
Abstract
Spatially heterogeneous synapse loss is a characteristic of many psychiatric and neurological disorders, but the underlying mechanisms are unclear. Here, we show that spatially-restricted complement activation mediates stress-induced heterogeneous microglia activation and synapse loss localized to the upper layers of the medial prefrontal cortex (mPFC) in male mice. Single cell RNA sequencing also reveals a stress-associated microglia state marked by high expression of the apolipoprotein E gene (Apoehigh) localized to the upper layers of the mPFC. Mice lacking complement component C3 are protected from stress-induced layer-specific synapse loss, and the Apoehigh microglia population is markedly reduced in the mPFC of these mice. Furthermore, C3 knockout mice are also resilient to stress-induced anhedonia and working memory behavioral deficits. Our findings suggest that region-specific complement and microglia activation can contribute to the disease-specific spatially restricted patterns of synapse loss and clinical symptoms found in many brain diseases.
Similar content being viewed by others


Introduction
Synapse loss is a central mechanism that plays an important role in the pathogenesis of many psychiatric and neurological diseases1,2,3,4,5,6. Neuroimaging and neuropathological studies have revealed disease-specific and reproducible spatial patterns of synapse loss in many brain disorders7,8,9,10. In some neuropsychiatric diseases, severe synapse loss can occur in remarkably small brain regions, while nearby regions are spared. For example, postmortem studies of schizophrenic patients show a consistent loss of dendritic spines in layer 3 of the dorsolateral prefrontal cortex (PFC)11,12, while spine density in deeper PFC layers13 and other cortical regions14 are unaffected. Studies of rodent models of depression also show that dendritic spines in the upper layers of the medial PFC (mPFC) are lost in response to stress, while spines in the deeper mPFC layers remain unaffected15,16. Despite the prevalence of region-specific synapse loss in brain diseases, the pathophysiological processes involved remain unclear.
Microglia are dynamic brain-resident macrophages that constantly survey their environment and respond to a wide variety of pathological insults. Single cell-RNA sequencing studies have revealed that under physiological conditions, microglia adopt diverse regional states dictated by their local environment17,18,19,20. A large number of pathological and spatially heterogeneous microglial transcriptomic states have also been described diseases such as Alzheimer’s disease21,22,23, multiple sclerosis19,24,25, and chronic pain26. These diverse microglia states are likely to perform key homeostatic functions in the healthy brain and play critical pathogenic roles in the diseased brain. While several signaling pathways have been shown to induce specific pathological microglial states27,28,29,30, whether these mechanisms act in a spatially-restricted manner remain unclear.
Stress is a major risk factor for psychiatric diseases, and extended periods of stress can directly cause affective disorders such as major depressive disorder (MDD)31, post-traumatic stress disorder (PTSD)32,33, and anxiety disorders32,33. Extended periods of stress also cause synapse loss in the PFC, an effect which is mediated by glucocorticoid stress hormones. Excessive secretion of cortisol is found in 50% of depressed patients34, and levels of cortisol are positively correlated to depressive behavior in nonhuman primates35. In humans, elevated salivary, hair and serum cortisol levels are all correlated with poorer working memory performance and lower brain volumes36,37,38,39, suggesting that prolonged exposure to cortisol can lead to changes in brain structure. Chronic stress is known to increase microglia activation in the PFC40,41, and can also induce complement activation in the brain42,43,44, suggesting that microglial phagocytosis of synapses may mediate stress-induced synapse loss. However, previous studies have assumed that stress-induced microglia activation occurs homogeneously throughout the PFC. Because the complement cascade is a secreted and extracellular signaling system, it was unclear whether this signaling pathway can result in spatially-restricted microglia activation and synapse loss in the brain.
Here, we provide evidence that chronic stress causes heterogeneous microglia activation in the mPFC resulting in layer-specific synapse loss and stress-associated behavioral deficits in male mice. We show that multiple chronic stress protocols cause localized complement activation specifically in the upper layers of the mouse mPFC. Complement activation in layer 2/3 leads to microglial phagocytosis of VGlut2 synapses and synapse loss specifically in those cortical layers, while synapses in deeper mPFC layers are spared. Single-cell RNA (scRNA-seq) of microglia from cortices of stressed mice also reveals an Apoehigh transcriptomic state upregulated in the stressed brain and localized to layer 2/3 of the mPFC. In complement C3 knockout (KO) mice, we found that chronic stress no longer induces the Apoehigh microglia state. C3 KO mice are also resilient to stress-induced microglia phagocytosis of synapses and synapse loss, and do not exhibit stress-induced anhedonia and working memory deficits. Our data shows that spatially restricted complement activation can drive heterogeneous activation of microglia to cause layer-specific synapse loss in the stressed mPFC.
Results
Multiple stress protocols cause layer-specific complement activation in the mPFC
The classical complement cascade is a major signaling pathway that mediates microglial phagocytosis of synapses45,46,47. Complement component C3, the main effector molecule in the complement cascade, is deposited onto synapses and recognized by microglial CR3 receptors for phagocytosis45. To determine whether chronic stress induces heterogeneous complement activation, we subjected male C57BL/6 J mice to the 21-day chronic corticosterone (CORT) administration protocol (Fig. 1A), and assessed complement activation in the mPFC by staining for C3 with a KO-verified antibody (Supplemental Fig. 1E). We focused on the prelimbic cortex of the mPFC (Fig. 1B), a region known to be especially sensitive to the effects of stress48. Surprisingly, we found that CORT treatment causes a layer-specific increase in C3 deposition in layer 2/3 of the mPFC but not layer 5/6 (Fig. 1C–E). In addition to the mPFC, we found that chronic CORT causes C3 deposition in other brain regions, such as the hippocampus CA1, the paraventricular nucleus (PVN), the lateral habenula (LHb), and the bed nucleus of the stria terminalis (BNST) (Supplementary Fig. 1A–D). We also examined C1q deposition after chronic CORT, and found that stress does not increase C1q deposition in the mPFC (Supplementary Fig. 2), suggesting that the classical complement pathway is not involved in stress responses, and that the alternative, lectin, or non-canonical pathways may be involved. The effects of stress on neuroimmune processes can vary depending on the stressor used49, raising the question of whether layer-specific complement activation is a core feature of diverse stressors. In order to determine whether layer-specific complement activation also occurs in other stressors, we assessed C3 deposition in two other commonly used stress protocols, chronic restraint stress (CRS), and chronic unpredictable stress (CUS) (Fig. 1A). Both of these protocols engage the fast autonomic nervous system (ANS) response, in addition to the slow hypothalamic-pituitary-adrenal (HPA) axis hormonal response that the CORT protocol mimics. Both of these protocols also provide different patterns of CORT responses. CRS is expected to cause an initial large increase in CORT levels which habituates over time, whereas CUS is designed to cause a nonhabituating increase in CORT levels. Surprisingly, both CRS and CUS cause a layer-specific increase in C3 deposition similar to that found in chronic CORT administration (Fig. 1F–K), despite differences in the physiological systems engaged and patterns of CORT induced by these protocols. Therefore, multiple chronic stress protocols lead to layer-specific C3 deposition in the mPFC, suggesting that layer-specific complement activation is a core feature of chronic stress.

A Schematic depicting timeline for chronic corticosterone (CORT) stress, chronic restraint stress (CRS), and chronic unpredictable stress (CUS) protocols. B Low-magnification image depicting layers of the medial prefrontal cortex. Stained for VGlut2 (red) and DAPI (blue). Scale bar 200 µm. C Control/CORT: ow magnification C3 deposition in mPFC. Top panel shows C3 channel (white), bottom panel shows merged C3 (red) and DAPI (blue) channels. Scale bar 50 µm. D Control/CORT: Layer 2/3 C3 deposition. Quantitation (left), representative image (right). Analyzed by two-tailed unpaired t-test, t(20) = 2.733, *p = 0.0128, n = 11 control and 11 CORT mice. Scale bar 5 µm. E Control/CORT: Layer 5 C3 deposition. Quantitation (left), representative image (right). Analyzed by two-tailed unpaired t-test, t t(17) = 0.4277, p = 0.6742, n = 10 control and 9 CORT mice. Scale bar 5 µm. F Control/CRS: low magnification C3 deposition in mPFC. Low magnification image showing laminar C3 deposition in mPFC in control/CRS mice. Top panel shows C3 channel (white), bottom panel shows merged C3 (red) and DAPI (blue) channels. Scale bar 50 µm. G Control/CRS: Layer 2/3 C3 deposition. Quantitation (left), representative image (right). Analyzed by two-tailed unpaired t-test, t(10) = 3.919, **p = 0.0029, n = 7 control and 5 CRS mice. Scale bar 5 µm. H Control/CRS: Layer 5 C3 deposition. Quantitation (left), representative image (right). Analyzed by two-tailed unpaired t-test, t(11) = 0.8459, p = 0.4157, n = 7 control and 6 CRS mice. Scale bar 5 µm. I Control/CUS: low magnification C3 deposition in mPFC. Top panel shows C3 channel (white), bottom panel shows merged C3 (red) and DAPI (blue) channels. Scale bar 50 µm. J Control/CUS: Layer 2/3 C3 deposition. Quantitation (left), representative image (right). Analyzed by two-tailed unpaired t-test, t(11) = 3.801, **p = 0.0029, n = 6 control and 7 CUS mice. Scale bar 5 µm. K Control/CUS: Layer 5 C3 deposition. Quantitation (left), representative image (right). Analyzed by two-tailed unpaired t-test, t(12) = 0.9315, p = 0.3699, n = 6 control and 8 CUS mice. Scale bar 5 µm. A–K All data shown as mean ± s.e.m. Source data provided in Source File.
Stress causes layer-specific microglial synapse phagocytosis and synapse loss
Complement activation is known to lead to microglial activation and phagocytosis of synapses45. To further characterize microglia in the context of chronic stress, we chose to perform all further experiments with the CORT stress protocol, because CORT is the principal murine stress hormone, and CORT levels are increased by all rodent stress protocols50,51,52. Furthermore, direct CORT administration causes synapse losses and behavioral deficits in rodents53,54,55,56,57, and glucocorticoid receptor blockade in rodents block stress-induced microglia activation, neuronal remodeling, and behavioral deficits57, indicating that CORT is necessary and sufficient for the neurobiological effects of chronic stress. To assess microglial activation, we stained brain slices for the microglial cytoplasmic marker Iba1 and the lysosomal marker CD68 (Fig. 2A, Supplementary Fig. 3), reconstructed high-resolution 3D images of individual microglia, and quantitated the percentage of Iba1 volume occupied by CD68 lysosomes. We found that the CD68 content of microglia is increased by chronic CORT administration only in layer 2/3 (Fig. 2B), while microglia in layer 5 are not affected (Fig. 2C). Previous studies have shown that chronic CORT administration causes synapse loss in the mPFC53,54,55. Furthermore, live imaging studies indicate that stress-induced synapse loss in the mPFC is due to an increase in the rate of synapse elimination54,58,59, as opposed to a decrease in the rate of synapse formation, suggesting that microglial synapse phagocytosis may be involved. To assess microglial synapse phagocytosis in the mPFC of chronically stressed mice, we stained their brain slices for Iba1, CD68, and the presynaptic marker VGlut2. We chose VGlut2 because studies from our lab and others have shown that microglia preferentially phagocytose this population of synapses45,46,60. We quantitated the 3D colocalization of the three markers Iba1/CD68/VGlut2 as a measure of microglial synapse phagocytosis (Fig. 2D), and found that chronic CORT administration strongly increases microglial synapse phagocytosis in layer 2/3 of the mPFC, but not in layer 5/6 (Fig. 2E, F).

A Orthogonal view of lysosomal marker CD68 (red) within Iba1-positive microglia (blue). Scale bar 7 µm. B CD68 occupancy in mPFC L2/3. Two-tailed unpaired t-test, t(18) = 4.839, ***p = 0.0001, n = 10 control and 10 CORT mice. C CD68 occupancy in mPFC L5. Two-tailed unpaired t-test, t(17) = 1.015, p = 0.3243, n = 10 control and 9 CORT mice. D Microglial engulfment. Left: Representative image of Iba1-positive microglia (green) with CD68-positive lysosomes (blue) and VGlut2 (magenta), scale bar 5 µm. Right: Corresponding 3D reconstruction. Inset scale bar 1 µm. E VGlut2 engulfment in mPFC L2/3. Two-tailed unpaired t-test, t(18) = 4.24, ***p = 0.0005, n = 10 control and 10 CORT mice. F VGlut2 engulfment by microglia in mPFC L5. Two-tailed unpaired t-test, t(17) = 2.006, p = 0.061, n = 10 control and 9 CORT mice. G VGlut2/PSD95 synapse density in mPFC L2/3. Representative images and quantitation of colocalized VGlut2 (magenta) and PSD95 (green) puncta. Circles indicate colocalized puncta. Scale bar 5 µm. Two-tailed unpaired t-test, t(16) = 2756, *p = 0.0141, n = 9 control and 9 CORT mice. H VGlut2/PSD95 synapse density in mPFC L5. Two-tailed unpaired t-test, t(15) = 0.2651, p = 0.7945, n = 9 control and 8 CORT mice. I VGlut1/PSD95 synapse density in mPFC L2/3. Representative images and quantitation of colocalized VGlut1 (magenta) and PSD95 (green) puncta. Circles indicate colocalized puncta. Scale bar 5 µm. Two-tailed unpaired t-test, t(17) = 0.2565, p = 0.8006, n = 10 control and 9 CORT mice. J VGlut1/PSD95 synapse density in mPFC L5. Two-tailed unpaired t-test, t(17) = 0.3788, p = 0.7095, n = 10 control and 9 CORT mice. K VGAT/gephyrin synapse density in mPFC L2/3. Representative images and quantitation of colocalized VGAT (magenta) and gephyrin (green) puncta. Circles indicate colocalized puncta. Scale bar 5 µm. Two-tailed unpaired t-test, t(21) = 0.4483, p = 0.6585, n = 12 control and 11 CORT mice. L VGAT/gephyrin synapse density in mPFC L5. Two-tailed unpaired t-test, t(18) = 0.5949, p = 0.5593, n = 11 control and 9 CORT mice. M Representative image of c-fos (red) and DAPI (blue) staining in mPFC after TOR working memory task. Scale bar 50 µm. N c-Fos quantitation in mPFC L2/3 after TOR task. Two-tailed unpaired t-test, t(17) = 2.618, *p = 0.0180, n = 9 control and 10 CORT mice. O c-Fos quantitation in mPFC L5 after TOR task. Two-tailed unpaired t-test, t(17) = 0.6393, p = 0.5311, n = 9 control and 10 CORT mice. A–K All data shown as mean ± s.e.m. Source data provided in Source File.
To determine if the increase in microglial synapse phagocytosis leads to a decrease in synapse density in the stressed mPFC, we stained brain slices for three types of synapses: 1) thalamocortical synapses marked by colocalized VGlut2 presynaptic and PSD95 postsynaptic markers, 2) corticocortical synapses marked by colocalized VGlut1 presynaptic and PSD95 postsynaptic markers, and 3) inhibitory synapses marked by colocalized VGAT presynaptic and gephyrin postsynaptic markers. We found that chronic CORT administration results in a layer-specific reduction of VGlut2/PSD95 synapses in layer 2/3, but not layer 5 (Fig. 2G, H), while the densities of VGlut1/PSD95 and VGAT/gephyrin synapses are unchanged in all cell layers (Fig. 2I–L, Supplementary Fig. 4). These findings suggest that microglial synapse phagocytosis induced by chronic CORT administration specifically targets thalamocortical synapses in layer 2/3 of the mPFC, while sparing corticocortical and inhibitory synapses.
The activity of the thalamic projections to the PFC is known to be required for working memory performance, where it acts to sustain the delay-period activity of PFC neurons, which are the neuronal substrates of working memory maintenance61,62,63. Chronic stress is also known to impair thalamic inputs to the mPFC and working memory performance in mice56,57,64. To determine whether stress-induced loss of synapses in the mPFC results in a layer-specific reduction of functional activation of neurons during working memory maintenance, we utilized the temporal object recognition (TOR) working memory task, which has previously been shown to be impaired by chronic stress in mice56,57. We assessed the number of c-fos positive neurons in the mPFC shortly after the completion of the TOR task, and found a significant reduction in layer 2/3 of the mPFC from CORT mice, but not in layer 5/6 (Fig. 2M–O). Together, these data indicate that chronic CORT causes heterogeneous microglia activation in the mPFC, leading to layer-specific synapse loss.
Stress induces an Apoe high microglia state enriched in endocytotic genes
To characterize the heterogeneous states of microglia in the stressed brain, we performed scRNA-seq on microglia isolated from the brains of CORT and control mice. To isolate single-cell microglia from the gray matter of the mouse cortex, we used a cold mechanical dissociation protocol that was specifically optimized for the isolation of microglia with minimal ex vivo transcriptional alterations (Supplementary Fig. 5A)19,65. After dissociation, microglia were sorted from the single-cell suspension with fluorescence-activated cell sorting (FACS) based on the expression of Cd45, Cd11b, and Cx3cr1 (Supplementary Fig. 5B–G). The resulting cells were processed for single-cell RNA sequencing, and sequenced to a depth of about 50,000 reads/cell. We processed 4 replicates each for the control and CORT conditions, with 4-5 mice per replicate. After quality control, we retained a total of 81,773 cells from all replicates for downstream analysis.
We then performed unsupervised clustering analysis on the single scRNA-seq dataset, revealing 5 clusters across all conditions and replicates (Fig. 3A, Supplementary Figs. 6A,B and 7A,B). We annotated each cluster with a marker gene that shows the highest cluster-specific expression relative to all other clusters (Supplemental Fig. 6A,B). The largest cluster expresses most genes in common with all other clusters, with only a modestly enriched expression of the Fcrls gene, which is a canonical marker for microglia cells. Previous reports suggest that adult microglia are largely homogeneous and are mostly in the homeostatic state18,19,65, and together with the lack of cluster-specific gene expression, suggest that the Fcrlshigh cluster are microglia in the homeostatic state. The second largest cluster is marked by expression of the Apoe gene, with a nearly 32-fold increase in expression of Apoe over other clusters, as well as increases in the expression of several Ms4a genes (Supplemental Dataset 1). Differential abundance analysis shows that chronic CORT administration significantly increases the number of microglia in the Apoehigh cluster while decreasing the number of microglia in the Fcrlshigh cluster (Fig. 3B, C). In addition to the Fcrlshigh and Apoehigh cell clusters, we also identified several smaller cell clusters, which are largely unchanged by chronic CORT administration (Fig. 3B, C), many of which have already been identified in previous microglia scRNA-seq studies65,66,67, suggesting that they are microglia states present in basal conditions. These clusters include an interferon-responsive state marked by expression of Ifitm3 and other interferon-responsive genes65,67, and a chemokine-expressing state marked by Ccl4 and other chemokine expression (Fig. 3A, Supplemental Fig. 6A)65,66. We also identified a microglia state marked by expression of the mitochondrial Taco1 gene. All cell clusters express canonical microglial genes such as C1qa, Cx3cr1, P2ry12, and Tmem119 (Supplemental Fig. 6C), indicating that they are bona fide microglia states. A very small percentage of non-microglial cells were also recovered in the dissociation procedure as previously described19, consisting mainly of brain border macrophages marked by expression of F13a1, Mgl2, Ms4a7, and Pf4 (Supplemental Fig. 6D), as well as infiltrating monocytes marked by expression of Ccr2 and Ly6c2 (Supplemental Fig. 6E). All microglia clusters show extremely low expression of the C3 gene (Supplemental Figure 7C), suggesting that stress-induced C3 deposition is mediated by other cell types. To identify the genes induced by chronic CORT administration, we performed pseudobulk differential expression gene (DEG) analysis, which also showed a CORT-induced increase in expression of the Apoe gene (Fig. 3E). A gene set enrichment analysis (GSEA) on the DEGs showed that genes associated with increased cell motility and activation, as well as genes associated with innate immune responses, are the major classes of microglial genes upregulated by chronic CORT administration (Fig. 3F).

A UMAP plot of 81,773 microglia isolated from the cerebral cortex of control and CORT mice, 4 replicates per condition, n = 4–5 mice per replicate. In total, 5 clusters of microglia were identified. All cell clusters are annotated with the marker gene that shows the highest cluster-specific expression relative to all other clusters. B Microglia cluster proportions for each of the 4 replicates of both the CORT and groups. C Differential abundance test for each microglia cluster performed with Speckle, showing that chronic CORT administration causes a significant decrease in the abundance of the Fcrlshigh cluster (two-tailed unpaired t-test, t(2) = −3.85, p = 0.00202, n = 4 samples) and a significant increase in the abundance of the Apoehigh cluster (two-tailed unpaired t-test, t(2) = 3.84, p = 0.00206, n = 4 samples). Data shown as means ± s.e.m. D Heat map showing the top genes that are upregulated in each cluster relative to all other clusters. E Volcano plot of genes identified by pseudobulk DEG analysis comparing the ctrl and CORT groups. Selected genes with log2-fold change > 1 and p < 10e−4 are labeled. P values adjusted for multiple comparisons as performed by the Libra run_de function. F Dot plot showing top 10 biological function GO terms identified by gene set enrichment analysis (GSEA) of DEGs from pseudobulk DEG analysis. GO terms are arranged in order of increasing adjusted p value. P values adjusted for multiple comparisons as performed by the clusterProfiler enrichGO function. A–F Source data provided in Supplementary Dataset 1.
Complement activation drives formation of Apoe high microglia state
Next, we wanted to determine if layer-specific complement activation resulted in layer-specific induction of Apoehigh microglia in the mPFC. We performed RNAscope in situ hybridization (ISH) to quantitate Apoe expression in mPFC microglia, probing for Cx3cr1 mRNA as a microglia marker, and Apoe mRNA as the readout (Fig. 4A). We found that in the mPFC, chronic CORT administration increases microglial Apoe mRNA levels in layer 2/3 but not layer 5/6 (Fig. 4B, C). Furthermore, C3 deletion abolishes the layer-specific increase in microglial Apoe expression (Fig. 4B, C). These data suggest that complement activation drives the formation of the Apoehigh microglia state.

A Representative images of RNAscope in situ hybridization for Apoe mRNA (red) expression in Cx3cr1-positive microglia (green), with nuclei labeled with DAPI (blue). Scale bar 20 µm. B Quantitation of Apoe mRNA per DAPI nucleus in layer 2 mPFC of wildtype and C3−/− control/CORT mice, normalized to WT control. Data analyzed by two-way ANOVA, followed by Sidak’s post-hoc test comparing means within genotype. F(1,13) = 42.10, ****p < 0.0001 (genotype x stress interaction), n = 3–6 mice. Data plotted as mean ± s.e.m. Source data provided in Source File. C Quantitation of Apoe mRNA per DAPI nucleus in layer 5 mPFC of wildtype and C3−/− control/CORT mice, normalized to WT control. Data analyzed by two-way ANOVA, followed by Sidak’s post-hoc test comparing means within genotype. F(1,13) = 1.10, p = 0.2960 (genotype x stress interaction), n = 3–5 mice. Data plotted as mean ± s.e.m. Source data provided in Source File.
Complement activation drives stress-induced synapse loss and behavioral deficits
To determine if C3 deletion leads to a lack of stress-induced microglial and synaptic responses, we next assessed VGlut2/PSD95 synapse density and microglial phagocytosis of VGlut2 synapses in the mPFC of control and CORT-treated C3−/− mice (Fig. 5A, B). We found that mice lacking C3 were protected against CORT-induced microglial phagocytosis of synapses and synapse loss in layer 2/3 (Fig. 5C–F). These data indicate that stress-induced layer-specific changes in microglial synapse phagocytosis and synapse density are abolished in the C3−/− mice.

A Representative images of VGlut2 (magenta) and PSD95 (green) synapses colocalization in layer 2/3 mPFC from wildtype and C3−/− control/CORT mice. Circles indicate colocalized pre- and postsynaptic puncta. Scale bar 5 µm. B Representative images of microglial VGlut2 synapse engulfment in layer 2/3 mPFC from wildtype and C3−/− control/CORT mice. Representative IHC image (left) with corresponding 3D reconstruction (inset, right) showing localization of engulfed VGlut2 (magenta) in CD68-positive lysosomes (blue) in Iba1-positive microglia (green). Scale bar 5 µm for IHC image, scale bar 1 µm for inset. C Quantitation of colocalized VGlut2 & PSD95 synaptic puncta in layer 2/3 mPFC of wildtype and C3−/− control/CORT mice. Data analyzed by two-way ANOVA, followed by Sidak’s post-hoc test comparing means within genotype. F(1,29) = 4.518, p = 0.0422 (genotype x stress interaction), n = 9 wildtype control, 9 wildtype CORT, 8 C3−/− control, and 7 C3−/− CORT, **p 0.0079. Data plotted as mean ± s.e.m. D Quantitation of microglial VGlut2 engulfment in layer 2/3 mPFC of wildtype and C3−/− mice. Data analyzed by two-way ANOVA, followed by Sidak’s post-hoc test comparing means within genotype, F(1,30) = 12.87, p = 0.0012 (genotype x stress interaction), n = 9 wildtype control, 9 wildtype CORT, 8 C3−/− control, and 8 C3−/− CORT, ****p < 0.0001. E Quantitation of colocalized VGlut2 & PSD95 synaptic puncta in layer 5 mPFC of wildtype and C3−/− control/CORT mice. Data analyzed by two-way ANOVA, F(1,25) = 0.2419, p = 0.6271, n = 8 wildtype control, 7 wildtype CORT, 7 C3−/− control, and 7 C3−/− CORT mice. F Quantitation of microglial VGlut2 engulfment in layer 5 mPFC of wildtype and C3−/− mice. Data analyzed by two-way ANOVA, F(1,26) = 0.3373, p = 0.5664, n = 8 wildtype control, 8 wildtype CORT, 7 C3−/− control, and 7 C3−/− CORT mice. C–F All data shown as mean ± s.e.m. Source data provided in Source File.
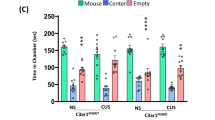
Finally, we wanted to determine if C3 deletion also leads to the rescue of behavioral deficits. We assessed C3−/− and wild-type mice in two behavioral tasks, the SPT task for anhedonia, and the TOR task for working memory (Fig. 6A, B). We found that wildtype mice, but not C3−/− mice, were susceptible to CORT-induced anhedonia in the SPT task (Fig. 6C). In addition, we found that wildtype mice but not C3−/− mice were susceptible to CORT-induced impairments in working memory. Specifically, we found that control-treated wildtype mice and CORT/control-treated C3−/− mice were able to remember objects they have recently encountered, as shown by their preferential exploration of the old and more unfamiliar object. In contrast, CORT-treated wildtype mice had impaired working memory and explored both old and recent objects equally (Fig. 6D, E). Therefore, our data indicate that C3−/− mice are resilient against stress-induced anhedonia and working memory deficits. Taken together, our findings indicate that stress-induced layer-specific complement activation in the mPFC leads to heterogeneous microglia activation, layer-specific synapse loss and behavioral deficits.

A Timeline of CORT administration and behavioral assays (temporal object recognition “TOR” and sucrose preference test “SPT”). B Schematic diagram of TOR assay (left), with an image of 3D printed objects used for assay (right). C Quantitation of sucrose preference of wildtype and C3−/− control/CORT mice. Analyzed by two-way ANOVA, followed by Sidak’s post-hoc test comparing means within genotype, F(1,52) = 4.481, p = 0.0391 (genotype x stress interaction), n = 14 wildtype control, 17 wildtype CORT, 13 C3−/− control, and 13 C3−/− CORT mice, wildtype control vs. wildtype CORT **p = 0.0017, C3−/− control vs. C3−/− CORT p = 0.9298. D Object exploration time during test phase of TOR. Data analyzed by three-way ANOVA, followed by uncorrected Fisher’s LSD test, F(1,100) = 5.758, p = 0.0183 (object x genotype x stress interaction), n = 26 wildtype control, 26 wildtype CORT, 25 C3−/− control, and 27 C3−/− CORT mice, adjusted p-values: wildtype control old vs. wildtype control recent **p = 0.0018, wildtype CORT old vs. wildtype CORT recent p = 0.8376, C3−/− control old vs. C3−/− control recent *p = 0.0116, C3−/− CORT old vs. C3−/− CORT recent ***p = 0.0004. Bar graphs showing the discrimination index of wildtype and C3−/− control/CORT mice, quantifying relative preference for the old object. Analyzed by two-way ANOVA, followed by Sidak’s post-hoc test comparing means within genotype, F(1,100) = 5.784, p = 0.0180 (genotype x stress interaction), n = 26 wildtype control, 26 wildtype CORT, 25 C3−/− control, and 27 C3−/− CORT mice, wildtype control vs. wildtype CORT *p = 0.0167, C3−/− control vs. C3−/− CORT p = 0.7820. C–E All data shown as mean ± s.e.m. Source data provided in Source File.
Discussion
In this study, we show that chronic stress causes spatially restricted complement activation in the upper cortical layers of the mPFC in male mice, resulting in local microglia synapse phagocytosis and synapse loss, as well as anhedonia and working memory deficits. Single cell transcriptomics of the stressed cortex show a dramatic upregulation of the Apoe gene in many microglia, with Apoehigh microglia also localized to layer 2/3 of the mPFC. Deletion of the C3 gene blocks the stress-induced layer-specific increases in microglial synapse phagocytosis, synapse loss, and Apoehigh microglia population in layer 2/3 of the mPFC. Mice lacking the C3 gene are also resilient against stress-induced anhedonia and working memory deficits. Our findings highlight the role of spatially restricted microglia activation in the brain in mediating the neurological symptoms of stress-associated diseases, such as depression, and reveals the potential role of localized complement and microglia activation in causing region-specific synapse loss in other brain diseases.
The PFC plays an important role in the complex cognitive control of many behaviors, including the regulation of emotion and working memory. During brain development, the PFC is the last region to mature, with an extended developmental period spanning decades in humans68,69, making the PFC vulnerable to many environmental and genetic insults. Recent studies indicate that the upper layers of the PFC are preferentially affected in many psychiatric and neurological diseases. Neuropathological studies of brains from schizophrenic patients show a consistent loss of dendritic spines in layer 3 of the dorsolateral PFC11,12, while spine density in deeper PFC layers13 and other cortical regions14 are unaffected. A transcriptomics study of brain cells from autistic patients has also shown that specific sets of genes enriched in neurons and microglia in the upper layers of the PFC are correlated with clinical severity70. In Alzheimer’s disease (AD), postmortem brain studies have revealed high Aβ plaque densities in PFC layers 2/371,72,73, and increased numbers of reactive astrocytes in the same layers74. Here, we show that layer 2/3 of the mPFC is selectively vulnerable to stress-induced synapse loss as a result of layer-specific complement and microglia activation. Our findings suggest that the selective vulnerability of the upper layers of the PFC in many brain diseases is based on a propensity for runaway complement activation, although the exact molecular mechanisms and cell types involved remain to be determined.
Many studies have shown that microglia are exquisitely sensitive to the environment. A recent single-cell transcriptomic study of microglia has shown that in the somatosensory cortex, microglia adopt layer-specific transcriptional states that are determined by signals from laminarly distributed pyramidal neurons17. Therefore, a straightforward mechanism for layer-specific complement activation is the stress-induced increase in the expression of complement components or activators in a particular neuronal or astrocytic layer75 in the mPFC. Alternatively, a stress-induced decrease in the expression of complement inhibitors46,76,77 within the upper layers can also lead to regional complement activation. In addition, inputs into the mPFC are known to be layer-specific78, and stress-induced signals emanating from specific presynaptic terminals that are laminarly distributed may also trigger layer-specific complement and microglia activation. In addition, chronic stress has also been shown to increase blood-brain barrier permeability79,80, which may also allow peripheral C3 in blood to enter the brain. Importantly, our findings constrain the possible upstream mechanisms to those that operate in a layer-specific manner. The above hypothetical mechanisms are not mutually exclusive and may span more than one cell type, and further investigation is required to fully elucidate the mechanisms involved.
In addition to showing that stress induces layer-specific complement and microglia activation, our scRNA-seq data also revealed a stress-induced Apoehigh microglia state. We speculate that the Apoehigh microglia population phagocytoses synapses in layer 2/3 of the mPFC based on two lines of evidence: 1) both the Apoehigh microglia and the microglia shown immunohistologically to be phagocytosing synapses are localized to layer 2/3, and 2) both the Apoehigh microglia and the microglia that phagocytose synapses are ablated by C3 gene deletion. However, we acknowledge that these lines of evidence are indirect. We are unable to directly use apoE protein as an immunohistological marker for Apoehigh microglia because microglia also phagocytose extracellular apoE, rendering apoE staining unreliable as a marker for Apoehigh microglia. We were also unable to identify additional genes that simultaneously label the entirety of the Apoehigh microglia population and were detectable by high-quality antibodies, which precluded the use of alternative markers. Furthermore, the ISH process for detecting Apoe transcripts is moderately destructive on brain slices, and renders them unsuitable for the synapse phagocytosis assay. Therefore, as a result of technical issues, we are unable to directly demonstrate a synapse phagocytosis function for the Apoehigh microglia. Both Apoehigh and microglia mediating synapse phagocytosis are relatively small populations of microglia, and it is possible that they are separate populations of microglia in layer 2/3. In addition, it is possible that the Apoehigh microglia population may perform other functions, such as cytokine secretion. Therefore, further work is required to elucidate the biological functions of the stress-induced Apoehigh microglia state.
Numerous scRNA-seq studies have shown that microglia adopt disease-associated transcriptomic states. Intriguingly, many of these states include the upregulation of the Apoe gene. Examples of Apoe-associated microglial states include the disease-associated microglia (DAMs) found in AD21,27, human AD microglia (HAMs)81, the microglial neurodegenerative phenotype (MGnD) found in mouse models of AD/ALS/MS24, microglia found in peripheral nerve injury and chronic pain26, activated response microglia (ARMs) found in aged mice and an AD mouse model82, microglia inflamed in MS (MIMS)25, and a microglia state associated with injury19. Compared to these disease-associated microglial states, the stress-induced Apoehigh microglia upregulate relatively few genes, consisting mainly of the Apoe and Ms4a genes (Supplemental Dataset 1). The upregulation of microglial Apoe in a wide variety of diseases suggests that Apoe upregulation is part of a generic microglial response to tissue damage. Interestingly, early studies in stress biology describe the stress response as a “general adaptation syndrome” that occurs in diverse diseases83,84. In this study, we show that the stress hormone corticosterone induces an Apoehigh microglia state, suggesting that the Apoe component of multiple disease-associated microglial states may be a result of stress hormone release in those diseases.
Stress is also known to aggravate or trigger the occurrence of other neurological and psychiatric diseases. For example, stress is known to trigger episodes of psychosis in schizophrenic patients85,86 and attacks in multiple sclerosis patients87, as well as accelerate the progression of AD88,89,90. All of these diseases are associated with aberrant complement activation in the brain25,91,92,93,94,95,96,97,98. Our findings suggest that stress may aggravate these diseases through additive complement and microglia activation. We also show that only VGlut2 synapses in layer 2/3 mPFC are phagocytosed by microglia, while VGlut1 and VGAT synapses are spared, indicating that even within a region of complement activation, specific microcircuits may be selectively impaired. These findings may be relevant when assessing the neural circuits involved in various complement-associated brain diseases.
Neuroimaging studies have revealed disease-specific and reproducible patterns of gray matter volume (GMV) reductions in many brain diseases, including depression99,100,101, schizophrenia102, and other major psychiatric diseases103,104. A more recent study has showed that substantial heterogeneity also exists in the individual pattern of GMV changes in patients diagnosed with the same psychiatric disorder, and suggests that individual variation in disease symptoms may be a result of this heterogeneity103. Other studies have also shown that elevated salivary, hair and serum cortisol levels are all correlated with lower brain volumes38,39, and that stressful life events leave a replicable pattern of brain volume reductions that is predictive of current and future anxiety105. While GMV is a complex measure that encompasses glia, vasculature and neuronal dendritic and synapse volumes, longitudinal studies show a lifetime trajectory of GMV loss106,107 that temporally correlates with neuropathological studies of dendritic spine loss due to increased synapse pruning during adolescence and early adulthood108,109, suggesting that synapse loss is a major contributor to GMV reductions. Given the large variation in complement activity found in the human population110, genetic polymorphisms in complement components and regulators may account for the individual variation in GMV reductions observed in psychiatric patients. Our study suggests that lifetime adversity can leave spatially heterogeneous patterns of brain volume reductions through heterogeneous complement and microglia activation, and that regional regulation of these processes play an important role in the heterogeneity of neuropsychiatric diseases.
Methods
Animal care
All animal work was approved by the UT Health Institutional Animal Care and Use Committee (IACUC), in accordance with animal protocol number 20150052AR. Euthanasia of mice were performed by exposure to carbon dioxide for at least 2 min, and visually confirming cessation of breathing. C3−/− mice (Jackson Laboratory stock 003641) were backcrossed to C57BL/6 J for more than 10 generations before use. Colonies were maintained in climate-controlled rooms within the AAALAC-accredited UT Health animal facility. Mice were group-housed (3–4 per cage) under a 14-h light/10-h dark cycle at 75 F with 30–70% humidity. Animals provided ad libitum access to food and water. Subjects were used at 3–5 months of age for experiments. Clear red-tinted tunnels (Bioserv, K3322) were placed in all cages for enrichment and transport purposes. To reduce distress during transfer, mice were habituated to tunnel handling with daily 3–5 min tube rides for 1 week prior to behavioral studies. Behavioral assays were conducted either during the last 4 h of the light cycle (temporal object recognition) or throughout the dark cycle (sucrose preference test). Only male mice were used in this study.
Chronic corticosterone (CORT) stress protocol
Mice received either corticosterone-infused (CORT) drinking water (Sigma 27840, corticosterone dissolved at 10 mg/ml in 100% ethanol, then mixed with facility-provided drinking water to a final concentration of 0.1 mg/ml corticosterone and 1% ethanol) or control drinking water (1% ethanol) for 21 days, as described54. Both CORT and control water were prepared in standard bottles using autoclaved drinking water from the animal facility and replaced weekly.
Chronic restraint stress (CRS) protocol
Mice were subjected to 4 h of restraint stress daily for 21 days, as described54. Restraint tubes were made from 50 mL canonical tubes (Falcon, #2098) with drilled holes for air circulation. Restraint tubes were placed inside the home cage. Control mice were handled twice daily, 4 h apart, for 5 min each. Tubes were cleaned with 70% ethanol between uses.
Chronic unpredictable stress (CUS) protocol
We used a protocol that had been shown in multiple studies to cause stress-induced circuitry changes in mice111,112. Mice were subjected to two different stressors per day, once in the morning and once in the afternoon, for 10 days. Stressors included: 15 min tail pinch, 2 hr tube restraint, 30 min elevated platform, 10 min inescapable footshock (0.7 mA, 2 s, 10 total shocks), 24 hr wet bedding with 45° cage tilt, and 24 hr light exposure. Control mice were handled twice daily. CUS mice were singly housed for the duration of the stress protocol, while control mice were group-housed.
Immunohistochemistry and microscopy
Mice were deeply anesthetized with Avertin (0.5 mg/g) for transcardial perfusion with ice-cold PBS and 4% paraformaldehyde (PFA). Brains were rapidly extracted, post-fixed for 2 hrs at 4 °C with gentle rocking, then cryoprotected in 30% sucrose in PBS for 48–72 h. Brains were sliced on a sliding-freezing microtome (Leica) into 20 μm sections (for synapse and complement staining) or thick 40 μm sections (for microglia staining). Wash, block and antibody solutions were made from PBS (pH 7.4) with either 0.15% TX-100 (0.15% PBT) or 0.3% PBT, depending on slice thickness. Free-floating sections were washed 2x in PBT (10 min/wash), post-fixed in 4% PFA (15 min) and briefly rinsed in PBS (5 min) while rocking at room temperature (RT). Sections were blocked in 10% normal donkey/goat serum (Jackson Immunoresearch) at RT for 2 hr, then incubated in primary antibodies in 1% blocking buffer overnight at 4 °C with gentle rocking. Once equilibrated to RT, sections were washed 4x (10 min/wash), then incubated in secondary for 2 hr. Sections were washed 2x in PBT (5 min/wash), 3x in PBS (10 min/wash) then floated onto glass slides. To remove lipofuscin autofluorescence in microglia which may confound synapse detection113, slides were treated with TrueBlack Lipofuscin Autofluorescence Quencher (Biotium, 23007), rinsed 3x in PBS (3 min/wash), then cover-slipped with Fluoromount-G (SouthernBiotech, 0100-01). Slides were stored in 4° and imaged within 1 week.
The following primary antibodies were used for staining: goat anti-C3 (1:1000, MP Biomedicals, cat#: 855730), rabbit anti-Iba1 (1:500, Fuji Film, cat#: 019-19741), rat anti-CD68 (1:500, Bio-Rad, cat#: MCA1957), guinea pig anti-VGlut2 (1:1000, Synaptic Systems, cat#: 135 404), rabbit anti-VGlut1 (1:1000, Synaptic Systems, cat#: 135 302), mouse anti-PSD95 K28/43, IgG2a (1:500, Neuromab, cat#: 75-028), guinea pig anti-VGAT (1:500, Synaptic Systems, cat#: 131 004), mouse anti-gephyrin, IgG1 (1:500, Synaptic systems, cat#: 147 021), rabbit anti-cFOS (1:500, Synaptic systems, cat#: 226-008).
The following secondary antibodies were used for staining: donkey Anti-Goat IgG A488 (1:500, Jackson, cat#: 705-545-147), donkey Anti-Rabbit IgG A488 (1:500, Jackson, cat#: 711-545-152), cy3-AffiniPure Donkey Anti-Rat IgG H&L (1:500, Jackson, cat#: 712-165-153), goat Anti-Guinea Pig IgG Polyclonal CF-488A (1:500, Biotium, cat#: 20017-1), cy5-AffiniPure Donkey Anti-Guinea Pig IgG H&L (1:500, Jackson, cat#: 706-175-148), goat Anti-Rabbit IgG H&L Cy5 (1:500, Abcam, cat#: ab6564), goat anti-Mouse IgG2a A555 (1:500, Thermo Fisher Scientific, cat#: A-21137), goat anti-Guinea Pig IgG H&L A647 (1:500, Thermo Fisher Scientific, cat#: A-21450), goat anti-Mouse IgG1 A488 (1:500, Thermo Fisher Scientific, cat#: A-21121).
Sections were imaged using a Zeiss AxioObserver inverted microscope equipped with a Zeiss Apotome.2 module. Images were acquired using a Zeiss 63x/1.4 oil Plan-Apochromat objective (synapse density, microglial activation and engulfment, high-resolution C3), or Zeiss 20x Plan-Neofluar objective (low-magnification C3 staining). Confocal microscopy was carried out with an Olympus FV-1000 confocal microscope with an Olympus Plan Apo 60x/1.3 NA oil immersion objective. Identical imaging and analysis parameters were used for each experimental group.
Image analysis for C3 deposition and synapse densities
ImageJ (NIH) was used to quantify synapse densities and C3 deposition. Individual channels were first Z-projected with summation over a depth of 2 µm, followed by background subtraction (8 px rolling ball radius) and Gaussian filter (sigma = 0.8). The threshold was empirically determined for each channel and applied consistently to all groups. Watershed was applied to the binarized image. For synapse density, colocalized puncta were revealed by multiplying the individual pre & post-synaptic channels using the image calculator tool. The Analyze Particles tool was used to quantify the number of colocalized synaptic puncta and individual C3 deposits.
While we used an anti-C3 antibody that binds to both intact and cleaved C3, total C3 staining is well-correlated with C3 activation in situ, because intact C3 is washed out during perfusion while cleaved C3 remains covalently bound to cell membranes and proteins. In the developing brain, transient increases in C3 staining in a brain region coincide with increases in microglial activation and phagocytosis of synapses46,114. In addition, viral overexpression of the complement inhibitor Crry has been shown to decrease total C3 staining95, indicating that C3 staining with an antibody that detect all forms of C3 is sensitive to local C3 activation.
Image analysis for microglial lysosome content and synapse phagocytosis
Reconstructions of microglia were performed using high-resolution confocal-quality images of Iba1-positive cells using Imaris Software (version 9.7.1, Bitplane, Concord MA), as previously described45,46. Using the image processing tool, background subtraction was first performed (sigma = 1 for VGlut2, CD68; sigma = 60 for Iba1), followed by the application of a gaussian filter (sigma = 0.144). All surfaces were rendered with 0.1 µm smoothing. Iba1-positive cells were reconstructed first, and disconnected processes were unified with the cell body to create a single surface representing one cell. To render CD68, a mask was then applied to isolate the CD68 signal only within the reconstructed microglia. Another mask was then applied to isolate the VGlut2 signal only within the reconstructed CD68-positive lysosomes within microglia. Volumes were recorded for each reconstructed surface. For microglial lysosome content, the percent of CD68 occupancy within microglia was calculated using the formula: (volume of CD68/volume of Iba1+ cell)*100. For microglial engulfment of VGlut2, the percent of VGlut2 within microglial lysosomes was calculated using the formula: (volume of VGlut2 within lysosome/volume of Iba1+ cell)*100.
Quantification of c-fos positive cells
Mice were sacrificed exactly 1 h after completion of the TOR test phase. Brain tissue was collected and processed as described above with slight modifications. In short, 40 µm-thick brain sections were fixed in ice-cold 100% methanol for 5 min, washed in 0.3% PBT for 15 min, then blocked in 1% BSA 5% NDS in 0.3% PBT for 2 h at RT. Sections were incubated in primary antibody in blocking buffer overnight (rabbit anti-c-fos, 1:1000, Synaptic Systems 226-008), washed 5x with PBT, then incubated in secondary antibody (donkey anti-rabbit A488, Jackson, catalog #: 711-545-152) for 2 hr at RT. Sections were imaged on a Zeiss AxioObserver inverted microscope equipped with Apotome.2 module using a Zeiss 20x Plan-Neofluar objective. Analysis of c-fos positive cells was automated using an ImageJ macro by applying the following sequence on 200 µm × 200 µm ROIs: gaussian filter (sigma = 2), threshold (1000,65535), convert to binary mask, watershed, analyze particles.
Microglia single cell dissociation and FACS
Microglia single cell dissociation was performed using the cold mechanical dissociation procedure which minimizes ex vivo activation of microglia as previously described19,65. Brains were collected from avertin-anesthetized control and CORT mice after transcardiac perfusion of ice-cold HBSS, and the cerebral cortices were dissected out and minced after removal of white matter. Pieces of cortices were then Dounce homogenized for 20 strokes with a loose rotating pestle, and then strained through a 70 µm strainer. Cells were washed and pelleted in HBSS, and then resuspended in 40% Percoll. Cells were spun at 500 g in the cold for 1 hr, and the pelleted cells were washed in HBSS and resuspended in FACS buffer for staining. Cell suspensions were incubated with Fc block reagent (1:50 dilution) for 15 min on ice, and then the following antibodies were added from a master mix: DAPI (1:10,000), Cd11b-PE (1:50), CD45-APC-Cy7 (1:100), Cx3cr1-APC (1:250). Cell suspensions were further incubated for 30 min on ice, and then washed twice and resuspended in FACS buffer. Cells were then immediately transported to the Flow Cytometry Core Facility on ice for sorting. Sorting was performed with the following gating strategy (Supplementary Fig. 1): whole cells versus debris (forward scatter (FSC-A) vs side scatter (SSC-A)), singlets vs aggregates (SSC-H vs SSC-A, and FSC-H vs FSC-A), live vs dead (DAPI vs SSC-A), Cd45high (APC-Cy7-CD45 vs SSC-A), Cx3cr1high and Cd11bhigh (APC-CX3CR1 vs PE-CD11b).
Single cell library generation and sequencing
After sorting in the cold, cells were put on ice and immediately transported to the Genome Sequencing Core Facility for 10X Genomics single-cell capture and sequencing with the Chromium 3′ library v3.1 kit, following manufacturer’s instructions. Approximately 10,000 cells were targeted per sample for single-cell sequencing. The single-cell libraries were then sequenced on an Illumina NovaSeq6000 sequencer, targeting at least 500 M reads per sample, for an average sequencing depth of 50,000 reads per cells. Cell Ranger v7.0.1 (10X Genomics) was used for sample demultiplexing, barcode processing, unique molecular identifiers filtering, gene counting, and mapping to the mouse mm10-2020-A (10X Genomics) reference genome.
scRNA-seq clustering analysis
Single-cell clustering analysis was performed with the R package Seurat v5.0.3115. For initial quality filtering, genes expressed in more than 5 cells and cells with greater than 10% mitochondrial gene expression were kept. The SCTransform v2 algorithm116 from Seurat was then used to normalize the counts in each sample. After filtering, a total of 81,177 microglia were retained for downstream analysis.
Cells from all samples were integrated with Canonical Correlation Analysis (CCA) with Seurat. Principal Components Analysis (PCA) was then performed with the integrated dataset, and the top 20 principal components (PCs) were chosen for clustering based on an elbowplot. Clustering was performed using the Seurat FindNeighbors function with the original Louvain algorithm for 20 PCs, and the FindClusters function was used at a resolution of 0.05. Uniform Approximation and Projection (UMAP) dimensionality reduction was performed to visualize the cell clusters with the RunUMAP and DimPlot functions. Marker genes for clusters were identified using the FindAllMarkers function with the Wilcoxon rank-sum test. The top-expressing genes in each cell cluster was plotted as a heat map using the DoHeatMap function. Differential abundance analysis of the cell clusters between control and CORT conditions were performed with the speckle R package117. Pseudobulk analysis of differentially expressed genes (DEGs) between the control and CORT conditions were performed with Libra118 with the edgeR-LRT algorithm, and the results were plotted in a volcano plot using the EnhancedVolcano R package119. The R package clusterProfiler120 was used to perform gene set enrichment analysis (GSEA). The list of stress-upregulated DEGs from the pseudobulk DEG analysis (logFC > 1 and adjusted p value < 10e−4) was used as input to the enrichGO function of clusterProfiler with the following parameters: keyType = “SYMBOL”, OrgDb = org.Mm.eg.db, ont = “BP”, pvalueCutoff = 0.05, pAdjustMethod = “BH”. The top 10 enriched biological process (BP) gene ontology (GO) terms were ranked in order of increasing adjusted p value, and plotted as a dot plot.
We also performed an alternative abbreviated scRNA analysis with the same dataset except without CCA integration. The same workflow was performed as above, except that CCA integration was not performed, and the FindClusters function was applied at a resolution of 0.05 to generate 5 clusters. The unintegrated analysis resulted in slightly different microglia cell clusters (Supplementary Fig. 8A), although the Fcrls and Apoe clusters were still generated, and both were still the only cell clusters found to be statistically different between the Ctrl and CORT conditions based on differential abundance analysis (Supplementary Fig. 8B,C). The marker genes for the cell clusters of the non-integrated analysis are presented in Supplementary Dataset 2.
RNAscope ISH
RNAscope™ Multiplex Fluorescent V2 assay (ACDBio, 323136) was carried out according to manufacturer’s instructions, with some modifications. Briefly, brains from PBS-perfused mice were rapidly dissected and snap-frozen in liquid nitrogen. Brains were sliced using a cryostat into 16um sections and adhered onto super-frost plus slides (Electron Microscopy Sciences, 71869-20). Slides were kept on dry ice during slicing and then promptly stored at −80 °C in a sealed box until use. Immediately upon removal from the freezer, slides were plunged into fresh ice-cold 4% PFA for 15 min, rinsed 2x with PBS, then dehydrated in serial dilutions of ethanol (50%, 75%, 100%, 100%). Slides were then air-dried for 5 min, and a PAP pen was used to create a hydrophobic barrier around the tissue. For all subsequent steps, slides were moved to the HybEZ Humidity Control Tray with wet humidifying paper for treatment/incubation, and washed in coplin jars with gentle rocking (2 min per wash). At room temperature (RT), sections were treated with hydrogen peroxide for 10 min, washed 3x in distilled water, treated with Protease IV for 20 min, then washed 3x with PBS. Probes (Mm-CX3CR1-C2, cat no. 314221-C2 and ApoE-C3, cat no. 313271-C3) were hybridized to tissue at 37 °C for 2 hr, rinsed 3x in wash buffer, and stored in 5x saline sodium citrate (5x SSC) buffer overnight at RT. Slides were rinsed 1x with wash buffer before experiment continuation. Subsequent amplification & HRP steps were performed according to manufacturer’s instructions. HRP-C2 and HRP-C3 were labelled with Opal 520 (1:750, Akoya FP1487001KT) and Opal 570 (1:1500, Akoya FP1488001KT), respectively. Slides were counterstained with DAPI, cover slipped with Prolong Gold (Cell Signaling Technology #9071), stored in 4 °C and imaged within 1 week.
Images were acquired with a Zeiss AxioObserver equipped with an Apotome.2 module using a Zeiss 40x Plan-Neuofluar objective. Images were analyzed with the ImageJ/Fiji software. Maximal projection images (summation) were prepared from the Z-stacks. The DAPI channel was binarized and the “Fill Holes” and “Watershed” functions were applied before the “Analyze Particle” function (pixel size 10-Inf, circularity 0-Inf) was used to create ROIs of nuclei. The Cx3cr1 channel was processed with the “Subtract Background” (rolling ball radius 50 px) function, and then thresholded and binarized to isolate Cx3cr1 cells. The Apoe channel was processed with the “Subtract Background” (rolling ball radius 50 px) function, and then thresholded to isolate Apoe puncta. DAPI ROIs that had areas that were >25% covered by Cx3cr1 puncta were deemed microglia, and the “Analyze Particles” function was applied within the ROI to determine the Apoe content (area x mean intensity, or integrated density). Identical thresholds were applied to all experimental groups in each staining batch, which contains balanced numbers of control and CORT mice. Apoe content was normalized to the average WT control Apoe content in each staining batch. A few data points notably deviate from the group mean, likely due to differential drying of RNAscope slides. We performed outlier testing with the Grubb’s test, and removed one outlier each from the layer 2 WT-Ctrl, WT-Cort, and C3-KO-Cort groups.
Sucrose preference test (SPT) task
Sucrose preference test was conducted using a 1% sucrose solution (Sigma S9378-1KG). Bottles were made from 50 mL conical “iTubes” with low-aldehyde odor (Stellar Scientific, T50-600) custom fitted with 2.5” sipper tubes (Fisher, NC1636707) and rubber stoppers (VWR, 76293-684) to minimize leakage. Sucrose and control water were prepared using autoclaved drinking water from the animal facility. Sucrose water and control water were sterile filtered before aliquoting into conical tubes. Mice were simultaneously habituated to sucrose and control water bottles for 3 consecutive days prior to testing. Bottles were switched in positions once daily to avoid side preference. On the test day, mice were separated into individual housing and deprived of water for the last 4 h of the light cycle. Sucrose and water bottles were weighed at the beginning and end of the dark cycle. Percent sucrose preference was calculated with the following formula: ((sucrose consumption - water consumption) / total consumption) x 100.
Temporal Object Recognition (TOR) task
TOR was conducted in an 80 × 80 × 30 cm white acrylic open field arena, with white opaque wall inserts separating the arena into (4) 40 × 40 × 30 cm height square chambers (Conduct Science). Objects were 3D printed from natural PLA filament (3D printer files provided by Maithe Arruda-Carvalho121). Two types of objects were printed: steps and domes. To minimize intrinsic preference of mice for either object, both objects were made of the same material, and were designed to be equal in surface area. Objects were also tested in pilot studies to verify that mice had no intrinsic preference for either object, and counterbalanced between phase 1 and phase 2 as described below. Objects were centered midway along the inside of the external chamber walls, equidistant from the corners. To prevent movement, objects were affixed to the chamber floor using a small piece of BluTack adhesive putty (a fresh piece was used every round for every mouse). Chambers and objects were cleaned with 15% ethanol between each mouse and phase to minimize odor carryover. All equipment was cleaned with 70% ethanol at the end of the test session. Mice were habituated to each quadrant (chamber) of the arena, 5 min per chamber, for two consecutive days prior to testing. The TOR procedure consisted of three 5-min phases (phase 1, phase 2 and test) with 1 hr inter-phase intervals. Mice were returned to home cage between phases. During phase 1, mice were placed in the arena with a pair of identical objects (either 2 steps or 2 domes). After a 1 hr delay, mice were reintroduced to the same chamber containing a different pair of identical objects for phase 2. During the test phase, mice were reintroduced to the same chamber containing one object from phase 1 (old object) and one object from phase 2 (recent object). Sessions were recorded using an infrared camera (OEM Cameras, DFK 33UX290) with a 3–8 mm focal length lens (OEM Cameras, T3Z0312CS-MPIR) fixed to the ceiling directly above the arena. Videos were analyzed using Noldus Ethovision tracking software (Noldus Ethovision XT, Version 11.5). Behavioral analysis during the test phase was restricted to the first 20 s of total interaction time with objects, as previously described121. The object discrimination index was calculated using the following formula: old object interaction time / total object interaction time.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw single cell RNA sequencing data has been deposited in the NCBI Gene Expression Omnibus (GEO) database under accession GSE272813. Source data are provided with this paper.
Code availability
Code required to replicate the single cell RNA sequencing analysis can be found at Github [https://doi.org/10.5281/zenodo.13732420].
References
Thomas, M. S. C., Davis, R., Karmiloff-Smith, A., Knowland, V. C. P. & Charman, T. The over-pruning hypothesis of autism. Dev. Sci. 19, 284–305 (2016).
State, M. W. & Šestan, N. The Emerging Biology of Autism Spectrum Disorders. Science 337, 1301–1303 (2012).
Kang, H. J. et al. Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat. Med. 18, 1413–1417 (2012).
Feinberg, I. Schizophrenia: Caused by a fault in programmed synaptic elimination during adolescence? J. Psychiatr. Res. 17, 319–334 (1982).
Garey, L. J. et al. Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J. Neurol. Neurosurg. Psychiatry 65, 446–453 (1998).
Selkoe, D. J. Alzheimer’s disease is a synaptic failure. Science 298, 789–791 (2002).
Osimo, E. F., Beck, K., Reis Marques, T. & Howes, O. D. Synaptic loss in schizophrenia: a meta-analysis and systematic review of synaptic protein and mRNA measures. Mol. Psychiatry 24, 549–561 (2019).
Holmes, S. E. et al. Lower synaptic density is associated with depression severity and network alterations. Nat. Commun. 10, 1529 (2019).
Duric, V. et al. Altered expression of synapse and glutamate related genes in post-mortem hippocampus of depressed subjects. Int. J. Neuropsychopharmacol. Off. Sci. J. Coll. Int. Neuropsychopharmacol. CINP 16, 69–82 (2013).
Pickett, E. K. et al. Region-specific depletion of synaptic mitochondria in the brains of patients with Alzheimer’s disease. Acta Neuropathol. (Berl.) 136, 747–757 (2018).
Arnsten, A. F. T., Woo, E., Yang, S., Wang, M. & Datta, D. Unusual Molecular Regulation of Dorsolateral Prefrontal Cortex Layer III Synapses Increases Vulnerability to Genetic and Environmental Insults in Schizophrenia. Biol. Psychiatry 92, 480–490 (2022).
Dienel, S. J., Schoonover, K. E. & Lewis, D. A. Cognitive Dysfunction and Prefrontal Cortical Circuit Alterations in Schizophrenia: Developmental Trajectories. Biol. Psychiatry, S0006-322301072-1. https://doi.org/10.1016/j.biopsych.2022.03.002 (2022).
Kolluri, N., Sun, Z., Sampson, A. R. & Lewis, D. A. Lamina-specific reductions in dendritic spine density in the prefrontal cortex of subjects with schizophrenia. Am. J. Psychiatry 162, 1200–1202 (2005).
Glantz, L. A. & Lewis, D. A. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch. Gen. Psychiatry 57, 65–73 (2000).
Qiao, H., et al. Dendritic Spines in Depression: What We Learned from Animal Models. Neural Plast. 2016. https://doi.org/10.1155/2016/8056370 (2016).
Duman, C. H. & Duman, R. S. Spine synapse remodeling in the pathophysiology and treatment of depression. Neurosci. Lett. 601, 20–29 (2015).
Stogsdill, J. A., et al. Pyramidal neuron subtype diversity governs microglia states in the neocortex. Nature, 1–7. https://doi.org/10.1038/s41586-022-05056-7 (2022).
Li, Q. et al. Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron 101, 207–223.e10 (2019).
Hammond, T. R. et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 50, 253–271.e6 (2019).
Tan, Y.-L., Yuan, Y. & Tian, L. Microglial regional heterogeneity and its role in the brain. Mol. Psychiatry 25, 351–367 (2020).
Keren-Shaul, H. et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290.e17 (2017).
Mathys, H. et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337 (2019).
Olah, M. et al. Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat. Commun. 11, 6129 (2020).
Krasemann, S. et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47, 566–581.e9 (2017).
Absinta, M., et al. A lymphocyte-microglia-astrocyte axis in chronic active multiple sclerosis. Nature. https://doi.org/10.1038/s41586-021-03892-7 (2021).
Tansley, S. et al. Single-cell RNA sequencing reveals time- and sex-specific responses of mouse spinal cord microglia to peripheral nerve injury and links ApoE to chronic pain. Nat. Commun. 13, 843 (2022).
Wang, S. et al. TREM2 drives microglia response to amyloid-β via SYK-dependent and -independent pathways. Cell 185, 4153–4169.e19 (2022).
Ennerfelt, H., et al. (2022). SYK coordinates neuroprotective microglial responses in neurodegenerative disease. Cell, S0092-867401249-1.
Yin, Z., et al. Identification of a protective microglial state mediated by miR-155 and interferon-γ signaling in a mouse model of Alzheimer’s disease. Nat. Neurosci., 1–12. https://doi.org/10.1038/s41593-023-01355-y (2023).
Kaya, T., et al. CD8+ T cells induce interferon-responsive oligodendrocytes and microglia in white matter aging. Nat. Neurosci., 1–12. https://doi.org/10.1038/s41593-022-01183-6 (2022).
Hammen, C. Stress and depression. Annu. Rev. Clin. Psychol. 1, 293–319 (2005).
McEwen, B. S. & Akil, H. Revisiting the Stress Concept: Implications for Affective Disorders. J. Neurosci. 40, 12–21 (2020).
Smoller, J. W. The Genetics of Stress-Related Disorders: PTSD, Depression, and Anxiety Disorders. Neuropsychopharmacology 41, 297–319 (2016).
Parker, K. J., Schatzberg, A. F. & Lyons, D. M. Neuroendocrine aspects of hypercortisolism in major depression. Horm. Behav. 43, 60–66 (2003).
Qin, D.-D. et al. Prolonged secretion of cortisol as a possible mechanism underlying stress and depressive behaviour. Sci. Rep. 6, 30187 (2016).
Lee, B. K. et al. Associations of salivary cortisol with cognitive function in the Baltimore memory study. Arch. Gen. Psychiatry 64, 810–818 (2007).
Pulopulos, M. M. et al. Hair cortisol and cognitive performance in healthy older people. Psychoneuroendocrinology 44, 100–111 (2014).
Geerlings, M. I. et al. Salivary cortisol, brain volumes, and cognition in community-dwelling elderly without dementia. Neurology 85, 976–983 (2015).
Echouffo-Tcheugui, J. B. et al. Circulating cortisol and cognitive and structural brain measures: The Framingham Heart Study. Neurology 91, e1961–e1970 (2018).
Yirmiya, R., Rimmerman, N. & Reshef, R. Depression as a Microglial Disease. Trends Neurosci 38, 637–658 (2015).
Wohleb, E. S., Terwilliger, R., Duman, C. H. & Duman, R. S. Stress-Induced Neuronal Colony Stimulating Factor 1 Provokes Microglia-Mediated Neuronal Remodeling and Depressive-like Behavior. Biol. Psychiatry 83, 38–49 (2018).
Zhang, M.-M. et al. IL-1R/C3aR signaling regulates synaptic pruning in the prefrontal cortex of depression. Cell Biosci 12, 90 (2022).
Wang, J., et al. Microglia-dependent excessive synaptic pruning leads to cortical underconnectivity and behavioral abnormality following chronic social defeat stress in mice. Brain. Behav. Immun. https://doi.org/10.1016/j.bbi.2022.12.019 (2022).
Crider, A. et al. Complement component 3a receptor deficiency attenuates chronic stress-induced monocyte infiltration and depressive-like behavior. Brain. Behav. Immun. 70, 246–256 (2018).
Schafer, D. P. et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705 (2012).
Cong, Q., Soteros, B. M., Wollet, M., Kim, J. H. & Sia, G.-M. The endogenous neuronal complement inhibitor SRPX2 protects against complement-mediated synapse elimination during development. Nat. Neurosci. 23, 1067–1078 (2020).
Stevens, B. et al. The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178 (2007).
Arnsten, A. F. T. Stress signalling pathways that impair prefrontal cortex structure and function. Nat. Rev. Neurosci. 10, 410–422 (2009).
Haykin, H. & Rolls, A. The neuroimmune response during stress: A physiological perspective. Immunity 54, 1933–1947 (2021).
Shoji, H. & Miyakawa, T. Differential effects of stress exposure via two types of restraint apparatuses on behavior and plasma corticosterone level in inbred male BALB/cAJcl mice. Neuropsychopharmacol. Rep. 40, 73–84 (2020).
Gong, S. et al. Dynamics and Correlation of Serum Cortisol and Corticosterone under Different Physiological or Stressful Conditions in Mice. PLOS ONE 10, e0117503 (2015).
Ding, J. X., Rudak, P. T., Inoue, W. & Haeryfar, S. M. M. Physical restraint mouse models to assess immune responses under stress with or without habituation. STAR Protoc 2, 100838 (2021).
Wellman, C. L. Dendritic reorganization in pyramidal neurons in medial prefrontal cortex after chronic corticosterone administration. J. Neurobiol. 49, 245–253 (2001).
Moda-Sava, R. N. et al. Sustained rescue of prefrontal circuit dysfunction by antidepressant-induced spine formation. Science 364, eaat8078 (2019).
Anderson, R. M. et al. Prolonged corticosterone exposure induces dendritic spine remodeling and attrition in the rat medial prefrontal cortex. J. Comp. Neurol. 524, 3729–3746 (2016).
Yuen, E. Y. et al. Repeated Stress Causes Cognitive Impairment by Suppressing Glutamate Receptor Expression and Function in Prefrontal Cortex. Neuron 73, 962–977 (2012).
Horchar, M. J. & Wohleb, E. S. Glucocorticoid receptor antagonism prevents microglia-mediated neuronal remodeling and behavioral despair following chronic unpredictable stress. Brain. Behav. Immun. 81, 329–340 (2019).
Chen, C.-C., Lu, J., Yang, R., Ding, J. B. & Zuo, Y. Selective activation of parvalbumin interneurons prevents stress-induced synapse loss and perceptual defects. Mol. Psychiatry 23, 1614–1625 (2018).
Liston, C. & Gan, W.-B. Glucocorticoids are critical regulators of dendritic spine development and plasticity in vivo. Proc. Natl. Acad. Sci. 108, 16074–16079 (2011).
Cong, Q. et al. C1q and SRPX2 regulate microglia mediated synapse elimination during early development in the visual thalamus but not the visual cortex. Glia 70, 451–465 (2022).
Bolkan, S. S. et al. Thalamic projections sustain prefrontal activity during working memory maintenance. Nat. Neurosci. 20, 987–996 (2017).
Schmitt, L. I. et al. Thalamic amplification of cortical connectivity sustains attentional control. Nature 545, 219–223 (2017).
Parnaudeau, S. et al. Inhibition of mediodorsal thalamus disrupts thalamofrontal connectivity and cognition. Neuron 77, 1151–1162 (2013).
Jett, J. D., Bulin, S. E., Hatherall, L. C., McCartney, C. M. & Morilak, D. A. Deficits in cognitive flexibility induced by chronic unpredictable stress are associated with impaired glutamate neurotransmission in the rat medial prefrontal cortex. Neuroscience 346, 284–297 (2017).
Marsh, S. E. et al. Dissection of artifactual and confounding glial signatures by single-cell sequencing of mouse and human brain. Nat. Neurosci. 25, 306–316 (2022).
Festa, B. P. et al. Microglial-to-neuronal CCR5 signaling regulates autophagy in neurodegeneration. Neuron https://doi.org/10.1016/j.neuron.2023.04.006 (2023).
Escoubas, C. C. et al. Type I interferon responsive microglia shape cortical development and behavior. BioRxiv Prepr. Serv. Biol. https://doi.org/10.1101/2021.04.29.441889 (2023). 2021.04.29.441889.
Kolk, S. M. & Rakic, P. Development of prefrontal cortex. Neuropsychopharmacology 47, 41–57 (2022).
Petanjek, Z. et al. Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proc. Natl. Acad. Sci. 108, 13281–13286 (2011).
Velmeshev, D. et al. Single-cell genomics identifies cell type–specific molecular changes in autism. Science 364, 685–689 (2019).
Rogers, J. & Morrison, J. H. Quantitative morphology and regional and laminar distributions of senile plaques in Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 5, 2801–2808 (1985).
Pearson, R. C., Esiri, M. M., Hiorns, R. W., Wilcock, G. K. & Powell, T. P. Anatomical correlates of the distribution of the pathological changes in the neocortex in Alzheimer disease. Proc. Natl. Acad. Sci. USA. 82, 4531–4534 (1985).
Duyckaerts, C. et al. Laminar distribution of neocortical senile plaques in senile dementia of the alzheimer type. Acta Neuropathol. (Berl.) 70, 249–256 (1986).
Marutle, A. et al. 3H-deprenyl and 3H-PIB autoradiography show different laminar distributions of astroglia and fibrillar β-amyloid in Alzheimer brain. J. Neuroinflammation 10, 90 (2013).
Bayraktar, O. A. et al. Astrocyte layers in the mammalian cerebral cortex revealed by a single-cell in situ transcriptomic map. Nat. Neurosci. 23, 500–509 (2020).
Yin, C. et al. ApoE attenuates unresolvable inflammation by complex formation with activated C1q. Nat. Med. 25, 496–506 (2019).
Zhou, J. et al. The neuronal pentraxin Nptx2 regulates complement activity and restrains microglia-mediated synapse loss in neurodegeneration. Sci. Transl. Med. 15, eadf0141 (2023).
Anastasiades, P. G. & Carter, A. G. Circuit organization of the rodent medial prefrontal cortex. Trends Neurosci 44, 550–563 (2021).
Menard, C. et al. Social stress induces neurovascular pathology promoting depression. Nat. Neurosci. 20, 1752–1760 (2017).
Sántha, P. et al. Restraint Stress-Induced Morphological Changes at the Blood-Brain Barrier in Adult Rats. Front. Mol. Neurosci. 8, 88 (2015).
Srinivasan, K. et al. Alzheimer’s Patient Microglia Exhibit Enhanced Aging and Unique Transcriptional Activation. Cell Rep 31, 107843 (2020).
Frigerio, C. S. et al. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Aβ Plaques. Cell Rep 27, 1293–1306.e6 (2019).
Selye, H. A Syndrome produced by Diverse Nocuous Agents. Nature 138, 32–32 (1936).
Selye, H. Stress and the general adaptation syndrome. Br. Med. J. 1, 1383–1392 (1950).
Birley, J. L. & Brown, G. W. Crises and life changes preceding the onset or relapse of acute schizophrenia: clinical aspects. Br. J. Psychiatry J. Ment. Sci. 116, 327–333 (1970).
Nuechterlein, K. H. et al. The vulnerability/stress model of schizophrenic relapse: a longitudinal study. Acta Psychiatr. Scand. Suppl. 382, 58–64 (1994).
Mohr, D. C., Hart, S. L., Julian, L., Cox, D. & Pelletier, D. Association between stressful life events and exacerbation in multiple sclerosis: a meta-analysis. BMJ 328, 731 (2004).
Sotiropoulos, I. et al. Stress Acts Cumulatively To Precipitate Alzheimer’s Disease-Like Tau Pathology and Cognitive Deficits. J. Neurosci. 31, 7840–7847 (2011).
Harvanek, Z. M., Fogelman, N., Xu, K. & Sinha, R. Psychological and biological resilience modulates the effects of stress on epigenetic aging. Transl. Psychiatry 11, 1–9 (2021).
Madore, C., Yin, Z., Leibowitz, J. & Butovsky, O. Microglia, Lifestyle Stress, and Neurodegeneration. Immunity 52, 222–240 (2020).
Sekar, A. et al. Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183 (2016).
Hong, S. et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716 (2016).
Shi, Q. et al. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 9, eaaf6295 (2017).
Hammond, J. W., et al. Complement-dependent synapse loss and microgliosis in a mouse model of multiple sclerosis. Brain. Behav. Immun. https://doi.org/10.1016/j.bbi.2020.03.004 (2020).
Werneburg, S. et al. Targeted Complement Inhibition at Synapses Prevents Microglial Synaptic Engulfment and Synapse Loss in Demyelinating Disease. Immunity 52, 167–182.e7 (2020).
Bourel, J. et al. Complement C3 mediates early hippocampal neurodegeneration and memory impairment in experimental multiple sclerosis. Neurobiol. Dis. 160, 105533 (2021).
Sellgren, C. M. et al. Patient-specific models of microglia-mediated engulfment of synapses and neural progenitors. Mol. Psychiatry 22, 170–177 (2017).
Yilmaz, M. et al. Overexpression of schizophrenia susceptibility factor human complement C4A promotes excessive synaptic loss and behavioral changes in mice. Nat. Neurosci. 24, 214–224 (2021).
Hellewell, S. C. et al. Profound and reproducible patterns of reduced regional gray matter characterize major depressive disorder. Transl. Psychiatry 9, 1–8 (2019).
Hibar, D. P. et al. Cortical abnormalities in bipolar disorder: an MRI analysis of 6503 individuals from the ENIGMA Bipolar Disorder Working Group. Mol. Psychiatry 23, 932–942 (2018).
Grieve, S. M., Korgaonkar, M. S., Koslow, S. H., Gordon, E. & Williams, L. M. Widespread reductions in gray matter volume in depression. NeuroImage Clin 3, 332–339 (2013).
Gur, R. E., Turetsky, B. I., Bilker, W. B. & Gur, R. C. Reduced Gray Matter Volume in Schizophrenia. Arch. Gen. Psychiatry 56, 905–911 (1999).
Segal, A., et al. Regional, circuit and network heterogeneity of brain abnormalities in psychiatric disorders. Nat. Neurosci., 1–17. https://doi.org/10.1038/s41593-023-01404-6 (2023).
Opel, N. et al. Cross-Disorder Analysis of Brain Structural Abnormalities in Six Major Psychiatric Disorders: A Secondary Analysis of Mega- and Meta-analytical Findings From the ENIGMA Consortium. Biol. Psychiatry 88, 678–686 (2020).
Holz, N. E., et al. A stable and replicable neural signature of lifespan adversity in the adult brain. Nat. Neurosci., 1–10. https://doi.org/10.1038/s41593-023-01410-8 (2023).
Bethlehem, R. a. I., et al. Brain charts for the human lifespan. Nature, 1–11. https://doi.org/10.1038/s41586-022-04554-y (2022).
Brouwer, R. M. et al. Genetic variants associated with longitudinal changes in brain structure across the lifespan. Nat. Neurosci. 25, 421–432 (2022).
Huttenlocher, P. R. Synaptic density in human frontal cortex - developmental changes and effects of aging. Brain Res 163, 195–205 (1979).
Rakic, P., Bourgeois, J. P., Eckenhoff, M. F., Zecevic, N. & Goldman-Rakic, P. S. Concurrent overproduction of synapses in diverse regions of the primate cerebral cortex. Science 232, 232–235 (1986).
Harris, C. L., Heurich, M., Cordoba, S. Rde & Morgan, B. P. The complotype: dictating risk for inflammation and infection. Trends Immunol 33, 513–521 (2012).
Lei, Y. et al. SIRT1 in forebrain excitatory neurons produces sexually dimorphic effects on depression-related behaviors and modulates neuronal excitability and synaptic transmission in the medial prefrontal cortex. Mol. Psychiatry 25, 1094–1111 (2020).
Zhu, X. et al. Distinct thalamocortical circuits underlie allodynia induced by tissue injury and by depression-like states. Nat. Neurosci. 24, 542–553 (2021).
Stillman, J. M. et al. Lipofuscin-like autofluorescence within microglia and its impact on studying microglial engulfment. Nat. Commun. 14, 7060 (2023).
Kopec, A. M., Smith, C. J., Ayre, N. R., Sweat, S. C. & Bilbo, S. D. Microglial dopamine receptor elimination defines sex-specific nucleus accumbens development and social behavior in adolescent rats. Nat. Commun. 9, 3769 (2018).
Stuart, T. et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21 (2019).
Choudhary, S. & Satija, R. Comparison and evaluation of statistical error models for scRNA-seq. Genome Biol 23, 27 (2022).
Phipson, B. et al. propeller: testing for differences in cell type proportions in single cell data. Bioinformatics 38, 4720–4726 (2022).
Squair, J. W. et al. Confronting false discoveries in single-cell differential expression. Nat. Commun. 12, 5692 (2021).
Blighe, K., Rana, S. & Lewis, M. EnhancedVolcano: publication-ready volcano plots with enhanced colouring and labeling, (2018).
Yu, G., Wang, L.-G., Han, Y. & He, Q.-Y. clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 16, 284–287 (2012).
Cruz-Sanchez, A. et al. Developmental onset distinguishes three types of spontaneous recognition memory in mice. Sci. Rep. 10, 10612 (2020).
Acknowledgements
We thank Dr. M. Arruda-Carvalho (U. Toronto) for 3D printer files for TOR objects. This work is supported by the National Institute of Neurological Disorders and Stroke (NINDS) grant (R01NS112389) and the National Institute of Mental Health (NIMH) grant (R21MH132078) to G.M.S., the National Institute on Deafness and other Communication Disorders (NIDCD) grants (R01DC018797, R01DC019371) to J.H.K, the National Institute of Mental Health (NIMH) grant (R01MH053851) to D.A.M., and the NIMH grant (R21MH113899) to F.R.C. Imaging was performed at UTHSA Optical Imaging Facility, which is supported by a grant from the NCI (P30CA54174). FACS was performed at the Flow Cytometry Shared Resource at UTHSA, which is supported by an NCI grant (P30CA054174) to the Mays Cancer Center, a Cancer Prevention and Research Institute of Texas (CPRIT) grant (RP210126), a National Institutes of Health (NIH) Shared Instrument grant (S10OD030432), and support from the Office of the Vice President for Research at UTHSA. RNA sequencing was performed at the UTHSA Genome Sequencing Facility, which is supported by an NCI grant (P30CA054174), an NIH Shared Instrument grant (S10OD030311), and a CPRIT core facility award (RP220662). Q.C. is supported by a Natural Science Foundation of China grant (No. 32200778), a Natural Science Foundation of Jiangsu Province grant (No. BK20220494), a Suzhou Medical and Health Technology Innovation Project grant (No. SKY2022107), a Clinical Research Center of Neurological Disease in The Second Affiliated Hospital of Soochow University grant (No. ND2022A04), and a State Key Laboratory of Drug Research grant (No. SKLDR-2023-KF-05). L.S. is supported by a Postgraduate Research & Practice Innovation Program of Jiangsu Province grant (No. KYCX24_3335).
Author information
Authors and Affiliations
Contributions
G.M.S. and B.M.S. designed the study. H.T. and B.M.S. carried out most of the anatomical and behavioral experiments. H.T.,H.C., and J.G. assisted with anatomical and behavioral experiments. M.W. and J.H.K. assisted with neuronal functional experiments. J.B.L. assisted with scRNA-seq analysis. D.A.M. and F.R.C. assisted with the design of the stress protocols. L.S. and Q.C. assisted with manuscript revision experiments. G.M.S. supervised the project. G.M.S., H.T. and B.M.S. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tillmon, H., Soteros, B.M., Shen, L. et al. Complement and microglia activation mediate stress-induced synapse loss in layer 2/3 of the medial prefrontal cortex in male mice. Nat Commun 15, 9803 (2024). https://doi.org/10.1038/s41467-024-54007-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-54007-5
This article is cited by
-
Regulatory T cells promote microglia-mediated synapse engulfment and functional recovery via the OPN-CD74 axis after spinal cord injury in mice
Journal of Neuroinflammation (2025)
-
NADPH alleviates LPS-induced neuropathology and depression-like behaviors by suppressing microglial inflammatory response
Translational Psychiatry (2025)
-
Opposing roles of microglial and macrophagic C3ar1 signaling in stress-induced synaptic and behavioral changes
Molecular Psychiatry (2025)
-
Tactile stimulation reverses painful stimuli outcomes via PVN-VTA oxytocin circuitry and dopaminergic regulation
Communications Biology (2025)
