
Abstract
The composition of tricarboxylic acid cycle metabolites in the external environment of cells determines vital physiological functions, including nutrient and mineral absorption, inflammation, and cellular energy management. Here, we study how the transport of external metabolites into the cells functions as an independent metabolic pathway that controls cellular energy. We show that liver cells orchestrate simultaneous fluxes of glucose and the omnipotent metabolite citrate across the cell membrane, acting as a first line metabolic pathway that responds to nutrient availability. Using functional mapping and gene silencing, we delineate the underlying molecular mechanism showing that the liver citrate transporter (NaCT) interacts with glucose transporters (Glut) and the anion transporters. The interaction is mediated by a specific region of the NaCT protein to reciprocally regulate the transport functions. Our findings describe an independent mechanism that coordinates external metabolites and glucose balance, thus driving key energy management processes in response to nutrient availability in the liver.
Similar content being viewed by others


Introduction
The coordination of metabolic and bioenergetics processes between different cells and tissues is quintessential for the proper function of the organs, especially under changing physiological conditions that alter the metabolic demand. Therefore, metabolic information needs to be exchanged between tissues and cells via networks of senso-regulatory proteins that orchestrate cellular metabolic functions in a process termed metabolic communication. The homeostasis of the tricarboxylic acid (TCA) cycle metabolites, citrate, and succinate, is essential for forming metabolic crosstalk between different organs. This includes the extracellular composition of metabolites, the exometabolites, required to maintain intracellular bioenergetic processes and simultaneously control whole body physiological functions regulated by citrate/succinate. Succinate is critical for mitochondrial energy production but is also a significant regulator of inflammation and blood pressure1,2,3. Citrate, which is the focus of this study, is an omnipotent metabolite. Citrate acts as a regulator of glycolysis, a precursor for fatty acid (FA) metabolism, and a significant physiological Ca2+ buffer4,5. Therefore, the intracellular and extracellular availability of citrate and succinate is essential for bioenergetics and other key physiological functions. Serum citrate and succinate concentrations are determined by gut absorption of dietary or microbiota-produced metabolites6, cellular production, and renal reabsorption3. Citrate and succinate are negatively charged under physiological conditions and, therefore, must be actively transported across cell membranes by transport proteins. Yet, the role of citrate/succinate transporters in forming inter-cellular networks that mediate metabolic communication to coordinate bioenergetics processes is unknown.
Several members of the SLC13 transporters mediate Na+-dependent citrate/succinate transport in many cells, including hepatocytes, gut epithelia, renal proximal tubule epithelia, and macrophages7,8,9. In hepatocytes and neurons, the major citrate transporter is SLC13A5 (NaCT), which is associated with the SLC13A5 deficiency disorder characterized by epilepsy10. In humans and mice, impaired NaCT expression and function profoundly affect citrate and glucose homeostasis11,12,13. Particularly, in the slc13a5 KO mice, glucose (or 2-deoxyglucose) uptake into muscle cells and osteoblasts is elevated12,14. Moreover, a dose-dependent decrease in extracellular glucose levels was monitored following the application of slc13a5 inhibitor to the osteoblasts14. These observations indicate that the expression of slc13a5 is associated with increased glucose fluxes. During fasting periods, NaCT expression is upregulated by glucagon stimulation of the CREB transcription factor, which also elevates the expression of specific glucose transporters of the Glut family11,15. However, how glucose and citrate homeostasis is locally co-regulated in response to changing nutrient availability is unknown. The answer to this question is of paramount physiological importance since cells must promptly respond to specific metabolic conditions like low energy or nutrient deficiency. It has been reported that the cellular energy sensor, AMP kinase (AMPK), is activated under nutrient deprivation conditions, low energy availability, or metformin treatment16, thus affecting the expression of citrate and glucose transporters17. Nevertheless, intracellular sensors react to extracellular changes already reflected within the cell and may respond too late. Hence, robust membrane mechanisms that respond rapidly to local physiological changes by maintaining metabolic homeostasis and bioenergetics are required as a first line of defense from uncontrolled intracellular metabolic alterations. These mechanisms may also exchange metabolic information with other cells through signaling by controlling extracellular and serum concentrations of specific metabolites. Therefore, we asked how citrate/succinate and glucose transporters control cellular bioenergetics in conjunction with extracellular homeostasis of citrate, glucose, succinate, and other metabolites.
In mice and Drosophila Melanogaster, NaCT deletion mimics caloric restriction, which promotes prolonged life span and elevates energy expenditure12,18. These findings coincide with the function of cytoplasmic citrate, transported by NaCT, as a key intermediate used by the enzyme ATP citrate lyase to produce cytoplasmic acetyl CoA in the FA synthesis pathway4. Moreover, cytoplasmic citrate is utilized for FA synthesis and acts as an allosteric modulator of a significant regulatory enzyme of glycolysis, phosphofructokinase-1 (PFK-1). However, the functional role of extracellular citrate uptake in regulating glycolysis and mitochondrial metabolism is poorly understood.
Extracellular glucose for the glycolytic flux is delivered into the cells by the Glut transporters that mediate glucose uptake. Glut-1 is highly ubiquitous and mediates basal glucose uptake, for example, across the blood-brain barrier and in hepatocytes. Other Gluts, for example, Glut-2 and Glut-4, determine the glucose homeostasis. Glut-2 is insulin-insensitive, thus acting as a glucose sensor that reflects extracellular glucose levels within the intracellular environment, which is necessary in tissues like the liver and pancreas. Moreover, Glut-2 mediates glucose efflux in hepatocytes that produce glucose almost exclusively in the gluconeogenesis pathway19. On the other hand, Glut-4 is insulin-sensitive and determines glucose homeostasis in cells that depend on insulin regulation to govern their metabolic state, e.g., muscle cells and adipocytes. These functions show that Glut transporters are traditionally considered the couriers of glucose that are merely controlled by other determinants like intracellular glucose sensors, insulin, or gene expression elements. Moreover, the mechanisms that coordinate the transmembrane fluxes of glucose and other metabolites and nutrients to optimize bioenergetics processes are not well understood. Here, we show that the transport of glucose and citrate in primary hepatocytes responds to glucose availability in a synchronized manner, which is controlled by a local NaCT/Glut interaction that reciprocally regulates the transport function.
We have previously reported that NaCT and other SLC13 transporters interact with members of the SLC26 family of Cl−/anion/oxalate transporters via regulatory interaction mediated by the H4c helix on transmembrane domain 4 (TMD4) of SLC13s20. Several human SLC13A5 deficiency-causing mutations have been identified on TM 4: an H4a residue (Tyr 82) impairs the citrate transport activity, likely due to impaired dimerization21; and two H4c mutations, namely, H106R and L111R, that may affect NaCT conformational changes22,23. We show that the Glut/NaCT interaction is mediated by the NaCT-H4c domain to drive synchronized glucose and citrate transport. This interaction controls the transport function of NaCT and is the underlying mechanism of regulatory interaction with Glucose transporters and bicarbonate transporters via dynamic intra-molecular interaction with the catalytic domain. Importantly, this mechanism tightly controls cellular metabolism.
Results
Citrate and glucose transport functions are synchronized in response to glucose availability in hepatocytes
Deletion or inhibition of NaCT in mice elevates extracellular glucose uptake into muscle and osteoblasts12,14. In hepatocytes, the transported citrate is metabolized for glucose production in gluconeogenesis11 or as a glucose substitute for energy production. To investigate how citrate and glucose transporters are regulated to control citrate and glucose homeostasis, we monitored ex vivo citrate and glucose transport in primary mouse hepatocytes under glucose starvation followed by glucose satiation conditions (Fig. 1A–C). We found that the uptake of 3H-citrate (Fig. 1B) and 3H-2-deoxyglucose (3H-2-DG) (Fig. 1C) into primary hepatocytes increased in response to prior glucose starvation of the cells for 4, 6, 8, and 10 h. In addition, we applied citrate and glucose/2-DG to hepatocytes that were glucose starved for 8 h and monitored a fast decrease in 2-DG uptake after 30, 120, and 240 min (Fig. 1C, empty black squares). Concomitantly, the increased uptake rate of citrate was halted in these hepatocytes (Fig. 1B, empty black squares). We repeated this experiment by monitoring citrate and 2-DG uptake in human THLE2 hepatocyte cultures, showing similar trends of citrate and glucose uptake in response to glucose availability (Supplementary Fig. 1A, B). In addition, we monitored citrate uptake for 15 min after 4, 6, and 8 h of glucose starvation, but in this set of experiments, we added glucose to the uptake solution (which contains 3H-citrate). As shown in Supplementary Fig. 1C, the increase in citrate uptake was stopped after 6 h of starvation, likely due to the presence of glucose during the uptake measurement (in contrast to the measurements in Fig. 1B and Supplementary Fig. 1A). These findings indicate that the citrate and glucose transporters synchronize in response to glucose availability in hepatocytes. To determine whether the primary hepatic citrate transporter, NaCT, regulates glucose transport, we monitored 2-DG uptake following 8 h of glucose starvation in the presence or absence of glucose and the human NaCT inhibitor (inh), BI01383298, in THLE2 hepatocytes. The results in Fig. 1D indicate that NaCT inhibition dose-dependently elevates glucose (2-DG) uptake in the absence and presence of glucose, suggesting that NaCT function is essential for down-regulating glucose transport to synchronize citrate and glucose transport. We also monitored 2-DG uptake into human HepG2 cells in the presence or absence of the NaCT inhibitor—the results in Supplementary Fig. 1D, E suggest that the synchronization between citrate and glucose transport in human HepG2 cells is also disrupted, as the net 2-DG uptake change (Δ 2-DG) in response to glucose re-application is ~30% lower in the presence of NaCT inhibitor. To determine the role of endogenous NaCT expression in regulating glucose transport in vivo, we injected mice with either NaCT-siRNA to silence NaCT or with Scr-siRNA as a control (the protocol is described in Fig. 1E). The NaCT siRNA sequence was initially tested in vitro and compared to a sequence that was previously published (NaCT siRNA*24) (Supplementary Fig. 1F). To determine NaCT silencing efficiency in vivo, we monitored NaCT expression in primary hepatocytes isolated from siRNA-injected mice and non-fasting blood glucose (Fig. 1F, G). We also monitored glucose uptake (Fig. 1H) and citrate uptake (Supplementary Fig. 1G) using the isolated primary hepatocytes. We found that NaCT silencing by NaCT-siRNA (Fig. 1F) significantly lowered non-fasting blood glucose levels relative to the glucose levels before injection (Fig. 1G). This is explained by the high levels of glucose transport that we monitored in mouse hepatocytes from NaCT siRNA-injected mice (Fig. 1H). Importantly, NaCT silencing also abolished the response of glucose transport to glucose availability that was retained in hepatocytes from Scr-injected mice. The NaCT siRNA treatment lowered the citrate uptake after 8 h of glucose starvation and abolished the response of citrate transport to glucose availability (Supplementary Fig. 1G). In contrast to non-injected WT mice, the increase in citrate uptake by hepatocytes from Scr siRNA-injected mice was attenuated after 10 h, likely due to the effects of the experimental procedure, but further study is required to determine the cause of this observation. To investigate how NaCT affects the synchronized citrate and glucose transport and glucose homeostasis, we expressed NaCT in mammalian cells (HEK293-T). Subsequently, we monitored the glycolytic function using the Seahorse analyzer glycostress protocol in the presence of low glucose (2.5 mM) and the presence or absence of 1 mM citrate (as indicated in Fig. 1I and Supplementary Fig. 2). We found that the glycolytic function was significantly lower in cells expressing NaCT. However, the inhibition of glycolysis by recombinant NaCT expression was largely independent of citrate.

A The experimental regime of primary mouse hepatocyte isolation, culture, and uptake assays. Created in BioRender. Ohana, E. (2025) https://BioRender.com/6lu0mpn. Analysis and summary of 3H-citrate (B) or 3H-2DG (C) uptake in primary mouse hepatocytes after 4,6, 8, and 10 h of glucose starvation (closed circles) or after 8 h of starvation and application of 10 mM citrate and 5 mM glucose for 30, 120, and 240 min (open squares). Radiolabeled metabolite uptake times are indicated above the figure. The data presented in (B and C) was collected simultaneously for each culture isolated from seven different mice in seven independent experiments. N is number of uptake measurements relative to the average of 4 h starvation in individual wells containing a similar number of hepatocytes. D The uptake of 2DG was monitored in THLE2 hepatocytes following 8 h glucose starvation in the presence of citrate (Cit), in the presence or absence of glucose (Glc) or human NaCT inhibitor (inh), as indicated. N is the number of uptake measurements in individual wells. E The experimental regime of siRNA injection for the experiments described in (F–H), hepatocyte isolation, culture, and uptake assays were performed as described in (A). Created in BioRender. Ohana, E. (2025) https://BioRender.com/zht4zvi. F Western blot analysis of NaCT expression in mouse hepatocytes isolated from siRNA-injected mice, as indicated (lysates from a different mouse in each lane, NaCT expression was not decreased in NaCT siRNA injected mouse 5-red). G Analysis of non-fasting blood glucose measurements from mice injected with Scr or NaCT siRNA as indicated (5 and 6 mice in the Scr or NaCT siRNA groups, respectively). N represents the number of mice. H Analysis and summary of 3H-2DG uptake in primary mouse hepatocyte after 4, 8, and 10 h of glucose starvation (closed circles) or after 8 h of starvation and application of 10 mM citrate and 5 mM glucose for 30, and 120 min (open squares) in mice injected with either scrambled siRNA (Scr) (3 mice) or NaCT-siRNA (5 mice). N is the number of uptake measurements in individual wells. I A seahorse glucostress analysis in mammalian HEK293-T cells transfected with either NaCT or GFP (control). The measurements were performed in the presence or absence of 1 mM citrate, as indicated. Statistical analysis of the data is in Supplementary Fig. 2. N is the number of individual HEK293 culture wells for which glucostress measurements were performed. Statistical analysis in Fig. 1 was performed using one-way ANOVA, followed by Tukey’s multiple comparison test to determine significant differences between groups.
Based on these findings, we asked how the expression of NaCT (or other SLC13s) is altered in tumors since cancer cells are infamous for their “super glycolysis” function due to the “Warburg effect”. Using the GEPIA2 online tool, we analyzed the gene expression of NaCT (SLC13A5), SLC13A2, and Glut-1 in different cancer types25. The results in Supplementary Fig. 3 show that the expression of NaCT (SLC13A5) and SLC13A2 are significantly downregulated in the liver, brain, and other cancer types as opposed to Glut-1 upregulation. Therefore, downregulation of SLC13 gene expression may play a critical role in maintaining cancer cell glucose metabolism and survival.
Our findings thus far indicate that NaCT controls glycolysis via the regulation of glucose uptake and synchronization between glucose and citrate transport. However, the glycolytic flux is not inhibited by the citrate accumulation, which is transported by recombinant NaCT.
NaCT deficiency-associated mutations in the NaCT-H4c regulatory domain alter cellular metabolism
To delineate the molecular mechanism of synchronized glucose and citrate transport regulation, we hypothesized that NaCT membrane expression, conformational changes during the turnover cycle, or both affect the regulatory interaction of NaCT with partner transporters to control glucose homeostasis. Therefore, we investigated how cellular bioenergetics is driven by residues in the regulatory NaCT-H4c domain that determines NaCT function via interaction with partner proteins20. First, we focused on two mutations in the regulatory H4c domain, H106R and L111R (Fig. 2A), which are associated with NaCT deficiency23,26 (ClinVar: H106R ID–967719, L111R ID–812784). To test how the disease-associated mutations H106R and L111R affect NaCT function, we generated and expressed the mutations in Xenopus oocytes. The disease-causing mutations H106R and L111R abolish NaCT function monitored by electrophysiology in Xenopus oocytes expressing either WT or mutant NaCT (Fig. 2B–E). The biotinylation analysis in Fig. 2F and the fluorescent imaging in Supplementary Fig. 4 show that several H4c mutations retained NaCT membrane expression except L111R. Thus, the L111R loss-of-function is likely due to the abolishment of its membrane expression. On the other hand, the membrane trafficking of H106R is prominent but eliminates NaCT function. The function of another mutation in position 111 (L111A) also impaired NaCT function. The location and orientation of H106 suggest that it plays a pivotal role in protein function and stabilization. To test that, we generated several mutations in position 106. As indicated in Fig. 2B and Supplementary Fig. 5A, all H106 mutations eliminated NaCT function, thus indicating that H106 is essential for NaCT expression and activity. These findings suggest that specific H4c residues are crucial and differential in controlling NaCT function and expression.

A The structure of human NaCT shows the intracellular H4c (blue) and H6b (cyan) domains and the proximate catalytic HPin (green). The H106 and L111 residues that have mutations that are associated with SLC13A5 deficiency are shown in red. B Summary and C representative traces of electrophysiological measurements in Xenopus oocytes expressing either WT or H106R mutations. D Summary and E representative traces of NaCT currents in Xenopus oocytes expressing either WT or L111R and L111A mutations. In (B) and (D), N represents the current measurement in a single oocyte. F A representative WB analysis of biotinylation assay (1 of 2 biological replicates) showing the membrane expression of H4c residues mutations, including H106R and L111R. G Glycostress protocol performed using HEK293-T cell cultures expressing either WT, H106R, or L111R NaCT constructs. N is the number of individual HEK293 culture wells for which glucostress measurements were performed. Statistical analysis of the seahorse measurements is in Supplementary Fig. 5B–E. Data are presented as mean values +\− SEM. Statistical analysis was performed using Student’s t-test or as described in Supplementary Fig. 5.
Therefore, we used the Seahorse analyzer to monitor the effect of citrate uptake on the glycolytic function and mitochondrial respiration in cells expressing NaCT, L111R, and H106R in the presence of 1 mM extracellular citrate, as indicated. The expression of the dysfunctional disease-causing mutation L111R resulted in low inhibition of glycolysis compared to that of WT NaCT. In contrast, H106R expression inhibited glycolysis similarly to WT NaCT (Fig. 2G and Supplementary Fig. 5B–E). Furthermore, mitochondrial respiration was significantly inhibited by NaCT, but the effects of H106R and L111R were not statistically significant and showed a similar trend to that of glycolysis (Supplementary Fig. 5F–L). These findings and those described in Fig. 1 suggest that NaCT expression and function drive citrate and glucose homeostasis. Hence, impaired NaCT function, as in NaCT deficiency, is expected to hamper citrate and glucose metabolism. Notably, the high expression of recombinant NaCT(H106R) loss-of-function mutation is sufficient to inhibit glycolysis in contrast to endogenous NaCT function, which is essential for glucose uptake (Fig. 1D).
The mechanism of glucose transport regulation by NaCT is revealed by differential effects of functional mutations in the H4c regulatory domain on glycolysis
Next, we investigated whether NaCT interaction with partner proteins regulates the fluxes of glucose and other metabolites in hepatocytes. Therefore, in contrast to the dysfunctional disease-causing mutants, we now focused on NaCT H4c mutations that retain NaCT expression and function but were reported to impair protein-protein interactions with the anion/HCO3− transporter SLC26A6. If these NaCT mutations control glycolysis, they can be used as robust tools to study how NaCT interacts with other transporters to control metabolism. Thus, before investigating the mutants’ direct effects on glucose transport, we studied how H4c mutations in key residues regulate the hepatic SLC26 family member, SLC26A1, that mediates anion/HCO3− transport27,28,29. This potential mechanism is of high physiological relevance for identifying common molecular determinants of NaCT that mediate regulatory interaction with other metabolite transporters in the sinusoidal membrane of hepatocytes30,31. We monitored endogenous Na+-dependent citrate uptake in hepatocellular carcinoma cells (HepG2) using a radiolabeled 3H-citrate flux assay (Supplementary Fig. 6A). To determine the Na+-dependent activity, we monitored citrate uptake at each concentration in the absence of Na+, which was subtracted from similar measurements in the presence of Na+. The cells were either transfected with SLC26A1 or treated with the human NaCT inhibitor in the presence of different citrate concentrations. The results in Supplementary Fig. 6A indicates that SLC26A1 inhibits endogenous NaCT-mediated Na+-dependent citrate uptake, which is also strongly inhibited by the human NaCT inhibitor BI01383298. To monitor real-time Na+-dependent citrate co-transport in mammalian cells, we devised a new protocol described in Supplementary Fig. 6B, using real-time fluorescent imaging in cells pre-loaded with the Na+-sensitive dye, ING-2. We found that in the presence of citrate, the Na+ uptake is dramatically elevated in NaCT-expressing HEK293-T cells compared to control due to Na+/citrate-coupled cotransport mediated by NaCT (Supplementary Fig. 6B, C). Using the new protocol, we also monitored the inhibition of NaCT in the presence of SLC26A1.
The physiological phenotypes of impaired NaCT function and citrate homeostasis differ between humans and mice (see introduction). To study NaCT regulation by both mouse and human SLC26A1 isoforms, we transfected HEK293-T cells with the functional human NaCT (hNaCT) either individually or together with human hSLC26A1 or mouse mSlc26a1. We then monitored citrate uptake using a radiolabeled 3H-citrate flux assay and protein-protein interaction by coimmunoprecipitation (CoIP). The results suggest that hSLC26A1 and mSlc26a1 inhibit NaCT-mediated citrate uptake (Supplementary Fig. 6D) and interact with NaCT (Supplementary Fig. 6E, F). Our findings thus far indicate that SLC26A1 and NaCT interact to control citrate homeostasis.
Next, we asked whether the SLC26A1/NaCT interaction regulates citrate uptake to modulate bioenergetics. We monitored glycolysis in cells expressing SLC26A1, NaCT, or both (Supplementary Fig. 6G). Our results indicate that the inhibitory effect of recombinant NaCT expression on glycolysis is not affected by SLC26A1 attenuation of citrate transport via interaction with NaCT.
Our previous findings showed that H4c domain residues are involved in the interaction between SLC13 and SLC26 transporters. Specifically, conserved mutations in the H4c region, namely, K107A and R112A (residue location described in Fig. 3A), impair renal SLC13A2-SLC26A6 proteins interaction and retain transport function3. Hence, we monitored the effect of mutations in H4c residues on hepatic SLC26A1 regulation of NaCT. First, we utilized electrophysiological measurements in Xenopus oocytes expressing NaCT(WT), NaCT(K107A), NaCT(R112A), or SLC26A1, as indicated in Fig. 3B, C (summary and traces). Importantly, both K107A and R112A mutations impaired the inhibition of NaCT-mediated citrate transport by SLC26A1. Second, we utilized a real-time fluorescent imaging protocol similar to that described in Supplementary Fig. 6B to monitor NaCT function in HEK293-T cells expressing NaCT(WT), NaCT(K107A), NaCT(R112A), or SLC26A1, as indicated (Fig. 3D, E, summary, and traces). Our results using fluorescent measurements were highly similar to those of the electrophysiological measurements, suggesting that K107 and R112 in the H4c domain play a key role in NaCT inhibition by SLC26A1. Finally, we monitored the effects of the K107A and R112A mutations on glycolysis and mitochondrial respiration. The results in Fig. 3F and Supplementary Fig. 5B–E indicate that while both mutations retain citrate transport function, K107A inhibits glycolysis, while R112A inhibition is significantly lower. Furthermore, the effects of K107A and R112A on mitochondrial respiration were not statistically significant but indicated a similar trend to that of glycolysis (Supplementary Fig. 5F–L). Together, the results in Figs. 2, 3 and Supplementary Fig. 5 suggest that H4c residues, which mediate regulatory interaction with SLC26 transporters, control glucose metabolism and mitochondrial bioenergetics. However, glucose metabolism is not regulated by NaCT interaction with SLC26A1 in cells that express the recombinant transporters (Supplementary Fig. 6G).

A The structure of human NaCT shows H4c, H6b, and HPin. The K107 and R112 residues are highlighted within the H4c domain (inset). B Summary and C Traces of NaCT currents measured in Xenopus oocytes injected with NaCT-WT, K107A, R112A, F128A (dysfunctional), or SLC26A1, as indicated. N represents the current measurement in a single oocyte. D Representative traces and E Summary of NaCT function in mammalian HEK293-T cells transfected with NaCT(WT), K107A, R112A, or SLC26A1 (as in B and C). Real-time fluorescent measurements were performed in cells loaded with the ING-2 Na+-sensitive dye using the protocol described in the figure and the methods section. N represents the average changes in fluorescence relative to NaCT(WT) in the cells that were cultured on a single cover slip. F Glycostress protocol performed using HEK293-T cell cultures expressing either control, WT(NaCT), K107A, or R112A constructs. Statistical analysis of the seahorse measurements is in Supplementary Fig. 5B–E. Data are presented as mean values +\− SEM. Statistical analysis was performed using one-way ANOVA followed by Tukey’s multiple comparison test.
NaCT interacts with Glut transporters to inhibit glucose uptake and control cellular bioenergetics
Next, we hypothesized that NaCT may regulate glucose homeostasis via interaction with Glut transporters mediated by H4c residues. Therefore, we first monitored the membrane trafficking of different NaCT-H4c domain mutations in isolated Xenopus oocyte membranes under conditions similar to that of the electrophysiological functional assays (Fig. 4A). The expression of the disease-causing H106R and L111R was similar to the expression we monitored in mammalian cells (Fig. 2F). In addition, we measured citrate uptake in HEK293-T cells by 3H-citrate flux assay (Fig. 4B) and monitored the function of endogenous glucose transporters in these cells using 3H-2-DG uptake assay (Fig. 4C, red squares). Then, we plotted the glycolytic flux measurements from Figs. 2G and 3F, as described in the figure legend (Fig. 4C, blue squares). The results in Fig. 4A–D show that the effect of NaCT and mutations on glucose uptake correlates with their effect on metabolic processes, suggesting that the control of glycolysis by NaCT is primarily through the regulation of glucose uptake. On the other hand, the expression of NaCT and the different mutations correlate with their citrate transport function. Specifically, the inhibitory effect of the NaCT(L111R) mutation on glucose uptake and metabolism is lower than that of the NaCT(WT), probably due to the impaired membrane expression and function of this mutation. The inhibition of glucose uptake and metabolism by the NaCT(K107A) mutation is robust since it retains membrane trafficking and function (compared to NaCT(WT)). However, the two other mutations show unexpected effects—R112A, which is fully functional and expressed at the membrane (Fig. 4A and Supplementary Fig. 4)—shows low regulation of glucose uptake and metabolism. On the other hand, the dysfunctional H106R, expressed on the membrane, inhibits glucose uptake, glycolysis, and mitochondrial respiration. To identify which endogenous glucose transporter is regulated in our system, we treated the cells with the Glut-1 inhibitor BAY-876 (TOCRIS), which completely abolished glucose uptake into the cells (Fig. 4E). This coincides with previous reports that Glut-1 is a major endogenous glucose transporter in HEK293 cells32,33.

A–D are aligned (dash line) to show the expression, function, and metabolic outcome of NaCT WT and mutations. A A representative WB analysis showing the expression of NaCT in membranes isolated from Xenopus oocytes injected with NaCT and the indicated mutants. Densitometry of 3 independent experiments and statistical analysis are shown in Supplementary Fig. 11. B 3H-citrate transport measurements in HEK293-T cells transfected with WT NaCT or the indicated mutations. C 3H-2-DG uptake (red) and a re-plot of the glycolytic flux measurements at 33 min (glucose application) from Figs. 2G and 3F (blue, N is the number of glycostress measurements in individual wells) show a similar trend. Statistical analysis was performed using one-way ANOVA followed by Dunnet’s T3 multiple comparison test. D A re-plot of the ECAR measurements in Figs. 2G and 3F at 33 min (glucose application—closed blue squares), and 40 min (oligomycin application—open blue squares). Statistical analysis was performed using one-way ANOVA followed by Tukey’s multiple comparison test. A re-plot of the OCR measurements in Supplementary Fig. 5F at 14 min (baseline—closed red circles) and 53 min (FCCP application—open red circles). E 3H-2-DG uptake monitored in HEK293-T cells in the presence or absence of 10 µM Glut-1 inhibitor BAY-876. F 3H-2-DG uptake into HEK293-T is inhibited in cells transfected with WT (NaCT). The inhibition is attenuated in cells expressing the R112A mutation. G NaCT currents monitored in Xenopus occytes expressing NaCT, NaCT(R112A), or GLUT1, as indicated. N represents the current measurement in a single oocyte. H Representative CoIP of endogenous NaCT and Glut-1 in HepG2 hepatocytes. Densitometry of 3 independent experiments and statistical analysis are shown in Supplementary Fig. 11. I Immunofluorescence analysis in human HepG2 cells showing co-localization (merged panel) of the endogenous NaCT (green) and Glut-1 (red). J Representative WB blot analysis of a CoIP assay performed in HEK293-T cells transfected with NaCT WT, mutants, and Glut-1, as indicated. Densitometry of 3 independent experiments and statistical analysis are shown in Supplementary Fig. 11. K Alphafold2 multimer prediction of the interaction between NaCT dimer and Glut-2 monomer showing potential interaction mediated by NaCT R112. L NaCT-mediated 3H-citrate uptake in the presence of Glut-1 and Glut-2. In (B), (C, red), (E), (F), and (L), the N is the number of uptake measurements in individual wells. Data are presented as mean values +\− SEM.
To determine the reciprocal effect of NaCT and Glut-1 on citrate/glucose transport, we monitored 3H-2-DG uptake in HEK293-T cells, which highly express endogenous Glut-1, transfected with either WT NaCT or the R112A mutation in the H4c domain (Fig. 4F). In addition, we monitored NaCT activity using electrophysiological measurements in Xenopus oocytes expressing NaCT WT or R112A in the presence or absence of Glut-1, as indicated (Fig. 4G). Interestingly, we found that NaCT and Glut-1 reciprocally inhibit the citrate and glucose transport function. We also showed that the R112A mutation impairs the reciprocal regulation. Indeed, R112A is the only functional H4c mutation that we tested, which retains membrane expression and function but impairs the inhibition of glycolysis by NaCT (Fig. 3F).
Then, we monitored NaCT-Glut-1 interaction by CoIP either for the endogenous transporters in HepG2 cells or following the expression of the transporters in HEK293-T cells. As shown in the western blot analyses, we monitored the interaction between the endogenous NaCT and Glut-1 in human hepatocytes (HepG2) (Fig. 4H), supported by clear colocalization of these transporters in specific membrane regions (Fig. 4I). Moreover, we monitored the interaction between NaCT and Glut-1 in HEK293-T cells transfected with these transporters (Fig. 4J). The interaction between Glut-1 and NaCT was significantly attenuated by the NaCT(R112A) mutation, suggesting that it is essential for NaCT-Glut-1 binding. Notably, R112A is also crucial for NaCT interaction with SLC26 transporters. Hence, to determine whether NaCT regulates other Glut family members via interactions, we used Alphafold2 to predict the putative structure of the NaCT-Glut-2 interaction (Fig. 4K). Intriguingly, our analysis suggests that the intracellular NaCT site of interaction is at the H4c region and involves potential interaction with R112 and not with K107. The putative Glut site of interaction with R112 is the Intracellular Helix 4 (ICH4) domain. To predict the potential dynamics of interaction during the Glut turnover cycle, we superimposed the structures of Glut3 outward-facing (PDB: 4ZWB) and Glut-1 inward-facing (PDB: 4PYP) conformations on the predicted model of NaCT-Glut2 interaction forcing overlay on stationary helices (which are relatively stable during the Glut turnover cycle)—the structure prediction in Supplementary Fig. 7 indicates that while other intracellular helices of the bundle are dynamic, ICH4 is relatively stable and can maintain interaction with NaCT throughout the turnover cycle. Nevertheless, additional molecular dynamics and structural analyses are required to determine the dynamic nature of this interaction. Furthermore, we monitored the effect of citrate uptake in cells co-expressed with NaCT and either Glut-1 or Glut-2. We found that both Glut-1 and Glut-2 inhibit NaCT-mediated citrate transport (Fig. 4L). The results in Fig. 4 indicate that NaCT and Glucose transporters interact to control cellular metabolism primarily via reciprocal regulation of citrate and glucose transport. This mechanism may synchronize citrate and glucose uptake, as we monitored in hepatocytes (Fig. 1). Our putative model and data suggest that R112 is an intracellular residue, which is essential for regulatory interaction with Glut. However, an additional interaction site is predicted closer to the extracellular region of NaCT and Glut transporters and remains to be investigated.
The H4c domain that harbors NaCT deficiency-associated mutations interacts with the H6b domain to form a regulatory interface
To study the mechanism by which the H4c domain that harbors the disease-associated mutations interacts with partner proteins to regulate metabolism, we initially focused on the conserved K107 residue3,20. This is based on previous findings showing that K107 mutations retain NaCT function and expression but hamper functional regulation by partner proteins. The NaCT structure reveals that K107–E305–K308 form a salt bridge that mediates interaction between the H4c and H6b helices (Fig. 5A and ref. 23). Therefore, we generated several charge-swap mutations (K107E, E305K, K308E) and found that the function of K107E and E305K eliminated NaCT activity, while K308E activity is retained (Fig. 5B, C). Moreover, we generated the double mutation, K107E/E305K, to swap the charges between H4c(K107) and H6b(E305) interacting residues, respectively, which resulted in ~ three-fold gain-of-function compared to the single mutated proteins (Fig. 5B, C). To determine the role of K308, we generated a triple mutation (K107E/E305K/K308E), which also resulted in gain-of-function, suggesting that the charge in position 308 is essential for stabilizing the link between H4c and H6b, as described in Fig. 5B, C. This is further strengthened by the gain-of-function we monitored for the E305K/K308E double mutation. Moreover, we found that the interaction between H4c and H6b via the K107, E305, and K308 triad is crucial for regulation by SLC26 transporters since the K107E/E305K mutation is not inhibited by SLC26 transporters (Fig. 5B). The attenuation of NaCT inhibition by SLC26A6 is due to loss of interaction, as shown in the CoIP analysis (Fig. 5D, statistical analysis in Supplementary Fig. 11) monitoring interaction with K107E/E305K, K107E/E305K/K308E or E305K/K308E. All the mutations described in Fig. 5 retain membrane trafficking (Supplementary Fig. 4). These findings indicate that H4c and H6b form a regulatory interface that controls NaCT function and is stabilized by charged residues (Fig. 5E). The location of the H4c-H6b regulatory interface near the inner leaflet of the plasma membrane facing the intracellular region makes it ideal for interaction with and regulation by intracellular determinants.

A The K107-E305-K308 triad (orange) is highlighted in the NaCT structure (H4c-light blue, H6b-cyan, HPin-green). B Summary and C representative traces of NaCT currents in Xenopus oocytes expressing different K107, E305, and K308 mutants. D Representative WB blot analysis of a CoIP assay performed in HEK293-T cells transfected with NaCT WT, SLC26A6 WT (A6), and NaCT mutants, as indicated. The red arrow indicates the size of SLC26A6 in the coIP and blot. Densitometry and statistical analysis are shown in Supplementary Fig. 11. E Schematic summary of different K107-E305-K308 triad mutations and their functional outcome (from B and C). Data are presented as mean values +\− SEM. Statistical analysis was performed using one-way ANOVA followed by Tukey’s multiple comparison test.
The H4c/H6b domain acts as a modulatory interface, likely via simultaneous interaction with scaffold helices, catalytic helices, and partner proteins
To determine how the H4c-H6b domain controls NaCT activity and cellular metabolism, we functionally mapped the NaCT-H4c and -H6b helices by generating alanine and other mutations, as described in Fig. 6A–E (color-coded as detailed in the figure caption). Our functional mapping of H4c and H6b residues indicates that this region affects a wide dynamic range of protein function and expression, ranging from inactive to gain-of-function mutations compared to WT (Fig. 6E, F). More specifically, mutations at each of the helices separately show a similar dynamic range, suggesting that both are critical for NaCT function (Supplementary Fig. 8). Based on our functional map of the H4c/H6b domains and previous dynamic models of SLC13 transporters34,35, we posit that H4c and H6b residues may interact with residues of the catalytic HPin during the transport cycle. Therefore, we focused on three residues of the catalytic HPin domain, which are predicted to interact with H4c or H6b (F128, M147, and I151). To this end, we mutated these residues to alanine. As indicated in Supplementary Fig. 9A–C, the function of NaCT was significantly attenuated for F128A, M147A, and I151A compared to WT NaCT. However, since sodium ion binding stabilizes the HPin disordered structure crucial for NaCT function36, we cannot exclude the option of these mutants affecting the secondary structure and stability of HPin helices.

A–D Different NaCT structure representations show the functional map of the H4c/H6b domain (light green—full function, dark green—less than 25% loss-of-function, orange—more than 50% loss-of-function, red—more than 75% loss-of-function). E Summary of NaCT currents monitored in Xenopus oocytes injected with H4c/H6b mutants. The color codes of the columns was used for functional mapping in all NaCT structural representations. F The currents shown in (E) are re-plotted by current intensity (high to low order) to demonstrate the full dynamic range determined by H4c/H6b residues (inset—ribbon representation of H4c/H6b functional mapping). The results in Fig. 2B and D, Supplementary Figs. 5A, 8B and E, and 9B and D, are presented in Fig. 6E and F.
Finally, we aimed to identify other H6b and H4c residues which may mediate SLC26 regulation of NaCT. Therefore, we utilized electrophysiological measurements of NaCT or mutants in the presence or absence of the SLC26A6 transporter, showing that SLC26A6 inhibits NaCT. Together with our previous findings, we conclude that SLC26A6 regulates all three succinate/citrate transporters of the SLC13 family, namely, SLC13A2, SLC13A3, and SLC13A5 (NaCT)5,6,20. As shown in Supplementary Fig. 9D, the additional mutations we tested in both H4c and H6b retained regulation by SLC26A6.
Based on our functional mapping of the H4c/H6b region, we identified three major clusters of residues (Figs. 6 and 7). In the first cluster (cluster I, Fig. 7A), we mapped several residues with a significant reduction in protein activity or expression. These residues are located within the “elbow” region, which was suggested to provide structural stability and optimize protein function (Supplementary Fig. 10A)35. Mutations in residues within the second cluster (cluster II, Fig. 7B) resulted in a significant functional decrease or loss of function. Cluster II residues may interact with dynamic HPin residues during the turnover cycle (including the three HPin residues in Supplementary Fig. 9B, C). This cluster of residues may act as a hydrophobic “rail,” which facilitates HPin movement via hydrophobic interactions. The third cluster (cluster III, Fig. 7C, surface representation in the inset) includes residues that retain significant protein function. Notably, the residues of cluster III face the protein surface. The location of essential H4c residues described in this manuscript are shown in Fig. 7D, specifically, H106 and L111 that carry loss-of-function and disease-associated mutations (cluster II) and the functional K107 (triad) and R112 (cluster III) that mediate protein-protein interactions with Glut and SLC26 transporters.

A Cluster I—mutations in the “elbow” likely impair protein stability and function. B Cluster II—mutations in H4c, H6b, and HPin that may interact during the turnover cycle and significantly impair protein function. C, D Cluster III—mutations that retain protein function and face the surface of the protein. All residues are color-coded as in Fig. 6.
These findings indicate that the H4c\H6b acts as a regulatory junction that can respond to intracellular signals, such as interaction with partner proteins via cluster III residues, by regulating the catalytic HPin domain dynamics (cluster II) to control NaCT activity and reciprocally regulate Glut and SLC26. Ultimately, this molecular mechanism is essential for controlling metabolic fluxes in hepatocytes.
Discussion
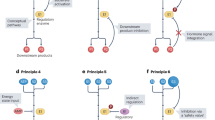
Many metabolites utilized intracellularly for canonical metabolic pathways are found in the extracellular environment and the serum, termed exomatbolites. Therefore, the mechanisms that control the transmembrane flux of metabolites into the cells can act as an independent metabolic pathway that determines the cellular availability of metabolites and the bioenergetic outcome. In this study, we have delineated the molecular mechanism of a new metabolic modulation pathway governed by transport protein interactions. This molecular pathway controls the cellular homeostasis of multiple metabolites, including citrate, glucose, and potentially other ions such as succinate, oxalate, bicarbonate, and sulfate, which are transporterd by NaCT, Glut, or SLC26A1. Specifically, we found that the H4c/H6b domain of SLC13 transporters acts as a regulatory interface that can integrate intracellular signals to control citrate and glucose homeostasis (Fig. 8). The NaCT/Glut interaction likely serves as the first line of rapid metabolic regulation that determines citrate/glucose homeostasis by controlling the intracellular and extracellular availability of the metabolites. This is achieved by selective control of glucose/citrate/succinate transport into the cells. Therefore, NaCT/Glut is expected to act prior to intracellular sensors such as glucokinase or other enzymes that sense downstream effects of metabolite availability, such as AMPK signaling and gene expression regulators. The physiological function of the NaCT/Glut interaction may affect two major tissues in which NaCT and Glut-1, Glut-2, or Glut-3 are highly expressed—the liver and the brain37,38.

Created in BioRender. Ohana, E. (2025) https://BioRender.com/remkqr9.
In the liver, NaCT, SLC26A1, and Glut-1/Glut-2 are expressed on the sinusoidal membrane of hepatocytes30,39,40, where the expression of Glut-1 and Glut-2 is insulin/glucagon insensitive. Under fasting conditions, glucagon upregulates NaCT expression via activation of the CREB transcription factor, which results in elevated citrate uptake11. Previous studies indicated that glucagon upregulates NaCT-mediated citrate uptake in starved rats11 and that NaCT is downregulated by the AMPK signaling pathway17. Namely, AMPK functions as a glucose-sensing mechanism that is activated in response to low-glucose availability and low energy production to increase glucose uptake via Glut. Indeed, metformin, which activates AMPK, downregulates NaCT and aggravates symptoms in SLC13A5 deficiency patients17,41. These observations describe potential changes in either Glut or NaCT individually in response to low glucose conditions. However, since citrate can be utilized as an alternative carbon source when glucose is low, the activity of the Glut and NaCT transporters should be monitored when both glucose and citrate are available. In hepatocytes, Glut-2 mediates the influx of glucose for energy production and the efflux of glucose generated by gluconeogenesis. Our results show a synchronized citrate and glucose transport in the presence of extracellular citrate, which is elevated in response to glucose starvation and reversed by re-application of glucose (Fig. 1B, C). These findings suggest that the cell rapidly synchronizes citrate and glucose uptake to control their transport in response to glucose availability. Indeed, we describe an intimate regulatory mechanism of citrate and glucose transporters, mediated by an interaction between the transporters that likely responds to extracellular metabolite availability before the changes are reflected inside the cells. As shown in Fig. 1, the citrate and glucose transporters synchronously responded to the application of glucose within minutes. Nevertheless, previous studies show that the gene expression and promoter activity of NaCT respond after several hours11, as we monitored in response to glucose starvation. In the long term, the NaCT\Glut interaction is likely to be affected by the membrane expression of the individual transporters. We found that in vivo silencing of NaCT by siRNA injection attenuated non-fasting blood glucose and elevated glucose transport function in hepatocytes (Fig. 1E–H). We did not monitor fasting glucose since it may counter NaCT silencing to elevate NaCT expression as observed in rats11. Moreover, as shown in Fig. 4A and C, the inhibitory effect of NaCT (L111R) on Glucose uptake and glycolysis is abolished due to impaired trafficking to the membrane. In agreement with our findings, it was previously reported that Slc13a5 deletion or inhibition resulted in elevated glucose uptake in other cell types, namely, muscle cells and osteoblasts12,14.
The NaCT/Glut interaction is mediated by the H4c-H6b regulatory array to determine their function (Fig. 4F–L). As a result, NaCT and Glut-1 reciprocally inhibit glucose and citrate transport, consequently attenuating the glycolytic flux. This mechanism can determine the intracellular and extracellular homeostasis of glucose and citrate to synchronize their uptake into individual hepatocytes (Fig. 1). Furthermore, if the NaCT/Glut interaction determines and responds to extracellular nutrient availability, it can potentially synchronize the metabolic activity of multiple cells in the tissue to optimize their metabolic function.
In the brain, NaCT and Glut-1 are not expressed in similar regions. NaCT is mainly found in neurons, which predominantly express Glut-3, while Glut-1 is expressed in astrocytes and endothelial cells that form the blood-brain barrier38. Therefore, the central role of Glut-1 is the delivery of glucose across the blood-brain barrier. Indeed, Glut-1 deficiency shares similar symptoms with slc13a5 deficiency42. These include epileptic seizures, developmental delay, and impaired glucose homeostasis in the cerebrospinal fluid (CSF). Under these pathological conditions, impaired glucose homeostasis in conjunction with hampered citrate homeostasis in the brain are significant potential causes for the neurological symptoms of slc13a5 deficiency. Therefore, the localization of both NaCT and Glut-3 on the plasma membrane of neurons suggests a prospective role for NaCT in driving neuronal citrate and glucose homeostasis similar to that in hepatocytes. Notably, Glut-1 deficiency can be treated by a ketogenic diet that shifts the metabolic burden from impaired glucose metabolism towards fatty acid oxidation43. However, the effectiveness of a ketogenic diet is very limited for slc13a5 deficiency patients and may even aggravate their symptoms21,44. Indeed, our findings suggest opposite effects of NaCT and Glut-1 deficiency. While impaired Glut-1 is expected to lower transepithelial flux of glucose, hampered NaCT will relieve inhibition and lead to elevated glucose flux and attenuated citrate flux. Another aspect that should be considered is the distribution of Glut at different developmental stages. The expression of Glut-1 in the brains of P10 rats is mainly in parenchymal cells but is highly expressed in the vascular endothelial cells before P10 and later45. Therefore, if similar changes in Glut transporters occur in humans, the impaired regulation of Glut-1 by NaCT during early stages may cause developmental defects, thus causing SLC13A5 deficiency symptoms. This may suggest that early metabolic intervention during pregnancy or post-partum may also improve SLC13A5 deficiency prognosis.
Notwithstanding, SLC13A5 deficiency could be the result of a hampered metabolic communication in the liver-brain axis, suggesting that the orchestrated function of NaCT and glucose transporters in both organs is impaired, rather than the function of these transporters in the brain alone. Both citrate and succinate are crucial in mediating metabolic communication between organs, microbes, and host cells, indicating that these metabolites have additional functions beyond bioenergetics. For example, intestinal cells deliver microbiota-generated succinate to macrophages for immuno-metabolic control of inflammation6. This is achieved by SLC13 transporters, which mediate succinate uptake across the intestinal epithelium and by macrophages. In the kidney, the circulating citrate is constantly reabsorbed by the kidney SLC13s (NaDC-1 and NaDC-3) in the proximal tubule to maintain normal citrate homeostasis in the serum, but also in the urinary filtrate where citrate protects against calcareous kidney stone formation due to its Ca2+-chelating capacity3,5. Epithelial cells in the kidney, gut, and many other organs express SLC13 transporters, SLC26 transporters, and glucose transporters. Therefore, the mechanism we describe is likely a model system for a global metabolic communication system in the body. This system comprises different members of these transporter families performing different metabolic functions required by the cells and organs in which they are expressed. As we show in Fig. 5B, C, in Supplementary Fig. 9D and as reported by Khamaysi et al.20, NaCT is regulated by SLC26A6 similarly to its regulation by SLC26A1, attesting to the high modularity of this system. The SLC13-Glut or SLC13-SLC26 interaction modularity may also play a crucial role in cancer cell metabolism. The gene expression regulation of Glut-1 and Glut-3 (or Glut-2 in hepatocellular carcinoma) in cancer cells is dramatically alleviated, resulting in increased expression for high glucose uptake to maintain cell survival and tumor progression46. Our analysis of human gene expression using the GEPIA2 tool indicates a reciprocal change in Glut-1 and SLC13 gene expression. Indeed, the expression of Glut-1 is generally elevated or not changed in most of the cancer types we tested and accompanied by a significant downregulation of SLC13A2 and SLC13A5 expression (Supplementary Fig. 3). This will likely impair the interaction between Glut and SLC13s, which is predicted to increase glucose uptake and support cancer cell survival, even if Glut expression is not elevated. Therefore, the Glut-SLC13 regulatory interaction is a potential target for cancer treatment.
Based on the meticulous functional mapping (Figs. 5–7), we identified the H4c-H6b domain as a major regulatory array that controls NaCT function in a wide dynamic range and also affects NaCT stability and expression. As shown in Fig. 7, we identified three amino acid clusters in the H4c-H6b domain that act to either mediate interaction with partner proteins, stabilize the protein structure or interact with the catalytic domain. Hence, our functional mapping indicates that the H4c-H6b is a regulatory hub that integrates intracellular signals like protein-protein interactions with Glut or SLC26 transporters via residues of cluster III that face the intracellular surface of the protein (Fig. 7C, D). Subsequently, these interactions may affect the binding between the H4c-H6b domain and the catalytic HPin domain mediated by residues of HPin and cluster II (Fig. 7B and Supplementary Fig. 9B, C), thus regulating the transport function. In the mammalian citrate/succinate transporters NaCT, NaDC-1, and NaDC-3, the sequence of H4c is highly conserved, and that of H6b is moderately conserved (Supplementary Fig. 10B, C). On the other hand, the structural conservation of H4c and H6b is relatively high between mammalian NaCT and the bacterial LaINDY. In another bacterial homolog, vcINDY, the H6b region does not maintain a helical structure35. Therefore, H4c is crucial for transporter function, and the H4c-H6b interface had likely evolved to provide efficient metabolic regulation.
Methods
Animal care and use
All the work on Xenopus laevis and mice was approved by the Institutional Animal Care and Use Committee of the Ben Gurion University of the Negev (approval No. IL-83-10-2019(C) and BGU337-06-2024D). Xenopus laevis were purchased from Xenopus 1, Corp. 5654 Merkel Rd. Dexter, MI. 48130 and housed at the BGU preclinical facility, Israel. C57Bl/6JOlaHsd mice were purchased from Harlan, Israel. Mice were housed at the BGU preclinical facility. Rodent care practices are under sterile conditions, with sterile supplies. Rodent housing conditions are: 12:12 light:dark cycles at 20–24 °C and 30–70% relative humidity. Animals are fed autoclaved rodent chow and have free access to reverse osmosis filtered water. Rodents are housed in individually ventilated cages.
In vivo and in vitro silencing of NaCT using siRNA
The following siRNA sequences were used to silence NaCT expression:
NaCT siRNA sense, 5′ G.A.U.A.A.U.G.A.A.C.U.U.C.G.U.U.G.G.A.dT.dT 3′; NaCT siRNA antisense, 5′ P.U.C.C.A.A.C.G.A.A.G.U.U.C.A.U.U.A.U.C.dT.dT 3′; Non targeting siRNA (Scr) sense, 5′ A.U.C.G.U.A.C.G.U.A.C.C.G.U.C.G.U.A.U.dT.dT 3′; Non targeting-siRNA (Scr) antisense, 5′ A.U.A.C.G.A.C.G.G.U.A.C.G.U.A.C.G.A.U.dT.dT 3′; NaCT siRNA* sense, 5′ C.C.A.C.A.G.A.G.U.G.C.A.C.A.A.G.U.A.A.dT.dT 3′; NaCT siRNA* antisense, 5′ U.U.A.C.U.U.G.U.G.C.A.C.U.C.U.G.U.G.G.dT.dT 3′.
All siRNAs were obtained from the ON-TARGETplus siRNA collection (Dharmacon). To validate NaCT silencing in vitro, AML12 mouse hepatocyte cells (ATCC, Virginia, USA) were transfected with either scrambled control siRNA, a previously reported NaCT siRNA* sequence24, or the newly designed NaCT siRNA described here for the first time. Transfections were performed using the TransIT-X2 Dynamic Delivery System (Mirus Bio, Wisconsin, USA) at final siRNA concentrations of 50 nM and 100 nM, according to the manufacturer’s instructions. Cells were lysed 120 h post-transfection for protein extraction and analysis.
For in vivo NaCT silencing, we used a modified sequence of these siRNA sequences based on the siGENOME platform, as described in Fig. 1E. Specifically, male mice (1 month old) received tail vein injections of either NaCT siRNA or scrambled control siRNA (Scr) once a week for seven consecutive weeks. The siRNAs were complexed with in vivo-jetPEI (Polyplus, Illkirch, France) for delivery. Blood glucose levels were measured using a standard portable glucometer from tail blood collected three days after injection, every other week. At week 8, mice were euthanized by CO₂ inhalation, and livers were harvested for hepatocyte isolation. Portions of the cell population were lysed in lysis buffer for protein extraction only when hepatocyte preparations yielded over 12 × 10⁶ viable cells. The remaining viable hepatocytes (12 × 106 cells) were maintained in culture for subsequent uptake assays, as described later. The in vivo and in vitro NaCT silencing efficiencies were assessed by Western blotting. Samples were lysed in ice-cold buffer containing 1× PBS, 10 mM Na⁺ pyrophosphate, 50 mM NaF, 1 mM Na⁺ orthovanadate, 1% Triton X-100, and a protease inhibitor cocktail (Roche), followed by sonication (SONICS, CT, USA). Proteins were separated on 8% SDS-PAGE gels and transferred to nitrocellulose membranes (GE Whatman, Pittsburgh, PA). Membranes were probed with primary antibodies against NaCT (LS-Bio, CA, USA; 1:1000), Calreticulin (Affinity BioReagents, CO, USA; 1:500), and Actin (Sigma; 1:30,000). Detection was performed using HRP conjugated goat anti-rabbit secondary antibody (ThermoFisher; 1:2000) and visualized with the ChemiDoc Imaging System (Bio-Rad). Densitometric analysis was performed using ImageJ software.
Plasmid construction, mutagenesis, and cRNA preparation
We used the human NaCT in pC4-TOPO (NCBI accession no. BC_104795), the mouse SLC26A6 (NCBI accession no. NM_134420) in the pCMV6- AC-mKate vector, the human SLC26A1 (NCBI accession no. NM_006516.3) in the pcDNA3.1-C-(k)DYK. The human NaCT was subcloned into both pCMV6-AC-Myc-His and pCMV6-AC-mKate. All site-directed mutants were generated with QuikChange Lightning site-directed mutagenesis kit (Agilent, Santa Clara, CA). All constructs, including the in-house generated site-specific mutants, were verified by sequencing and immunoblotting the protein products. The genes in pC4-TOPO were linearized using relevant restriction enzymes and transcribed in vitro with T7 mMessage mMachine ultra (Thermo Fisher Scientific). A list of plasmids can be found in Supplementary Table 3.
Injection of oocytes and membrane preparation
Oocytes were obtained by a partial ovariectomy of female Xenopus laevis (Xenopus 1, Dexter, MI). Briefly, the frogs were anesthetized, and follicle cells were removed in an OR-2 calcium-free medium. The defolliculated oocytes were washed with OR-2 calcium-free medium, and healthy oocytes in stages V to VI were identified, collected under binoculars, and maintained overnight at 18 °C in an ND96 solution containing 96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2,1 mM MgCl2, and 5 mM HEPES, pH 7.5. Then, 32 nl of the different cRNA were injected into the oocytes using a Nanoliter 2010 injector (World Precision Instruments, Inc., Sarasota, FL). Similar volumes and concentrations (4 g/l) of cRNA or water were mixed to inject equal amounts of cRNA per oocyte. The oocytes were incubated at 18 °C in an ND96 solution with pyruvate and antibiotics and were studied 48–96 h after cRNA injection. The crude membrane fraction was collected 2–5 days following cRNA injection. Batches of 15–20 oocytes were homogenized in 1 ml of HEDP buffer (100 mM HEPES, 1 mM EDTA, 5ul of protease inhibitor, pH 7.6). The supernatant was carefully pipetted after centrifugation of the homogenates onto 7 ml of ice-cold 15% sucrose in HEDP buffer using Beckman polyallomer centrifuge tubes. The samples were centrifugated at 175,000 × g for 90 min using the Beckman SW41Ti rotor. The membrane pellets were then solubilized in a sample buffer for subsequent Western blot analysis. A list of cell lines and primary cells can be found in Supplementary Table 2.
Voltage and current measurement in oocytes
Voltage and current recordings were performed with a two-electrode voltage clamp. The current was recorded with a Warner Instrument Corporation amplifier model OC-725C (Hamden, CT) and digitized via an A/D converter (Digidata 1550A; Axon Instruments, Inc.). The electrodes were backfilled with a 3 M KCl solution. During measurements, two channels were used to record and control the membrane potential. The data were analyzed using the Clampex 10 system (Axon Instruments, Inc.). The following solutions were used as indicated in the figures: standard HEPES buffered ND96 oocyte regular medium. 10 mM Na+-citrate was added to the solutions, as indicated in the figures.
Western blotting, co-immunoprecipitation, and biotinylation
For Western blotting and co-immunoprecipitation, cell lysates were prepared by incubating the cells with an ice-cold lysis buffer containing PBS, 10 mM Na+-pyrophosphate, 50 mM NaF, 1 mM Na+-orthovanadate, 1% Triton X-100, and a mixture of protease inhibitors (Roche). For CoIP, the extracts were incubated overnight at 4 °C with primary antibody (1 µg/100 µl anti-HisX6 tag monoclonal antibody, Thermo Fisher Scientific; 1 µg/100 µl anti-tRFP, Evrogen; 1 µg/50 µl of anti-Glut1, Cell Signaling). All samples were subsequently incubated with G Sepharose beads, and protein-bead complexes were collected by centrifugation followed by three washes. Subsequently, the samples were incubated at 37 °C for 30 min in SDS-containing sample buffer and transferred to nitrocellulose membranes. Then, the membranes were incubated with either anti-HisX6 (1:1000) or anti-tRFP (1:3000) antibodies overnight at 4 °C or with HRP conjugated anti-Flag antibody (1:1000) (Sigma–Aldrich) for 1 h at room temperature (RT) as indicated. On the next day, the membranes were exposed to either goat anti-mouse (1:1000) or goat anti-rabbit (1:2000) IgG HRP conjugated antibodies (Thermo Fisher Scientific) for 1 h at RT. Protein enrichment by pull-down assays was tested for the antibodies used in this work (Supplementary Fig. 12).
To monitor human NaCT surface expression, we used a modified biotinylation assay. Briefly, transfected cells were washed and incubated with EZ-Link Sulfo-NHS-SS-Biotin (0.5 mg/ml: Thermo Fisher Scientific) for 30 min on ice. The biotin was quenched with 50 mM glycine, and lysates were prepared with the lysis buffer described above. Finally, 50 μl of a 1:1 slurry of immobilized neutravidin beads (Thermo Fisher Scientific) were added to 0.4 ml of cell extracts and incubated for 2 h at 4 °C. The beads were washed with a binding buffer, and proteins were released with 50 μl of SDS-loading buffer. The extracts were loaded onto 10% Tris-glycine SDS-PAGE gels, which were subsequently transferred onto a nitrocellulose membrane and probed with anti-NaCT mAb (Santa Cruz Biotechnology, Dallas, TX) diluted at 1:500. The membranes were then incubated with a secondary goat anti-mouse IgG peroxidase conjugate antibody (Thermo Fisher Scientific), diluted at 1:1000. Densitometric analysis was performed using ImageJ software for biotinylated and input immunoblot images. The ratio between the densitometry values (biotin/input) for each lane was averaged between independent experiments. A list of antibodies can be found in Supplementary Table 1.
Hepatocyte isolation and culturing
Hepatocytes were isolated from adult male C57BL/6 mice (1-3 months) using a two-step collagenase perfusion method. Initially, the liver was perfused through the abdominal inferior vena cava, followed by cutting the hepatic portal vein. The liver was then washed with Hanks’ calcium and magnesium-free buffer for 6 min to remove blood. After blood removal, the calcium-free buffer was replaced with a liver digest media containing collagenase buffer (0.5 mg/ml) and perfused for an additional 7 min. Throughout the entire procedure, a perfusion rate of 5 ml/min and a temperature of approximately 37 °C were maintained for both solutions. Following perfusion with the collagenase solution, the liver was promptly excised, and the gall bladder and remnants of the diaphragm were removed. The liver was transferred to a sterile Petri dish containing plating medium. The cells were filtered through a 70-micron strainer and washed via low-speed centrifugation at 500 × g for 5 min at 4 °C. Subsequently, the cells were centrifuged for an additional 10 min after diluting the medium with Percoll (1:1). Finally, the cells were centrifuged again under the same conditions as the initial centrifugation. Cell viability and yield were assessed using trypan blue exclusion. The isolated hepatocytes were seeded onto collagen-coated 24-well plates, with 300,000 cells per well. Plating medium (10% FBS, Na-pyruvate 2 mM, 2% penicillin-streptomycin, 1 µM dexamethasone, 2 µM insulin in DMEM with glucose) was added. After 2 h, the plating medium was replaced with maintenance media (0.2% BSA in DMEM with glucose). The cells were then incubated for 8 h at 37 °C and 5% CO2. The attached cells were incubated for an additional 8 h in serum-free low-glucose culture media (DMEM supplemented with 0.75 g glucose, 0.5% fatty acid-free BSA, 2% penicillin-streptomycin, and 10 mM HEPES, pH 7.4). Then, the cells were incubated for an additional 8 h in serum- and glucose-free media (DMEM supplemented with 0.5% fatty acid-free BSA, 20 mM sodium lactate, 2 mM sodium pyruvate, and 10 mM HEPES, pH 7.4).
Metabolite uptake measurements
In primary hepatocytes: The cell cultures were washed twice with wash buffer (25 mmol/L HEPES [pH 7.4], 140 mmol/L NaCl, 5 mmol/L KCl, 1.8 mmol/L CaCl2, 0.8 mmol/L MgSO4, and with or without 5 mmol/L glucose). Subsequently, cells were further incubated at RT in wash buffer supplemented with 10 mM citrate and 1 µCi of 3H-citrate per 1 ml of wash buffer solution for either 15, 30, 120, or 240 min as indicated in the figures. For 2-DG uptake, the cells were washed twice with glucose-containing or glucose-free wash buffer and then incubated at RT with “cold” 10 mM Na+-citrate, 2.5 mM 2-DG, and 2 µCi of 3H-2-DG per 1 ml of solution for 30 min. After incubation, the cells were washed twice. Background measurements were determined in the presence of unlabeled 10 mM citrate or 2.5 mM 2-DG. The data was analyzed in CPM for the counted cell numbers (300,000 cells/well) or presented as percent of control for each day of experiments.
In cell-line cultures: HEK293-T cells were transfected with the relevant plasmids using the TransIT-X2 transfection reagent. On the day of the experiment, the cells were incubated with wash buffer containing 140 mM NaCl, 5 mM KCl, 10 mM HEPES, and 5 mM Na+-citrate at pH 7.4. After a 60 min incubation period, the cells were washed with the same wash buffer, supplemented with 5 mM Na+-citrate, 2.5 mM 2-Deoxy-D-glucose, and 1 µCi of 3H-2-DG per 1 ml of solution. The cells were then incubated for an additional 30 min. Subsequently, the cells were washed twice with wash buffer containing 2.5 mM 2-DG.
The human THLE2 or HepG2 cells were counted, seeded (300,000 cells/well), and incubated for 8 h in serum-free low-glucose culture media, followed by additional 8 h in serum- and glucose-free media, as described earlier for primary mouse hepatocyte cultures. The cells were then washed twice in the presence or absence of glucose and incubated at RT with 2 µCi of 3H-2-DG per 1 ml of solution for 30 min with or without 10–500 µM of the SLC13A5 inhibitor (BI01383298), as indicated. After incubation, the cells were washed twice with glucose-free wash buffer containing 10 mM Na+-citrate and 2.5 mM 2-DG.
Finally, in all cultures, the cells were lysed in 0.5 ml of 1 M NaOH, and the cell lysates were transferred to scintillation vials containing 0.25 ml of 2 M HCl. The radioactivity was determined by liquid scintillation counting using a Packard 1900CA TRI-CARB analyzer. The data was analyzed as CPM/cell following cell counting for each well.
Immunofluorescence and confocal imaging
HepG2 cells were cultured overnight on glass coverslips, washed in 1X PBS, and fixed with ice-cold Methanol for 5 min. Then, the cells were washed and incubated with blocking solution containing 0.1% Triton X-100, 3% donkey serum, and 2% BSA in PBS for 1 h at RT. Next, the samples were incubated with anti-NaCT (Santa Cruz Biotechnology, Dallas, TX) diluted at 1:25 and anti-Glut-1 (Cell Signaling) diluted at 1:50 for 1 h at RT. After 3 wash cycles with PBST (0.1% Triton X 100 in PBS), the samples were incubated with Alexa 488 conjugated donkey anti-mouse and Cy3-conjugated donkey anti-rabbit antibodies (Jackson Immunoresearch Inc.) for 1 h at RT. After 3 wash cycles with PBST, the samples were mounted on glass slides and incubated overnight. In addition, HEK293-T cells were cultured, fixed, and incubated with primary antibodies, as described above. After 3 wash cycles, the samples were incubated with Alexa488-conjugated donkey anti-mouse and Cy5-conjugated goat anti-rabbit antibodies (Jackson Immunoresearch Inc.) for 1 h at RT. The samples were analyzed using a Nikon Ti-E inverted confocal laser scanning microscope or a Zeiss LSM880 confocal laser scanning microscope. Images were acquired using NIS Elements imaging software version 4.40 or using Zen software, respectively. A list of antibodies can be found in Supplementary Table 1.
Metabolic measurements
HEK293-T cells were cultured in fetal bovine serum (Biological Industries, Israel, or GIBCO) containing DMEM (Biological Industries, Israel) and transfected with NaCT WT or different H4c domain mutants on a 6-well plate. Then, 48 h post-transfection, cells were transferred to a Seahorse 96-well microplate (Agilent, Santa Clara, CA) and incubated overnight. Glycolysis (extracellular acidification rate) and mitochondrial respiration (Oxygen consumption rate) of the cells were monitored using a Seahorse XFe96 Analyzer (Agilent), according to manufacturer’s instructions. Before the experiment, the cell media was substituted with an assay media: Seahorse XF DMEM (Agilent) supplemented with 2 mM l-glutamine (Biological Industries, Beit Haemek, Israel) and 1 mM sodium citrate (Sigma). For mitochondrial respiration measurements, 2.5 mM glucose (Sigma) and 1 mM sodium pyruvate (Sigma) were added to the media. This was followed by incubation for 60 min at 37 °C (in a non-CO2incubator) before loading into the XFe96 analyzer (Agilent). The injected compounds were loaded into cartridge ports with oxygen probes (20–25 μL/port), and the cartridge was calibrated. For glycolysis measurements, the following were injected: Glucose at a final concentration of 2.5 mM, then oligomycin (Sigma) at a final concentration of 1.5 μM and, finally, the glycolysis inhibitor 2-DG, 50 mM (Santa Cruz Biotechnology). For mitochondrial respiration measurements, the following were injected: Oligomycin at a final concentration of 1.5 μM, then carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone) FCCP (Sigma) at a final concentration of 1.5 μM and, finally, 0.5 μM Antimycin (Agilent)/Rotenone (Agilent). After each seahorse experiment, the cell number in each well was measured by monitoring the fluorescence of DAPI staining, 0.1 μg/ml (Sigma), using a plate reader (Infinite M200 PRO, TECAN).
Protein models
The structure prediction of NaCT and Glut interaction was performed using the Alphafold2 multimer software via the Google Colab online interface47. The protein sequences used for the prediction are human NP_808218.1 (human NaCT) NP_000331.1 human (Glut-2). The human NaCT structures (PDB_ID:7JSK) and Alphafold model images were generated and visualized using UCSF ChimeraX or Chimera 1.1648.
Real-time intracellular Na+ imaging
The imaging system consisted of an Eclipse Ti inverted microscope (Nikon, Tokyo, Japan), a PE-4000 LED monochromator (CoolLEd, Andover, UK), and a Hamamatsu flash 4.0LT camera (Hamamatsu Photonics, Hamamatsu City, Japan). Fluorescence images were acquired and analyzed with NIS-Elements software. HEK293-T cells expressing recombinant NaCT or other proteins were cultured on coverslips, were incubated for 1 h at RT with 1.5 μg of the ING2-AM Na+ sensitive dye (Ion biosciences), and washed with regular solution (140 mM NaCl, 10 mM HEPES, 10 mM glucose, 5 mM KCl, 1 mM MgCl2, 1 mM CaCl2, and pH was adjusted to 7.4) for 20 min on stage. Next, regular solution was replaced with a citrate solution (regular solution was adjusted to 110 mM NaCl and 10 mM Na+ citrate) or with a Na+-free solution (regular solution was adjusted to 140 mM N-methyl-D-glucamine (NMDG)). The osmolarity of all solutions was adjusted to 310 mOsm with the major salt.
Quantification and statistical analysis
Prism 10.0 Software (GraphPad Inc., San Diego, CA, USA) was used for statistical data analyses with a two-tailed Student’s t-test. We assessed normal distribution for three or more groups using Q–Q plot analysis (Supplementary Fig. 11) followed by the appropriate ANOVA multiple comparison tests, as indicated in the Figure legends. No statistical methods were used to predetermine sample size.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data are provided with this paper.
References
Mills, E. & O’Neill, L. A. Succinate: a metabolic signal in inflammation. Trends Cell Biol. 24, 313–320 (2014).
Toma, I. et al. Succinate receptor GPR91 provides a direct link between high glucose levels and renin release in murine and rabbit kidney. J. Clin. Investig. 118, 2526–2534 (2008).
Khamaysi, A. et al. Systemic succinate homeostasis and local succinate signaling affect blood pressure and modify risks for calcium oxalate lithogenesis. J. Am. Soc. Nephrol. 30, 381–392 (2019).
Bhutia, Y. D., Kopel, J. J., Lawrence, J. J., Neugebauer, V. & Ganapathy, V. Plasma membrane Na(+)-coupled citrate transporter (SLC13A5) and neonatal epileptic encephalopathy. Molecules 22, 378 (2017).
Ohana, E., Shcheynikov, N., Moe, O. W. & Muallem, S. SLC26A6 and NaDC-1 transporters interact to regulate oxalate and citrate homeostasis. J. Am. Soc. Nephrol. 24, 1617–1626 (2013).
Fremder, M. et al. A transepithelial pathway delivers succinate to macrophages, thus perpetuating their pro-inflammatory metabolic state. Cell Rep. 36, 109521 (2021).
Pajor, A. M. Molecular properties of the SLC13 family of dicarboxylate and sulfate transporters. Pflug. Arch. 451, 597–605 (2006).
Markovich, D. & Murer, H. The SLC13 gene family of sodium sulphate/carboxylate cotransporters. Pflug. Arch. 447, 594–602 (2004).
Bergeron, M. J., Clemencon, B., Hediger, M. A. & Markovich, D. SLC13 family of Na(+)-coupled di- and tri-carboxylate/sulfate transporters. Mol. Asp. Med. 34, 299–312 (2013).
Thevenon, J. et al. Mutations in SLC13A5 cause autosomal-recessive epileptic encephalopathy with seizure onset in the first days of life. Am. J. Hum. Genet. 95, 113–120 (2014).
Neuschafer-Rube, F. et al. The mammalian INDY homolog is induced by CREB in a rat model of type 2 diabetes. Diabetes 63, 1048–1057 (2014).
Birkenfeld, A. L. et al. Deletion of the mammalian INDY homolog mimics aspects of dietary restriction and protects against adiposity and insulin resistance in mice. Cell Metab. 14, 184–195 (2011).
Milosavljevic, S. et al. Untargeted metabolomics of Slc13a5 deficiency reveal critical liver-brain axis for lipid homeostasis. Metabolites 12, 351 (2022).
Dirckx, N. et al. A specialized metabolic pathway partitions citrate in hydroxyapatite to impact mineralization of bones and teeth. Proc. Natl. Acad. Sci. USA 119, e2212178119 (2022).
Kim, M. O. et al. PKA and cAMP stimulate proliferation of mouse embryonic stem cells by elevating GLUT1 expression mediated by the NF-kappaB and CREB/CBP signaling pathways. Biochim. Biophys. Acta 1820, 1636–1646 (2012).
Herzig, S. & Shaw, R. J. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 19, 121–135 (2018).
Kopel, J. et al. The hepatic plasma membrane citrate transporter NaCT (SLC13A5) as a molecular target for metformin. Sci. Rep. 10, 8536 (2020).
Rogina, B., Reenan, R. A., Nilsen, S. P. & Helfand, S. L. Extended life-span conferred by cotransporter gene mutations in Drosophila. Science 290, 2137–2140 (2000).
Navale, A. M. & Paranjape, A. N. Glucose transporters: physiological and pathological roles. Biophys. Rev. 8, 5–9 (2016).
Khamaysi, A., Aharon, S., Eini-Rider, H. & Ohana, E. A dynamic anchor domain in slc13 transporters controls metabolite transport. J. Biol. Chem. 295, 8155–8163 (2020).
Klotz, J., Porter, B. E., Colas, C., Schlessinger, A. & Pajor, A. M. Mutations in the Na(+)/citrate cotransporter NaCT (SLC13A5) in pediatric patients with epilepsy and developmental delay. Mol. Med. 22, 310–321 (2016).
Jaramillo-Martinez, V., Ganapathy, V. & Urbatsch, I. L. A home run for human NaCT/SLC13A5/INDY: cryo-EM structure and homology model to predict transport mechanisms, inhibitor interactions and mutational defects. Biochem. J. 478, 2051–2057 (2021).
Sauer, D. B. et al. Structure and inhibition mechanism of the human citrate transporter NaCT. Nature 591, 157–161 (2021).
Brachs, S. et al. Inhibition of citrate cotransporter Slc13a5/mINDY by RNAi improves hepatic insulin sensitivity and prevents diet-induced non-alcoholic fatty liver disease in mice. Mol. Metab. 5, 1072–1082 (2016).
Tang, Z., Kang, B., Li, C., Chen, T. & Zhang, Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 47, W556–W560 (2019).
Turro, E. et al. Whole-genome sequencing of patients with rare diseases in a national health system. Nature 583, 96–102 (2020).
Karniski, L. P. et al. Immunolocalization of sat-1 sulfate/oxalate/bicarbonate anion exchanger in the rat kidney. Am. J. Physiol. 275, F79–F87 (1998).
Wu, M. et al. Extracellular Cl(-) regulates human SO4 (2-)/anion exchanger SLC26A1 by altering pH sensitivity of anion transport. Pflugers Archiv. Eur. J. Physiol. 468, 1311–1332 (2016).
Markovich, D. Physiological roles of mammalian sulfate transporters NaS1 and Sat1. Arch. Immunol. Ther. Exp. (Warsz.) 59, 113–116 (2011).
Gopal, E. et al. Expression and functional features of NaCT, a sodium-coupled citrate transporter, in human and rat livers and cell lines. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G402–G408 (2007).
Quondamatteo, F. et al. Localization of the sulfate/anion exchanger in the rat liver. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G1075–G1081 (2006).
Castro, M. A., Angulo, C., Brauchi, S., Nualart, F. & Concha, I. I. Ascorbic acid participates in a general mechanism for concerted glucose transport inhibition and lactate transport stimulation. Pflug. Arch. 457, 519–528 (2008).
Zambrano, A. et al. Cytokine stimulation promotes increased glucose uptake via translocation at the plasma membrane of GLUT1 in HEK293 cells. J. Cell Biochem. 110, 1471–1480 (2010).
Mulligan, C. et al. The bacterial dicarboxylate transporter VcINDY uses a two-domain elevator-type mechanism. Nat. Struct. Mol. Biol. 23, 256–263 (2016).
Sauer, D. B. et al. Structural basis for the reaction cycle of DASS dicarboxylate transporters. eLife 9, e61350 (2020).
Sauer, D. B. et al. Structural basis of ion - substrate coupling in the Na(+)-dependent dicarboxylate transporter VcINDY. Nat. Commun. 13, 2644 (2022).
Kopel, J. J., Bhutia, Y. D., Sivaprakasam, S. & Ganapathy, V. Consequences of NaCT/SLC13A5/mINDY deficiency: good versus evil, separated only by the blood-brain barrier. Biochem. J. 478, 463–486 (2021).
Holman, G. D. Structure, function and regulation of mammalian glucose transporters of the SLC2 family. Pflug. Arch. 472, 1155–1175 (2020).
Brzica, H. et al. The liver and kidney expression of sulfate anion transporter sat-1 in rats exhibits male-dominant gender differences. Pflug. Arch. 457, 1381–1392 (2009).
Karim, S., Adams, D. H. & Lalor, P. F. Hepatic expression and cellular distribution of the glucose transporter family. World J. Gastroenterol. 18, 6771–6781 (2012).
Kopel, J., Grooms, A., Ganapathy, V. & Clothier, J. Metformin, valproic acid, and starvation induce seizures in a patient with partial SLC13A5 deficiency: a case of pharmaco-synergistic heterozygosity. Psychiatr. Genet. 31, 32–35 (2021).
Castellotti, B. et al. Screening of SLC2A1 in a large cohort of patients suspected for Glut1 deficiency syndrome: identification of novel variants and associated phenotypes. J. Neurol. 266, 1439–1448 (2019).
Kass, H. R., Winesett, S. P., Bessone, S. K., Turner, Z. & Kossoff, E. H. Use of dietary therapies amongst patients with GLUT1 deficiency syndrome. Seizure 35, 83–87 (2016).
Hardies, K. et al. Recessive mutations in SLC13A5 result in a loss of citrate transport and cause neonatal epilepsy, developmental delay and teeth hypoplasia. Brain 138, 3238–3250 (2015).
Devaskar, S., Zahm, D. S., Holtzclaw, L., Chundu, K. & Wadzinski, B. E. Developmental regulation of the distribution of rat brain insulin-insensitive (Glut 1) glucose transporter. Endocrinology 129, 1530–1540 (1991).
Ancey, P. B., Contat, C. & Meylan, E. Glucose transporters in cancer - from tumor cells to the tumor microenvironment. FEBS J. 285, 2926–2943 (2018).
Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022).
Pettersen, E. F. et al. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Acknowledgements
This work was supported by the Israeli Science Foundation (ISF grant no. 592/21) to E.O. by a science forefront grant sponsored by the Ministry of Science & Technology, Israel (No. 0002069) to E.O. and Dr. Miriam Amiram. We thank Mr. Yehonatan Krenzler for his technical assistance.
Author information
Authors and Affiliations
Contributions
N.Y. designed and conducted experiments; A.Khamaysi. designed and conducted experiments; L.S. designed and performed experiments; A.Keshset designed and conducted experiments; M.T. isolated primary hepatocytes; M.F. conducted experiments; H.E.R. designed and conducted experiments; E.O. designed the study and wrote the paper. All authors analyzed the data that they obtained and contributed to the writing and critical reading of the article.
Corresponding author
Ethics declarations
Competing interests
The patent application “Succinate-Binding Polypeptides and Use Thereof” (US011332510B2) by E.O. may be related to this work. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jan Hengstler, Da-Neng Wang, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yehoshua, N., Khamaysi, A., Shimshilashvili, L. et al. Regulatory interaction between metabolite transporters coordinates glucose and exometabolite fluxes to drive bioenergetics. Nat Commun 16, 6819 (2025). https://doi.org/10.1038/s41467-025-62103-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62103-3
