
Abstract
Ovarian cancer has the highest mortality rate among gynecologic tumors worldwide, with unclear underlying mechanisms of pathogenesis. RNA-binding proteins (RBPs) primarily direct post-transcriptional regulation through modulating RNA metabolism. Recent evidence demonstrates that RBPs are also implicated in transcriptional control. However, the role and mechanism of RBP-mediated transcriptional regulation in tumorigenesis remain largely unexplored. Here, we show that the RBP heterogeneous ribonucleoprotein L (hnRNPL) interacts with chromatin and regulates gene transcription by forming phase-separated condensates in ovarian cancer. hnRNPL phase separation activates PIK3CB transcription and glycolysis, thus promoting ovarian cancer progression. Notably, we observe that the PIK3CB promoter is transcribed to produce a non-coding RNA which interacts with hnRNPL and promotes hnRNPL condensation. Furthermore, hnRNPL is significantly amplified in ovarian cancer, and its high expression predicts poor prognosis for ovarian cancer patients. By using cell-derived xenograft and patient-derived organoid models, we show that hnRNPL knockdown suppresses ovarian tumorigenesis. Together, our study reveals that phase separation of the chromatin-associated RBP hnRNPL promotes PIK3CB transcription and glycolysis to facilitate tumorigenesis in ovarian cancer. The formed hnRNPL-PIK3CB-AKT axis depending on phase separation can serve as a potential therapeutic target for ovarian cancer.
Similar content being viewed by others


Introduction
RNA-binding proteins (RBPs) play crucial roles in a wide variety of physiological and pathological processes by regulating nearly all aspects of RNA metabolism including RNA splicing, degradation and translation1. Emerging studies have shown that RBPs tightly associated with chromatin are also widely involved in transcriptional control2,3. Chromatin immunoprecipitation sequencing (ChIP-seq) analysis by the ENCODE project demonstrated that various RBPs are more enriched at euchromatin than heterochromatin and strikingly at gene promoters3. These chromatin-associated RBPs (ChaRBPs) participate in transcription through partnering with transcription factors or accessory RNA4,5,6,7. Transcription by RNA polymerases is the basis of gene expression and occurs at highly compartmentalized nuclear sites8. Transcription factors, transcription co-activators, the transcriptional machinery and the RNA products of transcription form transcriptional condensates through liquid-liquid phase separation (LLPS) to orchestrate transcriptional dynamics9,10. The transcriptional co-activator BRD4 and the transcriptional Mediator complex undergo phase separation and are distributed at super enhancer clusters to modulate gene transcription9. Enhancer RNAs induced by estrogen recruit multiple proteins to form enhancer RNA-ribonucleoprotein complexes in chromatin regions and phase separates to activate transcription of downstream genes11, suggesting the implication of interplay between RNA and RBP phase separation in gene transcription.
Cancer cells often rely on the “Warburg effect”, an enhanced glycolytic process, which is highly dependent on the PI3K/AKT pathway12. PIK3CB, a key component of PI3K, induces the phosphorylation of glycolytic enzymes PFKFB2 and PKM213. PIK3CB is mutated or amplified in various cancers13. N6-methyladenosine RNA methylation-induced PIK3CB up-regulation followed by AKT signaling activation promotes PTEN-deficient pancreatic cancer progression14. However, the role and the underlying mechanism of RBP phase separation-mediated transcriptional regulation in glycolysis remain poorly understood.
In this study, we demonstrate that the heterogeneous ribonucleoprotein hnRNPL forms phase-separated condensates and promotes ovarian cancer progression. hnRNPL acts as a ChaRBP, binding to the PIK3CB promoter and activates its transcription. Up-regulation of PIK3CB by hnRNPL condensation induces PI3K/AKT signaling and promotes glycolysis in ovarian cancer cells. The promoter-associated RNA paPIK3CB enhances hnRNPL phase separation. Thus, these findings demonstrate that hnRNPL phase separation can serve as a promising target for ovarian cancer treatment.
Results
hnRNPL is located on chromatin and undergoes LLPS
To better understand gene regulation mediated by ChaRBPs, we defined ChaRBPs as those RBPs identified as chromatin-associated proteins in all four of K562 human erythroleukemia cells, THP-1 human monocytic leukemia cells, HEK293T cells and embryonic stem cells based on two prior proteomic screenings15,16. A total of 23 ChaRBPs were acquired, and 9 of these proteins are members of hnRNP family (Supplementary Fig. 1a–c). A previous study showed that hnRNPL regulates gene transcription17, however, the detailed mechanisms were not fully clarified. Thus, we focused on hnRNPL and detected its localization in ovarian cancer cells through nucleoplasmic separation and immunofluorescence staining assays. The results revealed that hnRNPL was localized on chromatin as condensates in ovarian cancer cells but dispersed in the nucleus in normal ovarian epithelium cells (Fig. 1a, b and Supplementary Fig. 1d). hnRNPL was predicted to exhibit a high capacity of LLPS (Supplementary Fig. 1e). Consistently, treatment with phase separation inhibitor 1,6-hexanediol significantly abrogated hnRNPL condensation in ovarian cancer cells (Supplementary Fig. 1f), indicating that hnRNPL might undergo phase separation in cells. Moreover, we performed immunofluorescence staining coupled with three-dimension imaging to examine the condensation of hnRNPL and found that hnRNPL formed condensates in the nucleus of ovarian cancer cells (Fig. 1c). We then tagged endogenous hnRNPL with the green fluorescent protein Clover in ovarian cancer cells using a CRISPR-Cas9 genome editing system and observed that the endogenous hnRNPL also formed discrete foci in live cells (Fig. 1d and Supplementary Fig. 1g, h). To confirm the LLPS property of hnRNPL in vitro, we purified recombinant wild-type hnRNPL protein or hnRNPL fused with mCherry or EGFP from E. coli BL21 (DE3) cells. Droplet fusion assays showed that all these recombinant hnRNPL proteins could fuse to form spherical liquid-like droplets (Fig. 1e, f and Supplementary Fig. 1i). Next, the liquid nature of the droplets formed by mCherry- or EGFP-tagged hnRNPL was examined by fluorescence recovery after photobleaching (FRAP) assays. In mCherry- or EGFP-tagged hnRNPL droplets, the bleached areas started recovery soon after photobleaching with stabilization after 2-3 min (Supplementary Fig. 1j, k). Consistently, in vivo FRAP assays confirmed condensation of mCherry- or EGFP-tagged hnRNPL in ovarian cancer cells (Fig. 1g, h).

a Immunofluorescent staining of hnRNPL in OVCAR3,SKOV3 ovarian cancer cells and HOSEpic normal ovarian cells. The right panel displays a plot illustrating the colocalization of hnRNPL-GFP and DAPI and Pearson correlation coefficient analysis. Scale bar, 5 μm. n = 5 independent experiments. Box plots show the distribution of data across groups. The central line within the box represents the median value. The upper and lower edges of the box represent the 75th and 25th percentiles, respectively (Interquartile Range, IQR). The whiskers extend from the box to the maximum and minimum values within 1.5 times the interquartile range (IQR), with any data points beyond this range considered as outliers. b Quantitative analysis of hnRNPL puncta in three cell lines (n = 50 cells for each group). c The three-dimensional (3D) imaging of the intracellular condensate formation of GFP-tagged hnRNPL protein in OVCAR3 cells. Scale bar, 5 μm. d Schematic of Clover green fluorescent tag knockin in OVCAR3 cells using the CRISPR-Cas9 system and representative images of endogenous expression of hnRNPL. Scale bar, 5 μm. e In vitro droplets coalescence of hnRNPL. Phase separation was induced by diluting the salt concentration from 500 mM to 100 mM. f In vitro droplets coalescence of C-terminally mCherry-tagged hnRNPL. Scale bar, 5 µm. g Representative images of fluorescence recovery and plot of fluorescence intensity of GFP-hnRNPL droplets in OVCAR3 cells after photobleaching over time. The red curves represent the mean of the normalized fluorescence intensity in the photobleached region in distinct droplets (n = 5 independent experiments). Scale bar, 2 µm. h Representative images of fluorescence recovery and plot of fluorescence intensity of mCherry-hnRNPL droplets in OVCAR3 cells after photobleaching over time. The red curves represent the mean of the normalized fluorescence intensity in the photobleached region in distinct droplets (n = 5 independent experiments). Scale bar, 5 µm. i Droplet formation of hnRNPL at different protein concentrations with the addition of 5% PEG4000 in buffer with the salt concentration from 50 mM to 400 mM. Scale bar, 10 µm. Data are shown as means ± S.D. Data are representative of at least three independent experiments (a–c, e, f, i). Source data are provided as a Source Data file.
Protein LLPS can sense external stress such as salt concentration, pH and temperature, contributing to the maintenance of cellular homeostasis18,19,20. Thus, we next evaluated the effect of these external stressors on hnRNPL droplet formation. Droplets were formed at hnRNPL protein concentration as low as 5 μM in KCl solution at 50 mM (Fig. 1i). Increased KCl concentration gradually disrupted hnRNPL droplet formation (Fig. 1i). Meanwhile, a turbid solution of hnRNPL formed at 4 °C and became transparent at 37 °C for 30 min, and it then became turbid again at 4 °C (Supplementary Fig. 2a), indicating that hnRNPL LLPS is affected by temperature. We also observed that hnRNPL formed droplets at pH 7.5, whereas either increased or decreased pH would disrupt the droplet formation (Supplementary Fig. 2b). Cellular stresses, including heat shock and sodium arsenite treatment, can induce gene regulation through different mechanisms. Thus, the effect of these cellular stresses on hnRNPL phase separation in ovarian cancer cells was assessed, and we found that neither heat shock nor sodium arsenite treatment affected hnRNPL phase separation (Supplementary Fig. 2c). In addition, post-translational modifications have also been demonstrated to be involved in protein LLPS. hnRNPL was predicated to undergo post-translational modifications through the PhosphoSitePlus database, though without more validation in ovarian cancer. We examined the effect of post-translational modifications on hnRNPL phase separation by using several post-translational modification inhibitors, and few changes of hnRNPL condensates were observed in ovarian cancer cells upon treatment with post-translational modification inhibitors, except Benzyl-α-GalNAc, an O-GlcNAcylation inhibitor. O-GlcNAcylation inhibition decreased hnRNPL condensation formation but promoted the smaller foci of hnRNPL in ovarian cancer cells (Supplementary Fig. 2d, e), aligning with the notion that O-GlcNAcylation disfavours protein condensate formation21,22,23,24,25. Collectively, these results suggest hnRNPL undergoes LLPS and forms dynamically exchanged condensates in cells.
Prolines in IDR2 contribute to the LLPS behavior of hnRNPL
To explore the critical regions that contribute to hnRNPL LLPS, we analyzed its sequence using IUPred226. hnRNPL contains two predicted intrinsically disordered regions (IDRs): glycine-rich IDR1 and proline-rich IDR2 (Fig. 2a). The LLPS properties of proteins are mainly controlled by the glycine-rich or proline-rich sequences in the IDR of proteins27,28. Therefore, we constructed mutants of hnRNPL deleted for glycine in IDR1 (IDR1delG) or deleted for proline in IDR2 (IDR2delP). The purified mutated hnRNPL proteins tagged with EGFP were purified from BL21 (DE3) cells (Fig. 2b). Akin to wild-type hnRNPL, IDR1delG formed spherical liquid-like droplets, and FRAP assays revealed that IDR1delG protein maintained the LLPS property (Fig. 2c–e). However, IDR2delP protein failed to form spherical liquid-like droplets (Fig. 2c), suggesting that proline-rich IDR2 but not IDR1 is responsible for hnRNPL condensation. To further confirm this, we produced a mutant of hnRNPL (IDR2FUS) where IDR2 was replaced by the IDR sequence of FUS protein that determines phase separation properties of FUS (Fig. 2b). The results showed that IDR2FUS exhibited intact phase separation capability in protein fusion and FRAP assays (Fig. 2f, g). In ovarian cancer cells, IDR1delG and IDR2FUS proteins formed phase-separated puncta similar to wild-type hnRNPL, while IDR2delP was dispersed in the nucleus and cytoplasm (Fig. 2h). Adding a nuclear localization sequence (NLS) to IDR2delP (IDR2delP-NLS) restored its nuclear localization but not condensation ability (Fig. 2h). To gain further insight of hnRNPL phase separation in live cells, we performed optogenetic droplet assays to demonstrate hnRNPL condensation based on light dependent oligomerization of cryptochrome 2 protein (Cry2). To this end, wild-type and IDR-deleted hnRNPL-mCherry-Cry2oligo fusion proteins were overexpressed in cells, respectively (Fig. 2i). After blue light illumination, wild-type hnRNPL, IDR1delG and IDR2FUS fusion proteins but not IDR2delP fusion protein fused into larger droplets (Fig. 2j), strengthening the LLPS-like condensation of hnRNPL in vivo. Taken together, these data suggest that the proline sequences of IDR2 contribute to hnRNPL condensation.

a Prediction of intrinsic disorder tendency of hnRNPL by IUPred2A (https://iupred2a.elte.hu/). Scores above 0.5 indicate disorder. b Schematic diagrams of wild-type hnRNPL (WT) and its mutant constructs (IDR1delG, IDR2delP, and IDR2FUS). c Droplet formation of C-terminally EGFP-tagged hnRNPL variants of WT, IDR1delG, IDR2delP, and IDR2FUS. Scale bar, 50 µm. d In vitro droplets coalescence of C-terminally EGFP-tagged IDR1delG variant of hnRNPL. e Images and quantification of droplet formation by IDR1delG variant of hnRNPL after photobleaching (n = 5 independent experiments). Scale bar, 5 µm. f Droplets coalescence of IDR2FUS in vitro. g Images and quantification of droplets formed by IDR2FUS after photobleaching (n = 5 independent experiments). Scale bar, 5 µm. h Representative images of exogenously expressed GFP-hnRNPL and each variant in OVCAR3 cells and SKOV3 cells. Scale bar, 5 µm. Statistical analysis of the LLPS puncta number of each variant is shown in the right panel (n = 50 cells for each group). i Schematic diagram of the opto-droplets system. j Representative images depicting the blue light-induced clustering of hnRNPL-WT, hnRNPL-IDR1delG, hnRNPL-IDR2delP and, hnRNPL-IDR2FUS in cells. Stimulation was performed using a 488 nm laser. Data are shown as means ± S.D. Data are representative of at least three independent experiments (c, d, f, j). Scale bar, 5 µm. Source data are provided as a Source Data file.
hnRNPL phase separation facilitates ovarian cancer progression
We next investigated the effect of phase separation on hnRNPL function in cancer cells. Wild-type hnRNPL and its mutants (IDR1delG, IDR2delP, and IDR2FUS) were individually overexpressed in ovarian cancer cells (Supplementary Fig. 3a), and their effects on cell growth, migration and invasion were assessed. CCK-8 and colony formation assays showed that hnRNPL overexpression significantly promoted ovarian cancer cell proliferation (Fig. 3a, b). Deletion of the proline sequences in IDR2, but not the glycine sequences in IDR1, limited the ability of hnRNPL to accelerate cell proliferation (Fig. 3a, b). Notably, the IDR2FUS mutant, where IDR2 was replaced with the IDR of FUS, retained the ability to enhance ovarian cancer cell growth (Fig. 3a, b). Transwell assays revealed that similar to wild-type hnRNPL, IDR1delG and IDR2FUS overexpression facilitated ovarian cancer cell migration and invasion, whereas no differences in cell migration and invasion between IDR2delP and the control group were observed (Fig. 3c and Supplementary Fig. 3b). Moreover, subcutaneous tumor experiments in mice showed that hnRNPL overexpression significantly accelerated tumor growth of ovarian cancer cells (Fig. 3d–f). In contrast, mice injected with cells expressing IDR2delP showed remarkably attenuated tumor growth, whereas IDR1delG and IDR2FUS mutants maintained robust tumor growth (Fig. 3d–f). These results suggest that the LLPS property plays a crucial role in hnRNPL promoting tumorigenesis of ovarian cancer. Moreover, loss-of-function results showed that hnRNPL depletion significantly hindered ovarian cancer cell growth and metastasis (Supplementary Fig. 3c–f, 4a). Subcutaneous tumor experiments in mice showed that hnRNPL knockdown significantly suppressed tumor growth (Fig. 3g–i). In vivo metastasis assays also revealed that hnRNPL deficiency suppressed the metastatic foci formation of ovarian cancer cells within the abdominal cavity (Supplementary Fig. 4b, c). We also evaluated the effect of hnRNPL deficiency on the sensitivity of ovarian cancer cells to chemotherapy drugs including cisplatin and paclitaxel and found that hnRNPL deficiency slightly increased the sensitivity of ovarian cancer cell to cisplatin but not paclitaxel (Supplementary Fig. 4d).

a–c CCK-8 assay (a), colony formation assay (b), and transwell assay (c) were performed to evaluate the growth, migration, and invasion of ovarian cancer cells with overexpression of hnRNPL variants of WT, IDR1delG, IDR2delP, and IDR2FUS compare with control ovarian cancer cells with pcDNA3.1-mCherry. d–f OVCAR3 ovarian cells with overexpression of hnRNPL variants of WT, IDR1delG, IDR2delP, and IDR2FUS or control were subcutaneously injected into nude mice. Tumors were harvested at day 24, photographed (d), and tumor volumes were measured every 3 days (e) and weighed (f). n = 5 mice for each group. g–i OVCAR3 ovarian cells with or without hnRNPL knockdown were subcutaneously injected into nude mice. Tumors were harvested at day 24, photographed (g), and tumor volumes were measured every 3 days (h) and weighed (i). n = 10 mice for each group. j-l CCK-8 assay (j), colony formation assay (k), and transwell assay (l) were performed to evaluate the growth, migration, and invasion of hnRNPL-depleted ovarian cancer cells with overexpression of hnRNPL variants of WT, IDR1delG, and IDR2delP. m–o hnRNPL-depleted OVCAR3 cells with overexpression of hnRNPL variants of WT, IDR1delG, and IDR2delP or control were subcutaneously injected into nude mice. Tumors were harvested at day 24, photographed (m), and tumor volumes were measured every 4 days (n) and weighed (o). n = 10 mice for each group. p Representative images of PDOs from two ovarian cancer patients upon hnRNPL knockdown (n = 3 independent experiments). Scale bar, 400 µm. q Representative images of PDOs from two ovarian cancer patients with overexpression of wild-type hnRNPL or its variants of IDR1delG, and IDR2delP (n = 3 independent experiments). Scale bar, 400 µm. Data are shown as means ± S.D. p value was calculated by one-way ANOVA test with multiple comparisons. Data are representative of at least three independent experiments (a–c, j–l). Source data are provided as a Source Data file.
To further assess the tumor-promoting effect of hnRNPL in ovarian cancer depending on phase separation, we knocked out hnRNPL in ovarian cancer cells and re-expressed either wild-type hnRNPL or its mutants (Supplementary Fig. 4e). hnRNPL knockout significantly suppressed ovarian cancer cell growth and metastasis, which was rescued by re-expressing wild-type hnRNPL or IDR1delG but not IDR2delP (Fig. 3j–l). Consistently, in subcutaneous tumor models, reintroducing wild-type hnRNPL or IDR1delG, but not IDR2delP, restored tumor growth in hnRNPL-depleted ovarian cancer cells (Fig. 3m–o). In addition, hnRNPL knockdown significantly reduced the formation of ovarian cancer patient-derived organoids (PDOs) (Fig. 3p). Conversely, overexpressing wild-type hnRNPL or IDR1delG facilitated the growth of ovarian cancer PDOs, whereas IDR2delP failed to promote the growth of ovarian cancer PDOs (Fig. 3q). Together, these results suggest that phase separation is indispensable for hnRNPL promoting ovarian cancer progression.
hnRNPL activates the transcription of PI3K-AKT pathway-associated genes
Given that hnRNPL interacted with chromatin in ovarian cancer cells (Supplementary Fig. 1d), we performed CoBATCH sequencing29, involving the chromatin immunoprecipitation followed by the high-throughput sequencing (ChIP-seq) to map genome-wide distribution of hnRNPL on chromatin. The results showed that hnRNPL bound 6287 loci which included 5937 genes (Fig. 4a). The binding sites were mainly distributed in the promoter, intron and intergenic regions (Fig. 4b). RNA sequencing (RNA-seq) was conducted in OVCAR3 ovarian cancer cells following hnRNPL knockdown, identifying 1480 differentially expressed genes (DEGs), including 651 up-regulated and 829 down-regulated genes (Fig. 4c and Supplementary Fig. 5a). Integrated analysis of CoBATCH sequencing and RNA-seq revealed that hnRNPL bound to the promoters of 417 DEGs, among which 321 were down-regulated and 96 were up-regulated (Fig. 4d). Cumulative curve analysis revealed that genes bound by hnRNPL were prone to be down-regulated upon hnRNPL knockdown (Fig. 4e). Higher enrichment of hnRNPL was observed at the promoters of down-regulated genes compared with up-regulated genes or non-regulated genes upon hnRNPL knockdown, suggesting hnRNPL binds to gene promoters to augment the transcription (Fig. 4f).

a Heatmap showing the hnRNPL-bound peaks in ovarian cancer cells. b Distribution of hnRNPL-bound peaks on different regions across chromatin. c Differentially expressed genes upon hnRNPL depletion in OVCAR3 cells by RNA-seq were shown as the volcano plot. p value was calculated by R package DESeq2. d Overlapping analysis of down-regulated genes upon hnRNPL knockdown and hnRNPL-bound genes. e Cumulative distribution of the fold changes (log2) in hnRNPL-depleted cells for hnRNPL-bound genes or not bound. p value was calculated by a two-sided Mann-Whitney U-test. f Distribution of hnRNPL-bound peaks on genes which were up-regulated, down-regulated or not in ovarian cancer cells upon hnRNPL knockdown. g Functional enrichment analysis of 321 genes described in (f). p value was calculated by “clusterProfiler” KEGG analysis. h hnRNPL binding sites across genes belonging to PI3K-AKT signaling pathway-associated genes. i Relative RNA level changes of PI3K-AKT signaling pathway-associated genes in OVCAR3 ovarian cancer cells upon hnRNPL knockdown. n = 3 independent experiments. j ChIP-qPCR detecting the interaction between hnRNPL and the promoters of target genes. n = 3 independent experiments. k Overlapping analysis of hnRNPL binding sites and RNA Pol II binding sites at the chromatin in ovarian cancer cells. l Correlation analysis of hnRNPL binding sites with RNA Pol II binding sites at the chromatin in ovarian cancer cells. m IGV plots showing RNA Pol II enrichment at promoters of hnRNPL targets in control and hnRNPL-depleted ovarian cancer cells. Data are shown as means ± S.D. p value was calculated by one-way ANOVA test with multiple comparisons (i) and unpaired two-sided Student’s t-test (j). Source data are provided as a Source Data file.
Next, we performed a functional enrichment analysis of the 321 down-regulated DEGs targeted by hnRNPL. Multiple tumor-associated pathways were identified, with the PI3K-AKT signaling pathway ranking first (Fig. 4g). Sixteen potential genes targeted by hnRNPL were involved in the PI3K-AKT signaling pathway, and hnRNPL ChIP-seq confirmed that hnRNPL bound to their promoters (Fig. 4h and Supplementary Fig. 5b). To validate the findings of PI3K-AKT signaling pathway gene regulation and promoter interaction by hnRNPL, we performed RT-qPCR and ChIP-PCR and found that among the 16 examined genes, 14 genes were down-regulated after hnRNPL knockdown in OVCAR3 and SKOV3 cells, and the promoters of eight genes were bound by hnRNPL (Fig. 4i, j and Supplementary Fig. 5c). hnRNPL has been reported to be associated with the RNA Pol II transcriptional complex to regulate gene transcription17. Immunofluorescence staining and co-localization analysis revealed that hnRNPL was co-localized with RNA Pol II (Supplementary Fig. 5d). Co-IP assays also showed that hnRNPL interacted with RNA Pol II independently on RNA in ovarian cancer cells (Supplementary Fig. 5e). Subsequently, we performed RNA Pol II ChIP-seq in control and hnRNPL-depleted ovarian cancer cells. The results showed a strong correlation between hnRNPL and RNA Pol II binding sites on chromatin (Fig. 4k and l). Moreover, hnRNPL knockdown deceased the binding of RNA Pol II on chromatin genome-widely (Supplementary Fig. 5f, g). Integrated analysis of hnRNPL ChIP-seq in control ovarian cancer cells, RNA Pol II ChIP-seq and RNA-seq in hnRNPL-depleted ovarian cancer cells revealed that enrichment of RNA Pol II at the promoters of 152 DEGs (113 down-regulated and 39 up-regulated) induced by hnRNPL depletion was deceased (Supplementary Fig. 5h). These down-regulated genes, including PIK3CB, EGFR, RXRA and CDK6, were enriched in the PI3K-AKT signaling pathway (Supplementary Fig. 5i), and RNA Pol II enrichment at the promoters of these genes was shown in Fig. 4m. These results demonstrate that hnRNPL activates the transcription of genes within the PI3K-AKT signaling pathway in ovarian cancer cells.
hnRNPL phase separation enhances glycolysis through activating PIK3CB transcription
Mammals express four catalytic isoforms of PI3K: PIK3CA, PIK3CB, PIK3CG, and PIK3CD. Among the four isoforms, only PIK3CA and PIK3CB are expressed ubiquitously in cells. Each catalytic isoform forms a dimer with a regulatory subunit that controls the activity of the complex. Many distinct PIK3CA-activating mutations have been identified in human tumors. Although PIK3CB has been also implicated in a variety of cancers13, how it is regulated in cancers remains largely unexplored. Thus, we next investigated whether hnRNPL regulates PIK3CB transcription in ovarian cancer. Correlation analysis revealed that hnRNPL expression was significantly positively correlated with PIK3CB expression in ovarian cancer, although a weaker positive correlation was shown in the TCGA ovarian cancer database (Fig. 5a and Supplementary Fig. 6a). Next, we next sought to validate the transcriptional regulation of PIK3CB by hnRNPL. The results of hnRNPL ChIP-seq showed that hnRNPL bound to the promoter (700 bp upstream of transcription start site (TSS)) of PIK3CB (Fig. 4h). To further map hnRNPL binding sites on the PIK3CB promoter, we performed ChIP-qPCR by using primers targeting different regions of the promoter (Supplementary Fig. 6b). The results showed that hnRNPL immunoprecipitated the regions of −300-−700 and +50-−100 of the PIK3CB promoter, but failed to bound other regions (Supplementary Fig. 6c). Then, the dual luciferase reporter assays were performed by using reporter vectors containing the wild-type PIK3CB promoter (pro-WT, 1 kb upstream of the TSS) or mutants with deletions of the −300 to −700 region (pro-mut1) or the TSS to −100 region (pro-mut2) (Fig. 5b). hnRNPL overexpression increased the luciferase activity driven by the pro-WT reporter but not the pro-mut reporters (Supplementary Fig. 6d). In contrast, hnRNPL knockdown in ovarian cancer cells reduced the luciferase activity by the pro-WT reporter but not pro-mut reporters (Fig. 5c). Furthermore, depletion of hnRNPL significantly decreased RNA Pol II enrichment at the promoter of PIK3CB, consistent with RNA Pol II ChIP-seq results in hnRNPL-depleted ovarian cancer cells (Figs. 4m and 5d).

a Spearman’s correlation analysis between mRNA levels of hnRNPL and PIK3CB according to the GEO ovarian cancer cohorts. b Schematic diagram of the luciferase reporters carrying wild-type or mutated PIK3CB promoter. c Analysis of luciferase activities of the reporters carrying wild-type or mutated PIK3CB promoters normalized to renilla luciferase activity in ovarian cancer cells with knockdown of hnRNPL. d Effect of hnRNPL loss on RNA Pol II enrichment at the promoter of PIK3CB. e Effect of wild-type or mutated hnRNPL overexpression on the PIK3CB mRNA level. f ChIP assays were performed to evaluate the enrichment of wild-type or mutated hnRNPL at the PIK3CB promoter in OVCAR3 cells by using the antibody specific to hnRNPL. g Schematic diagram of PI3K-AKT regulated glycolytic signaling pathway. h Western blot of PIK3CB and PI3K-AKT signaling pathway-associated proteins in ovarian cancer cells upon overexpression of wild-type or mutated hnRNPL. i Glucose content and lactate production in the culture medium were monitored in ovarian cancer cells upon overexpression of wild-type or mutated hnRNPL. j ECAR levels were determined in wild-type ovarian cancer cells and ovarian cancer cells with overexpression of hnRNPL variants of WT, IDR1delG, IDR2delP by the Seahorse assays. Data are shown as means ± S.D. p value was calculated by one-way ANOVA test with multiple comparisons (c, e, f, i, j) and unpaired two-sided Student’s t-test (d). Data are representative of at least three independent experiments (c–f, h–j). Source data are provided as a Source Data file.
To confirm whether the LLPS activity is essential for hnRNPL activating PIK3CB expression, we expressed wild-type hnRNPL, IDR1delG or IDR2delP in ovarian cancer cells, respectively. As expected, overexpression of both wild-type hnRNPL and IDR1delG significantly promoted PIK3CB expression (Fig. 5e). However, IDR2delP failed to induce PIK3CB expression (Fig. 5e). Dual fluorescent reporter assays showed that the impaired phase-separated IDR2delP exerted little effect on the luciferase activity in cells transfected with the pro-WT reporter (Supplementary Fig. 6e). Furthermore, loss of the LLPS property in IDR2delP abrogated the interaction of hnRNPL with PIK3CB promoter in ChIP assays (Fig. 5f), highlighting the necessity of LLPS activity for the hnRNPL-PIK3CB promoter interaction.
PIK3CB is an essential component of PI3K that induces the phosphorylation of key glycolytic enzymes PFKFB2 and PKM213. We thus next examined the effect of hnRNPL phase separation on the phosphorylation of PFKFB2 and PKM2 as well as glycolysis in ovarian cancer cells (Fig. 5g). Overexpression of either wild-type hnRNPL or IDR1delG enhanced the phosphorylation of PFKFB2 and PKM2, whereas IDR2delP overexpression failed to induce their phosphorylation (Fig. 5h). Notably, the glucose level was decreased upon wild-type hnRNPL or IDR1delG overexpression in ovarian cancer cells (Fig. 5i), while lactate was increased upon wild-type hnRNPL or IDR1delG overexpression in ovarian cancer cells (Fig. 5i). Nevertheless, IDR2delP overexpression did not affect glucose or lactate levels (Fig. 5i). We also measured the ATP content in ovarian cancer cells overexpressing wild-type or mutated hnRNPL. ATP levels were increased upon overexpression of wild-type hnRNPL, IDR1delG or IDR2FUS in ovarian cancer cells (Supplementary Fig. 6f). In contrast, disruption of hnRNPL phase separation failed to increase the ATP content in ovarian cancer cells (Supplementary Fig. 6f). In addition, extracellular acidification rate (ECAR) was measured by using the Seahorse system to evaluate glycolysis levels. Results showed that wild-type hnRNPL or IDR1delG, but not IDR2delP, enhanced glycolysis and glycolytic capacity (Fig. 5j). These results indicate that hnRNPL phase separation promotes PIK3CB transcription and glycolysis in ovarian cancer cells.
Promoter-associated PIK3CB RNA (paPIK3CB) facilitates hnRNPL phase separation
ChaRBPs bind to chromatin in either an RNA-dependent or RNA-independent manner to regulate gene expression2,5,7. hnRNPL was found to interact with chromatin in an RNA-dependent manner by nucleoplasmic separation assays (Supplementary Fig. 1d). ChIP assays also revealed that RNase treatment decreased the enrichment of hnRNPL at the promoters of genes including IRS1, CDK6, PIK3CB and PCK2 (Fig. 6a). Promoter-associated non-coding RNAs transcribed from promoter regions can regulate gene transcription14. To determine whether paPIK3CB RNA transcribed from the PIK3CB promoter recruited hnRNPL, RNA immunoprecipitation (RIP) was performed using the antibody specific to hnRNPL. The results showed that hnRNPL bound to paPIK3CB RNA (Fig. 6b). We also examined the expression of paPIK3CB RNA in ovarian cancer cells and found that paPIK3CB RNA was elevated in ovarian cancer cells compared normal ovarian epithelium cells (Supplementary Fig. 7a). To confirm that paPIK3CB RNA was transcribed by RNA Pol II, we treated ovarian cancer cells with the specific RNA Pol II inhibitor α-Amanitin and found that RNA Pol II inhibition significantly decreased the expression of paPIK3CB RNA (Supplementary Fig. 7b). To confirm the size of paPIK3CB RNA, we performed PCR assays by using a set of primers across the promoter of PIK3CB (Supplementary Fig. 7c). Compared with the genomic DNA as the template where all PCR products were detected, only the region across −200-−1200 bp upstream the TSS of PIK3CB was detected when using the cDNA as the template, suggesting that paPIK3CB RNA with the length of approximately 1 kb (Supplementary Fig. 7d). Additionally, we identified the transcript by hnRNPL RIP-PCR assays (Supplementary Fig. 7e).

a RNase-ChIP assays were performed to confirm that hnRNPL bound to the promoters of IRS1, CDK6, PIK3CB and PCK2 depending RNAs. b RIP-PCR detecting the interaction between hnRNPL and the promoter-associated RNA paPIK3CB by using the antibody specific to hnRNPL in wild-type ovarian cancer cells. c Schematic diagram of the RRM-deleted hnRNPL constructs (RRM1-1del, RRM1-2del and RRM5del). d, e The interaction of the RRM-deleted hnRNPL constructs with paPIK3CB was examined by RIP assays. f Colocalization between wild-type hnRNPL or RRM-mutants and paPIK3CB RNA was examined in ovarian cancer cells under confocal microscopy by RNA FISH and IF assays. Scale bar, 5 µm. g Schematic of products from hnRNPL eCLIP-qPCR across paPIK3CB RNA. h eCLIP-qPCR by using an antibody specific to hnRNPL was performed in OVCAR3 ovarian cancer cells. i Schematic of wild-type paPIK3CB RNA and its three deletion mutants. j, k RIP assays were performed by using an antibody specific to hnRNPL in OVCAR3 ovarian cancer cells with overexpression of wild-type paPIK3CB RNA and its three deletion mutants. hnRNPL immunoprecipitated was assessed by western blot (j), and interaction between hnRNPL and wild-type paPIK3CB RNA or its mutants was detected by RT-qPCR (k). l Relative RNA expression of PIK3CB was detected upon paPIK3CB was knocked down by ASOs. m ChIP asssys detecting the enrichment of hnRNPL at the promoter of PIK3CB upon paPIK3CB was knocked down. n Representative images and quantitative analysis of hnRNPL puncta in ovarian cancer cells upon paPIK3CB was knocked down by ASOs (n = 50 cells for each group). Scale bar, 5 µm. o Representative images and quantitative analysis of hnRNPL puncta in ovarian cancer cells with RNase treatment (n = 50 cells for each group). Scale bar, 5 µm. p Droplet formation of hnRNPL-EGFP under the influence of different concentrations of CY3-labeled RNA. Scale bar, 50 µm. q Analysis of fluorescent area of EGFP and CY3 signal in each image (n = 3 independent experiments). Data are shown as means ± S.D. p value was calculated by one-way ANOVA test with multiple comparisons (a, i, n) and unpaired two-sided Student’s t-test (e, h, k, m, o). Data are representative of at least three independent experiments (a, b, d–f, h, k–p). Source data are provided as a Source Data file.
hnRNPL contains three RNA recognition motifs (RRMs). To clarify the domain in hnRNPL that mediates its interaction with paPIK3CB RNA, we constructed the mutants with individual RRM deletion, respectively, followed by RIP assays (Fig. 6c). Deletion of RRM1-1, but not other domains, markedly decreased the interaction between hnRNPL and paPIK3CB RNA (Fig. 6d, e). Consistent with this result, co-localization between hnRNPL and paPIK3CB RNA was observed in ovarian cancer cells under confocal microscopy, which was disrupted upon deletion of RRM1-1 (Fig. 6f). We also found that the RRM1-1 mutation, but not other mutants, disrupted the condensation formation of hnRNPL in ovarian cancer cells (Supplementary Fig. 7f). Moreover, we performed hnRNPL enhanced cross-linking immunoprecipitation (eCLIP) assays followed by RT-qRCR to confirm the regions of paPIK3CB RNA bound by hnRNPL in ovarian cancer cells. The results revealed that hnRNPL bound to the 351-523 and 800–1000 regions (Fig. 6g, h). Since hnRNPL is known to bind CA repeats motif30,31,32, we analyzed paPIK3CB RNA sequences and identified three potential hnRNPL-binding regions (Fig. 6i). Then, we constructed the mutants of paPIK3CB RNA with the deletion of the individual potential hnRNPL-binding region. RIP-PCR assays showed that deletion of the 366-386 region or the 826-848 region significantly decreased interaction between paPIK3CB RNA and hnRNPL, consistent with eCLIP-PCR results (Fig. 6j, k).
Subsequently, paPIK3CB RNA was targeted by the antisense oligodeoxynucleotides (ASOs) to further verify whether paPIK3CB RNA was involved in PIK3CB regulation by hnRNPL. The results showed that knockdown of paPIK3CB RNA led to a reduction in PIK3CB expression (Fig. 6l and Supplementary Fig. 7g). Moreover, knockdown of paPIK3CB RNA by ASOs moderately reduced the foci formation of hnRNPL as well as its enrichment at the promoter of PIK3CB (Fig. 6m, n). RNase treatment extensively abrogated hnRNPL condensation in ovarian cancer cells (Fig. 6o), suggesting that hnRNPL forms condensates in an RNA-dependent manner in live cells. We also observed that overexpression of wild-type hnRNPL or IDR1delG, but not IDR2delP, increased the enrichment of hnRNPL at the promoter of PIK3CB in paPIK3CB RNA-proficient ovarian cancer cells, whereas overexpression of wild-type hnRNPL or IDR1delG failed to promote the enrichment of hnRNPL at the promoter of PIK3CB in paPIK3CB RNA-deficient ovarian cancer cells (Supplementary Fig. 7h), suggesting that hnRNPL recruitment to the promoter of PIK3CB depends on paPIK3CB RNA. Considering that hnRNPL interacted with RNA Pol II, we then examined whether hnRNPL was involved in RNA Pol II condensation. However, knockdown of hnRNPL only induced a marginal decrease of RNA Pol II condensation (Supplementary Fig. 7i). In addition, either in vitro-synthesized paPIK3CB RNA or the total RNA extracted from ovarian cancer cells facilitated the formation of hnRNPL droplets, whereas tRNAs did not affect hnRNPL droplet formation, suggesting that paPIK3CB RNA contributes to hnRNPL phase separation (Fig. 6p, q and Supplementary Fig. 7j). Thus, these results demonstrate that paPIK3CB RNA promotes hnRNPL phase separation to regulate PIK3CB transcription.
The hnRNPL-PIK3CB-AKT axis promotes ovarian cancer progression
PI3K-AKT pathway is an important pathway that promotes cancer progression13. Loss of PIK3CB significantly suppressed the growth, migration and invasion of ovarian cancer cells (Supplementary Fig. 8a–e). Re-expression of PIK3CB in ovarian cancer cells with PIK3CB down-regulation induced by hnRNPL knockdown rescued the cell growth, migration, and invasion (Supplementary Fig. 8f–j). These results suggest a role for the PI3K-AKT pathway in the effect of hnRNPL on ovarian cancer cell growth, migration, and invasion. To further explore the potential role of the hnRNPL-PIK3CB-AKT axis in ovarian cancer cells, we employed SC79, a PI3K-AKT pathway agonist, as well as TGX221 and PI-103, PI3K-AKT pathway inhibitors. SC79 partially reversed the decrease in phosphorylation levels of PFKFB2 and PKM2 in hnRNPL-depleted ovarian cancer cells, and restored the cell growth, migration and invasion (Fig. 7a, b and Supplementary Fig. 9a, b). In contrast, TGX221 of PI-103 treatment in hnRNPL-overexpressed ovarian cancer cells mitigated the up-regulation of PFKFB2 and PKM2 phosphorylation levels as well as malignant phenotypes of ovarian cancer cells (Fig. 7c, d and Supplementary Fig. 9c, d). Additionally, SC79 treatment suppressed glucose consumption, lactate production and ATP production in hnRNPL-depleted ovarian cancer cells (Fig. 7e, g). Conversely, TGX221 and PI-103 reversed the enhanced glycolysis in hnRNPL-overexpressed ovarian cancer cells (Fig. 7f, g). The Seahorse assays also confirmed the critical functions of the hnRNPL-PIK3CB-AKT axis in promoting glycolysis in ovarian cancer cells (Fig. 7h–k).

a Ovarian cancer cells were transfected with hnRNPL for 48 h. Then, cells were treated with TGX221 (5 μM) or PI-103 (5 μM), or DMSO as the negative control for 24 h for western blot. b CCK-8 assay detecting the growth of ovarian cancer cells described in (a). c Glucose content and lactate production were monitored in ovarian cancer cells described in (a). d hnRNPL was knocked down in ovarian cancer cells and then treatment with SC79 (5 μM) or DMSO as the negative control for 24 h was performed for western blot. e CCK-8 assay detecting the growth of ovarian cancer cells described in (d). f Glucose content and lactate production were monitored in ovarian cancer cells described in (d). g ATP content was measured in ovarian cancer cells described in (a) and (d). h, i ECAR levels were determined in ovarian cancer cells described in (a) by the Seahorse assays. j, k ECAR levels were determined in ovarian cancer cells described in (d) by the Seahorse assays. l Immunohistochemical staining assays detecting the protein expression of hnRNPL in ovarian cancer and ovarian epithelium tissues. Scale bar, 200 μm. m The overall survival and progression-free survival of patients with ovarian cancer with different hnRNPL levels according to the TCGA ovarian cancer database. n The overall survival, disease-free survival, and disease-specific survival of patients with ovarian cancer with hnRNPL gene alternations according to the TCGA ovarian cancer database. Data are shown as means ± S.D. p value was calculated by one-way ANOVA test with multiple comparisons (b, c, e–g, i, k) and two-sided Log-rank (Mantel-Cox) test (m, n). Data are representative of at least three independent experiments (a–k). Source data are provided as a Source Data file.
Finally, we examined the expression of hnRNPL and PIK3CB in fresh ovarian cancer tissues and fallopian tube tissues from patients and found that both hnRNPL and PIK3CB were increased in human ovarian cancer (Supplementary Fig. 10a). Immunohistochemical staining assays showed a positive correlation between hnRNPL and PIK3CB expression (Supplementary Fig. 10b). Tissue microarray results revealed that hnRNPL was up-regulated in ovarian cancer (Fig. 7l). hnRNPL expression was also associated with tumor grades, FIGO stages, and omental invasion in ovarian cancer patients, but not with platinum sensitivity (Supplementary Fig. 10c). Survival analysis revealed that high expression of hnRNPL was strongly associated with poor prognosis for ovarian cancer patients (Fig. 7m and Supplementary Fig. 10d). Similarly, PIK3CB was elevated in ovarian cancer, and its higher expression predicted worse prognosis for ovarian cancer patients (Supplementary Fig. 10e–h). Additionally, hnRNPL was highly amplified in various cancers, including ovarian cancer (Supplementary Fig. 10i). Alteration of hnRNPL gene status predicted worse outcomes for ovarian cancer patients (Fig. 7n). Thus, these results demonstrate hnRNPL is up-regulated in ovarian cancer, and the formed hnRNPL-PIK3CB-AKT axis plays a critical role in promoting ovarian cancer progression.
Discussion
In this study, we demonstrate that hnRNPL undergoes LLPS both in vivo and in vitro. hnRNPs are composed of RNA-binding motifs and auxiliary domains, including the low-complexity domain (LCD)33. The LCD has been recognized as the IDR that contributes to protein condensation34. Multiple hnRNP proteins phase separate through their IDRs35,36,37. FUS, also known as hnRNPP2, is one of the most extensively explored phase-separated proteins in neurodegenerative diseases35. Phase separation of hnRNP A1 via its LCDs promotes stress granules assembly and drives pathological fibrillization34. hnRNPL contains two IDRs, but only IDR2 mutation significantly abrogated its condensation, highlighting the critical role of IDR2 in hnRNPL phase separation. Replacing of hnRNPL IDR2 with the IDR sequence of FUS protein that determines phase separation properties of FUS preserves hnRNPL phase separation. Glycine and proline in the IDR are critical for protein LLPS38,39,40,41,42. Mutation of glycine residues in the LCD of FUS decreases its droplet fusion in vitro38. Mutation of prolines in the IDR of TDP-43 leads to severe protein aggregation and disrupts phase separation of TDP-4343. Intriguingly, deletion of proline residues in IDR2 but not glycine residues in IDR1 abrogates hnRNPL phase separation, indicating distinct roles for these amino acids in phase separation across specific proteins. Temperature also influences protein phase separation44,45, as evidenced by the evolutionarily conserved low-complexity protein BuGZ, which forms larger droplets as temperature increases46. Similarly, raising the temperature from 4 °C to 37 °C promotes droplet formation of YTHDF family proteins47. However, unlike BuGZ and YTHDF family proteins, whose phase separation follows a lower critical solution temperature profile47,48, hnRNPL LLPS belongs to the upper critical solution temperature, suggesting a distinct effect of temperature on LLPS of different proteins with unexplored mechanisms.
hnRNPL regulates RNA metabolism by affecting RNA splicing, decay and translation32. hnRNPL also binds to integrin and extracellular matrix genes and enhances their expression by directly interacting with RNA Pol II independent of RNA17. In constrast, during skeletal myoblast differentiation, enhancer RNAs induced by MyoD can recruit hnRNPL and activate gene transcription49. hnRNPL is also recruited by a muscle-specific long non-coding RNA lncFAM to gene promoters thus modulating gene transcription and contributing to myogenesis50. We demonstrate that hnRNPL, acting as a ChaRBP dependent on a paRNA, binds to chromatin in ovarian cancer. RNA molecules are implicated in diverse biomolecular condensates, including nuclear speckles, paraspeckles, and stress granules51. Previous studies reported that the synergistic action of RNAs contributed to the formation of RNA Pol II transcriptional complex condensates, promoting gene transcriptional activity52,53. A model has been proposed in which different RNAs produced at early steps in transcription initiation and elongation exert promoting and suppressive effects on condensation formation, respectively53. Consistently, we demonstrate that a paRNA promotes hnRNPL phase separation and regulates gene transcription. The interplay between RNA and RBP phase separation opens a realm of gene regulation.
Protein aggregates are a main hallmark of neurodegenerative diseases, with the transition from reversible LLPS to irreversible aggregation observed for TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia, respectively35,43. Phase separation has been linked to tumorigenesis. Condensation of the RBP AKAP95 mediates oncogenic alternative splicing to promote breast cancer54. hnRNPL has been implicated in several cancers through interplaying with mRNA and non-coding RNAs and regulating RNA metabolism55,56,57,58. However, whether the role of hnRNPL in cancer depends on phase separation was unexplored in these studies. Our investigation demonstrates that a promoter-associated RNA enhances hnRNPL phase separation, activating gene transcription. Disruption of hnRNPL phase separation significantly abrogated its tumor-promoting function in ovarian cancer. Thus, the role of phase separation in hnRNPL-mediated RNA metabolic regulation in cancer warrants further investigation. Notably, hnRNPL is involved in other diseases, including schemic stroke and lung fibrosis59,60, but its role in neurodegenerative diseases remains largely unexplored. Thus, LLPS may offer insights into the role of hnRNPL in these diseases.
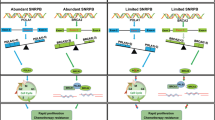
Further exploration of the pathological functions of protein phase separation may provide a foundation for therapeutic interventions for diseases. Traditionally, drug design and characterization heavily relied on the structures of underlying protein targets. However, LLPS introduces an approach for traditionally undruggable intrinsically disordered proteins61,62. Through a physicochemical mechanism, drugs targeting phase separation can modulate it by focusing on disordered regions, and structural domains, or modulating deformation63,64. Emerging technology such as proteolysis-targeting chimeras and the high-throughput screening method named DropScan have also been developed to target protein phase separation. However, several questions remain to be addressed for specific protein phase separations65,66. More investigations are needed to develop targeted drugs against hnRNPL phase separation and evaluate their effect when combined with existing PI3K-AKT pathway inhibitors or chemotherapy drugs such as cisplatin in the future, which might hold the promise of benefiting ovarian cancer patients. In conclusion, our study reveals that the proline-rich sequence in the IDR contributes to the LLPS property of hnRNPL. paPIK3CB RNA binds to the RRM of hnRNPL and promotes its phase separation in ovarian cancer cells. hnRNPL phase separation facilitates ovarian cancer progression via PIK3CB transcription to enhance glycolysis (Fig. 8). The hnRNPL-PIK3CB-AKT axis, driven by phase separation, could be a promising target for ovarian cancer therapy.

In ovarian cancer cells, a non-coding RNA transcribed from the PIK3CB promoter recruits hnRNPL and promotes its condensation, which activates PIK3CB transcription and downstream AKT signaling pathway, thus promoting glycolysis and cancer progression.
Methods
Ethical statement
Animal studies were approved by the Institutional Animal Care and Use Committee of Chongqing Medical University and were conducted according to institutional guidelines. Fresh ovarian cancer tissue and normal ovarian epithelial tissue specimens were taken from patients between August 30, 2020, and March 1, 2021. All of these tissue specimens were pathologically validated, and all study participants aged from 31 to 81 years gave written informed consent. The study was approved by the Ethical Review Committee of the Third Affiliated Clinical Hospital of Chongqing Medical University (202125) and the Ethical Review Committee of Army Specialty Medical Center (2022374).
Antibodies and chemicals
Antibodies used were as follows: hnRNPL (ab6106, Abcam), FLAG (ab205606, Abcam), mCherry (HA500049, HUABIO), myc (M192-3, MBL), PIK3CB (ab151549, Abcam), p-AKT (9271S, CST), p-PFKBF2 (13064S, CST), p-PKM2 (DF2975, Affinity), snRNP70 (ab83306, Abcam), H3 (17168-1-AP, Proteintech), HSP90 (13171-1-AP, Proteintech), RNA Pol II (ab238146, Abcam), GAPDH (60004-1-Ig, Proteintech), β-actin (66009-1-Ig, Proteintech).
Chemicals used in this study include: PI-103 (S1038, Selleckchem), TGX-221 (S1169, Selleckchem), ATP assay kit (A095-2-1, Nanjing Jiancheng Bioengineering Institute), Glucose content test kit (ADS-W-TDX002, AIDISHENG), Lactic acid content test kit (ADS-W-T009-96, AIDISHENG), TransDetect Double-Luciferase Reporter Assay Kit (FR201-02, TransGen), Versatile DNA/siRNA transfection reagent (101000046, Polyplus), Seahorse XFe24 FluxPak mini (102342-100, Agilent), Seahorse XF Glycolysis Stress Test Kit (103020-100, Agilent), ClonExpress MultiS One Step Cloning Kit (C113, Vazyme), TIANprep midi Plasmid kit (DP106, TIANGEN), TIANamp Genomic DNA kit (DP304, TIANGEN), Magna ChIP A/G Chromatin Immunoprecipitation kit (9003, CST).
Plasmids
pLKO.1 (#10878), psPAX2 (#12260) and pMD2.G (#12259) plasmid vectors were purchased from Addgene Company. pCMV6 plasmid vector was purchased from OriGene. Complementary sense and antisense oligonucleotides encoding shRNAs targeting hnRNPL were synthesized and cloned into pLKO.1 vector. The sequences of oligonucleotides were shown in Supplementary materials. hnRNPL-WT, hnRNPL-IDR1delG, hnRNPL-IDR2delP, and hnRNPL-IDR2FUS expression plasmids were constructed via cloning the corresponding sequence into pCDH-GFP-puro-3×flag, pcDNA3.1-mCherry, and pcDNA3.1-3×flag vector, respectively. hnRNPL-RRM1-1del, hnRNPL-RRM1-2del, hnRNPL-RRM5del and PIK3CB expression plasmids were constructed via cloning corresponding sequence into pcDNA3.1-3×flag vector, respectively. pHR-mCh-Cry2WT was a gift from Clifford Brangwynne (101221, Addgene; http://n2t.net/addgene:101221; RRID: Addgene, 101221). pHR-hnRNPL-IDR2FUS-mCh-Cry2WT, pHR-hnRNPL-WT-mCh-Cry2WT, pHR-hnRNPL-IDR1delG-mCh-Cry2WT, pHR-hnRNPL-IDR2delP-mCh-Cry2WT expression plasmids were constructed via cloning corresponding sequence into pHR-mCh-Cry2WT vector, respectively. LentiCRISPR V2 vector and pUC19 plasmids are gifts from Dang Lab (Chongqing Medical University).
Cell culture and transfection
HEK293T cells were purchased from ATCC (American Type Culture Collection). All cell lines were stored in liquid nitrogen for the long term. HEK293T was cultured in DMEM medium (GIBCO) containing 10% fetal bovine serum (Gemini) and 1% penicillin/streptomycin (GIBCO). SKOV3 and OVCAR3 cells were cultured in RPMI1640 medium (GIBCO) containing 10% fetal bovine serum and 1% penicillin/streptomycin. All cells were cultured in a cell incubator at 37 °C and 5% CO2. HEK293T, SKOV3, and OVCAR3 cells were transfected with Versatile DNA/siRNA transfection reagent following the manufacturer’s instructions.
Lentivirus production and infection
Lentiviral vectors were co-transfected into HEK293T cells with packaging vectors psPAX2 and pMD2.G using jetPRIME transfection reagent. Infectious lentivirus particles were harvested at 48 h after transfection.
RNA isolation and quantitative real-time PCR
Total RNA was extracted using Trizol (Invitrogen) according to the manufacturer’s instructions. cDNA used for RT-qPCR was conducted from total RNA using HiScript II Q RT SuperMix for qPCR (Vazyme). RT-qPCR was performed by SYBR qPCR Master Mix (Vazyme) and the Bio-rad CFX96 real-time PCR system (Biorad). Primers used in RT-qPCR were shown in Supplementary Data 1.
Protein purification
The expression of recombinant MBP-tagged or His-tagged proteins in E.coli strain BL21 (DE3) was induced at 16 °C using 0.5 mM IPTG. Bacteria were harvested and resuspended in the lysis buffer (50 mM Tris-HCl pH 8.5, 2 M KCl, 5 % glycerol, 10 mM MgCl2, 1 mM DTT, and 1 mM PMSF) and then lysed by high-pressure homogenization followed by centrifugation at 4 °C for 60 min. For His-tagged proteins, the supernatant was purified using a Ni2+-Sepharose affinity column (GE Healthcare) and eluted in an elution buffer consisting of 50 mM Tris-HCl pH 8.5, 250 mM NaCl, and 250 mM Imidazole. For MBP-tagged proteins, the supernatant was loaded onto dextrin sepharose (MBP Trap HP, GE Healthcare), and bound proteins were eluted with the elution buffer (50 mM Tris- HCl 8.5, 2 M KCl, 5 % glycerol, 10 mM MgCl2, 50 mM maltose) and concentrated into 500 µL using an Amicon Ultra Centrifugal Filter Unit. Then the target protein was purified by heparin sepharose (HiTrap Heparin HP, GE Healthcare) and size-exclusion chromatography (Superdex 200, GE Healthcare).
Western blot
The total protein of cells and tissues was obtained within RIPA lysis buffer (Beyotime, China) containing 1% PMSF and quantified by BCA detection kit (Solarbio, China). Proteins were separated with 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, USA). The membrane was blocked in 5% skim milk for 1 h and incubated with the primary antibodies overnight at 4 °C. The primary antibodies are as follows: hnRNPL (1:1000, ab6106, Abcam), FLAG (1:1000, ab205606, Abcam), mCherry (1:1000, HA500049, HUABIO), myc (1:1000, M192-3, MBL), PIK3CB (1:1000, ab151549, Abcam), p-AKT (1:1000, 9271S, CST), p-PFKBF2 (1:1000, 13064S, CST), p-PKM2 (1:1000, DF2975, Affinity), snRNP70 (1:1000, ab83306, Abcam), H3 (1:1000, 17168-1-AP, Proteintech), HSP90 (1:1000, 13171-1-AP, Proteintech), RNA Pol II (1:1000, ab238146, Abcam), GAPDH (1:1000, 60004-1-Ig, Proteintech), β-actin (1:1000, 66009-1-Ig, Proteintech). The next day, the membrane was incubated with the corresponding secondary antibody, goat anti-rabbit IgG-HRP (1:10000, PR30011, Proteintech) and goat anti-mouse IgG-HRP (1:10000, PR30012, Proteintech), at room temperature for 1 h, The positive band was then detected with chemiluminescence using a Western Blotting Imaging System.
Cell proliferation assay
Cell proliferation was measured by Cell Counting Kit-8 (CCK-8) and colony forming assays. For CCK-8 assay, the transfected cells were placed in 96-well plates with a density of 2 × 103 cells/well and cultured in 100 μL growth medium containing 10% FBS overnight. After 0, 24, 48, 72, and 96 h of incubation, 10 μL of CCK-8 solution (K1018, APExBIO) was added to each well and incubated at 37 °C for 2 h. The absorbance value (optical density, OD) at a wavelength of 450 nm and 630 nm was measured on a microplate spectrophotometer (Bio-Rad, USA). For colony formation assay was performed with 2 × 103 cells in 6-well plates and change the culture medium every 4 days. After 10 days of growth, the cells were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet. Finally, the colonies were washed with PBS and counted under a light microscope.
RNA immunoprecipitation (RIP)
Cells were lysed in RIP buffer supplemented with DTT, PIC and 0.2 U/L RNase inhibitor (Invitrogen, LT-02241) and 5% of supernatant was collected as input sample. hnRNPL antibody (ab6106, Abcam) or mouse IgG antibody (12-371, Millipore) were added into the lysis of control ovarian cancer cells for RIP at 4 °C overnight. FLAG antibody (ab205606, Abcam) or mouse IgG antibody (12-371, Millipore) were added into the lysis of wild-type or mutated hnRNPL overexpressing ovarian cancer cells for RIP at 4 °C overnight. Next day, 50 μL washed protein G beads (Invitrogen, 10004D) were added to the mixture and rotated at 4 °C for 4 h. After washed 5 times with 5 min, 10% of IP samples were collected for quality control using immunoblotting. The coprecipitated RNAs within beads and input RNAs were extracted in TRIzol reagent (Invitrogen) and ethanol-precipitated with glycogen (Invitrogen). Then, the enriched RNA was reverse-transcribed and amplified through RT-qPCR. The results were calculated as fold enrichment from specific antibody versus non-specific IgG.
Immunoprecipitation-free single-cell ChIP-seq
The ChIP-seq was performed following described with minor modifications through the combinatorial barcoding and targeted chromatin release (CoBATCH)29. Briefly, OVCAR3 cells were dissociated with 0.25% trypsin for 3 min at 37 °C and washed twice with 0.1% BSA DEPC-PBS, then fixed in 4% paraformaldehyde on ice for 5 min, followed by quenching with Tris-HCl. Fixed cells were washed by cold PBS three times, and the liquid was removed after centrifugation at 600 g for 5 min at 4 °C. Cell pellets were added 1 mL 30% methanol and could be stored at 80 °C. Frozen pellets were resuspended by 0.1% BSA DEPC-PBS and washed twice with Wash Buffer (20 mM HEPES, 150 mM NaCl, 0.5 mM Spermidine, 1× protease inhibitor cocktail (Beyotime, P1006), 1 mM PMSF and 10 mM sodium butyrate). Then, the Con-A beads were washed with 1 mL Binding Buffer (1 M HEPES, 1 M KCl, 1 M CaCl2, 1 M MnCl2) and were then resuspended with adequate Binding Buffer and applied to the cell mixture. The cell-bead mixture was incubated for 10 min and collected by the magnet stand. Next, cells were resuspended with adequate antibody buffer (0.5 M EDTA, 0.01% Digitonin-Wash Buffer) supplemented with 1× protease inhibitor cocktail, PMSF, 10 mM sodium butyrate and 0.1% Triton X-100 containing hnRNPL antibody (1:50, Abcam, ab6106) by gently flicking. The cell-antibody mixture was incubated at 4 °C for 2 h. Cells were washed twice with Dig-Wash Buffer (with 0.1% Triton X-100) and finally suspended with Dig-Wash Buffer (with 0.1% Triton X-100, 1× protease inhibitor cocktail, PMSF and 10 mM sodium butyrate) containing individual combinatorial barcoded 3 μg/mL PAT-T5 and 3 μg/mL PAT-T7 (PAT-MEAB, from Aibin He lab, Peking University). The cells were incubated at 4 °C for 1 h and washed with Dig-Wash Buffer with 0.1% Triton X-100 three times to get rid of free PAT-MEA/B. The reaction next was activated by suspending the cells with Reaction Buffer (10 mM TAPS-NaOH, 5 mM MgCl2, 10% DMF supplemented with 1× protease inhibitor cocktail, PMSF 10 mM sodium butyrate), followed by gently flicking and incubated at 25 °C for 1 h. The reaction was terminated with 40 mM EDTA, further incubated at RT for 15 min, and added cold Sort Buffer (PBS/2% BSA/2 mM EDTA) to each well. Cells were passed through a 30 mm cell strainer to remove cell clumps, and were incubated at 55 °C for 3 h for reverse crosslinking and incubated at 85 °C for 15 min to deactivate proteinase K with 1.8% Triton X-100.
PCR amplification was performed by addition of 25 mM i5 index primer, 25 mM i7 index primer (Aibin He Lab, Peking University), 2× KAPA master mix and ddH2O to the adequate CoBATCH cell lysis, and the reaction was set up by incubation at 98 °C for 2 min, 10-15 cycles of amplification (98 °C for 20 s, 65 °C for 30 s, 72 °C for 1 min), and final 72 °C extension for 5 min. After PCR amplification, the library was purified with XP beads once. Ultimately, the libraries were sequenced with paired-end average depth of 150-bp reads on HiSeq X-ten or NovaSeq 6000 platform (Illumina).
Immunofluorescence staining
Transfected cells were placed on 24-well plate or glass-bottom 35 mm dishes and fixed with 4% paraformaldehyde for 15 min, and permeabilized with 0.2% Triton X-100 for 10 min, then washed with PBS three times and blocked with 1% bovine serum albumin (BSA) for 1 h at room temperature. Primary antibodies against FLAG (1:100, ab205606, Abcam), mCherry (1:100, HA500049, HUABIO), PIK3CB (1:100, ab151549, Abcam) were added and incubated overnight at 4 °C. After being washed three times with PBS, Alex Fluor 594 Goat Anti-Mouse IgG (H + L) (ab150115, Abcam) or Alex Fluor 488 Goat Anti-Mouse IgG (H + L) (ab150077, Abcam) secondary antibody was incubated in the dark for 1 h at room temperature. DAPI (ab104139, Abcam) staining was performed for 5 min to visualize the cell nucleus. Finally, the cells were observed and photographed under a fluorescence confocal microscope (Leica TCS SP8).
Luciferase assay
HEK293T cells were co-transfected with the pGL3-hnRNPL-promoter (WT, mut1 (deletion of -700–300 bp from the transcription start site) or mut2 (-100 bp from the transcription start site)) and wild-type or mutated hnRNPL plasmids, or a vector control. Control or hnRNPL-depleted OVCAR3 and SKOV3 cells were transfected with the pGL3-hnRNPL-promoter (WT, mut1, or mut2) plasmids. Cells were collected at 48 h post-transfection. Luciferase assay was performed using the Double-Luciferase Reporter Assay Kit (TransGen, FR201-01-V2) following the manufacturer’s protocol. Firefly and renilla luciferase activities in cell lysates were measured using the SYNERGY microplate reader system (BioTek).
Immunohistochemistry
Paraffin sections were deparaffinized and dehydrated as routine methods. After the antigen was retrieved, endogenous peroxidases (ZSGB-BIO, PV-9001) were inactivated using citric acid buffer (ZSGB-BIO, ZLI-9064, pH 6.0). After washing three times with PBS, sections were blocked with goat serum (ZSGB-BIO, ZLI-9021) at 37 °C for 30 min. Next, hnRNPL antibody (Proteintech, 1:100) or PIK3CB antibody (Proteintech, 1:100) was dispensed onto each section and incubated overnight at 4 °C. The next day, after being washed, the sections were incubated with goat anti-rabbit IgG (ZSGB-BIO, PV-9001) at 37 °C for 40 min. Sections were then washed thoroughly, followed by developing color using DAB reagent (ZSGB-BIO, ZLI-9018). The sections were counterstained with hematoxylin, decolorized through hydrochloric acid and ethanol, dehydrated, and then mounted. The comprehensive score was calculated independently by two professional pathologists.
Fluorescence recovery after photobleaching (FRAP)
Droplets were formed in vitro by diluting the stock protein to a final concentration of 10 µM in a buffer with 5% PEG4000, 50 mM Tris-HCl, pH 7.4, 100 mM KCl. FRAP experiments were undertaken using an A1R+ confocal microscope (Nikon) with a 60× oil-immersion objective. For FRAP of hnRNPL droplets, spots of 1 mm diameter in droplets were photobleached with 70% laser power for 1 s using a 488 nm laser. Time-lapse images were acquired after bleaching at 1 s intervals. For cell imaging, the 70% maximum laser power of the 561 nm laser is used to bleach the point regions. Recovery was recorded at a rate of 2 s / interval for 79 cycles. Pictures were analyzed in Nikon software, and FRAP recovery curves were calculated and plotted using Prism software.
To account for background and photobleaching effects during acquisition, we used the mean intensity values of the bleaching zone (BL), background area (BG) and reference signal area (REF) to calculate the adjusted BL (BL _ corr) for each acquisition frame using the formula:
BL _corr 2(t) was further normalized to estimate the 100% fluorescence intensity.
Finally, a single exponential-like recovery curve was produced by comparing the normalized fluorescence intensity values to time.
OptoIDR assay
hnRNPL-WT, hnRNPL-IDR1delG, hnRNPL-IDR2delP and hnRNPL-IDR2FUS were cloned into pHR-mCh-Cry2WT (101221, addgene). HEK293T cells were plated at 70% fusion 24 h before transfection. Plasmids were transfected into cells using PEI reagent, and after 48 h the transfected cells were transferred to confocal dishes for subsequent experiments. Images were taken with the Nikon A1R confocal microscopy, using a 63× oil objective (zoom = 4). Cells were imaged by two laser wavelengths (488 nm for Cry2oligo activation /561 nm for mCherry imaging). Using 561 nm laser takes one image was taken before fluorescent stimulation, droplet formation was induced with 488 nm light for 10 seconds, and other images were taken every 4 seconds under 561 nm fluorescence.
Endogenously tagged cell line generation
The CRISPR-Cas9 knock-in assay was used to generate clover-tagged hnRNPL in the OVCAR3 cell line by homologous recombination. The guide RNA for targeting hnRNPL was used: 5’-CACCGTTAGGAGGCGTGCTGAGCAG-3’ (PAM: AGG). Repair templates were cloned into a pUC19 vector, two 499-bp homology arms, and a clover tag for the insert. Cells were co-transfected with 1 µg CRISPR-v2 vector and 1 µg donor repair templates using PEI reagent. Cells with the highest fluorescence values were sorted for monoclonal culture using flow cytometry and verified by PCR.
Establishment of hnRNPL knockout cell lines
The knockout cell lines were generated by the CRISPR/Cas9 technology. In brief, sgRNA#1 (GTCTTTCTGCACCACCACAA) and sgRNA#2 (TTTACAAATAAAATGCCCCG) to target hnRNPL were cloned into the pCRISPR-LvSG06-puro (Gene Copoeia, China). For hnRNPL knockout in OVCAR3 and SKOV3 cells, 2 μg each of the LvSG06-hnRNPL-KO plasmid was transfected into cells cultured in 6 cm dishes at a cell density of 60-70% using jet PRIME (Polyplus, France). Forty-eight hours after transfection, cells were screened with puromycin.
Glycolysis metabolism assay
Glycolysis metabolism was assessed based on glucose content, lactate production, and adenosine triphosphate (ATP) generation. Glucose content, lactate, and ATP production were quantified using the Glucose Assay kit (ADS-W-TDX002, AIDISHENG, Jiangsu, China), Lactate Assay kit (ADS-W-T009-96, AIDISHENG, Jiangsu, China), and ATP Assay kit (A095-2-1, Nanjing Jiancheng Bioengineering Institute), respectively. The experimental procedures were conducted in accordance with the instructions provided by each kit. Briefly, cells were transfected with plasmids for 48 h or treated with DMSO or indicated chemicals, and subsequently, the levels of ATP, glucose, and lactate concentrations in the cultured media were measured.
Extracellular acidification rate (ECAR)
ECAR was measured in the Seahorse XF Glycolysis Stress Test Kit (103020-100, Agilent) using the Seahorse XFe24 Analyser (Agilent Technologies) following the manufacturer’s instructions. Briefly, Transfected or control human ovarian cancer cells were plated on XFp cell culture plates. Cells were washed and incubated in base medium (Agilent Technologies) at 37 °C for 1 h. All samples were tested in triplicate, and ECAR were normalized to the total protein.
Chromatin immunoprecipitation (ChIP) and ChIP-seq
ChIP experiments were performed using the Magna ChIP A/G Chromatin Immunoprecipitation kit, following the manufacturer’s protocol. Briefly, we performed protein-DNA cross-linking by incubating 106 cells in 1% formaldehyde for 10 min at 37 °C. Micrococcal nuclease was used to digest the isolated nuclei, which was then sonicated to generate DNA/protein fragments of 150-900 bases (sample was vortex mixed every 3 min) and sonication was performed to nuclear lysate. For immunoprecipitation, 2 μg of antibody (hnRNPL antibody (Abcam, ab6106), RNA Pol II antibody (ab238146, Abcam) or mouse IgG antibody (12-371, Millipore)) was added to each sample, followed by incubation for 4 h at 4 °C. After immunoprecipitation, chromatin-protein complexes were eluted from protein G magnetic beads and reverse-cross-linked. DNA was purified for PCR or sequencing.
Enhanced cross-linking and immunoprecipitation (eCLIP)
eCLIP experiments were performed as described in the previous study with modifications67. Briefly, 20 million cells for UV at 254 nm crosslinking per sample, snap frozen, and then lysed prior to treatment with DNase and RNase A. 50 μL of the sample was used as input, and the remainder was processed for eCLIP. For immunoprecipitation, 10 μg of hnRNPL antibody (Abcam, ab6106), or mouse IgG antibody (12-371, Millipore) was added to each sample, followed by incubation rotated overnight at 4 °C. After immunoprecipitation, RNA-protein complexes were eluted from protein G magnetic beads. RNA was purified for RT-qPCR.
Ovarian cancer organoid culture
Ovarian cancer tissues was minced into small pieces sizing below 3 mm3 and were washed with AdDF + ++ (Advanced DMEM/F12 + 1% penicillin-streptomycin + Glutamax + 10 mM HEPES) and set aside for 5 min to allow tissue pieces to settle down. The supernatants were collected and centrifuged at 1000 rpm for 5 min to obtain ovarian cancer cell precipitation. The cell precipitation was resuspended in 10 mg/mL cold Cultrex Growth Factor Reduced BME Type 2 (3533-010-02, Trevigen), and 40 μL drops of BME-cell suspension were allowed to solidify on pre-warmed 24-well suspension culture plates (M9312, Greiner) at 37 °C for 30 min. Upon completion of gelation, 500 μL of organoid medium was added to each well, and the plates were transferred to 37 °C, 5% CO2 incubators. During culture, the medium was refreshed every 3–4 days.
Cultivated organoids were seeded into a 24-well culture plate. Organoid culture medium formulated with an appropriate amount of virus suspension and polybrene was used to replace the original culture medium for incubation at 37 °C for 24 h. After centrifugation, the supernatant was removed. Cells was resuspended and seeded into culture plate with matrix adhesive in 37 °C incubator for 15 min, then organoid culture medium was added for cultivation.
Animal studies
Mice were bred at the animal facility specific pathogen-free of Chongqing Medical University, and housed under standard conditions (standard diet and water ad libitum) at 23 °C, 50% hygrometry, with 12 h light and 12 h dark cycles. 107 cells were suspended in 100 μL PBS and subcutaneously injected into the right armpit of the nude mice at age of 6 weeks. The tumor was measured from the 7th day, once every 3 or 4 days. The tumor volume was calculated with the following formula: V (mm3) = 0.5 × length (mm) × width2 (mm2). After 21–24 days, mice were sacrificed, and the xenograft tumors were used for measurement. Tumor size did not exceed the maximal size (1.5 cm length, width, height) permitted by ethics committee. Studies were approved by the Institutional Animal Care and Use Committee of Chongqing Medical University.
Sequencing data analysis
For RNA-seq, all samples were sequenced using an Illumina HiSeq X Ten platform with a paired-end 150-bp read length. All reads were mapped to the Homo sapiens genome (Ensembl GRCh38.p5) by TopHat2 (v2.1.1) using default settings. Read counts were quantified using HTseq (v2.2.1). Log fold change of differential expression genes was computed with DESeq2 (version 1.26.0).
For ChIP-seq data, reads were aligned to the Homo sapiens genome (Ensembl GRCh38.p5) using the Eland aligner (Illumina) with the multi-option. Binding peaks for hnRNPL were identified using MACS software with default parameters or software.
Statistical analysis
Data and error bars are presented as mean ± standard deviation (S.D.) as indicated. Statistical significance was conducted using unpaired two-sided Student’s t-test for two-sample comparisons or one-way analysis of variance (ANOVA) test with multiple comparisons. Survival curves were generated according to the Kaplan-Meier method and compared using the log-rank test. The Spearman’s rank correlation test was performed to examine the correlations between the relative expression of hnRNPL and PIK3CB. All data were analyzed using GraphPad Prism 8.0 software or R version 4.3.0.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
RNA-seq and ChIP-seq data that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) database under the accession codes GSE289102, GSE289074 and GSE289075 as well as the Sequence Read Archive under the BioProject PRJNA988254, PRNA988255 [https://www.ncbi.nlm.nih.gov/bioproject/PRJNA988255/] and PRJNA1071467. The human cancer data were derived from GEO using the accession codes GSE143987, GSE30161 and GSE26712 as well as the TCGA Research [http://cancergenome.nih.gov/]. Survival analysis data were derived from the Kaplan-Meier Plotter database [https://kmplot.com/analysis/] and cBioPortal database [http://www.cbioportal.org/]. Unless otherwise stated, all data supporting the results of this study can be found in the article, supplementary, and source data files. Source data are provided with this paper.
References
Hentze, M. W., Castello, A., Schwarzl, T. & Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 19, 327–341 (2018).
Xiao, R. et al. Pervasive chromatin-rna binding protein interactions enable RNA-based regulation of transcription. Cell 178, 107–121.e118 (2019).
Van Nostrand, E. L. et al. A large-scale binding and functional map of human RNA-binding proteins. Nature 583, 711–719 (2020).
Fan, H. et al. The nuclear matrix protein HNRNPU maintains 3D genome architecture globally in mouse hepatocytes. Genome Res 28, 192–202 (2018).
Zhou, X. et al. Qki regulates myelinogenesis through Srebp2-dependent cholesterol biosynthesis. Elife 10, e60467 (2021).
Shin, S. et al. Qki activates Srebp2-mediated cholesterol biosynthesis for maintenance of eye lens transparency. Nat. Commun. 12, 3005 (2021).
Bi, X. et al. RNA Targets Ribogenesis Factor WDR43 to chromatin for transcription and pluripotency control. Mol. Cell 75, 102–116.e109 (2019).
Iborra, F. J., Pombo, A., Jackson, D. A. & Cook, P. R. Active RNA polymerases are localized within discrete transcription “factories’ in human nuclei. J. Cell Sci. 109, 1427–1436 (1996).
Sabari, B. R. et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958 (2018).
Boija, A. et al. Transcription factors activate genes through the phase-separation capacity of their activation domains. Cell 175, 1842–1855.e1816 (2018).
Nair, S. J. et al. Phase separation of ligand-activated enhancers licenses cooperative chromosomal enhancer assembly. Nat. Struct. Mol. Biol. 26, 193–203 (2019).
Icard, P. et al. How the Warburg effect supports aggressiveness and drug resistance of cancer cells?. Drug Resist Updat 38, 1–11 (2018).
Fresno Vara, J. A. et al. PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev. 30, 193–204 (2004).
Yan, B. X. & Ma, J. X. Promoter-associated RNAs and promoter-targeted RNAs. Cell Mol. Life Sci. 69, 2833–2842 (2012).
Ren, Y. et al. A global screening identifies chromatin-enriched RNA-binding proteins and the transcriptional regulatory activity of QKI5 during monocytic differentiation. Genome Biol. 22, 290 (2021).
Shao, W. et al. Phase separation of RNA-binding protein promotes polymerase binding and transcription. Nat. Chem. Biol. 18, 70–80 (2022).
Li, J., Chen, Y., Tiwari, M., Bansal, V. & Sen, G. L. Regulation of integrin and extracellular matrix genes by HNRNPL is necessary for epidermal renewal. PLoS Biol. 19, e3001378 (2021).
Wang, B. et al. Liquid-liquid phase separation in human health and diseases. Signal Transduct. Target Ther. 6, 290 (2021).
Gibson, B. A. et al. Organization of chromatin by intrinsic and regulated phase separation. Cell 179, 470–484.e421 (2019).
Banani, S. F., Lee, H. O., Hyman, A. A. & Rosen, M. K. Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 18, 285–298 (2017).
Nosella, M. L. et al. O-Linked-N-acetylglucosaminylation of the RNA-binding protein EWS N-Terminal low complexity region reduces phase separation and enhances condensate dynamics. J. Am. Chem. Soc. 143, 11520–11534 (2021).
Chen, Y. et al. O-GlcNAcylation determines the translational regulation and phase separation of YTHDF proteins. Nat. Cell Biol. 25, 1676–1690 (2023).
Yuzwa, S. A. et al. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat. Chem. Biol. 8, 393–399 (2012).
Gambetta, M. C. & Müller, J. O-GlcNAcylation prevents aggregation of the Polycomb group repressor polyhomeotic. Dev. Cell 31, 629–639 (2014).
Marotta, N. P. et al. O-GlcNAc modification blocks the aggregation and toxicity of the protein α-synuclein associated with Parkinson’s disease. Nat. Chem. 7, 913–920 (2015).
Mészáros, B., Erdos, G. & Dosztányi, Z. IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res 46, W329–w337 (2018).
Qamar, S. et al. FUS Phase Separation Is Modulated by a Molecular Chaperone and Methylation of Arginine Cation-π Interactions. Cell 173, 720–734.e715 (2018).
Li, P. et al. Phase transitions in the assembly of multivalent signalling proteins. Nature 483, 336–340 (2012).
Wang, Q. et al. CoBATCH for high-throughput single-cell epigenomic profiling. Mol. Cell 76, 206–216.e207 (2019).
Hui, J., Stangl, K., Lane, W. S. & Bindereif, A. HnRNP L stimulates splicing of the eNOS gene by binding to variable-length CA repeats. Nat. Struct. Biol. 10, 33–37 (2003).
Auerbach, K. G. Lactation educators/consultants. J. Am. Diet. Assoc. 86, 1740 (1986).
Gu, J., Chen, Z., Chen, X. & Wang, Z. Heterogeneous nuclear ribonucleoprotein (hnRNPL) in cancer. Clin. Chim. Acta 507, 286–294 (2020).
Geuens, T., Bouhy, D. & Timmerman, V. The hnRNP family: insights into their role in health and disease. Hum. Genet 135, 851–867 (2016).
Molliex, A. et al. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163, 123–133 (2015).
Hofweber, M. et al. Phase Separation of FUS Is Suppressed by Its Nuclear Import Receptor and Arginine Methylation. Cell 173, 706–719.e713 (2018).
Marenda, M., Lazarova, E. & Gilbert, N. The role of SAF-A/hnRNP U in regulating chromatin structure. Curr. Opin. Genet Dev. 72, 38–44 (2022).
Martin, E. W. et al. Interplay of folded domains and the disordered low-complexity domain in mediating hnRNPA1 phase separation. Nucleic Acids Res 49, 2931–2945 (2021).
Rhine, K. et al. ALS/FTLD-Linked Mutations in FUS glycine residues cause accelerated gelation and reduced interactions with wild-type FUS. Mol. Cell 80, 666–681.e668 (2020).
Conicella, A. E. et al. TDP-43 α-helical structure tunes liquid-liquid phase separation and function. Proc. Natl Acad. Sci. USA 117, 5883–5894 (2020).
Zhang, J. et al. Myristoylation-mediated phase separation of EZH2 compartmentalizes STAT3 to promote lung cancer growth. Cancer Lett. 516, 84–98 (2021).
Babu, M., Favretto, F., Rankovic, M. & Zweckstetter, M. Peptidyl prolyl isomerase a modulates the liquid-liquid phase separation of proline-rich IDPs. J. Am. Chem. Soc. 144, 16157–16163 (2022).
Zhao, Z. et al. Galectin-3 N-terminal tail prolines modulate cell activity and glycan-mediated oligomerization/phase separation. Proc. Natl Acad. Sci. USA 118, e2021074118 (2021).
Hallegger, M. et al. TDP-43 condensation properties specify its RNA-binding and regulatory repertoire. Cell 184, 4680–4696.e4622 (2021).
Martin, E. W. et al. Valence and patterning of aromatic residues determine the phase behavior of prion-like domains. Science 367, 694–699 (2020).
Falahati, H. & Wieschaus, E. Independent active and thermodynamic processes govern the nucleolus assembly in vivo. Proc. Natl Acad. Sci. USA 114, 1335–1340 (2017).
Jiang, H. et al. Phase transition of spindle-associated protein regulate spindle apparatus assembly. Cell 163, 108–122 (2015).
Ries, R. J. et al. m(6)A enhances the phase separation potential of mRNA. Nature 571, 424–428 (2019).
Woodruff, J. B. Phase separation of BuGZ promotes aurora A activation and spindle assembly. J. Cell Biol. 217, 9–10 (2018).
Zhao, Y. et al. MyoD induced enhancer RNA interacts with hnRNPL to activate target gene transcription during myogenic differentiation. Nat. Commun. 10, 5787 (2019).
Chang, M. W. et al. Enhanced myogenesis through lncFAM-mediated recruitment of HNRNPL to the MYBPC2 promoter. Nucleic Acids Res 50, 13026–13044 (2022).
Bauer, K. E., de Queiroz, B. R., Kiebler, M. A. & Besse, F. RNA granules in neuronal plasticity and disease. Trends Neurosci. 46, 525–538 (2023).
Asimi, V. et al. Hijacking of transcriptional condensates by endogenous retroviruses. Nat. Genet 54, 1238–1247 (2022).
Henninger, J. E. et al. RNA-mediated feedback control of transcriptional condensates. Cell 184, 207–225.e224 (2021).
Li, W. et al. Biophysical properties of AKAP95 protein condensates regulate splicing and tumorigenesis. Nat. Cell Biol. 22, 960–972 (2020).
Chang, C. et al. Intron Retention of DDX39A Driven by SNRPD2 is a crucial splicing axis for oncogenic MYC/spliceosome program in hepatocellular carcinoma. Adv. Sci. (Weinh.) 11, e2403387 (2024).
Lv, J. et al. HNRNPL induced circFAM13B increased bladder cancer immunotherapy sensitivity via inhibiting glycolysis through IGF2BP1/PKM2 pathway. J. Exp. Clin. Cancer Res 42, 41 (2023).
Wu, Q. et al. The m6A-induced lncRNA CASC8 promotes proliferation and chemoresistance via upregulation of hnRNPL in esophageal squamous cell carcinoma. Int J. Biol. Sci. 18, 4824–4836 (2022).
Wang, H. et al. lncRNA SNHG6 promotes hepatocellular carcinoma progression by interacting with HNRNPL/PTBP1 to facilitate SETD7/LZTFL1 mRNA destabilization. Cancer Lett. 520, 121–131 (2021).
Jiang, Z. & Jiang, Y. Circular RNA CircPDS5B impairs angiogenesis following ischemic stroke through its interaction with hnRNPL to inactivate VEGF-A. Neurobiol. Dis. 181, 106080 (2023).
Xu, P. et al. hnRNPL-activated circANKRD42 back-splicing and circANKRD42-mediated crosstalk of mechanical stiffness and biochemical signal in lung fibrosis. Mol. Ther. 30, 2370–2387 (2022).
Dang, C. V., Reddy, E. P., Shokat, K. M. & Soucek, L. Drugging the ‘undruggable’ cancer targets. Nat. Rev. Cancer 17, 502–508 (2017).
Enders, L. et al. Pharmacological perturbation of the phase-separating protein SMNDC1. Nat. Commun. 14, 4504 (2023).
Zhu, G. et al. Phase separation of disease-associated SHP2 mutants underlies MAPK Hyperactivation. Cell 183, 490–502.e418 (2020).
Wang, S. et al. Targeting liquid-liquid phase separation of SARS-CoV-2 nucleocapsid protein promotes innate antiviral immunity by elevating MAVS activity. Nat. Cell Biol. 23, 718–732 (2021).
Shi, Y. et al. BRD4-targeting PROTAC as a unique tool to study biomolecular condensates. Cell Discov. 9, 47 (2023).
Wang, Y. et al. Dissolution of oncofusion transcription factor condensates for cancer therapy. Nat. Chem. Biol. 19, 1223–1234 (2023).
Van Nostrand, E. L. et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods 13, 508–514 (2016).
Acknowledgements
This work was sponsored by the National Natural Science Foundation of China (82072886 to P.Y., 32101164 to Y.Y., and 82273156 to P.Y.), the Natural Science Foundation of Chongqing, China (CSTB2022NSCQ-MSX0897 to T.L. and CSTB2023NSCQ-MSX0338 to Y.Y.), Program for Youth Innovation in Future Medicine, Chongqing Medical University (W0058 to P.Y.), and Postdoctoral Natural Science Foundation of Chongqing Science and Technology Bureau (cstc2021jcyj-bshX0055 to Y.W.).
Author information
Authors and Affiliations
Contributions
P.Y. and T.L. conceived of and designed the experiments. P.Y., T.L. and W.W. supervised the study. F.Q (Fig. 1a, Supplementary Fig. 5d, and Supplementary Fig. 7f, i) and C.Y. (Fig. 1b–i, Fig. 2a–j, Fig. 6o–q, Supplementary Fig. 1e–p, Supplementary Fig. 2a–e, and Supplementary Fig. 4o–q) performed protein phase separation experiments in vivo and vitro. F.Q. (Fig. 3a–f, j–l, Fig. 7a–e, Supplementary Fig. 3a–f, Supplementary Fig. 4a, d, e, Supplementary Fig. 8 and Supplementary Fig. 9), Y.R. (Fig. 3g–i, m-o and Supplementary Fig. 4b, c), Y.Y. (Fig. 3p, q) conducted the cell proliferation and metastasis experiments. F.Q. (Fig. 5j, Fig. 7g–k and Supplementary Fig. 6f) and Y.W. (Fig. 5i and Fig. 7c, f) performed glycolysis experiments. Y.W. and H.W. performed ChIP-seq experiments. Y.W., F.Q., Y.R. and F.L. performed the luciferase, RIP, RNA FISH and ChIP experiments. Q.W. (Fig. 4a–h, k–m, Fig. 7m, n, Supplementary Fig. 5a, b, f-i and Supplementary Fig. 10h, i) and Jie X. (Fig. 5a, Supplementary Fig. 1a–c, Supplementary Fig. 6a and Supplementary Fig. 10c–g) analyzed the data of next-generation sequencing or public databases. Y.T. prepared the samples for RNA sequencing and conducted a part of cell proliferation and metastasis experiments. Q.L. (Supplementary Fig. 10i) performed the tissue microarray experiments. Xin L. (Supplementary Fig. 5e) conducted IP experiments. Xiaoyi L., X.Z., D.Y., W.W., Jing X., Y.Y. and C.C. helped analyze the data. T.L., F.Q., Y.W., and C.Y. wrote the draft manuscript. T.L. edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Bin Liu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Qin, F., Wang, Y., Yang, C. et al. hnRNPL phase separation activates PIK3CB transcription and promotes glycolysis in ovarian cancer. Nat Commun 16, 4828 (2025). https://doi.org/10.1038/s41467-025-60115-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-60115-7
