
Abstract
Gemcitabine combined with cisplatin is the first-line chemotherapy for advanced cholangiocarcinoma, but drug resistance remains a challenge, leading to unsatisfactory therapeutic effect. Here, we elucidate the possibility of chemotherapy regimens sensitized by inhibiting succinylation in patients with cholangiocarcinoma from the perspective of post-translational modification. Our omics analysis reveals that succinylation of PDHA1 lysine 83, a key enzyme in the tricarboxylic acid cycle, alters PDH enzyme activity, modulates metabolic flux, and leads to alpha-ketoglutaric acid accumulation in the tumor microenvironment. This process activates the OXGR1 receptor on macrophages, triggering MAPK signaling and inhibiting MHC-II antigen presentation, which promotes immune escape and tumor progression. Moreover, we show that inhibiting PDHA1 succinylation with CPI-613 enhances the efficacy of gemcitabine and cisplatin. Targeting PDHA1 succinylation may be a promising strategy to improve treatment outcomes in cholangiocarcinoma and warrants further clinical exploration.
Similar content being viewed by others


Introduction
Cholangiocarcinoma, ranking as the second most prevalent primary hepatic malignancy following hepatocellular carcinoma, is known for its aggressive invasiveness and grim prognosis1,2. Despite chemotherapy remaining the standard first-line treatment for cholangiocarcinoma over the past decade, patient survival and treatment efficacy have been suboptimal, mainly due to chemotherapy resistance3,4. This highlights the urgent need for a deeper understanding of the molecular pathways regulating cholangiocarcinoma progression and sensitizing it to chemotherapy.
Metabolic reprogramming, a hallmark of cancer, is intricately linked to post-translational modifications (PTMs)5,6. In recent years, the role of acyl modifications, including succinylation, in tumorigenesis has garnered considerable attention7. Succinylation, which occurs on lysine residues of PTM8, plays a key role in several core metabolic pathways, such as the tricarboxylic acid (TCA) cycle and glucose metabolism9,10, and has been shown to regulate tumor progression in various cancers11,12,13. Recent studies have identified the impact of succinylation on PDHA1, a key enzyme in the TCA cycle. PDHA1 catalyzes the conversion of pyruvate to acetyl-CoA, thereby linking glycolysis with the TCA cycle14,15. Acetylation of PDHA1 has been extensively studied and shown to influence the composition of the pyruvate dehydrogenase complex and contribute to the Warburg effect, a common metabolic feature of cancer cells16,17. Additionally, disruption of PDH activity is associated with increased chemotherapy resistance, positioning PDHA1 as a crucial target in cancer therapy18,19.
Endogenous metabolites are implicated in modulating cellular processes, such as immune homeostasis and tumor development20,21. Cancer cell metabolism will further deteriorate the living environment of immune cells, and metabolic changes in the microenvironment are involved in tumor immune sensitivity22. Increasing evidence indicates that metabolic reprogramming is linked to the polarization state of macrophages, and TCA cycle as well as its endogenous metabolites are vital in M1/M2 macrophage activation23,24,25,26,27,28,29. Alpha-ketoglutaric acid (α-KG) is a TCA cycle intermediate that serves to produce ATP. Several recent studies have shown the importance of α-KG in the tumor microenvironment (TME) for macrophage polarization, delivery of α-KG reprogramming the macrophage phenotype from pro-inflammatory M1 to anti-inflammatory and pro-repair M2 phenotype by modulating energy metabolism in the TCA cycle30, supplementing the cell-permeable dimethyl-α-KG significantly restores M2 activation during porphyromonas gingivalis infection31, and melatonin has anti-inflammatory properties by transporting the exosome α-KG to macrophages and leads to an increased ratio of M2 to M1 macrophages32. Moreover, α-KG coordinates the M1/M2 phenotype balance of alveolar macrophages through immune response regulators (PPARγ or mTORC1), thereby influencing septic ALI/ARDS23.
In this work, we investigate PDHA1 K83 succinylation and its impact on tumor metabolism in cholangiocarcinoma. We show that succinylation of PDHA1 at lysine 83 enhances PDHA1 activity, driving metabolic reprogramming that leads to the accumulation of α-KG in the TME. This accumulation of α-KG activates the OXGR1 receptor on macrophages, triggering the MAPK signaling pathway, which inhibits macrophage antigen presentation and promotes immune suppression. Furthermore, we explore the therapeutic potential of using CPI-613 to inhibit PDHA1 succinylation, and consider that it may enhance the sensitivity of cholangiocarcinoma to gemcitabine and cisplatin. These investigations contribute to our understanding of how PDHA1 succinylation modulates tumor-immune crosstalk and present targeted succinylation modification as a promising strategy to overcome chemotherapy resistance in cholangiocarcinoma.
Results
PDHA1 K83 is highly succinylated in cholangiocarcinoma and closely relates to survival
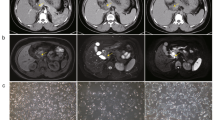
To elucidate the role of key metabolic enzyme succinylation in the progression of cholangiocarcinoma, our study comprehensively analyzed the succinylome of proteins from 10 human cholangiocarcinoma tissues and adjacent non-tumor samples. We discovered a significant upregulation of succinylation at PDHA1 lysine 83 in tumor tissues, marking it as the most pronounced modification observed (Fig. 1a, b). To determine if PDHA1 K83 succinylation occurs in vitro, we engineered mutations at K83 and other potential succinylation sites (K63, K244, K277, K313, K385) identified in the UniProt database to arginine (R), effectively mimicking desuccinylation. Notably, the K83R mutation substantially reduced PDHA1 succinylation (Supplementary Fig. S1a, b). Subsequent experiments confirmed PDHA1 K83 as the principal succinylation site in cholangiocarcinoma by utilizing Flag-tagged PDHA1 WT and K83R in QBC939 and RBE cholangiocarcinoma cell lines, which lack endogenous PDHA1 expression. The succinylation levels were significantly diminished in PDHA1 K83R compared to WT, underscoring the specific role of K83 (Supplementary Fig. S1c–i). A specific antibody developed for PDHA1 K83 succinylation, whose specificity was validated, enabled in situ detection of this modification (Supplementary Fig. S1j, k). Immunohistochemical analysis of a tissue microarray with 74 cholangiocarcinoma samples demonstrated elevated PDHA1 K83Succ levels in tumor tissues relative to adjacent normal tissues (Fig. 1c), and higher levels of PDHA1 K83Succ were associated with poorer patient prognosis, highlighting its potential as a prognostic marker (Fig. 1d). Importantly, the increased PDHA1 K83 succinylation observed in cholangiocarcinoma was not attributable to PDHA1 overexpression, which suggests a regulatory mechanism intrinsic to tumor biology (Supplementary Fig. S1l–n). Additionally, immunofluorescence (IF) (Fig. 1e) and Western blot analyses of clinical patient samples and cholangiocarcinoma cell lines further verified that PDHA1 K83Succ was predominantly expressed in cholangiocarcinoma cells, confirming their role as the primary source of PDHA1 K83Succ (Supplementary Fig. S1o, p).

a Upregulated genes revealed by succinylation proteomic analysis of cholangiocarcinoma and adjacent non-tumor tissues (n = 10). b Mass spectrometry identified the succinylation modification site K83 on PDHA1. c Quantification of PDHA1 K83Succ immunostaining performed to compare paired samples (n = 74). d Kaplan–Meier analysis of OS curves based on PDHA1 K83Succ expression. PDHA1 K83Succ expression associated with poor prognosis in CCA patients. e Representative immunofluorescence staining of CCA and adjacent normal tissues (n = 7). PDHA1 K83Succ (Red), DAPI (Blue). And statistical chart of PDHA1 K83Succ mean gray values. Scale bars: 50 μm. f Schematic illustrating transposon- and CRISPR-Cas9-(p53)-based injection of vectors into mice via the tail vein to generate tumors resembling human CCA. The transposon-based vector overexpressing KrasG12D can express luciferase. Created in BioRender. Zhang, N. (2025) https://BioRender.com/y78d999. Bioluminescence images and signal of C57BL/6 mice after plasmid injection on day 7 and day 28 (n = 10 biologically independent samples). g Front and back representative images of livers from (f). Scale bars: 1 cm. h Kaplan–Meier survival curve of C57BL/6 mice treated as in (f). i Tumor incidence of C57BL/6 mice treated as in (f). j Schematic illustrating transposon- and CRISPR-Cas9-(Pten)-based injection of vectors into mice via the tail vein to generate tumors resembling human CCA. The transposon-based vector overexpressing KrasG12D can express luciferase. Created in BioRender. Zhang, N. (2025) https://BioRender.com/j69i397. Bioluminescence images and signal of C57BL/6 mice after plasmid injection on day 7 and day 28 (n = 8 biologically independent samples). k Front and back representative images of livers from (j). Scale bars: 1 cm. l Kaplan–Meier survival curve of C57BL/6 mice treated as in (j). m Tumor incidence of C57BL/6 mice treated as in (j). Data are presented as mean values ± SD, and P values were calculated using two-tailed unpaired Student’s t-tests (c, e, f, j) or log-rank test (d, h, l). HR hazard ratio, WT wild type. Source data are provided as a Source Data File.
Results from a series of gain-and-loss-of-function experiments indicated that PDHA1 K83 desuccinylation did not affect PDHA1 proliferation in vitro, yet significantly curtailed tumor growth in vivo within a complex TME (Supplementary Figs. S2 and S3a–d). Notably, immunodepletion of CD8+T cells effectively negated the growth suppression seen in tumors with desuccinylated PDHA1 in these immunocompetent mice (Supplementary Fig. S3e–g), emphasizing the critical role of effector CD8+ T cells in mediating the impact of PDHA1 succinylation on tumor growth. However, it is essential to consider that subcutaneous xenograft models cannot replicate the comprehensive hepatic microenvironment. Consequently, we utilized a well-documented murine model of cholangiocarcinoma, combining p53/Pten deletions with KrasG12D overexpression33. Through hydrodynamic injection of plasmids into 5-week-old C57BL/6 male mice, bioluminescence imaging performed on day 7 post-injection confirmed equivalent luciferase expression across the groups, indicating uniform injection and expression efficacy. The experimental mice injected with PT3-PDHA1 K83R demonstrated a significant decline in luciferase expression by day 28 post-injection compared to the control group injected with PT3-PDHA1 WT (Fig. 1f, j). Furthermore, PDHA1 K83Succ levels were markedly lower in the spontaneous tumors of PDHA1 K83R mice compared to their WT counterparts (Supplementary Fig. S3h, i). Remarkably, four (4/10) of the p53 knockout experimental mice were tumor-free within 80 days, while the majority of the control mice (10/10) developed tumor burdens that led to death, with a median survival time of 42.5 days (Fig. 1g–i). Similar results were observed in Pten knockout mice, where PDHA1 K83 desuccinylation prolonged the median survival time; three (3/8) of the Pten knockout experimental mice remained tumor-free for 80 days, whereas all control mice (8/8) succumbed to tumor burdens, with a median survival time of 41.5 days (Fig. 1k–m). Collectively, these findings affirm that PDHA1 K83 succinylation significantly promotes CCA tumorigenesis, highlighting its pivotal role in the progression of cholangiocarcinoma.
Single-cell and Bulk RNA-seq analyses together reveal that PDHA1 K83 succinylation inhibits macrophage antigen presentation function
To illustrate the TME characteristics influenced by PDHA1 K83 succinylation, we conducted single-cell RNA sequencing (scRNA-seq) on tissue samples from a sg-p53 spontaneous mouse model of cholangiocarcinoma harboring either PT3-PDHA1-WT or K83R mutations. Post quality control, analysis proceeded with 15,830 cells. Gene expression was normalized, followed by dimensionality reduction and clustering using t-distributed stochastic neighbor embedding (tSNE) (Fig. 2a). Cells were classified into eight distinct types based on expression of specific marker genes: Macrophages (11,000 cells, 69.5%, marked by Cd68, C1qa, C1qb); Neutrophils (1439 cells, 9.1% marked by S100a9, S100a8, Lst1); Dendritic Cells (DCs) (1665 cells, 10.5%, marked by Cd209a, Ccr7, Clec10a); T cells (501 cells, 3.2%, marked by Cd3d, Cd3e, Cd3g); Endothelial/Fibroblasts (Endo/Fibro) (85 cells, 0.5%, marked by Col4a1, Vwf, Eng); Epithelial Cells (255 cells, 1.6%, marked by Krt18, Krt8, Clu); B cells (75 cells, 0.5%, marked by Cd79a, Cd79b, Ms4a1) and Proliferative Cells (810 cells, 5.1%, marked by Mki67, Top2a). Heatmaps (Fig. 2b) and Dot Plots (Supplementary Fig. S4a) further validated the cell type classification. Notably, macrophages constituted the largest proportion in both groups (Fig. 2c). Bulk RNA-seq from spontaneous tumors also indicated a significant enrichment of macrophage-specific markers, as identified in Wiki Pathways (Supplementary Fig. S4b–d). Consequently, we concentrated on macrophages for further analysis, performing tSNE specifically on this cell cluster (Fig. 2d). Macrophages were subdivided into five types using established markers: C1qc+ Macrophages (marked by C1qa, C1qb, C1qc), Ly6c+ Macrophages (marked by Ly6c2, Ly6e, Ccr2, S100a4), Chil3+ Macrophages (marked by Chil3, Lcn2, Gal), Cxcl9+ Macrophages (marked by Cxcl9, Cxcl10, Cd40, Batf2, Gbp2), and SPP1+ Macrophages (marked by Spp1, Arg1, Thbs1, Trem2). Each type corresponded to different immune phenotypes, chemokine profiles, and maturation states. Further verification of cell type accuracy was provided through Dot Plot (Fig. 2e) and UMAP (Supplementary Fig. S4f) analyses.

a tSNE (t-Distributed Stochastic Neighbor Embedding) plots of 15,830 high-quality single cells from spontaneous cholangiocarcinoma mice. b Heatmaps displaying marker genes for each cell type. c Histogram showing the proportion histogram of the eight major subtypes WT (left) and K83R (right). d tSNE plots of five different macrophage subgroups. e Dot plots showing the expression of marker genes for each cell type. f Quantitative RT-PCR analysis of mRNA levels of Cd74, H2-Ab1, H2-Aa, Cxcl9, Cxcl10 changes from PDHA1 WT or PDHA1 K83R spontaneous cholangiocarcinoma mouse tumors isolated CD45+CD11b+F4/80+ macrophages (n = 3 biologically independent samples). g Flow cytometry analysis of CD45+CD11b+F4/80+MHCII+ cells expression in mice with PDHA1 WT or PDHA1 K83R after hydrodynamic tail vein injection (n = 5 biologically independent samples). h Representative mIHC staining of spontaneous cholangiocarcinoma mouse tumors. MHCII (Red), CD86 (Green), PDHA1 K83Succ (Yellow) and DAPI (Blue). Scale bars: 20 μm. i Quantification of PDHA1 K83Succ+ and CD86+MHCII+ cells in PDHA1 WT and PDHA1 K83R spontaneous cholangiocarcinoma mouse tumors (n = 5 biologically independent samples). j Representative mIHC staining of human CCA samples using antibodies against HLA-DR (Red), CD86 (Green), PDHA1 K83Succ (Yellow) and DAPI (Blue). Scale bars: 20 μm. k Pearson correlation between the levels of PDHA1 K83Succ+ and CD86+HLA-DR+ in human CCA samples (n = 20). Data are presented as mean values ± SD, and P values were calculated using two-tailed unpaired Student’s t-tests (f, g, i) or two-tailed correlation (k). Mat matrix, Macro macrophages, WT wild type. RNA-seq data is in the Genome Sequence Archive database and source data are provided as a Source Data File.
The composition of various immune cells in the TME revealed that PDHA1 K83R mutation inhibits M2 macrophage polarization while promoting M1 polarization, thereby exerting antitumor effects (Fig. 2f, g and Supplementary Fig. S5a–e). Gene Ontology (GO) analysis of RNA sequencing data indicated a significant upregulation in antigen presentation processes and immune progression in PDHA1 K83R mutants compared to WT, emphasizing its role in immunomodulation (Supplementary Fig. S5f). The recognition of macrophage polarity between CXCL9 and SPP1 controls the cellular network in cancer as evidenced by previous studies34. Consequently, focusing on M1 polarization, particularly the CXCL9+ macrophage subset, we explored its contribution to antitumor immunity. KEGG pathway enrichment analysis further underscored the involvement of CXCL9+ macrophages in antigen presentation (Supplementary Fig. S5g). Moreover, multiplex immunohistochemistry (mIHC) on spontaneous tumors showed greater infiltration of CD86+MHCII+ cells in PDHA1 K83R mice compared to WT (Fig. 2h, i). Extending these findings to human relevance, we evaluated PDHA1 K83Succ, CD86, and HLA-DR expression in cholangiocarcinoma tissues. mIHC analysis revealed higher proportions of CD86+HLA-DR+ cells in patients with lower PDHA1 K83Succ levels (Fig. 2j), aligning with mouse model data where CD86+HLA-DR+ expression inversely correlated with PDHA1 K83Succ in clinical specimens (Fig. 2k). This highlights PDHA1 K83 succinylation’s inhibitory effect on macrophage antigen-presentation. In the T cell compartment, distinct subsets were identified: CD4+T, CD8+T, Proliferative T, and NK T cells. Notably, mice harboring the PDHA1 K83R mutation showed increased infiltration of CD4+T and CD8+T cells, suggesting that desuccinylation of PDHA1 K83 enhances macrophage MHC II-mediated antigen presentation, thus facilitating an effective immune response (Supplementary Fig. S6).
PDHA1 K83 succinylation modification increases α-KG in the tumor microenvironment
To investigate the impact of PDHA1 succinylation on macrophage antigen presentation within the TME, we devised a co-culture system of macrophages and cholangiocarcinoma cells (Fig. 3a). Our results demonstrated that the PDHA1 K83R mutation significantly improved the antigen-presenting capability of THP-1 cells (Fig. 3b). Given PDHA1’s pivotal role in linking glycolysis and the TCA cycle, we hypothesized that essential metabolites released into the TME are critical in this process. Supporting this, recent studies highlight the significant effect of metabolic intermediates on macrophage polarization and function35,36. The TCA cycle serves as a central metabolic node for the metabolism of proteins, lipids, and carbohydrates37,38, playing a vital role in tumor metabolic reprogramming. A functional TCA cycle depends on the continuous and balanced replenishment of its key metabolites, many of which exit the mitochondria to act as precursors for biosynthesis or to participate in signaling pathways37,39. Consequently, our focus shifted to examining the role of tumor-derived metabolites in modulating the antigen-presenting function of macrophages, which appears to be directly influenced by key metabolic enzymes and their substrates.

a Schematic diagram of THP-1 macrophages and cholangiocarcinoma cells co-culture. Created in BioRender. Zhang, N. (2025) https://BioRender.com/c42h873. After 24 h, macrophages were used for flow analysis, and CM was used for metabolite detection (n = 5 biological replicates). b Flow cytometry analysis of HLA-DR+ MFI values after co-culturing THP-1 and the CM of QBC939 sgPDHA1 with reconstituted expression of PDHA1 WT or PDHA1 K83R after 24 h (n = 5 biological replicates). c Heatmap representing metabolite changes in tumor WT CM compared with K83 CM was obtained after 24 h culture of tumor cells (n = 5 biological replicates). d Schematic diagram of the TCA cycle metabolism in cancer. e Metabolite secretion of TCA cycle metabolic substrates α-KG, succinate, fumarate, malate and citrate changes in CM from QBC939 sgPDHA1 with reconstituted expression of PDHA1 WT or PDHA1 K83R. Analysis was performed after 24 h (n = 5 biological replicates). f Flow cytometry analysis of HLA-DR+ MFI values of THP-1 in co-culture with tumor CM. Analysis was performed after 24 h (n = 5 biological replicates). g PDH enzyme activity of QBC939 sgPDHA1 with reconstituted expression of PDHA1 WT and PDHA1 K83R (n = 5 biological replicates). Activity is represented as the rate in ΔOD/min. h Western blot analysis showed the expression of PDHA1 pS293, PDHA1 K83Succ changes in QBC939 sgPDHA1 with reconstituted expression of PDHA1 WT, PDHA1 K83R, PDHA1 S293A and PDHA1 K83R S293A. The samples derive from the same experiment but different gels for PDHA1 pS293, PDHA1 K83Succ, PDHA1 and α-Tubulin were processed in parallel. The experiment was repeated three times with similar results. i PDH enzyme activity of QBC939 sgPDHA1 with reconstituted expression of PDHA1 WT, PDHA1 K83R, PDHA1 S293A and PDHA1 K83R S293A (n = 5 biological replicates). Activity is represented as the rate in ΔOD/min. j CM α-KG concentration of QBC939 sgPDHA1 with reconstituted expression of PDHA1 WT, PDHA1 K83R, PDHA1 S293A and PDHA1 K83R S293A (n = 5 biological replicates). Data are presented as mean values ± SD, and P values were calculated using two-tailed unpaired Student’s t-tests. Mac macrophages, CM conditioned medium, MFI mean fluorescence intensity. Source data are provided as a Source Data File.
To delineate specific metabolite alterations potentially responsible for inhibiting macrophage antigen presentation within the TME, we compared the metabolomic profiles of PDHA1 WT conditioned medium (CM) and PDHA1 K83R CM (Supplementary Fig. S7). Our analysis identified critical TCA cycle metabolites, including α-KG, succinate, and malate, which were secreted in response to PDHA1 succinylation. Notably, this secretion could be reversed by the desuccinylation of PDHA1 (Fig. 3c–e), with α-KG secretion being most significantly reduced by the PDHA1 K83R mutation. To further explore the influence of these metabolites, we augmented the extracellular media with them and observed that only the addition of α-KG led to decreased MHC-II expression (Supplementary Figs. S8 and S9a–e). Additionally, introducing α-KG to K83R CM sufficiently impaired macrophage antigen presentation in THP-1 cells (Fig. 3f) and bone marrow-derived macrophages (BMDMs) co-cultures (Supplementary Fig. S9f). The inhibitory effect of α-KG on macrophage antigen presentation was corroborated by in vivo experiments (Supplementary Fig. S9g). Collectively, these findings confirm that α-KG accumulation characterizes the immunosuppressive TME and suggest that reduced α-KG levels resulting from tumor-derived PDHA1 K83 desuccinylation could drive changes in macrophage antigen presentation.
To validate the accumulation mechanisms of α-KG within the TME, we measured α-KG levels in both tumor cells and CM, observing reduced levels following PDHA1 K83 desuccinylation (Supplementary Fig. S10a, b). These findings prompted us to explore the potential mechanisms by which PDHA1 K83 desuccinylation elevates α-KG levels. We further confirmed that PDHA1 succinylation-mediated regulation of α-KG generation in a PDH-dependent manner by performing metabolic flux analysis using [U-13C6] glucose and [U-13C5] glutamine. We noted a significant increase in M2 α-KG enrichment in PDHA1 WT compared to PDHA1 K83R using [U-13C6] glucose (Supplementary Fig. S10c, d). However, M5 a-KG synthesis by [U-13C5] glutamine showed no difference between the groups (Supplementary Fig. S10e, f). Consequently, we concluded that PDHA1 succinylation enhances α-KG production by modulating PDH activity. It is well-documented that PDH activity is crucial for controlling the TCA cycle40. In our experiments, a marked reduction in PDH activity was observed in cells expressing the PDHA1 K83R mutant compared to WT (Fig. 3g), with no impact on protein stability (Supplementary Fig. S11a). Previous studies indicate that phosphorylation of PDHA1 at S293 is a negative regulator of PDH complex activation, which is dependent on dephosphorylation of PDHA141,42. We observed increased phosphorylation at PDHA1 S293 upon desuccinylation of K83 (Supplementary Fig. S11b). Notably, the absence of S293 phosphorylation prevented any decrease in PDH activity or α-KG content in CM associated with PDHA1 K83 desuccinylation (Fig. 3h–j and Supplementary Fig. S11c, d), confirming that PDH activity is contingent on the succinylation status of PDHA1 K83. Overall, succinylation at PDHA1 K83 inhibits S293 phosphorylation, thereby enhancing PDH activity and promoting the release of α-KG into the TME.
α-KG-OXGR1-MAPK signaling pathway inhibits macrophage antigen presentation function
To further elucidate the impact of α-KG on macrophage function, we conducted RNA-seq analysis comparing gene expression in THP-1 cells co-cultured with or without exogenous α-KG-enriched CM. The analysis revealed that α-KG accumulation suppressed expression of immune response genes, such as HLA-DRA, HLA-DPA1, and HLA-DQA1, which are crucial for the MHC-II antigen presentation capability of macrophages (Fig. 4a, b and Supplementary Fig. S12a, b). Previous studies have demonstrated that LPS-induced downregulation of HLA-DR involves activation of ERK and p38 MAPK pathways43,44, and DcR3 inhibits HLA-DR expression through activation of ERK and JNK pathways45. We explored whether α-KG could similarly modulate MHC-II expression via MAPK activation. Gene Set Enrichment Analysis (GSEA) indicated significant activation of the MAPK signaling pathway by elevated α-KG levels in the TME (Fig. 4c). Furthermore, adding varying concentrations of α-KG to the co-cultured macrophages led to ERK phosphorylation and reduced expression of antigen presentation-related proteins HLA-DR and CD74 (Fig. 4d and Supplementary Fig. S12c, d). These findings suggest that α-KG modulates MHC-II molecule expression through ERK phosphorylation. However, the precise mechanisms by which α-KG activates the MAPK signaling pathway to influence the antigen-presenting function of macrophages warrant further investigation.

a Gene Ontology (GO) analysis of RNA-seq after 24 h co-culture of tumor CM with or without α-KG (8 μM) and THP-1 cells. b Volcano plot to depict the downregulated HLA-DRA, HLA-DPA1, and HLA-DQA1 expression changes of immune response observed in the RNA-seq data. c Gene Set Enrichment Analysis (GSEA) indicating that positive regulation of MAPK cascade was significantly upregulated after α-KG accumulation. d Western blot analysis showed the expression of the MAPK pathway and HLA-DR, CD74 changes after 24 h co-culture (n = 3 biological replicates). The samples derive from the same experiment but different gels for p-ERK, ERK, and HLA-DR, another for CD74 and p-P38, another for p-JNK, JNK, and another for P38 and α-Tubulin were processed in parallel. e Diagram of interaction between α-KG and OXGR1 protein using molecular docking. f Western blot analysis showed the expression of p-ERK, HLA-DR, and CD74 changes after 24 h co-culture (n = 3 biological replicates). The samples derive from the same experiment but different gels for p-ERK, ERK, and HLA-DR, another for CD74, and another for OXGR1 and α-Tubulin were processed in parallel. g Schematic diagram of THP-1 with or without OXGR1 depletion and tumor CM with α-KG (0, 8 μM). Created in BioRender. Zhang, N. (2025) https://BioRender.com/a10o519. After 24 h, macrophages were used for flow analysis. h Flow cytometry analysis of HLA-DR+ MFI values from data (g) (n = 4 biological replicates). i Schematic diagram of subcutaneous tumor treatment. Created in BioRender. Zhang, N. (2025) https://BioRender.com/j80q160. And representative images of vehicle, montelukast, α-KG or combined on day 26 (n = 5 biologically independent samples). j Tumor growth of LTP-C9 with overexpression of PDHA1 WT were injected into C57BL/6 mice (n = 5 biologically independent samples), with vehicle, montelukast, α-KG or combined treatment, beginning on day 14. k Tumor volume from data (i) (n = 5 biologically independent samples). l Tumor weight from data (i) (n = 5 biologically independent samples). Data are presented as mean values ± SD, and P values were calculated using two-tailed unpaired Student’s t-tests (d, f, h, k, l) or 2way ANOVA (j). Each Western blot was independently repeated three times. Mac macrophages, CM conditioned medium, MFI mean fluorescence intensity. Source data are provided as a Source Data File.
Literature indicates that α-KG serves as a ligand for the 2-oxoglutarate receptor1 (OXGR1), activating this surface receptor and initiating a G protein-coupled signaling cascade leading to transcriptional modifications46. Molecular docking analysis revealed a binding affinity score of −5.6 kcal/mol between α-ketoglutarate and LCK, suggesting a robust interaction (Fig. 4e). To validate this interaction, we performed OXGR1 knockdown experiments in macrophages (Supplementary Fig. S13a, b). Results showed that OXGR1 knockdown negated the impact of α-KG in CM on ERK phosphorylation and downregulation of MHC-II related molecules in macrophages (Fig. 4f–h and Supplementary Fig. S13c–f). Additionally, montelukast, a known inhibitor of OXGR147,48. When used it in subcutaneous mouse tumors, enhanced MHC-II expression (Supplementary Fig. S13g) and showed anticancer activity exclusively in immune-competent models, unaffected by external α-KG supplementation (Fig. 4i–l, Supplementary Fig. S13h–k). In summary, these findings demonstrate that α-KG in the TME engages the OXGR1 receptor, activates a G-protein-coupled signaling pathway, leads to ERK phosphorylation, and suppresses MHC-II class molecule expression, thereby diminishing the antigen-presenting function of macrophages.
DLST is the writer for PDHA1 K83 succinylation modification
The discovery of various lysine acylations raises questions about their dynamic regulation and the enzymes responsible49,50. Acyltransferases play a significant role in succinylation by coupling acyl-CoA to substrates, which may facilitate lysine acylation as a secondary reaction9. Using immunoprecipitation (IP) and liquid chromatography-tandem mass spectrometry (LC-MS/MS), we identified the succinyltransferase DLST as interacting with PDHA1 and promoting its succinylation (Supplementary Fig. S14a, b). This interaction was further validated through co-immunoprecipitation (Supplementary Fig. S14c), GST pull-down (Supplementary Fig. S14d, e) and immunofluorescence assays (Fig. 5a). Furthermore, the ectopic expression of DLST, but not other succinyltransferases such as HAT1, KAT2A or CPT1A, promotes succinylation of PDHA1 (Fig. 5b). To determine if DLST succinylates PDHA1 at K83, we transfected Flag-PDHA1 WT and Flag-PDHA1 K83R and found that the K83R mutant failed to be succinylated by DLST (Fig. 5c). Notably, DLST expression correlated positively with PDHA1 K83 succinylation in cholangiocarcinoma samples. (Supplementary Fig. S14f–j). DLST deletion reduced PDHA1 K83 succinylation, increased S293 phosphorylation, and decreased both PDH activity and α-KG levels in CM (Fig. 5d–g and Supplementary Fig. S15a–d). Moreover, CM from cells with reduced PDHA1 succinylation increased HLA-DR expression in co-cultured macrophages (Fig. 5h and Supplementary Fig. S15e). We confirmed DLST’s specific role in succinylating PDHA1 at K83 by showing that the K83R mutation prevented this modification (Fig. 5i, j and Supplementary Fig. S15f, g).

a Immunofluorescence analysis to detect colocalization of PDHA1 and DLST in HCCC9810 and RBE cells. Scale bars: 20 μm. b Western blot analysis of input and anti-FLAG immunoprecipitates (IPs) derived from HEK293T cells transfected with FLAG-PDHA1 and HA-tagged EV, HAT1, KAT2A, CPT1A, DLST. The samples derive from the same experiment but different gels for Succ-K, another for HA, another for Flag, and another for α-Tubulin were processed in parallel. c Western blot analysis of input and anti-FLAG immunoprecipitates (IPs) derived from HEK293T cells transfected with FLAG-PDHA1-EV, FLAG-PDHA1 WT, FLAG-PDHA1 K83R, and HA-DLST. The samples derive from the same experiment but different gels for Succ-K, another for HA, another for Flag, and another for α-Tubulin were processed in parallel. d Western blot analysis of DLST changes from QBC939 cells. e Western blot analysis of PDHA1 K83Succ, PDHA1 pS293, PDHA1 changes from QBC939 cells after DLST depletion. The samples derive from the same experiment but different gels for PDHA1 K83Succ, PDHA1 pS293, and DLST, another for PDHA1, and another for α-Tubulin were processed in parallel. f PDH enzyme activity of QBC939 cells after DLST depletion (n = 5 biological replicates). Activity is represented as the rate in ΔOD/min. g CM α-KG concentration of QBC939 cells after DLST depletion (n = 3 biological replicates). h Flow cytometry analysis of HLA-DR+ MFI values after co-culturing the CM of QBC939 cells after DLST depletion and THP-1 after 24 h (n = 5 biological replicates). i Western blot analysis of PDHA1 pS293 and PDHA1 K83Succ changes from QBC939 sgPDHA1 with reconstituted expression of FLAG-PDHA1 WT and FLAG-PDHA1 K83R with or without DLST. The samples derive from the same experiment but different gels for PDHA1 pS293, PDHA1 K83Succ, and DLST, another for PDHA1, and another for α-Tubulin were processed in parallel. j CM α-KG concentration of QBC939 sgPDHA1 with reconstituted expression of FLAG-PDHA1 WT and Flag-PDHA1 K83R with or without DLST (n = 5 biological replicates). k Western blot analysis of PDHA1 K83Succ, PDHA1 pS293 changes from QBC939 sgPDHA1 with reconstituted expression of FLAG-PDHA1 WT cell line used CPI-613 (0, 50, 100 μM). The samples derive from the same experiment but different gels for PDHA1 K83Succ, PDHA1 pS293, another for PDHA1 and Actin were processed in parallel. l CM α-KG concentration of QBC939 sgPDHA1 with reconstituted expression of FLAG-PDHA1 WT cell line used CPI-613 (0, 50, 100 μM) (n = 5 biological replicates). m Flow cytometry analysis of HLA-DR+ MFI values after co-culturing the CM of QBC939 sgPDHA1 with reconstituted expression of FLAG-PDHA1 WT cell line used CPI-613 (0, 50, 100 μM) (n = 5 biological replicates). Data are presented as mean values ± SD, and P values were calculated using two-tailed unpaired Student’s t-tests. Each Western blot was independently repeated three times. CM conditioned medium, MFI mean fluorescence intensity. Source data are provided as a Source Data File.
PDH and OGDH complexes are both E1-E2-E3 dehydrogenases. PDH complex contains pyruvate dehydrogenase (PDHA1 And PDHB1), dihydrolipoyl acetyltansferase (DLAT), and dihydrolipoyl dehydrogenase (DLD)51. OGDH complex contains OGDH, DLST, and DLD. Both DLST and DLAT contain lipoyl groups and both can therefore be inhibited by CPI-613. It was thought that these complexes may interact in the mitochondria so it would be possible for the DSLT of OGDHc to succinylate PDHA152. Interestingly, the application of the DLST inhibitor CPI-613 not only inhibited PDHA1 K83 succinylation but also increased PDHA1 S293 phosphorylation and decreased α-KG levels (Fig. 5k, l). In co-culture experiments, CPI-613 enhanced MHC-II antigen presentation in macrophages (Fig. 5m). To test CPI-613’s specificity, we mutated PDHA1 K83 to glutamate (E), mimicking succinylation13. CPI-613 reduced α-KG levels in WT but not in the mutant (Supplementary Fig. S15h, i). These results highlight DLST’s direct role in PDHA1 succinylation and CPI-613’s potential in modulating this modification and the associated tumor immune microenvironment.
Inhibiting PDHA1 K83 succinylation significantly improves the survival of cholangiocarcinoma
With breakthroughs in theory and technology, a series of mature targeted drugs have been widely used in the treatment of cholangiocarcinoma, such as IDH1 inhibitors, etc. However, the single-drug regimen of targeted drugs cannot further extend the survival of CCA patients, but targeted drugs can improve the inhibitory immune microenvironment by participating in immune regulation. This insight underpins the rationale for integrating gemcitabine and cisplatin with targeted therapies to bolster their collective antitumor effects and yield superior clinical outcomes53,54,55,56. In advanced cases, the dual regimen of gemcitabine (Gem) and cisplatin (Cis) remains the cornerstone of first-line therapy, but expanding this to triplet regimens including immunotherapies is poised to transform treatment paradigms57,58,59.
CPI-613, a lipoic acid analog approved for specific cancers, disrupts mitochondrial metabolism by targeting the TCA cycle enzymes PDHA1 and KGDH (DLST), which has proven effective in preclinical models60,61. Our research explored whether CPI-613 could enhance the effects of Gem and Cis in cholangiocarcinoma. We employed a C57BL/6 subcutaneous tumor model, treating mice with CPI-613, Gem and Cis individually or in combination. The combination therapy notably improved outcomes, reducing tumor mass more effectively than single treatment (Fig. 6a–d). In a spontaneous cholangiocarcinoma model (Fig. 6e), treatments significantly reduced bioluminescence from cancer cells (Fig. 6f, g). Impressively, in the combination therapy group, a higher proportion of mice remained tumor-free compared to single-agent groups, demonstrating extended survival. Four (4/10) of the combined treatment group mice, one (1/10) of the CPI-613 and two (2/10) of the Gem+Cis treatment group were tumor-free within 90 days, with the median survival time of 79.5 days, 53.5 days, 53.5 days, respectively, while the majority of the vehicle treatment group mice (10/10) developed tumor burdens that led to death, with a median survival time of 41.5 days (Fig. 6h–j). Flow cytometry revealed that the combination therapy notably increased the presence of immune cells such as MHCII+ macrophages, CD4+T, CD8+T cells, pointing to an invigorated immune response (Fig. 6k–m and Supplementary Fig. S16a–c). Cholangiocarcinoma, known for its immunosuppressive microenvironment, shows limited response to immunotherapy. Targeting PDHA1 K83 succinylation, we explored enhancing the immune landscape by upregulating MHC II genes, and the potential synergy of CPI-613 with anti-PD1 therapy. In a mouse subcutaneous tumor model using CPI-613, anti-PD1, and their combination, both single treatment reduced tumor size, but the combination therapy significantly enhanced efficacy, underscoring CPI-613’s role in cholangiocarcinoma treatment (Supplementary Fig. S16d–h). These findings underscore the potential of CPI-613 to synergize with chemotherapeutic agents and immunotherapy, offering a promising strategy to counter the immunosuppressive microenvironment of cholangiocarcinoma and enhance patient outcomes.

a Schematic diagram of subcutaneous tumor treatment. Created in BioRender. Zhang, N. (2025) https://BioRender.com/g40g507. b Tumor growth of LTP-C9 with overexpression of PDHA1 WT were injected into C57BL/6 mice (n = 6 biologically independent samples), with vehicle, CPI-613, Gem+Cis or combined treatment, beginning on day 14, CPI-613 and vehicle were administered daily, Gem+Cis were administered on experimental days (ED) 1, 5, 9, and 13. c Representative images of tumor volumes from vehicle, CPI-613, Gem+Cis or combined treatment on day 30 (n = 6 biologically independent samples). d Tumor volume and weight from data (b) (n = 6 biologically independent samples). e Schematic diagram of spontaneous tumor treatment. Created in BioRender. Zhang, N. (2025) https://BioRender.com/v16u276. f Bioluminescence images of C57BL/6 mice after plasmid injection on day 7, and mice on day 28 after administration of vehicle, CPI-613, Gem+Cis or combined treatment. g Bioluminescence signal from data (e) (n = 10 biologically independent samples). h Front and back representative images of livers from (e). Scale bars: 1 cm. i Kaplan–Meier survival curve of C57BL/6 mice treated as in (e) (n = 10 biologically independent samples). j Tumor incidence from data (e). k Flow cytometry analysis of CD45+CD11b+F4/80+MHCII+ cells expression of vehicle, CPI-613, Gem+Cis or combined treatment (n = 5 biologically independent samples). l Flow cytometry analysis of CD45+CD3+CD4+T cells expression of vehicle, CPI-613, Gem+Cis or combined treatment (n = 5 biologically independent samples). m Flow cytometry analysis of CD45+CD3+CD8+T cells expression of vehicle, CPI-613, Gem+Cis or combined treatment (n = 5 biologically independent samples). Data are presented as mean values ± SD, and P values were calculated using two-tailed unpaired Student’s t-tests (d, g, k–m), 2way ANOVA (b) or log-rank test (i). Gem gemcitabine, Cis cisplatin. Source data are provided as a Source Data File.
Discussion
Chemotherapy resistance in cholangiocarcinoma remains a critical challenge, compelling further exploration into metabolic and signaling pathways influencing treatment outcomes. Succinylation of metabolic enzymes is emerging as a significant area of interest, given its impact on cancer metabolism and cell signaling50,62,63. For instance, studies have demonstrated that inhibiting SUCLG2 K93 succinylation mitigates mitochondrial dysfunction and tumor growth in lung cancer12, whereas succinylation of GLS K311 promotes pancreatic cancer cell proliferation64. Our focus is on the role of succinylation in modulating chemotherapy resistance in cholangiocarcinoma, particularly through profiling succinylation patterns in PDHA1 at K83—a key enzyme in the TCA cycle.
Our findings reveal that PDHA1 K83 succinylation is integral to the TME, it affects PDH enzyme activity by the phosphorylation of PDHA1 at S293, leading to metabolic reprogramming of the TCA cycle, as well as increases the concentration of the TME metabolic intermediate, α-KG. And significantly inhibits the antigen presentation function of macrophages. Despite metabolomic analysis revealing increases in other metabolites in the CM of cholangiocarcinoma with PDHA1 K83 succinylation modification, but the most pronounced was the high expression of α-KG, making it a dominant molecule in the cancer cell CM. By tracing [U-13C6] glucose, we confirmed that succinylation boosts α-KG production via PDH activity, although the precise mechanisms behind its excessive accumulation warrant further exploration.
It has been reported that α-KG produced by glutamine promotes M2 polarization via the Jmjd3 signaling pathway and inhibits M1 macrophage function in an HDAC-dependent manner by suppressing the NF-κB pathway65. Consistent with previous research findings, our study confirms that tumor-derived α-KG inhibits macrophage MHC-II antigen presentation function. Exercise-induced α-KG, through stimulation of muscle hypertrophy and lipid breakdown via the OXGR1 receptor66, suggests that cancer-derived α-KG might also activate the macrophage surface receptor OXGR1. Our research demonstrates that when the macrophage surface receptor OXGR1 is specifically knocked down by OXGR1 shRNA, the inhibitory effect of α-KG on macrophage antigen presentation is abolished. Through RNA-seq and Western blot analysis verification, we found significant changes in the MAPK pathway. It has been reported that LPS downregulates HLA-DR expression by activating ERK and p38MAPKs, and DcR3 activates the MAPK pathway to downregulate HLA-DR expression44. Therefore, α-KG may activate the MAPK signaling pathway, thereby regulating the expression of the HLA-DR antigen presentation molecule, playing a role in immunosuppressive tumor microenvironments. Our study also showed that the gradient addition of α-KG activates the MAPK pathway, downregulating the expression of mRNA levels such as HLA-DR. It has been reported that ITA interacts with the orthosteric site of OXGR1, triggering downstream signaling pathways, including calcium ion release and activation of ERK phosphokinase67. In mice with intact immune systems, blocking OXGR1 with montelukast enhanced macrophage MHCII antigen presentation and slowed tumor growth, an effect that external KG supplementation could not reverse. Our research indicates that α-KG activates the MAPK pathway, affecting HLA-DR expression and contributing to an immunosuppressive environment.
Through mass spectrometry and immunoprecipitation (IP) assays, our study identifies DLST as the succinyltransferase that enhances PDHA1 succinylation, markedly enhancing its modification. We found that succinylation of PDHA1 is effectively inhibited by CPI-613, a succinyltransferase inhibitor, which leads to decreased α-KG levels in the TME and restores the antigen-presenting capability of macrophage MHC class II molecules. This finding highlights the pivotal role of PDHA1 K83 succinylation in modulating the tumor immune microenvironment in cholangiocarcinoma and positions its inhibition as a strategic target for therapeutic intervention. Although CPI-613 did not meet its primary endpoint in the Phase III AVENGER 500 trial in pancreatic cancer with the FOLFIRINOX regimen68, it is important to consider its application in different contexts. Gemcitabine combined with cisplatin remains the standard first-line treatment for CCA, with proven efficacy. Our study explores the potential of CPI-613 to enhance chemotherapy sensitivity by inhibiting mitochondrial metabolism. This approach may potentially offer effective therapeutic avenues, especially when used in combination with established chemotherapies. However, ongoing and future clinical trials are essential to fully determine the viability and scope of CPI-613 as a therapeutic option in CCA and other malignancies.
In conclusion, this study not only elucidates the role of α-KG in regulating tumor-immune interactions but also highlights the potential of targeting metabolic modifications to overcome chemotherapy resistance in cholangiocarcinoma. Further, we explore the molecular basis of cholangiocarcinoma progression from the perspective of succinylation modifications, complements the gaps in explaining cholangiocarcinoma chemotherapy from the perspective of metabolic reprogramming and post-translational modifications and validates the possibility of succinylation modifications in sensitizing chemotherapy regimens.
Methods
The research methodologies adhered to the principles outlined in the World Medical Association’s Declaration of Helsinki and its subsequent amendments. Furthermore, the Ethics Committee of The First Affiliated Hospital of the University of Science and Technology of China (USTC) conducted a review and granted approval for this research.
Clinical cholangiocarcinoma patients
Cholangiocarcinoma and adjacent normal tissues were obtained from 20 patients at the First Affiliated Hospital of the University of Science and Technology of China. The study protocol was approved by the Institutional Review Board of the First Affiliated Hospital of the University of Science and Technology of China, and written informed consent was obtained from each patient. Detailed clinical data of the patients involved are summarized in Supplementary Table S1, including 13 males and 7 females with the age range 50–85 years old. Additionally, a cholangiocarcinoma tissue microarray HIBDA180SU01 was purchased from Shanghai Outdo Biotech Company.
Cell lines and cell culture
QBC939, HiBEpic, HuCCT1, TFK-1, CCLP1, KMBC, HEK293T, RAW264.7 and LTP-C9 cells were cultured in DMEM medium (Sigma) supplemented with 10% FBS (Sigma), 1% penicillin/streptomycin (Gibco). RBE, HCCC9810 and THP-1 cells were cultured in RPMI1640 medium (Sigma) supplemented with 10% FBS (Sigma), 1% penicillin/streptomycin (Gibco). QBC939 was kindly provided by Prof. Shuguang Wang (Third Military Medical University, Chongqing, China). Normal primary human biliary epithelial cells (HiBEpic, Cat#5100) were gained from ScienCell Research Laboratories. HuCCT1 and TFK-1 cell lines were kindly provided by the Cancer Cell Repository of the Tohoku University in Japan. CCLP1 and KMBC cell lines were provided by Prof. Tongsen Zheng (Harbin Medical University, China). HEK293T (CRL-3216) were purchased from ATCC. RBE (CL-0191), HCCC9810 (CL-0095), THP-1 (CL-0233) and RAW264.7 (CL-0190) cell lines were purchased from Procell Life Science&Technology. LTP-C9 was gifted from Prof. Ming Kuang (SYSU). All cells were maintained in a humidified incubator with 5% CO2 at 37 °C.
Plasmids
The plasmids pcDNA3.1-HA and pcDNA3.1-FLAG were provided by Prof. Qingsong Hu (USTC). pLKO.1-puro, psPAX2 and pMD2.G were gifts from Prof. Mian Wu (USTC). CMV-SB13 was obtained from Prof.Amaia Lujambio (ISMMS). KrasG12D cDNA were amplified and cloned into the pT3-EF1A-IRES-Luc vector. pX330p53 (59910) and pX330Pten (59909) were purchased from Addgene. PDHA1 cDNA and its site-directed mutagenesis were cloned into the pcDNA3.1-FLAG vector. HAT1, KAT2A, CPT1A, and DLST were constructed into pcDNA3.1-HA vector. For spontaneous tumor murine ICC, PDHA1 cDNA was inserted into the PX330, replacing sg-P53 and sg-Pten. The sequences for sgRNAs, shRNAs used in this study are listed in Supplementary Table S2.
Cell transfection, and virus infection
Cell transfection was performed using Lipofectamine 3000 (Thermo). For virus infection, viruses were generated by transfecting HEK293T cells with psPAX2, pMD2.G, and PLKO.1 or pCMV-3xflag. The virus particles were then collected for infection. Cells were selected with appropriate concentrations of puromycin (APExBIO).
Single-cell cloning and establishment of stable cell lines
To establish stable PDHA1 knockout cell lines, cells were diluted to obtain single clones, with approximately 100 clones cultured in 10 cm dishes for one week. Knockout cells expressing green fluorescence were identified using a fluorescence microscope. Fluorescent colonies were marked and individually picked into 96-well plates. Selected clones were expanded over one month. Protein levels were assessed by Western blot analysis to confirm complete PDHA1 knockout. Cell lines with complete PDHA1 knockout are used for subsequent experiments. PDHA1-depleted cells were transfected with FLAG-tagged wild-type PDHA1 (FLAG-PDHA1 WT) or the K83R mutant (FLAG-PDHA1 K83R) to generate stable rescue cell lines.
Animal
Wild-type C57BL/6 and nude mice were sourced from Shanghai SLAC Laboratory Animal while NSG mice was procured from Shanghai Model Organisms. The mice were housed at the First Affiliated Hospital of USTC and underwent a comprehensive examination before conducting the experiments to ensure their well-being and adaptation to the laboratory environment. The mice were maintained under the natural day and night conditions, ensuring that the mice have a consistent circadian rhythm. The ambient temperature in the housing facility was kept constant at 22 ± 2 °C. The relative humidity in the housing environment was maintained at 50 ± 10%. Animals were humanely euthanized upon showing symptoms or when tumor volume reached a diameter of 2 cm or greater, in accordance with survival curve and end-stage analysis protocols. This practice was endorsed as a humane endpoint by the Animal Ethics Committee of the First Affiliated Hospital of USTC.
Subcutaneous tumor
3–5 million cells per sample were injected subcutaneously into 5-week-old male nude mice, NSG mice or C57BL/6 mice. After about 4 ~ 5 weeks, the tumors were isolated for analysis. The tumor volume was calculated as Length × Width × Width × 0.5. Tumor burden did not exceed the recommended size. Tumor volume was measured with a caliper every 3 to 4 days. When the volume of the xenograft tumor reached approximately 500 mm3, mice were randomly selected and grouped for subsequent antibody or drug treatment. For antibody injections, anti-CD8 mAb (Bioxcell, BE0117) (200 μg/mouse, twice a week) injection was started one week before tumor implantation. For drug treatment, montelukast (5 mg/kg) was administered by intraperitoneal injection. Mice were ad libitum fed ordinary chow and water with 2% alpha-ketoglutarate. For combination treatment, CPI-613 (25 mg/kg), Gem (200 mg/kg) +Cis (5 mg/kg), anti-PD1 mAb (5 mg/kg) and IgG2a (5 mg/kg) were administered by intraperitoneal injection.
Spontaneous models of ICC by hydrodynamic tail-vein injection
The method of establishing the spontaneous mouse ICC models was described in previous studies69, 15 µg PT3-EF1α-KrasG12D-IRES-Luc, 15 µg pX330-sg-p53 or pX330-sg-Pten, 15 µg px330-PDHA1 WT (or PDHA1 K83R), and 5.5 µg CMV-SB13 were dissolved in 2 ml of saline and injected into the tail vein within 5 to 7 s in 5-week-old mice70. Mice were intraperitoneally injected with fresh luciferin (150 mg/kg) (Gold Bio) and imaged after 5 min. In vivo bioluminescence imaging to quantify liver tumor burden was performed using the IVIS Spectrum system. The luciferase signal was quantified using Living Image software, then background signals were subtracted for calculation.
Drug treatment
Alpha-ketoglutarate (M1277), malate (M1314), CPI613 (A4333), Gem (A8437), and Cis (A8321) were obtained from APExBIO. Dimethyl succinate (820150) was purchased from Merck. Montelukast (HY-13315A) was obtained from MCE. Cycloheximide (S7418) was gained from Selleck. Anti-PD1 mAb (BE0146) and IgG2a (BE0089) were received from Bioxcell.
Isolation of BMDMs
Sterilization required scissors forceps instruments. The mice were killed by spinal cord dislocation and soaked in 75% ethanol. Bone marrow-derived cells were aseptically collected from the leg bones of 6 to 8-week-old C57BL/6 mice and washed with RPMI-1640. The bone marrow was placed on ice, washed off the bone marrow cells, filtered with a 70 μm filter, and centrifuged at 300 g/min for 3 min. Red blood cell lysate droplets were added to bone marrow cells, left at room temperature for 5 min, neutralized with PBS, filtered with a 70 μm filter, and centrifuged at 300 g for 5 min. BMDMs were cultured in RPMI-1640 medium containing 10% FBS and 10 μg/ml M-CSF (PM1008, APExBIO) for 5–7 days, and subsequent experiments were conducted after the cells had fully adhered cultured at 37 °C and 5% CO2.
Immunohistochemistry
Tissue sections were baked in an oven for 30 min, deparaffinized in xylene, hydrated through graded alcohols, and subjected to antigen retrieval using Antigen Unmasking Solution (Vector Labs). Then, they were preincubated with 10% goat serum and incubated with a primary antibody overnight at 4 °C. After washing with PBST, sections were treated with a biotinylated secondary antibody (Vector Labs) for 1 h, followed by ABC reagent (Vector Labs) for 30 min, and visualized using a diaminobenzidine (DAB) kit (Vector Labs). And the slides were counterstained with hematoxylin (Sigma). For staining quantification, ImageJ was used to analyze the digital pathological images of antigen expression. The Histochemistry score (H-Score) was used for cholangiocarcinoma tissue microarray’s quantification. Based on the H-Score, patients were divided into two groups: a low expression group (H-Score below the median) and a high expression group (H-Score above the median) to compare the overall survival of the patients.
Multiplex immunohistochemistry staining
Tissue sections underwent deparaffinization followed by treatment with antigen unmasking solution (Vector Labs). Then, they were preincubated with 10% goat serum 30 min and incubated with a primary antibody overnight at 4 °C. After washing with PBST, the tissue sections were incubated with SignalStain® Boost IHC detection Reagent (Cell Signaling Technology) for 30 min in a moist, light-protected environment. The fluorescein tyramide signal amplification staining (1:100) was used to see each target. Before proceeding to the next step, an antigen retrieval procedure that uses microwaves to remove excess antibodies was performed, following heating of the slides with Antigen Unmasking solution (Vector Labs). At the very end, 4′,6-diamidino-2-phenylindole (DAPI; Vector Labs) was employed to visualize the nuclei within the cells. Axio Imager 2 microscope (Carl Zeiss, Germany) was used to obtain the images and quantified using Image J.
Immunofluorescence
Cells are fixed with 4% paraformaldehyde for 20 min, followed by permeabilization with 0.2% Triton X-100 in PBS for 10 min. After blocking with PBS containing 0.05% Tween 20 and 1% bovine serum albumin (Sigma) for 1 h at room temperature, cells are blocked with 5% BSA for 30 min, then incubated with primary antibodies overnight at 4 °C. Then, cells are incubated with secondary antibodies for 1 h at room temperature. Finally, an antifade reagent with 4′6 diamidino-2-phenylindole (DAPI; Vector Labs) was used. All images were acquired using a confocal laser scanning microscope (Zeiss LSM 800).
Immunoprecipitation
For exogenous immunoprecipitation assay, cells were collected and lysed in lysis buffer (20 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.1% Triton X-100) supplemented with a protease inhibitor cocktail on ice for 30 min. After sonication and centrifugation, the cell supernatant was collected and incubated with magnetic beads (88837 and A36797, Thermo Fisher Scientific) in mild rotation at 4 °C for 4–6 h. After washing three times with lysis buffer, the beads were boiled and assessed by Western blot analysis.
For endogenous immunoprecipitation assay, cells were collected and lysed in lysis buffer) supplemented with a protease inhibitor cocktail. Cell lysates were incubated with Co-IP–grade antibodies with gentle rotation at 4 °C overnight. The next day, antibodies were pulled down with magnetic beads (88802, Thermo Fisher Scientific) at room temperature with rotation for 3 h. After washing the beads three times with lysis buffer, the beads were boiled and assessed by Western blot analysis.
Protein purification
GST/GST-fusion and His-fusion plasmids were transformed into competent BL21 cells.
Protein purification was carried out using the GST-tag protein purification kit (Beyotime, P2262) and His-tag protein purification kit (Beyotime, P2226) as per the manufacturer’s instructions. Cells were lysed via ultrasonic fragmentation after overnight culture. The supernatant was collected by centrifugation, incubated with BeyoGold™ GST-tag or His-tag Purification Resin at 4 °C for 1 h and then washed with wash buffer. Finally, the fused proteins were eluted using elution buffer for subsequent analysis.
GST pull-down
GST fusion proteins (10 μg/15 μl) and His fusion proteins (10–20 μg) were incubated in lysis buffer (pH 7.4, 150 mM NaCl, 1 mM EDTA, 5% glycerol, 0.1% Triton X-100, 50 mM Tris-HCl, 1 mM DTT) supplemented with a protease inhibitor cocktail at 4 °C for 4 h. After washing thrice with wash buffer (pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.2% Triton X-100, 50 mM Tris-HCl, 1 mM DTT) also supplemented with a protease inhibitor cocktail, proteins were centrifuged, boiled, and analyzed by Western blot analysis.
Western blot analysis
Whole cell lysates were prepared using cell lysis buffer (Beyotime Biotechnology). Protein concentrations were quantified using the BCA Protein Assay (Thermo Scientific), and equal amounts of proteins were subjected to SDS-polyacrylamide gel electrophoresis and transferred onto PVDF membranes (Qiagen). After blocking in 5% non-fat milk in PBST for 1 h, membranes were incubated with the following primary antibodies at specified concentrations overnight at 4 °C to detect target proteins. Secondary antibodies were used for incubation at room temperature for 1 h. Target bands were detected using ECL Western blotting Substrate (Thermo) and visualized with a ChemiDoc imaging system (BioRad).
Antibodies
The antibodies were as follows: anti-Succinyllysine Mouse mAb (419, 1:1000 for Western blot), anti-Succinyllysine Rabbit pAb (401, 1:1000 for Western blot), anti-Succinyl-PDHA1(K83) Rabbit pAb (Cat#CM0401, Lot#ZC492M726P1P-ET2, 1:1000 for Western blot, 1:200 for IHC, IF and mIHC) are from PTM BioLab, Co. Ltd. Anti-DYKDDDDK Tag Rabbit mAb (14793, 1:1000 for Western blot), anti-α-Tubulin (2144, 1:1000 for Western blot), anti-CD86 Rabbit mAb (91882, 1:1000 for Western blot, 1:200 for mIHC), anti-DLST Rabbit mAb (11954, 1:1000 for Western blot, 1:500 for IHC and 1:200 for IF), anti-CD206/MRC1 Rabbit mAb (24595, 1:1000 for Western blot, 1:200 for IHC), anti-β-Actin Antibody (4967, 1:1000 for Western blot), anti-Phospho-Erk1/2 Rabbit mAb (4370, 1:1000 for Western blot), anti-MAPK Rabbit mAb (4695, 1:1000 for Western blot), anti-Phospho-p38 Rabbit mAb (4511, 1:1000 for Western blot), anti-p38 MAPK Rabbit mAb (8690, 1:1000 for Western blot), anti-Phospho-SAPK/JNK Rabbit mAb (4668, 1:1000 for Western blot), anti-SAPK/JNK (9252, 1:1000 for Western blot), anti-Phospho-STAT1 Rabbit mAb (9167, 1:1000 for Western blot), anti-Phospho-NF-kB Rabbit mAb (3033, 1:1000 for Western blot), anti-Phospho-AKT Rabbit mAb (4060, 1:1000 for Western blot), anti-HA-Tag Rabbit mAb (3724, 1:1000 for Western blot), anti-His rabbit mAb (2365, 1:1000 for Western blot), anti-GST rabbit mAb (2622, 1:1000 for Western blot) are from Cell Signaling Technology. Anti-PDHA1 Rabbit mAb (ab168379, 1:1000 for Western blot), anti-PDHA1 Mouse mAb (ab110330, 1:1000 for Western blot, 1:200 for IHC and IF), anti-HLA-DR Rabbit mAb (ab92511, 1:1000 for Western blot, 1:200 for mIHC), anti-CD74 Rabbit mAb (ab108393, 1:1000 for Western blot) are from Abcam. Anti-MHCII (14-5321-82, 1:1000 for Western blot, 1:200 for mIHC) from Thermo Fisher, anti-phospho-PDHA1-S293 Rabbit mAb (AP1022, 1:1000 for Western blot) and anti-OXGR1 Rabbit pAb (A15431, 1:1000 for Western blot) are from Abclonal. Anti-phospho-PDHA1-S232 mAb (81491-1-RR, 1:1000 for Western blot) and anti-phospho-PDHA1-S300 mAb (29583-1-AP, 1:1000 for Western blot) are from Proteintech.
For multiplex immunohistochemistry staining and immunofluorescence, Alexa Fluor 488 conjugated goat anti-mouse IgG (A11001), Alexa Fluor 488 conjugated goat anti-rabbit IgG (A11008). Alexa Fluor 555 conjugated goat anti-mouse IgG (A21422) and Alexa Fluor 555 conjugated goat anti-rabbit IgG (A21428) are from Thermo Fisher Scientific.
For flow antibodies, zombie (423106, 1:1000 for FC), anti-mouse CD45 (103138, 1:250 for FC), anti-mouse F4/80 (123114, 1:250 for FC), anti-mouse CD11b (101208, 1:250 for FC), anti-mouse MHCII (107606, 1:250 for FC), anti-mouse CD206 (141708, 1:250 for FC), anti-mouse CD3 (100248, 1:250 for FC), anti-mouse CD4 (100406, 1:250 for FC), anti-human MHCII (327006, 1:250 for FC), anti-mouse CD8a (100734, 1:250 for FC), anti-mouse CD11c (117305, 1:250 for FC), anti-mouse NK-1.1(156507, 1:250 for FC) are from Biolegend. CoraLite® Plus 488-conjugated Osteopontin Polyclonal antibody (CL488-22952, 1:100 for FC) is from Proteintech.
Colony formation assay
One thousand cells were seeded in 6 cm dishes. The medium was changed every three days, and cell status was monitored. After two weeks, the medium was discarded, the cells were washed with PBS (three times), fixed with methanol for 20 min, and stained with crystal violet for 30 min. After three times of PBS cleaning, pictures were taken and analyzed by image J.
CCK8 assay
Cells were seeded at 1000 per well in a 96-well plate with 200 µl of medium. On each measurement day, 90 µl of medium and 10 µl of Cell Counting Kit-8 reagent (DOJINDO) were added, followed by 1 h incubation in the dark at 37 °C. Absorbance was then measured at 450 nm using a microplate reader.
PDH activity assay
The PDH enzyme activity microplate assay kit (Abcam, ab109902) was used to measure PDH activity of cells. Cells were homogenized to determine protein concentrations by BCA Protein Assay kit (Thermo Scientific) and supernatants were used for PDH assay using manufacturer’s protocol.
α-KG quantification
After 24 h of cell culture, cells or conditioned medium (CM) were collected for α-Ketoglutarate (α-KG) detection using the Elabscience α-KG assay kit (E-BC-F047). For cell samples: 1 × 106 cells were homogenized in 200 μl saline (0.9% NaCl), centrifuged at 4 °C, 12,000 × g for 15 min, and the supernatant was collected. The supernatant was then passed through a 50 KD ultrafiltration tube and centrifuged at 4 °C, 12,000 × g for 15 min. The filtrate was collected for analysis. For CM samples: the supernatant was filtered using a 50 KD ultrafiltration tube at 4 °C, 12,000 × g for 15 min, and the filtrate was collected for analysis. The fluorescence values were measured using a fluorescence microplate reader at an excitation wavelength of 535 nm and emission wavelength of 587 nm, following the experimental procedure.
Preparation of conditioned medium and macrophage coculture assay
To prepare the conditioned medium, start by treating cells with fresh serum-free medium. After 24 h, collect the cell supernatant, and centrifuge it at 1000 × g for 10 min at 4 °C. Finally, filter the supernatant using a 0.22 µm filter (SLGP033R, Merck). For the macrophage coculture assay, differentiated THP1, RAW264.7 and isolated BMDMs were treated with homologous CM for 24 h. Macrophages were collected for quantitative RT-PCR, WB, and FCM analyses.
Flow sorting and flow cytometry analysis
In flow sorting experiments, mouse tumor single-cell suspensions were stained with anti-CD45, anti-CD11b, and anti-F4/80 antibodies. Flow cytometry gate strategy images are shown in Supplementary Fig. 17a. Macrophages were sorted for RNA extraction and qRT-PCR analysis.
In mouse experiments, single-cell suspensions were obtained from tissues treated with Miltenyi Biotec reagent kit. Mouse specimens extracted were immediately washed with phosphate-buffered saline (PBS) and divided into two equal parts. One part was used for preparing single-cell suspensions, and the other for subsequent flow cytometry experiments. Tissue digestion was performed in 15 ml centrifuge tubes, and the cell suspension was filtered through a 70 µm filter. It was then centrifuged at 200 g for 6 min at 4 °C to separate dead and red blood cells. Cells were washed twice and resuspended in PBS containing 0.5% bovine serum albumin (BSA, Sigma). After treatment with red blood cell lysis buffer, cells were incubated with corresponding antibodies on ice for 30 min. Following fixation and permeabilization, the suspension was incubated on ice and stained with antibodies in the dark. Single-cell suspensions were prepared from cells in culture and mouse tumors, stained with antibodies, and analyzed on a Cyto FLEX (Beckman Coulter) flow cytometer, and the representative flow cytometry gate strategy images are shown in Supplementary Fig. 17b–h.
In cells experiment, collected the co-cultured macrophages, washed them twice with pre-cooled PBS (500 g, 5 min, 4 °C), discarded the supernatant, and resuspended the cells in Staining Buffer (PBS containing 1% BSA) to a density of 1 × 106 cells/100 μl. Added the labeled antibodies and incubated at room temperature in the dark for 1 h. After incubation, washed the cells twice with pre-cooled Staining Buffer (500 g, 5 min, 4 °C), discarded the supernatant, and resuspended the cells in 500 μl Staining Buffer. Filtered the cell suspension through a 70 μm strainer and proceeded to flow cytometry analysis. Used the CytoFLEX flow cytometer for detection, and analyzed the data with FlowJo (version 10.8.1). The representative flow cytometry gate strategy images are shown in Supplementary Fig. 18.
RNA isolation and quantitative real-time PCR
Total RNA was isolated using the Gene JET RNA Purification Kit (Thermo Fisher) following the manufacturer’s recommendations, and cDNA was synthesized using Prime Script RT Master Mix (Takara). Quantitative PCR (qPCR) was performed using TB Green Premix Ex Taq II (Takara). Semi-quantitative PCR was conducted with Phanta Max Super-Fidelity DNA Polymerase (Vazyme). β-Actin was used as the internal control for mRNA, and the primer sequences used are listed in Supplementary Table 2. Amplification was performed with an initial 10 min denaturation at 95 °C, followed by 40 cycles of 15 s at 95 °C and 1 min at 60 °C. The fold change in gene expression was calculated as: Fold change = 2(−ΔΔCt).
Molecular docking
The LCK protein structure was acquired from the Protein Data Bank (PDB ID: 6PDJ), and the 3D structure of alpha-ketoglutaric acid was sourced from PUBCHEM. Using the MMFF94 force field, the energy of alpha-ketoglutaric acid was minimized to optimize its conformation for docking. Molecular docking was conducted with Auto Dock Vina 1.2.3 software71. We prepared the LCK protein by stripping water molecules and extraneous ions using PyMol 2.5.5, ensuring a clean binding site. A docking box was set to cover the entire protein to allow comprehensive spatial sampling. The conversion of protein and ligand structures into the PDBQT format, necessary for docking, was done using ADFRsuite 1.0372. We set the global search exhaustiveness to 32 to ensure thorough sampling. The docking result with the highest binding affinity was selected for further analysis. Visualization and analysis of the docking results were performed using PyMol 2.5.5.
Single-cell gene expression quantification and subcluster detection of spontaneous mice tumor
Fresh single-cell suspensions were prepared according to the 10× Genomics platform’s protocols (10× Genomics, Pleasanton, CA), ensuring immediate processing to maintain cell viability. Sequencing libraries were constructed following standard protocols and sequenced on the NovaSeq 6000 system to ensure high-throughput coverage. Initial raw data processing was conducted using the Seurat package (version 4.4.0), which facilitated rigorous quality control. Cells displaying extreme transcript counts—either below 500 or above 40,000 unique molecular identifiers (UMIs), or expressing genes in counts outside the range of 300 to 7000—were excluded. Additionally, cells with over 25% of UMIs originating from mitochondrial DNA were removed to prevent bias associated with cellular stress or apoptosis. The remaining high-quality gene expression matrices were normalized for total cellular UMI counts to mitigate depth variability across samples. Data were further scaled (scale factor = 10,000) adjusting for total UMI counts and mitochondrial gene content. Variable genes were identified using stringent criteria to highlight the most significant contributors to variability. Principal Component Analysis was employed on these genes, selecting the top 20 principal components to guide the clustering using Seurat’s Find Clusters function at a resolution of 0.3. Dimensionality reduction was visualized using tSNE to delineate distinct cell populations, ensuring a comprehensive analysis of cellular heterogeneity.
The sequencing of proteomics
Our study conducted a comprehensive proteomic analysis on protein extracts from 10 pairs of human cholangiocarcinoma and adjacent non-tumor tissues. Protein samples were equilibrated in concentration using lysis buffer and subjected to trichloroacetic acid (TCA) precipitation with 20% TCA, followed by centrifugation at 4500 × g for 5 min at 4 °C. The supernatant was discarded, and the pellet was washed 2–3 times with cold acetone before drying. The dried pellet was resuspended in 200 mM triethylammonium bicarbonate (TEAB) and sonicated. Trypsin digestion was performed at a 1:50 enzyme-to-protein ratio overnight. The digested samples were then reduced with 5 mM dithiothreitol at 56 °C for 30 min and alkylated with 11 mM iodoacetamide in the dark at room temperature for 15 min. Peptides were solubilized in IP buffer and incubated with succinyl-lysine-specific resin (PTM-402, PTM Bio) overnight at 4 °C on a rotator. Post-binding, the resin was washed with IP buffer and deionized water, and peptides were eluted with 0.1% trifluoroacetic acid, vacuum dried, and desalted using C18 ZipTips. Liquid chromatography-mass spectrometry analysis was performed on a NanoElute high-performance liquid chromatography system. Mobile phase A consisted of 0.1% formic acid and 2% acetonitrile, while phase B was 0.1% formic acid in acetonitrile. The gradient ran as follows: 6–22% B over 44 min, 22–30% B over the next 12 min, ramping up to 80% B in the following 2 min, and holding at 80% B for the last 2 min at a flow rate of 450 nl/min. Data were analyzed using MaxQuant version 1.6.6.0 against the Homo sapiens database (20,366 sequences), incorporating a decoy strategy to assess false discovery rates and removing contaminants. Search settings included trypsin/P as the cleavage enzyme, up to 4 missed cleavages, a minimum peptide length of 7, and a maximum of 5 modifications per peptide. Parent ion and fragment ion mass tolerances were set at 20 ppm. Fixed modification was carbamidomethyl (C), and variable modifications included acetylation of protein N-termini, methionine oxidation, and lysine succinylation. Label-free quantification was employed, maintaining identification and peptide-spectrum match FDRs at 1%.
Metabolite extraction and chromatography-mass spectrometry analysis
To accurately quantify metabolites, we first allowed samples to thaw in an ice-water bath, followed by a 30-s vortex mixing. We then transferred a 100 μl aliquot from each sample into an Eppendorf tube, to which we added 300 μl of methanol, vortexed for 60 s, and sonicated for 15 min in an ice-water bath. Subsequently, samples were incubated at −40 °C for one hour and centrifuged at 16,000 × g at 4 °C for 15 min. The supernatants were collected, vacuum dried, and reconstituted in 160 μl of water. The reconstituted samples were vortexed and filtered through a membrane filter prior to being transferred to vials equipped with inserts for high-performance ion chromatography-mass spectrometry/mass spectrometry (HPIC-MS/MS) analysis73. For HPIC, we used a Thermo Scientific Dionex ICS-6000 system equipped with Dionex IonPac AS11-HC (2 × 250 mm) analytical and AG11-HC (2 mm × 50 mm) guard columns. The mobile phase A comprised 100 mM NaOH in water, and mobile phase D was ultrapure water. An additional solvent system delivered 2 mM acetic acid in methanol, which mixed with the effluent before entering the electrospray ionization (ESI) source at a flow rate of 0.15 ml/min. Column and autosampler temperatures were maintained at 30 °C and 4 °C, respectively, with an injection volume of 5 μl. Mass spectrometric analysis was conducted using an AB SCIEX 6500 QTRAP+ triple quadrupole mass spectrometer equipped with an ESI interface. Ion source settings were as follows: IonSpray Voltage at −4500 V, temperature at 450 °C, Ion Source Gas1 and Gas2 both at 45 psi, and Curtain Gas at 30 psi. We optimized multiple reaction monitoring (MRM) parameters via flow injection analysis of individual analyte standards into the mass spectrometer, selecting the most sensitive Q1/Q3 transitions and adjusting collision energies accordingly. The most sensitive transitions were designated as ‘quantifiers’ for quantitation, with additional transitions serving as ‘qualifiers’ to confirm analyte identities74. Calibration standards were prepared by serial dilution of a mixed standard solution and analyzed using the same HPIC-MRM-MS methodology. The calibration curve was constructed by plotting analyte peak areas (y) against concentrations (x, nmol/L) and fitting the data using a least squares regression with 1/x weighting to achieve the highest accuracy and correlation coefficient (R2). Calibration points not meeting 80–120% accuracy were excluded from the analysis.
Labelled metabolite measurement by GC-MS
Seeding 1 million cells into a 6 cm dish, after cells adhere, the original culture medium was completely discarded, and the cells were washed twice with phosphate-buffered saline (PBS). Subsequently, 2 ml of 13C-labeled medium was added to each dish. After a 3-h incubation period, the cells were rapidly frozen using liquid nitrogen and stored on dry ice for subsequent analysis. For metabolite extraction, cells were ground under liquid nitrogen and resuspended in 1 ml of cold (−40 °C) 50% aqueous methanol. Samples were kept on dry ice for 30 min to ensure thorough cooling, followed by thawing on ice. Next, 0.4 ml of chloroform was added to each sample, which was then vortexed for 30 s. Samples were centrifuged at 10,000 × g for 15 min at 4 °C. The aqueous phase was carefully transferred to new 1.5 ml microcentrifuge tubes and the solvents were evaporated under vacuum. The dried extracts were stored at −80 °C until further analysis. For GC-MS analysis, metabolites were derivatized by first adding 70 μl of pyridine to the dried samples, followed by incubation at 85 °C for 20 min. After this initial step, 30 µl of N-tert-butyldimethylsilyl-N-methyltrifluoroacetamide (Sigma) was added, and samples were incubated for an additional 60 min at 85 °C. Post-incubation, samples were centrifuged at 10,000 × g for 15 min at 4 °C, and the supernatant was transferred to autosampler vials. GC/MS was then performed to analyze the isotopic labeling and concentration of metabolites. Metabolites were quantified based on the total ion count peak area, using standard curves generated from reference standards processed alongside the experimental samples. To determine the extent of 13C labeling, the mass distribution for known fragments of metabolites was extracted from the appropriate chromatographic peak75. These fragments included those representing the entire carbon skeleton of the metabolite, those lacking the alpha carboxyl carbon, and, in some cases for amino acids, only the backbone minus the side chain. For each fragment, isotopomer distributions were calculated by measuring the mass intensities from the lightest isotopomer (M0, no heavy isotopes) up to M6, reflecting successive incorporations of labeled carbon. These distributions were normalized by dividing the sum of the intensities from M0 to M6, providing a comprehensive profile of 13C incorporation into cellular metabolites.
IP-MS
Cultivate QBC sg-PDHA1 Flag-PCDH and Flag-WT cell lines, and proceed with cell lysis by adding protease and phosphatase inhibitors. Perform IP experiments as previously described, with Western blot confirming successful pull-down of PDHA1. First, the sample digestion: After reduction and alkylation treatment, trypsin is added to the sample at a mass ratio of 1:50 and digested for 20 h at 37 °C. The digested products are desalted, lyophilized, and reconstituted in 0.1% FA solution, then stored at −20 °C for future use. Mass spectrometry analysis: Solvent A is a 0.1% formic acid aqueous solution, and solvent B is a 0.1% formic acid acetonitrile aqueous solution (84% acetonitrile). After equilibrating the chromatographic column with 95% solvent A, samples are loaded onto the Trap column by an autosampler. A 0.5-h gradient is applied. Mass-to-charge ratios of peptides and their fragments are collected as follows: after each full scan, 20 fragment spectra (MS2 scans) are collected. Data analysis: The raw files from mass spectrometry are searched against the appropriate database using Mascot 2.2 software, resulting in the identification of proteins.
RNA-seq
Our methodology commenced with the preparation of mRNA libraries from 1 μg of total RNA per sample. Initially, poly(A) mRNA was isolated using Oligo (dT) beads, followed by fragmentation through the application of divalent cations under high-temperature conditions. Random primers were employed for priming, leading to the synthesis of the first and second strands of cDNA. Subsequent steps involved the purification of double-stranded cDNA, which was then processed for end repair and dA-tailing in a single reaction. This preparation was further subjected to T-A ligation to attach adaptors on both ends. Size selection of adaptor-ligated DNA was performed using DNA Clean Beads. The library construction was completed by amplifying each sample through PCR using P5 and P7 primers. The PCR products were validated to ensure library integrity. Libraries, each bearing a unique index, were pooled and sequenced on an Illumina HiSeq, Illumina NovaSeq, or MGI2000 platform, utilizing a 2 × 150 paired-end (PE) configuration as per the manufacturer’s guidelines. Quality Control76: Technical sequences including adapters, polymerase chain reaction (PCR) primers, and their fragments, along with low-quality bases (below a quality threshold of 20), were removed. Raw data in fastq format were processed using Cutadapt (v1.9.1) with parameters set for a phred cutoff of 20, an error rate of 0.1, adapter overlap of 1 bp, a minimum length of 75, and a maximum proportion of Ns set at 0.1%, to obtain high-quality clean data. Alignment: Reference genome sequences and gene model annotation files were downloaded from genome databases such as UCSC, NCBI, or ENSEMBL. The reference genome was indexed using Hisat2 (v2.2.1). Clean data were then aligned to the reference genome using Hisat2. Expression analysis: Transcript data in fasta format, derived from known gff annotation files, were indexed accordingly. HTSeq (v0.6.1) was employed to estimate gene and isoform expression levels from the paired-end clean data. Differential expression analysis was conducted using DESeq2, a Bioconductor package that models data based on the negative binomial distribution. This analysis includes estimates of dispersion and log fold changes with data-driven prior distributions, and genes with an adjusted p-value (Padj) ≤ 0.05 were considered differentially expressed. Gene Ontology Analysis: GO Seq (v1.34.1) was utilized to identify Gene Ontology (GO) terms annotating a list of enriched genes, focusing on those with a significant Padj of ≤ 0.05, indicating a noteworthy enrichment in specific biological processes77.
Gene set enrichment analysis (GSEA)
GSEA was performed using the software package (version 3.0) provided by the Broad Institute. Gene expression data were preprocessed to filter out low expression genes and normalized. Gene sets from the Molecular Signatures Database (MSigDB) were used as reference sets. Enrichment analysis (Enrichment Score, ES) is calculated by comparing the ranks of genes within a gene set to their ranks across the entire dataset. The significance of ES is evaluated through permutation analysis (n = 1000).
Statistical analysis
Western blot analysis using Image lab (version 5.1), Flow cytometry analysis using CytExpert (version 2.4) and FlowJo (version 10.8.1), Image analysis using ZEN lite (version 3.1) and Image J (version 1.8), RNA-seq analysis using GSEA (version 3.0) and GO Seq analysis (version 1.34.1), Molecular docking analysis using PyMol (version 2.5.5), Single-cell using Seurat R package (version 4.4.0) and IP-MS analysis using Mascot (version 2.2).
Statistics and reproducibility
The sample size for the animals was not determined using a statistical method. Instead, the selection was guided by our familiarity with the experimental models commonly employed in cell biology and animal research. All data shown were obtained from at least three biological independent experiments with similar results. Upon confirmation that the values followed a normal distribution, a two-tailed Student’s t-test was applied to determine the significance of differences between two independent sample groups. Pearson correlation analysis was used to determine the correlation between two variables. Survival rates were analyzed using the Kaplan–Meier method (log-rank test). Statistical analyses were conducted using GraphPad Prism software (version 9.5). All p values are indicated in the figures. The schematic illustration images were created with BioRender.com with publication licenses.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The single-cell RNA-seq data generated by mice in this study have been deposited in the Genome Sequence Archive database under accession code CRA017390, and the bulk RNA-seq data generated by mice in this study have been deposited in the Genome Sequence Archive database under accession code CRA017777. RNA-seq data of the cell lines in this study have been deposited in Genome Sequence Archive Human database under accession code HRA007972. The MS data of immunoprecipitation against PDHA1 were deposited in PRIDE (Project accession: PXD060056). The sequencing of proteomics data was deposited in PRIDE (Project accession: PXD060091). Source data for the figures and Supplementary Information are provided as a Source Data file. Source data are provided with this paper.
References
Brindley, P. J. et al. Cholangiocarcinoma. Nat. Rev. Dis. Prim. 7, 65 (2021).
Banales, J. M. et al. Cholangiocarcinoma: state-of-the-art knowledge and challenges. Liver Int. 39, 5–6 (2019).
Valle, J. et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 362, 1273–1281 (2010).
Okusaka, T. et al. Gemcitabine alone or in combination with cisplatin in patients with biliary tract cancer: a comparative multicentre study in Japan. Br. J. Cancer 103, 469–474 (2010).
Li, W., Li, F., Zhang, X., Lin, H. K. & Xu, C. Insights into the post-translational modification and its emerging role in shaping the tumor microenvironment. Signal Transduct. Target Ther. 6, 422 (2021).
Faubert, B., Solmonson, A. & DeBerardinis, R. J. Metabolic reprogramming and cancer progression. Science 368, eaaw5473 (2020).
Shi, H. et al. A glimpse into novel acylations and their emerging role in regulating cancer metastasis. Cell Mol. Life Sci. 81, 76 (2024).
Zhang, Z. et al. Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 7, 58–63 (2011).
Carrico, C., Meyer, J. G., He, W., Gibson, B. W. & Verdin, E. The mitochondrial acylome emerges: proteomics, regulation by sirtuins, and metabolic and disease implications. Cell Metab. 27, 497–512 (2018).
Rardin, M. J. et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 18, 920–933 (2013).
Yang, X. et al. SHMT2 desuccinylation by SIRT5 drives cancer cell proliferation. Cancer Res. 78, 372–386 (2018).
Hu, Q. et al. SUCLG2 regulates mitochondrial dysfunction through succinylation in lung adenocarcinoma. Adv. Sci. 10, e2303535 (2023).
Li, X. et al. Lysine-222 succinylation reduces lysosomal degradation of lactate dehydrogenase a and is increased in gastric cancer. J. Exp. Clin. Cancer Res. 39, 172 (2020).
Kaplon, J. et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 498, 109–112 (2013).
Jun, S. et al. The requirement for pyruvate dehydrogenase in leukemogenesis depends on cell lineage. Cell Metab. 33, 1777–1792.e1778 (2021).
Chen, J. et al. Compartmentalized activities of the pyruvate dehydrogenase complex sustain lipogenesis in prostate cancer. Nat. Genet. 50, 219–228 (2018).
Xu, S. et al. Butyrate induces apoptosis by activating PDC and inhibiting complex I through SIRT3 inactivation. Signal Transduct. Target. Ther. 2, 16035 (2017).
Cevatemre, B., Ulukaya, E., Dere, E., Dilege, S. & Acilan, C. Pyruvate dehydrogenase contributes to drug resistance of lung cancer cells through epithelial mesenchymal transition. Front. Cell Dev. Biol. 9, 738916 (2021).
Deng, L., Jiang, A., Zeng, H., Peng, X. & Song, L. Comprehensive analyses of PDHA1 that serves as a predictive biomarker for immunotherapy response in cancer. Front. Pharmacol. 13, 947372 (2022).
DeBerardinis, R. J. & Chandel, N. S. Fundamentals of cancer metabolism. Sci. Adv. 2, e1600200 (2016).
Wu, J. Y. et al. Cancer-derived succinate promotes macrophage polarization and cancer metastasis via succinate receptor. Mol. Cell 77, 213–227.e215 (2020).
Cheng, J. et al. Cancer-cell-derived fumarate suppresses the anti-tumor capacity of CD8(+) T cells in the tumor microenvironment. Cell Metab. 35, 961–978.e910 (2023).
Liu, M. et al. α-Ketoglutarate modulates macrophage polarization through regulation of PPARγ transcription and mTORC1/p70S6K pathway to ameliorate ALI/ARDS. Shock 53, 103–113 (2020).
Lampropoulou, V. et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. 24, 158–166 (2016).
Wang, N., Liang, H. & Zen, K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front. Immunol. 5, 614 (2014).
Akram, M. Citric acid cycle and role of its intermediates in metabolism. Cell Biochem. Biophys. 68, 475–478 (2014).
Mills, E. L., Kelly, B. & O’Neill, L. A. J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 18, 488–498 (2017).
Bénit, P. et al. Unsuspected task for an old team: succinate, fumarate and other Krebs cycle acids in metabolic remodeling. Biochim. Biophys. Acta 1837, 1330–1337 (2014).
Pearce, E. L. & Pearce, E. J. Metabolic pathways in immune cell activation and quiescence. Immunity 38, 633–643 (2013).
Li, M. et al. α-Ketoglutaric acid reprograms macrophages by altering energy metabolism to promote the regeneration of small-diameter vascular grafts. ACS Biomater. Sci. Eng. 11, 518–530 (2025).
Yu, S., Ding, L., Liang, D. & Luo, L. Porphyromonas gingivalis inhibits M2 activation of macrophages by suppressing α-ketoglutarate production in mice. Mol. Oral. Microbiol 33, 388–395 (2018).
Liu, Z., Gan, L., Zhang, T., Ren, Q. & Sun, C. Melatonin alleviates adipose inflammation through elevating α-ketoglutarate and diverting adipose-derived exosomes to macrophages in mice. J. Pineal Res. 64, e12455 (2018).
Martin-Serrano, M. A. et al. Novel microenvironment-based classification of intrahepatic cholangiocarcinoma with therapeutic implications. Gut 72, 736–748 (2023).
Bill, R. et al. CXCL9:SPP1 macrophage polarity identifies a network of cellular programs that control human cancers. Science 381, 515–524 (2023).
Wu, J. Y. et al. Cancer-derived succinate promotes macrophage polarization and cancer metastasis via succinate receptor. Mol. Cell. 77, 213–227.e5 (2020).
Runtsch, M. et al. Itaconate and itaconate derivatives target JAK1 to suppress alternative activation of macrophages. Cell Metab. 34, 487–501.e488 (2022).
O’Neill, L., Kishton, R. & Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 16, 553–565 (2016).
Pålsson-McDermott, E. & O’Neill, L. Targeting immunometabolism as an anti-inflammatory strategy. Cell Res. 30, 300–314 (2020).
Pavlova, N. & Thompson, C. The emerging hallmarks of cancer metabolism. Cell Metab. 23, 27–47 (2016).
Cai, Z. et al. Phosphorylation of PDHA by AMPK drives TCA cycle to promote cancer metastasis. Mol. Cell 80, 263–278.e267 (2020).
Li, X. et al. Mitochondria-translocated PGK1 functions as a protein kinase to coordinate glycolysis and the TCA cycle in tumorigenesis. Mol. Cell 61, 705–719 (2016).
Osman, A. & van Loveren, H. Phosphoproteomic analysis of mouse thymoma cells treated with tributyltin oxide: TBTO affects proliferation and energy sensing pathways. Toxicol. Sci. 126, 84–100 (2012).
Park, M. Y. et al. Scutellarein inhibits LPS-induced inflammation through NF-κB/MAPKs signaling pathway in RAW264.7 cells. Molecules. 27, 3782 (2022).
Yao, Y. et al. ERK and p38 MAPK signaling pathways negatively regulate CIITA gene expression in dendritic cells and macrophages. J. Immunol. 177, 70–76 (2006).
Chang, Y. C. et al. Epigenetic control of MHC class II expression in tumor-associated macrophages by decoy receptor 3. Blood 111, 5054–5063 (2008).
Grimm, P. et al. Integrated compensatory network is activated in the absence of NCC phosphorylation. J. Clin. Investig. 125, 2136–2150 (2015).
Guerrero, A. et al. α-Ketoglutarate upregulates collecting duct (Pro)renin receptor expression, tubular angiotensin II formation, and Na(+) reabsorption during high glucose conditions. Front. Cardiovasc. Med. 8, 644797 (2021).
Cárdenas, P. et al. OXGR1-dependent (Pro)Renin receptor upregulation in collecting ducts of the clipped kidney contributes to Na(+) balance in goldblatt hypertensive mice. Int. J. Mol. Sci. 25, 10045 (2024).
He, Y. et al. Lysine butyrylation of HSP90 regulated by KAT8 and HDAC11 confers chemoresistance. Cell Discov. 9, 74 (2023).
Ma, Q. et al. Acetyltransferase P300 regulates glucose metabolic reprogramming through catalyzing succinylation in lung cancer. Int. J. Mol. Sci. 25, 1057 (2024).
Echeverri Ruiz, N. P. et al. Dynamic regulation of mitochondrial pyruvate metabolism is necessary for orthotopic pancreatic tumor growth. Cancer Metab. 9, 39 (2021).
Gibson, G. E. et al. Alpha-ketoglutarate dehydrogenase complex-dependent succinylation of proteins in neurons and neuronal cell lines. J. Neurochem. 134, 86–96 (2015).
Ioka, T. et al. Randomized phase III study of gemcitabine, cisplatin plus S-1 versus gemcitabine, cisplatin for advanced biliary tract cancer (KHBO1401- MITSUBA). J. Hepatobiliary Pancreat. Sci. 30, 102–110 (2023).
Kelley, R. K. et al. Pembrolizumab in combination with gemcitabine and cisplatin compared with gemcitabine and cisplatin alone for patients with advanced biliary tract cancer (KEYNOTE-966): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 401, 1853–1865 (2023).
Shi, G. M. et al. Toripalimab combined with lenvatinib and GEMOX is a promising regimen as first-line treatment for advanced intrahepatic cholangiocarcinoma: a single-center, single-arm, phase 2 study. Signal Transduct. Target. Ther. 8, 106 (2023).
Dong, X. et al. Triple therapy in biliary tract cancers: GemOX plus immune checkpoint inhibitor in combination with lenvatinib or NGS-guided targeted therapy. J. Cancer Res. Clin. Oncol. 149, 1917–1927 (2023).
Valle, J. et al. Cisplatin and gemcitabine for advanced biliary tract cancer: a meta-analysis of two randomised trials. Ann. Oncol. 25, 391–398 (2014).
Woods, E., Le, D., Jakka, B. K. & Manne, A. Changing landscape of systemic therapy in biliary tract cancer. Cancers. 14, 2137 (2022).
Benson, A. B. et al. Hepatobiliary cancers, version 2.2014. J. Natl Compr. Canc Netw. 12, 1152–1182 (2014).
Alistar, A. et al. Safety and tolerability of the first-in-class agent CPI-613 in combination with modified FOLFIRINOX in patients with metastatic pancreatic cancer: a single-centre, open-label, dose-escalation, phase 1 trial. Lancet Oncol. 18, 770–778 (2017).
Pardee, T. S. et al. A phase I study of CPI-613 in combination with high-dose cytarabine and mitoxantrone for relapsed or refractory acute myeloid leukemia. Clin. Cancer Res. 24, 2060–2073 (2018).
Liu, Z., Wang, R., Wang, Y., Duan, Y. & Zhan, H. Targeting succinylation-mediated metabolic reprogramming as a potential approach for cancer therapy. Biomed. Pharmacother. 168, 115713 (2023).
Mu, R. et al. Role of succinylation modification in thyroid cancer and breast cancer. Am. J. Cancer Res. 11, 4683–4699 (2021).
Tong, Y. et al. SUCLA2-coupled regulation of GLS succinylation and activity counteracts oxidative stress in tumor cells. Mol. Cell 81, 2303–2316.e2308 (2021).
Liu, P. S. et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 18, 985–994 (2017).
Yuan, Y. et al. Exercise-induced α-ketoglutaric acid stimulates muscle hypertrophy and fat loss through OXGR1-dependent adrenal activation. Embo J. 39, e103304 (2020).
Zeng, Y. R. et al. The immunometabolite itaconate stimulates OXGR1 to promote mucociliary clearance during the pulmonary innate immune response. J. Clin. Invest. 133 e160463 (2023).
Philip, P. A. et al. A phase III open-label trial to evaluate efficacy and safety of CPI-613 plus modified FOLFIRINOX (mFFX) versus FOLFIRINOX (FFX) in patients with metastatic adenocarcinoma of the pancreas. Future Oncol. 15, 3189–3196 (2019).
O’Dell, M. R. et al. Kras(G12D) and p53 mutation cause primary intrahepatic cholangiocarcinoma. Cancer Res. 72, 1557–1567 (2012).
Liu, F., Song, Y. & Liu, D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 6, 1258–1266 (1999).
Eberhardt, J., Santos-Martins, D., Tillack, A. F. & Forli, S. AutoDock vina 1.2.0: new docking methods, expanded force field, and python bindings. J. Chem. Inf. Model. 61, 3891–3898 (2021).
Ravindranath, P. A., Forli, S., Goodsell, D. S., Olson, A. J. & Sanner, M. F. AutoDockFR: advances in protein-ligand docking with explicitly specified binding site flexibility. PLoS Comput. Biol. 11, e1004586 (2015).
Dunn, W. B. et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 6, 1060–1083 (2011).
Wiklund, S. et al. Visualization of GC/TOF-MS-based metabolomics data for identification of biochemically interesting compounds using OPLS class models. Anal. Chem. 80, 115–122 (2008).
van Winden, W. A., Wittmann, C., Heinzle, E. & Heijnen, J. J. Correcting mass isotopomer distributions for naturally occurring isotopes. Biotechnol. Bioeng. 80, 477–479 (2002).
Wang, L., Wang, S. & Li, W. RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185 (2012).
Harris, M. A. et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 32, D258–D261 (2004).
Acknowledgements
We thank Prof. Ming Kuang (SYSU) for providing the LTP-C9 cell line, Prof. Amaia Lujambio (ISMMS) for providing the CMV-SB13 vector, Prof. Qingsong Hu (USTC) for the gift of pcDNA3.1-HA and -FLAG and Prof. Mian Wu (USTC) for giving the pMD2.G and psPAX2. This study was supported by National Natural Science Foundation of China (Grant No.82102705/Y.L., No. U19A2008/L.L., NO.81972307/L.L., No.82272787/J.W., No.82373232/L.L., No.82473189/Y.L. and No.82303259/Y.F.L.). National Key R&D Program of China (Grant No.2019YFA0709300/L.L.). Noncommunicable Chronic Diseases-National Science and Technology Major Project (Grant No.2023ZD0507500/Y.L.). Natural Science Foundation of Anhui Province (2208085J47/J.W., 2408085QH255/L.S.). Provincial Key R&D Program of Anhui Province (202204295107020025/L.L., 202204295107020022/Y.L.). The Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB0940201/L.L.). Scientific Research Project of Anhui Provincial Department of Education (Grant No.2022AH030125/Y.L.). Beijing Xisike Clinical Oncology Research Foundation (Grant No. Y-JS2019-014/J.W.). Research Funds of Centre for Leading Medicine and Advanced Technologies of lHM (Grant No.2023IHM01031/Y.L.). USTC Research Funds of the Double First-Class Initiative (YD9110002048/J.W., YD9110002050/Y.L., and YD9110002068/L.S.). Program for Innovative Research Team of The First Affiliated Hospital of USTC (CXGG01/L.L.). Figures 1f, j, 3a, 4g, j, 6a, e and Supplementary Figs. 13h and 16d created with BioRender.com and got the publication and licensing Rights.
Author information
Authors and Affiliations
Contributions
Conceptualization, N.Z., L.S., S.Z., C.J., T.C.; Methodology, S.Z., N.Z., Y.F.L., L.S., S.L., Q.C.; Software, C.J., K.M., S.L.; Validation, J.Y., W.Z., X.G., T.C.,X.L, Y,Z; Formal Analysis, N.Z., C.C., X.G.,Q.Z.; Investigation, J.C., Y.Z, S.T.; Writing - Original Draft, N.Z., L.S.; Writing - Review & Editing, S.Z., L.S., C.J.; Visualization, L.S., C.W.; Funding Acquisition, L.S., Y.F.L, J.W., Y.L., L.L.; Resources, J.W., Y.L., L.L.; Supervision, J.W., Y.L., L.L.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Taro Hitosugi, who coiewed with Yetong Huang, Nicholas Denko, Jia Fan and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, N., Sun, L., Zhou, S. et al. Cholangiocarcinoma PDHA1 succinylation suppresses macrophage antigen presentation via alpha-ketoglutaric acid accumulation. Nat Commun 16, 3177 (2025). https://doi.org/10.1038/s41467-025-58429-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58429-7
This article is cited by
-
Metabolic reprogramming in cancer: signaling pathways and therapeutic targets
Molecular Cancer (2026)
-
Targeting cuproptosis for treatment and prognostic assessment in endometrial cancer
Cancer Cell International (2025)
-
SIRT5-mediated desuccinylation of MTHFD2 enhances chemoresistance in breast cancer cells by reducing therapy-induced senescence
Communications Biology (2025)
-
Inflammation and immunity in liver homeostasis and disease: a nexus of hepatocytes, nonparenchymal cells and immune cells
Cellular & Molecular Immunology (2025)
