
Abstract
Developing targeted treatment for glioblastoma is crucial but challenging. Herein, we propose a size-variable self-feedback nanomotor system, utilizing the unique high-calcium microenvironment of glioblastoma to prevent its progression through mitochondrial mineralization. It comprises three components: a self-feedback degradable lipid shell (containing nitric oxide-releasing lipid and nitric oxide-responsive degradable lipid), a motion nanomotor core (containing L-arginine derivatives and carboxyl-rich zwitterionic monomers for Ca2+ recruitment), and curcumin (inhibiting Ca2+ efflux). Nitric oxide-releasing lipid can be catalyzed by inducible nitric oxide synthase to release nitric oxide, triggering nitric oxide-responsive degradable lipid degradation. Initially, the larger nanomotors (~ 500 nm) penetrate the blood-brain barrier via chemotaxis towards glioblastoma microenvironment. During chemotaxis, the lipid shell gradually degrades, releasing smaller nanomotor core (~50 nm), which can target mitochondria and recruit Ca2+ to induce mitochondrial mineralization together with curcumin, inhibiting glioblastoma progression. This work may provide a glioblastoma-specific treatment strategy.
Similar content being viewed by others


Introduction
Glioblastoma (GBM) is one of the most aggressive and deadly brain tumors, classified by the World Health Organization as the most malignant glioma (Grade IV)1. The standard treatment for GBM typically involves a combination of surgical resection followed by radiotherapy and/or chemotherapy. However, the characteristic infiltrative growth of GBM makes it difficult to eradicate surgically and may damage critical brain functional areas. The efficacy of first-line chemotherapeutic agents such as temozolomide (TMZ) is often limited by the inherent resistance of tumor cells and severe systemic toxicity (including hematologic toxicity and thrombocytopenia associated with long-term and high-dose administration)2. The presence of the blood-brain barrier (BBB) also prevents most drugs entering the tumor cells. Although radiotherapy has shown good cytotoxic effects, its low selectivity and specificity can also cause irreversible damage to normal tissues. Consequently, the median survival for GBM patients remains low at 14.6–20.5 months, with a 5-year survival rate of less than 10%3. Emerging modalities such as electro-fields therapy4 and focused ultrasound therapy5 have also shown promising advancements in solid tumor therapy in clinic or pre-clinical settings. In situ drug delivery strategies for GBM (convection-enhanced delivery (CED), implantation of slow-release devices) circumvent the BBB by delivering the drug directly to the tumor or postoperative residual cavity, achieving high local drug exposure and reducing systemic toxicity. However, higher operational requirements, the need for more complex equipment, unpredictable and heterogeneous drug distribution, and susceptibility to localized brain tissue injury, edema, or inflammation may limit their application6. Therefore, developing targeted therapeutic strategies specifically for GBM has emerged as a critical area of research.
The unique microenvironment of GBM provides valuable insights for the design of targeted therapeutic strategies (Supplementary Table 1). GBM is characterized by an immunosuppressive tumor microenvironment, often referred to as a “cold” tumor, which has spurred researchers to actively explore immunotherapeutic approaches for GBM. These include tumor vaccines1,7,8, immune checkpoint inhibitors (ICIs)9, and chimeric antigen receptor T-cell (CAR-T) therapies10, etc. However, the efficacy of immunotherapy is affected by individual patient variability, leading to insufficient or excessive immune responses that limit its broad application11. In addition to immunotherapy, molecular targeted therapies have been developed to target cellular molecules that promote the proliferation and differentiation of GBM, such as anti-angiogenic therapy12,13 and tyrosine kinase inhibitors14,15. However, their effectiveness is often limited by factors such as drug resistance in tumor cells and high recurrence rates following treatment16. Gene therapy represents another promising approach, targeting specific genes involved in the proliferation, migration, invasion, apoptosis, and angiogenesis of malignant glioma cells, including gene editing corrections using CRISPR-Cas9, or suppressing immune-suppressive genes expressed in brain tumors to reprogram the tumor immune microenvironment3. However, challenges such as the poor stability of nucleic acids in vivo, variable transfection efficiency, and high treatment costs have restricted its widespread application3. Therefore, there is a pressing need to find new therapeutic strategies specifically for GBM. In fact, beyond the aforementioned characteristics, the intracellular Ca2+ in GBM is significantly higher than that of normal tissues, and this elevated Ca2+ level is crucial for tumor progression17,18. Mitochondria, which contain abundant Ca2+ (≈ 570 nM), are key organelles for the storge and regulation of intracellular Ca2+17,19,20,21.
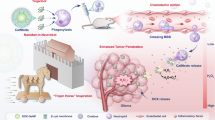
In this work, we propose a therapeutic strategy that induces mitochondrial mineralization in tumor cells within GBM to inhibit tumor growth, thereby achieving effective treatment of GBM with minimal side effects on normal tissues (Fig. 1). Specifically, we developed size-variable self-feedback nanomotors comprising a self-propelled nanomotor core, a degradable lipid shell for self-feedback, and the drug curcumin (Cur), which inhibits Ca2+ efflux. Firstly, L-arginine (L-Arg) derivatives and zwitterionic monomer 3-[[2- (methacryloxy) ethyl] dimethylammonium] propionate (CBMA) were used to synthesize PAC nanomotors (PAC NMs) via free radical polymerization reaction. These PAC NMs served as the core of the nanomotor composite system. PAC NMs could target mitochondria via chemotaxis towards the high concentration of inducible nitric oxide synthase (iNOS), and recruit cytoplasmic Ca2+ through their abundant surface carboxyl groups, thereby increasing the local mitochondrial Ca2+ concentration and inducing mitochondrial mineralization. To prevent the carboxyl groups on PAC NMs from being occupied by Ca2+ in the bloodstream and losing their ability to recruit Ca2+, self-feedback liposomes were used to encapsulate the PAC NMs along with curcumin (Cur), to construct the nanomotor composite system NO-Lip@PAC@Cur NMs. The self-feedback liposomes contained nitric oxide (NO)-releasing lipid (NOR) and NO-responsive degradable lipid (NOD). The self-feedback mechanism operates as follows: NO-Lip@PAC@Cur NMs could effectively target GBM via chemotaxis towards highly expressed iNOS in the GBM microenvironment. During this chemotactic process, NO released from NOR reacts with the o-phenylenediamine group in NOD to form an amide-derived benzotriazole group. This compound spontaneously hydrolyzes, leading to the gradual degradation of the liposomes (~ 500 nm). This process releases the encapsulated PAC NMs (~ 50 nm) and Cur. The released PAC NMs recruit Ca2+ to mitochondria, while Cur inhibits Ca2+ efflux, thereby enhancing mitochondrial mineralization, inducing mitochondrial dysfunction, and ultimately triggering tumor cell death.

Schematic diagram of (a) preparation of NO-Lip@PAC@Cur and its schematic diagram in response to NO degradation and (b) NO-Lip@PAC@Cur selectively inducing mitochondrial mineralization for the treatment of GBM. By Figdraw.
Results
Preparation of NO-Lip@PAC NMs and their degradation behavior in response to NO
NO-Lip@PAC NMs were composed of a nanomotor core (PAC NMs) and a self-feedback degradable lipid shell. Firstly, PAC NMs were prepared. The monomer was prepared according to the method reported in the literature22,23. L-Arg was combined with methacrylic anhydride to form N-methacryloyl-L-arginine (M-Arg) (Supplementary Fig. 1). Both 1H-NMR and 13C-NMR confirmed the successful preparation of M-Arg (Supplementary Figs. 2 and 3)22,23,24. Next, the zwitterionic monomers CBMA and M-Arg were reacted by radical polymerization to obtain PAC NMs, enriched with carboxyl and guanidine groups (Supplementary Fig. 4)25. Transmission electron microscope (TEM) images demonstrated that the synthesized PAC NMs have a uniform size of approximately 50 nm (Fig. 2a). According to the dynamic light scattering (DLS) test, the hydration diameter of PAC NMs was 128.7 nm (Fig. 2b). The structure of PAC NMs was further characterized by Fourier transform infrared spectroscopy (FTIR) spectra. As shown in Supplementary Fig. 5, there were amino II band of M-Arg (1530 cm−1) in PAC, amino I band of BAC (1650 cm−1), and C = O (1716 cm−1) in CBMA. Meantime, the disappearance of the characteristic peak of the C = C double bond that originally belonged to the cross-linker indicates the reaction of free radical polymerization occurred, confirming the successful synthesis of PAC NMs26,27.

a TEM images (Scale bar: 500 nm), (b) DLS and (c) Zeta potential of different samples (I: PAC, II: NO-Lip, III: NO-Lip@PAC; n = 3 independent samples). d Representative confocal laser scanning microscope (CLSM) images of NO-Lip@PAC (Scale bar: 50 μm, green: DiO labeled NO-Lip, red: Cy5 labeled PAC; n = 3 independent samples); Representative DLS changes of NO-Lip@PAC NMs incubated with (e) PBS and (f) 100 μM H2O2 for different times (n = 3 independent samples). g Representative TEM images of NO-Lip@PAC NMs incubated with PBS and 100 μM H2O2 for different times (Scale bar: 500 nm; red arrows indicate PAC released by lipid degradation; n = 3 independent samples). h Representative CLSM images of NO-Lip@PAC incubated with HUVECs and Gl261 cellular lysates incubated for different times (Scale bar: 10 μm, green: DiO labeled Lip or NO-Lip; Red: Cy5 labeled PAC; Purple arrows indicate PAC released by lipid degradation; n = 3 independent samples). Data in (b and c) were presented as mean ± SD. Source data are provided as a Source Data file.
Subsequently, we used a liposomal shell to protect the PAC NMs, thereby preventing the saturation of the carboxyl groups in the PAC NMs by Ca2+ during blood circulation. The liposome portion was composed of NOR, NOD, 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine- n[methoxy(polyethylene glycol)−2000 (DSPE-PEG2000) and cholesterol. NOR was synthesized through an amide reaction (Supplementary Fig. 6)28, using 1,2-distearoyl-sn-glycerin-3-phosphate ethanolamine (DSPE) and N-Boc-N’-(2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl)-L-arginine (Boc-Arg(Pbf)-OH) as raw materials, with 1-Hydroxybenzotriazole (HOBT) serving as the activating agent. This process yielded the intermediate product Boc-Arg(Pbf)-OH-DSPE. Trifluoroacetic acid (TFA) was then used to remove p-toluene sulfonyl (Tos-protecting group) and tert-butoxy carbonyl (Boc-protecting group) from Boc-Arg(Pbf)-OH-DSPE, resulting in the formation of NOR, which contains free guanidino groups. The Boc-Arg(Pbf)-OH-DSPE and NOR were characterized using NMR spectrometry (1H-NMR, 13C-NMR), all characteristic peaks from H and C were labeled in Supplementary Figs. 7–10, indicating that the structure of the obtained product aligned with the target structure reported in the literature28. In addition, peaks corresponding to a molecular weight of 904 Da were observed in the mass spectrometry (MS) analysis (Supplementary Fig. 11), consistent with the theoretical value of 904 Da. NOD was prepared by using tetra decanoic acid and 1,2-diaminobenzene as the reaction materials, 2-(6-Chloro-1-H-benzotriazole-1-yl)−1,1,3,3-tetramethylaminium hexafluorophosphate (HCTU) as the active agent, the specific synthetic route was shown in Supplementary Fig. 12, The results of 1H-NMR, 13C-NMR and MS proved the successful synthesis of NOD (Supplementary Figs. 13–15)29.
Subsequently, NO-Lip@PAC NMs were prepared using the thin-film hydration method with NOD, NOR, DSPE-PEG2000, cholesterol and PAC NMs as raw materials. The TEM image showed that the size of the synthesized NO-Lip NMs was about 450.0 nm. After loading PAC NMs in the hydrophilic core, a large number of PAC NMs were found to overlap with the liposomes, suggesting that the liposomes may have encapsulated the PAC NMs (Fig. 2a). The DLS results showed that the hydration diameter of NO-Lip NMs was about 502.2 nm, and the hydration diameter did not change much after loading PAC NMs, which was about 583.3 nm (Fig. 2b). Subsequently, the zeta potential of different samples was measured. The potential values of PAC NMs and NO-Lip NMs were − 9.3 and − 7.9 mV, respectively. The potential value of NO-Lip@PAC NMs decreased to − 19.2 mV (Fig. 2c). To further demonstrate the successful preparation of NO-Lip@PAC NMs, the phospholipid bilayer membrane of NO-Lip@PAC NMs was labeled with the lipophilic dye DiO, PAC NMs were labeled with Cy5, and the red fluorescence of PAC NMs overlapped with the green phospholipid bilayer membrane of DiO-labeled liposomes (Fig. 2d).
The degradation behavior of NO-Lip@PAC NMs in response to NO was detected. The guanidine group in NOR can react with ROS/iNOS in cancer cells to produce NO. Next, NO released from NOR can react with the o-phenylenediamine group in NOD to form an amide-derived benzotriazole group29,30, which spontaneously hydrolyzes and leads to the slow degradation of liposomes and the release of encapsulated PAC NMs (Supplementary Fig. 16). Thus, the performance of NO-Lip@PAC NMs responding to NO degradation was verified by monitoring the changes in DLS of NO-Lip@PAC NMs incubated in PBS or simulated tumor environment with a high concentration of ROS (100 μM H2O2) in different times. As shown in Fig. 2e, f, the DLS of NO-Lip@PAC NMs in PBS remained relatively stable, mostly maintaining 500–600 nm. However, following incubation with H2O2, NO-Lip@PAC NMs began to degrade gradually, with a peak at around 160.0 nm. This indicated that the smaller PAC NMs were gradually released from the liposomes, and the quantity of degraded liposomal fragments increased with prolonged incubation time. In addition, the TEM images showed the degradation behavior of NO-Lip@PAC NMs more visually (Fig. 2g). The morphology and size of NO-Lip@PAC NMs were basically unchanged in the PBS environment, and the degradation of liposomes started at about 0.5 h after the incubation, and the released PAC NMs can be observed at 1 h (red arrows), and the liposomal structure was completely lost at about 4 h. To better simulate the tumor cell environment, DiO-labeled liposomes and Cy5-labeled PAC NMs were used. NO-Lip@PAC NMs were incubated with normal and cancer cellular lysate. As shown in Fig. 2h and Supplementary Fig. 17, after 4 h of incubation with normal cellular lysates, the red fluorescence of PAC NMs in NO-Lip@PAC NMs still co-localized well with the green phospholipid bilayers of DiO-labeled liposomes. In contrast, in the tumor cellular lysate, the green and red fluorescence began to separate, with the red fluorescence of the PAC NMs appearing independently (purple arrows). The above data demonstrated that NO-Lip@PAC NMs can generate NO in response to the high concentration of ROS/iNOS in the tumor environment and degrade to release the encapsulated PAC NMs.
Then, the degradation profiles were evaluated by monitoring turbidity changes. The absorbance values of liposomes at 660 nm were investigated under PBS and simulated tumor high concentration of ROS (100 and 500 μM H2O2). As shown in Supplementary Fig. 18, Lip@PAC maintained stable relative turbidity in both 100 μM and 500 μM H2O2 solutions over 6 h, indicating that it does not degrade at high concentrations of H2O2. In contrast, the relative turbidity of NO-Lip@PAC in the 100 μM H2O2 solution gradually decreased, with 50.5% degradation occurring within 2.5 h, progressing to 73.9% degradation at 6 h. The degradation rate in the 500 μM H2O2 environment was slightly higher than that in the 100 μM H2O2, with 49.5% degradation occurring at 2 h and 84.2% degradation achieved by 6 h.
The motion ability of NO-Lip@PAC NMs and their chemotactic behavior in static and dynamic environments
It has been proved before that the highly expressed iNOS in the tumor microenvironment can act as a chemoattractant to induce the chemotactic effect of the nanomotor, thereby facilitating their effective targeting of tumor tissues31,32,33. In this section, we investigated the motion behavior of PAC NMs, NO-Lip NMs, and NO-Lip@PAC NMs in normal and tumor cellular environments. Firstly, CBMA and N-methacryloyl-L-lysine (M-Lys, without guanidino) were used to prepare PLC NPs as the control group, which were structurally similar to PAC NMs but without motion ability (Supplementary Figs. 19–22). TEM image showed that the morphology and size of PLC NPs were similar to those of PAC NMs (Supplementary Fig. 23). As shown in Supplementary Fig. 24, characteristic peaks such as the amino group of L-Lys (1526 cm−1), the amino group of BAC (1630 cm−1), and the C = O bond in CBMA (1720 cm−1) appeared in FTIR, proving the successful preparation of PLC NPs26,27.
First, the content of iNOS in bEnd.3 cells and different concentrations of Gl261 cells was detected (Supplementary Fig. 25). The concentration of iNOS in bEnd.3 cells (1 × 105) was about 3.61 μM, whereas Gl261 cells (1 × 105) was about 12.38 μM. When the density of Gl261 cells increased from 1 × 103 to 1 × 106, the amount of intracellular iNOS gradually increased, suggesting that the consistency and stability of iNOS concentration could be maintained by controlling the number of cells. As shown in Supplementary Figs. 26, 27 and Supplementary Movie 1–4, the motion displacement of PLC NPs in bEnd.3 and Gl261 cellular environment was relatively small, with an average speed distribution ranging from 0.5 to 1.6 μm s−1, indicating Brownian motion. PAC NMs showed Brownian motion in bEnd.3 cellular environment, and obvious motion displacement was observed in Gl261 cellular environment, with an average speed distribution of 1.7–5.0 μm s−1, displaying enhanced Brownian motion. Furthermore, the mean square displacement (MSD), which describes the average of the squares of a particle’s displacement from its initial position during motion, reflects the motion state of the particles34. Therefore, we investigated particle motion in different environments by analyzing MSD (Supplementary Fig. 28). The diffusion coefficients of PLC in bEnd.3 and Gl261 cellular environments were 0.33 and 0.29 μm2/s, respectively, indicating Brownian motion35,36,37. For PAC NMs, the diffusion coefficient was 0.26 μm2/s in the bEnd.3 cellular environment but increased significantly to 3.8 μm2/s in the Gl261 cellular environment35,36,37, indicating Brownian motion in normal cells and enhanced Brownian motion in cancer cells. To investigate the motion behavior of NO-Lip NMs, non-nanomotor Lip NPs (consisting of DSPE, DSPE-PEG2000, and cholesterol) were constructed as control group. As shown in Supplementary Figs. 29, 30 and Supplementary Movie 5–8, the Lip NPs displayed Brownian motion in bEnd.3 and Gl261 cellular environment with an average speed distribution of 0.5–1.5 μm s−1. The NO-Lip NMs showed Brownian motion in bEnd.3 cellular environment, and had a larger movement displacement in Gl261 cellular environment with an average speed distribution of 2.3–3.8 μm s−1, indicating enhanced Brownian motion. The MSD results further support this conclusion (Supplementary Fig. 31). Based on the above results, the motion behaviors of Lip@PAC NPs and NO-Lip@PAC NMs were further investigated, as shown in Fig. 3a, b and Supplementary Fig. 32 and Supplementary Movie 9, 10, the Lip@PAC NPs were showed Brownian motion in bEnd.3 and Gl261 cellular environment, with an average speed distribution of 0.5–1.5 μm s−1. The observed phenomenon may be attributed to the encapsulation of PAC NMs within liposomes, which lack guanidine groups on their surface to serve as a power source for the movement of nanomotors. As shown in Fig. 3c, d, Supplementary Fig. 32 and Supplementary Movie 11, 12, NO-Lip@PAC NMs displayed Brownian motion in bEnd.3 cellular environment, and a significant displacement was observed in the Gl261 cellular environment, with an average speed distribution of 1.9–3.7 μm s−1. As shown in Supplementary Fig. 33, the diffusion coefficients of Lip@PAC NPs in bEnd.3 and Gl261 cellular environments were 0.26 and 0.32 μm2/s, respectively, indicating Brownian motion35,36,37. For NO-Lip@PAC NMs, the diffusion coefficient was 0.28 μm2/s in the bEnd.3 cellular environment, but it increased significantly to 3.1 μm2/s in the Gl261 cellular environment35,36,37, indicating Brownian motion in normal cells and enhanced Brownian motion in cancer cells.

Normalized motion trajectories (n = 20 independent samples) and motion speed distribution plots (n = 50 independent samples) of Lip@PAC NPs in (a) bEnd.3 and (b) Gl261 cellular environment (Supplementary Movie 9, 10); Normalized motion trajectories (n = 20 independent samples) and motion speed distribution plots (n = 50 independent samples) of NO-Lip@PAC NMs in (c) bEnd.3 and (d) Gl261 cellular environment (Supplementary Movie 11, 12). e Schematic of the Y-channel model, by Figdraw. f Representative fluorescence images (Scale bar: 1000 μm) and (g) fluorescence quantification of NO-Lip@PAC NMs in Y-channel regions (ii) and (iii) at different times (n = 3 independent samples). h Representative fluorescence images of different samples at the exit of the Ψ-shaped microfluidic channel (Scale bar: 200 μm) and (i) the corresponding fluorescence quantification in the presence of bEnd.3 cellular lysates (n = 3 independent samples). j Representative fluorescence images of different samples at the exit of the Ψ-shaped microfluidic channel (Scale bar: 200 μm) and (k) the corresponding fluorescence quantification in the presence of bEnd.3 and Gl261 cellular lysates (n = 3 independent samples). Data in (g) were presented as mean ± SD. Significance was calculated via a two-tailed unpaired Student’s t test in (g). Source data are provided as a Source Data file.
The motion life of the outer shell (NO-Lip NMs), inner core (PAC NMs) and NO-Lip@PAC NMs in Gl261 cellular lysate were investigated. As shown in Supplementary Fig. 34a, b, the average speed of PAC NMs decreased from 2.9 μm s−1 (6 h) to 1.6 μm s−1 (24 h). This diminished motility could be attributed to the enzymatic depletion of L-Arg in the nanomotors. As shown in Supplementary Fig. 34c, d, the average speed of NO-Lip NMs decreased from 3.5 μm s−1 (2 h) to 1.8 μm s−1 (6 h). The shortened motility lifetime was not only related to the depletion of L-Arg, but also to its ability to response to NO degradation. The initial motility of NO-Lip@PAC NMs depends on L-Arg on the liposome shell. As NO accumulates, it causes the liposome shell to degrade and release the loaded PAC NMs, at which time the motility comes from L-Arg on the PAC NMs. We labeled the lipid shell of NO-Lip@PAC NMs with DiO to produce green fluorescence (named DiO-NO-Lip@PAC NMs), the core PAC of NO-Lip@PAC NMs with Cy5 to produce red fluorescence (named NO-Lip@PAC-Cy5 NMs), and separately studied the movement behavior of the shell (NO-Lip) and core (PAC) after incubation with Gl261 cellular lysate for different times. As shown in Supplementary Fig. 34e–h, when incubated with Gl261 cellular lysate for 2 h, DiO-NO-Lip@PAC NMs exhibited enhanced Brownian motion with a speed of 3.4 μm s−1, which was comparable to the speed of NO-Lip@PAC-Cy5 NMs (3.3 μm s−1), indicating that the movement at this time depended on L-Arg on the liposome shell. When incubated with Gl261 cellular lysate for 4 h, the movement speed of NO-Lip@PAC-Cy5 NMs decreased to 2.6 μm s−1, comparable to that of DiO-NO-Lip@PAC NMs (2.7 μm s−1). When the incubation time was extended to 6 h, as the lipid bilayer degraded, the speed of DiO-NO-Lip@PAC NMs decreased to 2.0 μm s−1, while NO-Lip@PAC-Cy5 NMs continued to exhibit a speed of 3.5 μm s−1, indicating that the movement capacity at this time originated from L-Arg on the PAC NMs.
Next, we employed a straight single-channel model to evaluate the nanomotors’ chemotactic responses under varying conditions. As shown in Supplementary Fig. 35, an iNOS gradient was established within the straight channel. DiL-labeled Lip@PAC or NO-Lip@PAC was introduced into reservoir (i), and the motion videos of different samples were recorded at position “a” to analyze their motion behaviors (Supplementary Fig. 36). As shown in Supplementary Fig. 37 and Supplementary Movie 13, when reservoir (ii) contained bEnd.3 cellular lysate-embedded gel, both Lip@PAC and NO-Lip@PAC exhibited minimal displacement at position “a”, with a speed of 1.0 μm s−1, exhibit non-directional Brownian motion. In contrast, when reservoir (ii) was loaded with Gl261 cellular lysate-embedded gel, NO-Lip@PAC displayed significantly enhanced Brownian motion, with a speed of 3.3 μm s−1 and both moved toward the higher iNOS concentration, exhibiting directional chemotactic movements. To further quantify migration persistence, the chemotaxis index (CI), defined as the ratio of total displacement to path length, was calculated33. Notably, NO-Lip@PAC in the iNOS gradient demonstrated the highest CI value (~ 0.5), surpassing both Lip@PAC and NO-Lip@PAC in the absence of a chemotactic gradient. These results confirmed that the iNOS gradient drives directional migration of NO-Lip@PAC, highlighting its chemotactic specificity.
Subsequently, we investigated the chemotactic behavior in static environments with an iNOS concentration gradient through Y-shaped channels. As shown in Supplementary Fig. 38, the iNOS gradient was established in the Y-shaped channels31,32,33,38. As shown in Fig. 3e, regions (i), (ii) and (iii) contained samples, bEnd.3 or Gl261 cellular lysate gels, respectively. The chemotaxis behavior of NO-Lip@PAC NMs was characterized by recording fluorescence images of regions (ii) and (iii) at different times. The fluorescence intensity of the Lip@PAC NMs in regions (ii) and (iii) did not differ significantly, the fluorescence quantification illustrated the same results (Supplementary Fig. 39). As shown in Figs. 3f and g, the fluorescence intensity in regions (ii) and (iii) gradually increased with time, but the fluorescence intensity in region (iii) was significantly higher than that in region (ii), suggesting that NO-Lip@PAC NMs tended to accumulate more in tumor cells with high concentrations of iNOS. These results suggested that due to the presence of L-Arg in the liposomes, the NO-Lip@PAC NMs could autonomously enrich to sites with higher iNOS concentration and showed good chemotactic performance.
In addition, we established a dynamic microfluidic model to investigate the chemotactic ability of nanomotors in a flow state (Supplementary Fig. 40). To simulate the capillary blood flow rate, the volume flow rate of the microfluidic syringe pump was controlled to be about 0.6 mL/h39. When inlets (i), (iii) were pumped with bEnd.3 cellular lysates, and inlet (ii) was pumped with Lip@PAC NPs or NO-Lip@PAC NMs, there was no fluid shift, fluorescence quantification results showed that the fluorescence intensity of Lip@PAC and NO-Lip@PAC was comparable in both sides (Figs. 3h, i and Supplementary Fig. 41). When inlets (i), (ii) and (iii) were pumped with Gl261 cellular lysate, different samples and bEnd.3 cellular lysate, respectively, the fluorescence signals of the NO-Lip@PAC NMs fluid could be observed to shift towards the Gl261 cellular lysate side (Fig. 3j). The fluorescence quantification results showed that the NO-Lip@PAC NMs were shifted towards the Gl261 cellular lysate side (3.0 times higher than the side of bEnd.3 cellular lysate), indicating that they could diffuse from channel (ii) to the channel with higher iNOS concentration (Fig. 3k).
The ability of NO-Lip@PAC NMs to cross the BBB in vitro and the mitochondrial targeting performance
To evaluate the ability of nanomotors crossing the BBB at the cellular level, in vitro BBB model was established using a transwell model containing a porous membrane (Fig. 4a). The upper chamber of the transwell model was inoculated with bEnd.3 cells to simulate a dense BBB layer (which was formed after 10 days of culture)40,41 and the lower chamber was inoculated with Gl261 cells to simulate GBM, forming an environment with an iNOS concentration gradient. Then we added Lip@PAC NPs and NO-Lip@PAC NMs (50 µg mL−1) to the upper chamber respectively, and incubated for 6 h. The fluorescence intensity of the samples in the upper and lower chambers of the transwell model were detected by CLSM. Meantime, we collected the liquid and cells in the lower chamber and quantitatively detected the amount of samples in each part. As shown in the CLSM images of bEnd.3 cells in the upper chamber (Fig. 4b and Supplementary Fig. 42), DiL-labeled Lip or NO-Lip colocalized well with Cy5-labeled PAC NMs, indicating minimal of Lip@PAC NPs and NO-Lip@PAC NMs before crossing the BBB. The separation of red and green fluorescence in Gl261 cells treated with NO-Lip@PAC NMs group suggests lipid degradation in the tumor environment, which cannot be found in Lip@PAC NPs treated groups. In addition, the red fluorescence intensity in bEnd.3 cells in the upper chambers treated with NO-Lip@PAC NMs was lower than that treated with Lip@PAC NPs (Fig. 4c), while Gl261 cells in the lower chambers had stronger red fluorescence signals, and fluorescence quantification showed that their intensity was 8.7 times higher than that of Lip@PAC NPs (Figs. 4c and d). The transport ratio in Fig. 4e showed that the transport efficiency of Lip@PAC NPs was only about 13.7%, while that of NO-Lip@PAC NMs reached about 63.5% (4.6 times higher than that of Lip@PAC). When Gl261 cells in the lower chamber were pretreated with iNOS inhibitor (NG-Monomethyl-L-arginine, L-NMMA) for 24 h. The penetration efficiency of NO-Lip@PAC decreased to 21.7% (Supplementary Fig. 43), which confirmed that iNOS concentration gradient-driven chemotaxis was the NO-Lip@PAC to achieve efficient BBB penetration. Next, we used CLSM to check the structural integrity of BBB after treatment with different samples. As shown in Supplementary Fig. 44, the cell layer that had formed a dense BBB, there was no significant change in the BBB cellular structure treated with Lip@PAC NPs or NO-Lip@PAC NMs for 24 h. Meanwhile, FITC-dextran (FD-4, 4000 Da) --- a polysaccharide composed of fluorescein isothiocyanate coupled to dextran---was used as a marker to identify BBB leakage, if leakage occurred, we would expect to observe stronger FD-4 fluorescence signals in the lower chamber42. As shown in Fig. 4f and Supplementary Fig. 45, compared to the control group, the upper compartment of bEnd.3 cells treated with Lip@PAC NPs and NO-Lip@PAC NMs remained tightly connected, with no significant increase in the intensity of FD-4 fluorescence detected in the lower compartment. This further demonstrated that NO-Lip@PAC NMs penetrated the BBB through chemotactic effects on the iNOS concentration gradient, rather than disrupting the integrity of BBB.

a Schematic diagram of the transwell model, by Figdraw. b Representative CLSM images of bEnd.3 cells and Gl261 cells of the transwell system treated with different samples for 6 h (blue: nucleus, red: Cy5-labeled PAC, green: DiL-labeled Lip or NO-Lip; Scale bar: 50 μm; n = 3 independent samples); Quantitative analysis of red and green fluorescence in the (c) upper and (d) lower compartments (I: Control, II: Lip@PAC, III: NO-Lip@PAC; n = 3 independent samples); (e) BBB transport efficiency of different samples (I: Lip@PAC, II: NO-Lip@PAC; n = 3 independent samples); (f) Paracellular permeability of FD-4 treated with different samples for 24 h (I: Control, II: Lip@PAC, III: NO-Lip@PAC; n = 3 independent samples); (g) Representative CLSM images of Gl261 cells treated with different samples for different time (blue: nucleus, green: DiL-labeled Lip or NO-Lip, red: Cy5-labeled PAC; Scale bar: 50 μm; n = 3 independent samples); (h) Representative CLSM images of mitochondrial co-localization treated with different samples for 2 h and corresponding fluorescence curves (I: PAC, II: PLC, III: Lip@PAC, IV: NO-Lip@PAC; Blue: nucleus, green: mitochondria, red: Cy5-labeled different samples; Scale bar: 50 μm; n = 3 independent samples). i Schematic illustration of using the Y-channel model to assess the chemotaxis of different samples towards mitochondria, By Figdraw. Representative fluorescence images of (j) PLC NPs and (l) PAC NMs at different times in Y-channel regions (ii) and (iii) (Scale bar: 1000 μm) and fluorescence quantification of (k) PLC and (m) PAC (n = 3 independent samples). Data in (c, d, e, f, k and m) were presented as mean ± SD. Significance was calculated via two-tailed unpaired Student’s t test in (c, d, e, k and m); and one-way ANOVA with Tukey multiple comparisons tests in (f). Source data are provided as a Source Data file.
Further, we evaluated whether NO-Lip@PAC NMs could degrade and release PAC NMs after crossing the BBB. We used DiL to label Lip or NO-Lip, Cy5 to label the PAC NMs, respectively. Different samples were co-incubated with Gl261 cells for varying durations to observe the release of PAC NMs. As shown in Fig. 4g and Supplementary Figs. 46 and 47, when Lip@PAC NPs and NO-Lip@PAC NMs were co-incubated with Gl261 cells for 2 h, the perinuclear showed more green fluorescence, representing the outer liposomes, while only minimal red fluorescence representing the PAC NMs. With the extension of the incubation time to 6 h, the red fluorescence representing PAC NMs in the NO-Lip@PAC NMs treated group can be observed, and the intensity of red fluorescence was 1.5 times of that at 2 h. As the incubation time was extended to 12 h, the red fluorescence was gradually enhanced (2.6 times of that at 2 h), while the green fluorescence was gradually reduced, which confirmed that NO-Lip@PAC NMs could gradually degrade and release their contents in response to the self-released NO. This phenomenon was not observed in Lip@PAC NPs treated groups. The ability of PAC NMs to target mitochondria was further assessed. Studies have shown that the concentration of iNOS in cancer cell mitochondria was much higher than that in normal cell mitochondria43,44,45. We hypothesized that PAC NMs could accumulate around mitochondria due to their chemotactic effects on iNOS. As shown in Fig. 4h and Supplementary Fig. 48, we used Cy5 to label PAC, and the red fluorescence of PAC NMs in NO-Lip@PAC NMs overlapped more with the green fluorescence of mitochondria, suggesting better mitochondrial targeting. Next, we investigated the iNOS induced chemotaxis kinetics of the nanomotors through Y-shaped channels (Fig. 4i). The regions (i), (ii) and (iii) comprising different samples, mitochondria lysate, or other organellar lysate gels, respectively. As shown in Fig. 4j and k, the fluorescence in regions (ii) and (iii) gradually increased over time, regions (ii) and (iii) of the PLC NPs did not differ much, and fluorescence quantification also illustrated the same result. The fluorescence in region (ii) of the PAC NMs group is higher than that in region (iii), suggesting that the PAC NMs tended to aggregate more in mitochondria (Fig. 4l, m). These results demonstrated that the guanidino group in PAC NMs could drive them to enrich autonomously to the sites with higher iNOS concentration, exhibiting effective chemotactic performance toward mitochondria.
Evaluation of the ability to induce mitochondrial mineralization in Gl261 cells by NO-Lip@PAC@Cur NMs
The previous results confirmed that NO-Lip@PAC NMs displayed good chemotaxis behavior in the tumor microenvironment, and the NO generated during this process promoted the degradation of liposomes, leading to the release of PAC NMs. The released PAC NMs can be further chemotactically targeted to mitochondria, and the carboxyl groups on their surface can recruit Ca2+ from the cytoplasm46,47,48, leading to an increase in the local Ca2+ concentration around the mitochondria, and thus inducing their mineralization. To further enhance the effect of mitochondrial mineralization, we also loaded Cur in the lipid hydrophobic region to inhibit Ca2+ efflux49,50,51,52 and constructed NO-Lip@PAC@Cur NMs. We prepared NO-Lip@PAC@Cur NMs with different drug-lipid ratios and tested their encapsulation rate and drug loading to explore the optimal incorporation conditions of Cur. As shown in Supplementary Fig. 49, the encapsulation rate of Cur gradually decreased and the drug loading amount increased with higher drug-lipid ratios. However, its stability decreased and liposomes appeared to agglomerate. On the one hand, this may be due to the low absolute value of the zeta potential (less than 30 mV). On the other hand, excessive Cur insertion into the phospholipid bilayer may disrupt the original tight arrangement of phospholipid molecules, resulting in changes in the fluidity and permeability of the membrane. (Supplementary Fig. 50). Considering this, we chose a drug-lipid ratio of 1:10 for subsequent experiments, achieving an encapsulation rate of 73.8% and a drug loading capacity of 7.6%. The TEM images showed that NO-Lip@PAC@Cur had a diameter of about 500 nm. The hydrated particle size was about 545.0 nm, and the zeta potential was − 14.0 mV (Supplementary Fig. 51). As shown in Supplementary Fig. 52a, b, the DLS and zeta potential of NO-Lip@PAC@Cur NMs remained unchanged within 7 days, with values of 598.8 nm and − 13.9 mV, respectively. Additionally, the optical images in Supplementary Fig. 52c indicate that no significant precipitation was observed in PBS after 7 days of incubation, demonstrating their good stability.
Subsequently, we investigated in detail whether the addition of Cur could play a synergistic role with NO to improve intracellular Ca2+ levels. Firstly, intracellular NO levels were detected using an NO probe. As shown in Fig. 5a and Supplementary Fig. 53, Gl261 cells treated with NO-Lip@PAC and NO-Lip@PAC@Cur exhibited significantly stronger NO fluorescence signals (2.7 and 2.9 times that of the control group, respectively), which were significantly higher than those observed in the other groups. In contrast, the NO fluorescence signals in bEnd.3 cells treated with different samples did not show significant differences, likely due to the lower concentration of iNOS to react with the nanomotors and produce NO. Next, we examined the intracellular Ca2+ content using inductively coupled plasma-Mass Spectrometry (ICP-MS). Figure 5b showed that the Ca2+ content of Cur-treated Gl261 cells increased to 0.30 μmol, which was comparable to the Ca2+ content in the NO-Lip@PAC-treated cells (0.27 μmol). This increase is likely due to the Cur or high concentration of NO causing endoplasmic reticulum stress and releasing more Ca2+ into the cytoplasm, a decrease in endoplasmic reticulum Ca2+ leads to Ca2+ influx from the extracellular space19,53,54,55. Cells treated with NO-Lip@PAC@Cur exhibited an even higher intracellular Ca2+ content (0.46 μmol), which was 2.7 times higher than that of the control group, suggesting that the inhibition of Ca2+ exocytosis by Cur can further increase the intracellular Ca2+ levels, confirming that Cur and NO can exert a synergistic effect to enhance the mineralization effect.

a Representative fluorescence image of intracellular NO (Scale bar: 50 μm; n = 3 independent samples). b Intracellular Ca2+ concentration (n = 3 independent samples). c Ca2+ concentration in mitochondria and other organelles of Gl261 cells (n = 3 independent samples). d Representative bio-TEM images of Gl261 cells (and II are control, III and IV are NO-Lip@PAC@Cur; Scale bar in I and III are 5 μm, Scale bar in II and IV are 2 μm; n = 3 independent samples); Representative TEM-Mapping of Gl261 cellular mitochondria after treated with (e) control and (f) NO-Lip@PAC@Cur (Scale bar: 400 nm; n = 3 independent samples). g Representative CLSM images of cytoplasmic Ca2+ and mitochondrial Ca2+ and the fluorescence distribution curves along selected lines (indicated by the white lines in the images; Blue: nucleus, green: Fluo-4-labeled cytoplasmic Ca2+, red: Rhod-2-labeled mitochondrial Ca2+; Scale bar: 20 μm; n = 3 independent samples). h Changes in mitochondrial membrane potential (red: JC-aggregates, green: JC-1 monomers; Scale bar: 50 μm, n = 3 independent samples) and (i) the corresponding red/green fluorescence ratios (n = 3 independent samples). j Intracellular ATP concentrations (n = 3 independent samples). k Cellular activity of Gl261 cells after treated with different NO-Lip@PAC@Cur concentrations (n = 3 independent samples). l Cellular activity of bEnd.3 and Gl261 cells treated with different samples (200 μg mL−1) (n = 3 independent samples). The names corresponding to the samples in (a, g, h, i, j and l) are (I) Control, (II) Lip@PLC, (III) NO-Lip@PAC and (IV) NO-Lip@PAC@Cur, while samples in (b, c) are (I) Control, (II) Cur, (III) Lip@PLC, (IV) NO-Lip@PAC and (V) NO-Lip@PAC@Cur. Data in (b, c, i–l) were presented as mean ± SD. Significance was calculated via one-way ANOVA with two-tailed LSD multiple comparisons test in (b) (Gl261), j–l and two-tailed Dunnett T3 multiple comparisons test in (b) (bEnd.3), (c, i). Source data are provided as a Source Data file.
To assess intracellular mitochondrial mineralization, Gl261 cells were lysed after treated with different samples, and mitochondria and other organelles were collected. Then the Ca2+ concentration was detected using ICP-MS. As shown in Supplementary Fig. 54, the Ca2+ content in mitochondria was 0.17 μmol at 6 h and increased to 0.33 μmol after extending the incubation time to 36 h, which was comparable to 0.36 μmol at 48 h. In addition, to determine that mitochondrial damage was caused by mineralization rather than Ca2+ overload. We added a control group--A23187 (a chemical reagent commonly used to induce mitochondrial Ca2+ overload)56,57. As shown in Fig. 5c and Supplementary Fig. 55, the mitochondrial Ca2+ content in Cur-treated cells was 0.18 μmol, which was comparable to that in the A23187 and NO-Lip@PAC treated-group (0.20 μmol and 0.19 μmol, respectively), but the concentration was much lower than that in the NO-Lip@PAC@Cur group (0.36 μmol, which was 6.2 times higher than the control group), suggesting that Cur can cause mitochondrial calcium overload, but cannot induce mitochondrial mineralization. This result indicates that mitochondrial mineralization requires a higher level of Ca2+ concentration than Ca2+ overload, Cur, NO-Lip@PAC, and A23187 were able to trigger Ca2+ overload, but did not reach the level of mineralization, while NO-Lip@PAC @Cur can recruit more Ca2+ to gather around mitochondria, which can trigger mitochondrial mineralization.
To visually observe mitochondrial calcification, NO-Lip@PAC@Cur-treated Gl261 cells were sectioned, and intracellular mitochondrial structure was observed using Bio-TEM. As shown in Fig. 5d and Supplementary Fig. 56, the cell membranes of Gl261 cells treated with NO-Lip@PAC remained largely intact, with most mitochondria retaining their structural integrity, only a small number of mitochondria exhibited structural abnormalities. The cell membrane of Gl261 cells treated with NO-Lip@PAC@Cur exhibited significant disruption compared to the untreated group, and the morphology of the mitochondria was notably altered. In addition, the TEM-mapping images of the mitochondria in the NO-Lip@PAC@Cur group and the control group were compared, and the mitochondria in the NO-Lip@PAC@Cur group had stronger Ca fluorescence signals in the mitochondria (Fig. 5e and f). Subsequently, Ca2+ in the cytoplasm was labeled with green fluorescence using Flou-4, Ca2+ in the mitochondria was labeled with red fluorescence using Rhod-2, and the co-localization was observed by CLSM. As shown in Fig. 5g and Supplementary Figs. 57 and 58, the green and red fluorescence intensities of the Lip@PAC group did not differ from those of the control group. The green and red fluorescence of the NO-Lip@PAC group were much higher than those of the control group (2.4- and 1.9-times higher, respectively), indicating that NO can induce endoplasmic reticulum stress, leading to the release of Ca2+ and an increase in Ca2+ levels in the cytoplasm. PAC NMs can recruit Ca2+ around mitochondria, resulting in an increase in mitochondrial Ca2+ levels. The green and red fluorescence intensities of the NO-Lip@PAC@Cur group were 2.6 and 1.4 times higher than those of the NO-Lip@PAC group, respectively, indicating that Cur could inhibit Ca2+ efflux in the cytoplasm and further increase the Ca2+ levels in the cytoplasm and mitochondria. Co-localization analysis showed that the red fluorescence and green fluorescence in the NO-Lip@PAC@Cur NMs group had a higher degree of overlap, suggesting that the mineralization process occurred in the mitochondria. Further, in order to prove that the Ca2+ accumulated around mitochondria are insoluble calcium salts, we used Alizarin Red S to stain cells, which can form red complexes by specifically chelating insoluble calcium salts (such as calcium phosphate), which is a classic method to evaluate cellular calcium deposition58. As shown in Supplementary Fig. 59, cells treated with Lip@PLC, A23187 and NO-Lip@PAC was similar to that of the control group, and the production of red complexes was not observed in the cells, which may be because although they can induce the mitochondrial Ca2+ overload, the concentration did not reach the formation of insoluble calcium salts, so the signal was weak. And the formation of red complex was obviously observed in the cells treated with NO-Lip@PAC@Cur, which indicated that the NO-Lip@PAC@Cur can induce mitochondria to form insoluble calcium salts, confirming the occurrence of mitochondrial mineralization.
Subsequently, the effects of mineralization on mitochondrial function were explored in detail. The stability of mitochondrial membrane potential (MMP) is a prerequisite for mitochondria to maintain normal physiological functions, and the JC-1 dye has been widely used to assess MMP. Under normal conditions, JC-1 accumulates in the mitochondrial stroma and formed aggregates that emit red fluorescence (529 nm); when the mitochondrial transmembrane potential was impaired and depolarized, JC-1 was released from mitochondria at a reduced concentration, and then JC-1 was a monomer and emitted green fluorescence (585 nm)59,60. As shown in Fig. 5h, i and Supplementary Fig. 60, JC-1 showed strong red fluorescence and only a small amount of green fluorescence in both the control and Lip@PLC NPs group, whereas the red JC-1 aggregates in the NO-Lip@PAC@Cur NMs group were converted in large quantities to green JC-1 monomers. In addition, we assessed the effect of different control groups on the behavior of adenosine triphosphate (ATP) generation in tumor cells. As shown in Fig. 5j, the intracellular ATP concentration decreased from 8.9 to 2.4 µM after NO-Lip@PAC@Cur NMs treatment compared to the control group. Apparently, NO-Lip@PAC@Cur NMs induced mitochondrial calcification, effectively depolarized the cellular mitochondrial membranes, leading to mitochondrial dysfunction and inhibition of intracellular ATP production.
The impact of the mitochondrial mineralization process on cellular activity was also explored. As shown in Figs. 5k, 3-(4,5-dimethylthiazol-2-yl)-2,5-biphenyl tetrazolium bromide (MTT) results showed that NO-Lip@PAC@Cur NMs had a concentration-dependent cytotoxicity against Gl261 cells, as the concentration increased from 50 to 800 μg mL−1, the cell activity decreased from 80.2% to 24.5%. In addition, the cytotoxicity of different samples on Gl261 and bEnd.3 cells was compared (Fig. 5l), the cell viability of Gl261 cells in the NO-Lip@PAC NMs group (200 μg mL−1) was 74.6%, while the viability decreased to 44.5% when Cur was loaded, indicating that mitochondrial mineralization in tumor cells resulted in significant cytotoxicity effects. Meanwhile, the viability of bEnd.3 cells treated with NO-Lip@PAC@Cur NMs (200 μg mL−1) was 83.8%, with no significant damage to normal cells, due to the absence of overexpressed iNOS in normal cells, which did not undergo mitochondrial mineralization.
Targeting ability of NO-Lip@PAC NMs in GBM model mice
To investigate the in vivo targeting ability of NO-Lip@PAC NMs, we established a GBM model in mice through orthotopic implantation of Gl261-Luc cells. The establishment of the GBM model was determined by in vivo bioluminescence 10 days after in situ inoculation of Gl261-Luc cells (Supplementary Fig. 61). Subsequently, Lip@PAC NPs and NO-Lip@PAC NMs were injected intravenously and their fluorescence images were captured using an in vivo imaging system (IVIS) various time points post-injection (Fig. 6a). As shown in Fig. 6b–d, significant fluorescence aggregation signals could be observed in GBM model mice at 1 h after intravenous injection of NO-Lip@PAC, indicating its ability to rapidly penetrate the BBB. The fluorescence intensity of brain tumor in the NO-Lip@PAC group continued to increase over time, reaching a peak at 12 h after injection, with an intensity 3.6 times greater than that of the Lip@PAC group (Fig. 6e). The fluorescence signal gradually weakened thereafter. It is noteworthy that the fluorescence signal in the brain tumor region of the Lip@PAC group was weak and basically disappeared after 48 h. In contrast, a significant fluorescence signal remained detectable in the NO-Lip@PAC group at 48 h (Fig. 6e), confirming its ability to maintain stable retention in the brain tumor region of the Lip@PAC group was weak and basically disappeared after 48 h. In contrast, a significant fluorescence signal remained detectable in the NO-Lip@PAC group at 48 h (Fig. 6e), confirming its ability to maintain stable retention in the brain microenvironment for at least 48 h. These findings were further validated through ex vivo organ imaging (Fig. 6f, g and Supplementary Fig. 62). After 12 h of intravenous injection, the Cy5 fluorescence intensity of the brain tissue of GBM mice in the NO-Lip@PAC group was significantly higher than that in 7 h, demonstrating its notable specificity for targeting brain tumors. The fluorescence intensity of the NO-Lip@PAC group was still 3.7 times that of the Lip@PAC group at 24 h after injection, which fully reflected its active targeting characteristics driven by the iNOS concentration gradient. In addition, ex vivo organ imaging showed that the both samples were mainly cleared by liver and kidney metabolism (Supplementary Fig. 63 and 64). Further, we collected different organs and examined the proportion of different samples to determine the targeting efficiency in different organs. The results showed that the NO-Lip@PAC NMs group accumulated about 29.3% ID/g in the brain, which was 3.6 times higher than that of the Lip@PAC NPs group (Fig. 6h). To further observe the distribution of the nanomotor fluorescence signals in the tumor tissues, whole brain tissues were cry-sectioned and stained using immunofluorescence techniques. FITC-CD31 was used to label the vascular endothelium, DAPI was used to label the nucleus, and red fluorescence was derived from Cy5-labeled nanomotor. Equal area region of interest (ROI) was selected perpendicular to the blood vessel and used as the starting point, red fluorescence signals within the ROIs were quantitatively analyzed to evaluate the permeability of different samples from the blood vessel to the GBM. As shown in Fig. 6i and j, the chemotactic NO-Lip@PAC NMs showed the strongest fluorescence intensity and depth of penetration in the GBM. Obvious red fluorescence was still observed at the distal end of the brain vessels (~ 300 μm), indicating that NO-Lip@PAC NMs could effectively cross the BBB and penetrate deeply into the tumor tissues.

a Schematic diagram of the assessment of the targeting ability of different samples in a GBM model mouse; In vivo fluorescence imaging of mice injected with (b) PBS, (c) Lip@PAC or (d) NO-Lip@PAC at various time points (n = 5 independent samples). e Quantification of the mean fluorescence intensities of the brain sites from mice in (b–d) at various time points (I: PBS, II: Lip@PLC, III: NO-Lip@PAC, n = 5 independent samples). f Cy5 fluorescence imaging of the brain after intravenous injection of different samples for 24 h (n = 5 independent samples; Scale bar: 1 cm) and (g) the corresponding quantitative analysis (I: Control, II: Lip@PLC, III: NO-Lip@PAC, n = 5 independent samples). h Quantitative analysis of different sample accumulations in major organs, expressed as injected dose per gram of tissue (% ID/g) (n = 5 independent samples). i Representative CLSM images of brain tumors in mice (Blue: nucleus, green: FITC-CD31, red: Cy5-labeled different samples; Scale bar: 500 μm) and (j) red fluorescence distribution curves (I: PBS, II: Lip@PLC, III: NO-Lip@PAC) along selected lines (indicated by white lines in the images). Data in g and h were presented as mean ± SD. Significance was calculated via a two-tailed unpaired Student’s t test in (g, h). Source data are provided as a Source Data file.
Antitumor efficacy of NO-Lip@PAC@Cur NMs in GBM model mice
Before evaluating the therapeutic effects in vivo, we quantified the levels of Cur in circulating blood at different time intervals after intravenous injection of free Cur and NO-Lip@PAC@Cur in healthy SD rats. As shown in Supplementary Fig. 65, NO-Lip@PAC@Cur can prolong the circulation time of Cur in the body. Then, their therapeutic efficacy in vivo was further evaluated. In this section, we chose TMZ, a first-line drug for clinical treatment of GBM, as a control for chemotherapeutic agents. Successful brain tumor construction was confirmed by in vivo bioluminescence imaging on day 11 after Gl261-Luc cell transplantation. Thereafter, mice (female) were randomly divided into 6 groups and sham-operated groups, and different drugs were intravenously every two days (Fig. 7a). As shown in Fig. 7b, c and Supplementary Table 2, in vivo bioluminescence imaging and quantification of total radiant brightness showed that the bioluminescence signal in the PBS group continued to enhance over time, and was strongest at the end of treatment (62.2 times of the intensity at the initial moment), suggesting that the tumor was growing rapidly. The intensity of the mouse brain decreased following treatment with free TMZ or NO-Lip@PLC@Cur treatment, suggesting mild inhibition of tumor growth (49.1 and 31.6 times of the intensity at the initial moment, respectively). In contrast, the anti-GBM growth effect was sequentially enhanced in the PAC@Cur and Lip@PAC@Cur groups, and the bioluminescent signals were significantly decreased (20.1 and 20.7 times of the intensity at the initial moment, respectively). NO-Lip@PAC@Cur can significantly inhibit tumor growth, with the intensity at 7.9 times the initial moment after treatment. Kaplan-Meier survival curves showed that NO-Lip@PAC@Cur significantly prolonged the survival of the GBM model mice, with a median survival time (MST) of 49 days, whereas PAC@Cur and Lip@PAC@Cur groups were both 39 days, which was slightly longer than that of the TMZ and NO-Lip@PLC@Cur groups (both 37 days), while the PBS control group was only 33 days (Fig. 7d). Further, dissected tumors from different treatments were stained with hematoxylin and eosin (H&E) to determine the destruction of tumor cells. The H&E results showed that the tumor area in the NO-Lip@PAC@Cur group was smaller than that in the other treatment groups (Fig. 7e and Supplementary Fig. 66). The Brain tumor area in the PBS group was the largest at the end of treatment, accounting for 41.8% of the whole brain. The brain tumor area in the free TMZ-treated mice was reduced and accounted for 31.3% of the whole brain. In contrast, the anti-GBM growth effect was enhanced in the PAC@Cur, Lip@PAC@Cur and NO-Lip@PLC@Cur group, with brain tumor area accounting for 22.0%, 25.3% and 25.0% of the whole brain in mice, respectively. NO-Lip@PAC@Cur was able to significantly inhibit the growth of brain tumors, which accounted for 9.5% of the whole brain at the end of treatment in mice. TUNEL and Ki67 immunofluorescence were used to label apoptotic and proliferating cells in the tumor tissue (Fig. 7e–g). Quantitative analysis showed that mice treated with NO-Lip@PAC@Cur had the highest rate of apoptotic cells in their tumors (~ 48.0%) and the lowest proportion of positive tumor cells proliferating (~ 10.0%). These differences demonstrated that NO-Lip@PAC@Cur was able to inhibit the growth of GBM by killing tumor cells through inducing mitochondrial mineralization in brain tumor cells.

a Treatment protocols for orthotopic brain-GBM-tumor-bearing models. By Figdraw. b Representative IVIS spectrum images and (c) quantified signal intensity (n = 6 independent samples per group); (d) Kaplan-Meier survival curves (n = 8 independent samples) of GBM model mice with different treatments. e Representative H&E (Scale bar: 4 mm), TUNEL, and Ki67 staining images (n = 6 independent samples; Scale bar: 500 μm) of dissected brain tissues at the end of treatment. f Tunel and (g) Ki67 quantitative fluorescence analysis; (I: sham, II: PBS, III: TMZ, IV: PAC@Cur, V: Lip@PAC@Cur, VI: NO-Lip@PLC@Cur, VII: NO-Lip@PAC@Cur, n = 6 independent samples). h Representative Bio-TEM images of brain tumor (I: PBS, II: magnification of I, III: NO-Lip@PAC@Cur, IV: magnification of III, Scale bar of I and III: 5 μm; n = 6 independent samples; Scale bar of II and IV: 200 nm); Representative TEM-Mapping (P and Ca) of brain tumor cellular mitochondria after treated with (i) control and (j) NO-Lip@PAC@Cur (n = 6 independent samples; Scale bar: 200 nm). Data in (c, f and g) were presented as mean ± SD. Significance was calculated via one-way ANOVA with two-tailed LSD multiple comparisons test in (c), and two-tailed Dunnett T3 multiple comparisons test in (f and g). Source data are provided as a Source Data file.
Safety is another key issue in the treatment of glioblastoma. The biocompatibility and systemic response of the different agents were assessed by hemolysis rate and erythrocyte morphology analysis, weight changes in mice, hematological and histopathological tests. Results showed no significant damage to erythrocytes with the different samples, indicating that the samples have good blood compatibility (Supplementary Figs. 67 and 68). The mice in the PBS and TMZ groups showed significant weight loss during treatment, which may be attributed to the fact that chemotherapeutic agents inevitably damage normal tissues and their functions during treatment, leading to weight loss. In contrast, the NO-Lip@PAC@Cur NMs did not observe a significant weight loss trend in mice during treatment (Supplementary Fig. 69). High-dose TMZ treatment leads to bone marrow suppression61,62. The TMZ treatment group resulted in a significant increase in bone marrow vacuoles (Supplementary Fig. 70) and a decrease in blood cell counts (Supplementary Fig. 71), suggesting that free TMZ may have side effects on bone marrow. Blood biochemistry data showed that alanine aminotransferase (ALT), aspartate aminotransferase (AST), lactate dehydrogenase (LDH) and serum albumin concentration (ALB) indices were increased in the TMZ group, suggesting that the TMZ treatment may cause some damage to the liver function of mice (Supplementary Fig. 72). In contrast, the hematological parameters and H&E staining results of mice treated with NO-Lip@PAC@Cur NMs indicated that the liver and kidney tissues of mice did not show significant pathological damage (Supplementary Fig. 73).
In order to observe mitochondrial calcification more visually, the brain tumor tissues of mice in the PBS and NO-Lip@PAC@Cur groups were sectioned, and the structures of cells and mitochondria within the tumor tissues were observed using Bio-TEM. As shown in Fig. 7h, compared with the tumor cells in the PBS group, cell membranes in the NO-Lip@PAC@Cur group were broken, nuclei were fragmented, and mitochondrial structures were lost, indicating cell apoptosis. TEM-Mapping results showed that mitochondria in the NO-Lip@PAC@Cur group had stronger Ca fluorescence signals (Fig. 7i and j), further demonstrating that NO-Lip@PAC@Cur could inhibit tumor growth by inducing mitochondrial mineralization in brain tumor cells. In addition, in order to verify whether there is tumor calcification in vivo, von Kossa staining was carried out on the tumor tissues of each group58. As shown in Supplementary Fig. 74, the brain tumor tissue in the NO-Lip@PAC@Cur group showed dark brown areas, indicating that there was calcification in the tumor. The other groups were similar to the PBS group, showing the same dark red color as the normal tumor. These results suggest that the apoptosis of tumor cells is mainly due to mitochondrial mineralization rather than Ca2+ overload.
We further investigated the efficacy of NO-Lip@PAC@Cur NMs in situ GBM model mice (male). As shown in Supplementary Fig. 75a–c, quantitative analysis of in vivo bioluminescence imaging and total radioluminescence intensity revealed that the bioluminescence signal in the PBS group continued to increase over time and reached its peak at the end of treatment (54.4 times of the initial radioluminescence intensity), indicating rapid tumor growth. Following free TMZ treatment, the fluorescence intensity of representative tumors in the mouse brain decreased, indicating mild inhibition of tumor growth (40.6 times of the initial radioluminescence intensity). In contrast, the anti-GBM growth effect was enhanced in the NO-Lip@PAC@Cur group, with a significant reduction in bioluminescence signals (only 9.6 times of the initial radioluminescence intensity). At the end of treatment, the brain tumor area was the largest in the PBS group, accounting for 39.6% of the entire brain. Brain tumor area in mice treated with free TMZ were reduced, accounting for 26.1% of the entire brain. In contrast, NO-Lip@PAC@Cur significantly inhibited brain tumor growth, with tumor area accounting for 9.0% of the entire brain at the end of treatment in mice (Supplementary Fig. 75d, e). Mice in the PBS and TMZ groups experienced significant weight loss during treatment, which may be attributed to the inevitable damage caused by chemotherapy drugs to normal tissues and their functions during treatment. In contrast, no significant weight loss trend was observed in mice treated with the NO-Lip@PAC@Cur NMs during treatment (Supplementary Fig. 75f). The therapeutic efficacy of NO-Lip@PAC@Cur NMs in C57BL/6 male mice was similar to that in C57BL/6 female mice.
In summary, we successfully developed size-variable self-feedback nanomotors, NO-Lip@PAC@Cur NMs, and verified their therapeutic efficacy against GBM. The lipid shell of NO-Lip@PAC NMs degraded in response to the NO generated during chemotactic targeting to the GBM microenvironment, thereby releasing the loaded Cur and PAC NMs. The released NO induced endoplasmic reticulum stress and subsequent Ca2+ release, while Cur inhibited Ca2+ efflux. This dual action resulted in an increase in the intracellular Ca2+ concentration (2.7 times higher than that in the control group). In addition, PAC NMs targeted mitochondria via chemotaxis, where the carboxyl groups recruited Ca2+ from the cytoplasm, thereby increasing the local mitochondrial Ca2+ concentration to 6.2 times higher than that in the control group. Fluorescence imaging and TEM-mapping of Ca2+ in tumor cell mitochondria revealed that mitochondria in the control group. Fluorescence imaging and TEM-mapping of Ca2+ in tumor cell mitochondria revealed that mitochondria in the NO-Lip@PAC@Cur NMs-treated group exhibited stronger fluorescence signals. This indicates that NO-Lip@PAC@Cur NMs effectively induced mitochondrial dysfunction, loss of mitochondrial membrane potential, and a significant decrease in ATP-generating capacity from 8.9 µM to 2.4 µM. In vivo experiments demonstrated that NO-Lip@PAC NMs could recognize the highly expressed iNOS in the tumor microenvironment, cross the BBB, and accumulate in brain tumors. Specifically, the accumulation in brain tissues of GBM model mice reached 29.3% ID/g, which was 3.6 times higher than that of the Lip@PAC NMs group. The treatment results showed that NO-Lip@PAC@Cur NMs effectively inhibited the growth of the tumor by inducing mitochondrial mineralization and subsequent tumor cell death (the brain tumor area accounted for 9.5% of the whole brain in the NO-Lip@PAC@Cur NMs group at the end of the treatment, which was 22.5% of that in the PBS group). Compared with the chemotherapeutic drug TMZ, NO-Lip@PAC@Cur NMs exhibited favorable biocompatibility. At the end of the treatment, the weight of the mice remained basically unchanged, and blood biochemistry and hematology indices were within normal ranges. In addition, histological analysis using H&E staining revealed no significant damage to major organs. Given that tumor cell mitochondria are crucial for their growth and proliferation, the strategy of specifically targeting mitochondrial mineralization in tumor cells is expected to provide valuable ideas for designing therapeutic strategies specifically for GBM. Due to the easy accumulation of NMs in organs such as the liver and kidneys, the injected dose is often limited, which in turn restricts the delivery efficiency of the drug to brain tissue. To overcome this bottleneck, a promising strategy is to use composite targeting systems, such as the combination of molecular recognition targeting and microenvironment recognition targeting, which is expected to direct more drug molecules across the BBB to enter the brain tissue, thereby enhancing drug accumulation concentrations in the brain while reducing systemic exposure and limiting potential liver and kidney toxicity.
Methods
Cell culture
The mouse glioma cell line Gl261 (ORC0929) and Gl261-Luc (ORC0929LUC) were purchased from Shanghai Aorui cell Biotechnology CO., Ltd.; the mouse brain microvascular endothelial cells (bEnd.3, CL-0598) and human umbilical vein endothelial cells (HUVECs, CP-H082) were purchased from Procell Life Science & Technology CO., Ltd. Gl261 and Gl261-Luc cells were cultured in complete culture medium containing 89% v/v high-sugar Dubelcco’s modified Eagle’s medium (DMEM with 4.5 g L−1 D-glucose, Jiangsu Keygen BioTECH Corp., CO., Ltd.), 10% v/v fetal bovine serum (SKU: SP011010500, Sperikon Life Science & Biotechnology CO., Ltd.), and 1% v/v penicillin-streptomycin mixture (Jiangsu Ketu Biotechnology Co., Ltd, RG-CE-8) in complete culture medium. bEnd.3 and HUVECs cells was cultured in 89% v/v high-sucrose DMEM (containing 4.5 g L−1 D-glucose), 10% v/v fetal bovine serum (LONSERA. Suzhou Shuangru Biotechnology Co., Ltd), and 1% v/v penicillin-streptomycin (Jiangsu Ketu Biotechnology Co., Ltd, RG-CE-8) mixture in complete culture medium. All the cells were cultured in a humidified atmosphere that contained 5% CO2 at 37 °C. When not in use, cell cryopreservation with cell saving (PB180438, Procell Life Science & Technology CO., Ltd.) at − 80 °C.
iNOS content in different cells
The bEnd.3 (1.0 mL, 1 × 105 cells mL−1) or Gl261 cells (1.0 mL, 103, 104, 105 or 106 cells mL−1) were broken by sonication, and the supernatant was collected after centrifugation at 1006 × g for 10 min. The iNOS concentration was detected using the mouse iNOS ELISA research kit (Jiangsu Meimian Industrial Co., Ltd, MM-0454M2).
Motion behaviors of different samples
To analyze motion behavior, bEnd.3 or Gl261 cells (1.0 mL, 1 × 105 cells mL−1) was collected, 200.0 μL of RIPA (Biosharp, BL504A) was added, and the mixture was shaken for 15 min to ensure complete lysis. Then, 20.0 μL of Cy5- or DiO-labeled different samples was added to the confocal dish. The motion behavior of different samples was recorded using the 100 × objective of an inverted fluorescence microscope (Micro-shot MF53-N), and the motion trajectories of the different samples were marked using the tracking plug-in of the Fiji software. The trajectories of 50 particles were randomly selected to calculate the average speed of nanomotors and analyze the speed distribution histogram.
For the assessment of the motion lifetime of different samples, Cy5- or DiO-labeled samples were incubated with Gl261 cellular lysate and recorded using the 100 × objective of an inverted fluorescence microscope (Micro-shot MF53-N) at different time points. And the motion trajectories of the different samples were marked using the tracking plug-in of the Fiji software. The trajectories of 5 particles were randomly selected to calculate the average speed of nanomotors.
iNOS content in straight or Y-shaped channels
To determine the iNOS content in the straight or Y-shaped channel, 25.0 μL of agarose solution was mixed with an equal volume of bEnd.3 or Gl261 cellular lysate in the storage chamber (ii) and (iii), respectively. And then the Y-shaped channel was transferred to 4 °C to wait for the agarose to form a gel. Subsequently, it was filled with 300.0 μL of PBS (Jiangsu Ketu Biotechnology Co., Ltd, RG-CE-10) and after standing at 4 °C for 15 min, samples were collected from different locations of the Y-shaped channel. The iNOS concentration was detected using the mouse iNOS ELISA research kit (Jiangsu Meimian Industrial Co., Ltd, MM-0454M2).
Collective chemotaxis behaviors of nanomotors in the Y-shaped channel
To observe the collective chemotaxis behavior of nanomotors, a Y-shaped glass substrate microchannel was used. The main channel was 1.0 cm in length, 0.4 cm in width, and the branch channel was 0.7 cm in length and 0.3 cm in width. The concentration gradient of the chemoattractant was created by different types of cell lysates that were placed in reservoirs (II) or (III) in the branched channel. Briefly, 5.0 mg of agarose was completely dissolved in 0.5 mL of PBS at 90 °C, and 50.0 μL of Gl261 or bEnd.3 cellular lysate was added when the melted agarose was cooled to room temperature, but not solidified. And then, it was transferred to 4 °C for gelation. Before assessing the chemotactic motion of the nanomotors, the Y-shaped channel was prefilled with 300.0 μL PBS and quiescence for 10 min. Then 50.0 μL of Cy5-labeled nanomotors was gently dropped into the reservoir (I). The fluorescence microscopes of the reservoir (II) and (III) were captured with an inverted fluorescence microscope (Micro-shot MF53-N), equipped with 4 × objective, at specific times. The corresponding fluorescence intensity was quantified using ImageJ.
Dynamic chemotaxis behaviors of nanomotors in the microfluidic channel
The three-inlet one-outlet glass substrate microfluidic channel with dimensions of 2.2 cm (length) × 1.5 mm (width) × 0.3 mm (height) was used to evaluate the dynamic chemotaxis of nanomotors. Among them, the diluent of Gl261 cellular lysate (lysate: PBS = 1:4, v/v) flowed through inlet (I), the Cy5-labeled nanomotors in PBS was flowed through inlet (II), and the dilution of bEnd.3 cellular lysate (lysate: PBS = 1:4, v/v) was flowed through the inlet (III). The flow velocity of each channel was controlled at 0.6 mL h−1. The video was captured continuously for 5 min (1 frame per second) at the position near to outlet using an inverted fluorescence microscope (10 × objective). The fluorescence intensity of Cy5 perpendicular to the flow direction was measured using ImageJ. Moreover, the dilution of bEnd.3 cell lysate (lysate: PBS = 1:4, v/v) flowed through both inlets (I) and (III) as control, while the flow of inlet (II) was not replaced.
Detection of intracellular Ca2+ concentration
Gl261 cells (1.0 mL, 1 × 105 cells mL−1) were inoculated into a 6-well plate and incubated overnight. Then, different samples (Lip@PLC, NO-Lip@PAC, NO-Lip@PAC@Cur, 1.0 mL, 200 μg mL−1 in DMEM or Cur, 1.0 mL, 15 μg mL−1 in DMEM) were added and incubated for 48 h. The cells were washed three times with PBS to remove free samples. Cells were collected with a cell scraper and placed in a reactor with nitric acid (1.0 mL) and H2O2 (1.5 mL), which was nitrated in a vacuum oven at 180 °C for 1.5 h. The above system was subsequently volume-determined to 5.0 mL, and the intracellular Ca2+ concentration was measured using inductively coupled plasma-Mass Spectrometry (ICP-MS, IRIS Intrepid II).
Detection of Ca2+ concentration in mitochondria
Gl261 cells (1 × 108 cells) were inoculated into T175 cell culture flasks and incubated overnight. Then, different samples (Lip@PLC, NO-Lip@PAC, NO-Lip@PAC@Cur, 20.0 mL, 200 μg mL–1 in DMEM or Cur, 20.0 mL, 15 μg mL−1 in DMEM, A23187, 20.0 mL, 5 μM, Cat No. 50403ES03; Yeasen, Shanghai, China) were added and incubated for 48 h. The cells were washed three times with PBS to remove free samples. Cells were then collected, and mitochondria were extracted according to the instructions of the mitochondrial isolation kit (C3601, Beyotime Biotechnology). The mitochondria and other organelles were placed in a reactor with nitric acid (1.0 mL) and H2O2 (30%, 1.5 mL), which was nitrated in a vacuum oven at 180 °C for 1.5 h. The above system was subsequently volume-determined to 5.0 mL, and the Ca2+ concentration in mitochondria was measured using ICP-MS.
Detection of intracellular NO release
Gl261 cells (1.0 mL, 1 × 105 cells mL−1) were inoculated into a 6-well plate and incubated overnight. Then different samples (1.0 mL, 200 μg mL−1 in DMEM, Lip@PLC, NO-Lip@PAC, or NO-Lip@PAC@Cur) were added and incubated for 6 h. 3-Amino, 4-aminomethyl-2’,7’-difluorescein, diacetate (DAF-FM DA, NO fluorescent probe, 5 µM, Beyotime Institute of Biotechnology, China) was incubated with the cells for 30 min, and images were taken using an inverted fluorescence microscope (Micro-shot MF53-N), their fluorescence being quantified using ImageJ.
Detection of mitochondrial membrane potential
Gl261 cells (1.0 mL, 1 × 105 cells mL−1) were inoculated in confocal dishes and incubated overnight. Then different samples (1.0 mL, 200 μg mL−1 in DMEM, Lip@PLC, NO-Lip@PAC, or NO-Lip@PAC@Cur) were added and incubation for 24 h. The cells were washed three times with PBS to remove free material. Mitochondria was labeled with JC-1 (Beyotime biotechnology, C2006) and the nucleus was labeled with Hoechst 33342 (C0031, Beijing Solarbio Science & Technology Co., Ltd) in blue fluorescence. Cells were fixed with 4% paraformaldehyde. Fluorescence images were taken with CLSM, and the fluorescence intensity of the acquired images was analyzed using ImageJ software.
Intracellular ATP concentration assay
Gl261 cells (1.0 mL, 1 × 105 cells mL−1) were inoculated into a 6-well plate and incubated overnight. Then different samples (1.0 mL, 200 μg mL−1 in DMEM, Lip@PLC, NO-Lip@PAC, or NO-Lip@PAC@Cur) were added and incubation for 24 h. The cells were washed three times with PBS to remove free samples. Cells were then collected, and intracellular ATP concentration was detected according to the instructions for use of the ATP Assay Kit (S0026, Beyotime Biotechnology).
Evaluation of cell viability in vitro
Gl261 cells or bEnd.3 cells (200.0 μL, 1 × 105 cells mL−1) were inoculated in 96-well plates and incubated overnight. The cell cultures were then removed and 200.0 μL of different concentrations in DMEM (0, 50.0, 100.0, 200.0, 400.0 and 800.0 μg mL−1) of NO-Lip@PAC@Cur were added and incubated for 24 h. The cells were incubated with 3-(4,5-dimethylthiazol-2-yl)-2,5-dibenzenetetrazolium bromide (MTT, ApexBio Technology Co., Ltd.) colorimetric method to assess cell viability.
To compare the in vitro cell viability of different nanomotors, Gl261 cells (200.0 μL, 1 × 105 cells mL−1) were inoculated in 96-well plates and incubated overnight. Then, different samples (1.0 mL, 200 μg mL−1 in DMEM, Lip@PLC, NO-Lip@PAC, or NO-Lip@PAC@Cur) were added and incubated for 24 h. Cell viability was assessed by the MTT colorimetric assay described above.
In vitro mitochondrial mineralization detection
For Alizarin Red S staining, Gl261 cells (1.0 mL, 1 × 105 cells mL−1) were inoculated in confocal dishes and incubated overnight. After removal of the medium, 1 mL of Lip@PLC, NO-Lip@PAC, NO-Lip@PAC@Cur (200.0 µg mL−1) or A23187 (5 μM) dispersed in fresh medium was added and incubated for 48 h. After removal of the culture medium, and the cells were then washed and fixed with 95% ethanol. Subsequently, the cells were stained with Alizarin Red S (1 mL, Beyotime Biotechnology) for 30 min. After the extra staining solution was washed away by ultrapure water, the stained cell samples were observed under a light microscope.
Detection of pharmacokinetics
Male SD rats (Jiangsu Wukong Biotechnology Co., LTD.) were used for the pharmacokinetic determination of NO-Lip@PAC@Cur NMs. SD rats (1 per cage) were housed in standard ventilated cages at 21 °C, with free access to water. The lighting intensity in the housing room was 15–20 lx, with a light-dark cycle of 10 h/14 h, and relative humidity was maintained at 50%–60%. Briefly, Cur (1.0 mL, 15.0 mg mL−1) and NO-Lip@PAC@Cur (1.0 mL, 15.0 mg mL−1) were injected through the tail vein, and 500 μL blood was drawn from the orbits of the rats at different times using capillary glass tubes soaked with sodium heparin. After centrifugation at 698 × g for 15 min, the supernatant was obtained, and the absorbance of curcumin was measured at 420 nm using a Multi-Function Measuring Instrument (Infinite® E Plex), and the content of the samples was calculated using a standard curve of absorbance of Cur or NO-Lip@PAC@Cur concentration.
Establishment of glioblastoma (GBM) model mice
Female C57BL/6 mice (6–8 weeks old) were purchased from Hangzhou Ziyuan Laboratory Animal Technology Co., Ltd. All animal experiments were conducted under the supervision and guidance of the Ethical Review Committee for Laboratory Animal Welfare of Nanjing Normal University (Nanjing, China, approval No. IACUC-20220901-1 and 20200802). C57BL/6 mice (5 per cage) were housed in standard ventilated cages at 21 °C, with free access to water. The lighting intensity in the housing room was 15–20 lx, with a light-dark cycle of 10 h/14 h, and relative humidity was maintained at 50–60%. To establish the GBM model mice, Gl261-Luc cells (106 cells in 8.0 μL Corning® Matrigel® Matrix) were slowly injected into the brain using a brain stereotaxic instrument (RWD Life Science Co., Shenzhen) positioned to (1.8 mm,0.6 mm, 2 mm depth) using the fontanel point of origin, where the craniotomy operation was kept consistent provided that the mice without cell injection were referred to as the sham-operated group as a control. During the operation, the mice were anesthetized by inhalation of 1–5% isoflurane mixed with oxygen. 10 days later, all the mice were intraperitoneally injected D-Luciferin potassium salt (150 mg kg−1 in PBS, MedChemExpress, Cat. No. HY-12591B), 15 min later, bioluminescent imaging was performed immediately using the in vivo image system (IVIS Spectrum, Aniview 600 Multi-mode Animal Imaging, Guangzhou Biolight Biotechnology Co.).
In vivo targeting and tumor permeability
To assess the in vivo targeting ability of different samples, 200.0 μL of Cy5-labeled different samples (2.0 mg mL−1, Lip@PAC or NO-Lip@PAC) was injected intravenously. Notably, 200.0 μL of PBS was injected as a control. Fluorescence imaging (excitation filter: 620 nm, emission filter: 670 nm) was performed at different times by IVIS Spectrum in vivo. Euthanizing mice at different times, and the brain tissue and major organs (heart, liver, spleen, lung, and kidney) were collected for fluorescence imaging using IVIS Spectrum (excitation filter: 620 nm, emission filter: 670 nm). The in vivo targeting ability of different samples was quantified using Living Image software. Subsequently, the above whole brain tissues were subjected to coronal frozen sections. The cell membranes of cerebrovascular endothelial cells were labeled with rabbit anti-mouse CD31 antibody and Alexa Fluor 488-coupled goat anti-rabbit IgG, and the nucleus were labeled with DAPI, respectively. Tissue sections were blocked, and fluorescence imaging was performed using CLSM. Normal brain tissues and GBM tissues were distinguished by cell density, and the GBM permeability of different nanomotors was analyzed by ImageJ software.
In vivo delivery efficiency of different samples
To investigate the in vivo delivery efficiency and biodistribution of the nanomotors, 200.0 μL of different samples (2.0 mg mL−1, Lip@PAC or NO-Lip@PAC) was injected intravenously. and 200.0 μL of PBS was injected as a control. After 24 h, the mice were euthanized, and the brain tissue and major organs (heart, liver, spleen, lung, and kidney) were collected and weighed. Then 1.0 mL RIPA (Biosharp, BL504A) lysis buffer was added to every 100.0 mg of tissue, and tissue homogenate was prepared using the tissue grinder (frequency: 70 Hz, time: 8 min, run times: 8 times). The homogenate was centrifuge (4 °C, 1006 × g, 10 min) and the supernatant was collected. The supernatant was diluted, and the fluorescence intensity was measured using a multifunctional enzyme-linked immunosorbent assay reader. The injection dose percentage (% ID/g) of each group of samples was calculated based on the sample concentration fluorescence intensity standard curve and the initial injection dose.
In vivo anti-GBM efficacy and biosafety evaluation of different samples
On the 11th, 13th, 15th, 17th, 19th, 21st and 23rd day after injection of Gl261-Luc cells into the skull of mice, 200.0 μL different samples (2.0 mg mL−1, PAC@Cur, Lip@PAC@Cur, NO-Lip@PLC @Cur, NO-Lip@PAC@Cur and temozolomide (TMZ, Beyotime Biotechnology)) were injected intravenously. Notably, the sham group received no injections and 200.0 μL PBS was injected as a control. The growth of the glioma was monitored by IVIS Spectrum at specific times. The mice were sacrificed after treatment, and the brain tissues were taken for paraffin-embedding and coronal sectioning, which were performed with hematoxylin-eosin (H&E) staining and Ki-67 immunohistochemical staining, respectively. The main tissues, including the heart, liver, spleen, lung, kidney and bone marrow, were collected and fixed with 4% paraformaldehyde. Then the tissues were embedded in paraffin and stained with H&E staining to evaluate the histopathological changes after different treatments. The blood samples were obtained from each group for biochemical analysis and routine blood examination. The biochemical analysis was measured by the automatic biochemical analyzer. The routine blood examination was performed by a blood cell analyzer. Then, 8 additional mice were used in the survival experiments and survival curves were obtained using the Kaplan-Meier method.
Statistical analysis
Differences between the two groups were analyzed by a two-sample t test. Differences among multiple groups were analyzed using two-tailed unpaired Student’s t test and one-way ANOVA with two-tailed LSD, Dunnett T3 or Tukey multiple-comparisons tests. All statistical analyses were performed on Excel v.2019, SPSS v.16.0 and GraphPad Prism v.9.0.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data generated or analyzed during this study are available within the Article, its Supplementary Information file, and the Source data file. Source data is available for Figs. 2b, c, 3a–d, g, i, k, 4c–f, h, k, m, 5b, c, i–l, 6e, g, h, j, 7c, d, f, g and Supplementary Figs. 5, 18, 23–26, 28, 29, 31, 33–35, 37–39, 41, 43, 47, 49–55, 58, 65–67, 71, 72, and 75. Source data are provided in this paper.
References
Hamad, A., Yusubalieva, G. M., Baklaushev, V. P., Chumakov, P. M. & Lipatova, A. V. Recent developments in glioblastoma therapy: oncolytic viruses and emerging future strategies. Viruses 15, 547 (2023).
Yalamarty, S. S. K. et al. Mechanisms of resistance and current treatment options for glioblastoma multiforme (GBM). Cancers 15, 2116 (2023).
Karlsson, J., Luly, K. M., Tzeng, S. Y. & Green, J. J. Nanoparticle designs for delivery of nucleic acid therapeutics as brain cancer therapies. Adv. Drug Deliv. Rev. 179, 113999 (2021).
Stupp, R. et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. JAMA. 318, 2306–2316 (2017).
Idbaih, A. et al. Safety and feasibility of repeated and transient blood–brain barrier disruption by pulsed ultrasound in patients with recurrent glioblastoma. Clin. Cancer Res. 25, 3793–3801 (2019).
D'Amico, R. S., Aghi, M. K., Vogelbaum, M. A. & Bruce, J. N. Convection-enhanced drug delivery for glioblastoma: a review. J. Neurooncol. 151, 415–427 (2021).
Ma, R., Li, Z., Chiocca, E. A., Caligiuri, M. A. & Yu, J. The emerging field of oncolytic virus-based cancer immunotherapy. Trends Cancer 9, 122–139 (2023).
Xu, B. et al. An oncolytic virus expressing a full-length antibody enhances antitumor innate immune response to glioblastoma. Nat. Commun. 12, 5908 (2021).
Zhang, H. et al. Boost infiltration and activity of T cells via inhibiting ecto-5′-nucleotidase (CD73) immune checkpoint to enhance glioblastoma immunotherapy. ACS Nano 18, 23001–23013 (2024).
Kim, G. B. et al. High-affinity mutant Interleukin-13 targeted CAR T cells enhance delivery of clickable biodegradable fluorescent nanoparticles to glioblastoma. Bioact. Mater. 5, 624–635 (2020).
Suijkerbuijk, K. P. M., van Eijs, M. J. M., van Wijk, F. & Eggermont, A. M. M. Clinical and translational attributes of immune-related adverse events. Nat. Cancer 5, 557–571 (2024).
Wang, H. et al. The challenges and the promise of molecular targeted therapy in malignant gliomas. Neoplasia 17, 239–255 (2015).
Keunen, O. et al. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc. Natl. Acad. Sci. USA 108, 3749–3754 (2011).
Batchelor, T. et al. A phase II trial of AZD2171 (cediranib), an oral pan-VEGF receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. J. Clin. Oncol. 25, 18 (2007).
Batchelor, T. T. et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell 11, 83–95 (2007).
O’Hare, P. et al. Resistance, rebound, and recurrence regrowth patterns in pediatric low-grade glioma treated by MAPK inhibition: a modified delphi approach to build international consensus-based definitions-international pediatric low-grade glioma coalition. Neuro Oncol. 26, 1357–1366 (2024).
Chang, X. et al. Precise starving therapy via physiologically dependent photothermal conversion promoted mitochondrial calcification based on multi-functional gold nanoparticles for effective tumor treatment. Adv. Funct. Mater. 33, 2303596 (2023).
Fan, M. et al. Structure and mechanism of the mitochondrial Ca2+ uniporter holocomplex. Nature 582, 129 (2020).
Giorgi, C., Marchi, S. & Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 19, 713–730 (2018).
Liew, S. S. et al. Smart design of nanomaterials for mitochondria-targeted nanotherapeutics. Angew. Chem. Int. Ed. 60, 2232–2256 (2021).
Guo, X. et al. Intelligent gold nanoparticles for synergistic tumor treatment via intracellular Ca2+ regulation and resulting on-demand photothermal therapy. Chem. Eng. J. 433, 133850 (2022).
Huang, X. & Huang, Y. Solubilization of organic compounds by arginine-derived polymers. Chin. Chem. Lett. 26, 636–640 (2015).
Kim, Y., Binauld, S. & Stenzel, M. H. Zwitterionic guanidine-based oligomers mimicking cell-penetrating peptides as a nontoxic alternative to cationic polymers to enhance the cellular uptake of micelles. Biomacromolecules 13, 3418–3426 (2012).
Bersani, S. et al. Star-like oligo-arginyl-maltotriosyl derivatives as novel cell-penetrating enhancers for the intracellular delivery of colloidal therapeutic systems. Bioconjug. Chem. 23, 1415–1425 (2012).
Fang, H. et al. In situ polymerization of zwitterions on therapeutic proteins to enable their effective oral delivery. ACS Nano 17, 1128–1143 (2023).
Wu, D. Q. et al. Synthesis and characterization of arginine-NIPAAm hybrid hydrogel as wound dressing: in vitro and in vivo study. Acta Biomater. 65, 305–316 (2018).
Li, T. et al. Chemotactic nanomotor for multimodal combined therapy of glioblastoma. Sci. China Chem. 67, 1277–1288 (2024).
Zhou, S. et al. Dendritic lipopeptide liposomes decorated with dual-targeted proteins. Biomater. Sci. 10, 7032–7041 (2022).
Liu, Q., Hu, J., Whittaker, M. R., Davis, T. P. & Boyd, B. J. Nitric oxide-sensing actuators for modulating structure in lipid-based liquid crystalline drug delivery systems. J. Colloid Interf. Sci. 508, 517–524 (2017).
Hu, J. et al. Biomimetic polymers responsive to a biological signaling molecule: nitric oxide triggered reversible self-assembly of single macromolecular chains into nanoparticles. Angew. Chem. Int. Ed. 53, 7779–7784 (2014).
Li, T. et al. A universal chemotactic targeted delivery strategy for inflammatory diseases. Adv. Mater. 34, 2206654 (2022).
Chen, H. et al. A nitric-oxide driven chemotactic nanomotor for enhanced immunotherapy of glioblastoma. Nat. Commun. 14, 941 (2023).
Zhang, H. et al. Dual-responsive biohybrid neutrobots for active target delivery. Sci. Robot. 6, eaaz9519 (2021).
Ma, X. et al. Enzyme-powered hollow mesoporous Janus nanomotors. Nano Lett. 15, 7043–7050 (2015).
Peng, X. et al. Autonomous metal-organic framework nanorobots for active mitochondria-targeted cancer therapy. Sci. Adv. 9, eadh1736 (2023).
Yang, Z. et al. Ultrasmall enzyme-powered Janus nanomotor working in blood circulation system. ACS Nano 17, 6023–6035 (2023).
Sun, Z. et al. Self-propelled Janus nanocatalytic robots guided by magnetic resonance imaging for enhanced tumor penetration and therapy. J. Am. Chem. Soc. 145, 11019–11032 (2023).
Ye, Y. et al. Apoptotic tumor DNA activated nanomotor chemotaxis. Nano Lett. 21, 8086–8094 (2021).
Meyer, M. F. et al. Lack of a difference in increased capillary blood cell velocity in the skin over proximal interphalangeal joints between rheumatoid arthritis and osteoarthritis. Microvasc. Res. 70, 1–6 (2005).
Chelliah, S. S., Paul, E. A. L., Kamarudin, M. N. A. & Parhar, I. Challenges and perspectives of standard therapy and drug development in high-grade gliomas. Molecules 26, 1169 (2021).
Lin, K.-H. et al. Enhancing anticancer effect of gefitinib across the blood-brain barrier model using liposomes modified with one α-helical cell-penetrating peptide or glutathione and tween 80. Int. J. Mol. Sci. 17, 1998 (2016).
Natarajan, R., Northrop, N. & Yamamoto, B. Fluorescein Isothiocyanate (FITC)-dextran extravasation as a measure of blood-brain barrier permeability. Curr. Protoc. Neurosci. 79, 9.58.1–9.58.15 (2017).
Escames, G. et al. Attenuation of cardiac mitochondrial dysfunction by melatonin in septic mice. Febs J. 274, 2135–2147 (2007).
Hatano, Y. & Toda, H. Modulation of opioid actions by nitric oxide signaling. Anesthesiology 110, 166–181 (2009).
Sun, M. et al. Specific generation of nitric oxide in mitochondria of cancer cell for selective oncotherapy. Nano Res. 15, 5273–5278 (2022).
Jiang, Z. et al. Versatile polymer-initiating biomineralization for tumor blockade therapy. Adv. Mater. 34, e2110094 (2022).
Liu, Y. et al. Acidity-triggered transformable polypeptide self-assembly to initiate tumor-specific biomineralization. Adv. Mater. 35, e2203291 (2023).
Wu, J. et al. Bioinspired tumor calcification enables early detection and elimination of lung cancer. Adv. Funct. Mater. 31, 2101284 (2021).
Zheng, P. et al. A multichannel Ca2+ nanomodulator for multilevel mitochondrial destruction-mediated cancer therapy. Adv. Mater. 33, 2007426 (2021).
Xu, L. et al. Enhanced intracellular Ca2+ nanogenerator for tumor-specific synergistic therapy via disruption of mitochondrial Ca2+ homeostasis and photothermal therapy. ACS Nano 12, 6806–6818 (2018).
Banerjee, S. & Chakravarty, A. R. Metal complexes of curcumin for cellular imaging, targeting, and photoinduced anticancer activity. Acc. Chem. Res. 48, 2075–2083 (2015).
Wang, S. et al. Core-satellite polydopamine-gadolinium-metallofullerene nanotheranostics for multimodal imaging guided combination cancer therapy. Adv. Mater. 29, 1701013 (2017).
Sun, J. et al. Dithiol-activated bioorthogonal chemistry for endoplasmic reticulum-targeted synergistic chemophototherapy. Angew. Chem. Int. Ed. 61, e202213765 (2022).
Jiang, W. et al. Nitric oxide induces immunogenic cell death and potentiates cancer immunotherapy. ACS Nano 16, 3881–3894 (2022).
Chu, X. et al. Nitric oxide modulating calcium store for Ca2+-initiated cancer therapy. Adv. Funct. Mater. 31, 2008507 (2021).
Zhang, L. et al. Cytosolic TGM2 promotes malignant progression in gastric cancer by suppressing the TRIM21-mediated ubiquitination/degradation of STAT1 in a GTP binding-dependent modality. Cancer Commun. 43, 123–149 (2023).
Zhang, Z. et al. CGI1746 targets σ1R to modulate ferroptosis through mitochondria-associated membranes. Nat. Chem. Biol. 20, 699–709 (2024).
Ma, Z., Zeng, P., Zhai, T., Zhao, Y. & Liang, H. In situ mitochondrial biomineralization for drug-free cancer therapy. Adv. Mater. 36, 2310218 (2024).
Li, Y., Liu, J., Chen, Y., Weichselbaum, R. R. & Lin, W. Nanoparticles synergize ferroptosis and cuproptosis to potentiate cancer immunotherapy. Adv. Sci. 11, e2310309 (2024).
Xu, R. et al. Remodeling of mitochondrial metabolism by a mitochondria-targeted RNAi nanoplatform for effective cancer therapy. Small 20, e2305923 (2024).
Komotar, R. J., Wilson, D. A. & Connolly, E. S. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. Neurosurgery 57, 7–8 (2005).
Fei, H. et al. Gint4.T-siHDGF chimera-capped mesoporous silica nanoparticles encapsulating temozolomide for synergistic glioblastoma therapy. Biomaterials 306, 122479 (2024).
Acknowledgements
This work was supported by the Jiangsu Basic Research Center for Synthetic Biology Grant (No. BK20233003, C.M.), the National Natural Science Foundation of China (Nos. 52422306 (M.M.W.), 22275095 (M.M.W.), 22175096 (C.M.) and 22475103 (C.M.)), the Jiangsu Key Laboratory of Biofunctional Materials and the Jiangsu Collaborative Innovation Center of Biomedical Functional Materials.
Author information
Authors and Affiliations
Contributions
T.T.C. and M.M.W. conceived the project, designed the experiments, and prepared the manuscript. Y.D. and Y.J.W. contributed to movie processing. T.T.L. assisted with in vitro cell experiments, etc., while S.L.L. and X.X. assisted with animal experiments, etc. T.T.C. and Y.D. wrote the manuscript. M.M.W. and C.M. supervised and directed this project; All authors commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yingfeng Tu, Marc-Antoine Lauzon and the other anonymous reviewer for their contribution to the peer review of this work. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, T., Duan, Y., Wang, Y. et al. Size-variable self-feedback nanomotors for glioblastoma therapy via mitochondrial mineralization. Nat Commun 16, 8990 (2025). https://doi.org/10.1038/s41467-025-64020-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-64020-x
