
Abstract
Fecal microbiota transplantation (FMT) has shown efficacy for the treatment of ulcerative colitis but with variable response between patients and trials. The mechanisms underlying FMT’s therapeutic effects remains poorly understood but is generally assumed to involve engraftment of donor microbiota into the recipient’s microbiome. Reports of microbial engraftment following FMT have been inconsistent between studies. Here, we investigate microbial engraftment in a previous randomized controlled trial (NCT01545908), in which FMT was sourced from a single donor, using amplicon-based profiling, shotgun metagenomics, and culture-enriched metagenomics. Placebo samples were included to estimate engraftment noise, and a significant level of false-positive engraftment was observed which confounds the prediction of true engraftment. We show that analyzing engraftment across multiple patients from a single donor enhances the accuracy of detection. We identified a unique set of genes engrafted in responders to FMT which supports strain displacement as the primary mechanism of engraftment in our cohort.
Similar content being viewed by others

Introduction
Ulcerative colitis (UC) is a type of inflammatory bowel disease (IBD) of unknown etiology that is restricted to the colon1. UC is believed to arise from a disruption in the balance between the immune system and microbiota in a genetically susceptible individual2,3. Current standard medical treatments have focused on suppressing the immune response and are not always effective at controlling disease4. An alternative approach is to alter the microbial environment responsible for driving the immune response5. Fecal microbiota transplantation (FMT) has emerged as an increasingly popular approach to alter the colonic microbiota6 and is a standard therapy for patients with recurrent Clostridioides difficile infection (rCDI)7,8 and has also been evaluated in UC9,10,11,12.
We reported the first randomized controlled trial (RCT) of FMT for patients with active UC5. This RCT showed that the proportion of patients with active UC in which remission was induced after FMT (24%) was significantly higher than a placebo group (5%), with no difference in adverse events. This has been replicated by other researchers and a systematic review of 12 RCTs suggests there is moderate quality evidence that FMT can induce remission in active UC13. One of the donors involved in our trial was more successful at inducing remission than other donors, with 7 of the 9 responders receiving Donor B stool5. Our RCT did use single donors for each participant with only a small pool of donors overall5. We were therefore positioned to study donor effects, and we are building on our previous work by further investigating the microbial composition of patients who received FMT from Donor B compared to those who received placebo treatments to ask whether a specific group of microbes were engrafted following FMT, and to determine whether microbial engraftment is associated with remission after FMT.
Previous studies have reported microbial engraftment—the transfer of microbes from donor to patients—following FMT in various disease contexts, particularly in rCDI14,15,16,17,18,19. However, there are inconsistent results between studies. Even larger studies, which applied their bioinformatic workflows to multiple publicly available datasets and disease types20,21,22, reported disparate outcomes. To our knowledge, none of these studies have applied their analytical pipelines to placebo samples from RCTs sequenced with a metagenomic approach to assess background noise or establish thresholds for engraftment detection. However, placebo samples have been used to examine background noise in a defined consortium to treat rCDI23,24, and alternatively, samples from the same and different individuals have been used to estimate potential noise in colonization21,25. Moreover, culture-independent approaches often lack the sensitivity to detect low-abundance bacteria. Culture-enriched sequencing methods provide a more comprehensive view of the human microbiome than culture-independent sequencing, particularly for low-abundance bacteria, and past studies have shown the utility of this approach in capturing the diversity of intestinal26,27, lung28,29, skin30, and urine31 human microbiota.
To answer the question of whether specific groups of microbes are responsible for inducing remission in UC, we have therefore used three high-throughput sequencing approaches; culture-independent (direct) 16S rRNA gene amplicon sequencing, culture-independent (direct) shotgun metagenomic sequencing (DMG), and culture-enriched shotgun metagenomic sequencing (CEMG). Further, we asked whether observed engraftment truly reflected the donor microbiome’s influence, or if it was an artefact of low sampling resolution. To address this, we used a unique dataset, containing stool samples from patients receiving FMT from a single donor and those procured from patients undergoing placebo treatments. This multifaceted approach enables us to provide insights into the microbiome’s potential role in UC remission.
Results
Study design
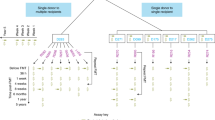
Our RCT involved 70 patients, with 36 undergoing FMT and 34 receiving a placebo5. At the trial’s conclusion, 9 patients in the FMT group (7 from Donor B) and 2 in the placebo group had entered remission (Fig. 1A). For an accurate assessment of microbial engraftment from Donor B, we revisited the 16S rRNA gene amplicon sequencing data with enhanced resolution by using amplicon sequence variants (ASV)32 instead of operational taxonomic units (OTUs). This reanalysis involved paired pre- and post-treatment samples from 20 FMT recipients of Donor B’s stool, and 31 placebo recipients (Fig. 1B). We selected all available responders and an equal number of non-responders from the Donor B FMT group (6 per group) and both responders and 10 non-responders (chosen at random) from the placebo groups for shotgun metagenomics. Additionally, shotgun metagenomic sequencing was applied to four longitudinal samples from Donor B (Fig. 1C). We built a comprehensive sequence library of Donor B’s samples via culture-enriched metagenomic sequencing, assembly, and annotations previously described29 (Fig. 1D, see Supplementary Information, Supplementary Fig. 1).

A The double-blind randomized control trial of FMT for UC patients5. B Direct 16S rRNA gene amplicon sequencing of stool was conducted for all patients and Donor B samples. Note that no baseline sample was available for one of the Donor B responders and this subject was excluded from our analysis. C Shotgun metagenomic sequencing was carried out for a subset of patients who received FMT from Donor B. 6 responders and 6 non-responders and an equivalent number of placebo treated patients. D The culture-enriched metagenomics workflow to build a comprehensive sequence database of Donor B’s gut microbiome. FMT fecal microbiome transplant, PLCA plate coverage algorithm. This figure includes artwork from Servier Medical Art https://smart.servier.com) licensed under a Creative Commons Attribution 4.0 Unported License (CC BY 4.0).
Microbial engraftment is present after FMT against a background of false positives
We have previously shown that culture-independent methods often underestimate microbial diversity in a stool sample compared to culture-enriched approaches26. If a patient has a donor feature below the detection limit at baseline but it is detected after FMT, this will appear as engraftment but would be a false positive. We refer to this as spurious engraftment. Here we compare measurements of engraftment in FMT recipients from a single donor and placebo recipients to estimate the extent of spurious engraftment at different sequencing resolutions, including 16S rRNA gene amplicons (ASVs), short-read metagenomics analysis at species (MetaPhlAn433), and strain (StrainPhlAn433) levels.
Conceptually, there should be no engraftment in the placebo group, and we hypothesized that spurious engraftment (false positives) would be minimal. Furthermore, we expect significant microbial engraftment to correlate with clinical remission after FMT. To investigate this, we utilized samples from Donor B in both 16S rRNA and shotgun metagenomics sequencing datasets, establishing a stringent cut-off to define apparent microbial engraftment. A microbe was only described as engrafted if its relative abundance was zero prior to FMT and rose above our established cut-off after FMT (detailed in Methods, Supplementary Fig. 2).
Initially, we investigated whether a clear difference exists between rates of apparent engraftment in the FMT and placebo groups. Our dataset, involving a single donor, allowed us to identify features that were apparently engrafted in multiple patients. We postulate that engraftments observed in multiple individuals are more likely to be the result of true microbial colonization and less reflective of individual-specific microbial changes during FMT. We identified donor ASVs, species, and strain markers that appeared to be engrafted in a given numbers of patients ranging from one, two, three, etc. from both FMT and placebo groups (Fig. 2A−C, top panels). We then computed the differences in numbers of each type of engraftment event for each dataset (donor vs placebo) and used permutation tests to determine whether the amount of apparent engraftment observed in patients who received FMT was significantly different from the amount observed in placebo (see Methods, Supplementary Table 1). The results showed that the FMT group had a significantly higher number of apparent engraftments than the placebo group (p-values presented in Fig. 2A−C, top panels), highlighting the likelihood that FMT leads to (at least some) true engraftment. However, a high number of spurious engraftments were also observed in the placebo group at ASV, species, and strain marker, even with our stringent cut-off, suggesting considerable noise (false positive) in microbial engraftment detection.

Detection of apparently engrafted ASVs from 16S rRNA gene sequencing (A), species predicted from shotgun metagenomic sequencing (B) and strain markers predicted from shotgun metagenomic sequencing (C). Top panels: Comparison of the number of apparently engrafted features observed in FMT vs. placebo samples. The x-axis shows the number of patients a feature is apparently engrafted present in, and the y-axis shows the number of features observed to be apparently engrafted in exactly that many patients within FMT (green) or placebo (gray) samples. Middle panels: Counting the number of apparently engrafted features that are unique to FMT samples (green), placebo samples (gray), or present in both (orange). The x-axis shows the number of patients a feature is apparently engrafted in, and the y-axis shows the number of features observed apparently engrafted in exactly that many patients. Bottom panels: The number of apparently engrafted features in FMT samples that are present in patients who responded to FMT treatment (Res,blue lines) and did not respond (NoRes, red lines). The differences between the points on the green and gray (left panels) or blue and red (right panels) lines were used to compute test statistics, with two-sided p values calculated using 1999 permutations by comparing the observed test statistic to the null distribution.
Next, we identified apparently engrafted donor ASVs, species, and strain markers that were unique to either the FMT or placebo groups, or shared between them (“overlap”), as shown in Fig. 2A−C (middle panels). The shotgun metagenomic dataset contained a stronger signal than the 16S dataset, showing that a higher proportion of apparently engrafted features are unique to the FMT group. Interestingly, the number of apparent engraftments in the unique-to-placebo group that were shared in more than one individual was reduced in species- and strain-level analysis. This indicates that looking for features that appear to engraft in multiple patients reduces the risk of detecting spurious engraftment. The number of apparently engrafted features that were observed in both FMT and placebo groups underscores the importance of including placebo sequencing in studies and reflects the fact that common microbial changes in individuals over time can mimic an engraftment signal.
Last, we explored whether microbial engraftment is associated with the response to FMT treatment. We compared the number of apparently engrafted features in responder and non-responder patients in the FMT group across ASV, species, and strain marker datasets (Fig. 2A−C, bottom panels). The 16S dataset included 14 non-responders and 6 responders, while the shotgun metagenomic datasets included the same 6 responders and a sub-set of non-responders (n = 6). Similar to the placebo vs. FMT comparisons described above, we calculated the differences in the number of engrafted features and weighted engraftment statistics (see Methods) and used permutation tests to compare the two groups. The results indicated no clear difference in the number of apparent engraftments between responders and non-responders Supplementary Table 1). Numbers of apparently engrafted features can be found in Supplementary Tables 2 and 3.
High resolution mapping of patient reads to Donor B MAGs to detect apparent genome engraftment
The results above indicate that detection of microbial engraftment may require greater microbial resolution than amplicon-based or read-based shotgun metagenomic sequencing. We hypothesized that co-assembling sequences from direct and culture-enriched metagenomic sequencing (CEMG) of the Donor B microbiota would improve gene and metagenome assembled genome (MAG) recovery from this donor compared to the commonly used direct metagenomic (DMG) approach. To generate the highest possible resolution assembly of Donor B’s microbiota, we built a comprehensive database using a co-assembly of four longitudinal DMG samples as well as our single CEMG sample (see Supplementary Information, Supplementary Fig. 1). We focused on 209 high quality MAGs out of a total of 447 bins in our databases. To track the engraftment of these MAGs after FMT, we mapped raw shotgun reads — subsampled to uniform sequencing depth — from the pre- and post-treatment samples of 24 patients (12 FMT recipients and 12 placebo-treated individuals, 48 samples in total) to these 209 MAGs.
To define the thresholds for MAG engraftment, we mapped raw sequence reads from our four Donor B samples to the Donor B MAG database. In each sample, we observed bimodal distribution in MAG coverage, with the modes at the extremities of the distribution. We reasoned that MAGs that were present in that sample were the ones that appeared in the upper mode, where at least 75% of the length of the MAG was covered at least 1x, and the MAGS that were absent from that sample were those that appeared in the lower mode, where no more than 20% of the length of the MAG was covered at least 1x. We therefore set these coverage values as our cutoffs for presence and absence, respectively (Supplementary Fig. 2).
We assessed the number of apparently engrafted MAGs in FMT recipients compared to placebo recipients, using methods like those in the read-based metagenomic analyses described above. Our goal was to determine if assembly-based assessments would reveal clear differences between the FMT and placebo groups. Consistent with previous results, we found a significant increase in the number of MAGs apparently engrafted in any given number of patients in the FMT group compared to placebo (Fig. 3A top panel), permutation test p = 0.008). We observed no significant differences in the numbers of engrafted MAGS, or the numbers of patients per engrafted MAG, when comparing FMT responders to non-responders (Fig. 3A bottom panel), permutation test p = 0.24).

A Top Panel: Comparison of apparently engrafted MAGs in patients who received FMT (n = 12) vs. placebo (n = 12) treatment. The x-axis displays number of patients in which a given MAG appears engrafted. A Middle panel: Counts of MAGs engrafted in only FMT (green), only Placebo (gray), and both (orange). A Bottom panel: MAGs engrafted in FMT patients, observed in responders (Res, n = 6, blue) and non-responders (NoRes, n = 6, red). The differences between the points on the green and gray (A, top panel;) or blue and red (A, bottom panel) lines were used to calculate the test statistics for the permutation tests, with two-sided p values calculated using 1999 permutations by comparing the observed test statistic to the null distribution. B All donor MAGs engrafted in at least one patient. Green represents MAGs exclusively engrafted in FMT recipients, gray indicates MAGs spuriously detected as engrafted in the Placebo group, and orange signifies MAGs detected in both FMT and Placebo treatment cohorts. MAGs are ordered on the x axis by which group(s) of patients they were apparently engrafted in (C). The family-taxonomy of donor MAGs engrafted in at least one patient.
Upon examining MAGs engrafted in one or more individuals, we found that 57% of engrafted MAGs (42 out of 74) were unique to patients who received and responded to FMT (Fig. 3A middle panel B), and most of these events were observed in 3 recipients (patients 4, 10, and 56). Lachnospiraceae, Ruminococcaceae, and Oscillospiraceae were the most abundant families among these MAGs (Fig. 3C). These findings suggest that microbial alterations in response to FMT are indicating that even genome-resolved metagenomic approaches to detect engraftment are also prone to false positives.
A signature of gene engraftment in patients who responded to FMT
Although culture enrichment allowed us to refine 209 MAGs from a single FMT donor, tracking MAGs omits information from the majority of assembled reads, which remain in bins or contigs excluded from MAGs. Since MAGs represent incomplete genomes34, a more comprehensive approach to investigating engraftment would focus on all donor genes.
We aimed to identify genes linked with engraftment and response to FMT. Using all Donor B contigs >2.5 kb long, we identified a total of 755,662 genes. We then used precise mapping of raw reads from all the patient samples (n = 48), to the Donor B gene database. We define apparently engrafted genes as those with ≤1x coverage over less than 25% of their length in the baseline sample, and ≥1x coverage over 90% or more of their length after treatment.
We employed the same methodology used in Fig. 2 to assess engraftment in the FMT and placebo groups. Our results revealed a significant difference in the number of apparently engrafted genes between patients treated with FMT and those receiving placebo (Fig. 4A top panel), permutation test p = 0.001). Consistent with all previous analyses, the number of apparently engrafted genes unique to the placebo group decreased as we restricted the count to genes apparently engrafted in increasing numbers of patients (Fig. 4A middle panel). Consistent with other datasets, we could not clearly see a difference in the number of engrafted genes between responders and non-responders within the FMT group (Fig. 4A bottom panel, permutation test p = 0.86).

A Counts of apparently engrafted genes in FMT and placebo groups. A Top panel: Examination of apparently engrafted genes in patients receiving FMT (n = 12) or placebo (n = 12) treatment. The x-axis shows the number of patients in which each gene appears to be engrafted, emphasizing common gene engraftment. A Middle panel: Discriminating donor gene engraftment patterns across all patients (n = 24) to distinguish unique engraftment in FMT (green), in Placebo (gray), and genes engrafted in both FMT and Placebo (orange). A Bottom panel: Comparison of apparently engrafted genes in FMT patients in responders (Res, n = 6, blue) and non-responders (NoRes, n = 6, red). The differences between the points on the green and gray lines (A, top panel)) or blue and red lines (A, bottom panel) were used to calculate the test statistics for the permutation tests, with two-sided p values calculated using 1999 permutations by comparing the observed test statistic to the null distribution. B Donor genes that engrafted in at least 3 patients are categorized as follows: genes engrafted in FMT patients but also present in the placebo group are shown in orange; genes unique to the placebo or FMT groups are depicted in gray and green, respectively. Among genes apparently engrafted only in FMT, those found in both responders and non-responders, as well as those unique to each group, are separately grouped on the x-axis. C Distribution of the genes apparently uniquely engrafted in 3 or more FMT responders in our Donor B assemblies. Among these genes, 333 were found in 32 metagenomic bins, 620 were present in 55 MAGs, and 535 genes were located on un-binned contigs (“UnBin”).
To enrich for legitimate engraftment events, we focused on genes engrafted in three or more patients. This provided a set of 45,419 genes. Among these, 56.9% (25,826) were engrafted in both FMT and placebo recipients (Fig. 4B, orange). Notably, 37.9% (17,230) were exclusive to FMT recipients (Fig. 4B, green), while only 5.2% (2363) were unique to the placebo group (Fig. 4B, gray). This final category, since it is known to be false positives, was not subject to further analysis. Genes unique to responders represented 3.3% (1488), compared to only 0.6% (283) in non-responders (Fig. 4B). To trace the origins of genes uniquely associated with FMT responders, we examined their presence in contigs grouped in MAGs and non-MAG bins. We found 620 genes within 55 unique MAGs, 535 in contigs not assigned to any bin (“UnBin”), and 333 in 32 non-MAG bins (Fig. 4C). These findings highlight the importance of our gene-based approach. Relying solely on genome detection strategies using MAGs would have prevented the identification of most of these genes.
Taxonomic specificity of commonly engrafted genes and distribution in an independent IBD patients
To further investigate the genes engrafted in at least 3 responders, we searched for these genes in 33,167 RefSeq genomes from all bacterial phyla, updated as of May 2023. Our objective was to ascertain whether these genes are widely distributed across various taxa or are specific to certain strains.
Our analysis revealed that 66 out of the 33,167 genomes contained the genes of interest, each with more than five genes per genome. These genomes were distributed across six bacterial families (Fig. 5A). Notably, genes identified in the families Bacteroidaceae were present in multiple species within their genera, suggesting these were widely shared between strains within the Bacteroides genus. By contrast, genes present in Lachnospiraceae and Ruminococcaceae, were more strain-specific within a species. The remaining families contained 11% of the detected genes. (Fig. 5B).

A We searched for genes commonly apparently engrafted in FMT responders in the RefSeq complete bacterial genomes (n = 33,167) database. The number of observed engrafted genes per genome is represented by green bars on the x-axis, while strains with at least 5 engrafted genes are displayed on the y-axis. Bacterial strains are grouped and color-coded on the y-axis according to their family. B To demonstrate the specificity of these genes within a genus, unique engrafted genes are represented on the x-axis and compared across the genomes depicted on the y-axis (shared with A). C Comparison of the number of genes commonly engrafted in responders that were found in healthy individuals (n = 56) and patients with UC (n = 76) and CD (n = CD). We utilized metagenomic reads from a publicly available dataset (PRJNA4000722935) that includes samples from healthy donors and IBD patients to explore the association of these genes with health status. Box plots show the median (center line), interquartile range (IQR) (box bounds), and whiskers extending to the smallest and largest values within 1.5 × IQR, with outliers excluded. Statistical significance was assessed using one-way ANOVA followed by Tukey’s post-hoc test, both of which were two-sided. D Comparison of the relative abundance of these commonly engrafted genes within the same cohort as (C), using the same statistical analysis.
Among the responder specific genes, 1415 were annotated as coding sequences (CDS) with a median length of 491 bp. Of these, 845 proteins were previously characterized or had predicted function by homology, while the remaining were hypothetical proteins with no predicted function. Among the characterized proteins, 593 were distributed across 19 diverse Clusters of Orthologous Groups (COG) categories. The most prevalent categories included Transcription, which accounted for 12.8% of these proteins, followed by Replication, Recombination, and Repair (12.3%), and Carbohydrate Transport and Metabolism (8%).
One notable observation (Supplementary Fig. 3) is that for any individual only a small portion of the microbiome features are shared between patient and donor. However, most of the donor’s microbiome features are shared with at least one subject. This is also true of engrafted features. This reflects the high individual variability in human gut microbiomes.
Expanding our investigation, we utilized a larger dataset of publicly available metagenomic samples (PRJNA4000722935), which included healthy individuals (n = 56), UC patients (n = 76), and patients with Crohn’s disease (CD) (n = 88). We mapped these reads to the responder specific genes to determine their presence and relative abundance in the samples. Intriguingly, we found a notable reduction in both the number and relative abundance of these genes in IBD patients compared to healthy individuals. (Tukey test, p-values in Fig. 5C, D, overall ANOVA p < 0.0001). These observations, derived from an independent and more extensive dataset, further underscore the potential significance of these genes in the context of IBD.
Discussion
FMT has emerged as a promising therapeutic option for UC, demonstrating efficacy in RCTs when compared to placebo13,36. However, the variability in patient response remains a critical challenge, underscoring the need to understand the factors influencing the success of FMT. Donor microbiota composition has been implicated in treatment outcomes, with studies reporting both donor-dependent efficacy and improved outcomes when pooling microbiomes from multiple donors5,18,37. In this study, we aimed to dissect the mechanisms underlying successful FMT by examining microbial engraftment in UC patients.
We examined the rates of apparent engraftment in both FMT and placebo groups. We observe a high rate of spurious engraftment (false positives) in the placebo group which confounds the prediction of true engraftment. This demonstrates that it is not sufficient for a feature to meet engraftment criteria of absence in a baseline and presence in both donor and post-FMT samples. Additionally, our data suggest that analyzing engraftment across multiple patients from a single donor enhances the accuracy of detection. It is possible that placebo engraftment represents the acquisition of strains from environmental sources. However, the probability that environmental sources of spurious engrafted features match donor is low.
Our analysis also underscores the necessity of integrating placebo group sequencing in investigation of post-FMT engraftment. Our data from the placebo controls demonstrated that a substantial amount of false positives detection occurs, and caution must be taken when interpreting results. These false positives arise because donor features in a patient (e.g., species/strains/MAGs/genes) may be present at baseline but below the level of detection. If these features increase and are detected at a second timepoint, then they will appear to be engrafted. By refining engraftment thresholds and employing rigorous controls, we aim to provide a clearer and more reliable framework for interpreting FMT outcomes in UC. This is not unexpected since even deep sequencing does not capture the full complement of microbes present. As we have shown previously26,28,29 and here (Supplementary Fig. 1), comprehensive culture methods enrich the number of features detected 3–4 fold.
The application of MAGs in studying microbial engraftment after FMT presents significant challenges. The inherent difficulties in accurately assembling MAGs from complex microbial communities result in the exclusion of a substantial portion of sequencing data (>50%). The non-MAG metagenomic data will include species present at lower abundance, plasmids, mobile genetic elements, and when multiple strains of a species are present - strain-specific genes. Using the define thresholds for donor MAG presence and absence in subjects, we observed 4 MAGs predicted to be engrafted in at least three FMT recipients, but only one of these, a Lachnospiraceae, was shared between 3 responders (Hominisplanchenecus faecis).
The clearest signals of engraftment in our study came from the gene dataset where specific sets of donor genes were predicted to be engrafted and associated with response in multiple FMT recipients. The gene-level analysis is consistent with most engraftment events being associated with strain displacement where a donor strain replaces a recipient strain of the same species. It is also possible that some engrafted genes may represent horizontal gene transfer events. Here we use strain in the context of the presence and absence of accessory genes which focuses on functional differences between strains. Approximately 60% of these genes were not found in MAGs even though our culture-enriched metagenomic increased the number of MAGS 4-fold and increased the size of most MAGs compared to standard metagenomics. Furthermore, MAGs containing these genes (n = 55) were not predicted to be engrafted. This reflects the incomplete nature of MAGs and the challenges of establishing the thresholds set for presence and absence of features (Supplementary Fig. 2).
Strains can be defined by single nucleotide polymorphisms in defined set of single core genes (e.g., StrainPhlAn). This is a powerful approach for tracking lineages over time and other evolutionary studies as it avoids the confounding effects of horizontal gene transfer. The strain-sharing inference pipeline within StrainPhlAn calculates pairwise distances between samples without distinguishing unique strain properties33. Applying this method to our data, we found a strong signature of engraftment (Supplementary Fig. 7) and only a small number of the total species-level bins passed the threshold to be included 54 out of 252 (Supplementary Fig. 8). However, this is a distance matrix of the distribution of strain markers within a species and it does not identify specific engrafted strains per se. While the results suggest that engraftment is occurring within a species, we can not identify a specific strain or marker associated with this approach. Other limitations of the strain-sharing inference pipeline for our specific question of identifying engrafted features are discussed in the supplementary methods.
Using a gene-centric methodology we identified specific sets of genes uniquely present in patients who responded to FMT treatment. Many of these genes are involved in diverse microbial metabolic pathways. However, a main challenge in interpreting these results arises from the limited annotations available in existing databases, and a significant proportion of the predicted genes (~25%) have no predicted function. Our study was limited by small number of subjects and responders in our trial. Despite these limitations, when examining these commonly engrafted genes with a larger cohort of IBD patients, we found that they were consistently depleted, suggesting a possible protective or beneficial role for these genes that is disrupted in this disease state.
Our analysis revealed a notable pattern of strain specificity among these genes, particularly in species within the Lachnospiraceae and Ruminococcaceae families. Interestingly, the same taxa were among the top engrafted bacteria in other feature types (Supplementary Fig. 9). This specificity suggests that certain strains of these species may be important for achieving the desired therapeutic outcomes of FMT. Conversely, in the Bacteroides genus, we observed that these genes are not confined to strains of specific species but are rather widespread across the genus (Fig. 5). As might be predicted from this distribution, there is relatively low level of prediction for Bacteroides MAG engraftment (Fig. 3). This distribution pattern could indicate a broader functional role for these genes within the Bacteroides or may be reflective of an accessory genome that is more widely shared across the family.
Strain level engraftment may also reflect direct strain-strain competition where donor strains that are more adept at outcompeting a patient’s own strains and may be independent of response to FMT. However, we do find donor genes specifically engrafted in multiple responders, and also show these genes are depleted in stool metagenomic samples from ulcerative colitis and Crohn’s disease patients compared to healthy controls (Fig. 5C, D). These observations provide support for a role of these genes in response to FMT.
The objective of this study was to attempt to identify engrafted features of the donor that were associated with clinical response in patients. The high level of false positive engraftment in the placebo group highlights one of the challenges in studying engraftment. This was independent of the methodological approach applied and is an inherent challenge amplicon of metagenomics studies of microbiomes. An interest in this field has been to replace FMT with defined consortia of microorganisms. For reasons discussed above, defining strains based on precise gene level engraftment may more accurately reflect functional requirements for response. A consortium of strains defined by gene-level engraftment might be more effective as a FMT alternative than a consortium based only on species considerations. By focusing on these key microbial players and their functional genes, and investigating their roles in health and disease, we can begin to piece together the intricate mechanisms through which the microbiota exerts their beneficial effects, paving the way for refined FMT strategies tailored to individual microbial compositions.
Methods
Study design and sample collection
The clinical study design and sample collection was as described previously5. Briefly, 70 active UC patients (Mayo score >=4 with an endoscopic Mayo score >=1) were randomly assigned to either 6 weeks of FMT (once per week; 50 mL, via enema, from healthy anonymous donor) or placebo (once per week; 50 mL water enema) in a double-blind randomized controlled trial. Stool samples were collected at baseline (before treatment), and during each week of the trial. Written informed consent for microbiome analysis was obtained by all participants. The clinical trial1 (NCT01545908) was approved by the Hamilton Health Sciences/McMaster University Research Ethics Board (REB # 11-600).
DNA extraction and 16S rRNA gene sequencing
Genomic DNA extraction and PCR amplification of the V3 region of 16S rRNA gene was conducted using previously described protocols5,38,39. Briefly, 0.2 g of fecal matter was mechanically homogenized using ceramic beads in 800 μL of 200 mM NaPO 4 (pH 8) and 100 μL of guanidine thiocyanate-EDTA-N-lauroyl sacosine. This was followed by enzymatic lysis of the supernatant using 50 μL of 100 mg/mL lysozyme, 50 μL of 10 U/μL mutanolysin, and 10 μL of 10 mg/mL RNase A for one hour at 37 °C. Then, 25 μL of 25% sodium dodecyl sulfate (SDS), 25 μL of 20 mg/mL proteinase K, and 75 μL of 5 M NaCl was added, and incubated for one hour at 65 °C. Supernatants were collected and purified through the addition of phenolchloroform-isoamyl alcohol (25:24:1; Sigma, St. Louis, MO, USA). DNA was recovered using the DNA Clean & Concentrator TM −25 columns, as per manufacturer’s instructions (Zymo, Irvine, CA, USA) and quantified using the NanoDrop (Thermofisher, Burlington, ON). After genomic DNA extraction, the V3 region of the 16S rRNA gene was amplified via PCR using these conditions per reaction well: Total polymerase chain reaction volume of 50 μL (5 μL of 10X buffer, 1.5 μL of 50 mM MgCl 2, 1 μL of 10 mM dNTPs, 2 μL of 10 mg/mL BSA, 5 μL of 1 μM of each primer, 0.25 μL of Taq polymerase (1.25 U/μL), and 30.25 μL of dH 2 O. Each reaction was divided into triplicate for greater efficiency. The primers used in this study were developed by Bartram et al.39. PCR conditions used included an initial denaturation at 94 °C for 2 min, followed by 30 cycles of 94 °C for 30 s, 50 °C for 30 s, 72 °C for 30 s, followed by a final elongation at 72 °C for 10 min. All samples were sequenced using an Illumina MiSeq platform at the McMaster Genomics Facility (Hamilton, Ontario, Canada). Samples were processed in batches, meaning not all samples were extracted and sequenced at the same time.
16S rRNA gene sequencing processing pipeline
Cutadapt40 v1.14 was used to filter and trim adapter sequences and PCR primers from the raw reads, using a quality score cut-off of 30 and a minimum read length of 100 bp. We used DADA232 v1.14.0 to resolve the sequence variants from the trimmed raw reads as follow. DNA sequences were trimmed and filtered based on the quality of the reads for each Illumina run separately. The Illumina sequencing error rates were detected, and sequences were denoised to produce ASV count table. The sequence variant tables from the different Illumina runs were merged to produce a single ASV table. Chimeras were removed and taxonomy was assigned using the DADA2 implementation of the RDP classifier against the SILVA database41 v1.3.2, at 50% bootstrap confidence.
The ASV, taxonomy, and clinical tables were all merged into one data object in R v4.2.0 using Phyloseq v1.40.0 package42.
Library preparation for shotgun metagenomic sequencing
We conducted direct shotgun metagenomic sequencing on 48 samples collected from 24 patients (12 patients who received FMT (6 responders and 6 non-responders) and 12 who received placebo (both responders and 10 non-responders)), at 2 time points each (baseline and 6 weeks after treatment), as well as 4 samples from Donor B. Genomic DNA was standardized to 5 ng/μL and sonicated to 500 bp. Using the NEBNext Multiplex Oligos for Illumina kit (New England Biolabs), DNA ends were blunted, adapter ligated, PCR amplified, and cleaned as per manufacturer’s instructions. Library preparations were sent to the McMaster Genome Facility and sequenced using the Illumina HiSeq platform, with a mean depth of approximately 18 million paired-end reads per sample.
Culture-enriched and independent metagenomics on Donor B samples
A fresh, anaerobic fecal sample was collected from Donor B. The collected sample was cultured using 33 different media, and incubation of plates anaerobically and aerobically resulted in 66 culture conditions for culture-enriched molecular profiling using a previously described protocol26. The list of media and culture conditions are described therein. 16S rRNA gene amplicon sequencing was conducted on plate pools of all 66 culture conditions. To determine a subset of plates that adequately represent the sample, the distribution of ASVs in the direct sequencing was compared to the culture-enriched sequencing using the PLCA algorithm29. Shotgun metagenomics was conducted on the 13 plate pools identified by the PLCA algorithm as representing the community. Genomic DNA was isolated from the all 13 selected plate pools and shotgun metagenomics conducted as previously described, with a mean depth of approximately 14 million paired-end reads per plate pool. Direct shotgun metagenomics was also conducted on the same fecal sample directly.
Comparison of the culture-enriched metagenomics with direct metagenomics data
To build a culture-enriched metagenomic library, raw shotgun sequences from the selected plate pools and the original fecal sample collected from Donor B for culturing were co-assembled as follows: Low-quality reads and sequencing primers were removed using Trimmomatic43 v0.38 with ‘LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36’ option. The reads were decontaminated for any human DNA using DeconSeq44 v0.4.3. These cleaned reads were then co-assembled using metaSPAde45 v3.10.1. Filtered raw reads were mapped to contigs (minimum length of 2.5 kb) using BWA-mem46 v0.7.17, and then binned with Metabat247 v2021. This was done for both the CEMG assembly and direct metagenomic sequencing (DMG) reads from the fecal sample, using identical methods. These datasets are referred to as CEMG and DMG, respectively.
The microbial compositions of DMG and CEMG datasets were then comprehensively evaluated using the following procedure: The single-copy core genes were identified within each bin using CheckM48 v1.1.2, any bin with minimum 70% completion and maximum 10% contamination was defined as a metagenome assembled genome (MAG). The shotgun reads were mapped to the assembled contigs to estimate sequence coverage for all contigs, Bins, and MAGs. We used BWA-mem46 to map reads to assembled contigs and the anvio pipeline49 to normalize the coverage to depth of sequencing. Detection values were calculated for each bin using anvio49. A detection value is defined as the proportion of a given MAG that is covered by at least one read; in other words, it estimates the proportion of MAG that recruited reads to it.
For taxonomic assignment of the metagenome-assembled genomes (MAGs), we utilized GTDB-Tk50 v202.0. We built a maximum-likelihood phylogenetic tree using the aligned protein sequences from GTDB with FastTree51. To compare the lengths of MAGs in DMG and CEMG, we selected MAGs based on their proximity in the phylogenetic tree. The total assembly length was determined by summing contig lengths, calculated with metaSPADE, in R. All figures were created and visualized using R version 4.0.3.
Shotgun metagenomic sequencing processing pipeline
Raw reads were filtered to remove low-quality sequences and decontaminate human-derived reads using KneadData. These filtered reads were then analyzed to identify species- and strain-level markers with MetaPhlAn4 and StrainPhlAn4, respectively. To profile the relative abundance of species, MetaPhlAn433 v4.0.4 was employed with the ‘-t rel_ab’ option. MetaPhlAn4 introduces the ability to perform strain-level profiling using non-aggregate marker information from the StrainPhlAn4 pipeline33. This method enables strain tracking and comparison across multiple samples. For strain tracking in this study, we used the ‘-t marker_ab_table’ option alongside the ‘--nreads’ parameter to specify the number of metagenomic reads. A default minimum Reads Per Kilobase (RPK) of 1 was applied to identify a marker as present.
We estimated strain-sharing inferences using a previously published workflow (Valles-Colomer et al.25) based on StrainPhlAn4 v4.0.433. Species-level markers, previously generated during the MetaPhlAn4 step, were extracted from all samples using the ‘sample2markers.py’ script. These markers, referred to as Species-level Genome Bins (SGBs), were used to infer strain-level variation.
For every species (SGB) detected in Donor B samples (n = 4), a database containing the marker genes was built using the ‘extract_markers.py’ script. Single Nucleotide Variant (SNV) profiling was performed for each Donor B species to generate phylogenetic trees using StrainPhlAn4. To enhance sensitivity for strain engraftment detection, we applied a set of predefined parameters (‘--sample_with_n_markers 20’, ‘--marker_in_n_samples 50’) to retain more samples and markers during SNV alignment. Pairwise phylogenetic distances for SGBs passing the SNV profiling step were calculated using the ‘tree_pairwisedists.py’ script.
We utilized pre-computed species-specific strain identity thresholds from Valles-Colomer et al.25 which were based on comparisons of inter- and intra-individual phylogenetic distances. These thresholds, publicly available on the MetaPhlAn GitHub repository, were used to identify strain engraftment events.
Filtered metagenomic reads from donor samples (n = 4) and culture-enriched plate pools (n = 13) were co-assembled using metaSPAdes45 v3.10.1 to create a comprehensive Donor B database. Contigs of at least 2.5 kb were annotated with Bakta52 v1.5.0 to identify genes. These annotated contigs were then binned with Metabat247 v2021, leveraging mapping data from all plate pools and Donor B samples. The quality of the resulting bins was evaluated using CheckM48 v1.1.2; bins with a completion rate of at least 70% and contamination of 10% or less were designated as metagenome-assembled genomes (MAGs). Taxonomic classification of MAGs was performed using GTDB-Tk50 v202.0.
All patient metagenomic samples (n = 48) were normalized to the lowest sequencing depth observed in the dataset (5.5 million paired-end reads) using seqtk53 v1.1.0 for random subsampling. These rarefied reads were mapped to the genes and contigs assembled from Donor B using BWA-mem46 v0.7.17, applying perfect match filtering options ‘-O 60 -E 10 -L 100’. Coverage information at 1x was calculated using samtools54 ‘coverage’.
A database of 33,167 RefSeq bacterial genomes, categorized as ‘assembly-level complete’ and downloaded as of May 2023, was compiled using a custom Python script. We identified a unique set of genes present exclusively in responder patients and used these as query sequences for BLAST searches against this database, employing BLAST55 v2.13.0 + . Hits were selected based on stringent criteria: percent identity of at least 90%, query coverage of at least 90%, and gene lengths of 100 base pairs or more. Genomes containing at least five such genes were visualized. The taxonomy of these genomes was determined using GTDB-Tk50 v202.0. To functionally annotate these genes, they were clustered at 90% protein identity using MMseqs256 v13.45111 and then profiled against KEGG, COG, and PFAM databases using eggNOG-mapper57 v 2.1.7 and database version of 5.0.2.
Additionally, a publicly available metagenomic dataset (PRJNA4000722935), comprising samples from 56 healthy subjects, 76 patients with ulcerative colitis (UC), and 88 patients with Crohn’s disease (CD), was downloaded from the Sequence Read Archive (SRA). These samples were mapped to the identified gene set using BWA-mem46 v0.7.17, applying strict matching filters (‘-O 60 -E 10 -L 100’). samtools54 ‘coverage’ was then used to quantify the presence of these genes in each sample and to calculate the percentage of reads mapped to these genes per sample.
Feature types
As described above, we used the 16S and shotgun metagenomic reads from our samples to generate five datasets, each consisting of a single feature type: 16S rRNA gene amplicon ASVs, species, strains, MAGs, and genes. The 16S ASVs are the only feature type that was generated using amplicon-based methods. The rest were based on the shotgun metagenomic reads. Within these remaining four, species and strains were read-based methods because they did not require assembly, while MAGs and genes were assembly-based, since they used contigs of assembled reads. We analyzed all five feature types to determine which method allowed us to minimize the spurious appearance of engraftment, while still having the sensitivity to detect true engraftment that may be happening.
Assessing engraftment
In order for us to call a feature “apparently engrafted”, it needed to meet three criteria: 1. It must be observed in the Donor B samples. 2. It must be absent from the patient’s pre-treatment sample. 3. It must be present in the patient’s post-treatment sample.
For all feature types, criterion 1 was met if the feature was observed at least once in any Donor B sample. The definitions of criteria 2 and 3 (absence and presence) depended on feature type. Our aim was to be quite strict with these criteria in order to minimize spurious identification of engraftment.
For the 16S ASVs, and strain and species marker feature types, a feature was deemed to be absent at baseline (criterion 2) if the patient’s pre-treatment sample contained that feature exactly 0 times. Even a single read assigned to a given feature in a baseline sample would remove it from consideration for engraftment in that patient. Criterion 3 (presence after treatment) was met if the feature’s relative abundance in the post-treatment sample surpassed a given threshold. The threshold varied between the three feature types, and in each case was chosen such that, when it was applied to Donor B’s own samples, it eliminated the bottom 20% of features from the merged datasets. The cutoffs were 1.3e04 relative abundance for 16S ASVs and strain markers, and 0.0072 for species markers. Supplementary Fig. 2 shows the presence vs. relative abundance histograms used to determine these cutoffs.
We additionally used the StrainPhlAn strain-sharing workflow25 to assess engraftment of strains. In this case, a strain was considered absent from a sample if its species-level genomic bin (SGB) pairwise distance was not below the pre-computed similarity threshold49 with any Donor B sample, and was considered present if its SGB pairwise distance was below the threshold. The results of this analysis can be found in the supplemental material.
For assembly-based feature types (genes and MAGs), presence and absence were determined by the proportion of the sequence with at least 1x coverage, rather than abundance. In order to identify reasonable cutoffs for MAGs, we mapped Donor B’s reads back to the database of Donor B assemblies, and then created a histogram showing the frequencies with which MAGs were covered at least 1x over different proportions of their length. We observed a u-shape in this histogram, where approximately 50% of the MAGs in each sample were covered 1x over at least 75% of their length, most intermediate values of coverage were present at very low frequency, and there was another peak of varying height when there was at least 1x coverage over less than 20% of the MAG’s length. We reason that MAGs that were present in a given sample are represented by the peak above 75% coverage, while MAGs that are absent from that sample can be seen in the lower peak. This threshold aligns with the theoretical minimum detectable genome overlap proposed by Lander and Waterman (1988)58. We called a MAG absent in a baseline sample if it was covered at least 1x over less than 20% of its length, and present in a post-treatment sample if it was covered at least 1x over at least 75% of its length. For genes, we used a 25% coverage criterion to define a gene as absent, and required its 1x coverage to be over at least 90% of its length in order to call it present after treatment. Both cutoffs were raised relative to the MAG cutoffs to allow for the fact that different genes which happen to share similar motifs can have quite similar DNA sequences over a substantial proportion of their lengths. Because we did not have empirical support for the gene cutoffs, we conducted a sensitivity analysis to ensure that our results were robust to changes in the absence and presence cutoffs (see Supplemental Information, Supplementary Fig. 4−6).
Within each patient, any features that failed to meet even one of the three criteria were removed from consideration for engraftment.
Statistical analysis of engraftment
Once apparently engrafted features were identified in all samples across all feature types, we sought to test whether patients who received FMT had higher levels of apparent engraftment than those who received placebo treatment. We constructed three test statistics, all of which we tested using the same permutation test. These three test statistics were the differences between FMT and placebo groups in: 1. the number of features engrafted at least once in the given treatment group (T1), 2. the number of engraftment events in that occurred in each treatment group (i.e., the count of engrafted features, weighted by the number of patients each feature was engrafted in) (T2), and 3. the number of engraftment events weighted by the number of patients each event occurred in (.e. the count of engrafted features, weighted by the square of the number of patients each feature was engrafted in) (T3). The reason for this third test statistic was that we reasoned that a feature that appears engrafted in multiple patients is less likely to have been a spurious signal of engraftment, and so we wanted to up-weight those features based on the number of individuals they were engrafted in. Looking at the graphs in Figs. 2A−C, 3A and 4A (top panels), the test statistics can be calculated as follows, where x is the number of patients a feature is engrafted in, and f(x) is the number of features engrafted in exactly that many patients:
Permutation tests were conducted by permuting the group membership of each patient 1999 times, and for each permutation calculating the number of engrafted features, engraftment events, and weighted engraftment events in each group, and taking their difference. The 2000th value in each permuted null distribution was the observed value. Two-tailed p-values were calculated by finding the quantile of the observed test statistic in the null distribution, taking its distance from 0 or 1, whichever was smaller, and doubling that value. This same method was used to compare the amounts of apparent engraftment in responder- vs. non-responder-patients within the FMT treatment group (Figs. 2A−C, 3A and 4A, bottom panels).
Once count tables or coverage values had been calculated for a given feature type, all data organization and analysis code were written in R v4.2.0 using the tidyverse59 collection of packages. Figures were generated in R using ggplot2 and tidytext and refined in Inkscape. R scripts are available at https://github.com/SShekarriz/UCFMT1.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Shotgun metagenomic sequences used in this study are available via the SRA under BioProject number PRJNA1220173. These samples include fecal samples from the donor and patients with ulcerative colitis including both FMT and placebo groups, as well as plate pool metagenomes from the donor.
Code availability
All code used for figure generation and statistical analyses is available at https://github.com/SShekarriz/UCFMT1.
References
Kappelman, M. D. et al. The prevalence and geographic distribution of Crohn’s disease and ulcerative colitis in the United States. Clin. Gastroenterol. Hepatol. 5, 1424–1429 (2007).
de Souza, H. S. P. & Fiocchi, C. Immunopathogenesis of IBD: current state of the art. Nat. Rev. Gastroenterol. Hepatol. 13, 13–27 (2016).
Hindryckx, P., Jairath, V. & D’Haens, G. Acute severe ulcerative colitis: from pathophysiology to clinical management. Nat. Rev. Gastroenterol. Hepatol. 13, 654–664 (2016).
Talley, N. J. et al. An evidence-based systematic review on medical therapies for inflammatory bowel disease. Am. J. Gastroenterol. 106, S2–S25 (2011).
Moayyedi, P. et al. Fecal microbiota transplantation induces remission in patients with active ulcerative colitis in a randomized controlled trial. Gastroenterology 149, 102–109.e6 (2015).
Peery, A. F. et al. AGA clinical practice guideline on fecal microbiota-based therapies for select gastrointestinal diseases. Gastroenterology 166, 409–434 (2024).
Khoruts, A., Staley, C. & Sadowsky, M. J. Faecal microbiota transplantation for Clostridioides difficile: mechanisms and pharmacology. Nat. Rev. Gastroenterol. Hepatol. 18, 67–80 (2021).
Khanna, S., Shin, A. & Kelly, C. P. Management of clostridium difficile infection in inflammatory bowel disease: expert review from the clinical practice updates committee of the AGA Institute. Clin. Gastroenterol. Hepatol. 15, 166–174 (2017).
Feng, J. et al. Efficacy and safety of fecal microbiota transplantation in the treatment of ulcerative colitis: a systematic review and meta-analysis. Sci. Rep. 13, 14494 (2023).
Pai, N. et al. Results of the first pilot randomized controlled trial of fecal microbiota transplant in pediatric ulcerative colitis: lessons, limitations, and future prospects. Gastroenterology 161, 388–393.e3 (2021).
Narula, N. et al. Systematic review and meta-analysis: fecal microbiota transplantation for treatment of active ulcerative colitis. Inflamm. Bowel Dis. 23, 1702–1709 (2017).
Jaramillo, A. P. et al. Effectiveness of fecal microbiota transplantation treatment in patients with recurrent clostridium difficile infection, ulcerative colitis, and Crohn’s disease: a systematic review. Cureus 15, e42120 (2023).
Liu, H., Li, J., Yuan, J., Huang, J. & Xu, Y. Fecal microbiota transplantation as a therapy for treating ulcerative colitis: an overview of systematic reviews. BMC Microbiol. 23, 371 (2023).
Smillie, C. S. et al. Strain tracking reveals the determinants of bacterial engraftment in the human gut following fecal microbiota transplantation. Cell Host Microbe 23, 229–240.e5 (2018).
Staley, C. et al. Durable long-term bacterial engraftment following encapsulated fecal microbiota transplantation to treat clostridium difficile infection. mBio 10, https://doi.org/10.1128/mbio.01586-19 (2019).
Aggarwala, V. et al. Precise quantification of bacterial strains after fecal microbiota transplantation delineates long-term engraftment and explains outcomes. Nat. Microbiol. 6, 1309–1318 (2021).
Deng, Z.-L. et al. Engraftment of essential functions through multiple fecal microbiota transplants in chronic antibiotic-resistant pouchitis—a case study using metatranscriptomics. Microbiome 11, 269 (2023).
Wilson, B. C. et al. Strain engraftment competition and functional augmentation in a multi-donor fecal microbiota transplantation trial for obesity. Microbiome 9, 107 (2021).
Chen-Liaw, A. et al. Gut microbiota strain richness is species specific and affects engraftment. Nature 637, 422–429 (2025).
Podlesny, D. et al. Identification of clinical and ecological determinants of strain engraftment after fecal microbiota transplantation using metagenomics. Cell Rep. Med. 3, 100711 (2022).
Ianiro, G. et al. Variability of strain engraftment and predictability of microbiome composition after fecal microbiota transplantation across different diseases. Nat. Med. 28, 1913–1923 (2022).
Schmidt, T. S. B. et al. Drivers and determinants of strain dynamics following fecal microbiota transplantation. Nat. Med. 28, 1902–1912 (2022).
Dsouza, M. et al. Colonization of the live biotherapeutic product VE303 and modulation of the microbiota and metabolites in healthy volunteers. Cell Host Microbe 30, 583–598.e8 (2022).
Menon, R. et al. Multi-omic profiling a defined bacterial consortium for treatment of recurrent Clostridioides difficile infection. Nat. Med. 31, 223–234 (2025).
Valles-Colomer, M. et al. The person-to-person transmission landscape of the gut and oral microbiomes. Nature 614, 125–135 (2023).
Lau, J. T. et al. Capturing the diversity of the human gut microbiota through culture-enriched molecular profiling. Genome Med. 8, 72 (2016).
Browne, H. P. et al. Culturing of ‘unculturable’ human microbiota reveals novel taxa and extensive sporulation. Nature 533, 543–546 (2016).
Sibley, C. D. et al. Culture enriched molecular profiling of the cystic fibrosis airway microbiome. PLOS ONE 6, e22702 (2011).
Whelan, F. J. et al. Culture-enriched metagenomic sequencing enables in-depth profiling of the cystic fibrosis lung microbiota. Nat. Microbiol. 5, 379–390 (2020).
Myles, I. A. et al. A method for culturing Gram-negative skin microbiota. BMC Microbiol. 16, 60 (2016).
Hilt, E. E. et al. Urine is not sterile: use of enhanced urine culture techniques to detect resident bacterial flora in the adult female bladder. J. Clin. Microbiol. 52, 871–876 (2014).
Callahan, B. J. et al. DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Blanco-Míguez, A. et al. Extending and improving metagenomic taxonomic profiling with uncharacterized species using MetaPhlAn 4. Nat. Biotechnol. 41, 1633–1644 (2023).
Meziti, A. et al. The reliability of Metagenome-Assembled Genomes (MAGs) in representing natural populations: insights from comparing MAGs against isolate genomes derived from the same fecal sample. Appl. Environ. Microbiol. 87, e02593–20 (2021).
Franzosa, E. A. et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 4, 293–305 (2019).
Montrose, J. A., Kurada, S. & Fischer, M. Current and future microbiome-based therapies in inflammatory bowel disease. Curr. Opin. Gastroenterol. https://doi.org/10.1097/MOG.0000000000001027 (2024).
Costello, S. P. et al. Effect of fecal microbiota transplantation on 8-Week remission in patients with ulcerative colitis: a randomized clinical trial. JAMA 321, 156–164 (2019).
Whelan, F. J. et al. The loss of topography in the microbial communities of the upper respiratory tract in the elderly. Ann. Am. Thorac. Soc. 11, 513–521 (2014).
Bartram, A. K., Lynch, M. D. J., Stearns, J. C., Moreno-Hagelsieb, G. & Neufeld, J. D. Generation of multimillion-sequence 16S rRNA gene libraries from complex microbial communities by assembling paired-end illumina reads. Appl. Environ. Microbiol. 77, 3846–3852 (2011).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12 (2011).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2013).
McMurdie, P. J. & Holmes, S. phyloseq: an R Package for reproducible interactive analysis and graphics of microbiome census data. PLOS ONE 8, e61217 (2013).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Schmieder, R. & Edwards, R. Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLOS ONE 6, e17288 (2011).
Nurk, S., Meleshko, D., Korobeynikov, A. & Pevzner, P. A. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834 (2017).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Kang, D. D. et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359 (2019).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Eren, A. M. et al. Community-led, integrated, reproducible multi-omics with anvi’o. Nat. Microbiol. 6, 3–6 (2021).
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics 36, 1925–1927 (2020).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2 – approximately maximum-likelihood trees for large alignments. PLOS ONE 5, e9490 (2010).
Schwengers, O. et al. Bakta: rapid and standardized annotation of bacterial genomes via alignment-free sequence identification. Microb. Genom. 7, 000685 (2021).
Shen, W., Le, S., Li, Y. & Hu, F. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLOS ONE 11, e0163962 (2016).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinform. 10, 421 (2009).
Steinegger, M. & Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829 (2021).
Lander, E. S. & Waterman, M. S. Genomic mapping by fingerprinting random clones: a mathematical analysis. Genomics 2, 231–239 (1988).
Wickham, H. et al. Welcome to the Tidyverse. J. Open Sour. Softw. 4, 1686 (2019).
Acknowledgements
This work was supported by grants from Crohn’s Colitis Canada, and the Inflammation, Microbiome, and Alimentation: Gastrointestinal and Neuropsychiatric Effects (IMAGINE) Network CIHR grant. M.G.S. is supported by a Canada Research Chair. During some of this work S.S. was supported by Mitacs Elevate Fellowship and F.J. Whelan was supported by an Anne McLaren Fellow funded by the University of Nottingham and a Marie Skłodowska-Curie Individual Fellowship (grant agreement number 793818). The authors would like to thank Dr. Ben Bolker and the McMaster biodata lunch group, as well as Dr. Kevin Purbhoo, for their valuable insight and suggestions in the development of our statistical methods.
Author information
Authors and Affiliations
Contributions
S.S. is the primary author of this prepared manuscript and M.G.S is the corresponding author. S.S. and M.G.S. conceptualized the experimental outline. S.S. conducted all data analyses and wrote the manuscript. S.S. and J.C.S conceptualized statistical analyses. J.C.S. conducted permutations test and updated the statistical codes. S.S. and J.C.S. uploaded and reviewed GitHub codes. J.L., M.W., C.H.L. and P.M. conducted the clinical trial and collected patients’ information. J.L., L.R., and M.E.F. extracted DNA and conducted sequencing. J.T.L. conducted comprehensive cultured-enriched microbial profiling. F.J.W. developed the plate coverage algorithm and the culture-enriched sequencing strategy. All authors edited and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jeremiah Faith, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shekarriz, S., Szamosi, J.C., Whelan, F.J. et al. Detecting microbial engraftment after FMT using placebo sequencing and culture enriched metagenomics to sort signals from noise. Nat Commun 16, 3469 (2025). https://doi.org/10.1038/s41467-025-58673-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58673-x
