
Abstract
Long non-coding RNAs (lncRNAs) are pivotal regulators of cellular processes. Here we reveal an interaction between the lncRNA NORAD, noted for its role in DNA stability, and the immune related transcription factor STAT3 in embryonic and differentiated human cells. Results from NORAD knockdown experiments implicate NORAD in facilitating STAT3 nuclear localization and suppressing antiviral gene activation. In NORAD-deficient cells, STAT3 remains cytoplasmic, allowing STAT1 to enhance antiviral activity. Analysis of RNA expression data from in vitro experiments and clinical samples demonstrates reduced NORAD upon viral infection. Additionally, evolutionary conservation analysis suggests that this regulatory function of NORAD is restricted to humans, potentially owing to the introduction of an Alu element in hominoids. Our findings thus suggest that NORAD functions as a modulator of STAT3-mediated immune suppression, adding to the understanding of lncRNAs in immune regulation and evolutionary adaptation in host defense mechanisms.
Similar content being viewed by others


Introduction
In recent years, it has become apparent that RNA, specifically long non-coding RNAs (lncRNAs), can contribute to fine tune gene regulation pathways by diverse mechanisms1,2,3,4,5,6,7,8,9,10,11. Based on various studies it is evident that the functional unit of the lncRNA can be either its transcription process or the lncRNA itself12. In the latter case the lncRNA will often nucleate to condensates of ribonucleoprotein particles (RNPs), which enable efficient response to various cellular stresses, such as viral infections13. In a common scenario the transcription of a lncRNA regulates the expression of a nearby gene (positive or negative), while the resulting lncRNA transcript may be incidental to the transcription process. A classic example of such a mechanism is the Airn lncRNA in mice that was found to silence its neighboring gene Igf2r by interfering with the recruitment of RNA polymerase 2 to the gene promoter14. In many other cases lncRNAs were found to regulate transcription by directly recruiting or inhibiting regulatory proteins or indirectly via modulating the chromatin and nuclear architecture2. Acting via the aforementioned mechanisms, lncRNAs have been demonstrated to be implicated in stem cells and during reprograming and development15,16,17. The lncRNAs ES1 and ES2 were found to facilitate pluripotency and reprograming via serving as a modulator factor of the pluripotent marker SOX23,4,5. Other lncRNAs, such as DEANR1, were proposed to mark lineage differentiation via binding to germ layer specific TFs18.
In a recent study we identified an interaction between the Non-coding RNA Activated by DNA Damage (NORAD) and the Signal Transductor and Activator of Transcription 3 (STAT3) in the cytoplasm of human embryonic stem cells (hESCs)19. STAT3 is known for its different roles in inflammation, the immune system, embryonic development, and cancer and was previously shown to be associate with lncRNAs in diverse human and murine cell types8,9,10,11. The highly conserved, cytoplasmic lncRNA NORAD is most known for maintaining DNA stability. It does so by binding the RNA-binding protein (RBP) Pumilio at multiple sites across its transcript, through Pumilio Recognition Elements (PREs)20,21,22. This sequesters Pumilio into phase-separated RNP granules compartments23. In addition, an interaction between the 5’ region of NORAD and the RBP RBMX was shown to play a role in maintaining genome integrity24. While the PREs and 5’ regions of NORAD have been extensively studied, not much is known about the function of other regions in the NORAD transcript, specifically the relatively lowly conserved 3’ end, where we identified the interaction with STAT319.
In this study, we demonstrate that the interaction between STAT3 and the lncRNA NORAD is conserved in human cells and facilitates the nuclear translocation of unphosphorylated STAT3, leading to suppression of interferon-stimulated genes (ISGs). Knockdown (KD) of NORAD mimics an antiviral response, highlighting its role in innate immunity. Interestingly, this interaction is absent in murine cells, suggesting species-specific evolution. These findings reveal a novel mechanism by which NORAD modulates immune responses, potentially offering new insights into therapeutic strategies for viral infections and immune dysregulation.
Results
Knockdown of NORAD mimics viral response
Consistent with previous studies20,21 and our recent work suggesting that STAT3 interacts with NORAD outside of the nucleus, we found that NORAD localizes mostly to the cytoplasm of hESCs (Supplementary Fig. 1a). To test the possible function of its interaction with STAT3, we knocked down NORAD in hESCs. The downregulation was confirmed by RT-qPCR (Supplementary Fig. 1b) and its impact was assessed with RNA sequencing (RNA-seq). RNA-seq analysis revealed a significant upregulation of 29 genes (Padj < 0.05, FC > 2), 16 of which are ISGs , including ISG15, IFI6, MX1, IFITs, and OASs. Gene ontology (GO) analysis reinforced that NORAD KD activates the “defense response to viruses” pathway (Fig. 1a and Supplementary Data 1). Notably, KD of another lncRNA, GAS5, did not result in the activation of this pathway (Supplementary Fig. 1c). To explore whether the response is unique to pluripotent cells, we repeated the NORAD KD and following assays in fully differentiated foreskin fibroblasts (HFFs) (Fig. 1b and Supplementary Fig. 1d). In this case, it resulted in an even more robust upregulation of the antiviral response, observed from 8 h post-transfection and most prominently at 24 h (Fig. 1b, c and Supplementary Data 2). The number of ISGs that were induced in response to the KD in HFFs was significantly higher than in hESCs (69 genes, Padj < 0.05, FC > 2) (Fig. 1b). Consistently, in a proteomics analysis of HFF cells at 24 h following NORAD KD we observed a significant change in 122 proteins, 96 of them were upregulated upon the perturbation (Padj < 0.05, FC > 2). GO analysis clearly indicated that the upregulated proteins are significantly enriched for the viral defense pathways (e.g. type I interferon signaling pathway, defense response to viruses etc). As expected, the majority of the enriched proteins belong to the interferon stimulated gene family (Fig. 1d and Supplementary Data 3). Notably, 34 of the significantly upregulated ISG proteins were also significantly upregulated at the RNA level upon NORAD KD (Fig. 1e). Further, when analyzing RNA-Seq data from HCT116 cells in which NORAD was downregulated by CRISPRi 24, a similar upregulation of ISGs is observed up to 96 h following induction, most significantly at 24 h post-induction (see Supplementary Fig. 1e). Of note, most of the interferon genes themselves were not upregulated in any of these conditions (Fig. 1b, Supplementary Data 2 and 3)24. Remarkably, the changes in ISGs expression in the KD cells correlate with the changes in the same cell types infected by influenza A virus (IAV), though this correlation was weaker in hESCs (Fig. 1f) (data from Eggenberger et al.25). Moreover, the activation of the antiviral response in both IAV-infected and NORAD KD in HFFs was noticeably higher than in hESCs (Fig. 1f). This can be explained by the fact that ESCs do not utilize the canonical interferon immune response—rather, they have an alternate pathway based on RNAi25. A recent study detected a novel stem cell specific form of the Dicer protein (aviD) in mammalians, shown to provide protection against virus infection26. In addition, consistent with previous findings that human pluripotent and multipotent cells express a unique repertoire of ISGs critical for viral resistance27 we found a noticeable difference in the basal expression levels of a subset of ISGs between the non-treated HFFs and hESCs, as for example BST2 and MT1G (Supplementary Fig. 1f, Supplementary Data 4). Nevertheless, upon NORAD downregulation, other ISGs were activated in both pluripotent and differentiated cells (Fig. 1a, b). This suggests a functional role for NORAD in the transcriptional circuit regulating the interferon pathway.

a, b left: Volcano plots of RNA-seq data from NORAD KD versus control siRNA. In black, differentially expressed genes (Padj < 0.05, -2 > FC > 2), ISGs are marked in red. right: Gene ontology enrichment analyses of the differentially expressed genes upon NORAD KD. hESCs 72 h post siRNA transfection (n = 4) (a). HFFs 24 h post transfection ( n = 4) (b). c Heatmap representing the fold changes (FCs) in ISG expression levels at 8, 16, 24 h post transfection in HFFs, shown are only differentially expressed ISGs in at least one time point. d left: Volcano plot of proteomics data from NORAD KD versus control siRNA (n = 3). Differentially expressed genes are bolded (Padj < 0.05), ISGs are marked in red. right: Gene ontology enrichment analyses of the differentially expressed genes upon NORAD KD, sorted by enrichment score and colored by FDR q-value. e Log FCs of RNA (top) and protein (bottom) levels of differentially expressed ISGs from NORAD KD versus control siRNA, gray circles represent non-significant results. f Scatterplots demonstrating the correlations between FCs calculated for all expressed ISGs in IAV relative to mock-infected cells and the FCs calculated for ISG expression in NORAD KD in this study. top: Comparison between IAV infection in hiPSCs to siNORAD treatment in hESCs. bottom: Comparison between IAV infection to NORAD KD in HFFs. Source data are provided as a Source Data file.
To test this, we performed additional KD experiments followed by RT-qPCR. We found that KD of STAT3, as well as its double KD with NORAD, induced the expression of ISGs in a similar manner to NORAD KD (Fig. 2a). On the contrary, double KD of STAT1 and NORAD restrained ISGs expression, effectively rescuing the effect of NORAD’s absence (Fig. 2b). These results reinforce that the activation of the antiviral response caused by the reduction of NORAD is mediated by its interaction with STAT3, and requires STAT1 expression.

a, b RT-qPCR analysis of ISGs expression in double KD experiments in HFFs of STAT3 + NORAD (n = 2) and STAT1 + NORAD (n = 2) (a), siNORAD (red), siSTAT (light blue) and siSTAT + siNORAD (yellow) relative to siCTRL (b). c Western blot analysis of STAT3 and STAT1 protein expression and phosphorylation in HFFs 24 h post siRNA transfection. d, e Immunofluorescence (IF) visualization—upon NORAD KD compared to siCTRL samples in HFFs (16 h post siRNA transfection) In blue: nuclear staining by DAPI, STAT3 (d) STAT1 (e). f Box plots representing the ratios between the nucleus and cytoplasm IF signals (55 and 61 cells for STAT3 siCTRL and siNORAD, respectively and 71 and 64 cells for STAT1 siCTRL and siNORAD, respectively) showing significant differences between the different conditions (STAT3 p-value = 8.1 e-10, STAT1 p-value = 9.7e-9, two-sided Mann Whitney Wilcoxon test). g, h An IF visualization—upon NORAD KD compared to siCTRL samples in hESCs (72 h post siRNA transfection). In blue: nuclear staining by DAPI, STAT3 (g) STAT1 (h). i Box plots representing the ratios between the nucleus and cytoplasm IF signals (282 and 302 cells for STAT3 siCTRL and siNORAD, respectively and 59 and 67 cells for STAT1 siCTRL and siNORAD, respectively) showing significant differences between the different conditions (STAT3 p-value = 3.76e-23, STAT1 p-value = 0.0157, two-sided Mann Whitney Wilcoxon test). Source data are provided as a Source Data file.
NORAD affects STAT1/STAT3 balance in the nucleus
Next, we tested the effect of NORAD KD on STAT3 and STAT1 protein level, modifications, and localization. Overall, the levels of the transcriptionally active form of STAT3 (p-Y705) slightly increased (2.08 ± 0.33), while the total STAT3 protein level and its transcriptionally enhanced form (p-S727) did not considerably change (0.99 ± 0.19 and 1.2 ± 0.56, respectively). On the other hand, while the overall STAT1 protein did not change dramatically (1.74 ± 0.44) the levels of the modification associated with nuclear translocation and transcription activation (p-Y701) of STAT128 increased substantially (29.9 ± 1.82) (Fig. 2c, Supplementary Fig. 2a, and Source Data file). When looking at their cellular localization, we observed a significant shift upon NORAD KD—STAT3 was less present in the nucleus, whereas STAT1 nuclear levels increased (Fig. 2d–i). Unlike STAT1, STAT3’s nuclear localization could not be explained by changes in its phosphorylation.
An alternative path requires the interaction of unphosphorylated STAT3 with Importin-β1 (KPNB1)29. Previous RNA pulldown assays20,21,30 identified Importin-β1 as directly interacting with NORAD, though it was not found among the highly enriched interactors in Munschauer et al.24. In a co‐immunoprecipitation assay of STAT3 and Importin-β1 we show a two-fold reduction (0.48 ± 0.09) in the interaction upon NORAD KD (Fig. 3a, Supplementary Fig. 2b, c, and Source Data file). Notably, the interaction between STAT3 and its well-known partner STAT131 did not change upon NORAD KD (Fig. 3a, Supplementary Fig. 2b, c). We further sought to examine whether the change in the ratio between STAT3 and STAT1 in the nucleus influences their binding preference. We carried out Chromatin Immunoprecipitation (ChIP)-qPCR in HFFs, both with and without NORAD KD. To this end we selected a panel of ISGs that contain the Interferon-Sensitive Response Element (ISRE) consensus within their proximal promoter regions, 1 kb upstream to the Transcription Start Site (TSS) (Supplementary Fig. 3). ISRE is known to mediate the interferon pathway via binding to STAT1, usually in complex with STAT2, upon virus stimulation32. The panel of ISGs selected in this study (MX2, RSAD2, IFIT1, IFIT3, OAS1, OAS2), have been previously shown to be induced by STAT1 binding to their promoter upon interferon stimulation33. Consistent with studies suggesting that STAT3 can dimerize or compete with STAT1 in binding promoters of ISGs32,34,35,36 our ChIP analyses confirmed that all selected promoters bound STAT3 in WT HFFs (Fig. 3b top, and Source Data file). Following NORAD KD, our ChIP results revealed a significant decrease in the occupancy of STAT3 at the tested ISGs. Conversely, the binding of STAT1 to the same promoters was significantly increased in the KD cells (Fig. 3b bottom, and Source Data file). The observed change in the binding of STAT1 and STAT3 in response to NORAD KD aligns with the upregulation of these ISGs and is consistent with earlier studies showing that by binding to the promoters of ISGs, STAT3 inhibits their transcription, while STAT1 activates it. Altogether, we show that in the presence of NORAD, STAT3 interacts with Importin-β1 and is translocated to the nucleus, where it binds to ISG promoters and inhibits their transcription. Upon NORAD down-regulation, STAT3’s nuclear levels decrease, allowing STAT1 to bind in its place and activate the interferon pathway. Early activation of these ISGs increases the phosphorylation levels of STAT1, thereby enhancing the transcription of both these initially activated ISGs and many others. Together, this results in positive feedback that intensifies the antiviral response (Fig. 3c).

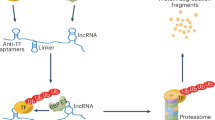
a Top: Western blot analysis of Co-IP experiment. HFF lysates from siNORAD and siCTRL samples immunoprecipitated by anti-STAT3. Protein-protein interactions were immunodetected by anti-Importin-β1, anti-STAT3 and anti-GAPDH. Bottom: Quantification of protein levels of STAT3 Co-IP. The graph shows the log2 FCs of protein levels between siNORAD and siCTRL in Input (yellow) and IP-STAT3 (red). (STAT3, Importin-β1, GAPDH n = 2, STAT1 n = 1). Upon NORAD KD, a significant reduction in the interaction between STAT3 and Importin-β1 is observed. b Binding enrichment of STAT3 (top) and STAT1 (bottom) to the promoters of six selected ISGs in siCTRL and siNORAD in HFFs. DNA binding levels were determined by ChIP-qPCR (n = 2). c Illustration of the proposed model for the role of NORAD-STAT3-interaction in regulating ISGs transcription. left: In normal cells with high NORAD levels, STAT3 interacts with Importin-β1 and is translocated to the nucleus, where it binds to ISGs promoters and represses their transcription. right: In NORAD-depleted cells, STAT3’s nuclear localization is reduced, resulting in decreased STAT3 occupancy at ISGs promoters. STAT3 reduction allows STAT1 to replace STAT3 at these promoters, leading to the activation of ISGs transcription and initiation of the interferon response. Source data are provided as a Source Data file.
NORAD is downregulated in viral infections
Building on our finding that NORAD downregulation triggers the antiviral response, we aimed to gauge NORAD’s involvement in viral infections. To begin, we conducted a general assessment of lncRNA expression in cells infected by three different viruses: SINV37, SARS-CoV-238 (RNA viruses), and HSV139 (DNA virus). We found 928 lncRNAs whose expression changed in at least one dataset, 60 of which changed in at least two, and only six were differentially expressed in all three infections. Out of these six lncRNAs, four were upregulated: SNHG1, SNHG12, SNHG15, and LINC00511, and two were downregulated: NORAD and its pseudogene HCG1121 (Fig. 4a, Supplementary Data 5).

a Heatmap of differentially expressed lncRNAs upon infection in SINV, SARS-CoV-2 and HSV1. b, c Analyses of NORAD expression levels in COVID-19 patients compared to healthy controls: UMAP colored by the patient label (green and red shades for Healthy control (HC) and COVID patients (Cov), respectively) (b), UMAP colored by the expression levels of ISG15 (top) and NORAD (bottom) (c). Quantification of the gene expression levels are demonstrated in box plots besides each UMAP. P-adj values are depicted above the plots (one-sided Mann-Whitney Wilcoxon test with Bonferroni correction). d, e Analysis of NORAD expression in cells infected by IAV compared to mock infected cells, based on single cell RNA-seq data. In (d) left: UMAP colored by hemagglutinin viral gene (IAV-HA), right: UMAP colored by the cell category. In (e) UMAPs colored by the expression levels of ISG15 (top) and NORAD (bottom). Quantification of the expression levels of the two factors are demonstrated in box plots besides each UMAP. P-adj values are depicted above the plots (two-sided Mann-Whitney Wilcoxon test with Bonferroni correction). As shown, NORAD levels are significantly decreased in the virus infected cells compared to mock and bystanders. Source data are provided as a Source Data file.
We next examined the effect of viral infection on NORAD based on clinical data. We analyzed single-cell RNA-seq data from nasopharyngeal samples of four COVID-19 patients and three healthy controls40, finding a significant decrease of NORAD expression in epithelial cells, which shows an inverse relation to ISG15 (Fig. 4b, c). A consistent significant reduction was also observed by Butler et al.41 in bulk RNA-seq data from a larger cohort of 173 COVID-19 positive patients relative to the expression in 407 negative controls (nasopharyngeal samples).
The downregulation of NORAD upon viral infection could be explained in two ways. Either it is caused directly by the penetration of the virus into the cell, or NORAD could be downregulated as part of the interferon pathway response. To address this question, we analyzed single-cell RNA-seq data from human lung cells infected by Influenza-A virus or a mock infection42. Based on the experimental setup, we classified the cells into two categories: ‘mock’ for control cells and ‘exposed’ for those where the virus was introduced to the culture. The exposed cells were then subdivided into two categories based on viral gene expression, as defined in Ramos et al.42: ‘infected’ cells and ‘bystander’ cells—uninfected but responsive to interferon secretion by the adjacent infected cells (Fig. 4d).
While both infected and bystander cells express similarly high levels of ISGs, NORAD was significantly reduced in the infected cells compared to mock and bystanders (Fig. 4e and Supplementary Fig. 4). Notably, NORAD promoter (defined as 3 kb upstream and downstream to the TSS) does not exhibit a binding motif of any of the STAT/ IRF family members and consistently its expression was not found to significantly change upon type I interferon treatment in 107 datasets (based on the interferome database43). Together this suggests that the downregulation of NORAD is likely a direct cellular response to the presence of the virus, rather than to the Interferon, which further induces the activation of the interferon pathway.
A novel function for NORAD in evolution
The role of NORAD lncRNA in DNA stability has been revealed to be highly conserved in mammals20,21,22. Several lncRNAs are known to play a role in regulating the immune response, both in humans and mice44,45,46,47. Upon demonstrating the involvement of NORAD in the defense against viruses in human, we were intrigued as to how this newly discovered function is conserved in mammals. To address this, we knocked down Norad in mouse embryonic fibroblasts (MEFs), followed by RNA-seq (Fig. 5a, b and Supplementary Data 6). While NORAD KD in human cells resulted in the upregulation of ISGs (Fig. 5b), Norad downregulation did not activate the transcription of ISGs in murine cells (Fig. 5a, b). Consistently, no interaction between Stat3 and Norad in MEF cells was detected in CLIP-qPCR (Fig. 5c and Supplementary Fig. 5a). As can be learned from exploring the sequence conservation along the NORAD transcript (based on the conservation track in UCSC genome browser that was calculated by PhyloP based on 30 mammals), the STAT3 binding site is markedly lowly conserved compared to the NORAD repeats21 (Fig. 6a). The latter point is further emphasized when observing the detailed multiple sequence alignment of the regions that align to the human binding site identified in our CLIP experiment (Supplementary Fig. 5b). Interestingly, we found that in humans the STAT3 binding site is located at ~100nts downstream to a primate-specific Alu element. To ask whether the presence of the Alu element influences the STAT3 binding regions we constructed a phylogenetic tree, taking as an input the STAT3 binding region of NORAD from 30 mammals (Fig. 6b and Supplementary Fig. 5b). Among the 30 mammals that were included in the phylogenetic analysis were 10 primates that contain the primate specific Alu element in NORAD in their genome (Alu+ primates), five primates that do not contain Alu in NORAD (Alu- primates) and 15 non-primate mammals (other mammals). As shown in Fig. 6b, the species represented in the phylogenetic tree that was constructed solely based on the sequences from the STAT3 binding region were split to three groups (Alu+ , Alu-, other mammals). We further predicted the RNA secondary structure of the sequences that were used to build the tree using RNAfold http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi. The predicted RNA secondary structures presented in the outer circle in Fig. 6b demonstrate that sequences from primates containing an Alu element fold to a similar stem-loop structure that is highly consistent with the experimentally solved structure of this region in human NORAD by COMRADES48, while the structures from other species were highly variable. To further quantify the observed differences and examine to what extent they are consistent at the three dimensional (3D) structural level we employed the simRNA modeling algorithm49. Next, we conducted an all-versus-all pairwise structural comparison analysis for all predicted 3D structures. As demonstrated in Fig. 6c the 3D structures predicted from the sequences of the STAT3 binding region from Alu+ species are more similar to each other than to any other species, with significantly lower RMSD values when compared to each other (excluding Chimp). These results strongly reinforce that the presence of an Alu element in the NORAD transcript of Alu+ primates renders a local change in the downstream region. We propose that the insertion of an Alu in the 3’ region of NORAD during primate evolution may have induced a change in the sequence and structure of this region, enabling an interaction with STAT3.

a qPCR analysis of Norad KD in MEF cells demonstrating that KD of Norad does not change the RNA levels of Stat3, Stat1, and Isg15 (n = 2). b A volcano plot of RNA-seq data from Norad KD versus control siRNA in MEF cells at 24 h post-transfection. Differentially expressed genes (Padj < 0.05, -2 > FC > 2) are in bold, ISGs are marked in red (n = 3). c The FCs of differentially expressed genes upon NORAD KD in HFFs compared to hESCs (left) and HFFs compared to MEFs (right) showing a high correlation between the undifferentiated and the fully differentiated human cells while no significant changes are detected in gene expression in MEFs. ISGs are marked in red. d CLIP-qPCR validation of STAT3 binding to NORAD in hESCs, HFFs and MEFs (n = 2). As demonstrated, there is no interaction between STAT3 and Norad in the mouse cells. Source data are provided as a Source Data file.

a Evolutionary conservation across the NORAD transcript, depicting weak sequence conservation at the STAT3 binding site. STAT3 binding site, Alu and NORAD repeats are illustrated in gray rectangles. b Phylogenetic tree based on the sequence of the STAT3 binding site (135 nts). RNA predicted structures (predicted by RNAfold) are presented. Bootstrap values above 0.7 (calculated based on 1000 trees) are represented in circles. Red, Blue and green colored branches depict sequences derived from primates containing Alu in NORAD, primates not encoding Alu and other mammals, respectively. c Heatmap of RMSD values representing pairwise structural comparisons of the predicted 3D structures of the STAT3 binding region on NORAD from the 30 organisms shown in supplementary Fig. 5b. The structural models were built based on the sequences around the core region, centered around the loop. d EMSA results for STAT3 binding to predicted binding site in Human (red), Rhesus (pink), Ma’s Night Monkey (blue) and Mouse (green) (n = 2). Source data are provided as a Source Data file.
To test this conjecture, we extracted the sequences that align to the STAT3 binding site on NORAD from four representative species, two Alu+ (Human and Rhesus) and two Alu- (Mouse and Ma’s night Monkey) and conducted an in-vitro binding assay, EMSA, with STAT3 protein, purified from human cells. Of note, STAT3 is 100% conserved between human and mouse. As demonstrated (Fig. 6d and Supplementary Fig. 5c), the EMSA results are highly consistent with the CLIP-qPCR experiment, reinforcing that STAT3 binds to the human sequence but not to the mouse sequence. Moreover, while the RNA sequences used in the EMSA experiments were derived from the region that does not harbor the Alu element, STAT3 binding was detected only to the sequences from Alu+ species (243 and 420 nM KD for the Human and Rhesus sequences, respectively) and not to the Alu- derived sequences (Ma’s night monkey and Mice). These results strongly support that the presence of the Alu in the NORAD transcript induced a change in the sequence downstream. Taken together we propose that the introduction of the Alu in the conserved lncRNA NORAD enabled the interaction between STAT3 and NORAD leading to the birth of a new mechanism in evolution for rapid activation of the interferon pathway.
Discussion
In this study, we propose a novel role for the lncRNA NORAD in regulating the innate immune response in human cells. These results are consistent with previous studies that have shown that other lncRNAs can act as regulators of the antiviral response through diverse mechanisms44,45,46,47. The involvement of a lncRNA in sensing and transducing a signal enables a highly efficient and quick cellular response to viral infections, without the need for protein metabolism. Here we have found that NORAD is required for the translocation of the unphosphorylated STAT3 to the nucleus via Importin-β1, where it binds to the promoters of ISGs and negatively regulates their transcription. We also observed that NORAD levels are reduced in human viral infections, both in cells infected in-vitro and in patients. This can be explained by recent studies that show that among other RNAs, NORAD is recruited to stress granules upon viral infection, where it is cleaved by RNaseL50,51,52 as part of the intrinsic cellular antiviral response. This is an extremely rapid reaction that inhibits viral replication without requiring changes in gene expression. Our findings indicate that NORAD downregulation releases the STAT3-mediated inhibition of ISGs, thus contributing to the activation of the innate immune response by STAT1. Collectively, these results suggest that the lncRNA NORAD may serve as a bridge between the intrinsic defense response and the more established STAT1-mediated innate immune response. The link that NORAD generates between the two pathways may lead to a more direct transcriptional activation of ISGs, bypassing many regulatory processes. This is particularly significant given that many viruses have evolved strategies to inhibit the classical activation of the interferon pathway, a key element of the host’s antiviral defense. This ‘shortcut’, or non-canonical activation, could circumvent these viral inhibitions, as it does not rely on the activation of IRFs and interferon secretion. Such a mechanism might expedite the response of the host defense system, providing a rapid reaction to viral threats while still invoking the robustness of the STAT1-mediated innate immune response. It is interesting to note that pluripotent cells do not normally activate the interferon pathway and have been shown to intrinsically express a subset of ISGs that grant them the required viral resistance27. Therefore, the upregulation of ISGs induced by NORAD KD in pluripotent and differentiated human cells reinforces that the new role suggested for NORAD does not rely on interferon stimulation.
Unlike protein-coding genes, the majority of lncRNAs have evolved under little to no selective constraints53. Thus, lncRNAs have the potential to better adapt to rapid changes in evolution compared to proteins. We found that the interaction between STAT3 and NORAD is mediated by a structured region in the 3’ domain of NORAD, downstream to the Alu element that is present only in hominoids. This led us to propose that the introduction of Alu changed the sequence and structure of the region downstream, which enabled STAT3 binding to NOARD, giving rise to a new function for NORAD in regulating the antiviral response in hominoids. It has been suggested that mammalian embryonic stem cells keep the interferon response silent as it interferes with their two defining characteristics; namely self-renewal and pluripotency25,27. Our identification of NORAD as the link between the interferon pathway and the intrinsic cellular response could provide a rationale for a reduced intensity of this intrinsic response in hESCs compared to mESCs. This may suggest a protective mechanism to mitigate the activation of the interferon response54.
Together, we postulate that a non-classical interaction between a lncRNA and a bona fide TF may have a key role in the rapid cellular response to external stimuli, adding an additional important layer of regulation in eukaryotes.
Methods
Cell culture
Human foreskin fibroblasts (HFFs)55 and Mouse embryonic fibroblasts (MEF) were cultured in Dulbecco’s modified eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), glutamine, and penicillin/streptomycin (pen/strep). The cells were maintained in a humidified incubator at 37 °C with 5% CO2 and passaged every 3–4 days by trypsinization and re-seeding at a split ratio of 1:3. Human embryonic stem cells (hESCs) were maintained in suspension in 82% DMEM/F-12, 15% KnockOut Serum Replacement, 3 mM L-glutamine, 0.1 mM β-mercaptoethanol, 1% non-essential amino acids, and 100 ng/ml FGF basic (bFGF), and pen/strep. The hESCs were separated into single cells by treatment with EGTA and TrypLE Express and then transferred to Matrigel-coated plates in mTeSR1 medium supplemented with 5 μM ROCK inhibitor. The cells were allowed to attach to the plates for at least 24 h before the start of the experiment.
Single-molecule FISH
Cells were fixed using 4% formaldehyde in PBS for 15 min at room temperature, followed by a rinse with 1X PBS. After rinsing, the samples were incubated with cold 70% ethanol at 4 °C for at least 2 h. Subsequently, cells were washed twice with wash buffer (10% Formamide, 2X SSC) for 5 min each. Custom Stellaris® FISH Probes recognizing NORAD and labeled with Quasar® 670, were purchased from Biosearch Technologies, Inc. (Petaluma, CA). Probe set sequences utilized in the experiments have been previously described in Tichon et al.21. The probes were diluted in UPW to a working solution of 2.5 µM. A volume of 5 µl of these probes was added to 45 µl of hybridization buffer (10% Dextran sulfate, 10% Formamide, 1 mg/ml E.coli tRNA, 2X SSC, 0.02% BSA, 2 mM Vanadyl-ribonucleoside complex). This mix was applied onto a clean piece of parafilm for each sample. The samples were carefully placed section-down onto a drop of the hybridization mix and incubated overnight at 37 °C. The next day, samples were washed once in wash buffer for 30 min at 37 °C. Concurrently, a solution of wash buffer with 1:200 of DAPI (10 µg/ml) was pre-warmed for 30 min together with the samples. The samples were then incubated in GLOX buffer (100 mM TRIS (pH 8.0), 2X SSC, 0.4% Glucose), allowed to stand for a few minutes, mounted with Prolong Gold Antifade Mountant, and immediately visualized on a Leica DMI8 inverted fluorescent microscope with an x68 oil-immersion objective. The dot detection was carried out automatically using Imaris software. RNA signal detection and segmentation were carried out using Imaris software. RNA spots of 0.3 µm XY diameter were detected using the built-in Spots function. The nuclear signal was masked and segmented using the built-in surface algorithm.
NORAD knockdown
HFFs, hESCs and MEFs were transfected with small interfering RNAs (siRNAs) using Lipofectamine RNAiMAX. TriFECTa kit DsiRNA Duplex (IDT) containing three RNAi duplexes were designed to specifically target the NORAD lncRNA. The cells were incubated with the siRNAs for 8,16, and 24 h for HFF, 24 h for MEF, and 72 h for hESC. After the incubation period, total RNA was purified using TRI-reagent. To evaluate the KD efficiency, RT-qPCR was performed using primers specific for the NORAD lncRNA and the reference gene GAPDH. STAT1 and STAT3 KD in HFFs was performed by transfecting cells with Ambion, Silencer® Select siRNAs using Lipofectamine RNAiMAX and then incubated for 72 h. For double KD, cells were first transfected with siSTAT1/3 and after 48 h were further transfected with siNORAD.
RNA sequencing
RNA sequencing was performed using the CEL-seq2 protocol56 and sequenced on an Illumina NextSeq550 or NextSeq2000 platforms. To analyze the CEL-seq2 RNA sequencing data, the reads were first processed using the cutadapt software57, then aligned to the reference genome using RNA-STAR58. HTSEQ count59 was used to generate raw count data, and the DESeq2 R package60 was used to conduct differential gene expression analyses. GO enrichment was analyzed using DAVID web server (https://david.ncifcrf.gov/summary.jsp) and visualized by the GOplot R package61.
Proteomics analysis
Cells were lysed in a solution containing 8.5 M Urea, 400 mM Ammonium bicarbonate, and 10 mM DTT. The lysates were sonicated twice at 90% power (10 s on, 10 s off, for 5 min total) and centrifuged at 10,000 g for 10 min. Protein concentrations were determined using Bradford readings. The samples were then reduced at 60 °C for 30 min, followed by alkylation with 35.2 mM Iodoacetamide in 100 mM Ammonium bicarbonate at room temperature for 30 min in the dark. Digestion was performed overnight at 37 °C using modified Trypsin in a solution of 1.5 M Urea and 66 mM Ammonium bicarbonate at an enzyme-to-substrate ratio of 1:50 (M/M). A second digestion was conducted for 4 h at 37 °C at a 1:100 (M/M) enzyme-to-substrate ratio. The resulting tryptic peptides were desalted using homemade C18 stage tips, dried, and re-suspended in 0.1% Formic acid. Tryptic peptides were analyzed by LC-MS/MS using an Exploris 480 mass spectrometer (Thermo) coupled with a Vanquish Neo UHPLC (Thermo-Fisher). Peptides were loaded in solvent A (0.1% Formic acid in water) onto a homemade capillary column (30 cm length, 75 µm inner diameter) packed with Reprosil C18-Aqua. Peptides were separated with a 6–34% linear gradient of solvent B (80% Acetonitrile, 0.1% Formic acid) over 180 min, followed by a gradient of 30–80% for 11 min, and then at 80% acetonitrile with 0.1% formic acid in water for an additional 11 min. The flow rate was maintained at 0.15 µl/min. Mass spectrometry was performed in positive ion mode with an m/z range of 350–1200. MS1 scans were acquired at a resolution of 120,000, followed by MS2 scans at a resolution of 15,000 using high collision dissociation (HCD) at 27% normalized collision energy. The 30 most abundant ions (charge >1) from the MS1 scan were selected for fragmentation. The intensity threshold for MS/MS triggering was set to 8×103, and a dynamic exclusion list was applied with a 30 s exclusion duration.
MS files were processed using MSFragger via FragPipe with default parameters62. downstream analysis was performed on Perseus63. The MaxLFQ values were log2 transformed and filtered to include proteins that were detected in at least two replicates. Missing values were imputated from normal distribution followed by a two-sample t-test. Permutation based FDR with S0 parameter of 0.1 were applied.
ChIP-qPCR
Chip-qPCR was performed with primers designed to the promoter regions of a selected panel of six ISGs, around the ISREs (as described in main text). Chromatin immunoprecipitation (ChIP) followed by quantitative PCR (qPCR) was performed to examine the DNA binding of STAT1 and STAT3. Cells were crosslinked with 1% formaldehyde for 10 min at room temperature and quenched with 125 mM glycine. Chromatin was lysed in FA lysis buffer (50 mM HEPES-KOH pH7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 1 mM PMSF) and sonicated by Sonics Vibracell VCX130 for 2.5 min (10 pulses of 15” on 10” off). Immunoprecipitation was performed using 3 μg antibody against STAT1 or STAT3 for 4 h and afterward incubated with 50 μl Protein G magnetic beads. Beads were washed in RIPA buffer (50 mM Tris-HCl pH8, 150 mM NaCl, 2 mM EDTA, 1% IGEPAL, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM DTT, 1 mM PMSF) and the immunoprecipitated DNA was eluted with proteinase K and purified by MN NucleoSpin Gel and PCR cleanup kit. qPCR was performed using primers specific to the ISGs and ACTB serving as an internal control. The qPCR reactions were performed in triplicate, and the enrichment of ISGs was calculated as the percent input after normalization to the total input DNA.
Western blot analysis
Total protein was extracted from cells using iCLIP lysis buffer (50 mM Tris-HCl pH 7.5, 100 mM NaCl, 1% IGEPAL, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM PMSF, 1% Phosphatase inhibitors cocktail 2, 1% Phosphatase inhibitors cocktail 3). Western blot analysis was performed using antibodies against STAT1, STAT3, phosphorylated STAT1 (pSTAT1) at residue 701, and phosphorylated STAT3 (pSTAT3) at residues 705 and 727. GAPDH was used as a loading control.
Immunofluorescence
Immunofluorescence staining was performed to visualize STAT3 and STAT1 proteins in hESCs and HFF cells. Cells were fixed in 4% formaldehyde for 15 min at room temperature. Cells were blocked and permeabilized with 5% normal goat serum and 0.3% Triton X-100 in PBS for 1 h at room temperature and incubated with primary antibodies against STAT1 or STAT3 overnight at 4 °C. Cells were washed with PBS and incubated with alexafluor 488-conjugated secondary antibody for 1 h at room temperature. Nuclei were stained with DAPI for 5 min at room temperature. Cells were mounted with Fluoromount-G and visualized using an LSM700 microscope (Zeiss). Images were analyzed using Fiji software. For each cell, the nuclear-to-cytoplasm fluorescence signal ratio was calculated individually. A square region of interest (ROI) was selected within the nucleus, and an equally sized ROI was placed in the cytoplasm directly outside the nucleus. The average fluorescence signal (Alexa Fluor 488) within each ROI was measured64. Nuclei were identified based on the DAPI signal, and regions showing Alexa Fluor 488 fluorescence, but lacking DAPI signal were considered cytoplasmic. This approach allowed for consistent and accurate measurement of nuclear and cytoplasmic fluorescence intensities across the sample.
Co-Immunoprecipitation
Co-IP was performed to analyze the interaction between STAT3 and Importin-β1 in HFFs. Cells were crosslinked with 1% formaldehyde for 10 min at room temperature and quenched with 125 mM glycine. Cells lysed in FA lysis buffer (50 mM HEPES-KOH pH7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 1 mM PMSF) then Immunoprecipitation was performed using 3 μg antibody against STAT3 and 50 μl Protein G magnetic beads. The immunoprecipitated proteins were eluted from the beads with NuPAGE LDS sample buffer and analyzed by western blotting using antibodies against STAT3 and Importin-β1. STAT1 was used as a positive control. GAPDH was used as a loading control of the input and as a negative control for Co-IP.
Electrophoretic mobility shift assay
RNA was synthesized in vitro using the MEGAscript kit and incubated at 37 °C for 16 h, followed by purification using the Quick-RNA Miniprep Kit. STAT3 protein was extracted from 50 million cells that were lysed using 1 ml iCLIP lysis buffer (50 mM Tris-HCl pH 7.5, 100 mM NaCl, 1% IGEPAL, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM PMSF, 1% Protease Inhibitor Cocktail III), and treated with 10 μl Turbo DNase. The cell lysate was then subjected to immunoprecipitation using 3 μg STAT3 antibody and 50 μl Protein G magnetic beads. Samples were washed 3 times with high salt buffer (50 mM Tris-HCl pH 7.5, 1 M NaCl, 1 mM EDTA, 1% IGEPAL, 0.1% SDS, 0.5% sodium deoxycholate). STAT3 was eluted by incubation with 0.2 M glycine at pH 2.6 for 10 min and immediately neutralized with 1 M Tris at pH 8.0. For the EMSA, binding reactions were performed in a buffer containing 20 mM HEPES (pH 7.9), 50 mM KCl, 1 mM DTT, 0.1 mM EDTA, 5% glycerol, and 0.05% IGEPAL, with STAT3 added at concentrations of 0, 150, 300, 600, and 1200 nM. RNA was denatured at 90 °C before being cooled on ice and added at 140 nM. The complexes were incubated at 37 °C for 30 min and then resolved on a 2.5% agarose gel containing SYBR Safe. Gel electrophoresis was conducted in 0.5x TB buffer (45 mM Tris base, 45 mM Boric acid) at 20V/cm for 7 min, and the complexes were visualized using a Li-Cor Odyssey FC system with an exposure of 1.5 min. Images were analyzed using ImageJ, and the fraction bound was calculated by the shifted signal divided by the total signal. The dissociation constant (Kd) values for Human and Rhesus were determined by fitting the binding data to a sigmoid curve using nonlinear regression in R with the nlsLM function from the minpack.lm package. For Mouse and Ma’s night monkey, polynomial regression was used to fit the binding data using the lm function in R. Graphs were made using R ggplot.
CLIP-qPCR with STAT3 in HFF, hESCs, and MEF
Cells from hESCs, HFF, and MEF underwent UV cross-linking at 254 nm (3.2 J/cm2 for hESCs and 1 J/cm2 for HFF and MEF) to stabilize RNA-protein interactions. Post-crosslinking, cells were lysed using 1 ml iCLIP lysis buffer (50 mM Tris-HCl pH 7.5, 100 mM NaCl, 1% IGEPAL, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM PMSF, 1% Protease Inhibitor Cocktail III), and treated with 10 μl Turbo DNase for DNA digestion. The cell lysate was then subjected to immunoprecipitation using 3 μg STAT3 antibody and 50 μl Protein G magnetic beads. Post-immunoprecipitation, samples were washed 3 times with high salt buffer (50 mM Tris-HCl pH 7.5, 1 M NaCl, 1 mM EDTA, 1% IGEPAL, 0.1% SDS, 0.5% sodium deoxycholate) and protein digested with Proteinase K, followed by RNA purification by phenol-chloroform extraction and the Zymo RNA Clean & Concentrator kit. The extracted RNA underwent reverse transcription using SuperScript III Reverse Transcriptase with random hexamers at 25 °C for 5 min, then at 55 °C for 50 min to produce cDNA. qPCR analysis of the cDNA, utilizing NORAD-specific primers, was conducted with GAPDH serving as an internal control. The ΔΔCt method was used for data analysis, determining the enrichment of NORAD RNA in STAT3 immunoprecipitates relative to input samples.
RNA-seq data analysis
Publicly available RNA-seq datasets from cells infected with three different viruses—SINV37, SARS-CoV-238, and HSV139—were reprocessed and analyzed. For each dataset, the raw counts were imported and handled using the DESeq2 R package60 for both normalization and differential expression analysis at each timepoint relative to control samples. Differentially expressed genes were defined as those with an adjusted p-value (Padj) < 0.05. We chose the most responding timepoint as the one with the highest number of significantly upregulated ISGs. The differentially expressed lncRNAs identified in each dataset were then compiled together to identify lncRNAs that were differentially expressed on two out of three datasets.
Single-cell RNA-seq data analysis for COVID-19 patients
Single-cell RNA-seq data were analyzed from Seq-Well libraries of nasal wash samples from COVID-19 patients, generated by Gao et al.40. Raw count data was filtered to eliminate potential dead cells and doublets by removing cells with an atypical number of detected genes (Specifically- <500 or >10,000)65. We also removed cells in which >33% of the reads were mapped to mitochondrial genes, since this may indicate cell stress66,67. Counts from the remaining cells were then normalized to the median counts of all cells. Epithelial cells were identified based on gene expression criteria (KRT7 > = 1 and VIM < 3). Within these epithelial cells, the expression levels of NORAD and ISG15 were compared between COVID-19 patients and healthy controls using a Mann–Whitney U-test.
Single-cell RNA-seq analysis of influenza A virus-infected cells
For the analysis of 10x Genomics single-cell RNA-seq data from Influenza-A virus and mock-infected human lung cells42 we adapted the Python pipeline previously used for COVID-19 patient samples with a few modifications. We first filtered cells that had <2500 or >5000 identified genes. Then, normalization was performed as described above, using median counts across all cells. The cells were then classified into three groups: (1) Mock-infected cells defined as in the original sequencing library. (2) Infected cells expressing the Influenza A virus PR8_NP gene above 90 reads. (3) Bystander cells that do not express the PR8_NP (below 90 reads). Differential expression analysis was performed using a Mann–Whitney U-test, and p-values were corrected for multiple testing using Bonferroni correction.
Phylogenetic analysis and RNA structure prediction
Sequences corresponding to the STAT3 binding site in human (hg38 chr20:36,046,781-36,046,915) were extracted using the UCSC Genome Browser. For primates, sequences were derived from the Mammals Multiz Alignment & Conservation (27 primates), for other mammals, sequences were obtained from the Vertebrate Multiz Alignment & Conservation (100 Species). Sequences extracted from the reverse strands were aligned using the T-Coffee web server (https://www.ebi.ac.uk/Tools/msa/tcoffee/). A phylogenetic tree was subsequently built using the BioNJ algorithm68, with confidence assessed through 1000 bootstrap replicates. The resulting tree was visualized using the iTOL tool69. Simultaneously, the RNA secondary structure of the STAT3 binding site was predicted using the Minimum Free Energy (MFE) function in RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi). The predicted structures were visualized in Forna (http://rna.tbi.univie.ac.at/forna). The final assembly of the figure, which included both the phylogenetic tree and predicted RNA structures, was conducted in Adobe Illustrator.
The three-dimensional (3D) structure of the RNA was predicted using the simRNA webserver. Secondary structure restraints were applied based on the secondary structure predicted earlier with RNAfold, excluding unpaired nucleotides at the sequence ends from the input. Predicted 3D structures were then compared using the Matchmaker tool in ChimeraX70, focusing on the core region centered around the loop (details are provided in the Source Data file). RMSD values were calculated for all atom pairs, and these values were visualized as a heatmap using the pheatmap R package.
Materials
The full list of oligonucleotides, primers, siRNAs, antibodies, reagents, chemicals, and cells used in this study are provided in Supplementary Data 7.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA-Seq data generated in this study have been deposited in the GEO database under accession code GSE241264. The mass spectrometry data generated in this study have been deposited in the PRIDE database under accession code PXD056160. All data are included in the Supplementary Information or available from the authors. The raw numbers for charts and graphs are available in the Source Data file whenever possible. Source data are provided with this paper.
References
Hudson, W. H. & Ortlund, E. A. The structure, function and evolution of proteins that bind DNA and RNA. Nat. Rev. Mol. Cell Biol. 15, 749–60 (2014).
Long, Y., Wang, X., Youmans, D. T. & Cech, T. R. How do lncRNAs regulate transcription? Sci. Adv. 3, eaao2110 (2017).
Ng, S.-Y., Johnson, R. & Stanton, L. W. Human long non-coding RNAs promote pluripotency and neuronal differentiation by association with chromatin modifiers and transcription factors. EMBO J. 31, 522–533 (2012).
Holmes, Z. E. et al. The Sox2 transcription factor binds RNA. Nat. Commun. 11, 1805 (2020).
Hou, L. et al. Concurrent binding to DNA and RNA facilitates the pluripotency reprogramming activity of Sox2. Nucleic Acids Res. 48, 3869–3887 (2020).
Ng, S.-Y., Bogu, G. K., Soh, B. S. & Stanton, L. W. The Long Noncoding RNA RMST Interacts with SOX2 to Regulate Neurogenesis. Mol. Cell 51, 349–359 (2013).
Luo, H. et al. HOTTIP-dependent R-loop formation regulates CTCF boundary activity and TAD integrity in leukemia. Mol. Cell 82, 833–851.e11 (2022).
Guo, H. et al. The aspirin-induced long non-coding RNA OLA1P2 blocks phosphorylated STAT3 homodimer formation. Genome Biol. 17, 24 (2016).
Smith, K. N., Starmer, J. & Magnuson, T. Interactome determination of a long noncoding RNA implicated in embryonic stem cell self-renewal. Sci. Rep. 8, 17568 (2018).
Wang, P. et al. The STAT3-binding long noncoding RNA lnc-DC controls human dendritic cell differentiation. Science (1979) 344, 310–313 (2014).
Zhang, J. et al. Long noncoding RNA TSLNC8 is a tumor suppressor that inactivates the interleukin‐6/STAT3 signaling pathway. Hepatology 67, 171–187 (2018).
Hentze, M. W., Castello, A., Schwarzl, T. & Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 19, 327–341 (2018).
Onoguchi-Mizutani, R. & Akimitsu, N. Long noncoding RNA and phase separation in cellular stress response. J. Biochem 171, 269–276 (2022).
Latos, P. A. et al. Airn transcriptional overlap, but not its LncRNA products, induces imprinted Igf2r silencing. Science 338, 1469–1472 (2012).
Guo, G. et al. Epigenetic resetting of human pluripotency. Development 144, 2748–2763 (2017).
Sarropoulos, I., Marin, R., Cardoso-Moreira, M. & Kaessmann, H. Developmental dynamics of lncRNAs across mammalian organs and species. Nature 571, 510–514 (2019).
Perry, R. B.-T. & Ulitsky, I. The functions of long noncoding RNAs in development and stem cells. Development 143, 3882–3894 (2016).
Yang, J. & Jiang, W. The role of SMAD2/3 in human embryonic stem cells. Front Cell Dev. Biol. 8, 653 (2020).
Dvir, S. et al. Uncovering the RNA-binding protein landscape in the pluripotency network of human embryonic stem cells. Cell Rep. 35, 109198 (2021).
Lee, S. et al. Noncoding RNA NORAD regulates genomic stability by sequestering PUMILIO proteins. Cell 164, 69–80 (2016).
Tichon, A. et al. A conserved abundant cytoplasmic long noncoding RNA modulates repression by Pumilio proteins in human cells. Nat. Commun. 7, 12209 (2016).
Kopp, F. et al. PUMILIO hyperactivity drives premature aging of Norad-deficient mice. Elife 8, e42650 (2019).
Elguindy, M. M. & Mendell, J. T. NORAD-induced Pumilio phase separation is required for genome stability. Nature 595, 303–308 (2021).
Munschauer, M. et al. The NORAD lncRNA assembles a topoisomerase complex critical for genome stability. Nature 561, 132–136 (2018).
Eggenberger, J., Blanco-Melo, D., Panis, M., Brennand, K. J. & tenOever, B. R. Type I interferon response impairs differentiation potential of pluripotent stem cells. Proc. Natl Acad. Sci. USA 116, 1384–1393 (2019).
Poirier, E. Z. et al. An isoform of Dicer protects mammalian stem cells against multiple RNA viruses. Science 373, 231–236 (2021).
Wu, X. et al. Intrinsic immunity shapes viral resistance of stem cells. Cell 172, 423–438.e25 (2018).
Sadzak, I. et al. Recruitment of Stat1 to chromatin is required for interferon-induced serine phosphorylation of Stat1 transactivation domain. Proc. Natl Acad. Sci. USA 105, 8944–8949 (2008).
Cimica, V., Chen, H.-C., Iyer, J. K. & Reich, N. C. Dynamics of the STAT3 transcription factor: nuclear import dependent on Ran and importin-β1. PLoS ONE 6, e20188 (2011).
Dodel, M. et al. TREX reveals proteins that bind to specific RNA regions in living cells. Nat. Methods 21, 423–434 (2024).
Niwa, H., Burdon, T., Chambers, I. & Smith, A. Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 12, 2048–60 (1998).
Levy, D. E. & Darnell, J. E. Stats: transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 3, 651–62 (2002).
Michalska, A., Blaszczyk, K., Wesoly, J. & Bluyssen, H. A. R. A positive feedback amplifier circuit that regulates interferon (IFN)-stimulated gene expression and controls type I and type II IFN responses. Front Immunol. 9, 1135 (2018).
Wang, W.-B., Levy, D. E. & Lee, C.-K. STAT3 negatively regulates type I IFN-mediated antiviral response. J. Immunol. 187, 2578–85 (2011).
Ho, H. H. & Ivashkiv, L. B. Role of STAT3 in type I interferon responses. Negative regulation of STAT1-dependent inflammatory gene activation. J. Biol. Chem. 281, 14111–8 (2006).
Wang, H. et al. STAT3 regulates the type I IFN-mediated antiviral response by interfering with the nuclear entry of STAT1. Int J. Mol. Sci. 20, 4870 (2019).
Garcia-Moreno, M. et al. System-wide profiling of RNA-binding Proteins uncovers key regulators of virus infection. Mol. Cell 74, 196–211.e11 (2019).
Wyler, E. et al. Transcriptomic profiling of SARS-CoV-2 infected human cell lines identifies HSP90 as target for COVID-19 therapy. iScience 24, 102151 (2021).
Wyler, E. et al. Single-cell RNA-sequencing of herpes simplex virus 1-infected cells connects NRF2 activation to an antiviral program. Nat. Commun. 10, 4878 (2019).
Gao, K. M. et al. Human nasal wash RNA-Seq reveals distinct cell-specific innate immune responses in influenza versus SARS-CoV-2. JCI Insight 6, e152288 (2021).
Butler, D. et al. Shotgun transcriptome, spatial omics, and isothermal profiling of SARS-CoV-2 infection reveals unique host responses, viral diversification, and drug interactions. Nat. Commun. 12, 1660 (2021).
Ramos, I. et al. Innate immune response to influenza virus at single-cell resolution in human epithelial cells revealed paracrine induction of interferon Lambda 1. J. Virol. 93, e00559–19 (2019).
Rusinova, I. et al. Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res. 41, D1040–6 (2013).
Ouyang, J. et al. NRAV, a long noncoding RNA, modulates antiviral responses through suppression of interferon-stimulated gene transcription. Cell Host Microbe 16, 616–626 (2014).
Nishitsuji, H. et al. Long noncoding RNA #32 contributes to antiviral responses by controlling interferon-stimulated gene expression. Proc. Natl Acad. Sci. USA 113, 10388–10393 (2016).
Shirahama, S. et al. Long noncoding RNA U90926 is crucial for herpes simplex virus type 1 proliferation in murine retinal photoreceptor cells. Sci. Rep. 10, 19406 (2020).
Agarwal, S. et al. The long non-coding RNA LUCAT1 is a negative feedback regulator of interferon responses in humans. Nat. Commun. 11, 6348 (2020).
Farberov, S. et al. Structural features within the NORAD long noncoding RNA underlie efficient repression of Pumilio activity. Nat. Struct. Mol. Biol. https://doi.org/10.1038/s41594-024-01393-5 (2024).
Magnus, M., Boniecki, M. J., Dawson, W. & Bujnicki, J. M. SimRNAweb: a web server for RNA 3D structure modeling with optional restraints. Nucleic Acids Res. 44, W315–W319 (2016).
Burke, J. M., Lester, E. T., Tauber, D. & Parker, R. RNase L promotes the formation of unique ribonucleoprotein granules distinct from stress granules. J. Biol. Chem. 295, 1426–1438 (2020).
Matheny, T., Van Treeck, B., Huynh, T. N. & Parker, R. RNA partitioning into stress granules is based on the summation of multiple interactions. RNA 27, 174–189 (2021).
Namkoong, S., Ho, A., Woo, Y. M., Kwak, H. & Lee, J. H. Systematic characterization of stress-induced RNA granulation. Mol. Cell 70, 175–187.e8 (2018).
Ulitsky, I. Evolution to the rescue: using comparative genomics to understand long non-coding RNAs. Nat. Rev. Genet. 17, 601–614 (2016).
Guo, Y. Utilization of different anti‐viral mechanisms by mammalian embryonic stem cells and differentiated cells. Immunol. Cell Biol. 95, 17–23 (2017).
Amit, M. et al. Human feeder layers for human embryonic stem cells. Biol. Reprod. 68, 2150–2156 (2003).
Hashimshony, T. et al. CEL-Seq2: sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol. 17, 77 (2016).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10 (2011).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Anders, S., Pyl, P. T. & Huber, W. HTSeq—a python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Walter, W., Sánchez-Cabo, F. & Ricote, M. GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics 31, 2912–2914 (2015).
Kong, A. T., Leprevost, F. V., Avtonomov, D. M., Mellacheruvu, D. & Nesvizhskii, A. I. MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat. Methods 14, 513–520 (2017).
Tyanova, S. et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016).
Pradhan, A. K., Shi, Q., Tartler, K. J. & Rammes, G. Quantification of astrocytic synaptic pruning in mouse hippocampal slices in response to ex vivo Aβ treatment via colocalization analysis with C1q. STAR Protoc. 3, 101687 (2022).
Luecken, M. D. & Theis, F. J. Current best practices in single‐cell RNA‐seq analysis: a tutorial. Mol. Syst. Biol. 15, e8746 (2019).
Ilicic, T. et al. Classification of low quality cells from single-cell RNA-seq data. Genome Biol. 17, 29 (2016).
Lun, A. T. L., McCarthy, D. J. & Marioni, J. C. A step-by-step workflow for low-level analysis of single-cell RNA-seq data with bioconductor. F1000Res 5, 2122 (2016).
Gascuel, O. BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 14, 685–695 (1997).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245 (2016).
Meng, E. C. et al. UCSF ChimeraX: tools for structure building and analysis. Protein Sci. 32, e4792 (2023).
Acknowledgements
This work was supported by the Israel Science Foundation (ISF), grant numbers 1182/16 and 1556/22 granted to YMG. We thank Nitsan Fourier from the Technion Genome Center for help and advice with RNA-Seq experiments, Nitsan Dahan, Yael Lupu-Haber from the Life Sciences & Engineering infrastructure center for support and aid with microscopy imaging. We thank Anna Argoetti for her help in generating the illustration in Fig. 3. Thanks to Oded Beja and Andrey Rozenberg and Fabian Glazer for help with phylogenetic analysis and Oded Kleifeld for advice on the proteomics experimental design. We thank the Smoler Proteomics Center at the Technion for the proteomic analysis.
Author information
Authors and Affiliations
Contributions
A.A. and Y.M.G. conceived of the project; A.A. perform all experiments and analyzed the data; D.S. and G.P. contributed to the immunofluorescence and Mass spectrometry; G.P. assisted with the Co-IP and CLIP-qPCR experiments; N.S. helped with the ChIP experiments; T.H. prepared RNA-seq libraries; A.A., H.B. and T.L. performed RNA-seq analysis; Y.M.G. supervised the project and acquired funding; A.A. and Y.M.G. wrote the manuscript, and T.H. and G.P. provided critical assessment to the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Thomas Aune and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Argoetti, A., Shalev, D., Polyak, G. et al. lncRNA NORAD modulates STAT3/STAT1 balance and innate immune responses in human cells via interaction with STAT3. Nat Commun 16, 571 (2025). https://doi.org/10.1038/s41467-025-55822-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-55822-0
