
Abstract
Stem cell-derived islet cell therapy can effectively treat type 1 diabetes, but its efficacy is hindered by low oxygen supply post-transplantation, particularly in subcutaneous spaces and encapsulation devices, leading to cell dysfunction. The response to hypoxia and effective strategies to alleviate its detrimental effects remain poorly understood. Here, we show that β cells within stem cell-derived islets gradually undergo a decline in cell identity and metabolic function in hypoxia. This is linked to reduced expression of immediate early genes (EGR1, FOS, and JUN), which downregulates key β cell transcription factors. We further identified genes important for maintaining β cell fitness in hypoxia, with EDN3 as a potent player. Elevated EDN3 expression preserves β cell identity and function in hypoxia by modulating genes involved in β cell maturation, glucose sensing and regulation. These insights improve the understanding of hypoxia’s impact on stem cell-derived islets, offering a potential intervention for clinical applications.
Similar content being viewed by others
Introduction
Type 1 diabetes (T1D) is a chronic autoimmune disorder characterized by the loss of insulin-producing β cells in the pancreas that are killed by autoreactive T cells. This destruction results in deficient insulin secretion, leading to high blood glucose levels and various long-term complications1,2,3. Current treatment options for T1D rely on exogenous insulin administration. However, cell replacement therapies utilizing human stem cell-derived islets (SC-islets) have emerged as a promising approach to restore glycemic homeostasis in patients4,5,6,7,8,9,10,11. Nevertheless, the success of SC-islet cell therapy relies on maintaining the survival and function of β cells post-transplantation6,12,13,14. One significant challenge in this regard is the inadequate oxygen (O2) supply encountered at the transplantation site15,16,17. The delivery of O2 to the transplanted cells is restricted due to slow passive diffusion from extra-arterial sources, particularly in subcutaneous (s.c.) sites18,19. Furthermore, this issue could be exacerbated by the encapsulation of SC-islets within devices that are intended to provide immune protection20,21,22,23.
Pancreatic β cells rely heavily on aerobic metabolism to generate adenosine triphosphate (ATP) for insulin secretion24. In the native pancreas, islets are densely vascularized, and the insulin-producing β cells experience a partial pressure of oxygen (pO2) of approximately 40–60 mmHg25. Studies show that the pO2 of islets in the transplantation sites, such as the kidney capsule, drops below 10 mmHg, equivalent to around 1-2% oxygen in the incubator26,27. Ischemia, which occurs post-transplantation, leads to endoplasmic reticulum (ER) stress and the production of reactive oxygen species (ROS) within the transplanted islets24,28,29,30. These factors can ultimately contribute to islet cell death16,31. β cells that survive in hypoxic conditions undergo a metabolic shift from aerobic glucose metabolism to anaerobic glycolysis, resulting in impaired functional capabilities32,33.
With advances in stem cell differentiation, functional SC-islets are successfully generated in vitro using a combination of small molecules and growth factors in incubators with a supply of 21% oxygen, approximately 160 mmHg5,7,34,35,36. The resulting β cells within SC-islets exhibit several characteristics resembling primary β cells found in human islets, including the expression of β-cell-specific markers, glucose-responsive insulin secretion, and the capability to reverse diabetes in animal models5,13,15,34. While making SC-islets that respond to glucose challenges represents a significant breakthrough, the final product is not an exact replica of a human islet. Differences between β cells within SC-islets and primary β cells from human islets include RNA transcripts34, epigenetic modifications37,38, enzymatic and mitochondrial function9. Although the impact of oxygen limitations on the survival and metabolic function of primary islets and SC-islets has been documented16,25,39,40,41,42,43,44,45,46, our understanding of how SC-islets respond to hypoxia at single-cell resolution and potential strategies to mitigate its effects is still lacking.
This study investigates the influence of hypoxia on SC-islets. We employed transcriptional profiling, including single-cell RNA sequencing and multimodal single-nucleus ATAC and RNA sequencing, to describe the molecular mechanisms in SC-islets subjected to hypoxia. Our findings show that, unlike other stem cell-derived endocrine cells, there is a progressive loss of cell identity and metabolic function in stem cell-derived β (SC-β) cells when exposed to hypoxia14. Through a comparative analysis of SC-islets under low and high oxygen environments, we identified genes that play a role in maintaining the fitness of SC-islets under low oxygen conditions. Notably, we demonstrate that overexpression of EDN3 helps preserve β-cell identity in hypoxic environments. Upregulations of EDN3 in SC-islets mitigate the effects of hypoxia by modulating genes involved in glucose sensing and insulin expression.
Results
Gradual loss of cell identity and metabolic function
After transplantation, cells undergo a stress response resulting from a lack of nutrients, accumulation of waste, and a hypoxic environment6. The complex in vivo environment presents challenges in understanding the impact of hypoxia on β cells. To uncouple the effect of hypoxia, we conducted a temporal analysis of SC-islets under different oxygen concentrations in vitro. SC-islets were differentiated in vitro using a six-stage protocol lasting for over a month in incubators supplied with 21% oxygen5,34. SC-islets were then challenged with three different oxygen levels (21%, 5%, and 2%) over a six-week period, followed by analysis with flow cytometry, immunofluorescent staining, single-cell RNA sequencing, multimodal single-nucleus RNA sequencing and single-nucleus ATAC sequencing (Fig. 1a). Experiments were performed in spinner flasks to ensure rapid liquid-gas equilibration. The subcutaneous space has a pO2 level (45 mmHg) equivalent to about 5% oxygen in incubator19. 2% oxygen is considered to be very hypoxic in culture. The proportion of SC-β cells (C-peptide+/NKX6.1+) remained stable during the six-week culture period in normoxic conditions (21%), indicating SC-β cells maintain their identity and population distribution even when cultured for a long period in vitro (Fig. 1b and Supplementary Fig. 1a). In contrast, a gradual decline in the percentage of C-peptide+/NKX6.1+ β cells was observed in 5% and 2% oxygen environments. Initially, approximately 55% of cells were identified as C-peptide+/NKX6.1+ β cells at day 0, but only 10% of the C-peptide+/NKX6.1+ β cells were maintained by week six (Fig. 1b and Supplementary Fig. 1a). In comparison, the populations of all endocrine cells in the SC-islets, including SC-β cells, stem cell-derived alpha cells (SC-α) and stem cell-derived enterochromaffin cells (SC-EC) remained relatively unchanged throughout the six-week period, in 5% or 2% oxygen (Fig. 1c and Supplementary Fig. 1b). Representative flow cytometry analysis at the six-week time point illustrated a decline in β-cell population, with 50% of C-peptide+/NKX6.1+ β cells under the 21% oxygen condition compared to a 10% under the 2% oxygen condition (Fig. 1d and Supplementary Fig. 1a). Representative immunofluorescent staining further substantiated the flow cytometry data at week 6 (Fig. 1e). There was no change in cell counts showing that minimal cell death occurs during hypoxia and changes in cell distributions are likely due to the change in gene expression (Supplementary Fig. 1c). A glucose stimulated insulin secretion (GSIS) assay showed that SC-islets remain functional in normoxia. However, insulin secretion was impaired after 1 week and exacerbated after 2 weeks in 5% oxygen. More importantly, SC-islets lost function only after 1 week in 2% oxygen. These data indicated impaired glucose-sensitive insulin secretion of SC-islets under hypoxia (Fig. 1f). Under hypoxia, SC-β cells stop expressing β cell-specific markers, notably insulin. To be clear, we did not observe changes in cell number during hypoxia. The changes refer to the loss of specific β-cell gene markers.

a Schematics of experiment setup. b Percentage of cells expressing NKX6.1 and C-peptide (SC-β cells) during 6 weeks’ culture in 21%, 5%, and 2% oxygen (n = 3 biological replicates). The data were expressed as means ± SEM and compared using one-way ANOVA followed by Tukey’s test. P = 0.221 (D35), P = 0.017 (D42). c Percentage of SC-endocrine cells during 6 weeks’ culture in 21%, 5%, and 2% oxygen (n = 3 biological replicates). The data were expressed as means ± SEM and compared using two-way ANOVA followed by Tukey’s test. P = 0.8546. d Flow cytometry plots of SC-islets cultured in 21%, 5%, and 2% oxygen for 4 weeks and stained with CPEP and NKX6.1. e Immunofluorescent staining of SC-islets cultured in 21%, 5%, and 2% oxygen for 4 weeks with antibodies (green, CPEP; purple, NKX6.1; gray, DAPI). f Static human insulin secretion of SC-islets cultured in 21%, 5%, and 2% oxygen for 1 week (left) and 2 weeks (right) in a glucose-stimulated insulin secretion (GSIS) assay (n = 8 biological replicates). The data were expressed as means ± SD and compared using one-way ANOVA followed by Tukey’s test. *P < 0.05; n.s., non-significant. Scale bar: 50 μm (e). Schematics of the experiment setup created in BioRender. Wang, K. (2025) https://BioRender.com/kpjb3o4.
Characterization of the regulatory response to hypoxia in SC-islets
The impact of hypoxia on SC-islets was investigated by single-cell RNA sequencing (scRNA sequencing). SC-islets were cultured in 21%, 5%, and 2% oxygen for 2 weeks and 4 weeks as indicated (Fig. 1a). Utilizing uniform manifold approximation and projection (UMAP) and unbiased clustering analysis, eight distinct cell clusters were identified and assigned cell identities based on endocrine gene markers (Fig. 2a). We identified three endocrine cell populations SC-α, SC-EC and SC-β that comprise most of the endocrine cell populations of the SC-islets. We also identified a small population of poly-hormonal cells marked by the expression of ghrelin-obestatin preproprotein gene, GHRL. UMAP visualization demonstrated that SC-α and SC-EC cells from different time points and oxygen levels clustered by cell identity (Fig. 2 b and c). On the other hand, SC-β cells showed significant changes to their distribution across both time points and oxygen levels. Expression of glucagon, tryptophan hydroxylase 1 (TPH1), and insulin confirmed the presence of SC-α cells, SC-EC cells, and SC-β cells, respectively (Fig. 2d). Cells expressing high levels of INS and NKX6.1 (SC-β INShigh) constituted most of the endocrine cell population (51%) in high oxygen levels (21%) but their levels significantly decreased with decreasing hypoxic conditions 5% oxygen level (17% − 2 W and 4 W) and 2% oxygen level (3% − 2 W and 4 W) (Fig. 2e). In opposition, cells cultured under hypoxic condition showed an increase in the number of NKX6.1+ cells that expressed basal levels of INS (NKX6.1+, SC-β INSlow). Transcriptionally, both the SC-β INSlow and NKX6.1+ cell populations were similar to the SC-β INShigh cell population (Fig. 2f). Moreover, both SC-β INSlow and NKX6.1+ cells expressed SC-β maturation markers (ITGA1, PCSK1), leading to identify these populations as SC-β cells with low insulin expression (Fig. 2g and Supplementary Fig. 2a, b). We did not observe significant changes in cell number during hypoxic conditions (Supplementary Fig. 1c). The combined ratios of SC-β INShigh and SC-β INSlow remain almost identical across the 2 W and 4 W time points and between different oxygen levels, constituting > 50% of the total cell populations.

a A uniform manifold approximation and projection (UMAP) visualization of SC-islets cultured in 2, 5, and 21% oxygen for 2 or 4 weeks, combining all the time points and all the conditions. Cells are colored according to their assigned cell type. b UMAP of cells colored according to acquisition time point. c UMAP of cells colored according to the oxygen level. d UMAP of cells colored according to the expression of endocrine-associated markers (SC-β, INS; SC-α, GCG; SC-enterochromaffin cells, TPH1). Colors in (d) are unique and refer to gene expression. a–d represent the same UMAP colored as indicated above. e The population ratios of SC-islets cultured in 2, 5, and 21% oxygen levels after 2 weeks (left) and 4 weeks (right). Vertical bars are colored according to their assigned cell type in (a). f The correlation map of expression profiles of the main cell types identified in (a). g Expression levels of cell markers across cell types identified in the SC-islet cell population under hypoxia. The shading indicates the mean expression of each gene in the group, and the diameter of dots denotes the percentage of cells expressing the gene. h KEGG pathway analysis of SC-β cells in response to hypoxia, upregulated pathways (blue) or downregulated pathways (red) in SC-β INShigh vs. SC-β INSlow cells. Gene Set Enrichment Analysis (GSEA) is applied to analyze KEGG pathways. Multiple comparison was adjusted by Benjamini-Hochberg method. i Heatmap of selected genes in the KEGG pathway analysis shown in (h).
We investigated the signaling pathways that respond when SC-β cells are exposed to hypoxia. Pathway analysis revealed an upregulation of glycolytic, hypoxia-inducible factor (HIF) and central carbon metabolism signaling pathway in SC-β cells under hypoxia (Fig. 2h). However, the insulin secretion pathway is downregulated in SC-β cells exposed to low oxygen (Fig. 2h). Genes involved in glycolysis including LDHA and PKM are upregulated in low oxygen (Fig. 2i), as are genes associated with the HIF pathway, including SLC2A1, and central carbon metabolism genes, (TPI1 and PEKP). Conversely, downregulation of genes involved in insulin secretion, including INS, PDX1, and CACNA1D, is observed in response to oxygen deprivation. In addition, genes related to regulating insulin secretion in response to external stimuli, including the cAMP signaling pathway (VAV3 and PTGER3), were downregulated in SC-β cells in hypoxia (Fig. 2i). These aforementioned genes are top-related within each pathway.
Cellular dynamics of SC-islets in response to hypoxia
We performed a velocity analysis of transcription to better understand the dynamics of SC-islets in response to hypoxia. The method examines both nascent and mature RNA, categorized by intronic and exonic reads, with a focus on the behavior of major cell clusters, including SC-α cells, SC-EC cells, and SC-β cells. Cells were grouped by expression of endocrine markers and their dynamics were analyzed under 2% oxygen over time (1, 3, 5 days and 2, 4 weeks) in response to hypoxia with a reference of SC-islets cultured in 21% oxygen (Fig. 3a). The time points were picked to mimic both acute (1, 3 and 5 days) and longer-term (2 and 4 weeks) adaptations of SC-islets under hypoxia. Using trajectory analysis, we mapped the transitions between different cell states and examined the velocity of these changes. This analysis reveals distinct cellular responses to hypoxia within the SC-islet cell populations. RNA velocity-derived vectors of SC-α cells exhibited no significant changes in their profile over time, indicating relative stability under hypoxic conditions. Similarly, SC-EC cells at different time points overlapped, suggesting that changes in these cells occur between different cell states rather than over time in response to hypoxia. While SC-α cells and SC-EC cells exhibited relatively stable profiles, SC-β cells showed dynamic changes in cell identity over time (Fig. 3a, b). Trajectory mapping of SC-β cells revealed non-overlapping cell populations at different time points, indicating a flow from one cell state to another in response to hypoxia. Similarly, the correlation between transcriptome of SC-β cells from different time points decreased as a function of time and had an overall lower correlation compared to SC-α, and SC-EC cells across all time points. It further supports that cells undergo a gradual change in SC-β cell identity during hypoxia not observed in the other endocrine cell types (Fig. 3c).

a RNA velocity visualized on UMAP of SC-islets cultured in 21% oxygen for 1 day or cultured in 2% oxygen for 1 day, 3 days, 5 days, 2 weeks, and 4 weeks. Cells are colored according to their assigned cell type. b UMAP of cell types shown in (a), colored according to acquisition time point. a, b represent the same UMAP colored as indicated above. c Correlation of expression profiles for major cell types, including SC-β cells, SC-α cells, and SC-enterochromaffin cells (SC-EC) in 2% oxygen, broken down by acquisition time point. d RNA velocity trajectories visualized on UMAP of SC-β cells cultured in 21% oxygen for 1 day or cultured in 2% oxygen for 1 day, 3 days, 5 days, 2 weeks, and 4 weeks. e Representative plots of low-to-high (bottom) and high-to-low (top) insulin trajectories. f UMAP of SC-β cells colored according to acquisition time point. g Representative gene expression visualized on UMAP of SC-β cells in response to hypoxia (INS, left; HIF1α, right). h Expression of selected marker genes as a function of latent time in (d). Dots show expression in single cells, sorted and shaded according to pseudotime order. The lines represent the regression fitted as a function of pseudotime. i Pathway enrichment of differential genes of SC-β cells in response to hypoxia. Gene Set Enrichment Analysis (GSEA) is applied to analyze KEGG pathways. Multiple comparison was adjusted by Benjamini-Hochberg method. j Principal component analysis (PCA) of proteome profiles of SC-β cells cultured in 2% oxygen for 1 day, 3 days, and 5 days compared to those of SC-β cells cultured in 21% oxygen (one-way ANOVA P < 0.01). (n = 3 biological replicates). k KEGG pathway analysis of proteome profiles of SC-β cells cultured in 2% oxygen for 5 days compared to that of SC-β cells cultured in 21% oxygen, downregulated pathways (blue) or upregulated pathways (red). Gene Set Enrichment Analysis (GSEA) is applied to analyze KEGG pathways. Multiple comparison was adjusted by Benjamini-Hochberg method. l Fold changes of protein expression of SC-β cells cultured in 2% oxygen for 1 day (red), 3 days (green), 5 days (orange) to protein expression of SC-β cells cultured in 21% oxygen; dot size represents statistical significance (one-way ANOVA) (n = 3 biological replicates). Proteins were selected as differentially expressed if they met the criteria of an adjusted p-value < 0.01 (one-way ANOVA) and a log fold change (LFC) > 1 at least at one time point (days 1, 3, or 5) under 2% oxygen conditions.
To gain more insight into the dynamics of SC-β cells, we conducted velocity analysis within this population. The velocity map demonstrated that most cells progressed chronologically from day 1 to four weeks of hypoxia challenge (Fig. 3d, e; low insulin branch). The SC-β cell population was characterized by gradually downregulating insulin expression and upregulating the hypoxia master regulator, HIF1A (Fig. 3f and g). Interestingly, a second trajectory was observed within the SC-β cell population, representing cells moving from a low insulin to high insulin state (Fig. 3d, e; high insulin branch). Further analysis shows a downregulation of INS and pathways associated with insulin processing, including oxidative proinsulin folding (ERO1B) and insulin secretion (SLC30A8 and CPE). Interestingly, genes involved in insulin secretion (SLC30A8 and CPE) appeared to be downregulated at a faster rate than insulin downregulation, suggesting that cell may lose their ability to release insulin before insulin production is halted. Whereas genes involved in protein quality control and ER stress, such as ERO1B share a similar decrease in the rate of transcription as INS (Fig. 3h, i).
A proteomic analysis was performed to complement the transcriptional study. We applied principal component analysis (PCA) to samples cultured in 21% oxygen for 1 day or cultured in 2% oxygen for 1, 3, and 5 days, to explore the relationships and patterns among the samples based on their overall protein expression profiles. The PCA plot revealed distinct clustering patterns, indicating that the samples from different conditions were clearly separated from each other (Fig. 3j). In addition, the biological replicates within each condition clustered closely together, indicating a high degree of reproducibility and consistency between replicates (Fig. 3j). Moreover, the first principal component, which accounted for 65% of variation between samples correlated with the progression of time in hypoxic conditions, with 21% D1 samples and 2% D5 samples separated the furthest apart. KEGG pathway analysis of the differentially expressed proteins in our study revealed upregulated proteins predominantly involved in glycolysis, HIF signaling pathway, and carbon metabolism (Fig. 3k). Conversely, downregulated proteins were associated with biosynthesis of amino acids and fatty acid (Fig. 3k). Mass spectrometry-based time course analysis including Day 1, Day 3 and Day 5 shows dynamics regulated by hypoxia by looking at proteins that show monotonic differential expression across time points. Proteins that showed a gradual increase in expression from Day 1 to Day 5, including LDHA and SYNPO, involved in anaerobic glycolysis and oxidative stress, respectively, indicate a progressive response to hypoxic conditions (Fig. 3l and Supplementary Fig. 3a–c). Conversely, proteins including ESYT1 display a steady decline in expression over time, indicating downregulation of insulin secretion during hypoxia47,48 (Fig. 3l). All together, these regulated proteins may represent an adaptation for cell survival under low oxygen levels.
TF regulatory network of SC-islets in response to hypoxia
To infer the gene regulatory network (GRN) involved in SC-islets response to hypoxia, a multi-omic analysis was performed to catch the transcriptional and chromatin features of SC-islets during hypoxia. SC-islets were cultured in 21% oxygen for 1 day, or cultured in 2% oxygen for 1 and 5 days, processed and sequenced using single-nucleus multi-omics, obtaining both gene expression (messenger RNA) and chromatin accessibility (ATAC) information for each cell. Analyzing gene expression identifies three distinct islet cell types, including SC-α cells, SC-EC cells, and SC-β cells (Fig. 4a). Cells exposed to hypoxia for 1 day and 5 days form a single group by RNA, not identified as a cohort by chromatin accessibility analysis. Whereas 2% D1 cells and 21% D1 cells showed significant overlap, indicating transcriptional regulation rather than changes in chromatin accessibility at early time points (Fig. 4b).

a UMAPs of single nuclei from SC-islets cultured in 2 and 21% oxygen for 1 or 5 days using either chromatin accessibility (ATAC) or gene (mRNA) information. Cells are colored according to their assigned cell type. b UMAP of cells colored according to experimental condition and acquisition time point. c UMAP of the modeled regulatory network of transcription factors. The color of nodes represents the relative gene expression of each gene in the model in 21% (D1) compared to 2% (D5). The size of each node represents the page centrality of each gene. The line colors represent the false-discovery rate (FDR) of the most significant interaction for each gene pair (FDR < 0.01). Transcription factors of interest are labeled in the UMAP. d Expression of selected marker genes along pseudotime ordering from (c). Dots show expression in single cells, sorted and shaded according to pseudotime order. The lines represent the regression fitted as a function of pseudotime. e A volcano plot of the enrichment of certain motifs peaks in 21% D1 vs 5% D5. f UMAP of SC-β cells (RNA expression-based) colored by condition. The middle row displays the expression levels of EGR1 and NEUROD1 in individual cells, while the bottom row shows the differential activity of motifs associated with EGR1 or NEUROD1 across individual cells. g Gene regulatory network inference of EGR1. The genes connected with black lines are predicted to be negatively regulated.
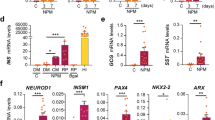
We integrated RNA expression data and DNA accessibility information37. A regulatory transcription factor network was constructed using a generalized linear model incorporating three key factors: expression value of genes differentially expressed between cells cultured in 21% versus 2% oxygen for one or five days (lfc > 0.5, p-adj < 0.01), the accessibility of identified transcription factor binding sites and the expression levels of the corresponding transcription factors, as measured by RNA levels. To visualize the gene regulatory network, we utilized a UMAP embedding based on the coefficients extracted from the fitted model. This uncovered distinct groups of transcription factors involved in different aspects of the cellular response to hypoxia. For example, a module of TFs including RFX3, SOX5, and PBX1 is downregulated during hypoxia and associated with glucose sensing and metabolism. β-cell-associated TFs critical for maintaining SC-β cell identity and maturation, including LMX1B, FOXP2, PAX4, BACH2, NKX6.1, and FOXO1, form another module of genes that is downregulated at the RNA level but showed increased motif accessibility (Fig. 4c and d). The results also revealed that many of these SC-β cell associated TFs directly or indirectly (second order) are regulated by Immediate Early Response (IER) genes EGR1, FOS and JUN (Fig. 4c). Pseudo time profiles of EGR1, FOS and JUN, showed downregulation in early stages of hypoxia, supporting the multimodal data set (Fig. 4c, d and Supplementary Fig. 4a). To elucidate which TFs play a role in the transcriptional changes during early onset of hypoxia, the differential chromatin accessibility of transcription factor-binding motif groups enriched in hypoxic SC-β cells compared to normoxic SC-β cells was examined. Transcription factor-binding motif groups enriched in hypoxic SC-β cells included the hypoxia master regulator, HIF1A, and IER genes EGR1, JUN1, and FOS binding motifs (Fig. 4e). In addition, hypoxic SC-β cells are enriched for binding motifs corresponding to known β-cell-associated transcription factors. For example, the binding motif for TCF7L2 and MAFA is enriched in hypoxic SC-β cells, though the expression of these transcription factors remained low compared to the control. We also observed reduced accessibility in the transcription factor binding motif involved in glucose-sensing-induced maturation, e.g., SIX2 and CLOCK.
EGR1 was central to many β-cell-associated transcription factors that were downregulated during hypoxia. Although the expression of EGR1 decreases, the accessibility of the motif peaks increases over time (Fig. 4f). Notably, the accessibility of the EGR1 motif binding sites exhibited an upward trend in response to hypoxia, indicating a potential regulatory role for EGR1 in the hypoxic response (Fig. 4f). Examining the local regulatory network of EGR1, suggests that it positively regulates pan-endocrine genes such as NEUROD1, PAX6, HNF4A, and LMX1B by modulating FOXO1 expression (Fig. 4g). Conversely, the accessibility of NEUROD1 motif binding sites showed a decreasing pattern in response to hypoxia (Fig. 4f).
Screening for genes important for SC-islet cell fitness in hypoxia
To identify genes that maintain SC-β cell identity during hypoxia, we devised a genome-scale screening strategy that identified known β cells mitigators of hypoxia, including GRID2. We identified genes differentially expressed in SC-β INShigh cells in low versus high oxygen conditions (lfc > 1, adj. p-value < 0.01) (Fig. 2a). To distinguish genes required for maintaining SC-β cell identity from genes differentially expressed during hypoxia, we only considered those that did not overlap with genes differentially expressed in low versus high oxygen conditions in the SC-β INSlow cohort (adj. p-value > 0.01). This approach identified genes which correlate with the maintenance of SC-β cell identity, including EDN3, DOK4, ASIC1, and GRID2 (Fig. 5b). To further validate the result, the expression level of these genes was determined as a function of latent time in our time course analysis. Many of the screen hits were downregulated as a function of latent time and INS expression in the low insulin branch, or upregulated and correlated with increased INS expression in the high insulin branch (Fig. 5c).

a Schematics of the experiment setup. b Upregulated genes of high insulin-expressing SC-β cells in low oxygen compared to that in high oxygen. The data were analyzed by a two-tailed Wilcoxon test and adjusted by the Benjamini-Hochberg method. c Dynamic gene expression during hypoxia challenge in the high-to-low (left) or low-to-high (right) insulin trajectories. Color bars showing the assigned latent time and the acquisition time are displayed on the bottom. Expression levels of individual genes were sorted by latent time and plotted as function of latent time and acquisition time (D1, D3, D5, 2 W, 4 W). d Flow cytometry plots of SC-islets control or modified cells with overexpression of indicated genes cultured in 21% and 2% oxygen for 2 weeks and stained with CPEP and NKX6.1 antibodies. e Quantitative analysis of CPEP+/NKX6.1+ cell percentage in (d) (n = 5 biological replicates). The data were expressed as means ± SD. Two-tailed Student’s t test was performed. Schematics of the experiment setup created in BioRender. Wang, K. (2025) https://BioRender.com/vxb9tmo.
We tested 12 genes that showed a nominal association (adj. p-value < 0.01) for the screen and lentiviral transduction was used to overexpress these genes to determine whether they could modify the hypoxic response (Supplementary Table 1 and Supplementary Fig. 5a). The genetically modified SC-islets were subjected to a 2-week exposure of low oxygen. And the expression of SC-β cell markers was assessed by flow cytometry (Fig. 5a). This screen encompassed twelve gene modifications (Supplementary Table 1). Flow cytometry data illustrated that nearly half of the SC-β cells in the control group lost their characteristic identity, as denoted by diminished expression of β cell markers C-peptide and NKX6.1 (Fig. 5d). A majority of the modified SC-islets, including those exhibiting overexpression of EDN3, ASIC1, DOK4, NAP1L5, or LGALS14 (Supplementary Fig. 5a), maintained their characteristic identity even after prolonged exposure to low oxygen (Fig. 5d). Quantitative assessment of the SC-β cell population before and after the hypoxia challenge revealed a preservation of SC-β cell identity following genetic modifications, specifically, the overexpression of EDN3, ASIC1, DOK4, NAP1L5, or LGALS14, contrary to the controls (Fig. 5e).
Overexpression of EDN3 in SC-islets mitigates hypoxia effect
We focused on the EDN3 gene, which encodes endothelin 3, a protein enriched in native pancreatic β cells (Fig. 6a, b and Supplementary Fig. 6a). To investigate the potential of EDN3 in mitigating hypoxia-induced effects, control SC-islets and SC-islets overexpressing EDN3 were subjected to a 4-week hypoxia challenge (2% oxygen) in vitro, followed by scRNA sequencing analysis (Fig. 6a, c). Unbiased clustering revealed eight distinct clusters, that included two SC-β cell clusters with high or basal expression of insulin (Supplementary Fig. 6b). EDN3 was highly expressed in the EDN3 overexpressed SC-islets compared to the control SC-islets (Fig. 6d and Supplementary Fig. 6c). After the 4-week hypoxia challenge, the control group exhibited only 14% of SC-β cells with high insulin expression, while approximately 40% of EDN3 overexpressed SC-β cells retained their cell identity and insulin expression. Comparing the insulin expressing SC-β cells, we found that insulin expression was also significantly higher in EDN3 overexpressed SC-β compared to control (Fig. 6e and Supplementary Fig. 6d). Expression of β cell markers, including IAPP and ITGA1, was better preserved in the EDN3 overexpressed SC-islet cell group under 2% oxygen compared to the control SC-islets under 2% oxygen (Fig. 6f). To note, PCSK1 expression is not noticeably preserved in the EDN3 overexpressed SC-islet cell group under 2% oxygen compared to the controls, which involved in the processing of proinsulin in islets. Moreover, EDN3 overexpressed SC-islets after hypoxia challenge presented a significant overlap with SC-β cells cultured in 21% oxygen and had a global gene expression profile closer to SC-β cells without hypoxia, compared to SC-β cells cultured in 2% oxygen for 4 weeks (Supplementary Fig. 6e). Many genes identified in our screen were upregulated in EDN3 overexpressing hypoxic SC-β cells, indicating that EDN3 is an upstream regulator of many target genes important for mitigating the effect of hypoxia, including GRID2, AQP10, LGALS14 and KLB (Fig. 6g).

a Schematics of the experiment setup. b EDN3 gene expression enrichment in native human tissue cell types. c A uniform manifold approximation and projection (UMAP) visualization of control SC-islets cultured in 2% or 21% oxygen or EDN3 overexpressed SC-islets cultured in 2% oxygen for 4 weeks. d Gene expression of EDN3 in SC-β cells (Ctrl) and EDN3 overexpressed SC-β cells (EDN3+). e Percentage of high insulin gene expression in SC-β cells (Ctrl) and EDN3 overexpressed SC-β cells (EDN3+) cultured in 2% oxygen for 4 weeks in vitro. f Dot plot of pancreatic β cell gene markers and gene expression in different conditions. g Dot plot of selected upregulated genes and downregulated genes in EDN3+ SC-β cells cultured in 2% oxygen compared to SC-β cells (Ctrl) cultured in 21% and 2% oxygen. h A uniform manifold approximation and projection (UMAP) visualization of SC-β cells and EDN3 overexpressed SC-β cells transplanted in NSG mice in the subcutaneous space for 1 week. i Gene expression of EDN3 in SC-β cells (Ctrl) and EDN3 overexpressed SC-β cells (EDN3+) transplanted in NSG mice in the subcutaneous space for 1 week (n = 2 biological replicates). The whiskers extend to the minimum and maximum values. The center line indicates the median. The box boundaries represent the 25th and 75th percentiles. j Cell type enrichment analysis of SC-β cells (Ctrl) and EDN3 overexpressed SC-β cells (EDN3+) transplanted in NSG mice in the subcutaneous space for 1 week. k A volcano plot of the differential gene expression of EDN3 overexpressed SC-β cells (EDN3+) and SC-β cells (Ctrl) transplanted in NSG mice in the subcutaneous space for 1 week. The data were analyzed by a two-tailed Wilcoxon test and adjusted by the Benjamini-Hochberg method. l Static human insulin secretion of control SC-islets (Ctrl) and EDN3 overexpressed SC-islets (EDN3+) cultured in 21% and 2% oxygen for 1 week in a glucose-stimulated insulin secretion (GSIS) assay (n = 5 biological replicates). The data were expressed as means ± SD. A Two-tailed Student’s t test was performed between the two groups. One-way ANOVA followed by Tukey’s test was performed between multiple groups. Schematics of the experiment setup created in BioRender. Wang, K. (2025) https://BioRender.com/t41ycxm.
To further test the efficacy of overexpressing EDN3 in maintaining β cell identity during hypoxia, control or EDN3 transduced SC-islets were imbedded in matrix and transplanted into immunodeficient non-diabetic mice in the dorsal subcutaneous space, which is considered hypoxic. One week post-transplantation, SC-islets were explanted and analyzed by scRNA sequencing (Fig. 6a, h). EDN3 was highly expressed in the EDN3 overexpressed SC-islets compared to the control SC-islets (Fig. 6i). We measured the similarity of the gene expression profiles of our samples to the expression profile of pancreatic β cells and converted that to an enrichment score for the control SC-β cells and EDN3 overexpressed SC-β cells (Fig. 6j). The analysis shows that EDN3 overexpressed SC-islets are transcriptionally more similar to pancreatic β cells compared to controls. EDN3 overexpressed SC-β cells upregulated genes associated with pancreatic β cell signatures, including INS (Fig. 6k). EDN3 overexpressed SC-β cells maintained significantly higher levels of insulin expression compared to control SC-β cells, after transplantation in the dorsal subcutaneous space for 1 week. Additionally, we found that EDN3 overexpressed SC-β cells upregulated critical genes associated with SC-β cell maturation markers, including the GSIS inhibitor G6PC2, TCF4, IGF1R, insulin-stabilizing zinc channel TMEM163, and the glucose-dependent transcription factor MIF, which is responsible for modulating insulin secretion. To evaluate whether EDN3 overexpression enhances the functional effects of SC-islets under hypoxic conditions, we conducted a GSIS assay on EDN3-overexpressing SC-islets exposed to 21% and 2% oxygen levels for one week, comparing them to control SC-islets. Under 21% oxygen, EDN3-overexpressing SC-islets remained functional, secreting significantly more insulin under high glucose conditions than low glucose, consistent with the control SC-islets (Fig. 6l). When exposed to 2% oxygen for one week, unlike control SC-islets, EDN3-overexpressing SC-islets retained glucose responsiveness, with significantly higher insulin secretion in response to high glucose (Fig. 6l). These results suggest that while EDN3 overexpression does not alter SC-islet function under normoxia, it provides functional benefits under hypoxic conditions. In conclusion, the overexpression of EDN3 in SC-islets can aid in the maintenance of β-cell identity and β-cell function during hypoxia.
Discussion
T1D treatments rely on exogenous insulin injections. Recently, cell replacement therapies using human stem cell-derived islets (SC-islets) offer an alternative with the potential to provide a functional cure49,50. However, the success of SC-islet cell therapy faces challenges including the supply of nutrients and oxygen following transplantation, in or out of an encapsulation device.
In the present study, SC-β cells exposed to hypoxic conditions undergo loss of function and identity. Prolonged hypoxia results in upregulation of the hypoxia master regulator, HIF-1α51,52,53,54,55, and glycolysis signaling, as well as a robust downregulation of the insulin processing and secretion pathways56. Treatment of islets with a HIF-1α inhibitor, PX-478, has been shown to improve their function in high-metabolic-demand-induced hypoxia57. The improved function is attributed to the damping of abnormal [Ca2+] oscillations at low glucose levels. In agreement, SC-islet downregulated multiple key genes in the cAMP pathway, which is involved in amplification of insulin secretion by calcium elevation58,59.
Divergent responses between transcriptomic and chromatin accessibility landscapes of SC-β cells revealed that temporal modulation of gene expression in response to hypoxia was discernible in the RNA level within 24 h after hypoxia induction. This finding shows that there is an immediate transcriptional adaptation to oxygen changes, whereas chromatin accessibility displays a delayed response37. It is also likely that other mechanisms, including protein interaction and phosphorylation, may play a role in the early response to hypoxia. Interestingly, many TFs associated with SC-β identity are downregulated in response to hypoxia. Expression of EGR1 is gradually reduced after hypoxia onset and is highlighted for its centrality in orchestrating the hypoxia-induced transcriptional program in SC-β cells60,61. A gene regulatory network (GRN) analysis showed that many TFs regulated by EGR1 during hypoxia are expressed in SC-β cells, including the pan-endocrine TFs FOXO1, NEUROD1, PAX6, HNF4A, and LMX1B. This points to a central role for EGR1 in the cellular response to hypoxia.
We identified genes that counteract the deleterious effects of hypoxia and overexpressed selected targets in SC-islets. Special attention is given to EDN362, a vasoactive secreted peptide that belongs to the endothelin family. EDN3 is enriched in native pancreatic islets63. Interestingly, EDN3-like peptides have been suggested for the treatment of metabolic disorders such as obesity and diabetes64. Hypoxic SC-β cells overexpressing EDN3 had a significantly higher expression of insulin and enhanced expression of β cell markers, many of which play a critical role in glucose sensing, insulin storage and release65,66,67. Among the most significantly differentially expressed genes, MIF stands out as a known regulator of glucose homeostasis68. MIF is released during insulin secretion and has an autocrine, glucose-dependent regulatory effect on insulin secretion. In pancreatic β cells, MIF expression is regulated by glucose, and its secretion potentiates the glucose-induced insulin secretion69. Collectively, our study showed that overexpression of EDN3 in SC-islets confers a protective role against the detrimental effects of hypoxia.
The main limitation of our study is that it remains unknown if EDN3 overexpression can improve the engraftment of SC-islets in a hypoxic transplantation site, such as the subcutaneous space70. The impact of EDN3 overexpression on SC-islet cell function in vivo needs to be further investigated. Furthermore, the in vivo environment is complicated, and the effects of EDN3 overexpression in combating other stressors71 warrants further investigation. Another notable limitation is the constrained availability of replicates in single-cell time course analysis. Nonetheless, when investigating small temporal intervals, changes observed within one time window are often sustained across subsequent windows, suggesting that robust temporal patterns can be discerned even in the absence of conventional biological replicates, provided sufficient data points are collected at closely spaced intervals. In addition, single-cell data analysis methods present inherent implementation challenges that require careful consideration. For example, while RNA velocity analysis, as applied in our study, is based on a theoretically sound concept72, it remains challenging to interpret in practice72,73. To address these limitations, we prioritized high-frequency sample collection, enabling a comprehensive understanding of dynamic processes over time.
Methods
Animals
Mice (6-8 weeks of age) NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (Jackson Laboratory, 005557) were purchased from the Jackson Laboratory. Mice were housed in pathogen-free conditions at Harvard University. All animal research was conducted under Harvard IACUC approval.
Stem cell culture and differentiation
Human embryonic stem cell maintenance and differentiation was carried out with Human Embryonic Stem Cells 8 (HUES8) following a six-stage differentiation protocol34. SC-islet cell differentiations were initiated 72 h after initial passage by aspirating mTeSR1 (STEMCELL Technologies, 85850) and replenished with stage- and day-specific media supplemented with the appropriate small molecules or growth factors34. All experiments involving human ES cells were approved by the Harvard University ESCRO (E00024) committee.
Magnetic enrichment using CD49a and reaggregation
SC-islets were magnetically enriched from SC-islet clusters using magnetic separation with anti-human CD49a PE-conjugated antibody (BD 559596, 1:100) and anti-PE UltraPure MACS microbeads for CD49a (Miltenyi 130-105-639, 1:2.5)34. Enriched cells were resuspended in S6 medium and seeded at 1 × 106 cells/ml in a 30 mL dispensable bioreactor (ABLE, ABBWVS03A-6) and incubated at 37 °C for 4–7 days to allow the cells to reaggregate. Enriched SC-islet cell clusters were fed with fresh Stage 6 (S6) medium every 2 days.
Hypoxia induction
SC-islets after enrichment and reaggregation were maintained in a 30 mL dispensable bioreactor to ensure gas equilibration and cultured in incubators (Eppendorf, C170i) with three different oxygen tensions-21%, 5%, and 2% O2. All cell culture was maintained in a humidified incubator at 5% CO2 and 37 °C. Tissue culture incubators were adapted to maintain different oxygen tensions. A nitrogen compressor was used to pulse N2 into incubators with a feedback loop to maintain the specified oxygen tension. All tissue culture handling was done outside of hypoxia incubators (at environmental oxygen tensions of 21% O2). Enriched SC-islets were fed with fresh S6 medium before hypoxia challenge and every other day during hypoxic incubation.
Flow cytometry
SC-islet cell clusters were collected, washed with PBS, dissociated with TrypLE (Gibco, 12563011) at 37 °C for 5 min and washed twice with S6 medium. Cell suspension was filtered through 40 μm strainer (Corning, 352340) and centrifuged at 350 g for 5 minutes. Cells were then washed, fixed, and permeabilized using Fixation/Permeabilization Kit (BD Biosciences, 554714) according to the manufacturer’s instructions. Fixed cells were then incubated in blocking buffer with rat anti-human C-peptide (DHSB, GN-ID4; 1:300) and mouse anti-human NKX6.1 (DHSB, F55A12; 1:100) overnight at 4 °C. Cells were then washed three times with blocking buffer, incubated with donkey anti-rat 488 (Life Technologies, A-21208; 1:400) and donkey anti-mouse 647 (Life Technologies, A-31571; 1:400) in blocking solution at room temperature for 1 h, washed three times and resuspended in blocking buffer. Samples were analyzed using an Attune NxT Flow Cytometer (Thermo Fisher Scientific) and FlowJo 10.7.1 (BD Biosciences).
Immunofluorescent staining
SC-islet cell clusters were collected, washed with PBS, fixed with neutral buffered formalin solution (Sigma Aldrich, HT501128), and stored in 4 °C. Then, clusters were washed with PBS, resuspended in HistoGel specimen processing gel (Thermo Fisher Scientific, HG-4000-012), transferred in tissue cassette, and embedded in paraffin. Samples were processed and sectioned by Harvard Stem Cell and Regenerative Biology (HSCRB) Histology-Immunohistochemistry Core Facility. The samples were sliced on a microtome at a thickness of 5 μm. To conduct immunofluorescent staining, the blank histological slides were deparaffinized followed by antigen retrieval as described before74,75. Nonspecific binding was blocked via incubation with 5% donkey serum (Sigma-Aldrich, S30-M) for 1 hour at room temperature. Sections were decanted and incubated with rat anti-human C-peptide (DHSB, GN-ID4; 1:300) and mouse anti-human NKX6.1 (DHSB, F55A12; 1:100) overnight at 4 °C. The sections were then washed and incubated with donkey anti-rat 488 (Life Technologies, A-21208; 1:400) and donkey anti-mouse 647 (Life Technologies, A-31571; 1:400) for 1 h at room temperature. Nuclei were labeled with DAPI, and slides were covered with fluorescent mounting medium (Sigma-Aldrich, F6057). Last, the sections were imaged through confocal microscopy Zeiss.Z2 with Apotome microscope (Zeiss), and analyzed using Zen Blue v2 (Zeiss).
In vitro glucose stimulated insulin secretion
The function of SC-islet clusters in vitro was evaluated by assessing glucose-stimulated insulin secretion (GSIS)76. To begin, the clusters were collected, washed twice in Krebs buffer (KRB), and then preincubated at 37 °C for 1 h in KRB containing a low glucose concentration of 2.8 mM. Subsequently, the clusters were subjected to a sequential treatment. This treatment consisted of alternating low-high KRB solutions containing glucose (with the high concentration being 20 mM). This was followed by depolarization using low KRB with 30 mM KCl. Each treatment lasted for 30 min, after which 100 μL of supernatant was collected. The concentration of human insulin in the supernatant was determined using the Human Ultrasensitive Insulin ELISA (ALPCO, 80-INSHUU-E01.1). To normalize the human insulin measurements, total cell counts were obtained by dispersing the clusters with TrypLE Express (Thermofisher, 12604013) and counting them using a Vi-Cell XR Cell Viability Analyzer (Beckman Colter).
Cell number count
To count the viable cell number of SC-islets cultured in incubators (Eppendorf, C170i) with three different oxygen tensions-21%, 5%, and 2% O2 for 1, 2, and 3 weeks, 1.5 ml of medium containing SC-islet clusters were collected from the bioreactor. SC-islet cell clusters were then dissociated into single cells using TrypLE Express. Total viable cells were quantified using the Vi-Cell XR Cell Viability Analyzer (Beckman Colter) following the manufacturer’s instructions with the Trypan Blue Dye Exclusion method.
Single-cell sample preparation and sequencing
SC-islet cell clusters were collected and dissociated into single cells as described before, quantified for viability using the Vi-Cell XR Cell Viability Analyzer (Beckman Colter), resuspended in PBS with 0.1% BSA at a concentration of ~ 1000 cells/μl and put on ice. Before proceeding, all samples were ensured to have > 90% viability to minimize dead cells carry over in sequencing. For the time course analysis and EDN3 in vitro testing, we used a single differentiation per condition, collecting cells at the appropriate time point for analysis (N = 1). We used two different differentiations for the EDN3 in vivo testing (N = 2). Single-cell samples were then processed using the Chromium 10x genomics instrument, with a target cell number of 10,000. The scRNA-seq libraries were created using the Chromium Single Cell 3’ V3 Library & Gel Bead Kit, following the manufacturer’s protocol. Single-cell encapsulation and library preparation were carried out as per the manufacturer’s guidelines. Libraries were then pooled and sequenced on the Illumina NovaSeq platform.
Processing and filtering of scRNA sequencing data
To obtain transcript count matrices, we employed Cell Ranger (version 7.1.0) with the default settings. This involved aligning the sequencing reads to the human genome and transcriptome (hg38, as provided by 10x Genomics, GRCh38-2020-A). The scRNA-seq data analysis pipeline utilized Scanpy (version 1.10.3)77 for dimensionality reduction, clustering, and visualization. Initially, individual sample output files from CellRanger Count were imported into Scanpy to generate a count matrix based on unique molecular identifiers (UMIs). To assess data quality, the percentage of mitochondrial genes was calculated for each sample in the merged dataset. Cells failing to meet the criteria of having at least 500 identified genes, 1000 UMI, or exceeding 25% of reads originating from mitochondrial genes were excluded from further analysis. Low-frequency genes that were detected in < 3 cells were also removed. Doublets were detected and removed using Scrublet (version 0.2.3) with default parameters. DecontX (version 1.2.0) was used to remove ambient RNA by running the DecontX function with default parameters on the raw counts matrix. After decontamination, the non-filtered and filtered expression matrices were retained for further analysis. The non-filtered matrix was used for cell clustering to preserve the full cellular information, while the filtered matrix (with ambient RNA removed) was used to analyze gene expression. Both expression matrices yielded similar results in follow-up analyses, indicating that the presence of ambient RNA did not significantly affect the clustering or gene expression conclusions. After the initial filtering steps, the dataset underwent normalization to address library size bias. This involved scaling the expression values to 10,000 counts per cell to account for variations in sequencing depth. Subsequently, the count-normalized expression matrix was log-normalized, and highly variable genes were identified based on dispersion and carried forward to the subsequent analysis steps. To address the influence of mitochondrial genes and UMI counts per cell, a simple linear regression was performed aimed to remove the effects of mitochondrial genes and UMI counts from the dataset. The data were further scaled to unit variance, and any values exceeding a standard deviation of 10 were clipped. Principal Component Analysis (PCA) was utilized to reduce the dimensionality of the data. In this step, the first 50 principal components, accounting for the majority variance in the dataset, were selected. A neighbor graph was constructed based on the PCA representation of the data and subsequently visualized using UMAP embedding.
Analysis of scRNA sequencing datasets
To identify related groups of cells, a neighborhood graph was created as described above. Subsequently, Leiden clustering was applied with the resolution parameter set to 0.2 for the analysis of the 2-week and 4-week datasets, comparing 2%, 5%, and 21% oxygen conditions, 0.17 for the time course analysis, and 0.25 for the EDN3 dataset analysis. This approach assigned cells into distinct clusters, which were then visualized using UMAP embedding. To assign cell types to each cluster, a list of established marker genes was used34. To identify differentially expressed genes across clusters or conditions, the corresponding log-fold ratios of expression levels were obtained, as well as the adjusted two-tailed Wilcoxon test p-values (using the Benjamini-Hochberg method). For testing differentially expressed genes (DEGs) in EDN3 overexpressing cells (EDN3+) versus control cells (Ctrl), we applied MAST (version 1.32.0) to improve robustness in identifying significant genes78. Subsequently, functional enrichment analysis was conducted using a Python implementation of the Gene Set Enrichment Analysis (GSEA) database-GSEApy (version 0.10.8). For transplanted samples, excess mice cells from host mice and non-endocrine cells were removed by filtering out cell clusters not expressing the human endocrine marker, CHGA.
Velocity analysis
To evaluate the islet cells states with respect to their order in hypoxia induction, we first used Velocyto (version 0.17.17) using default parameters to quantify the gene expression of unspliced and spliced transcripts79. Subsequently, scVelo (version 0.2.4) was used to remove low-quality counts with less than 20 spliced and unspliced counts80. Then the count-normalized expression matrix was log-normalized, and highly variable genes were identified based on dispersion. The 2000 most highly variable genes were used to reduce the dimensionality of the data by Principal Component Analysis (PCA). The first 30 principal components were subsequently used to construct a neighbor graph. To estimate the dynamics of gene expression, a steady-state ratio of pre-mature (unspliced) and mature (spliced) mRNA counts is fitted for each gene. This ratio represents a constant transcriptional state. The velocities of gene expression are then determined as the residuals obtained from this steady-state ratio using scVelo, the likelihood-based dynamical model. These velocities, which provide insights into the changes in gene expression levels in response to hypoxia, were visualized using a precomputed UMAP embedding graph81.
Trajectory analysis
To conduct a trajectory analysis of hypoxic SC-islets, we first inferred a latent pseudotemporal ordering of the cells. Using the splicing kinetics and RNA velocity estimation we obtained above, an efficient expectation-maximization framework is employed to infer the underlying gene-shared latent time. This latent time represents the continuous trajectory of cellular states. We then applied Leiden clustering to separate the SC-islet cell cluster into twelve distinct clusters. The resulting clusters were used to calculate a partition-based graph abstraction (PAGA) graph, which measures subgroup connectivity, and to draw a graph of possible transition paths between cell states within the SC-islet cell population. The strength of connectivity between clusters and their placement within the velocity field was used to assign the clusters into low-insulin and high-insulin branches. Gene dynamics along each branch was visualized by plotting log-normalized gene expression as a function of latent time and fitting the gene expression using a nonparametric lowness model.
Mass spectrometry sample preparation
Enriched SC-islets were reaggregated and maintained in 30 mL dispensable bioreactor in incubators supplied with 21% oxygen to form clusters. Then SC-islet cell clusters were challenged in incubators with 2% oxygen as described above. Clusters were collected in 1.5 ml low-adhesion microcentrifuge tubes after hypoxia induction for 1, 3, and 5 days. Clusters maintained in 21% oxygen for 1 day were also collected and used as controls. SC-islet cell clusters were washed with PBS, and the supernatant was removed after centrifuge. Then, samples were snap-frozen with liquid nitrogen.
Sample lysis and digestion for mass spectrometry
Cells were lysed in a lysis buffer composed of Covaris Protein Extraction Buffer (Covaris, Woburn, MA), protease and phosphatase inhibitor cocktail (ThermoFisher, Waltham, MA) using a Covaris S220 Focused Ultra-Sonicator. Lysate was transferred to a 1.5 mL centrifuge tube and then centrifuged at 16,000 × g for 20 min at 4 °C. Protein concentration of the supernatant solution was measured using a NanoDrop Spectrophotometer (ThermoScientific, Waltham, MA). Equal protein amounts from each sample were aliquoted and diluted to a total volume of 100 µL with 100 mM triethylammonium bicarbonate (TEAB) buffer. Proteins were reduced with 200 mM tris[2-carboxyethyl]phosphine (TCEP) at 55 °C for 1 h, and then alkylated with 375 mM iodoacetamide for 30 minutes in the dark. Proteins were precipitated overnight using cold acetone. Samples were centrifuged at 8000 relative centrifugal force (RCF) for 5 minutes at 4 °C, after which the acetone was completely removed. Precipitated proteins were redissolved in 100 mM TEAB and then digested with trypsin overnight at 37 °C.
TMT 16-plex labeling
TMT labeling for digested samples were performed according to manufacturer’s instructions (ThermoFisher, Waltham, MA). Briefly, TMT labeling reagents were dissolved with 20 µL of anhydrous acetonitrile and then added to the samples in equal volume. After one hour incubation at room temperature, the reaction was quenched with 5 µL of 5% hydroxylamine. Equal amounts of peptides from each sample were combined and dried in a SpeedVac.
ERLIC fractionation
After trypsin digestion and TMT labeling, the combined peptides were separated on an Agilent 1200 HPLC system (Santa Clara, CA) using a PolyWAX LP column (PolyLC, Columbis MD) 200 × 2.1 mm, 5 µm, 300 A running under ERLIC mode conditions (Electrostatic Repulsion Hydrophilic Interaction Chromatography). Peptides were separated across 90 min gradient from 0% buffer A (90% acetonitrile, 0.1% acetic Acid) to 75 % buffer B (30% acetonitrile, 0.1% formic Acid) with 20 fractions collected by time. Each fraction was dried in SpeedVac (Eppendorf, Germany) and resuspended in 0.1% formic acid solution before analysis by mass spectrometry.
LC-MS/MS analysis
Samples were analyzed by Q Exactive HF-X High Resolution Orbitrap (Thermo Fisher, Waltham, MA) coupled with Ultimate 3000 nanoLC (Thermo Fisher Scientific, Waltham, MA) at the Harvard Center for Mass Spectrometry. Peptides were first trapped on a trapping cartridge (300 µm × 5 mm PepMap™ Neo C18 Trap Cartridge, Thermo Fisher Scientific) prior to separation on an analytical column (µPAC, C18 pillar surface, 50 cm bed, Thermo Fisher Scientific). The LC gradient was as follows: 2−45% in mobile phase B (0.1% formic acid in acetonitrile) over 80 min and increased to 98% mobile phase B over 20 min at the flow rate of 300 nL· min−1. The mass spectrometer operated in data-dependent mode for all analyses. Electrospray positive ionization was enabled with a voltage at 2.1 kV. A full scan ranging from 400 to 1800 m/z was performed with a mass resolution of 12 × 104 and AGC target set to 3 × 106. The top fifteen most intensive precursor ions from each scan were used for MS2 fragmentation (normalized collision energy 32) at a mass resolution of 3.0 × 104 and AGC of 2e5. The dynamic exclusion was set at 50 s with a precursor isolation window of 1.2 m/z.
Data processing for mass spectrometry
Raw data was submitted for analysis in Proteome Discoverer 2.5 software (Thermo Fisher Scientific). The MS/MS Data was searched against the UniProt reviewed Homo sapiens database along with known contaminants such as human keratins and common lab contaminants. Sequest HT searches were performed using the following guidelines: a 10 ppm MS tolerance and 0.02 Da MS/MS tolerance; Trypsin digestion with up to two missed cleavages; carbamidomethylation (57.021 Da) on cysteine, TMTpro on peptide N-termini and lysine residue (+304.207 Da) were set as static modification; oxidation (+ 15.995 Da) of methionine set as variable modification; minimum required peptide length set to ≥ 6 amino acids. At least one unique peptide per protein group is required for identifying proteins. All MS2 spectra assignment FDR of 1% on both protein and peptide level was achieved by applying the target-decoy database search by Percolator.
Analysis of mass spectrometry data
Subsequent computational and statistical analyses was performed using established protocols in MATLAB software (Matlab 2023, Mathworks Ltd). Normalized protein counts were used to evaluate the statistical significance of differentially expressed proteins by one-way analysis of variance (ANOVA). Proteins with significant differential expression (adj. p-value < 0.01; Benjamini-Hochberg corrected), were used to perform a principal component analysis (PCA) and evaluate the similarity of samples within each time point with respect to other time points. The mean log-ratio value of normalized protein expression was calculated for cell cultured in 21% oxygen level and cell cultured in 2% oxygen levels for one, three, or five days. Proteins were selected as differentially expressed if they met the criteria of an adjusted p-value < 0.01 (one-way ANOVA with Benjamini-Hochberg correction) and a log fold change (LFC) > 1 at least at one time point (days 1, 3, or 5) under 2% oxygen conditions. Monotonically increasing or decreasing genes were identified and plotted. Volcano plots were generated using GraphPad Prism 9 (GraphPad Software). Proteins with significant differential expression (adj. p-val <0.01) were highlighted in red, and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway was analyzed using the Gene Set Enrichment Analysis (GSEA) database.
Single-nucleus sample preparation and sequencing
Single cells were obtained as described before, quantified for viability using the Vi-Cell XR Cell Viability Analyzer (Beckman Colter), resuspended in PBS with 0.1% BSA at a concentration of ~ 1000 cells/μl, and put on ice. Before proceeding, all samples were ensured to have > 90% viability to minimize dead cell carry over in sequencing. For the time course analysis, samples were collected from a single differentiation at the appropriate time points. Single-cell suspension samples were processed into nuclei according to the 10X Multiome ATAC + Gene Expression (GEX) protocol (CGOOO338). Single cells were lysed with chilled Lysis Buffer for 4 min, washed three times with wash buffer, and resuspended with 10x nuclei buffer at 3000–5000 nuclei/μl. Nucleus samples were checked for quality control with viability staining to ensure successful nuclei isolation. Nucleus samples were then processed using the Chromium 10x genomics instrument, with a target cell number of 7000–10,000. The 10x Single Cell Multiome ATAC + Gene Expression v1 kit was used according to the manufacturer’s instructions for library preparations. Libraries were then pooled and sequenced on the Illumina NovaSeq platform.
Processing of multi-omic datasets
Multi-omic sequenced files were demultiplexing and analyzed using Cell Ranger ARC v2.0. Genes were mapped and referenced using the human reference genome GRCh38. RStudio 1.3.1093 (version 4.0.3) was used to perform analyses. Datasets were analyzed using Seurat (version 4.01)82 and Signac (version 1.3.0)83. For ATAC data, peaks were called using MACS2 with default parameters, and the genomic positions were mapped and annotated with reference human genome EnsDb.Hsapeins.v86 and hg38. Subsequently, we generated a consolidated peak set by merging overlapping peaks derived from all the samples. The consolidated peak set was subjected to re-quantification, followed by normalization using term frequency-inverse document frequency (TF-IDF) normalization. Low-quality cells including doublets, dead cells and poor sequencing depth cells were removed by filtering out cells with low RNA counts (UMI < 1000) and low ATAC counts (nCount_ATAC < 25,000); high RNA counts ( > 40,000 − 50,000) and high ATAC counts ( > 40,000 − 50,000); nucleosome signal > 1 and transcription start site (TSS) enrichment < 2. Gene expression data was variance stabilized using SCTransform (v2 regularization) with default parameters, and then normalized for each cell by dividing by the total counts for that cell and then natural-log transformed using log1p.
Analysis of multi-omic datasets
To assign cell types in the integrated datasets, the gene expression matrix was used to identify variable genes using a loess span parameter when fitting the variance-mean relationship (VST method). The top two thousand variable genes were then used to perform a principal component analysis (PCA). The first 50 principal components were used to construct a nearest-neighbor graph, which was subsequently used to identify clusters of cells analysis using the FindClusters function with default parameters in Seurat (version 4.01). Cell types were assigned identity using the gene markers described above34. Cluster assignments were visualized through UMAP embedding. Similarity, for ATAC counts the most represented peaks were identified based on the total number of counts. We then used the RunSVD function with default parameters to utilize a Latent Semantic Indexing (LSI) analysis. The top 20 dimensions were used to generate a UMAP embedding map and visualize cell identities that were assigned using the gene expression data.
Calculation of motif enrichment scores in SC-islets was achieved by first assigning motif location to the accessible chromatin regions using the motif position frequency matrices from the JASPAR database (version 2020)84. Differential chromatin accessibility between cells cultured at 21% oxygen to those cultured for 5 days in 2% oxygen were assessed using the FindMarkers function for peaks detected in at least 5% of cells. We utilized Bonferroni-adjusted p-values to establish statistical significance, with a threshold set at a false discovery rate (FDR) less than 0.05. To visualize the activity of transcription factors, the scores of motifs in accessible regions were calculated for each cell using chromVAR and visualized on the UMAP embedding map of the SC-islet cell cluster.
Gene regulatory network (GRN) analysis
To infer the gene regulatory network of SC-islets cultured in hypoxic conditions, we utilized Pando (version 1.0.3)85 to identify genomic regulatory regions by intersecting peak regions with evolutionarily conserved regions from an alignment of 30 mammals using the PhastCons dataset (phastCons20). We then intersected these conserved regions with a curated list of transcription factor binding motifs provided by Pando using the find motifs function. We then used the infer grn function to infer a generalized linear model (GLM) of known transcription factors based on the peaks that we identified and genes differentially expressed between cells cultured in 21% oxygen compared to cells cultured in hypoxic condition for one or five days (p-adjusted < 0.01; LFC > 0.5). The GLM models the expression of each gene as a function of transcription factors expression values and the chromatin accessibility of their corresponding binding motifs. In the described context, the linear model is established to predict the log-normalized expression (Y) of a specific gene i.
This model is constructed based on all transcription factor (TF)-binding-site interactions within a particular genomic region where ε is the intercept or constant term in the model, accounting for any baseline expression level not explained by TF-binding interactions, βj: is the fitted coefficient for a specific \({{TF}}_{j}\), \({A}_{j}\) denotes the accessibility of the peak that overlaps the binding region of \({{TF}}_{j}\), and \({E}_{j}\) the expression value of \({{TF}}_{j}\). In summary, these coefficients indicate the strength of the relationship between the binding and expression of \({{TF}}_{j}\) and the expression of gene i. To visualize the GRN, a score matrix was defined for each TF based on the Pearson correlation between log-normalized expression of TF in the network and the fitted coefficient obtained from the GLM model for each pair of TFs85. A principal component analysis was performed on the score matrix, and the top 20 PCS were used to generate a UMAP.
Lentiviral transduction
The pLenti-based expression vectors containing a list of genes with selection (Supplementary Table 1) were constructed by Vector Builder. Lentiviral particles were generated by Vector Builder. SC-islets were enriched using magnetic separation with anti-human CD49a PE-conjugated antibody (BD 559596, 1:100) and anti-PE UltraPure MACS microbeads for CD49a (Miltenyi 130-105-639, 1:2.5), resuspended in fresh S6 medium supplied with polybrene (Santa Cruz Biotechnology, SC-134220; 1:1000) and 10 μM Y-27632 (Pepro Tech, 129382310MG) at a cell concentration of 2.5 million/ml in bioreactor, and transfected with 200 μl 109 concentrated lentiviral particles (final concentration: 2 × 107 virus/ml) for 2 days. After transduction, cells were collected, washed three times, and resuspended in fresh S6 medium. SC-islets with different modifications were cultured in incubators with 21% or 2% oxygen for further experiments.
Cell transplantation and retrieval
EDN3 overexpressed SC-islets were generated using lentiviral particles as described above. SC-islets transfected with control lentiviral particles were used as the control group. After SC-islets form clusters, 5 million EDN3 overexpressed SC-islets or control SC-islets were mixed with 100 μl low growth factor and phenol red-free Matrigel (Corning, #356255) on ice, and transferred into a syringe. NSG mice were anesthetized with isoflurane, and the right flank on the back of the mouse was shaved. The surgical area was sanitized with a povidone iodine swab and an ethanol swab. Then, EDN3 overexpressed SC-islets or control SC-islets in Matrigel were injected in the dorsal subcutaneous space in NSG mice. Each mice received 5 million SC-islets. After transplantation for one week, grafts were removed carefully and washed with PBS on ice. Then the grafts were transferred into gentleMACS™ C Tubes (Miltenyi Biotec, #130-093-237) and added with 25% Gentle Collagenase/Hyaluronidase (Stemcell Technologies, #07919) in DMEM. Further, grafts were lysed using gentleMACS™ Octo Dissociator with Heaters (Miltenyi Biotec, #130-096-427) using the manufacturer’s instructions.
Statistics
Unless otherwise stated, data were expressed as means ± SD. For comparisons between two groups, means were compared using two-tailed Student’s t tests. Comparisons between multiple groups were performed by analysis of variance (ANOVA), followed by Tukey’s post hoc analysis. All statistical analyses were performed using GraphPad Prism 9 (GraphPad Software). Adjusted p-values are indicated in the figures as ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005, and ∗∗∗∗p < 0.001.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Sequencing data that support the findings of this study have been deposited in the Gene Expression Omnibus, GSE263130 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE211533]. GRCh38 human reference was used. The protein mass spectrometry raw data of this study have been deposited in the ProteomeXchange (PXD062589). The data generated in this study are provided in the Source Data file. All other data supporting the findings of this study are available from the corresponding author upon reasonable request. Source data are provided in this paper.
References
DiMeglio, L. A., Evans-Molina, C. & Oram, R. A. Type 1 diabetes. Lancet 391, 2449–2462 (2018).
Warshauer, J. T., Bluestone, J. A. & Anderson, M. S. New frontiers in the treatment of type 1 diabetes. Cell Metab. 31, 46–61 (2020).
Roep, B. O., Thomaidou, S., van Tienhoven, R. & Zaldumbide, A. Type 1 diabetes mellitus as a disease of the β-cell (do not blame the immune system?). Nat. Rev. Endocrinol. 17, 150–161 (2021).
Melton, D. The promise of stem cell-derived islet replacement therapy. Diabetologia 64, 1030–1036 (2021).
Pagliuca, F. W. et al. Generation of functional human pancreatic β cells in vitro. Cell 159, 428–439 (2014).
Hogrebe, N. J., Ishahak, M. & Millman, J. R. Developments in stem cell-derived islet replacement therapy for treating type 1 diabetes. Cell Stem Cell 30, 530–548 (2023).
Rezania, A. et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol. 32, 1121–1133 (2014).
Siehler, J., Blöchinger, A. K., Meier, M. & Lickert, H. Engineering islets from stem cells for advanced therapies of diabetes. Nat. Rev. Drug Discov. 20, 920–940 (2021).
Balboa, D. et al. Functional, metabolic and transcriptional maturation of human pancreatic islets derived from stem cells. Nat. Biotechnol. 40, 1042–1055 (2022).
Ellis, C., Ramzy, A. & Kieffer, T. J. Regenerative medicine and cell-based approaches to restore pancreatic function. Nat. Rev. Gastroenterol. Hepatol. 14, 612–628 (2017).
Brusko, T. M., Russ, H. A. & Stabler, C. L. Strategies for durable β cell replacement in type 1 diabetes. Science 373, 516–522 (2021).
Maxwell, K. G., Kim, M. H., Gale, S. E. & Millman, J. R. Differential function and maturation of human stem cell-derived islets after transplantation. Stem Cells Transl. Med. 11, 322–331 (2022).
Du, Y. et al. Human pluripotent stem-cell-derived islets ameliorate diabetes in non-human primates. Nat. Med. 28, 272–282 (2022).
Cuesta-Gomez, N. et al. Characterization of stem-cell-derived islets during differentiation and after implantation. Cell Rep. 40, 111238 (2022).
Liang, Z. et al. Implantation underneath the abdominal anterior rectus sheath enables effective and functional engraftment of stem-cell-derived islets. Nat. Metab. 5, 29–40 (2023).
Faleo, G. et al. Mitigating ischemic injury of stem cell-derived insulin-producing cells after transplant. Stem Cell Rep. 9, 807–819 (2017).
Coronel, M. M., Geusz, R. & Stabler, C. L. Mitigating hypoxic stress on pancreatic islets via in situ oxygen generating biomaterial. Biomaterials 129, 139–151 (2017).
Pedraza, E., Coronel, M. M., Fraker, C. A., Ricordi, C. & Stabler, C. L. Preventing hypoxia-induced cell death in beta cells and islets via hydrolytically activated, oxygen-generating biomaterials. Proc. Natl. Acad. Sci. USA 109, 4245–4250 (2012).
Komatsu, H. et al. Posttransplant oxygen inhalation improves the outcome of subcutaneous islet transplantation: A promising clinical alternative to the conventional intrahepatic site. Am. J. Transplant. 18, 832–842 (2018).
Wang, K. et al. From Micro to Macro: The hierarchical design in a micropatterned scaffold for cell assembling and transplantation. Adv. Mater. 29, 1604600 (2017).
Einstein, S. A. et al. Hypoxia within subcutaneously implanted macroencapsulation devices limits the viability and functionality of densely loaded islets. Front. Transplant. 2, 1257029 (2023).
Desai, T. & Shea, L. D. Advances in islet encapsulation technologies. Nat. Rev. Drug Discov. 16, 338–350 (2017).
Papas, K. K., Leon, H. D., Suszynski, T. M. & Johnson, R. C. Oxygenation strategies for encapsulated islet and beta cell transplants. Adv. Drug Deliv. Rev. 139, 139–156 (2019).
Gerber, P. A. & Rutter, G. A. The role of oxidative stress and hypoxia in pancreatic beta-cell dysfunction in diabetes mellitus. Antioxid. Redox Signal. 26, 501–518 (2017).
Komatsu, H., Kandeel, F. & Mullen, Y. Impact of oxygen on pancreatic islet survival. Pancreas 47, 533–543 (2018).
Carlsson, P.-O., Palm, F., Andersson, A. & Liss, P. Markedly decreased oxygen tension in transplanted rat pancreatic islets irrespective of the implantation site. Diabetes 50, 489–495 (2001).
Moore, S. J., Gala-Lopez, B. L., Pepper, A. R., Pawlick, R. L. & Shapiro, A. J. Bioengineered stem cells as an alternative for islet cell transplantation. World J. Transplant. 5, 1 (2015).
Zheng, X. et al. Acute hypoxia induces apoptosis of pancreatic β-cell by activation of the unfolded protein response and upregulation of CHOP. Cell Death Dis. 3, e322–e322 (2012).
Emamaullee, J. A. & Shapiro, A. M. J. Interventional strategies to prevent β-cell apoptosis in islet transplantation. Diabetes 55, 1907–1914 (2006).
Smith, K. E. et al. Acute Ischemia induced by high-density culture increases cytokine expression and diminishes the function and viability of highly purified human islets of langerhans. Transplantation 101, 2705–2712 (2017).
Chen, C. et al. Hypoxia induces inflammatory response and cell death by promoting NLRP3 inflammsome activation via ROS-TXNIP axis in pancreatic &bgr; cells. Transplantation 102, S169 (2018).
Catrina, S.-B. & Zheng, X. Hypoxia and hypoxia-inducible factors in diabetes and its complications. Diabetologia 64, 709–716 (2021).
Cantley, J., Grey, S. T., Maxwell, P. H. & Withers, D. J. The hypoxia response pathway and β‐cell function. Diabetes Obes. Metab. 12, 159–167 (2010).
Veres, A. et al. Charting cellular identity during human in vitro β-cell differentiation. Nature 569, 368–373 (2019).
Russ, H. A. et al. Controlled induction of human pancreatic progenitors produces functional beta‐like cells in vitro. EMBO J. 34, 1759–1772 (2015).
Nair, G. G. et al. Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived β cells. Nat. Cell Biol. 21, 263–274 (2019).
Augsornworawat, P. et al. Single-nucleus multi-omics of human stem cell-derived islets identifies deficiencies in lineage specification. Nat. Cell Biol. 25, 904–916 (2023).
Alvarez-Dominguez, J. R. et al. Circadian entrainment triggers maturation of human in vitro islets. Cell Stem Cell 26, 108–122 (2020).
Cheng, K. et al. Hypoxia-inducible factor-1α regulates β cell function in mouse and human islets. J. Clin. Investig. 120, 2171–2183 (2010).
Stokes, R. A. et al. Hypoxia-inducible factor-1α (HIF-1α) potentiates β-Cell survival after islet transplantation of human and mouse islets. Cell Transpl. 22, 253–266 (2013).
Gerber, P. A. et al. Hypoxia lowers SLC30A8/ZnT8 expression and free cytosolic Zn2+ in pancreatic beta cells. Diabetologia 57, 1635–1644 (2014).
Wang, N. et al. Hypoxia-inducible factor-1 mediates pancreatic β-cell dysfunction by intermittent hypoxia. Am. J. Physiol. Cell Physiol. 319, C922–C932 (2020).
Bensellam, M. et al. Hypoxia reduces ER-to-Golgi protein trafficking and increases cell death by inhibiting the adaptive unfolded protein response in mouse beta cells. Diabetologia 59, 1492–1502 (2016).
Wang, X., Wang, K., Zhang, W., Qiang, M. & Luo, Y. A bilaminated decellularized scaffold for islet transplantation: Structure, properties and functions in diabetic mice. Biomaterials 138, 80–90 (2017).
Wang, L.-H. et al. An inverse-breathing encapsulation system for cell delivery. Sci. Adv. 7, eabd5835 (2020).
Wang, L.-H. et al. A bioinspired scaffold for rapid oxygenation of cell encapsulation systems. Nat. Commun. 12, 5846 (2021).
Xie, B., Nguyen, P. M. & Idevall‐Hagren, O. Feedback regulation of insulin secretion by extended synaptotagmin‐1. FASEB J. 33, 4716–4728 (2019).
Atla, G. et al. Genetic regulation of RNA splicing in human pancreatic islets. Genome Biol. 23, 196 (2022).
Butler, P. C. & Gale, E. A. M. Reversing type 1 diabetes with stem cell–derived islets: a step closer to the dream? J. Clin. Investig. 132, e158305 (2022).
Migliorini, A., Nostro, M. C. & Sneddon, J. B. Human pluripotent stem cell-derived insulin-producing cells: A regenerative medicine perspective. Cell Metab. 33, 721–731 (2021).
Gunton, J. E. Hypoxia inducible factors and diabetes. J. Clin. Investig. 130, 5063–5073 (2020).
Choudhry, H. & Harris, A. L. Advances in hypoxia-inducible factor biology. Cell Metab. 27, 281–298 (2018).
Keith, B. & Simon, M. C. Hypoxia-inducible factors, stem cells, and cancer. Cell 129, 465–472 (2007).
Lee, P., Chandel, N. S. & Simon, M. C. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat. Rev. Mol. Cell Biol. 21, 268–283 (2020).
Koh, M. Y., Darnay, B. G. & Powis, G. Hypoxia-associated factor, a novel E3-ubiquitin ligase, binds and ubiquitinates hypoxia-inducible factor 1α, leading to its oxygen-independent degradation. Mol. Cell. Biol. 28, 7081–7095 (2008).
Nomoto, H. et al. Activation of the HIF1α/PFKFB3 stress response pathway in beta cells in type 1 diabetes. Diabetologia 63, 149–161 (2020).
Ilegems, E. et al. HIF-1α inhibitor PX-478 preserves pancreatic β cell function in diabetes. Sci. Transl. Med. 14, eaba9112 (2022).
Gheni, G. et al. Glutamate acts as a key signal linking glucose metabolism to incretin/cAMP action to amplify insulin secretion. Cell Rep. 9, 661–673 (2014).
Campbell, J. E. & Newgard, C. B. Mechanisms controlling pancreatic islet cell function in insulin secretion. Nat. Rev. Mol. Cell Biol. 22, 142–158 (2021).
Yan, S.-F. et al. Tissue factor transcription driven by Egr-1 is a critical mechanism of murine pulmonary fibrin deposition in hypoxia. Proc. Natl. Acad. Sci. USA 95, 8298–8303 (1998).
Patel, N. & Kalra, V. K. Placenta growth factor-induced early growth response 1 (Egr-1) regulates hypoxia-inducible factor-1α (HIF-1α) in endothelial cells*. J. Biol. Chem. 285, 20570–20579 (2010).
Bloch, K. D., Eddy, R. L., Shows, T. B. & Quertermous, T. cDNA cloning and chromosomal assignment of the gene encoding endothelin 3*. J. Biol. Chem. 264, 18156–18161 (1989).
Oropeza, D. et al. Stage-specific transcriptomic changes in pancreatic α-cells after massive β-cell loss. BMC Genom. 22, 585 (2021).
Tamas, B. et al. EDN3-like peptides and uses thereof. US20130345392A1 (2012).
Xavier, G., da, S., Qian, Q., Cullen, P. J. & Rutter, G. A. Distinct roles for insulin and insulin-like growth factor-1 receptors in pancreatic beta-cell glucose sensing revealed by RNA silencing. Biochem. J. 377, 149–158 (2004).
Bahl, V. et al. G6PC2 controls glucagon secretion by defining the set point for glucose in pancreatic α cells. Sci. Transl. Med. 17, eadi6148 (2025).
Pound, L. D. et al. G6PC2: A negative regulator of basal glucose-stimulated insulin Ssecretion. Diabetes 62, 1547–1556 (2013).
Waeber, G. et al. Insulin secretion is regulated by the glucose-dependent production of islet β cell macrophage migration inhibitory factor. Proc. Natl. Acad. Sci. 94, 4782–4787 (1997).
Toso, C., Emamaullee, J. A., Merani, S. & Shapiro, A. M. J. The role of macrophage migration inhibitory factor on glucose metabolism and diabetes. Diabetologia 51, 1937–1946 (2008).
Aghazadeh, Y. et al. Microvessels support engraftment and functionality of human islets and hESC-derived pancreatic progenitors in diabetes models. Cell Stem Cell 28, 1936–1949 (2021).
Brielle, S., Gura, R. & Kaganovich, D. Imaging stress. Cell Stress Chaperones 20, 867–874 (2015).
Gorin, G., Fang, M., Chari, T. & Pachter, L. RNA velocity unraveled. PLoS Comput. Biol. 18, e1010492 (2022).
Zheng, S. C., Stein-O’Brien, G., Boukas, L., Goff, L. A. & Hansen, K. D. Pumping the brakes on RNA velocity by understanding and interpreting RNA velocity estimates. Genome Biol. 24, 246 (2023).
Wang, X. et al. Engineered immunomodulatory accessory cells improve experimental allogeneic islet transplantation without immunosuppression. Sci. Adv. 8, eabn0071 (2022).
Wang, X. et al. A nanofibrous encapsulation device for safe delivery of insulin-producing cells to treat type 1 diabetes. Sci. Transl. Med. 13, eabb4601 (2021).
Gerace, D. et al. Engineering human stem cell-derived islets to evade immune rejection and promote localized immune tolerance. Cell Rep. Med. 4, 100879 (2023).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Finak, G. et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 16, 278 (2015).
Manno, G. L. et al. RNA velocity of single cells. Nature 560, 494–498 (2018).
Bergen, V., Lange, M., Peidli, S., Wolf, F. A. & Theis, F. J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 38, 1408–1414 (2020).
Strunz, M. et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat. Commun. 11, 3559 (2020).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021).
Stuart, T., Srivastava, A., Madad, S., Lareau, C. A. & Satija, R. Single-cell chromatin state analysis with Signac. Nat. Methods 18, 1333–1341 (2021).
Schep, A. N., Wu, B., Buenrostro, J. D. & Greenleaf, W. J. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978 (2017).
Fleck, J. S. et al. Inferring and perturbing cell fate regulomes in human brain organoids. Nature 621, 365–372 (2023).
Acknowledgements
This work was supported by grants from the Harvard Stem Cell Institute (DP-0180-18-02), JDRF (5-COE-2020-967-M-N), and the JPB Foundation (award no. 1094) awarded to D.A.M. D.A.M. is the Distinguished Research Fellow at Vertex Pharmaceuticals. This research was performed while D.A.M. was on the faculty at Harvard University. We thank Harvard Stem Cell and Regenerative Biology (HSCRB) Histology-Immunohistochemistry Core Facility for the histological cross-sectioning. We thank the Bauer Core Facility at Harvard University for the technical support with next-generation sequencing (NGS). We thank Mei Chen and Steven Kolakowski at the Harvard Center for Mass Spectrometry for their assistance with the proteomics analyses. Some illustrations in the figures were made using BioRender.
Author information
Authors and Affiliations
Contributions
D.A.M. supervised the project. X.W. and S.B. conceived the experimental design. X.W., S.B. and D.A.M. wrote the original manuscript. All the authors (X.W., S.B., J.H.-R.K., N.K., D.B., R.P. and D.A.M.) reviewed and edited the manuscript. X.W., S.B., N.K. and J.H.-R.K. performed most of the experiments. X.W. and S.B. analyzed the data. X.W., S.B. and D.A.M. revised the paper.
Corresponding authors
Ethics declarations
Competing interests
D.A.M., J.H.-R.K. and D.B. are employees of Vertex Pharmaceuticals. All of the work described in this report was performed at Harvard University using research cell lines. Vertex was not involved in any aspect of the work. The authors declare no other competing interests.
Peer review
Peer review information
Nature Communications thanks Timo Otonkoski, Alexander Kleger, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, X., Brielle, S., Kenty-Ryu, J. et al. Improving cellular fitness of human stem cell-derived islets under hypoxia. Nat Commun 16, 4787 (2025). https://doi.org/10.1038/s41467-025-59924-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-59924-7
This article is cited by
-
Stable intracranial imaging of dura mater-engrafted pancreatic islet cells in awake mice
Nature Communications (2025)
