
Abstract
Acetate is the end product of alcohol metabolism. Acyl-CoA synthetase short-chain family member 2 (ACSS2) converts acetate to acetyl-CoA, involving metabolic pathways and epigenetic regulation. However, the function of ACSS2-mediated epigenetic control in alcoholic liver disease (ALD) remains incompletely understood. We demonstrate that alcohol downregulates hepatic ACSS2, causing acetate accumulation in the liver and serum. This disrupts iron metabolism and hepatic ferroptosis, triggering liver injury and inflammation. Mechanistically, ACSS2 binds CREB binding protein (CBP) to mediate histone acetylation and regulate hepcidin antimicrobial peptide 1/2 (HAMP1/2) transcription. ACSS2 deficiency downregulates HAMP1/2, causing systemic iron dyshomeostasis and ferroptosis, which is restored by overexpression of HAMP1/2. Iron chelators or ferroptosis inhibitors attenuates alcohol-induced liver injury in ACSS2-deficient mice. Our study uncovers the epigenetic mechanisms of ACSS2-mediated ferroptosis and its role in ALD progression.
Similar content being viewed by others

Introduction
Alcoholic liver disease (ALD) is a hepatic disease caused by alcohol abuse. Continuous alcohol abuse can produce alcoholic fatty liver, hepatitis, cirrhosis, and hepatocellular carcinoma1. The deleterious impacts of ethanol have been recognized, but the global level of alcohol consumption and incidence of ALD is rising2. Chronic alcohol abuse, alongside other risk factors, like smoking, diet, and physical inactivity, increases the risk of ALD3. However, no drug therapy for end-stage ALD has received approval, and liver transplantation is the only curative option4. Therefore, it is necessary to understand ALD pathogenesis, screen effective medicines and identify new therapeutic molecular targets.
Alcohol consumption has been demonstrated to increase circulating acetate5, but its mechanism of action remains unclear. Martino et al. demonstrated that acetate reprogrammed the gut microbiota during alcohol consumption, but targeting therapies for alcohol-related liver disease remains challenging6. Acyl-CoA synthetase short-chain family member 2 (ACSS2), critical Acyl-CoA synthetase short-chain (ACSS) family member, can convert acetate to acetyl coenzyme A (acetyl-CoA)7. Acetyl-CoA is an essential substrate for anabolic processes that is an acetyl group in epigenetic regulation. Any disruption in acetylation and deacetylation can contribute to transcriptional alteration, leading to various human diseases8. Many studies have reported that ACSS2-mediated acetylation regulation is related to various diseases, including tumors, fibrotic kidney disease, aging, neuropsychological disease relief, and memory management9,10,11,12,13. Ethanol is metabolized in hepatocytes and can be converted to acetate by aldehyde dehydrogenase4. However, ACSS2-mediated conversion of acetate to acetyl-CoA to ALD pathogenesis remains elusive and requires further investigation.
Iron is an essential trace element with crucial roles in various biological processes, including innate immunity, energy metabolism, nucleotide synthesis and DNA repair14. Iron metabolism disturbance is common in patients with chronic alcohol overconsumption15. Iron deficiency, with or without anemia, is associated with symptoms and complications with a dramatic negative influence on ALD patients16. Conversely, iron overload can cause liver injury by triggering oxidative stress, lipid peroxidation (LPO), and iron-dependent cell death. Ferroptosis is a new type of programmed cell death, triggered by excessive iron accumulation and LPO-induced damage17. Clinical studies have demonstrated that ALD patients exhibit diverse clinical manifestations associated with iron overload and ferroptosis18. Nevertheless, ferroptosis contributions to ALD are more extensive than our current understanding.
Hepcidin, a 25-amino-acid (aa) peptide hormone synthesized in hepatocytes, regulates iron absorption and distribution19. Many studies have revealed that acute and chronic alcohol exposure can disrupt systemic iron homeostasis by suppressing hepcidin mRNA and protein expression19,20,21. Hepcidin expression downregulation can increase intestinal iron absorption and iron release from its physiologic supplies by upregulating divalent metal transporter 1 (DMT1) and ferroprotein (FPN) expression22. A recent study determined that ethanol metabolism-related oxidative stress could inhibit hepcidin expression by suppressing CCAAT enhancer binding protein alpha (C/EBPα). Moreover, antioxidants like vitamin E and N-acetylcysteine mitigated hepcidin expression suppression by ethanol, suggesting that oxidative stress limits hepcidin expression following ethanol consumption23. Moreover, alcohol consumption can promote erythropoietin production24, which inhibits hepcidin expression by suppressing C/EBPα expression and the Smad1/5 signal pathway25. Therefore, targeting hepcidin-mediated iron homeostasis may be a target for preventing and treating ALD.
In this study, we explored the relationship between ACSS2 and hepcidin-mediated systemic iron homeostasis via multi-omics analysis. Genetic deletion of ACSS2 in hepatocytes aggravates ALD progress caused by alcohol consumption. In contrast, overexpression of hepatic ACSS2 elicited opposing effects. Mechanistic findings reveal that ACSS2 deficiency specifically results in reduced expression of hepcidin antimicrobial peptide 1/2 (HAMP1/2) via CBP-mediated histone acetylation, impairing systemic iron homeostasis. Our studies reveal a cross-link between ACSS2 and iron metabolism. This suggests an important role of ACSS2-mediated acetyl-CoA biosynthesis in epigenetically regulating iron homeostasis and hepatic ferroptosis, offering a target for ALD pharmacological intervention.
Results
ACSS2 is downregulated in ALD models
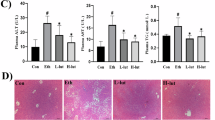
Alcohol consumption limits liver function and causes hepatocellular damage via ethanol metabolism-induced oxidative stress and inflammation. Ethanol undergoes oxidation to acetaldehyde and subsequently to acetate. Thereafter, acetate is converted into acetyl-CoA, a substrate for histone acetylation, lipid synthesis, and the tricarboxylic acid cycle26,27. We collected human blood samples from healthy subjects and ALD patients and measured serum acetate levels. The levels of acetate in the serum of ALD patients were significantly higher than healthy controls (Fig. 1a). Receiver operating characteristic curve analysis of acetate in healthy controls and ALD patients suggested that serum acetate were potential diagnostic markers of ALD (Supplementary Fig. 1a). As presented in Fig. 1b, we found that acetate levels in serum and liver were elevated in ethanol-fed (EF) mice compared to pair-fed (PF). The acetate metabolic state triggers the regulation of specific genes via ACSS1/2/3. To explore the association of ACSS1/2/3 and ALD, ACSS2/3 were found to be highly expressed in the liver tissue, while ACSS1 was nearly absent, as demonstrated by Human Protein Atlas database and western blot results (Supplementary Fig. 1b–d). Due to liver cell population heterogeneity, we analyzed single-cell sequencing data from public databases and detected the expression levels of ACSS1/2/3 in mouse primary hepatocytes (HEPs). These approaches demonstrated that ACSS2 was the most abundant in hepatocytes but not ACSS1/3 (Fig. 1c and Supplementary Fig. 1e, f). ACSS2 was abundantly expressed in primary hepatocytes but hardly detected expressed in Kupffer cells (KCs) and hepatic stellate cells (HSCs, Supplementary Fig. 1h). We compared the gene expression profiles of patients and an experimental animal model of ALD (GSE142530 and GSE137059) and determined that ACSS2 expression was decreased in the liver of ALD patients and an experimental animal model, but neither ACSS1 nor ACSS3 (Fig. 1d, e). We performed Mendelian Randomization (MR) analyses to demonstrate the potential causal relationship between single-nucleotide polymorphisms of the ACSS2 gene and ALD (Supplementary Fig. 1i). These results suggest that ACSS2 expression may be critical for ALD progression.

a Serum acetate levels in patients with ALD (n = 30 patients per group) and normal control (n = 10 samples per group). b Serum acetate levels in the mice with PF (n = 7 mice per group) and EF (n = 4 mice per group). Liver acetate levels in the mice with PF (n = 6 mice per group) and EF (n = 4 mice per group). c Single-cell RNA-seq analysis of ACSS2 levels in different liver cell types. d, e Heatmap indicating the DEGs in alcoholic hepatitis patients relative to normal controls and EF group relative to PF group from GEO database. f Representative images of the immunohistochemistry staining and quantitative analysis of ACSS2 in human liver sections from the normal (n = 6 samples per group), normal with alcohol history (n = 5 samples per group), alcohol-induced steatosis (n = 17 samples per group), and alcohol-induced steatohepatitis individuals (n = 18 samples per group). Scale bar: 1 mm. g Representative images of the immunofluorescence staining of ACSS2 (red), Alb (green), CD68 (green), α-SMA (green), and DAPI (blue) in the livers of mice with PF and EF (n = 4 mice per group). Immunofluorescence co-localization analysis was quantified respectively by ImageJ. Scale bars, 50 μm. h, i Western blot (n = 6 mice per group) and qPCR (n = 5 mice per group) analysis of relative ACSS2 protein and mRNA levels in the liver of mice with PF and EF. j, k Western blot ( j) and qPCR (k) analysis of relative ACSS2 protein and mRNA levels in MIHA cells exposed to EtOH of varied concentrations (n = 3 biological replicates per group). i–m Western blot analysis of relative ACSS1/2/3 protein levels in mouse HEPs exposed to EtOH of varied concentrations (n = 3 mice per group). m Relative Acss2 mRNA levels in mouse HEPs exposed to EtOH of varied concentrations were detected by qPCR analysis (n = 3 mice per group). Data are presented as the mean ± SD. The p-values were calculated by two-sided test and adjusted for p-values by the Benjamini–Hochberg method to control for false positives due to multiple comparisons (d, e), two-tailed Student’s t test (a–c, h, i) and one-way ANOVA (f, j–m). Source data are provided as a Source Data file.
To examine the association between ACSS2 expression and ALD, we performed immunohistochemistry staining for ACSS2 protein expression across 46 liver sections from individuals with normal controls (n = 6), normal controls but with alcohol history (n = 5), alcohol-induced steatosis (n = 17) and alcohol-induced steatohepatitis (n = 18). Statistical analysis of stained images indicated that hepatic ACSS2 was substantially downregulated in the patients of ALD compared to controls (Fig. 1f). We performed immunofluorescence and immunohistochemistry staining, western blot, and quantitative PCR (qPCR) assay to assess the expression alteration of ACSS2 EF mouse livers. Statistical analysis indicated that hepatic ACSS2 expression was significantly downregulated in EF mouse livers mice compared to PF, primarily in hepatocytes (Fig. 1g–i and Supplementary Fig. 2a). Non-parenchymal cell single-cell sequencing data from GSE166178 demonstrated that ACSS2 was not significantly altered in monocytes, T cells and endothelial cells in ALD mice compared to control mice (Supplementary Fig. 2b). Consistently, the protein and mRNA expression levels of ACSS2 were downregulated in a dose-dependent manner in ethanol (EtOH)-induced cells including MIHA, AML12, HepG2 and primary hepatocytes, while ACSS3 did not change significantly (Fig. 1j–m and Supplementary Fig. 2c–f). Interestingly, we observed a slight increase in ACSS1 expression following EtOH treatment, which may represent a compensatory response. To delineate the temporal dynamics of EtOH-induced ACSS2 suppression, we conducted in vitro experiments using AML12 cells treated with EtOH. After 24 h of EtOH exposure, the medium was replaced with EtOH-free medium, and ACSS2 levels were measured at 6, 12, 24, 48, and 72 h post-treatment. A time-dependent partial recovery of ACSS2 expression was observed starting 6 h after ethanol exposure, though expression levels remained significantly attenuated relative to controls at the 72 h (Supplementary Fig. 2g). Taken together, these data demonstrated that ACSS2 is decreased by alcohol.
Hepatocyte-specific ACSS2 deficiency exacerbates alcohol-induced liver injury and inflammation
To characterize the role of hepatic ACSS2 in ALD pathogenesis in vivo, we generated hepatocyte-specific Acss2-knockout mice (Acss2-HKO; Supplementary Fig. 3a). These mice were subjected EF for 10 days plus a binge (Fig. 2a). ACSS2 protein and mRNA levels were significantly reduced in the liver and primary hepatocytes of Acss2-HKO mice compared to Acss2-Flox mice (Supplementary Fig. 3b–e). After ethanol feeding, Acss2-HKO mice had a dramatically larger livers and increased ratios of liver-to-body weight as compared to Acss2-Flox group (Fig. 2b and Supplementary Fig. 3f). Moreover, serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were also increased in Acss2-HKO mice (Fig. 2c, d). H&E and Oil red O staining showed more severe liver injury and hepatic steatosis in Acss2-HKO mice than in Acss2-Flox mice. This was confirmed by an increase in hepatic triglyceride (TG), total cholesterol (TC), and free fatty acid (FFA, Fig. 2e–i).

a All female mice were adapted to the fluid diet by feeding the Lieber-DeCarli regular control diet for 5 consecutive days, and the model group were then fed with Lieber-DeCarli ethanol mice diet containing 5% (vol/vol) ethanol for 10 consecutive days and ethanol (5 g/kg) was administered by oral gavage on day 11 as shown in the diagram protocol was created with MedPeer (medpeer.cn). b Ratios of liver weight to body weight of Acss2-Flox and Acss2-HKO mice fed with an PF or EF diet (n = 8 mice per group). c, d Serum ALT (c) and AST (d) concentrations in the indicated groups of mice (n = 8 mice per group). e–g, Hepatic TG (e), TC (f), and FFA (g) content in the indicated groups of mice (n = 5 mice per group). h Representative H&E staining images in liver sections from the indicated groups of mice and quantification of relative vacuolar areas (n = 5 mice per group). Scale bar: 50 μm. i Representative Oil Red O staining images in liver sections from the indicated groups of mice and quantification of relative staining areas (n = 5 mice per group). Scale bar: 50 μm. j–m Serum levels of TNF-α ( j) and liver levels of TNF-α (k), IL-1β (l), and IL-6 (m) were detected by ELISA (n = 5 mice per group). n Relative mRNA levels of pro-inflammatory and chemokine molecules were detected by qPCR analysis in the liver of Acss2-Flox and Acss2-HKO mice fed with EF (n = 5 mice per group). o, p Representative images of the immunofluorescence staining and quantitative analysis of F4/80 (red) and CD68 (green) in the livers of mice from the indicated groups in which nuclei were stained with DAPI (blue, n = 5 mice per group). Scale bar: 50 μm. Data are presented as the mean ± SD. The p-values were calculated by two-way ANOVA. Source data are provided as a Source Data file.
Previous studies indicated that increased expression of proinflammatory cytokines and chemokines in ALD livers promote the recruitment and activation of different immune cells, including neutrophils and macrophages. To determine whether ACSS2 deficiency aggravates pro-inflammatory cytokine and chemokine induction in the livers of ALD, we performed qPCR, ELISA, immunofluorescence staining, and flow cytometry. We observed that ACSS2 deletion upregulated pro-inflammatory cytokines expression, including TNF-α, IL-1β, and IL-6, CXCL1, CXCL2, CCL2, CCL4, and CCR2, alongside intercellular and vascular cell adhesion molecules like ICAM-1 and VCAM-1 (Fig. 2j–n). Moreover, Acss2-HKO female mice livers exhibited a high percentage of infiltrated macrophages and neutrophils compared to Acss2-Flox female mice (Fig. 2o, p and Supplementary Fig. 3k–n). Taken together, these results suggested that hepatic ACSS2 deficiency exacerbates alcohol-induced liver injury and inflammation.
The levels of ethanol, acetaldehyde, acetate, and acetyl-CoA in serum and liver tissues were assessed using assay kits. ACSS2 deficiency did not affect the ethanol levels but significantly increased acetaldehyde and acetate levels (Supplementary Fig. 3g–i). Due to the absence of ACSS2, a critical enzyme for converting acetate to acetyl-CoA, the levels of acetyl-CoA were dramatically decreased in the liver and serum of Acss2-HKO mice (Supplementary Fig. 3j).
To further elucidate the specific effect by which ACSS2 contributes to ALD, we employed a 4-week chronic ethanol feeding model (Supplementary Fig. 4a). Our results demonstrated that, consistent with the short-term model, hepatocyte-specific ACSS2 deficiency significantly exacerbated liver injury in mice subjected to 4 weeks of ethanol feeding (Supplementary Fig. 4b–i).
Similar changes were observed in male mice. Compared to Acss2-Flox male mice, ACSS2 deletion induced significant liver-to-body weight ratio changes and liver injury in Acss2-HKO male mice fed with EF (Supplementary Fig. 5a–c). In addition, vacuoles and lipid accumulation in Acss2-HKO male mice livers were also significantly increased, determined by H&E staining, Oil red O staining, TG and TC quantification kits (Supplementary Fig. 5d–h). Since females are more sensitive to alcohol than males28, the latter experiments were all manipulated in females.
Administration of an ACSS2 inhibitor exacerbates alcohol-induced liver injury and inflammation
Having established that hepatic ACSS2 deficiency exacerbates alcohol-induced liver injury and inflammation, we then examined the effects of an ACSS2 inhibitor (ACSS2i) in mice ALD model (Supplementary Fig. 6a). As presented in Supplementary Fig. 6b–e, pharmacological inhibition of ACSS2 increased liver-to-body ratios, serum ALT, liver TG, and liver TC levels, indicating ACSS2 inhibition exacerbated alcohol-induced liver injury and hepatic steatosis. Moreover, treatment with ACSS2i aggravated alcohol-induced inflammation (Supplementary Fig. 6f–i). We found that ACSS2i also greatly promoted the production and secretion of inflammatory factors in the PF group (Supplementary Fig. 6f–h), potentially related to the association of ACSS2i with immune cell activation and infiltration29. Consistently, H&E and Oil red O staining also revealed that treatment with ACSS2i also promoted alcohol-induced hepatic steatosis (Supplementary Fig. 6j, k).
Hepatocyte-specific ACSS2 depletion aggravates alcohol-induced iron metabolism disorder
To assess the underlying mechanism by which hepatic ACSS2 deficiency exacerbated ALD, we conducted RNA sequencing (RNA-seq) to examine transcriptome alterations in Acss2-Flox/HKO mice with EF. Differentially expressed gene (DEG) analysis identified 607 differentially expressed genes, including 426 upregulated and 181 downregulated genes (Supplementary Fig. 7a). The Gene Ontology (GO) enrichment analysis of DEGs showed high clustering in iron metabolism, including iron ion transport and iron ion homeostasis (Fig. 3a), closely related to liver injury and inflammation. Importantly, we observed that iron metabolism-related genes like Hamp1/2 were significantly decreased as shown in the volcano map and heat map (Fig. 3b, c). Most of bodily iron comes from the heme recycling after phagocytosis of senescent red cells by macrophages, while portion comes from food iron absorption by the small intestine through DMT1. Iron is transported across the basement membrane of intestinal epithelial cells through FPN, released into the blood, and oxidized to Fe3+, which binds to transferrin (TF), and reaches the liver through the portal system. This bound iron is taken up by hepatocytes and stored as ferritin via the transferrin receptor (TFRC, Fig. 3d)30. HAMP1/2 encodes hepcidin, a peptide hormone synthesized and secreted by the liver, negatively regulating iron homeostasis by binding to FPN and promoting its internalization and degradation31.

a GO enrichment analysis of gene networks associated with biological processes in the liver of Flox-EF and HKO-EF mice. b Volcano plot of the differentially up-(red) or down-(bule) regulated genes between Flox-EF and HKO-EF groups. c Heatmap of DEGs related to iron metabolism in the liver of Flox-EF and HKO-EF mice (n = 3 mice per group). d Schematic of hepcidin-mediated systemic iron homeostasis was created with MedPeer (medpeer.cn). e Relative mRNA levels of genes related to iron metabolism in the liver of Flox-EF and HKO-EF mice were detected by qPCR analysis (n = 5 mice per group). f Western blot analysis of relative FPN protein levels in the liver of Flox-EF and HKO-EF mice (n = 3 mice per group). g–j Representative images of the immunohistochemistry staining and quantitative analysis of HAMP1/2 (g, n = 3 mice per group) in liver sections and FPN in the liver (h, n = 3 mice per group), duodenum (i, n = 5 mice per group) and spleen ( j, n = 5 mice per group) of the indicated groups of mice. Scale bar: 50 μm. k–m Representative images of DAB enhanced Prussian Blue staining and quantitative analysis of iron content in the liver (k), duodenum (l), and spleen (m) of the indicated groups of mice (n = 5 mice per group). Scale bar: 50 μm. n–q Levels of hepcidin and iron content in serum and liver of the indicated groups of mice (n = 5 mice per group). r, s Serum hepcidin (r) and iron (s) levels in the patients with ALD (n = 30 patients per group) and normal control (n = 10 samples per group). t–v, Correlation analyses were conducted between serum acetate and iron levels (t), acetate and hepcidin levels (u), as well as iron and hepcidin levels (v) in human samples (n = 40 samples). Data are presented as the mean ± SD. The p-values were calculated by two-sided test and adjusted for p-values by the Benjamini–Hochberg method to control for false positives due to multiple comparisons (b). The p-values were calculated by two-tailed Student’s t test (e, f, r, s), two-way ANOVA (g–q), and Spearman correlation analysis (t–v). Source data are provided as a Source Data file.
Many studies have shown that iron homeostasis dysregulation occurs in ALD32. Ethanol exposure can downregulate the hepcidin mRNA and protein levels to promote iron overload in hepatocytes20,23,33. The qPCR and immunohistochemistry staining results suggested that HAMP1/2 mRNA and protein levels were evidently decreased in the livers of Acss2-HKO mice compared to the Acss2-Flox mice fed with EF (Fig. 3e–g). Similarly, ACSS2 knockout mice on a control diet exhibited reduced hepatic HAMP1/2 expression compared to Acss2-Flox littermates (Supplementary Fig. 7c). We evaluated the association of ACSS2 and HAMP1/2 in a larger cohort (GSE137059). We identified that the mRNA level of Acss2 was positively correlated with Hamp2 expression (Supplementary Fig. 7b). Several other genes involved in iron metabolism (e.g., Pcbp1, Tfrc, Noca4, Scl39a14, and Trpml2) showed significant differences after ACSS2 depletion, resulting in aggravating the alcohol-induced iron disruption (Fig. 3e). Interestingly, hepatic ACSS2 deficiency had no effect on the mRNA level of Fpn (Fig. 3e), but significantly increased its protein level in the liver, spleen and duodenum (Fig. 3f and h–j). We observed that ACSS2 deficiency induced iron homeostasis disruption, reflected by reduced serum and liver hepcidin levels, increased serum and liver iron levels, decreased spleen and duodenum iron levels, and increased liver TFRC and TF expression levels (Fig. 3k–q and Supplementary Fig. 7d, e). As characterized in recent reviews, ferroptosis is regulated by iron metabolism, the GSH system, the BH4/CoQ10 system, and transcription factors30. To validate our findings, we employed qPCR to assess mRNA levels of other ferroptosis-related modulators in mouse livers. The levels of other ferroptosis-related modulators did not significantly change following ACSS2 knockout aside from arachidonate 5-Lipoxygenase (Alox5, Supplementary Fig. 7f). The increase in Alox5 in HKO-EF mice may be due to elevated LPO, activating lipoxygenase Alox5 by the peroxidation products of polyunsaturated fatty acids. Furthermore, the heatmap results indicate that ACSS2 deficiency significantly promotes hepatic inflammation infiltration in EtOH-fed mice, whereas no significant changes were observed in mice on a normal diet (Supplementary Fig. 7g, h).
Similarly, in-vitro experiments using siRNA-mediated ACSS2 knockdown in AML12 cells and primary hepatocytes revealed a significant reduction in Hamp1/2 mRNA levels, but no significant changes in protein levels (Supplementary Fig. 8a–d). This discrepancy may be attributed to the fact that mRNA undergoes various regulatory processes after transcription, such as splicing, editing, degradation, and stability regulation. These processes can affect mRNA stability and translation efficiency, thereby leading to less pronounced changes in protein levels.
To further determine the association between ACSS2-mediated ethanol metabolism and iron homeostasis in ALD patients, we examined the levels of hepcidin and iron in human serum. We observed that serum hepcidin significantly decreased, while iron dramatically increased in ALD patients (Fig. 3r, s). Acetate levels were positively correlated with iron levels and negatively correlated with hepcidin levels in human samples (Fig. 3t, u). Iron levels were also negatively correlated with hepcidin levels in human samples (Fig. 3v). These findings suggested that ACSS2 was significantly correlated with disrupted iron homeostasis.
Hepatocyte-specific ACSS2 deficiency exacerbates alcohol-induced ferroptosis
Above results have demonstrated that hepatic ACSS2 deficiency disrupts iron homeostasis, which may lead to hepatic iron deposition. Iron-mediated Fenton reaction and enzymatic oxygenation leading to LPO and toxic ROS, which are hallmarks of ferroptosis34. Therefore, we evaluated the antioxidant properties of ACSS2 depletion. The results showed that ACSS2 deficiency in female mice decreased glutathione (GSH) and superoxide dismutase (SOD) levels and increased ROS enrichment (Fig. 4a–d) in the liver. Under conditions rich in iron and ROS, polyunsaturated-fatty-acid–containing phospholipids in the cell membrane is prone to peroxidation and lipid peroxides formation, which accumulate in the cell membrane to destroy the membrane and induce cell death35. We investigated the oxidized lipid profile in the liver by targeted metabolomics (Fig. 4e). As presented in Fig. 4f, the oxidized lipid profile was significantly altered in Flox-EF and HKO-EF mice. Levels of metabolites including ±8-HDoHE, ±13-HDoHE, 13S-HODE, 9R-HETE, PGE1, and PGE2 were significantly increased in Acss2-HKO mice (Fig. 4g). We further demonstrated that ACSS2 deletion promoted LPO detected by C11-BODIPY staining (Fig. 4h) and significantly increased the lipid peroxides levels, including malondialdehyde (MDA), and 4-hydroxynonenal (4-HNE, Fig. 4i–k). To evaluate the effects of ACSS2 deficiency on mitochondrial ultrastructure, we employed transmission electron tomography to analyze the three-dimensional structure of mitochondria. As presented in Fig. 4l, we observed more severe mitochondrial atrophy, and the mitochondrial cell membrane was broken severely in the liver of Acss2-HKO mice. ACSS2 deficiency decreased hepcidin levels and increased iron levels in serum and liver, while also elevating 4-HNE, C11-BODIPY and LPO levels and decreased GSH levels in the liver of Acss2-HKO male mice (Supplementary Fig. 9a–h).

a–c Hepatic GSH (a), SOD (b), and ROS (c) content in mice of the indicated groups (n = 5 mice per group). d Representative images of DHE staining and quantitative analysis of ROS in mouse liver of the indicated groups (n = 5 mice per group). Scale bar: 50 μm. e Schematic representation of mouse liver subjected to metabolomics analysis was created with MedPeer (medpeer.cn). f Heatmap showing the expression of metabolites involved in oxidized lipids (n = 3 mice per group). g Levels of ±8-HDoHE, ±13-HDoHE, 13S-HODE, 9R-HETE, PGE1, and PGE2 in the liver of Flox-EF and HKO-EF mice (n = 3 mice per group). h Representative images of BODIPY 581/591 C11 staining and quantitative analysis of lipid peroxidation in the liver of the indicated groups of mice (n = 3 mice per group). Scale bar: 50 μm. i Representative images of immunohistochemistry staining and quantitative analysis of 4-HNE in the liver of the indicated groups of mice (n = 5 mice per group). Scale bar: 50 μm. j Hepatic MDA content in the indicated groups of mice (n = 5 mice per group). k Western blot analysis of relative 4-HNE levels in the liver of Flox-EF and HKO-EF mice (n = 6 mice per group). l Representative images showed by electron microscopy in the liver of Flox-EF and HKO-EF mice Scale bars: 2 μm (low-magnification images) and 0.5 μm (high-magnification images). Yellow arrows pointed to atrophic mitochondria. Red arrows indicated mitochondrial membrane disruption. m Cell death of AML12 cells transfected with Acss2 small interfering RNA (si-Acss2) or the scramble control (si-NC) and treated with EtOH (400 mM) was visualized by PI staining. n Proportion of dead cells was detected by flow cytometry in AML12 cells transfected with si-Acss2 or si-NC and treated with EtOH (400 mM, n = 3 biological replicates per group). o Relative cell viability was measured by CCK-8 assay in AML12 cells transfected with si-Acss2 or si-NC and treated with EtOH (400 mM, n = 8 biological replicates). Data are presented as the mean ± SD. The p-values were calculated by two-tailed Student’s t test (d, g–i, k) and two-way ANOVA (a–c, j, n, o). Source data are provided as a Source Data file.
Subsequently, to investigate cell death, we initially constructed a co-culture model of cells exposed to EtOH as presented in Supplementary Fig. 8e. We employed propidium Iodide (PI) staining by flow cytometry and immunofluorescence to detect dead cells and utilized the CCK-8 assay to measure cell viability. The gating strategy of PI+ cells was illustrated in Supplementary Fig. 8g. To establish the relationship between cell death and EtOH exposure, we treated AML12 cells exposed to EtOH with ferroptosis inhibitors (Fer-1, Lip-1, DFO), necroptosis inhibitor (Nec-1), and apoptosis inhibitor (Z-VAD), respectively. These inhibitors significantly reduced EtOH-induced cell death (Supplementary Fig. 8f). Moreover, our results also revealed that ACSS2 deficiency significantly promoted hepatocyte death (Fig. 4m–o). Taken together, our findings demonstrated that ACSS2 deficiency exacerbates alcohol-induced ferroptosis.
Administration of an ACSS2 inhibitor exacerbates alcohol-induced disorder of iron homeostasis and ferroptosis
Pharmacological inhibition of ACSS2 decreased HAMP1/2 expression levels and increased the iron levels in the serum and liver of mice fed EtOH (Supplementary Fig. 10a–f). Immunohistochemistry staining showed that the protein levels of TFRC and TF were induced in mice with EF and ACSS2i (Supplementary Fig. 10g, h). In addition, treatment administered ACSS2i dramatically promoted the alcohol-induced ferroptosis, as evidenced by lower GSH levels and higher LPO, 4-HNE, and ROS levels (Supplementary Fig. 10i–l). Taken together, these data suggested that pharmacological inhibition of ACSS2 exacerbates alcohol-induced ferroptosis.
ACSS2 overexpression in hepatocytes alleviates alcohol-induced liver injury and inflammation
Given that ethanol treatment decreases hepatic ACSS2 expression and hepatic ACSS2 deficiency exacerbates alcohol-induced liver injury, adeno-associated virus (AAV) carrying the Acss2 gene was injected to restore ACSS2 expression in hepatocytes (Fig. 5a). Transfection efficiency of the AAV-Acss2 was determined by western blot (Fig. 5b). We examined the effects of AAV-Acss2 transduction on liver injury, inflammation, and hepatic steatosis in ALD mouse models. As presented in Fig. 5c–h, ACSS2 gain-of-function considerably reduced liver-to-body ratios, serum ALT and AST levels, and liver TG and TC levels in EF mice relative to control. Moreover, ACSS2 overexpression significantly alleviated alcohol-induced liver injury, hepatic steatosis, and inflammation (Fig. 5i–o). These results were consistent when ACSS2 was overexpressed in Acss2-HKO mice. After ACSS2 supplementation, induced liver injury, steatosis, and inflammation caused by ACSS2 deficiency were dramatically reduced, and more significant than in Acss2-Flox mice (Fig. 5c–o).

a ACSS2 overexpression in hepatocytes were achieved by AAV-Acss2 administration. AAV-Ctrl was used as control. After 2 weeks, all mice were adapted to the fluid diet by feeding the Lieber-DeCarli regular control diet for 5 consecutive days, and the model group were then fed with Lieber-DeCarli ethanol mice diet containing 5% (vol/vol) ethanol for 10 consecutive days and ethanol (5 g/kg) was administered by oral gavage on day 11 as shown in the diagram protocol was created with MedPeer (medpeer.cn). b The efficacy of AAV-Acss2 was determined by western blot in the liver of mice injected with AAV-Acss2 and AAV-Ctrl (n = 6 mice per group). c Ratios of liver weight to body weight in Acss2-Flox and Acss2-HKO mice transduced with AAV-Acss2 or AAV-Ctrl and fed with PF or EF (n = 5 mice per group). d–g Serum ALT (d) and AST (e) concentrations, and hepatic TG (f) and TC (g) content in the indicated groups of mice (n = 5 mice per group). h Representative images of whole liver tissue from Acss2-Flox and Acss2-HKO mice transduced with AAV-Acss2 or AAV-Ctrl. i Representative H&E staining images in liver sections from the indicated groups of mice and quantification of relative vacuolar areas (n = 5 mice per group). Scale bar: 50 μm. j Representative Oil Red O staining images of liver sections from the indicated groups of mice and quantification of relative staining areas (n = 5 mice per group). Scale bar: 50 μm. k Representative images of the immunofluorescence staining and quantitative analysis of levels of CD68 in the livers of mice from the indicated groups in which nuclei were stained with DAPI (n = 5 mice per group). Scale bar: 50 μm. l–o Relative mRNA levels of F4/80 and Cd68 (l, m, n = 5 mice per group), Il-1β (n, n = 3 mice per group) and Vcam-1 (q, n = 3 mice per group) were detected by qPCR analysis in the livers of mice from the indicated groups. Data are presented as the mean ± SD. The p-values were calculated by two-tailed Student’s t-test. Source data are provided as a Source Data file.
ACSS2 overexpression in hepatocytes alleviates alcohol-induced disorder of iron homeostasis and ferroptosis
To determine whether ACSS2 overexpression mitigated the alcohol-induced disorder of iron homeostasis and ferroptosis, we evaluated the effects of AAV-Acss2 transduction on iron distribution, ferroptosis, and associated gene expression. We demonstrated that the mRNA and protein levels of HAMP1/2 were increased in ACSS2 overexpressed Flox and HKO mice (Supplementary Fig. 11a, b), but ACSS2 overexpression had no effect on the level of acetaldehyde in ACSS2 Flox and HKO mice (Supplementary Fig. 11c). Moreover, increasing ACSS2 could restore hepcidin and iron contents in the serum and liver (Supplementary Fig. 11d–g), decreasing the FPN protein levels in the liver (Supplementary Fig. 11h). This alleviated alcohol-induced ferroptosis as evidenced by higher GSH levels, and lower LPO, MDA, 4-HNE and ROS levels (Supplementary Fig. 11i–n). These findings suggested that ACSS2 expression levels negatively correlate with alcohol-induced ferroptosis.
ACSS2 regulates HAMP1/2 transcription via CBP-mediated histone acetylation
In mammalian cells, ACSS2 uses intracellular acetate to synthesize acetyl-CoA, a donor of acetyl groups for histone acetylation that drives gene expression via epigenetic regulation. ACSS2 may regulate HAMP1/2 expression through histone acetylation. To test this, we examined acetyl-CoA and histone acetylation levels in Acss2-Flox and HKO mice fed with EF. As illustrated in Supplementary Fig. 3j, a dramatic reduction of acetyl-CoA in the serum and liver of Acss2-HKO mice was observed. Moreover, we performed western blot to determine that ACSS2 deletion significantly reduced the acetylation levels of histone 3 (H3) and histone 4 (H4), including H3K9ac, H3K14ac, H3K18ac, H3K27ac, H4K5ac, H4K8ac, and H4K12ac (Fig. 6a, b).

a, b Western blot analysis of histones H3 and H4 related acetylated proteins in the AML12 cells transfected with si-Acss2 or si-NC (n = 3 biological replicates per group). c Western blot analysis of Co-IP of the Flag-ACSS2 with HA-CBP in the AML12 cells transfected with the vector of HA-CBP and Flag-ACSS2. d Western blot analysis of Co-IP of the Flag-ACSS2 with endogenous CBP in the AML12 cells transfected with the vector of Flag-ACSS2. e, f Western blot analysis of Co-IP of the endogenous ACSS2 with endogenous CBP in the AML12 cells. g Immunofluorescence staining and analysis of colocalization of CBP and ACSS2 in AML12 cells. h Schematic of ChIP-qPCR assay procedure was created with MedPeer (medpeer.cn). i, j ChIP-qPCR assays of the enrichment of CBP, H3K9ac and H4K8ac at Hamp1 and Hamp2 promoters in the AML12 cells transfected with si-Acss2 or si-NC (n = 3 biological replicates per group). IgG was used as the negative control and H3 was used as the positive control. k Schematic of the effect of ACSS2 and CBP complex on regulation of Hamp1/2. Data are presented as the mean ± SD. The p-values were calculated by two-tailed Student’s t-test. Source data are provided as a Source Data file.
Previous studies have demonstrated that ACSS2 is recruited with CBP to the promoter region of genes, supporting histone acetylation and gene expression. To determine whether ACSS2 directly binds to CBP in altering HAMP1/2 expression, we co-transfected the vector carrying Flag-ACSS2 and HA-CBP to AML12 cells. Co-immunoprecipitation (Co-IP) assays suggested that ACSS2 binds to CBP (Fig. 6c, d). Similarly, the Co-IP assay using an endogenous protein of AML12 cells confirmed binding between ACSS2 and CBP (Fig. 6e, f). Additionally, an immunofluorescence assay showed the co-localization of ACSS2 and CBP in AML12, primary hepatocytes and MIHA cells (Fig. 6g and Supplementary Fig. 12a, b). However, ACSS2 could not bind to other acetyltransferases, including lysine acetyltransferase 2B (PCAF) and lysine acetyltransferase 7 (KAT7, Supplementary Fig. 12c, d).
To explore the underlying mechanism by which the ACSS2-CBP complex regulates HAMP1/2 expression, we performed ChIP-qPCR using a specific CBP antibody to determine the binding of ACSS2-CBP complex on the promoter region of HAMP1/2 (Fig. 6h). ACSS2 bound to the −1781 to −1700 region of the HAMP1 promoter and the −1850 to −1724 region of the HAMP2 promoter (Fig. 6i, j). We carried out ChIP-qPCR using H3K9ac and H4K8ac antibodies. As illustrated in Fig. 6i, j, H3K9ac and H4K8ac were enriched in the HAMP1/2 promoter region. However, in AML12 cells with ACSS2 knockdown, the binding of CBP, H3K9ac, and H4K8ac was significantly reduced (Fig. 6i, j). These results suggested that ACSS2 combines with CBP coregulated acetylation at the HAMP1/2 promoter (Fig. 6k).
HAMP1/2 overexpression in hepatocytes alleviates alcohol-induced liver injury and inflammation in Acss2-HKO mice
To determine the contribution of HAMP1/2 in ACSS2 deficiency-exacerbated alcohol-induced liver injury, we increased HAMP1/2 content in hepatocytes by transducing mice with AAV encoding Hamp1/2, whose expression was controlled by the thyroxine-binding globulin promoter (Fig. 7a). We first measured the level of hepcidin in serum to determine the success of overexpression (Fig. 7b). As illustrated in Fig. 7c–f, HAMP1/2 overexpression significantly reduced the high liver-to-body ratio, serum ALT, hepatic TG, and TC levels in Acss2-HKO mice fed with EF. In addition, ACSS2 deficiency-exacerbated liver injury, steatosis, inflammation and ferroptosis were significantly reduced in Acss2-HKO mice injected with AAV-Hamp1/2 compared to Acss2-HKO mice injected with AAV-Ctrl (Fig. 7g–o). Notably, ACSS2 knockout in EF induced acetaldehyde accumulation, enabling the progression of alcohol-induced liver injury. To rule out the impact of acetaldehyde, overexpression of Hamp1/2 in Acss2-HKO mice did not influence the acetaldehyde levels in serum and liver (Fig. 7p, q). However, it significantly alleviated liver injury, steatosis, inflammation, and ferroptosis, suggesting that ACSS2 deficiency may particularly aggravate liver injury through HAMP1/2 mediated iron homeostasis.

a HAMP1/2 overexpression in hepatocytes was achieved by AAV-Hamp1/2 administration. AAV-Ctrl was used as control. After 2 weeks, all mice were adapted to the fluid diet by feeding the Lieber-DeCarli regular control diet for 5 consecutive days, and the model group were then fed with Lieber-DeCarli ethanol mice diet containing 5% (vol/vol) ethanol for 10 consecutive days and ethanol (5 g/kg) was administered by oral gavage on day 11 as shown in the diagram protocol was created with MedPeer (medpeer.cn). b Hepcidin levels in serum of Acss2-Flox and Acss2-HKO mice transduced with AAV-Hamp1/2 or AAV-Ctrl with EF diet (n = 5 mice per group). c Ratios of liver weight to body weight of the indicated groups of mice (n = 5 mice per group). d–f Serum ALT (d) concentrations, hepatic TG (e) and TC (f) content of the indicated groups of mice (n = 5 mice per group). g, h Representative H&E staining (g) and Oil Red O staining (h) images in liver sections from the indicated groups of mice and quantification of damaged area (n = 5 mice per group). Scale bar: 50 μm. i, j Representative images of the immunofluorescence staining and quantitative analysis of CD68 (i) and F4/80 ( j) in the liver from the indicated groups of mice in which nuclei were stained with DAPI (n = 5 mice per group). Scale bar: 50 μm. k Representative images of BODIPY 581/591 C11 staining (k), immunohistochemistry staining for 4-HNE (l), and DHE staining (m) in the liver from the indicated groups of mice (n = 5 mice per group). Scale bar: 50 μm. Quantitative analysis was also provided for each staining. n, o Hepatic MDA (n) and GSH (o) content in mice of the indicated groups (n = 5 mice per group). p, q Acetaldehyde levels in the serum and liver of the indicated groups of mice (n = 5 mice per group). Data are presented as the mean ± SD. The p-value was calculated by two-way ANOVA. Source data are provided as a Source Data file.
Administration of ferrostatin-1 and recombinant hepcidin ameliorates alcohol-induced liver injury in Acss2-HKO mice
To demonstrate that ACSS2 deficiency exacerbates alcohol-induced liver injury by modulating ferroptosis, we treated EF-fed Acss2-Flox/HKO mice with ferrostatin-1 (Fer-1) and recombinant hepcidin, two common ferroptosis inhibitors36 (Supplementary Fig. 13a). We observed that liver steatosis, inflammation, and ferroptosis were significantly reduced in Acss2-Flox/HKO mice following treatment with Fer-1 and hepcidin as evidenced by liver-body ratio, serum ALT and liver TG levels, H&E and Oil red O staining, liver CD68 and F4/80 immunofluorescence staining, liver C11-BODIPY, 4-HNE, and DHE staining and hepatic MDA and GSH levels (Supplementary Fig. 13b–n). Similarly, treatment with Fer-1 and hepcidin in Acss2-Flox/HKO mice did not change acetaldehyde levels in the serum and liver (Supplementary Fig. 13o, p), but significantly attenuated liver injury in Acss2-HKO mice fed with EF. To demonstrate that ACSS2 deficiency promotes cell death by modulating ferroptosis, we treated ACSS2-knockdown AML12 cells with ferroptosis inhibitors (Fer-1, Lip-1, DFO), a necroptosis inhibitor (Nec-1), and an apoptosis inhibitor (Z-VAD). Only the ferroptosis inhibitors (Fer-1, Lip-1, DFO) could rescue the exacerbated cell death caused by ACSS2 knockdown in EtOH-exposed cells. In contrast, the necroptosis inhibitor (Nec-1) and the apoptosis inhibitor (Z-VAD) failed to exert such a protective effect (Supplementary Fig. 14), suggesting that ACSS2 deficiency may aggravate alcohol-induced liver injury by modulating ferroptosis. Additionally, A systematic dose-response curve analysis demonstrated that ACSS2 knockdown lowered the threshold for EtOH-induced cell death (Supplementary Fig. 15a–c). Cell viability was selectively restored by ferroptosis inhibitors, but not by inhibitors targeting necroptosis and apoptosis (Supplementary Fig. 15e). Furthermore, in control cells, ferroptosis inhibitors exhibited significantly greater protection against ethanol-induced cell death compared to necroptosis and apoptosis inhibitors (Supplementary Fig. 15d). These findings demonstrate that ACSS2 specifically modulates ferroptosis, thereby influencing alcohol-induced injury.
Discussion
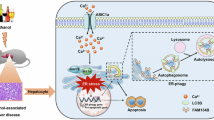
ACSS2, as a key molecule in acetate metabolism, can catalyze acetate to acetyl-CoA conversion, is considered an important intermediate metabolite in energy substrates metabolism and donates an acetyl group in epigenetic regulation. In this study, we identified ACSS2 as a potential negative regulator of ALD, independent of its typical role as an enzyme in fat synthesis. In vivo studies demonstrated that hepatocyte-specific deletion of ACSS2 and pharmacological inhibition of ACSS2 aggravated alcohol-induced liver injury and inflammation. Conversely, hepatocyte ACSS2 gain-of-function in mice with ALD elicited opposing effects. Mechanistically, ACSS2-mediated biosynthesis of acetyl-CoA from alcohol-derived acetate contribute to CBP-modulated histone acetylation and HAMP1/2 expression, regulating systemic iron homeostasis and inhibiting ferroptosis (Fig. 8).

ACSS2 interacts with the histone acetyltransferase CBP on the HAMP1/2 promoter, promoting histone acetylation and enhancing hepcidin transcription. In mice with ALD, downregulation of hepatic ACSS2 impairs this process, leading to reduced hepcidin secretion and disrupted iron homeostasis. Consequently, iron overload occurs in hepatocytes, promoting ferroptosis. The figure was created with MedPeer (medpeer.cn).
The global incidence of alcohol-associated disease has increased recently, especially in young people and women. Despite significant investments and research efforts, there are no approved therapies to treat alcohol-associated diseases beyond steroid. Iron overload is a hallmark of ALD32. Excessive alcohol can downregulate hepcidin expression and upregulate TFRC expression to promote iron overload in hepatocytes, causing ferroptosis33,37. However, direct supplementation of hepcidin may affect other aspects of systemic iron metabolism, such as inducing anemia or interfering with iron utilization in other organs19. Moreover, hepcidin expression is regulated by multiple signaling pathways, and direct intervention may face complex physiological and pathological mechanisms. Our study revealed that hepatic ACSS2 can regulate hepcidin expression through CBP-dependent histone acetylation, thereby alleviating iron overload and improving alcoholic liver injury. This finding expands the role of ACSS2 to the inhibition of iron homeostasis disorders and ferroptosis, holding promise as a potential therapeutic approach to address hepcidin deficiency in ALD. Compared with direct modulation of hepcidin, targeting ACSS2 may represent a more effective upstream intervention, as it can indirectly improve iron overload by influencing the transcriptional regulation of hepcidin. Future research can further explore its potential applications in clinical therapy.
ACSS2 has a predominantly lipogenic function, but increasing evidence suggests it also acts as a transcriptional enzyme in epigenetic regulation. Recent studies have demonstrated that ACSS2 promotes whole-body fat deposition through selective regulation of genes associated with lipid metabolism38, whereas deletion of ACSS2 can inhibit de novo fat synthesis to enhance renal function in multiple mouse models of kidney disease39. This appears to contradict our results. Our findings suggested that hepatocyte-specific ACSS2 knockout aggravated fat accumulation in the liver of mice with ALD, potentially due to ferroptosis and hepatocyte injury aggravation, leading to liver lipid metabolism disorders and thus fat deposition. A recent cell death and differentiation study from Dr. Zhang demonstrated that ACSS2 improves HFD-induced obesity in mice by promoting adipose tissue thermogenesis via SIRT1 regulation of PPARγ activity11. Both contradictory roles of ACSS2 in lipogenic functions may rely on subcellular localization in the cytosol and nucleus. The two well-established routes of nuclear-cytosolic acetyl-CoA pools are employed for lipid synthesis and histone acetylation. Cytosolic ACSS2 has long been associated with lipid synthesis by providing acetyl-CoA, a primary anabolic substrate. Nuclear ACSS2 can form complexes with transcription activators on the promoter of diverse gene sets to locally regulate histone modification, including acetylation and crotonylation, and activate gene expression. For instance, ACSS2 regulates not only hippocampal memory and cognition in a mouse model of Alzheimer’s disease by modulating histone acetylation12, as well as renal fibrosis by modulating histone crotonylation impacting macrophage activation and renal tubular cell senescence10. Intriguingly, our study demonstrated that ACSS2 is primarily localized in the nucleus and cytoplasm of hepatocytes in Pair-fed mice. Conversely, in the livers of EtOH-fed mice, the reduction in ACSS2 levels is largely due to the depletion of nuclear ACSS2 in hepatocytes. This suggests that ACSS2-mediated histone acetylation may be compromised. Consequently, we characterized the direct downstream target gene of ACSS2, HAMP1/2, by RNA-seq and confirmed its involvement in CBP-mediated histone acetylation at the HAMP1/2 promoter. The altered localization of ACSS2 is significant for the dependence of ACSS2 on histone acetylation to regulate gene expression. Interestingly, our study results indicate that the deletion of ACSS2 does not affect liver phenotype or ferroptosis in pair-fed mice, which may be attributed to compensatory effects. Furthermore, ACSS2 deficiency leads to reduced mRNA levels of Hamp1/2 under both alcohol-free and alcohol-containing conditions. The protein levels of HAMP1/2 are decreased only in the presence of alcohol by ACSS2 deficiency. This may be because ethanol affects the post-translational modification of HAMP1/2. Increased histone deacetylase HDAC activity reduced histone acetylation and inhibited hepcidin expression in hepatitis C40, which indicated that the level of hepcidin was affected by histone acetylation. In addition, HAMP1/2 overexpression and supplementation with hepcidin or Fer-1 ameliorated ferroptosis as well as liver injury in Acss2-HKO mice fed EtOH. ACSS2 may be a precise target for eliminating ethanol stress-induced dysregulation of iron homeostasis and ferroptosis.
It should be noted that ACSS2, an enzyme that can metabolize acetate into acetyl-CoA, may also play a crucial role in ethanol metabolism. Our findings suggested that deletion of ACSS2 induces acetaldehyde accumulation in the livers of mice with ALD, potentially due to the accumulation of acetate slowing acetaldehyde metabolism. Acetaldehyde, a toxic metabolite, is important in the pathogenesis of alcoholic liver injury. It has been shown that pharmacological activation of aldehyde dehydrogenase 2 reduces hepatic acetaldehyde levels and improves alcohol-induced hepatic steatosis in mice41. Conversely, Gao et al. demonstrated that aldehyde dehydrogenase 2 deficiency ameliorates ALD but increases hepatic acetaldehyde levels and aggravates liver inflammation and fibrosis in mice42. 4-methylpyrazole, an alcohol dehydrogenase inhibitor, can effectively inhibit the metabolism of ethanol to acetaldehyde43. However, it has also been reported to cause liver injury following low doses of 4-methylpyrazole in chronic alcoholic rats44. These contradictory views indicate that simply activating or inhibiting acetaldehyde dehydrogenase to reduce acetaldehyde production does not have a good therapeutic effect on alcohol-induced damage, and other potential acetaldehyde-independent mechanisms should be of interest. To determine whether ACSS2 protects against ALD not only by reducing acetaldehyde levels but also by modulating HAMP1/2-mediated ferroptosis, we identified no significant change in acetaldehyde content but still significant improvement in alcohol-induced liver injury in mice transduced with AAV-Hamp1/2 or supplemented with hepcidin and Fer-1. Moreover, alcohol induces high levels of acetate in the body5,45, while the role of acetate in ALD is not completely clear. We demonstrated that blockade of acetate metabolism by knocking out ACSS2 in the liver contributed to ALD progression. However, ACSS2 is generally considered as a target for cancer therapy and promotes the occurrence and development of tumors by regulating cell proliferation, gene expression, and energy metabolism46,47,48,49. For example, Miller et al. demonstrated that acetate enhanced tumor immunity, whereas ACSS2 inhibitors significantly improved chemotherapy efficacy in tumor mice29. This suggests that future anti-tumor or anti-ALD drug targets targeting ACSS2 should consider possible side effects in different diseases.
Our study revealed that ethanol-induced hepcidin downregulation was associated with ACSS2 depletion, which relied on CBP-mediated histone acetylation at the HAMP1/2 promoter. This study did not investigate the underlying mechanism of downregulation of ACSS2 in ALD. In addition, although our study demonstrated that overexpression of ACSS2 could alleviate alcohol-induced liver injury, the pharmacological activation of ACSS2 and its function was not investigated due to the absence of ACSS2 agonists.
In conclusion, our study demonstrated that hepatic ACSS2 inhibited ferroptosis by regulating iron metabolism and significantly ameliorated alcohol-induced liver injury. This study provides new insights into the role of ACSS2 in regulating systemic iron homeostasis in ALD and offers a potential therapeutic approach for the treatment of ALD.
Methods
Human samples
Human tissue of liver steatosis/steatohepatitis with a history of alcohol use (Lot No.2410095 and Lot No.2210241) were obtained from Xeno Tech LLC, which totally include 35 liver alcohol-associated hepatic steatosis/steatohepatitis samples and 11 control samples. Human serum samples were obtained from the remaining serum of subjects after physical examination in the Physical examination Center of the Second Affiliated Hospital of Chongqing Medical University. Among them, 30 patients were clinically diagnosed as ALD, and 10 healthy subjects were included. The clinical diagnostic criteria for ALD are as follows: 1. Long-term alcohol consumption history: Typically exceeding 5 years, with an ethanol intake of ≥40 g/day for men and ≥20 g/day for women; or a history of heavy drinking (> 80 g/day) within the past 2 weeks. 2. Elevated serum biomarkers: Increased levels of AST, ALT, gamma-glutamyl transferase (GGT), total bilirubin, prothrombin time, and mean corpuscular volume (MCV). An AST/ALT ratio > 2, elevated GGT, and increased MCV were observed. 3. Clinical symptoms: Symptoms are often non-specific, including asymptomatic presentations, or manifestations such as right upper quadrant distension, loss of appetite, fatigue, weight loss, and jaundice. Severe cases may present with spider angiomas, palmar erythema, and delirium. 4. Liver imaging techniques: a. Ultrasound diagnosis: Diffuse fatty liver was diagnosed if two of the following three abdominal ultrasound findings were present: (1) diffuse enhancement of near-field liver echo, stronger than that of the kidney; (2) gradual attenuation of far-field liver echo; and (3) unclear intrahepatic duct structures. b. CT diagnosis: Diffuse reduction in liver density, with a liver/spleen CT ratio ≤ 1.0. The liver/spleen CT ratio was used to assess the severity of ALD: mild (0.7 <ratio ≤ 1.0), moderate (0.5 <ratio ≤ 0.7), and severe (ratio ≤ 0.5). 5. Exclusion of other etiologies: Other causes of liver injury, such as active hepatotropic virus infection, drug-induced liver injury, toxic liver damage, and autoimmune liver diseases, were excluded. All procedures involving human specimen collection and application were approved by the Medical Ethics Committee of Hefei University of Technology (permission number: HFUT20240408001) following the principles of the Declaration of Helsinki. Written informed consent was obtained from the human participants or their family members. The clinical characteristics of participants are provided in Supplementary Tables 1 and 2.
Mice
Female/male mice aged 8 to 10 weeks were used for all experiments. Mice were housed in a specific pathogen-free environment with a 12-h light/dark cycle at 22 ± 2 °C. All animals handling procedures were approved by the Biomedical Research Ethics Committee of Hefei University of Technology (permission number: HFUT20220304008) and Chongqing Medical University (permission number: IACUC-CQMU-2023-0438). Hepatocyte-specific Acss2 deletion (Acss2-HKO) mice were generated using CRISPR/Cas9 technology. Briefly, CRISPR/Cas9 system and Donor were microinjected into the fertilized eggs of C57BL/6JGpt mice. Fertilized eggs were transplanted to obtain positive F0 mice which were confirmed by PCR and sequencing. A stable F1 generation mouse model (Acss2flox/flox)) was obtained by mating positive F0 generation mice with C57BL/6JGpt mice. The Acss2flox/flox mice mate with the Albumin-Cre mice (purchased from Jackson Laboratory, USA) and got the Acss2-HKO; Acss2flox/flox (Acss2-Flox) mice were used as a control group. Primers F1/R1 and F2/R2 were designed to amplify DNA fragments from the left and right loxp and floxed target regions, respectively. We extracted mouse genomic DNA from the toe of mice to detect whether loxp and cre were inserted simultaneously by PCR amplification. The primers used are listed in Supplementary Table 3.
Alcoholic liver disease (ALD) model
A short-term chronic ethanol feeding model was established in female/male mice referring to previous studies50. Initially, all mice were adapted to the fluid diet by feeding the Lieber-DeCarli regular control diet for 5 consecutive days. The model group were then fed with Lieber-DeCarli ethanol mice diet containing 5% (vol/vol) ethanol for 10 consecutive days. On day 11, ethanol (5 g/kg) was administered by gavage. The control group was given isocaloric Lieber-DeCarli regular control diet and gavage of isocaloric maltose dextrin solution. To establish a 4-week chronic ethanol feeding model, mice were fed a liquid diet containing ethanol for 4 weeks. Mice were sacrificed 9 h after the gavage with ethanol or maltose dextrin. Euthanasia was performed by controlled CO2 asphyxiation. Subsequently, livers and serum were collected and stored in liquid nitrogen.
ACSS2 inhibitor administration
10-week-old wild type C57BL/6J female mice were divided into PF, PF+ACSS2i, EF, EF+ACSS2i groups. All mice were acclimated to the Lieber-DeCarli regular control diet for 5 days as described above. The PF+ACSS2i and EF+ACSS2i groups were intraperitoneally injected with 15 mg/kg ACSS inhibitor daily during modeling51, while other mice were injected with the same volume of vehicle daily. ACSS2i was dissolved in a vehicle containing 5% DMSO, 40% PEG300, 5% Tween-80, and 50% sterilized distilled water. Mice were sacrificed 9 h after a gavage with ethanol or maltose dextrin, serum and liver tissues were collected for further testing.
Generation of hepatocyte-specific ACSS2 or Hamp1/2 overexpression mice
To achieve hepatocyte-specific ACSS2 or Hamp1/2 overexpression, recombinant AAV serotype 8 gene transfer vector carrying hepatocyte-specific promoter (TBG) and mouse Acss2 or Hamp1/2 sequence (AAV8-Acss2, AAV8-Hamp1/2) was injected into the tail vein of C57BL/6J mice. Mice injected with AAV8-TBG-null vector served as controls (AAV8-Ctrl). AAV injections were performed 2 weeks before the initiation of ethanol feeding.
Generation of recombinant hepcidin-2
Recombinant hepcidin-2 was synthesized by Tsingke Biotech (Beijing, China). Briefly, the signal peptide was excised and the range of 24–83 amino acids was expressed. The target fragment was subcloned into pET-28a-sumo vector by 5′ BamHI-3′ Hindlll. The recombinant vector was transformed into Escherichia coli BL21-DE3, and the recombinant fusion protein was induced by IPTG and identified. The recombinant fusion protein was purified by Ni2+ resin column and digested with sumo protease. His tag was purified by affinity chromatography, and the obtained samples were dialyzed and concentrated for detection and identification. The sequence (with tags) is as follows: MGSSHHHHHHSSGLVPRGSHMASMSDSEVNQEAKPEVKPEVKPETHINLKVSDGSSEIFFKIKKTTPLRRLMEAFAKRQGKEMDSLRFLYDGIRIQADQTPEDLDMEDNDIIEAHREQIGGSMTYLQQQMRQTTELQPLHGEESRADIAIPMQKRRKRDINFPICRFCCQCCNKPSCGICCEEWSHPQFEK.
Ferrostatin-1 and recombinant hepcidin-2 administration
10-week-old Acss2-Flox and Acss2-HKO females were adapted to the Lieber-DeCarli regular control diet for 5 days. The mice were then fed the Lieber-DeCarli ethanol diet and given daily intraperitoneal injection of either Fer-1 (10 mg/kg bodyweight)52 or recombinant hepcidin-253 (2 mg/kg bodyweight). The other mice received the same volume of vehicle daily. Fer-1 was dissolved in a solvent containing 5% DMSO, 40% PEG300, 5% Tween-80, and 50% sterilized distilled water. Recombinant hepcidin-2 was diluted in phosphate-buffered saline (PBS). Mice were sacrificed 9 h after a gavage with ethanol or maltodextrin, and serum and liver tissues were collected for further detection.
Mouse primary hepatocyte, Kupffer cell, and Stellate cell isolation/culture
Primary hepatocytes were isolated from mice. In short, after the mice were anesthetized, mice were perfused through the inferior vena cava with buffer A (Calcium and magnesium-free PBS containing 0.2 μM EGTA) followed by buffer B (30 mg of collagenase IV was dissolved powder in 50 mL of PBS containing calcium and magnesium), when the color of the liver changed from bright red to beige. And then, the liver was excised into ice-cold PBS and torn apart, shook the damaged liver cells to flow out, passed the suspension through a 70 mm cell filter and centrifuged at 100 × g for 5 min at 4 °C. Cell precipitations were resuspended with 36% percoll, and centrifuged at a differential speed of 800 × g for 10 min at 4 °C. The supernatant was discarded and the precipitate (HEPs) was resuspended in high sugar culture medium Dulbecco’s modified Eagle’s Medium (DMEM) containing 10% FBS and 1% penicillin-streptomycin. Inoculated on a cell culture dish and the culture medium was replaced after 2 h. The supernatant was centrifuged at 1400 × g for 15 min at 4 °C and resuspended using 15 mL of 25% Percoll solution. After centrifuging at 1400 × g for 20 min, three discrete layers were established, the top layer were HSCs and the middle layer was KCs. The cells from two layers were collected in separate tubes and Isolated cells were resuspended in PBS and centrifuged to remove the remaining percoll. HSCs were incubated in high sugar culture medium DMEM containing 20% FBS and 1% streptomycin penicillin, and KCs were incubated in high sugar culture medium DMEM containing 10% FBS and 1% streptomycin penicillin.
Cell lines
MIHA cells (Immortalized human hepatic cell line), HepG2 cells (human hepatocellular carcinoma cell line), HEK293T cells (human embryonic kidney cell line) and RAW264.7 cells (mouse monocytic macrophage cell line) were purchased from American Type Culture Collection and grown in DMEM. AML12 cell (Immortalized mouse hepatic cell line) was purchased from Procell and grown in AML12 Cell Complete Medium. MODE-K cells (mouse intestinal epithelial cell line) were purchased from Otwo Biotech and grown in RPMI-1640. All the cells maintained at 37 °C in a 5% CO2 humidified atmosphere and were tested and found to be mycoplasma-free. All culture media were supplemented with 10% fetal bovine serum (FBS) and 100 U/mL penicillin/streptomycin. In a 6-well Transwell system, AML12 cells were added to the upper chamber, while RAW264.7 cells and half of the cell slides coated MODE-K cells were placed in the lower chamber. After co-culturing in medium containing 400 mM EtOH for 2 days, AML12 cells were collected.
Bioinformatics analysis
We obtained RNA-seq datasets (GSE137059, GSE142530, GSE115469, and GSE166178) from the Gene Expression Omnibus (GEO) database with detailed information provided in the files titled “Supplementary Data files for the open RNA-seq databases information” and “Additional Supplementary Files Description”. To analyze differential gene expression, we used the limma package (version 3.26.8) in the R Bioconductor project. According to the criteria of log2|FoldChange| > 1 and an adjusted p value < 0.05, the significant DEGs were determined. The data were normalized and processed using the Seurat package for cell clustering and dimensionality reduction. CellMarkers2.0 system was employed to annotate the clusters, and UMAP was then used to visualize the cellular populations.
Mendelian randomization
We conducted MR analysis through the following steps: (1) Selection of instrumental variables: genetic variants significantly associated with the expression levels of ACSS2 were extracted from publicly available eQTL datasets. Specifically, we utilized the eQTL data for ACSS2 (available at https://gwas.mrcieu.ac.uk/datasets/eqtl-a-ENSG00000131069/), ensuring that the instrumental variables satisfied the three core assumptions: relevance, independence, and exclusion restriction. (2) Acquisition of outcome data: genetic variant data associated with outcome variables (ALD) were extracted from the FinnGen database. Specifically, liver disease data were sourced from the finn-b-K11_LIVER entry in the FinnGen database. The extracted data included SNP beta coefficients and their standard errors. (3) Data integration and analysis methods: using a standard two-sample MR analysis framework, data on exposure variables (ACSS2) and outcome variables (ALD) were integrated. Five methods were employed for MR analysis: MR-Egger, Weighted Median, Inverse Variance Weighting, Simple Mode, Weighted Mode. All methods were implemented using the TwoSampleMR R package. Multiple methods were used to validate the robustness of the results and assess the reliability of the inferred causal relationship. Heterogeneity tests were conducted to evaluate potential unmeasured pleiotropy between instrumental variables and outcomes. MR-Egger was used to estimate and correct for directional pleiotropy that could bias the results.
Small-interfering RNA transfection
Si-RNA targeting ACSS2 was sequenced as follows (5′-3′): sense-GGAGCAUUGUGGACAAAUATT; antisense- UAUUUGUCCACAAUGCUCCTT. When the cells were inoculated until the density reached 50%, the cells were transferred to fresh medium without antibiotics but supplemented with 10% FBS. si-NC/si-Acss2 was transfected into cells using Lipofectamine™ RNAiMAX transfection reagent. After 6 h, the medium was replaced with complete medium, and cells were cultured for 24 to 36 h before subsequent experiments.
Plasmid transfection
The FLAG-PCAF (#8941), FLAG-KAT7 (#86266) and HA-CBP (#16701) plasmids were obtained from Addgene. The FLAG-ACSS2 plasmid was obtained from Zhimin Liu’s laboratory (The University of Texas MD). Take a 15-cm cell culture dish as an example. A sterile 1.5 mL EP tube was taken and 500 μL of Opti-MEM™ serum-free medium was added, followed by 15 μg of the target plasmid and left at room temperature for 5 min. In another sterile 1.5 mL EP tube, 500 μL Opti-MEM™ and 15 μL Liposomal 2000 Transfection Reagent were added and left at room temperature for 5 min. Then the above two tubes were mixed and left at room temperature for 10 min. Eight milliliters Opti-MEM™ serum-free medium was added to the dishes, and the entire mixture in the EP tubes described above was added and replaced with complete medium after 10–12 h.
Histological analyses
Mouse liver tissues were collected, fixed in 4% paraformaldehyde for 24 h, embedded in paraffin, and sectioned. Paraffin sections were stained with hematoxylin and eosin (H&E) to detect liver morphological changes. Mouse liver tissues were dehydrated with 30% sucrose and cut into frozen sections, which were stained with Oil red O according to previous studies54 to detect lipid droplet aggregation. First, wash the sections with PBS. Stain the sections with a freshly prepared 0.5% Oil Red O solution in isopropanol for 15–30 min at room temperature. After staining, rinse the sections with 60% isopropanol to remove any unbound stain, followed by a thorough wash with PBS. Finally, counterstain the nuclei with hematoxylin for 1–2 min, rinse with water, and mount the sections with a coverslip using the glycerin gelatin. All sections were observed using a ZEISS Scope A1 microscope (Carl Zeiss AG, Oberkochen, Germany).
Immunohistochemistry
Paraffin sections were dewaxed in gradient, followed by antigen repair, membrane rupture with 0.1% Triton X-100 if the target protein was a membrane protein (optional step), and sections were treated with the enhanced endogenous peroxidase blocking buffer for 10 min at room temperature and blocked with blocking solution with 10% goat serum (reagent A, KGC3201-300, KeyGEN BioTECH, China) for 30 min at room temperature. The sections were incubated with primary antibodies at 4 °C overnight. The cells were washed 3 times with PBS for 2 min each time. Sections were added with one drop of booster (reagent B, KGC3201-300, KeyGEN BioTECH, China) and incubated for 30 min at room temperature. They were washed 3 times with PBS for 2 min each time. Sections were incubated with HRP polymer (reagent C, KGC3201-300, KeyGEN BioTECH, China) for 30 min at room temperature and washed twice with PBS for 2 min each. One drop of the above DAB color solution (reagent D, E, F, KGC3201-300, KeyGEN BioTECH, China) was added to the section, and the color was developed for 2 to 5 min at room temperature (the degree of color development could be mastered under the microscope). Rinse with tap water for 2 min; Sections were counterstained with hematoxylin, rinsed with tap water, and returned to blue with PBS. Sections were dried by dehydration with gradient alcohol, transparent with xylene, and sealed with neutral balsam. All sections were observed using a ZEISS Scop e A1 microscope (Carl Zeiss AG, Oberkochen, Germany).
Immunofluorescence
After dewaxing and hydration, the sections were immersed in 1 × sodium citrate antigen repair solution at 100 °C for 30 min. The cells cultured on 8-well chamber were washed with cold 1 × PBS and fixed with 4% paraformaldehyde for 20 min, the sections or cells were treated with 0.1% triton X-100 at room temperature for 20 min and blocked with 3% BSA at room temperature for 60 min. The samples were incubated with primary antibody overnight at 4 °C. After washing 3 times with PBS, the samples were incubated with fluorescent secondary antibody at room temperature for 120 min. Images were captured with confocal laser microscopy (Carl Zeiss AG, Oberkochen, Germany).
Flow cytometry
The appropriate amount of tissue was ground and filtered through a 200-mesh screen to obtain a single cell suspension. Erythrocytes in the cell suspension were lysed. Blood obtained in anticoagulated tubes was added proportionally to red blood cell lysate to remove red blood cells. The cells were then counted, and the corresponding antibodies were added and incubated on ice for 30 min before detection by flow cytometry. As a negative control, cells were stained with isotype-matched control antibodies. CytExpert 2.5 and Flowjo 10.0.7 softwares were used for analysis.
Transmission electron microscopy
The liver samples were fixed overnight at 4 °C in 2.5% glutaraldehyde solution, and then the samples were processed as described below. The fixative was decanted, and the samples were rinsed three times for 15 min each with 0.1 M, pH 7.0 phosphate buffer. The samples were fixed with 1% osmic acid solution for 1–2 h, the waste osmic acid solution was carefully removed, and the samples were rinsed three times for 15 min each with 0.1 M, pH 7.0 phosphate buffer. The samples were dehydrated with gradient concentrations of ethanol solution (including 30%, 50%, 70%, 80%, 90%, and 95%) for 15 min, and then treated with 100% ethanol for 20 min. The samples were treated with the mixture of embedding agent and acetone (VV-11) for 1 h, the mixture of embedding agent and acetone (VV-31) for 3 h, and the samples were treated with pure embedding agent overnight. Embedded samples were obtained by embedding the osmotically treated samples and heating them overnight at 70 °C. The samples were sectioned in LEICA EM UC7 ultramicrotome to obtain 70–90 nm sections. The sections were stained with lead citrate solution and dioxygen acetate solution for 5 min and then dried before being observed under microscope.
Mouse serum assay
The blood samples were allowed to stand at room temperature for at least 2 h, followed by centrifugation at 2000 × g for 20 min, and the serum in the supernatant was removed and stored at −80 °C. The levels of ALT and AST in serum were detected by automatic biochemical analyzer (Hitachi High-tech Corporation, Tokyo, Japan). Enzyme linked immunosorbent assay (ELISA) was used to measure the levels of serum TNF-α (CSB-E04741m, CUSABIO, https://www.cusabio.com/), IL-1β (CSB-E04639m, CUSABIO, https://www.cusabio.com/), IL-6 (SYP-M1543, Ruixinbio), hepcidin (E-EL-M0671, Elabscience) and acetyl-CoA (JM-11689M2, JINGMEI BIOTECHNOLOGY). Serum ethanol (RXWB0248, Ruixinbio), acetaldehyde (RXWB0251, Ruixinbio) and acetate (EOAC-100, BioAssay Systems) levels were determined by biochemical reagent kits according to the manufacturer’s protocol.
Mouse tissue assay
TNF-α, IL-1β, IL-6, hepcidin and acetyl-CoA contents in tissues were measured by ELISA. The contents of TG (KTB2200, Abbkine), TC (KTB2220, Abbkine), FFA (RXWB0836, Ruixinbio), MDA (S0131SB, Beyotime), GSH (A006-2-1, Nanjing Jiancheng Bioengineering Institute), LPO (A106-1-3, Nanjing Jiancheng Bioengineering Institute), SOD (RXWB0477, Ruixinbio), ROS (HR8835, Ruixinbio), ethanol, acetaldehyde and acetate in the tissues were determined by biochemical reagent kits according to the manufacturer’s instructions.
Measurements of serum iron and tissue non-heme iron
Serum iron was determined by a total iron colorimetric assay kit (E-BC-K772-M, Elabscience Biotechnology Co.,Ltd, China) according to the manufacturer’s instructions. Tissue iron content was determined by a nonheme iron detection kit (TC1000, LEAGENE, China) according to the manufacturer’s instructions.
DAB enhanced Prussian blue staining
Prussian Blue Iron Stain Kit (G1429) was obtained from Beijing Solarbio Science & Technology Co., Ltd. According to the manufacturer’s instructions, paraffin sections were deparaffinized and rehydrated routinely. Prussian blue working liquid was added to the sections and completely covered with tissue and incubated at 37 °C for 20 min in a wet box. After washing 3 times for 1 min in PBS, enhanced working liquid was added to the sections and completely covered with tissue and incubated at 37 °C for 20 min in a wet box. The sections were washed 3 times for 5 s and then incubated with the redyeing solution for 5 min. At the end, the sections were immersed in distilled water for 10 min, dehydrated with gradient ethanol, transparent with xylene, and sealed with neutral balsam. All sections were observed using a ZEISS Scope A1 microscope (Carl Zeiss AG, Oberkochen, Germany).
Propidium Iodide (PI) staining
Cells are harvested and washed with phosphate-PBS to remove any residual culture medium. The cell suspension is then resuspended in a binding buffer. The cell suspension is incubated with PI solution (typically 1–10 µM) for 15–30 min in the dark. The binding buffer is added to ensure optimal staining conditions. PI penetrates cells with damaged membranes, binding to DNA and emitting red fluorescence, which can be detected using a fluorescence microscope.
Cell Counting Kit-8 (CCK-8) assay
Cells were seed in a 96-well plate at the desired density and allow them to adhere overnight. After treatment, add 10 μL of CCK-8 solution to each well containing 100 µL of culture medium. Incubate the plate for 1 h at 37 °C. The absorbance was measured at 450 nm using a microplate reader.
RNA extraction and quantitative real-time PCR
Total RNA was extracted from liver samples or cultured cells used total RNA isolation reagent and reverse transcribed by HiScript II 1st Strand cDNA Synthesis Kit following the manufacturer’s protocols. AceQ qPCR SYBR Green Master Mix was used to detect corresponding PCR products, the CT value of the target gene was normalized. Primers used are listed in Supplementary Table 4 and all mRNAs were normalized to the level of β-actin expression.
Co-immunoprecipitation (Co-IP)
Cells were washed in ice-cold PBS for 3 times, and collected and lysed by RIPA buffer with protease inhibitors (PMSF and cocktail) on ice. 1 mg of protein were carried out for Co-IP using indicated primary antibodies (ACSS2, CBP) or Pierce™ Anti-DYKDDDDK Magnetic Agarose or Pierce™ Anti-HA Magnetic Beads, and then the immune complexes were washed for 3–5 times with RIPA buffer and measured by western blot.
DHE and BODIPY 581/591 C11 staining
Fresh tissues were removed and snap-frozen in liquid nitrogen into a freezing microtomy for sectioning. To detect the ROS levels in tissue, the sections were incubated with DHE (50102ES02, Yeasen, Shanghai, China) at 37 °C for 20 min. Intracellular LPO was assessed using BODIPY 581/591 C11 (HY-D1301, MedChemExpress, Monmouth Junction, NJ, USA) staining for 30 min, then washed twice with PBS. Confocal microscopy (Carl Zeiss AG, Oberkochen, Germany) was used to capture the images.
Chromatin immunoprecipitation (ChIP)-qPCR
ChIP assays were performed in AML12 cells using the SimpleChIP® Plus Enzymatic Chromatin IP Kit. Briefly, AML12 cells were cross-linked in 1% formaldehyde in DMEM medium for 10 min at room temperature. The nuclei were isolated, micrococcal nuclease was added and sonicated to digest the chromatin. Then the soluble chromatin was immunoprecipitated with IgG, H3 or indicated antibodies (CBP, H3K9ac, H4K8ac) incubated at 4 °C for 4 h to overnight with rotation. Protein G magnetic beads were added to each IP reaction and incubated for 2 h with rotation at 4 °C. After washing 4 times with washing buffer. Chromatin was eluted from antibody/protein G magnetic beads for 30 min at 65 °C with gentle vortexing. Reverse cross-linking was performed by adding 5 M NaCl and proteinase K and incubated for 2 h at 65 °C. Purified DNA. Quantitative PCR was performed using the primers listed in Supplementary Table 5.
Western blotting
Tissue or cell samples were lysed in RIPA buffer supplemented with protease inhibitors (PMSF and cocktail). A BCA protein assay kit was used to determine protein concentration. Protein was separated on SDS-PAGE gel and transferred to 0.45 μm NC membranes. Membranes were sealed at room temperature with 5% skim milk for 90 min and incubated with primary antibodies overnight at 4 °C. After washing three times with PBST, the membranes were incubated with secondary antibodies at room temperature for 90 min. Images were captured by automatic chemiluminescence gel imager (OI600, BIO-OI, Guangyi, China). Antibodies list can be found in Supplementary Table 6.
RNA-seq and data processing
Total RNA was extracted using total RNA isolation reagent according to the manufacturer’s instructions. RNA purity and quantification were identified using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA), and RNA integrity was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Transcriptome libraries were constructed using the VAHTS Universal V6 RNA-seq Library Prep kit according to the manufacturer’s instructions. Transcriptome sequencing was performed by Shanghai OE Biotech Co., Ltd. (Shanghai, China). Illumina Novaseq 6000 sequencing platform was used to sequence the library, and DESeq2 software was used to analyze the DEGs. Genes that met the threshold of p value < 0.05 and log2|foldchange| > 1 were defined as DEGs. The raw RNA-seq data generated in this study have been deposited in the GEO database under accession number GSE267638. To analyze the functions of (DEGs), we employed GO enrichment and Kyoto Encyclopedia of Genes and Genomes pathway analysis. In our statistical analysis, a pathway was statistically significant when the p-value was less than 0.05.
Oxidative Lipidomics assay by UHPLC-MS/MS
About 50 mg liver sample was weighed, then 600 μL extraction solution (80% methanol water, pre-cooled at −40 °C, containing isotope labeled internal standard mixture) was added and vortexed for 30 s. The samples were ground for 4 min at 35 Hz and sonicated for 5 min under an ice water bath. The grinding ultrasound step was repeated twice, and the samples were left at −40 °C for 1 h. After the samples were centrifuged at 13,000 × g for 15 min at 4 °C, 480 μL of supernatant was taken, 320 μL of ultrapure water was added, vortexed and mixed for 30 s, and then purified by Solid-Phase Extraction. Then the eluent was evaporated to dryness under a gentle stream of nitrogen and were reconstituted in 100 μL of 30% ACN/H2O (v/v). The reconstituted solution was vortexed for 30 s and homogenized at 60 Hz for 4 min and sonicated for 5 min in the ice-water bath. After centrifugation (1 min, 13,000 × g, and 4 °C), the reconstituted solution was transferred to an EP tube with a filter membrane. After centrifugation (15 min, 13,000 × g, and 4 °C), the clear supernatant was subjected to UHPLC-MS/MS analysis. The UHPLC separation was carried out using an ACQUITY Premier (Waters), equipped with a Waters ACQUITY UPLC BEH C18 column (150 × 2.1 mm, 1.7 μm, Waters). The mobile phase A was 0.01% formic acid in water, and the mobile phase B was 0.01% formic acid in acetonitrile. The column temperature was set at 50 °C. The auto-sampler temperature was set at 4 °C and the injection volume was 10 μL. A SCIEX Triple Quad™ 6500+ mass spectrometer (Sciex), equipped with an IonDrive Turbo V electrospray ionization interface, was applied for assay development. Typical ion source parameters were: Curtain Gas = 40 psi, IonSpray Voltage = −4500 V, temperature = 500 °C, Ion Source Gas 1 = 30 psi, Ion Source Gas 2 = 30 psi. SCIEX Analyst WorkStation Software (Version 1.6.3) and Multiquant 3.03 software were employed for MRM data acquisition and processing. The data that support the findings of this study have been deposited into CNGB Sequence Archive55 of China National GeneBank DataBase56 with accession number CNP0006975.
Reagents
The catalogs and sources of all reagents used in this study are given in Supplementary Table 6.
Statistical analysis
The data analysis was conducted by a technician who was blind to the experimental design. GraphPad Prism 9 was used for statistical analysis. Data are expressed as the mean ± SD from at least three independent experiments. Single factor comparison between the two groups using the unpaired two-tailed Student’s t test. One-way and two-way ANOVA with post-hoc tests were used for comparisons of more than two groups. Spearman correlation analysis was used to test whether the two variables were correlated. p < 0.05 and p < 0.01 were considered statistically significant. More than three independent experiments were performed to ensure the reproducibility of the results.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
There are no restrictions on data availability. Source data are provided with this paper. All data supporting the findings described in this manuscript are available in the article and in the Supplementary Information and from the corresponding author upon request. RNA-seq data have been deposited in the NCBI Gene Expression Omnibus under accession number GSE267638. The oxidative lipidomics data have been deposited into CNGB Sequence Archive of China National GeneBank DataBase with accession number CNP0006975. The illustrations in this manuscript were created with MedPeer [medpeer.cn] and Adobe AI.
References
Wu, X. et al. Recent advances in understanding of pathogenesis of alcohol-associated liver disease. Annu. Rev. Pathol. 18, 411–438 (2023).
Devarbhavi, H. et al. Global burden of liver disease: 2023 update. J. Hepatol. 79, 516–537 (2023).
Yan, C. et al. Pathogenic mechanisms and regulatory factors involved in alcoholic liver disease. J. Transl. Med. 21, 300 (2023).
Seitz, H. K. et al. Alcoholic liver disease. Nat. Rev. Dis. Prim. 4, 16 (2018).
Mascord, D., Smith, J., Starmer, G. A. & Whitfield, J. B. Effects of increasing the rate of alcohol metabolism on plasma acetate concentration. Alcohol Alcohol 27, 25–28 (1992).
Martino, C. et al. Acetate reprograms gut microbiota during alcohol consumption. Nat. Commun. 13, 4630 (2022).
Moffett, J. R., Puthillathu, N., Vengilote, R., Jaworski, D. M. & Namboodiri, A. M. Acetate revisited: a key biomolecule at the nexus of metabolism, epigenetics, and oncogenesis - Part 2: acetate and ACSS2 in health and disease. Front. Physiol. 11, 580171 (2020).
Wang, Y. et al. The multiple facets of acetyl-CoA metabolism: energetics, biosynthesis, regulation, acylation and inborn errors. Mol. Genet. Metab. 138, 106966 (2023).
Ling, R. et al. Acetyl-CoA synthetase 2(ACSS2): a review with a focus on metabolism and tumor development. Discov. Oncol. 13, 58 (2022).
Li, L. et al. Inhibition of ACSS2-mediated histone crotonylation alleviates kidney fibrosis via IL-1beta-dependent macrophage activation and tubular cell senescence. Nat. Commun. 15, 3200 (2024).
Chen, N. et al. ACSS2 controls PPARgamma activity homeostasis to potentiate adipose-tissue plasticity. Cell Death Differ. 31, 479–496 (2024).
Lin, Y. et al. ACSS2-dependent histone acetylation improves cognition in mouse model of Alzheimer’s disease. Mol. Neurodegener. 18, 47 (2023).
Bradshaw, P. C. Acetyl-CoA metabolism and histone acetylation in the regulation of aging and lifespan. Antioxidants (Basel) 10, 572 (2021).
Andrews, N. C. & Schmidt, P. J. Iron homeostasis. Annu. Rev. Physiol. 69, 69–85 (2007).
Ali, N., Ferrao, K. & Mehta, K. J. Liver iron loading in alcohol-associated liver disease. Am. J. Pathol. 193, 1427–1439 (2023).
Ioannou, G. N., Dominitz, J. A., Weiss, N. S., Heagerty, P. J. & Kowdley, K. V. The effect of alcohol consumption on the prevalence of iron overload, iron deficiency, and iron deficiency anemia. Gastroenterology 126, 1293–1301 (2004).
Stockwell, B. R. Ferroptosis turns 10: emerging mechanisms, physiological functions, and therapeutic applications. Cell 185, 2401–2421 (2022).
Ganne-Carrie, N. et al. Liver iron is predictive of death in alcoholic cirrhosis: a multivariate study of 229 consecutive patients with alcoholic and/or hepatitis C virus cirrhosis: a prospective follow up study. Gut 46, 277–282 (2000).
Nemeth, E. & Ganz, T. Hepcidin and iron in health and disease. Annu. Rev. Med. 74, 261–277 (2023).
Bridle, K. et al. Hepcidin is down-regulated in alcoholic liver injury: implications for the pathogenesis of alcoholic liver disease. Alcohol Clin. Exp. Res. 30, 106–112 (2006).
Ohtake, T. et al. Hepcidin is down-regulated in alcohol loading. Alcohol Clin. Exp. Res. 31, S2–S8 (2007).
Nemeth, E. & Ganz, T. Hepcidin-ferroportin interaction controls systemic iron homeostasis. Int. J. Mol. Sci. 22, 6493 (2021).
Harrison-Findik, D. D. et al. Alcohol metabolism-mediated oxidative stress down-regulates hepcidin transcription and leads to increased duodenal iron transporter expression. J. Biol. Chem. 281, 22974–22982 (2006).
Giglio, M. J., Santoro, R. C. & Bozzini, C. E. Effect of chronic ethanol administration on production of and response to erythropoietin in the mouse. Alcohol Clin. Exp. Res. 8, 323–325 (1984).
Wang, C. Y. et al. Smad1/5 is required for erythropoietin-mediated suppression of hepcidin in mice. Blood 130, 73–83 (2017).
Bjornson, E. et al. Stratification of hepatocellular carcinoma patients based on acetate utilization. Cell Rep. 13, 2014–2026 (2015).
Comerford, S. A. et al. Acetate dependence of tumors. Cell 159, 1591–1602 (2014).
Danpanichkul, P. et al. From shadows to spotlight: exploring the escalating burden of alcohol-associated liver disease and alcohol use disorder in young women. Am. J. Gastroenterol. 119, 893–909 (2024).
Miller, K. D. et al. Acetate acts as a metabolic immunomodulator by bolstering T-cell effector function and potentiating antitumor immunity in breast cancer. Nat. Cancer 4, 1491–1507 (2023).
Chen, J., Li, X., Ge, C., Min, J. & Wang, F. The multifaceted role of ferroptosis in liver disease. Cell Death Differ. 29, 467–480 (2022).
Camaschella, C., Nai, A. & Silvestri, L. Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica 105, 260–272 (2020).
Li, L. X., Guo, F. F., Liu, H. & Zeng, T. Iron overload in alcoholic liver disease: underlying mechanisms, detrimental effects, and potential therapeutic targets. Cell Mol. Life Sci. 79, 201 (2022).
Harrison-Findik, D. D. et al. Iron-mediated regulation of liver hepcidin expression in rats and mice is abolished by alcohol. Hepatology 46, 1979–1985 (2007).
Dixon, S. J. & Stockwell, B. R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 10, 9–17 (2014).
Yan, H. F. et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct. Target Ther. 6, 49 (2021).
Du, Y. & Guo, Z. Recent progress in ferroptosis: inducers and inhibitors. Cell Death Discov. 8, 501 (2022).
Dostalikova-Cimburova, M. et al. Role of duodenal iron transporters and hepcidin in patients with alcoholic liver disease. J. Cell Mol. Med. 18, 1840–1850 (2014).
Huang, Z. et al. ACSS2 promotes systemic fat storage and utilization through selective regulation of genes involved in lipid metabolism. Proc. Natl. Acad. Sci. USA 115, E9499–E9506 (2018).
Li, H. & Humphreys, B. D. Targeting de novo lipogenesis to mitigate kidney disease. J. Clin. Invest. 134, e178125 (2024).
Miura, K., Taura, K., Kodama, Y., Schnabl, B. & Brenner, D. A. Hepatitis C virus-induced oxidative stress suppresses hepcidin expression through increased histone deacetylase activity. Hepatology 48, 1420–1429 (2008).
Zhong, W. et al. Pharmacological activation of aldehyde dehydrogenase 2 by Alda-1 reverses alcohol-induced hepatic steatosis and cell death in mice. J. Hepatol. 62, 1375–1381 (2015).
Kwon, H. J. et al. Aldehyde dehydrogenase 2 deficiency ameliorates alcoholic fatty liver but worsens liver inflammation and fibrosis in mice. Hepatology 60, 146–157 (2014).
Blomstrand, R., Ellin, A., Lof, A. & Ostling-Wintzell, H. Biological effects and metabolic interactions after chronic and acute administration of 4-methylpyrazole and ethanol to rats. Arch. Biochem. Biophys. 199, 591–605 (1980).
Lindros, K. O., Sipponen, P., Pikkarainen, P., Turunen, U. & Salaspuro, M. Alcoholic liver damage is provoked by 4-methylpyrazole, which prolongs the influence of ethanol but reduces acetaldehyde levels. Alcohol Clin. Exp. Res. 3, 78–82 (1979).
Yamashita, H., Kaneyuki, T. & Tagawa, K. Production of acetate in the liver and its utilization in peripheral tissues. Biochim. Biophys. Acta 1532, 79–87 (2001).
Ciraku, L. et al. O-GlcNAc transferase regulates glioblastoma acetate metabolism via regulation of CDK5-dependent ACSS2 phosphorylation. Oncogene 41, 2122–2136 (2022).
Gu, D. et al. Hypoxia upregulating ACSS2 enhances lipid metabolism reprogramming through HMGCS1 mediated PI3K/AKT/mTOR pathway to promote the progression of pancreatic neuroendocrine neoplasms. J. Transl. Med. 22, 93 (2024).
Li, Z. et al. Acetyl-CoA synthetase 2: a critical linkage in obesity-induced tumorigenesis in myeloma. Cell Metab. 33, 78–93 e77 (2021).
Zhang, B. et al. ACSS2 enables melanoma cell survival and tumor metastasis by negatively regulating the Hippo pathway. Front. Mol. Biosci. 11, 1423795 (2024).
Bertola, A., Mathews, S., Ki, S. H., Wang, H. & Gao, B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat. Protoc. 8, 627–637 (2013).
Hao, F. et al. Butyrate enhances CPT1A activity to promote fatty acid oxidation and iTreg differentiation. Proc. Natl. Acad. Sci. USA 118, e2014681118 (2021).
You, Y. et al. FNDC3B protects steatosis and ferroptosis via the AMPK pathway in alcoholic fatty liver disease. Free Radic. Biol. Med. 193, 808–819 (2022).
Huang, Y. H. et al. Hepcidin protects against lipopolysaccharide-induced liver injury in a mouse model of obstructive jaundice. Peptides 35, 212–217 (2012).
Wang, M. et al. Glabridin ameliorates alcohol-caused liver damage by reducing oxidative stress and inflammation via p38 MAPK/Nrf2/NF-kappaB pathway. Nutrients 15, 2157 (2023).
Guo, X. et al. CNSA: a data repository for archiving omics data. Database 2020, baaa055 (2020).
Chen, F. Z. et al. CNGBdb: China National GeneBank DataBase. Yi Chuan 42, 799–809 (2020).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (U22A20272 and 82173807 to Y.D., 82370813 to W.H.), the CQMU program for Youth Innovation in Future Medicine (W0159 to W.H.), the Chongqing Natural Science Foundation (CSTB2023NSCQ-MSX0328 to W.H.), the Science and Technology Research Program of Chongqing Municipal Commission (KJQN202200443 to W.H.), the Young Scientists Fund of the National Natural Science Foundation of China (82300516 to Z.Y. and 82304503 to S.Z.), the Fundamental Research Funds for the Central Universities (WK9100000064 to Y.D.) and the USTC Research Funds of the Double First-Class Initiative (YD9110002089 to Y.D.). We are grateful to Professor Liwei Jiang for her invaluable guidance on flow cytometry.
Author information
Authors and Affiliations
Contributions
M.W., W.H. and Y.D. participated in research design; M.W. and X.W. performed major experiments; S.C., K.G., M.C. and R.P. assisted with the experimental operation. Z.F. and M.B.T. performed the bioinformatics analysis. F.G. assisted with Mendelian randomization analysis. M.W. and W.H. drafted the paper; W.H., Y.D., Q.M., C.L., Y.H. and Y.C. designed and supervised the studies; H.C. polished the manuscript; Y.D., W.H., S.Z. and Z.Y. provided the foundation; and all authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks John Moffett and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, M., Wen, X., Feng, Z. et al. ACSS2 protects against alcohol-induced hepatocyte ferroptosis through regulation of hepcidin expression. Nat Commun 16, 5491 (2025). https://doi.org/10.1038/s41467-025-61067-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61067-8
