
Abstract
Colloidal properties of nanoparticles are intricately linked to their morphology. Traditionally, achieving high-concentration dispersions of two-dimensional (2D) nanosheets has proven challenging as they tend to agglomerate or re-stack under increased surface contact and Van der Waals attraction. Here, we unveil an excluded volume effect enabled by 2D morphology, which can be coupled with electrostatic repulsion to synthesize high-concentration aqueous graphene dispersions. To achieve this, we designed a sequential process involving edge oxidation, bubble expansion and mechanical shearing, through which graphite flakes were exfoliated into aqueous dispersions with ~94.5 wt.% yield of few-layer graphene, high concentration exceeding 100 mg mL-1, long-term stability over ~550 days, and large-scale wet processability. Structural analysis and theoretical modeling suggested that the 2D morphology of the resultant graphene nanosheets facilitates inter-sheet repulsive excluded volume interactions, leading to a fractal jammed network structure composed of nanosheets and tactoids to prevent their agglomeration. This effect was further leveraged in a continuous stirred tank reactor for the pilot-scale production of concentrated graphene dispersions. Our study unveils the role of excluded volume effect in stabilizing 2D-material colloids for industrial production and processing.
Similar content being viewed by others
Introduction
Graphene and related two-dimensional (2D) materials with extraordinary physical and chemical properties hold great promise for a myriad of applications including electronics, energy, composites, and more1,2. After nearly two decades of research and development, transitioning these materials from research to industry has been receiving growing attention3,4. For large-scale practical applications, 2D materials are typically used in a solid dry form, yet the majority of their processing and manufacturing occurs in a dispersed state5,6. A crucial step of the commercialization of 2D materials is the scalable synthesis of high-concentration dispersions7,8,9,10, which can reduce the volume of liquids required in the material production and storage, saving energy and cost10. Moreover, a high concentration of 2D materials allows the utilization of their rheological properties including viscosity and shear-thinning behavior11,12, for their fabrication into desired forms (e.g., coatings13, films11, and fibers14) through various wet processing techniques.
Graphene can be produced in liquids through a top-down approach, involving the exfoliation of graphite into individual graphene nanosheets using mechanical15,16, chemical17,18, or electrochemical techniques5. However, the strong Van der Waals attraction between the exfoliated graphene nanosheets often leads to their reassembly into graphitic multilayers17,18. To address this issue, electrostatic repulsion has been widely employed by altering the properties of the dispersing medium (e.g., pH value18) or by functionalizing graphene with charged groups or molecules18,19,20. However, the concentration of graphene dispersions achieved using this method is often limited to below 5 mg mL-1 due to the increased likelihood of intersheet collisions at higher particle concentrations, reducing the effectiveness of electrostatic repulsion. On the other hand, steric stabilization methods have also been developed to physically prevent graphene agglomeration, involving the adsorption of bulky groups such as organic solvents7, ionic liquids8, or polymers9 onto the surface of graphene nanosheets. Although the latter method can achieve exfoliation and stabilization of graphene dispersions at high concentrations ranging from 30 to 100 mg mL-17,8,9, the use of toxic or expensive organic solvents, as well as foreign stabilizers, is undesirable for many applications. So far, achieving direct exfoliation of graphite into high-concentration graphene dispersions in a green and cost-effective medium, particularly in water, still remains a significant challenge.
Apart from the conventional enthalpic interactions (e.g., electrostatic, steric, and van der Waals interactions), the entropic interactions arising from the confinement or restriction of the motion of colloidal particles can also be critical to the overall stability and structure of colloidal systems21,22,23. As a type of entropic interaction, the excluded volume effect occurs because particles cannot simultaneously occupy the same space due to their finite size24,25. In concentrated suspensions, the excluded volume effect could be more pronounced as reduced particle distance would increase the likelihood of overlapping of their excluded volumes. As highlighted by previous theoretical studies, anisotropic particles have a larger excluded volume compared to isotropic particles26,27. Graphene, an atomic-thin nanosheet with high anisotropy, is thus expected to have a significant excluded volume. Research has shown that the excluded volume effect plays a crucial role in the phase behavior of graphene oxide dispersions, such as promoting the formation of liquid crystal phases in graphene oxide14,28,29 and maintaining the orientation of graphene oxide liquid crystals upon shearing30. However, it is still unclear whether and how the excluded volume effect can be utilized to enhance the stability of graphene colloids at high concentration.
In this study, we design a synthetic route that capitalizes on the excluded volume effect of 2D nanosheets to achieve the exfoliation of graphite into high-concentration aqueous dispersions of few-layer graphene. The exfoliation process is depicted in Fig. 1, involving the introduction of oxygen groups primarily at the edges of graphite flakes through chemical treatment and the expansion of graphitic layers using gas bubbles. The pre-treated graphite, serving as a precursor for mechanical exfoliation, yielded aqueous graphene dispersions with ~98 wt.% yield and high concentration exceeding 100 mg mL-1. It is crucial to emphasize that while chemical oxidation, bubble expansion, or mechanical shearing has been previously employed for graphene preparation either individually or in combination10,31,32,33,34,35,36,37,38,39,40, they typically resulted in low-concentration dispersions or high-concentration agglomerations in water medium. Through the sequential use of each technique, we successfully manipulate the aggregation structure of graphene nanosheets to avoid restacking, leading to high-concentration aqueous dispersions with exceptional stability lasting over years. This is attributed to the strong excluded volume repulsion effect intrinsic to 2D morphology, which, in conjunction with traditional electrostatic repulsion, results in jammed graphene network composed of nanosheets and tactoids to prevent their restacking. This work highlights the great potential of the entropic excluded volume of 2D nanostructures in stabilizing concentrated graphene colloids for large-scale production and wet processing.

The exfoliation process contains chemical pre-treatment (edge oxidization and bubble expansion) and mechanical shearing. In the resulting concentrated graphene dispersions, graphene tactoids are surrounded by the exfoliated nanosheets, forming a disordered jammed network structure, where the entropic excluded volume repulsion (marked by the ellipsoid region) couples with the enthalpic electrostatic repulsion to co-stabilize the graphene dispersions.
Results
Chemical pre-treatment of graphite
The raw graphite used had a main flake size of 4~16 μm (Supplementary Fig. 1). In the first step, we introduced oxygen groups to the edges of graphite flakes by reducing the oxidant usage. The mass ratio of potassium permanganate (KMnO4) to graphite was controlled at 1:1, significantly lower than the traditional Hummers’ method ratio (3:1)41, resulting in a low-level oxidation with a C/O atomic ratio of 5~6 (Supplementary Fig. 2 and Supplementary Table 1). Raman mapping spectra of oxidized graphite (OG) flakes (Fig. 2a) showed that the intensity ratio (ID/IG) of the D peak to G peak decreased from ~0.9 at the edge to ~0.2 in the interior (Fig. 2b), indicating that oxidative defects are mainly at the flake edge42,43. Conversely, at a 3:1 ratio of KMnO4 to graphite, ID/IG ratios remained above 1 at all sites (Supplementary Fig. 3), suggesting a homogeneous distribution of oxygen defects.

a, b Raman mapping of graphite upon chemical oxidization. The mass ratio of KMnO4 to graphite is 1:1 in a and 3:1 in b, respectively. The scale bar is 10 μm in a. c, d Scanning electron microscope (SEM) image of the OG. The scale bar is 5 μm. e X-ray diffraction (XRD) spectra of Oxide graphite (OG), bubble-expanded graphite (BEG) and non-bubble-expanded graphite (NBEG) after different reaction time. NBEG samples were prepared by immersing OG in phosphoric acid without the presence of Na2CO3 to avoid bubbling. The marked crystallization peaks corresponding to H2SO4-intercalated graphite (*, 22.3° for 002 plane)45,46, KMnO4-oxidized graphite (♦, 12.9° and 23.1°)45,46 and unreacted graphite domains (•, 26.4° for 002 plane).
In the second step, we mixed OG and sodium carbonate (Na2CO3), which were then added to concentrated phosphoric acid (H3PO4) to generate CO2 gas (Supplementary Fig. 4). Continuous emission of CO2 bubbles was observed for over three hours (Supplementary Fig. 5), which can be attributed to the high viscosity of the reaction system and the encapsulation of Na2CO3 particles by OG, reducing the reaction rate between Na2CO3 and the acid44. The generated bubbles also caused OG to float during the washing process. After bubbling treatment, the edges of the flakes exhibited cracking and expansion, with ripples appearing on their surfaces (Fig. 2c, d and Supplementary Fig. 6). Comparing the X-ray diffraction (XRD) curves of OG (Fig. 2e top), bubble-expanded graphite (BEG, Fig. 2e middle), and non-bubble-expanded graphite (NEBG, Fig. 2e bottom) revealed a progressive distortion in the crystallization regions of OG induced by the bubbling treatment. The OG samples show a diffraction pattern with multiple crystallization peaks (Fig. 2e top), which could be attributed to H2SO4-intercalated graphite (*, 22.3° for 002 plane)45,46, KMnO4-oxidized graphite (♦, 12.9° and 23.1°)45,46 and unreacted graphite domains (•, 26.4° for 002 plane). After 15 min bubbling treatment, all the above crystallization peaks no longer exist. Instead, a broad peak in the 15°~30° region appears with a new sharp peak at 23.8° superimposed on top (Fig. 2e middle). This sharp peak gradually attenuates and finally emerges into the broad peak after 305 mins bubbling, indicating the formation of an amorphous phase. In striking contrast, if no Na2CO3 was added to avoid bubbling, the crystallization peaks at 11.6°, 23.8° and 26.4° related with oxidized or unreacted graphite were always observable (Fig. 2e bottom).
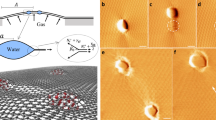
Molecular dynamics simulations further unveiled that the presence of oxygen groups on the graphene nanoslits, specifically the -COOH edge and -OH/-C-O-C- interior, lowered the energy barrier for HCO3- ions (protonated CO32-) to enter the nanoslits in acid solution environment (Fig. 3 and Supplementary Table 2), making it drop below the threshold of thermal fluctuation (<3 kcal mol-1, Fig. 3b). Consequently, HCO3- ions were observed to spontaneously enter the graphene nanoslits (Fig. 3a right and 3c). The reduction in barrier was attributed to hydrogen-bond interactions between HCO3- ions and the oxygen groups of the nanoslits (Fig. 3a right and Supplementary Fig. 7), compensating for the dehydration energy of HCO3- ions, thereby facilitating their entry and reaction with H+ ions to generate bubbles within the nanoslits. Given the high internal pressure of the confined bubbles, their rupture would release significant energy to expand the oxidized graphite and reduce the interlayer binding energy, making BEG a suitable precursor for mechanical exfoliation47,48.

a Molecular dynamics (MD) simulation model (left) and snapshoot (right) for HCO3- ions entering the graphene nanoslits decorated with -COOH edge and -OH/-C-O-C- interior. The red and white spheres indicate O and H atoms, respectively. The grey balls indicate C atoms in graphene nanoslits. The purple and blue balls indicate C and O atoms in HCO3- ions. b The potential of mean force (PMF) curves of HCO3- ions when entering the graphene nanoslits. The position of x = 0 is defined as the position of the C atom on the edge. The error bars in b represent the standard deviation of the mean. c The number (N) of HCO3- ions coordinated with the oxygen groups of graphene nanoslits as the modeling time increases.
Mechanical exfoliation of high-concentration graphene dispersions
The as-pretreated graphite was subjected to high-rate mechanical shearing in weakly alkaline water (pH = 9~10, Supplementary Fig. 8), which is chosen to ionize the oxygen groups to provide electrostatic repulsion between graphene nanosheets18. When the mass concentration of the graphite was 60 mg mL-1, concentrated graphene dispersions were obtained with a yield of ~98 wt.% (Fig. 4a, left). Dilute graphene dispersions exhibited the Tyndall effect, indicating the dispersed state of exfoliated graphene nanosheets (Fig. 4a, right). The chemical pre-treatment of the raw graphite is pivotal for achieving a high yield. Without chemical oxidization or bubble expansion, the exfoliation yield decreased to ~3 wt.% and ~35 wt.%, respectively (Fig. 4b). Notably, methylene blue (MB) adsorption tests revealed a significant increase in adsorption rate and amount of the concentrated graphene dispersions compared with OG and BEG (Fig. 4c). The high adsorption amount (420 mg g-1) of the resultant graphene dispersions indicated a specific surface area of 800~1100 m2 g-1, which corresponds to 1/2~1/3 of the theoretical specific surface area of monolayer graphene (~2630 m2 g-1), confirming effective exfoliation of graphite into few-layer graphene after high-rate shearing (Supplementary Fig. 9).

a) Photographs of shear-exfoliated graphene dispersions (~60 mg mL−1) and Tyndall effect of dilute graphene dispersion. b The yield of graphene prepared under different conditions. c Adsorption amount and adsorption rate of Methyl blue (MB) from OG, BEG and concentrated graphene dispersions. d X-ray photoelectron spectroscopy (XPS) C 1 s spectrum and e Scanning electron microscope (SEM) image of freeze-dried graphene dispersions. f Atomic force microscope (AFM) image and g the thickness and size distribution of graphene nanosheets. h Transmission electron microscopy (TEM) image and i High-resolution TEM images (left) and selected-area electron diffraction images (right) of graphene nanosheets. The scale bars are 1 μm in e and f, 100 nm in h, and 5 nm in i (left) and 5 nm−1 in i (right).
The chemical structure and morphology of the graphene nanosheets were investigated. The C 1s XPS spectrum of freeze-dried graphene dispersions shows a strong C=C peak at 284.7 eV, a weak peak from C–O bonds at 286.3 eV, and a long tail from O–C=O bonds at 290.5 eV (Fig. 4d). The C/O atomic ratio is 5-6, with oxygen groups primarily distributed along the nanosheet edges (Supplementary Fig. 10), similar to those in the pre-treated graphite precursor. SEM images of freeze-dried graphene dispersions depict a three-dimensional interconnected network structure composed of thin graphene nanosheets (Fig. 4e). Atomic force microscopy (AFM) histograms of graphene nanosheets on mica indicate a thickness of ~0.8 nm (Fig. 4f), which is close to that of monolayer reduced graphene oxide. Statistical analysis of over 100 nanosheets reveals that ~98% of graphene sheets are 1-2 layers thick (39% monolayer, 59% bi-layer), and the dominant lateral size is 1–3 μm (Fig. 4g and Supplementary Fig. 11). Transmission electron microscopy (TEM) images of graphene nanosheets exhibit edge-crumpled topography and a thickness of 1–3 layers (Fig. 4h, i and Supplementary Fig. 12). The selected-area electron diffraction images of graphene nanosheets show distinct bright hexagons (Fig. 4i), indicating the presence of 1–2 layered crystalline graphene nanosheets. The rotation angle between two hexagons is less than 10°, suggesting the disruption of the graphitic structure caused by bubble expansion.
The preparation process of graphene was further investigated under various experimental conditions. Using graphite flakes of larger lateral sizes (e.g., 400–550 μm, 90–150 μm, and 25-55 μm) as feedstock (Supplementary Fig. 13), the lateral sizes of the resulting graphene nanosheets remained below 2 μm (Supplementary Fig. 14), likely due to the high shearing rate that fragments the graphite flakes into smaller pieces. In this work, a high shearing rate was applied to induce turbulent flow (with a Reynolds number of ~1.5 × 104 for the system15), generating strong inertial forces to effectively exfoliate the oxidized graphite. Additionally, AFM measurements indicated that over 95% of the graphene nanosheets were well exfoliated into few layers (≤3, Supplementary Fig. 15), consistent with results obtained from 4-16 μm graphite flakes. Furthermore, when the ratio of KMnO4 to graphite was varied from 1:1 to 0.4:1 to 4:1, the lateral sizes of the graphene nanosheets continued to remain below 2 μm, with over 95% still exhibiting a thickness of less than 2 nm (Supplementary Fig. 16), despite variations in the amount of oxygen defects introduced. With reduced usage of KMnO4, the yield of graphene also decreased from ~94.5% to ~40% (Supplementary Fig. 17).
Stability and processability of graphene dispersions
We further examined the colloidal properties of the concentrated graphene dispersions. When graphene dispersions with a concentration of 60 mg mL-1 were sealed in glass vials and left undisturbed for 550 days, their concentration remained nearly unchanged among the storage period (Fig. 5a). The primary particle size, as measured by light scattering, also remained consistent at ~300 nm (Fig. 5b). Notably, the XRD curves of the graphene dispersions did not show the presence of the (002) peak at 26.5 degrees corresponding to the graphitic structure, while a broad hump extended to higher angles (Fig. 5c), which is typical of scattering from liquid water49, indicating the absence of graphene nanosheet re-stacking. These results imply that the high-concentration graphene dispersions are remarkably stable.

a The concentration change of 60 mg mL−1 graphene dispersions during storage. b The size distribution of the dilute graphene dispersions (0.1 mg mL−1) before and after 550 days’ storage of concentrated graphene dispersions (60 mg mL−1). c The XRD curves of 60 mg mL−1 graphene dispersions after different storage times. d The viscosity curves of concentrated graphene dispersions in the shear rate window of 10-1~102 s-1. e The changes of storage modulus and loss modulus of 60 mg mL-1 graphene dispersions upon constant shearing. In the shaded regions, the constant shearing (10 s-1 for 10 s and 50 s-1 for 10 s) is applied. In the white regions, the small oscillatory strain of 0.1% is used to monitor the modulus recovery. f Photograph of graphene films. The scale bar is 5 cm. g Cyclic voltammetry curves of graphene films-based supercapacitors under different scan rates. h Volumetric capacitance (black points) of graphene films-based supercapacitor electrode in 1 M H2SO4 at different charging rates, the comparison of volumetric capacitance at 1 A g-1 and 5 A g-1 current density with those of previously reported50,51,52,53,54,55,56,57,58,59,60,61,62. Red and blue points represent the previously reported volumetric capacitance values at 1 A g-1 and 5 A g−1, respectively. i Electrical conductivity and thermal conductivity of graphene films after 1000 °C annealing, and comparison with those of previously reported graphene films after 2000–3000 °C annealing63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82. The error bars in h and i represent the standard deviation of the mean.
We then investigated the rheological properties of concentrated graphene dispersions to explore their potential for wet processing11. In steady shearing tests, the viscosity of graphene dispersions at different concentrations exhibited a significant reduction within the tested shearing range (10-1~102 s-1, Fig. 4d) due to the motion of graphene nanosheets under shearing. In dynamic shearing tests, the storage modulus of graphene dispersions decreased compared to the loss modulus during continuous shearing, but quickly recovered to a value higher than the loss modulus within 1–2 seconds after shearing ceased (Fig. 5e). This behavior can be attributed to the rapid reconstruction of the broken graphene network, as observed in graphene oxide-based hydrogels11. The shear-thinning behavior and fast structural recovery of graphene dispersions can facilitate their flow and enable them to retain their shape after the cessation of the shearing, making them suitable inks for electromechanical sensing (Supplementary Figs. 18, 19) and various wet processing techniques including casting and printing11.
To exemplify their wet processability, we explored the fabrication of graphene dispersions into films. Through the blade casting and drying of the graphene dispersions (Supplementary Fig. 20), we successfully obtained a large-area graphene film, as depicted in Fig. 5f. XRD measurements unveiled the presence of both graphitic regions and sub-nanometer porous regions within the films (Supplementary Fig. 21), prompting further investigation for their potential as supercapacitor electrode50. The symmetric supercapacitor constructed using the graphene films exhibits electrical double-layer ion adsorption feature (Fig. 5g). The supercapacitor electrode (1.39 g cm-3 density and 3.16 g cm-2 areal mass) demonstrates a volumetric capacitance of ~400 F cm-3 at a charging rate of 0.2 A g-1 and 355 F cm-3 at a charging rate of 5 A g-1 in H2SO4 electrolyte (Fig. 5h and Supplementary Fig. 22). These values are highly competitive compared to other carbon-based materials used in electrical double-layer supercapacitors operating in aqueous electrolytes (see the scattering dots in Fig. 4h and the values in Supplementary Table 3)50,51,52,53,54,55,56,57,58,59,60,61,62. The exceptional volumetric capacitive performance can be attributed to the unique pore structure of graphene films enabling mixed electron/ion transport, coupled with good wetting properties to acid electrolytes and a minimal contribution of oxygen-related pseudo-capacitance (Supplementary Note 3 and Supplementary Fig. 23-24).
We also explored the further thermal annealing of the graphene films. Without thermal annealing, the electrical conductivity and thermal conductivity of the graphene films (20 μm thickness) were measured at 1.28\(\pm\)0.13 × 104 S m-1 and 56.24\(\pm\)12.41 W m-1 K-1 (Fig. 5i), respectively. Subjecting the graphene films to 1000 °C thermal annealing resulted in a significant increase in electrical conductivity to 1.89 ± 0.28×105 S m-1 and thermal conductivity to 635.02 ± 174.81 W m-1 K-1 (Fig. 5i). In previous studies, achieving such high levels of conductivity in the graphene films typically necessitates much higher thermal annealing temperatures (i.e., 2000–3000 °C, Fig. 4i and Supplementary Table 4)63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82. The XRD characterization shows that our graphene films are prone to graphitization upon both 1000 and 2800 °C annealing (Supplementary Fig. 25). The capability to accomplish graphitization at lower temperatures may be ascribed to the more accessible elimination of oxygen defects at the edges of graphene nanosheets in comparison to those in proximity to the centers, and is anticipated to substantially diminish the energy consumption.
The stability mechanism of graphene dispersions
To understand the colloidal stability of the concentrated graphene dispersions, we examined the packing structure of graphene nanosheets. Rheological test of graphene dispersions reveal a power-law relationship (Fig. 6a) of their storage modulus (G') and the critical yield strain (γc) with the mass concentrations (ω), suggesting the presence of fractal graphene network structure with a mass fractal dimension (df) of 1.72 (see details in Supplementary Fig. 26). This value is smaller than the reported ones (2~2.3) for graphene suspersions83,84, indicating the loose stacking of graphene nanosheets. Meanwhile, polarized optical microscopy images of the dispersions reveal the existence of microdomains with parallel-aligned nanosheets (Supplementary Fig. 27a–d), but the overall orientation of graphene nanosheets is isotropic, with a low orientation degree (<0.15) measured by small-angle X-ray scattering (SAXS, Supplementary Fig. 27e, f). Based on these results, we propose the packing structure depicted in Fig. 6b, where graphene tactoids are surrounded by exfoliated nanosheets, forming a disordered jammed network.

a Storage modulus and yield strain of graphene dispersions plotted versus the mass concentration. The storage modulus (G’ ) is a measure of a concentrated graphene dispersions’s ability to store elastic energy when it is subjected to deformation. Critical yield strain (γ) is the amount of strain (deformation) a concentrated graphene dispersions can withstand before it begins to deform plastically. The red dashed line represents the linear fit of the data points for critical yield strain versus mass concentration on a log-log scale. The blue dashed line represents the linear fit of the data points for storage modulus versus mass concentration on a log-log scale. b Schematic diagram of packing structure of graphene nanosheets in the dispersions. c The intensity (I(q)) of scattered X-rays as a function of the scattering vector (q) for graphene dispersions with different mass concentrations during SAXS (small-angle X-ray scattering) measurement. The black solid lines represent the linear fit of the data points on a log-log scale. d Orientation order degree of graphene dispersions at different concentrations for tactoids containing single-layer and eight-layer graphene. The inset is the 2D SAXS image of graphene dispersions (120 mg mL−1). The scale bar in inset image is 0.5 nm−1.
The SAXS analysis also reveals the morphology of graphene nanosheets in the dispersions. The scattering curves (I∼q-a) for graphene dispersions with different concentrations of 30-120 mg mL-1 exhibit a similar slope (-a) of 3.40–3.46 in the log-log scale within the q range of 0.005 to 0.1 Å-1 (Fig. 6c). These slopes fall within the 3–4 range, suggesting the formation of a surface fractal structure with a fractal dimension (ds = 6-a) of 2.54–2.6085. The uniform ds indicates a consistent surface morphology of graphene nanosheets across different dispersed concentrations. This implies that the graphene nanosheets in the dispersions exhibit minimal surface corrugation and no discernible agglomeration even at high concentrations, supporting their observed stability.
We further examined whether the proposed colloidal structure for the concentrated graphene dispersions could be thermodynamically stable. Using Onsager theory86, we calculated the normalized orientation distribution and orientation factor of the graphene dispersions (Fig. 6d and Supplementary Fig. 28). If tactoids are assumed not to exist (i.e., with a single layer of graphene nanosheet), the orientation factor increases rapidly with concentration, approaching close to 1 after a critical concentration. This suggests that the phase must be ordered within the concentration range we studied. Thus, the formation of an isotropic glassy phase is physically impossible at high concentrations if all graphene nanosheets are individual. When the tactoids are assumed with 8 graphene layers, the orientation could be isotropic even at high concentration of 120 mg mL-1. The calculated orientation factor is 0.12, which is consistent with that measured by SAXS. That is, the formation of thermodynamically stable isotropic phase is physically possible as observed in our experimental results. Similar isotropic glassy phases with locally ordered structures have been reported in highly concentrated laponite systems87.
We have integrated colloidal experiments with the modified DLVO model previously published to shed light on the stability mechanism of the concentrated graphene dispersions28. The excluded volume effect associated with the 2D morphology of graphene nanosheets is expected to generate additional repulsive inter-sheet interactions that supplement the electrostatic repulsion ((Supplementary Note 1 and Supplementary Figs. 29, 30), preventing the parallel-aligned nanosheets within the tactoids from approaching each other. Additionally, the formation of the jammed network can further enhance the kinetic stability of concentrated dispersions by sterically hindering the movement of graphene nanosheets (Supplementary Fig. 31). Overall, a combination of medium lateral size (~2.0 μm), small thickness (less than 3 nm), and a low zeta potential (less than −25 mV) are beneficial for high-concentrated dispersion and good stability of graphene nanosheets in water ((Supplementary Note 2 and Supplementary Figs. 32, 33).
Scalable production of concentrated graphene dispersions
To explore scalable production of concentrated graphene dispersions, we integrated multiple continuous stirred tank reactors arranged in series, enabling the continuous feeding, mixing, and shearing of graphite flakes and chemical reactants (Fig. 7a). The inset of Fig. 7a shows a batch of concentrated graphene dispersions (100 mg mL-1) produced from the pilot line. Raman spectra consistently exhibited an ID/IG intensity ratio within 0.17~0.27 (Fig. 7b and Supplementary Fig. 34), indicating high-quality graphene production and batch-to-batch reproducibility. Figure 7c underscores the productivity features of our approach compared to other industrial routes for graphene production in water, including chemical exfoliation, ultrasonication, mechanical exfoliation, and various combinations of these, across key metrics such as yield, throughput, concentration and dispersibility10,31,32,33,34,35,36,37,38,39,40. Impressively, our graphene production approach achieved an ultrahigh throughput of up to 300 g h-1, far surpassing values (<100 g h-1) reported in previous studies of graphene production (Fig. 7c and Supplementary Table 5)7,8,9.

a Illustrated diagram of the workflow for the large-scale production process. The right inset shows photographs of the concentrated graphene dispersions. b ID/IG peak intensity ratio of graphene dispersions produced from different batches. Data are presented as box plots (center line at the median, upper bound at 75th percentile, lower bound at 25th percentile) with whiskers at minimum and maximum values. Each dot represents one ID/IG value. c Overall comparison of the concentration, throughput and yield of graphene produced from this work and previous studies10,31,32,33,34,35,36,37,38,39,40. The values can be found in Supplementary Table 5. d Three key impact indicators and their components of producing 1 kg graphene in our route at industrial manufacture scale.
We conducted a life cycle assessment to evaluate the environmental impact of the entire production process (Supplementary Note 4 and Supplementary Tables 6, 7). We calculated three key impact indicators commonly used in life cycle assessments (Fig. 7d) We calculated three key impact indicators commonly used in life cycle assessments (Fig. 7d)88: cumulative energy demand (CED), global warming potential (GWP), and water consumption potential (WCP). Figure 6d illustrates the contributions to each indicator, with H3PO4 making the most significant impact due to its extensive use in our method. In the future, H3PO4 could be replaced with more environmentally friendly acids to enhance sustainability. Notably, the three impact indicators of our method are comparable to those of previous methods (CED: 816.3 vs 146-1879 MJ/kg; GWP: 65.8 vs 57-79 kg CO2; WCP: 3.6 vs. 4.8-13 m3, Supplementary Table 8), suggesting low environmental obstacles to the industrial-scale production of concentrated graphene dispersions89,90,91,92.
Discussion
In this work, we have developed a water-based exfoliation strategy to produce high-concentrated few-layer graphene dispersions with colloidal stability and wet processability. Traditionally, achieving high graphene concentrations necessitated the use of non-aqueous solvents or the addition of aqueous dispersants, relying on enthalpic interactions such as electrostatic and steric repulsion to stabilize graphene colloids. However, our approach circumvents these methods by harnessing the inherent excluded volume repulsion of 2D structures to stabilize the exfoliated graphene nanosheets. Importantly, we have showcased how entropic excluded volume repulsion can complement enthalpic electrostatic repulsion, enabling the simultaneous achievement of pilot-scale production and processing of graphene in a water medium without any additives. This has the potential to streamline the cost and process of graphene-based applications, aligning with the persistent pursuit of sustainable technologies in graphene manufacturing. Given that the substantial excluded volume effect originates from the 2D nanosheet morphology, the uncovered entropic stabilization mechanism is expected to have a far-reaching impact on the industrial production and processing of other 2D materials.
Methods
Pre-treatment of graphite
Graphite with different flake sizes (i.e., 4–16 μm, 25–55 μm, 90–150 μm and 400–550 μm) was treated by oxidization and bubbling. Firstly, raw graphite (30 g, 200 mesh) was added to the mixture of KMnO4 (30 g) and sulfuric acid (180 mL) under stirring in the ice bath. The stirring was kept under ambient conditions for 1 h. Secondly, Na2CO3 (30 g) was added to the above mixture under mechanical stirring. With the gradual addition of Na2CO3, the system changed from a viscous state to a semi-solid granular state. H3PO4 (420 mL) was added into the above-mixed system and mechanically stirred for another 3 h. The resultant slurry was filtered through a 200-mesh stainless steel sieve to collect the black solid products, which were further washed by a large amount of ice-water mixture several times and were used as the precursor for mechanical exfoliation.
Preparation of concentrated graphene dispersions
A certain amount of the above-treated graphite was mixed with ammonia water solution (pH = 10, 750 mL). Subsequently, the mixture was exfoliated under 15,000 rpm shearing for 1 h using a high-speed homogeneous disperser (Fluko FA40). The mixing head with a four-blade rotor fixed on the stator (rotor/stator gap is ~100 µm) was used to ensure adequate shearing. The temperature was kept at room temperature by using circulated cooling water during the whole shearing process. The measurement of the yield of graphene was according to previous work to facilitate the comparison10. A small portion of the exfoliated products were centrifugated under 342 g for 10 min and were re-dispersed in N-Methyl-2-pyrrolidone (NMP) through 5 min sonication. The resultant graphene dispersions were centrifuged at 62 g for 20 min to remove the unexfoliated graphite particles. The supernatant was collected and the sediment was re-dispersed in NMP and further centrifuged until the supernatant was colorless. The collected upper graphene dispersions were centrifuged at 466 g for 1 h to remove the solvent and dried in a vacuum oven at 60 °C for 48 h. The yield of graphene flakes was calculated by the mass of the dried graphene to the raw graphite. Thermogravimetric analysis was used to quantify the residual NMP and determine the mass of the dried graphene (Supplementary Fig. 35)93. The graphene mass in the NMP dispersion was also validated using UV-vis spectroscopy (Supplementary Fig. 36)93,94,95.
Data availability
The Source Data underlying the figures of this study are available with the paper. All raw data generated during the current study are available from the corresponding authors upon request. Source data are provided with this paper.
Code availability
The simulation codes used in this study are available from the corresponding authors on request.
References
Geim, A. K. & Novoselov, K. S. The rise of graphene. Nat. Mater. 6, 183–191 (2007).
Li, D. & Kaner, R. B. Materials science - Graphene-based materials. Science 320, 1170–1171 (2008).
Graphene on the pilot line. Nat. Mater. 20, 573 (2021).
Ren, W. C. & Cheng, H. M. The global growth of graphene. Nat. Nanotechnol. 9, 726–730 (2014).
Wang, S. et al. Electrochemical molecular intercalation and exfoliation of solution-processable two-dimensional crystals. Nat. Protoc. 18, 2814–2837 (2023).
Pinilla, S., Coelho, J., Li, K., Liu, J. & Nicolosi, V. Two-dimensional material inks. Nat. Rev. Mater. 7, 717–735 (2022).
Khan, U. et al. Solvent-exfoliated graphene at extremely high concentration. Langmuir 27, 9077–9082 (2011).
Matsumoto, M., Saito, Y., Park, C., Fukushima, T. & Aida, T. Ultrahigh-throughput exfoliation of graphite into pristine ‘single-layer’ graphene using microwaves and molecularly engineered ionic liquids. Nat. Chem. 7, 730–736 (2015).
Park, H. J. et al. Fluid-dynamics-processed highly stretchable, conductive, and printable graphene inks for real-time monitoring sweat during stretching exercise. Adv. Funct. Mater. 31, 2011059 (2021).
Dong, L. et al. A non-dispersion strategy for large-scale production of ultra-high concentration graphene slurries in water. Nat. Commun. 9, 76 (2018).
Xiong, Z. et al. A Dynamic graphene oxide network enables spray printing of colloidal gels for high‐performance micro‐supercapacitors. Adv. Mater. 31, 1804434 (2019).
Naficy, S. et al. Graphene oxide dispersions: tuning rheology to enable fabrication. Mater. Horiz. 1, 326–331 (2014).
Secor, E. B., Ahn, B. Y., Gao, T. Z., Lewis, J. A. & Hersam, M. C. Rapid and versatile photonic annealing of graphene inks for flexible printed electronics. Adv. Mater. 27, 6683–6688 (2015).
Xu, Z. & Gao, C. Graphene chiral liquid crystals and macroscopic assembled fibres. Nat. Commun. 2, 1–9 (2011).
Paton, K. R. et al. Scalable production of large quantities of defect-free few-layer graphene by shear exfoliation in liquids. Nat. Mater. 13, 624–630 (2014).
Nicolosi, V., Chhowalla, M., Kanatzidis, M. G., Strano, M. S. & Coleman, J. N. Liquid exfoliation of layered materials. Science 340, 1226419 (2013).
Tung, V. C., Allen, M. J., Yang, Y. & Kaner, R. B. High-throughput solution processing of large-scale graphene. Nat. Nanotechnol. 4, 25–29 (2009).
Li, D., Müller, M. B., Gilje, S., Kaner, R. B. & Wallace, G. G. Processable aqueous dispersions of graphene nanosheets. Nat. Nanotechnol. 3, 101–105 (2008).
Xu, Y., Bai, H., Lu, G., Li, C. & Shi, G. Flexible graphene films via the filtration of water-soluble noncovalent functionalized graphene sheets. J. Am. Chem. Soc. 130, 5856–5857 (2008).
Lotya, M. et al. Liquid phase production of graphene by exfoliation of graphite in surfactant/water solutions. J. Am. Chem. Soc. 131, 3611–3620 (2009).
Calvin, J. J., Brewer, A. S. & Alivisatos, A. P. The role of organic ligand shell structures in colloidal nanocrystal synthesis. Nat. Synth. 1, 127–137 (2022).
Je, K., Lee, S., Teich, E. G., Engel, M. & Glotzer, S. C. Entropic formation of a thermodynamically stable colloidal quasicrystal with negligible phason strain. Proc. Natl Acad. Sci. 118, e2011799118 (2021).
Lee, S., Vo, T. & Glotzer, S. C. Entropy compartmentalization stabilizes open host–guest colloidal clathrates. Nat. Chem. 15, 905–912 (2023).
Bolhuis, P. G., Louis, A. A. & Hansen, J. P. Influence of polymer-excluded volume on the phase-behavior of colloid-polymer mixtures. Phys. Rev. Lett. 89, 128302 (2002).
Redner, G. S., Hagan, M. F. & Baskaran, A. Structure and dynamics of a phase-separating active colloidal fluid. Phys. Rev. Lett. 110, 055701 (2013).
Saar, M. O. & Manga, M. Continuum percolation for randomly oriented soft-core prisms. Phys. Rev. E 65, 056131 (2002).
Petukhov, A. V., Tuinier, R. & Vroege, G. J. Entropic patchiness: Effects of colloid shape and depletion. Curr. Opin. Colloid Interface Sci. 30, 54–61 (2017).
Choi, G. M., Park, M., Jeong, S. Y. & Lee, H. S. Orientation effect on the rheology of graphene oxide dispersions in isotropic phase, ordered isotropic biphase, and discotic phase. J. Rheol. 65, 791–806 (2021).
Wen, Y. et al. Chemically modified graphene films with tunable negative Poisson’s ratios. Nat. Commun. 10, 2446 (2019).
Jiang, Y., Guo, F., Xu, Z., Gao, W. & Gao, C. Artificial colloidal liquid metacrystals by shearing microlithography. Nat. Commun. 10, 4111 (2019).
Tian, S. et al. Controllable edge oxidation and bubbling exfoliation enable the fabrication of high quality water dispersible graphene. Sci. Rep. 6, 34127 (2016).
Ding, J. H., Zhao, H. R. & Yu, H. B. A water-based green approach to large-scale production of aqueous compatible graphene nanoplatelets. Sci. Rep. 8, 5567 (2018).
Kovtyukhova, N. I. et al. Non-oxidative intercalation and exfoliation of graphite by Bronsted acids. Nat. Chem. 6, 957–963 (2014).
Hernandez, Y. et al. High-yield production of graphene by liquid-phase exfoliation of graphite. Nat. Nanotechnol. 3, 563–568 (2008).
Amiri, A. et al. Facile, environmentally friendly, cost effective and scalable production of few-layered graphene. Chem. Eng. J. 326, 1105–1115 (2017).
Dimiev, A. M., Ceriotti, G., Metzger, A., Kim, N. D. & Tour, J. M. Chemical mass production of graphene nanoplatelets in approximately 100% yield. ACS Nano 10, 274–279 (2016).
O’Neill, A., Khan, U., Nirmalraj, P. N., Boland, J. & Coleman, J. N. Graphene dispersion and exfoliation in low boiling point solvents. J. Phys. Chem. C. 115, 5422–5428 (2011).
Xu, S. et al. Reverse-Micelle-induced exfoliation of graphite into graphene nanosheets with assistance of supercritical CO2. Chem. Mater. 27, 3262–3272 (2015).
Zhang, P. et al. Delocalized electrochemical exfoliation toward high-throughput fabrication of high-quality graphene. Chem. Eng. J. 428, 131122 (2022).
Lotya, M., King, P. J., Khan, U., De, S. & Coleman, J. N. High-concentration, surfactant-stabilized graphene dispersions. ACS Nano 4, 3155–3162 (2010).
Hummers, W. S. Jr., & Offeman, R. E. Preparation of graphitic oxide. J. Am. Chem. Soc. 80, 1339 (1958).
An, X. et al. Stable aqueous dispersions of noncovalently functionalized graphene from graphite and their multifunctional high-performance applications. Nano Lett. 10, 4295–4301 (2010).
Morimoto, N. et al. Real-time, in situ monitoring of the oxidation of graphite: lessons learned. Chem. Mater. 29, 2150–2156 (2017).
Olea, A. F. & Thomas, J. Rate constants for reactions in viscous media: correlation between the viscosity of the solvent and the rate constant of the diffusion-controlled reactions. J. Am. Chem. Soc. 110, 4494–4502 (1988).
Dimiev, A. M. & Tour, J. M. Mechanism of graphene oxide formation. ACS nano 8, 3060–3068 (2014).
Storm, M. M., Johnsen, R. E. & Norby, P. In situ X-ray powder diffraction studies of the synthesis of graphene oxide and formation of reduced graphene oxide. J. Solid State Chem. 240, 49–54 (2016).
Shi, X., Xue, S., Marhaba, T. & Zhang, W. Probing internal pressures and long-term stability of nanobubbles in water. Langmuir 37, 2514–2522 (2021).
Zamborlini, G. et al. Nanobubbles at GPa pressure under graphene. Nano Lett. 15, 6162–6169 (2015).
Amann-Winkel, K. et al. X-ray and neutron scattering of water. Chem. Rev. 116, 7570–7589 (2016).
Díez, N. et al. High density graphene–carbon nanosphere films for capacitive energy storage. J. Mater. Chem. A 7, 6126–6133 (2019).
Zhang, L. et al. Steamed water engineering mechanically robust graphene films for high-performance electrochemical capacitive energy storage. Nano Energy 26, 668–676 (2016).
Li, Z. et al. Tuning the interlayer spacing of graphene laminate films for efficient pore utilization towards compact capacitive energy storage. Nat. Energy 5, 160–168 (2020).
Tao, Y. et al. Towards ultrahigh volumetric capacitance: graphene derived highly dense but porous carbons for supercapacitors. Sci. Rep. 3, 2975 (2013).
Ghaffari, M. et al. High-volumetric performance aligned nano-porous microwave exfoliated graphite oxide-based electrochemical capacitors. Adv. Mater. 25, 4879–4885 (2013).
Xu, Y. et al. Holey graphene frameworks for highly efficient capacitive energy storage. Nat. Commun. 5, 4554 (2014).
Jung, N. et al. Synthesis of chemically bonded graphene/carbon nanotube composites and their application in large volumetric capacitance supercapacitors. Adv. Mater. 25, 6854–6858 (2013).
Liu, Z. et al. A supercapacitor with ultrahigh volumetric capacitance produced by self-assembly of reduced graphene oxide through phosphoric acid treatment. J. Mater. Chem. A 8, 18933–18944 (2020).
Yan, J., Wang, Q., Lin, C., Wei, T. & Fan, Z. Interconnected frameworks with a sandwiched porous carbon layer/graphene hybrids for supercapacitors with high gravimetric and volumetric performances. Adv. Energy Mater. 4, 1400500 (2014).
Wu, Q. et al. Carbon-carbon dense network composite with hierarchical structure for additive-free and high volumetric performance supercapacitor. J. Power Sources 497, 229878 (2021).
Li, G. et al. Achieving ultrahigh volumetric energy storage by compressing nitrogen and sulfur dual-doped carbon nanocages via capillarity. Adv. Mater. 32, e2004632 (2020).
Liu, X. et al. Biomass-derived robust three-dimensional porous carbon for high volumetric performance supercapacitors. J. Power Sources 412, 1–9 (2019).
Long, C. L., Chen, X., Jiang, L. L., Zhi, L. J. & Fan, Z. J. Porous layer-stacking carbon derived from in-built template in biomass for high volumetric performance supercapacitors. Nano Energy 12, 141–151 (2015).
Shen, B., Zhai, W. & Zheng, W. Ultrathin flexible graphene film: an excellent thermal conducting material with efficient emi shielding. Adv. Funct. Mater. 24, 4542–4548 (2014).
Wang, N. et al. Tailoring the thermal and mechanical properties of graphene film by structural engineering. Small 14, e1801346 (2018).
Chen, S. et al. Scalable production of thick graphene film for next generation thermal management application. Carbon 167, 270–277 (2020).
Ding, J. et al. An ultrahigh thermal conductive graphene flexible paper. Nanoscale 9, 16871–16878 (2017).
Huang, Y. et al. Fabrication and molecular dynamics analyses of highly thermal conductive reduced graphene oxide films at ultra-high temperatures. Nanoscale 9, 2340–2347 (2017).
Zeng, Y. et al. Thermally conductive reduced graphene oxide thin films for extreme temperature sensors. Adv. Funct. Mater. 29, 1901388 (2019).
Kumar, P. et al. Large-area reduced graphene oxide thin film with excellent thermal conductivity and electromagnetic interference shielding effectiveness. Carbon 94, 494–500 (2015).
Ghosh, T. et al. Solution-processed graphite membrane from reassembled graphene oxide. Chem. Mater. 24, 594–599 (2012).
Li, Z., Xu, Z., Liu, Y., Wang, R. & Gao, C. Multifunctional non-woven fabrics of interfused graphene fibres. Nat. Commun. 7, 13684 (2016).
Liu, Z. et al. Wet-spun continuous graphene films. Chem. Mater. 26, 6786–6795 (2014).
Arshad, A., Jabbal, M., Yan, Y. Y. & Reay, D. A review on graphene based nanofluids: Preparation, characterization and applications. J. Mol. Liq. 279, 444–484 (2019).
Peng, L. et al. Ultrahigh thermal conductive yet superflexible graphene films. Adv. Mater. 29, 1700589 (2017).
Renteria, J. D. et al. Strongly anisotropic thermal conductivity of free-standing reduced graphene oxide films annealed at high Temperature. Adv. Funct. Mater. 25, 4664–4672 (2015).
Liu, Y. et al. Utilizing ammonium persulfate assisted expansion to fabricate flexible expanded graphite films with excellent thermal conductivity by introducing wrinkles. Carbon 153, 565–574 (2019).
Xin, G. et al. Large-area freestanding graphene paper for superior thermal management. Adv. Mater. 26, 4521–4526 (2014).
Karagiannidis, P. G. et al. Microfluidization of Graphite and Formulation of Graphene-Based Conductive Inks. ACS Nano 11, 2742–2755 (2017).
Park, S. et al. Colloidal suspensions of highly reduced graphene oxide in a wide variety of organic solvents. Nano Lett. 9, 1593–1597 (2009).
Song, N.-J. et al. Thermally reduced graphene oxide films as flexible lateral heat spreaders. J. Mater. Chem. A 2, 16563–16568 (2014).
Hou, Z. L. et al. Flexible graphene-graphene composites of superior thermal and electrical transport properties. ACS Appl. Mater. Interfaces 6, 15026–15032 (2014).
Wu, T., Xu, Y., Wang, H., Sun, Z. & Zou, L. Efficient and inexpensive preparation of graphene laminated film with ultrahigh thermal conductivity. Carbon 171, 639–645 (2021).
Poulin, P. et al. Superflexibility of graphene oxide. Proc. Natl Acad. Sci. 113, 11088–11093 (2016).
Barwich, S., Coleman, J. N. & Möbius, M. E. Yielding and flow of highly concentrated, few-layer graphene suspensions. Soft Matter 11, 3159–3164 (2015).
Bale, H. D. & Schmidt, P. W. Small-angle X-ray-scattering investigation of submicroscopic porosity with fractal properties. Phys. Rev. Lett. 53, 596–599 (1984).
Onsager, L. The effects of shape on the interaction of colloidal particles. Ann. N. Y. Acad. Sci. 51, 627–659 (1949).
Mourchid, A., Delville, A., Lambard, J., Lecolier, E. & Levitz, P. Phase diagram of colloidal dispersions of anisotropic charged particles: equilibrium properties, structure, and rheology of laponite suspensions. Langmuir 11, 1942–1950 (1995).
Li, X., Jin, H., Chan, Y., Guo, H. & Ma, W. Environmental impacts of graphene at industrial production scale and its application in electric heating technology. Resour. Conserv. Recycl. 199, 107250 (2023).
Arvidsson, R. Review of environmental life cycle assessment studies of graphene production. Adv. Mater. Lett. 8, 187–195 (2017).
Pizza, A., Metz, R., Hassanzadeh, M. & Bantignies, J.-L. Life cycle assessment of nanocomposites made of thermally conductive graphite nanoplatelets. Int. J. Life Cycle Assess. 19, 1226–1237 (2014).
Arvidsson, R., Kushnir, D., Sanden, B. A. & Molander, S. Prospective life cycle assessment of graphene production by ultrasonication and chemical reduction. Environ. Sci. Technol. 48, 4529–4536 (2014).
Cossutta, M., McKechnie, J. & Pickering, S. J. A comparative LCA of different graphene production routes. Green. Chem. 19, 5874–5884 (2017).
Backes, C. et al. Spectroscopic metrics allow in situ measurement of mean size and thickness of liquid-exfoliated few-layer graphene nanosheets. Nanoscale 8, 4311–4323 (2016).
Paton, K. R. & Coleman, J. N. Relating the optical absorption coefficient of nanosheet dispersions to the intrinsic monolayer absorption. Carbon 107, 733–738 (2016).
Khan, U., O’Neill, A., Lotya, M., De, S. & Coleman, J. N. High‐concentration solvent exfoliation of graphene. Small 6, 864–871 (2010).
Acknowledgements
This work was supported by the Natural Science Foundation of Guangdong Province (2024A1515030072, Z.X.), the Australian Research Council (FL180100029, D.L.), the National Natural Science Foundation of China (51773184 and U1810114, Y.S.; 12102324, K.Z.), the Shanxi Provincial Natural Science Foundation of China (201803D421081 and 20181102014, Y.S.). Zhiyuan Xiong thanks for the financial support from the South China University of Technology. Thank the helpful discussion with Dr. Yang Cao and Dr. Enlai Gao.
Author information
Authors and Affiliations
Contributions
Z.X. did the conceptualization. Z.X., L.S., Y.S., and D. Li. designed the experiments. L.S. conducted the preparation, characterization, and processing experiments of graphene. X.L. and W.M. conducted a life cycle assessment analysis. K.Z. performed molecular dynamics simulations. G.C. and H.L. completed the calculation of excluded volume-incorporated DLVO theory with the discussion from Z.X. J.L., K.O., and G.Y. conducted material characterization. Z.X., D. Li., L.S., Y.S., and H.L. co-wrote the manuscript with input from all authors. All authors have given approval to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Guqiao Ding, Lei Dong and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xiong, Z., Shen, L., Long, J. et al. Ultrahigh concentration exfoliation and aqueous dispersion of few-layer graphene by excluded volume effect. Nat Commun 15, 10807 (2024). https://doi.org/10.1038/s41467-024-55131-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-024-55131-y
This article is cited by
-
Carbon fibre production using an ecofriendly water-soluble precursor
Nature Communications (2025)
-
Low-temperature graphitization by amine-assisted combustion of graphite oxide
npj 2D Materials and Applications (2025)
