed via permanova (scikit-bio library in python (ver. 0.5.6) using 999 permutations). On Bray-Curtis and Aitchison distances independently, we sampled one child per family (n = 2024), and repeated the procedure ten times. The pseudo F was then used to compute R2 via the formula: “1 - (1 + ((F x (p − 1)) / (n - p)))−1”, where n is the number of data points, F is the pseudo-F, and p is the number of parameters.
Machine learning approaches
We used MetAML software (with the scikit-learn Random Forest algorithm)122, and we trained and tested MetaPhlAn 3 species relative abundances ASD-from-controls prediction capabilities (2154 ASD children being the positive class, and 1646 NTs being the negative class). The rest of the non-default parameters of the algorithm were: 1000 estimators (trees), a minimum of 10 samples per leaf, 10% of the whole feature space as input to each tree, entropy as the information gain criterion. To rule-out the effect of sex, age, depth, and family ID from the predictive power of the microbiome, 250 ASD male children, 250 ASD female children, 250 neurotypical male children, and 250 neurotypical female children were sampled randomly so that each individual was the only representative of his/her family. Before saving the dataset, anova was used to verify that differences in age and sequencing depth of the four groups were not significant (anova p > 0.01). The whole generation process was repeated a hundred times. For each of the a hundred sets, the described algorithm was then run in 10-fold, 10-times iterated cross-validation. The presented AUC are therefore an average over 10,000 tests. Species ranking was returned by a single ten-fold, ten-times iterated cross validation by MetAML, and is computed by averaging the features importances across the training folds to avoid overfitting.
Differential abundance analysis
Linear mixed models (python statsmodels library, function mixedlm, ver. 0.11.1) were run on each centered log-ratio (CLR) transformed MetaPhlAn 3 species, HUMAnN 3.0-derived MetaCyc pathway and gut-brain (GB) modules independently. Models were adjusted for sex, age in months, first 3 principal components of the genetic ancestry, read-depth, and blocked by (random intercept model) family ID. Models on species were run on 2154 ASD children and 1646 NTs. Models on pathways and GB modules were run on 2139 ASD children and 1646 NTs, and included alpha-diversity. Differential abundance was evaluated as the beta of the diagnosis with respect to the centered log-ratio transformed microbiome feature and by the Wald-q of the diagnosis corrected by false discovery rate over the whole set of features (1333 species, 724 pathways, 62 GB modules). Significance threshold used was set a priori to 0.005, used in species and in GB modules. Pathways are particularly collinear and tend to show extremely low p-values, so we ad-hoc choose to use for pathway q < 10−7. The method used to adjust the p-values was the Benjamini-Yekutieli (function fdrcorrection, from statsmodel.stats.multitest python library, ver. 0.11.1). Aerobicity and aero-tolerance were evaluated for the 108 differentially abundant species using DSMZ. The standardized ratio between the number of microbial reads and the total number of reads in each sample was used as a proxy for the oral cavity microbial load.
Polygenic risk score for ASD and association with microbiome features
We performed quality control with Plink1.9117 on batch 1, batch 2 and batches 3 human genotype calls, independently filtering out rare variants (MAF < 5%, --maf 0.05), variants with missing call in > 5% of the samples (--geno 0.05) and violating Hardy-Weinberg disequilibrium (--hwe 1e-6), and all samples with a missing genotype rate > 5% (–mind 0.05). Variants falling into regions with high recombination rate, in particular located in the Major Histocompatibility Complex (MHC)123,124, were removed from the PRS computation; we obtained a cohort composed of 7,824 samples and 5,981,273 SNPs. Variant association weights were derived from the most recent GWAS meta-analysis study on ASD125. Variants in strong linkage disequilibrium were removed from the summary stats using 1000 genomes as LD reference for clumping and default parameters for R2 and association p-value threshold. ASD Polygenic Risk Score (PRS) for each SPARK-WGS sample was computed using PRSice-2126. The p-value was optimized for ASD classification with the PRSice-2 algorithm; the best performing PRS included variants from the GWAS with a p < 0.1341, with a Negelkerke R2 = 0.6% (p = 8 × 10−9). The best PRS was used to fit a linear model associating the ASD risk score of the 3,800 children with CLR-transformed microbial species relative abundances adjusted for sex, age, ASD diagnosis, read depth, and the first three principal components of the genetic ancestry. We fitted a similar model excluding all non-caucasian samples (n = 2873), selected on the basis of the clustering on the ancestry PCAs into ethnic groups, and thus the ancestry components have been removed from this model.
Linear modelling of ASD-related instruments, full-scale IQ, NIAS items
Social Communication Questionnaire (SCQ), Repetitive Behaviors Scale (Revised) (RBS-R), Developmental Coordination Disorder Questionnaire (DCDQ), and Full Scale IQ were retrieved for 1750, 1724, 1468, and 1785 ASD children. Full Scale IQ was estimated via a machine learning algorithm developed to estimate cognitive levels from parent-reported data in large ASD cohorts59, which has shown an AUC of 0.876 when estimating severe cognitive impairment (IQ < 80), and validated over a set of 116 ASD children for which clinically-rated, full-scale IQ was determined by licensed clinical psychologists at SFARI via standardized methods59. Scores were associated with MetaPhlAn 3 species, HUMAnN 3 pathways, and GB modules using linear models adjusted for the same variables as in the differential abundance analysis. Associations with SCQ and DCDQ were also run adjusting by IQ. False discovery rates significance thresholds were set as before. Clinically-rated full-scale IQ was tested in a similar model including also the age at which the questionnaire was administered as covariate, and setting as significance threshold q < 0.2. We analyzed the Nine-Items ARFID Screen (NIAS)63 focusing on picky eating (Picky Factor from the NIAS) and on the “ARFID score” (derived from a model including the three NIAS factors and survey questionnaires on measures of eating behaviuors and GI symptoms). We retrieved the two scores for 291 ASD children. The 291 children were compared with the set of their 241 NTs by a mixed model identical to the differential abundance one. Linear models were then run on the ARFID score, on the Picky Factor, and on the full scale IQ considering only the same 291 ASD children this time. For the models based on the reduced sets of individuals, false discovery rate was set at 0.2 for species and GB modules, and at 0.05 for pathways. In all models the test used to assess the significance of a variable was the Wald test which equals a t-test of the beta coefficient over its standard error. To evaluate similarities across models, we pairwise correlated models’ beta coefficients and measured correlation using Spearman’s ρ and p.
Oral microbiome person-to-person strain sharing assessment and association with IQ
Strain-level analysis was performed from MetaPhlAn 3 output with StrainPhlAn ver. 3.0.1047. A total of 193 salivary species were present at sufficient coverage. StrainPhlAn was run with default parameters and –db mpa_v30_CHOCOPhlAn_201901 --phylophlan_mode fast, using mafft version 7.475127 to produce multiple-sequence alignments and RAxML version 8.1.15128. Species with <100 leafs in their strain-phylogeny were excluded, totalling 111 species. As described in ref. 64, pairwise genetic distances (GDs) normalised by total branch length from StrainPhlAn trees were used to compute person-to-person strain-sharing events using a custom version of the strain_transmission.py script (https://github.com/SegataLab/strain_transmission). Strain-sharing between two samples is based on strains of the same species present in the two samples being phylogenetically placed at a distance smaller than the 3rd percentile of the total genetic distance (GD) in the strain tree. Single nucleotide variant (SNV)-based methodologies were excluded for the reason that they are (a) less robust at low coverages to the noise introduced by the call of the consensus sequence, (b) dependent on the baseline variability of the specific marker (in contrast with phylogenetic methods, which can weight differentially a few invariant positions). In addition, phylogenetic methods are able to capture information relative to evolutionary models such as the synonymous vs nonsynonymous mutations. We evaluated person-to-person rates of sharing of microbiome strains as the total number of sharing events between two individuals divided by the number of common species between the two individuals among 111 species considered in this analysis. We annotated the ASD children according to their predicted IQ (3 classes: ≤70 (severe cognitive impairment),>70 and ≤85 (moderate cognitive impairment), >85 (neurotypical cognitive development))67. We then compared the between-individual strain-sharing rates of individuals from different classes of cognitive impairment with the NTs groups, with the mother, and with the father. Statistical differences were evaluated by Kruskal-Wallis test (scipy python library, ver. 1.4.1), post-hoc Dunn-test (Scikit-posthocs python library, ver. 0.6.7), and standardised mean difference (Pingouin python library, ver. 0.3.7)129.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Microbial raw sequencing reads for the healthy individuals in the SPARK-WGS cohort are available at European Nucleotide Archive at Bioproject PRJEB45799. MetaPhlAn 3 profiles, functional potential HUMAnN 3 profiles, and participant metadata for the 3,775 children in the cohort are available in the latest release of the curatedMetagenomicData R package (https://waldronlab.io/curatedMetagenomicData/) under the identifier ‘ManghiP_2024’. MetaPhlAn 3 profiles are available through Zenodo (https://zenodo.org/records/13986907). Approved researchers can obtain the SPARK population dataset described in this study (SFARI_SPARK_iWGS_v1.1) by applying at https://base.sfari.org.
References
Lord, C. et al. Autism spectrum disorder. Nat. Rev. Dis. Prim. 6, 5 (2020).
Posar, A., Resca, F. & Visconti, P. Autism according to diagnostic and statistical manual of mental disorders 5(th) edition: the need for further improvements. J. Pediatr. Neurosci. 10, 146–148 (2015).
Lai, M.-C., Lombardo, M. V. & Baron-Cohen, S. Autism. Lancet 383, 896–910 (2014).
Feliciano, P. et al. Exome sequencing of 457 autism families recruited online provides evidence for autism risk genes. NPJ Genom. Med. 4, 19 (2019).
Markram, K. & Markram, H. The intense world theory – a unifying theory of the neurobiology of Autism. Front. Hum. Neurosci. 4, 224 (2010).
Grandin, T. A personal perspective of Autism. In, Handbook of Autism and Pervasive Developmental Disorders (eds. Matson, J. L. & Sturmey, P.) 1609 (Springer, 2005).
Zablotsky, B., Black, L. I. & Blumberg, S. J. Estimated prevalence of children with diagnosed developmental disabilities in the United States, 2014-2016. NCHS Data Brief. 291, 1–8 (2017).
Fakhoury, M. Autistic spectrum disorders: a review of clinical features, theories and diagnosis. Int. J. Dev. Neurosci. 43, 70–77 (2015).
Sandin, S. et al. The heritability of Autism spectrum disorder. JAMA 318, 1182–1184 (2017).
Sanders, S. J. et al. Insights into Autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 87, 1215–1233 (2015).
Luo, Y. et al. A multidimensional precision medicine approach identifies an autism subtype characterized by dyslipidemia. Nat. Med. 26, 1375–1379 (2020).
Willsey, H. R., Willsey, A. J., Wang, B. & State, M. W. Genomics, convergent neuroscience and progress in understanding autism spectrum disorder. Nat. Rev. Neurosci. https://doi.org/10.1038/s41583-022-00576-7 (2022).
Gaugler, T. et al. Most genetic risk for autism resides with common variation. Nat. Genet. 46, 881–885 (2014).
An, J.-Y. et al. Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder. Science 362, eaat6576 (2018).
de la Torre-Ubieta, L., Won, H., Stein, J. L. & Geschwind, D. H. Advancing the understanding of autism disease mechanisms through genetics. Nat. Med. 22, 345–361 (2016).
Fu, J. M. et al. Rare coding variation provides insight into the genetic architecture and phenotypic context of autism. Nat. Genet. 54, 1320–1331 (2022).
Krishnan, A. et al. Genome-wide prediction and functional characterization of the genetic basis of autism spectrum disorder. Nat. Neurosci. 19, 1454–1462 (2016).
Searles Quick, V. B., Wang, B. & State, M. W. Leveraging large genomic datasets to illuminate the pathobiology of autism spectrum disorders. Neuropsychopharmacology 46, 55–69 (2020).
Consortium, T. H. M. P. & The human microbiome project consortium. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214 (2012).
Taniya, M. A. et al. Role of gut microbiome in Autism spectrum disorder and its therapeutic regulation. Front. Cell. Infect. Microbiol. 12, 915701 (2022).
Pulikkan, J., Mazumder, A. & Grace, T. Role of the gut microbiome in Autism spectrum disorders. Adv. Exp. Med. Biol. 1118, 253–269 (2019).
Mulle, J. G., Sharp, W. G. & Cubells, J. F. The gut microbiome: a new frontier in autism research. Curr. Psychiatry Rep. 15, 337 (2013).
Cryan, J. F. & Dinan, T. G. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 13, 701–712 (2012).
Collins, J., Borojevic, R., Verdu, E. F., Huizinga, J. D. & Ratcliffe, E. M. Intestinal microbiota influence the early postnatal development of the enteric nervous system. Neurogastroenterol. Motil. 26, 98–107 (2014).
Al-Asmakh, M., Anuar, F., Zadjali, F., Rafter, J. & Pettersson, S. Gut microbial communities modulating brain development and function. Gut Microbe. 3, 366–373 (2012).
McGuinness, A. J. et al. A systematic review of gut microbiota composition in observational studies of major depressive disorder, bipolar disorder and schizophrenia. Mol. Psychiatry https://doi.org/10.1038/s41380-022-01456-3 (2022).
Foster, J. A. Modulating brain function with microbiota. Science 376, 936–937 (2022).
Cryan, J. F. & Mazmanian, S. K. Microbiota-brain axis: context and causality. Science 376, 938–939 (2022).
Sampson, T. R. & Mazmanian, S. K. Control of brain development, function, and behavior by the microbiome. Cell Host Microbe. 17, 565–576 (2015).
Eltokhi, A., Janmaat, I. E., Genedi, M., Haarman, B. C. M. & Sommer, I. E. C. Dysregulation of synaptic pruning as a possible link between intestinal microbiota dysbiosis and neuropsychiatric disorders. J. Neurosci. Res. 98, 1335–1369 (2020).
Rutsch, A., Kantsjö, J. B. & Ronchi, F. The gut-brain axis: how microbiota and host inflammasome influence brain physiology and pathology. Front. Immunol. 11, 604179 (2020).
Kim, S. et al. Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature 549, 528–532 (2017).
Chen, Y., Xu, J. & Chen, Y. Regulation of neurotransmitters by the gut microbiota and effects on cognition in neurological disorders. Nutrients 13, 2099 (2021).
West, K. A. et al. Multi-angle meta-analysis of the gut microbiome in Autism spectrum disorder: a step toward understanding patient subgroups. Sci. Rep. 12, 17034 (2022).
Chavira, A., Wang, E.-J. & Mills, R. H. Meta-analysis of the autism gut microbiome identifies factors influencing study discrepancies and machine learning classification. bioRxiv https://doi.org/10.1101/2022.03.18.484910 (2022).
Morton, J. T. et al. Multi-level analysis of the gut-brain axis shows autism spectrum disorder-associated molecular and microbial profiles. Nat. Neurosci. 26, 1208–1217 (2023).
Sharon, G. et al. Human gut microbiota from Autism spectrum disorder promote behavioral symptoms in mice. Cell 177, 1600–1618.e17 (2019).
Yap, C. X. et al. Autism-related dietary preferences mediate autism-gut microbiome associations. Cell 184, 5916–5931.e17 (2021).
Qiao, Y. et al. Alterations of oral microbiota distinguish children with autism spectrum disorders from healthy controls. Sci. Rep. 8, 1597 (2018).
Abdulhaq, A. et al. Tongue microbiome in children with autism spectrum disorder. J. Oral. Microbiol. 13, 1936434 (2021).
Stewart Campbell, A. et al. Safety and target engagement of an oral small-molecule sequestrant in adolescents with autism spectrum disorder: an open-label phase 1b/2a trial. Nat. Med. 28, 528–534 (2022).
Diaz Heijtz, R., Gressens, P. & Swann, J. R. Targeting microbial metabolites to treat autism. Nat. Med. 28, 448–450 (2022).
Liu, X. et al. Sex differences in the oral microbiome, host traits and their causal relationships. iScience 26, 105839 (2022).
SPARK Consortium. SPARK: A US cohort of 50,000 families to accelerate Autism research. Neuron 97, 488–493 (2018).
Zhang, Y. et al. Genetic evidence of gender difference in autism spectrum disorder supports the female-protective effect. Transl. Psychiatry 10, 4 (2020).
Werling, D. M., Parikshak, N. N. & Geschwind, D. H. Gene expression in human brain implicates sexually dimorphic pathways in autism spectrum disorders. Nat. Commun. 7, 10717 (2016).
Beghini, F. et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. Elife 10, e65088 (2021).
Pasolli, E. et al. Accessible, curated metagenomic data through experimentHub. Nat. Methods 14, 1023–1024 (2017).
Chrisman, B. et al. The human ‘contaminome’: bacterial, viral, and computational contamination in whole genome sequences from 1000 families. Sci. Rep. 12, 1–9 (2022).
Walsh, I. et al. Author correction: DOME: recommendations for supervised machine learning validation in biology. Nat. Methods 18, 1409–1410 (2021).
Thomas, A. M. et al. Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat. Med. 25, 667–678 (2019).
Qiao, Y., Shi, H., Wang, H., Wang, M. & Chen, F. Oral health status of chinese children with Autism spectrum disorders. Front. Psychiatry 11, 398 (2020).
Kilian, M. et al. The oral microbiome - an update for oral healthcare professionals. Br. Dent. J. 221, 657–666 (2016).
Oyetola, E. O. et al. Salivary bacterial count and its implications on the prevalence of oral conditions. J. Contemp. Dent. Pract. 20, 184–189 (2019).
Choi, S. W., Mak, T. S.-H. & O’Reilly, P. F. Tutorial: a guide to performing polygenic risk score analyses. Nat. Protoc. 15, 2759–2772 (2020).
Rutter, M., Bailey, A., Lord, C., Cianchetti, C. & Fancello, G. S. SCQ: Social Communication Questionnaire. https://www.wpspublish.com/scq-social-communication-questionnaire.html (2007).
Wilson, B. N. et al. Psychometric properties of the revised developmental coordination disorder questionnaire. Phys. Occup. Ther. Pediatr. 29, 182–202 (2009).
Bodfish, J. W., Symons, F. J., Parker, D. E. & Lewis, M. H. Repetitive behavior scale–revised. PsycTESTS https://doi.org/10.1037/t17338-000 (2014).
Shu, C., Green Snyder, L., Shen, Y. & Chung, W. K. & SPARK Consortium. Imputing cognitive impairment in SPARK, a large autism cohort. Autism Res. https://doi.org/10.1002/aur.2622 (2021).
Buja, A. et al. Damaging de novo mutations diminish motor skills in children on the autism spectrum. Proc. Natl Acad. Sci. USA. 115, E1859–E1866 (2018).
Thomas, T. R. et al. Clinical autism subscales have common genetic liabilities that are heritable, pleiotropic, and generalizable to the general population. Transl Psychiatry 12, 247 (2022).
Koomar, T., Thomas, T. R., Pottschmidt, N. R., Lutter, M. & Michaelson, J. J. Estimating the prevalence and genetic risk mechanisms of ARFID in a large Autism cohort. Front. Psychiatry 12, 668297 (2021).
Zickgraf, H. F. & Ellis, J. M. Initial validation of the nine item avoidant/restrictive food intake disorder screen (NIAS): a measure of three restrictive eating patterns. Appetite 123, 32–42 (2018).
Valles-Colomer, M. et al. The person-to-person transmission landscape of the gut and oral microbiomes. Nature 614, 125–135 (2023).
Ianiro, G. et al. Variability of strain engraftment and predictability of microbiome composition after fecal microbiota transplantation across different diseases. Nat. Med. 28, 1913–1923 (2022).
Van Rossum, T., Ferretti, P., Maistrenko, O. M. & Bork, P. Diversity within species: interpreting strains in microbiomes. Nat. Rev. Microbiol. 18, 491–506 (2020).
Wolff, N., Stroth, S., Kamp-Becker, I., Roepke, S. & Roessner, V. Autism spectrum disorder and IQ - a complex interplay. Front. Psychiatry 13, 856084 (2022).
Valles-Colomer, M. et al. The neuroactive potential of the human gut microbiota in quality of life and depression. Nat. Microbiol 4, 623–632 (2019).
Sabit, H. et al. Gut microbiota metabolites in autistic children: an epigenetic perspective. Heliyon 7, e06105 (2021).
Kiser, D., Steemers, B., Branchi, I. & Homberg, J. R. The reciprocal interaction between serotonin and social behaviour. Neurosci. Biobehav. Rev. 36, 786–798 (2012).
Petersen, C. L., Davis, S. E. D., Patel, B. & Hurley, L. M. Social experience interacts with serotonin to affect functional connectivity in the social behavior network following playback of social vocalizations in mice. eNeuro 8, ENEURO.0247–20.2021 (2021).
Garbarino, V. R., Gilman, T. L., Daws, L. C. & Gould, G. G. Extreme enhancement or depletion of serotonin transporter function and serotonin availability in autism spectrum disorder. Pharmacol. Res. 140 85–99 (2019).
McDougle, C. J. et al. Effects of tryptophan depletion in drug-free adults with autistic disorder. Arch. Gen. Psychiatry 53, 993–1000 (1996).
Xiao, L. et al. Fecal microbiome transplantation from children with Autism spectrum disorder modulates tryptophan and serotonergic synapse metabolism and induces altered behaviors in germ-free mice. mSystems 6, e01343–20 (2021).
Lim, J. S., Lim, M. Y., Choi, Y. & Ko, G. Modeling environmental risk factors of autism in mice induces IBD-related gut microbial dysbiosis and hyperserotonemia. Mol. Brain 10, 14 (2017).
Tanaka, M. et al. Brain hyperserotonemia causes autism-relevant social deficits in mice. Mol. Autism 9, 60 (2018).
Roager, H. M. & Licht, T. R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 9, 3294 (2018).
Muller, C. L., Anacker, A. M. J. & Veenstra-VanderWeele, J. The serotonin system in autism spectrum disorder: from biomarker to animal models. Neuroscience 321, 24–41 (2016).
Israelyan, N. & Margolis, K. G. Serotonin as a link between the gut-brain-microbiome axis in autism spectrum disorders. Pharmacol. Res. 132, 1–6 (2018).
Gabriele, S., Sacco, R. & Persico, A. M. Blood serotonin levels in autism spectrum disorder: a systematic review and meta-analysis. Eur. Neuropsychopharmacol. 24, 919–929 (2014).
Ajram, L. A. et al. Shifting brain inhibitory balance and connectivity of the prefrontal cortex of adults with autism spectrum disorder. Transl. Psychiatry 7, e1137 (2017).
Selimbeyoglu, A. et al. Modulation of prefrontal cortex excitation/inhibition balance rescues social behavior in CNTNAP2 -deficient mice. Sci. Transl. Med. 9, eaah6733 (2017).
Zheng, Z., Zhu, T., Qu, Y. & Mu, D. Blood glutamate levels in Autism spectrum disorder: a systematic review and meta-analysis. PLoS One 11, e0158688 (2016).
Nisar, S. et al. Genetics of glutamate and its receptors in autism spectrum disorder. Mol. Psychiatry https://doi.org/10.1038/s41380-022-01506-w (2022).
Rojas, D. C., Becker, K. M. & Wilson, L. B. Magnetic resonance spectroscopy studies of glutamate and GABA in Autism: implications for excitation-inhibition imbalance theory. Curr. Dev. Dis. Rep. 2, 46–57 (2015).
Zhao, H. et al. GABAergic system dysfunction in Autism spectrum disorders. Front Cell Dev. Biol. 9, 781327 (2021).
Cochran, D. M. et al. Relationship among glutamine, γ-aminobutyric acid, and social cognition in Autism spectrum disorders. J. Child Adolesc. Psychopharmacol. 25, 314–322 (2015).
Gevi, F., Belardo, A. & Zolla, L. A metabolomics approach to investigate urine levels of neurotransmitters and related metabolites in autistic children. Biochim. Biophys. Acta Mol. Basis Dis. 1866, 165859 (2020).
Bowland, G. B. & Weyrich, L. S. The oral-microbiome-brain axis and neuropsychiatric disorders: an anthropological perspective. Front. Psychiatry 13, 810008 (2022).
DiCarlo, G. E. et al. Autism-associated variant in the SLC6A3 gene alters the oral microbiome and metabolism in a murine model. Front. Psychiatry 12, 655451 (2021).
Forsyth, A. et al. Children with autism spectrum disorder: Pilot studies examining the salivary microbiome and implications for gut metabolism and social behavior. Hum. Microb. J. 15, 100066 (2020).
Hicks, S. D. et al. Oral microbiome activity in children with autism spectrum disorder. Autism Res. 11, 1286–1299 (2018).
Olsen, I. & Hicks, S. D. Oral microbiota and autism spectrum disorder (ASD). J. Oral. Microbiol. 12, 1702806 (2020).
Jaber, M. A. Dental caries experience, oral health status and treatment needs of dental patients with autism. J. Appl. Oral. Sci. 19, 212–217 (2011).
Berding, K. & Donovan, S. M. Diet can impact microbiota composition in children with Autism spectrum disorder. Front. Neurosci. 12, 515 (2018).
Aleti, G. et al. Salivary bacterial signatures in depression-obesity comorbidity are associated with neurotransmitters and neuroactive dipeptides. BMC Microbiol. 22, 1–17 (2022).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Krueger, F. & Galore, T. A Wrapper Tool Around Cutadapt and FastQC to Consistently Apply Quality and Adapter Trimming to FastQ files. https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (2015).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592 (2023).
Darzi, Y., Falony, G., Vieira-Silva, S. & Raes, J. Towards biome-specific analysis of meta-omics data. ISME J. 10, 1025–1028 (2016).
Brito, I. L., Yilmaz, S., Huang, K., Xu, L. & Jupiter, S. D. Mobile genes in the human microbiome are structured from global to individual scales. Nature 535, 435–439 (2016).
Lassalle, F. et al. Oral microbiomes from hunter-gatherers and traditional farmers reveal shifts in commensal balance and pathogen load linked to diet. Mol. Ecol. 27, 182–195 (2018).
Castro-Nallar, E., Bendall, M. L. & Pérez-Losada, M. Composition, taxonomy and functional diversity of the oropharynx microbiome in individuals with schizophrenia and controls. PeerJ 3, e1140 (2015).
Ferretti, P. et al. Mother-to-infant microbial transmission from different body sites shapes the developing infant gut microbiome. Cell Host Microbe 24, 133–145.e5 (2018).
Ghensi, P. et al. Strong oral plaque microbiome signatures for dental implant diseases identified by strain-resolution metagenomics. NPJ Biofilms Microbiomes. 6, 47 (2020).
Shi, B. et al. Dynamic changes in the subgingival microbiome and their potential for diagnosis and prognosis of periodontitis. MBio 6, e01926–14 (2015).
Chu, D. M. et al. Maturation of the infant microbiome community structure and function across multiple body sites and in relation to mode of delivery. Nat. Med. 23, 314–326 (2017).
Olm, M. R. et al. Identical bacterial populations colonize premature infant gut, skin, and oral microbiomes and exhibit different in situ growth rates. Genome Res. 27, 601–612 (2017).
Herzig, A. F. et al. Evaluation of saliva as a source of accurate whole-genome and microbiome sequencing data. Genet. Epidemiol. 45, 537–548 (2021).
Pasolli, E. et al. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell 176, 649–662.e20 (2019).
Asnicar, F. et al. Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat. Commun. 11, 2500 (2020).
Asnicar, F., Weingart, G., Tickle, T. L., Huttenhower, C. & Segata, N. Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ 3, e1029 (2015).
1000 Genomes Project Consortium. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, 7 (2015).
Aitchison, J. The Statistical Analysis of Compositional Data, Vol. 416 (Springer, 1986).
Blanchet, F. G., Legendre, P. & Borcard, D. Forward selection of explanatory variables. Ecology 89, 2623–2632 (2008).
Legendre, P. & Anderson, M. J. Distance based redundancy analysis: testing multispecies responses in multifactorial ecological experiments. Ecol. Monographs 69, 1–24 (1999).
Hoffman, G. E. & Schadt, E. E. variancePartition: interpreting drivers of variation in complex gene expression studies. BMC Bioinforma. 17, 483 (2016).
Pasolli, E., Truong, D. T., Malik, F., Waldron, L. & Segata, N. Machine learning meta-analysis of large metagenomic datasets: tools and biological insights. PLoS Comput. Biol. 12, e1004977 (2016).
Price, A. L. et al. Long-range LD can confound genome scans in admixed populations. Am. J. Hum. Genet. 83, 132–135 (2008).
Weale, M. E. Quality control for genome-wide association studies. Methods Mol. Biol. 628, 341–72 (2010).
Grove, J. et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 51, 431–444 (2019).
Choi, S. W. & O’Reilly, P. F. PRSice-2: Polygenic risk score software for biobank-scale data. Gigascience 8, giz082 (2019).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Vallat, R. Pingouin: statistics in python. J. Open Source Softw. 3, 1026 (2018).
Acknowledgements
This work was supported by a grant from SFARI (Award 648614, E.D. and N.S.); by the European Research Council (ERC-STG project MetaPG-716575 and ERC-CoG project MicroTOUCH-101045015) to N.S.; by the European H2020 program (ONCOBIOME-825410 project and MASTER-818368 project) to N.S.; by the National Cancer Institute of the National Institutes of Health (1U01CA230551) to N.S.; by the Premio Internazionale Lombardia e Ricerca 2019 to N.S. We are grateful in particular to all of the families in SPARK, the SPARK clinical sites and SPARK staff. We appreciate obtaining access to the metagenomic raw data, the genetic data and the associated samples metadata on SFARI Base. The authors would like to thank Michael Whalen for his inspiring suggestion, which provided the starting point for this research. Approved researchers can obtain the SPARK population dataset described in this study (SFARI_SPARK_iWGS_v1.1) by applying at https://base.sfari.org.
Author information
Authors and Affiliations
Contributions
P.M., E.D., and N.S. conceptualized the study. M.Z., P.M., M.F., J.B.H., S.X., L.G.C., J.J.M., A.G.V., and N.V. coordinated data retrieval and preprocessing, which included the quality control and extraction of microbial reads. E.D., M.F., P.M., and S.P. blueprinted the statistical approaches. P.M. led the analyses on the association between oral microbiome features and cognitive and social deficits, and performed the majority of the analyses. P.M., M.Z., and D.G. performed microbiome functional, taxonomic, and strain-level profiling and corresponding analysis. S. Mattevi, S. Montalbano, S.C. and M.F. performed the human genomic analyses. L.W. provided key feedback on the validity of the statistical approaches. LA.G.S. provided feedback on the analysis of the ASD-related clinical phenotypes. M.V.C., A.M.T., V.H., and F.A. supervised all metagenomic analyses. E.D, and N.S. co-supervised the work. All the authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors do not have any competing interests to declare.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Manghi, P., Filosi, M., Zolfo, M. et al. Large-scale metagenomic analysis of oral microbiomes reveals markers for autism spectrum disorders. Nat Commun 15, 9743 (2024). https://doi.org/10.1038/s41467-024-53934-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-53934-7
This article is cited by
-
Human and bacterial genetic variation shape oral microbiomes and health
Nature (2026)
-
Human microbiome acquisition and transmission
Nature Reviews Microbiology (2025)
-
Beyond the gut: decoding the gut–immune–brain axis in health and disease
Cellular & Molecular Immunology (2025)
