
Abstract
All-perovskite tandem solar cells (PTSCs) demonstrate exceptional potential to surpass the Shockley-Queisser (SQ) theoretical limit. However, practical implementation faces critical challenges due to a self-reinforcing photothermal-mechanical degradation mechanism originating from multiscale physical couplings. In this study, a multifunctional polyamine ligand triphenyltriamine thiophosphate (TPTA) was introduced into the tin-lead (Sn-Pb) perovskite solution system to establish an I-Sn-N coordination-mediated lattice stabilization framework, and the photothermal-mechanical coupling path was cut off from multiple aspects such as suppressing periodic oscillations and regulating stress. Consequently, single-junction Sn-Pb perovskite solar cells (PSCs) achieve a power conversion efficiency (PCE) of 23.4% and retaining 94.9% of initial performance after 950 hours of maximum power point (MPP) tracking. When the device is integrated into the 2-terminal (2 T) tandem architecture, its PCE reaches a significant level of 29.6 % (certified PCE of 28.9%), and 93.4% of the initial efficiency can be maintained after 900 hours continuous operation.
Similar content being viewed by others


Introduction
By the detailed balance principle, mixed tin-lead (Sn-Pb) perovskite solar cells (PSCs) achieve an elevated radiative efficiency limit by extending their spectral utilization range, thereby more effectively harvesting photons across the solar spectrum1,2,3,4,5,6. This enhanced performance paves the way for perovskite tandem solar cells (PTSCs) incorporating tin-lead perovskites to overcome the Shockley-Queisser (SQ) limit, as their multi-junction architecture synergistically optimizes light absorption and carrier extraction across complementary bandgaps7,8,9. However, their practical deployment is severely constrained by a self-amplifying photothermo-mechanical degradation cycle arising from multiscale physical interactions10,11,12,13.
Under operational conditions, this self-amplifying degradation cascade involving light absorption14,15, thermal excitation16,17,18, and lattice deformation19,20. Light illumination (photonic field) generates high-density photogenerated carriers, which undergo non-radiative recombination at defect sites, releasing energy as localized heat (thermal field). This thermal energy triggers anisotropic lattice expansion and bond-length oscillations (mechanical field), which leads to stress concentration and damages the lattice structure. Particularly, it is more serious in the Sn-I framework with strong electron-phonon coupling of Sn2+. The resulting lattice strain propagates heterogeneously across Pb/Sn domains, accumulating interfacial stress that exceeds the fracture threshold of Sn-I bonds21. Subsequent bond rupture generates vacancies (e.g., VI, VSn) and halide migration pathways, amplifying defect density and further enhancing non-radiative recombination (thermal field intensification)22,23,24. Crucially, this light-thermal-mechanical degradation cycle drives irreversible lattice collapse and efficiency decay within hours22,25,26,27.
To disrupt this multiscale failure cascade, we propose a molecular-level intervention that effectively cuts off the photothermally mechanical degradation pathway. By introducing a multifunctional ligand triphenyltriamine thiophosphate (TPTA) into the perovskite solution, we establish a directional I-Sn-N coordination network reinforced by P = S-assisted multidentate anchoring. TPTA was rationally designed as a multifunctional additive incorporating a rigid tridentate architecture, where the P = S moiety synergizes with terminal triamine groups to enable simultaneous coordination with multiple Sn²⁺ centers. This multidentate interaction enhances complexation stability and spatial coverage during nucleation, thereby promoting uniform crystal growth and inhibiting Sn²⁺ oxidation. While both Sn²⁺ and Pb²⁺ are considered borderline acids, Sn²⁺ exhibits higher polarizability and a more chemically active 5 s² lone pair, giving it a greater affinity for soft donor atoms such as sulfur, which is supported by DFT-calculated charge redistribution and adsorption energies. In addition, the thiophosphoryl group’s structural rigidity aids in interfacial organization and improves bond durability under stress, collectively contributing to improved lattice stability and suppressed photothermal-mechanical degradation. Unlike conventional sulfur-based additives28,29,30, TPTA integrates dipole modulation, multi-site coordination, and structural robustness within a single molecular scaffold—an unexplored strategy in Sn–Pb perovskite engineering.
Combined experimental and theoretical analyses reveal that TPTA not only stabilizes the Sn²⁺ chemical environment but also homogenizes electron-phonon coupling across Pb/Sn sublattices. These synergistic effects enable Sn-Pb PSCs to achieve 23.4% efficiency with 94.9% of the initial PCE retained after 950 hours maximum power point (MPP) tracking and 29.6% (certified PCE of 28.9%) in two-terminal tandem architectures, retaining 93.4 % of the initial PCE after 900 h of MPP tracking, alongside exceptional stability under prolonged operational stress. This work resolves longstanding challenges in Sn-Pb perovskite stability, establishing a universal framework for designing high-efficiency, durable perovskite photovoltaics, advancing their transition from lab-scale to industry.
Results
To systematically investigate the relationship between molecular structure and additive functionality, we designed a series of triamine-terminated molecules—TAPB, TAPOB, and TPTA—with controlled variations in the central scaffold. By keeping the –NH₂ terminal groups constant as the primary coordination sites, this design enables isolated evaluation on how the central unit modulates dipole moment, electronic polarization, and additive–perovskite interactions. TAPOB, incorporating an ether bridge, increases the molecular dipole (1.42 D → 4.86 D), thereby enhancing local electrostatic interactions with Sn²⁺. Building upon this, TPTA introduces a thiophosphoryl (P = S) moiety, which not only further increases the dipole moment (to 5.13 D) but also provides a soft donor site (sulfur) with enhanced affinity toward Sn²⁺ due to its borderline acid–based compatibility4,31,32. In addition, the phosphorus atom adjacent to sulfur acts as an electron-withdrawing center, further polarizing the sulfur atom and reinforcing its coordination strength. The rigid tripodal architecture of TPTA allows for tridentate binding, offering spatially distributed, multidentate interactions that stabilize the perovskite lattice and suppress Sn²⁺ oxidation. These synergistic features render TPTA functionally superior to both TAPOB and TAPB, and justify its selection as the optimal perovskite solution additive in this work. This will be testified in the subsequent discussions.
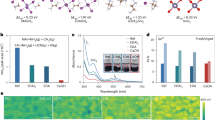
The electrostatic potential distributions of the different molecules obtained via calculations are shown in Fig. 1a. The dipole moments of the molecules presented in Fig. 1a follow the order μ(TAPΒ) < μ(TΑPΟΒ) < μ(ΤΡΤΑ). After the introduction of the P = S bond, μ(TPΤΑ) is the highest among the three molecules, reaching 5.13 Debye. The increase in dipole moment expects to enhance the molecular polarity and a more inhomogeneous charge distribution, thus inducing a stronger molecular interaction33,34. As an addition to the perovskite solution, TPTA has a stronger electrostatic attraction to metal cations (Sn 2+), and the increase of its dipole moment leads to enhanced charge transfer ability and improved defect passivation efficiency. This is further supported by DFT calculations (as shown in Supplementary Fig. 1). The calculation results indicate that the binding affinities of molecules with different central groups to the perovskite vary significantly. When TAPB, TAPOB, and TPTA interact with the perovskite, their adsorption energies are − 2.109 eV, − 2.328 eV, and − 2.525 eV, respectively. A more negative adsorption energy signifies a stronger binding interaction with the perovskite surface. This excellent binding strength can be attributed to the fact that P = S in TPTA greatly affects the electron cloud density of the terminal amine group and enhances the polarity of the molecule. In order to study the effects of different additive molecules on perovskites, differential charge density analysis was conducted35,36,37. The charge density distributions (Fig. 1b–d and Supplementary Fig. 2) show that TPTA exhibits the strongest interaction with Sn and shows the most significant electron cloud redistribution, attributed to the modification by the P = S bond. This can be attributed to the fact that Sn²⁺ possesses higher polarizability and a more chemically active 5 s² lone pair, rendering it a greater affinity for soft donor atoms such as sulfur. This strong coordination facilitates stable anchoring of the molecule onto the perovskite lattice and effectively stabilizes the chemical environment of Sn, which is beneficial for maintaining the strength of the Sn-I bonds and effectively preventing bond breakage under illumination.

a Chemical structures of TAPB, TAPOB, and TPTA, and the corresponding electrostatic potential diagram and dipole moment. b–d Differential charge density profiles of TAPB, TAPOB and TPTA interacting with perovskites. e, f FTIR spectra of TPTA, TPTA/PbI2 and TPTA/SnI2. g Sn 3 d XPS spectra of control, TAPB modified, TAPOB modified and TPTA modified films. h–k Raman spectra of the control, TAPB, TAPOB and TPTA modified films.
To further examine the effects of TAPB, TAPOB, and TPTA on SnI2 and PbI2, Fourier transform infrared (FTIR) spectroscopy was performed (Fig. 1e, f and Supplementary Fig. 3). It was found that when mixing with TAPB, no significant shift was observed in the N-H bending (overtone) and N-H stretching vibrations38, indicative of a weak interaction between TAPB and PbI2 or SnI2. In the mixed solution of TAPOB, SnI2 and PbI2, slight vibrational shifts of SnI2 and PbI2 were detected. In contrast, when TPTA was mixed with SnI2 and PbI2, both the N-H bending (overtone) and stretching vibrations shifted toward lower wavenumbers, with larger and more distinct redshifts observed in the presence of SnI2. These redshifts are likely due to the coupling interaction between TPTA and Sn2+ solvate species39, which disrupts intramolecular vibrations. In addition, a slight redshift was also observed in the P = S bond vibration after mixing with SnI2 and PbI2. This demonstrates that the introduction of the P = S bond in TPTA enhances the interaction strength between -NH2 and Sn2+ as P = S reshapes the electron cloud density of the terminal amino group, which effectively stabilizes the Sn chemical environment. This is consistent with the result of the charge density distribution mentioned earlier.
In order to more convincingly illustrate the interaction of additives with Sn and Pb, X-ray photoelectron spectroscopy (XPS) was performed to analyze the Sn 3 d and Pb 4 f spectra. Figure 1g and Supplementary Fig. 4 illustrate the binding energies of Sn 3 d and Pb 4 f orbitals. It is seen that the addition of TAPB, TAPOB, and TPTA reduced the binding energy of Sn 3d5/2 from 495.3 eV to 495.2 eV, 495.1 eV, and 494.8 eV, respectively, and the Sn 3d3/2 from 486.9 eV to 486.8 eV, 486.7 eV, and 486.4 eV, respectively. Whereas, the additives showed minimal effect on Pb, with the binding energy of the Pb 4 f orbital remained almost unchanged. This difference arises because the stereochemically active 5 s² lone pair on Sn²⁺ exhibits stronger positioning ability compared to the 6 s² lone pair on Pb²⁺. Consequently, Sn²⁺ is more susceptible to asymmetric ligand field interactions and more reactive towards soft donor atoms like sulfur (in the P = S group), consistent with HSAB theory. Furthermore, Sn²⁺ tends to segregate towards surfaces or grain boundaries in mixed Sn–Pb perovskites due to its lower formation energy, making it more accessible for surface complexation with additives5,30,40. This indicates a strong coordination between the additives and Sn, effectively stabilizing the chemical environment of the Sn site and reducing its chemical activity. Such stabilization enhances the Sn-I bond strength within the perovskite lattice, prevents bond dissociation, and inhibits the oxidation of Sn2+ to Sn4+.
Raman spectroscopy was employed to study how the central group-induced dipole moment variations affect bond strength and lattice framework stability41,42. By tracking the wavenumber shifts of Sn-I and Pb-I bonds under different illumination durations, the evolution of bond strength and the stability of these bonds were evaluated. Raman spectra of the Sn-Pb perovskite films at 0, 10, and 20 h of illumination elucidate the structural dynamics of the control and additive-treated films (as shown in Fig. 1h–k). The vibration peaks at 70–80 cm−1 and 115–125 cm−1 are assigned to Pb-I and Sn-I bonding modes, respectively. These peaks are indicative of the local bonding environment, as changes in vibrational frequency are directly associated with variations in bond strength and electron cloud distribution. In the control film, the Pb-I and Sn-I peaks were initially located at 74.0 cm−1 and 119.0 cm−1, respectively. Upon addition of TAPB, TAPOB, and TPTA, these Pb-I peaks shifted to 74.9, 75.6, and 77.0 cm−1, and Sn-I peaks to 119.4, 120.8, and 121.0 cm−1, respectively. These shifts suggest that modifying the central group alters the electronic environment surrounding of -NH2 and regulates its coordination capacity with Sn and Pb, thus redistributing the electron cloud and enhancing both bond polarity and vibrational frequency. After 10 and 20 h of illumination, the Sn-I peak of the control film shifted to lower wavenumbers by 1.0 cm−1 and 3.0 cm−1, respectively, and the Pb-I peak shifted by 0.8 cm−1 and 1.1 cm−1. It demonstrates that light excitation causes the elongation and thermal expansion of the bond, reducing the strength of the bond. Among the three additives, TPTA incorporation led to the smallest shifts in Sn-I and Pb-I peaks after 20 h, only 1.0 cm−1 and 0.3 cm−1, respectively. These results indicate that introducing the additive TPTA into the precursor solution effectively enhances the Sn-I bond and weakens the periodic light-induced oscillation of Sn-I. This bond enhancement helps preserve lattice symmetry, thereby preventing fragmentation of the Sn-I network, suppressing defect formation, and inhibiting ion migration, as will be discussed further.
The different electron-phonon coupling strengths of Pb2+ and Sn2+ induce lattice inhomogeneities, leading to local stress concentrations that are more pronounced than those in pure Sn-based systems. These stress concentrations reflect the intrinsic structural inhomogeneity of the material and render Sn-I bonds particularly vulnerable to breakage under illumination. To further investigate the effect of P = S-assisted polyamine anchoring on lattice vibrations in Sn-Pb perovskites, temperature-dependent photoluminescence (TD-PL) measurements were conducted on pristine and TPTA-incorporated narrow-bandgap (NBG) films. According to the Bose-Einstein distribution, the temperature-dependent linewidth broadening Γ(T) can be described by:
where \({\varGamma }_{{L}_{0}}\) represents the homogeneous temperature-dependent broadening, and the increase in PL spectral width is attributed to the interaction between LO phonons and electrons18,43,44,45. The electron-phonon coupling strength (γLO) was extracted by analyzing the full width at half maximum (FWHM) of PL spectra at different temperatures (Fig. 2a, b). As shown in Fig. 2c, the γLO of the control film is 2553.5 meV, indicating strong electron-phonon coupling and significant lattice disorder. In contrast, the film processed from precursor solution containing additives exhibits a much lower γLO value of 317.1 meV, suggesting that bond oscillations are suppressed and lattice vibrations are reduced. These results demonstrate that P = S-assisted polyamine anchoring in the TPTA-incorporated film significantly strengthens Sn-I bonding, improves lattice structural stability, and reduces phonon scattering. This finding is consistent with the Raman results, confirming that the addition of TPTA effectively suppresses thermal vibrations.

a, b Pseudo-color maps of temperature-dependent photoluminescence (TD-PL) spectra for the control and with TPTA films across a temperature range of 100–190 K. c Temperature dependence of the full width at half maximum (FWHM) of PL peaks for the respective films. d Stress statistics of control and TPTA modified perovskite films before and after 20 h of illumination, derived from Grazing incidence X-ray diffraction (GIXRD) analysis. e, f Integrated Differential Phase Contrast (IDPC) images, Geometric Phase Analysis (GPA) strain maps(exy), and Fast Fourier Transform (FFT) patterns of the perovskite lattice for (e) control and (f) TPTA modified films after 20 h of light exposure. g Arrhenius plots of conductivity for control, TAPB modified, TAPOB modified, and TPTA modified perovskite films. h, i Time-of-flight secondary ion mass spectrometry (TOF-SIMS) depth profiles of halide migration under light 40 hours for (h) pristine perovskite film and (i) perovskite film with TPTA. j Defect formation energies (Ef) of control, TAPB, TAPOB and TPTA with I, Pb and Sn vacancy defects (VI, VPb, VSn), I interstitial defects (Ii) and Pb interstitial defects (Pbi)on the perovskite surface.
X-ray diffraction (XRD) measurements under different illumination durations were further performed to investigate lattice evolutions (Supplementary Fig. 5a–d). In the control film, the (100) diffraction peak gradually shifts to lower angles and eventually disappears after 30 h of illumination, indicating light-induced lattice expansion. This expansion is mainly attributed to the accumulated Sn-I bond elongation and localized stress. While the incorporation of TAPB and TAPOB provides limited improvement in lattice retention, the TPTA-treated films exhibit excellent structural stability, maintaining a strong (100) peak even after prolonged illumination46. This indicates that TPTA effectively suppresses lattice distortion in Sn-Pb perovskites by reinforcing Sn-I bonds and alleviating photoinduced strain, thereby demonstrating superior lattice stability.
For a more intuitive study of the lattice distortion in thin films under illumination, grazing-incidence X-ray diffraction (GIXRD) was used to quantify stress evolution arisen from lattice expansion19,47,48 (Fig. 2d and Supplementary Fig. 5a–d). In the control film, the initial tensile stress was measured at 19 MPa, likely due to residual stress from film deposition, such as incomplete thermal relaxation or lattice mismatch between the perovskite layer and substrate. After 20 h of illumination, the tensile stress dramatically increased to 52 MPa, driven by light-induced bond elongation. These large stress increments highlight the tendency for stress accumulation and lattice framework instability. In contrast, the TPTA-incorporated film initially exhibited a compressive stress of − 15 MPa, which is attributed to the strong interaction of the P = S and -NH2 groups with Sn, allowing effective anchoring and lattice stabilization. Compressive stress plays a crucial role in stabilizing the perovskite lattice by suppressing lattice vibrations, inhibiting defect migration, reducing dynamic structural fluctuations, and improving thermodynamic stability. Moreover, compressive stress reinforces chemical bonding, rendering the lattice more resistant to external stimuli such as photon or thermal excitation49,50. Even after 20 h of illumination, the tensile stress of the film fabricated from the precursor solution containing TPTA remained at − 10 MPa. This excellent stress resistance indicates that TPTA effectively alleviates light-induced lattice oscillations, reduces stress generation, and helps maintain the integrity of the lattice.
To directly visualize structural degradation and stress accumulation in Sn-Pb perovskites under long-term illumination, transmission electron microscopy (TEM) was conducted using ultra-low-dose rate integrated differential phase contrast (IDPC) mode to avoid beam damage and enable high-resolution lattice imaging51,52,53. The TEM images reveal a stark difference in lattice stability between control and TPTA-incorporated films (Fig. 2e, f). After 20 hours of illumination, the control film exhibited pronounced structural deformation in IDPC imaging compared to the unexposed sample (Supplementary Fig. 7), directly confirming the lattice fragmentation caused by stress and strain intensification. Strain distribution analysis via geometric phase analysis (GPA) (Fig. 2e, f and Supplementary Fig. 8) further revealed localized shear strain accumulation (red-dominated regions) in the control film, ultimately driving lattice failure. In stark contrast, TPTA-contained films demonstrated exceptional lattice coherence and structural uniformity. IDPC characterization of TPTA-incorporated samples displayed well-aligned atomic arrangements, complemented by homogeneous strain profiles with negligible high-strain zones. In addition, fast fourier transform (FFT) patterns further exhibited diffuse, asymmetric diffraction spots of the control film after prolonged illumination, signaling crystallinity loss and disordered phase formation. Deviation from the ideal 90° diffraction angles in control films further corroborated symmetry breakdown and severe lattice distortion. Conversely, the film containing TPTA retained sharp, symmetric diffraction peaks aligned precisely at 90°, unequivocally preserving crystallinity and lattice symmetry under extended illumination. These findings underscore TPTA’s capability to mitigate strain propagation and stabilize lattice dynamics through its P = S-enabled multipoint anchoring mechanism, which functions as a “molecular spring” to sustain both structural integrity and optoelectronic performance in Sn-Pb perovskites.
Ion migration is conducive to the formation of VI and non-radiative recombination centers. By fitting the Arrhenius equation to the temperature-dependent conductivity results (as shown in Supplementary Fig. 9), the activation energy (Eaion) for iodide ion migration was extracted, revealing the interaction between stress modulation and ion dynamics45,54. As shown in the fitting of Fig. 2g, the untreated control film exhibits the lowest activation energy for iodide ion migration (Eaion = 347.3 meV), which can be explained by the weakening of Sn-I and Pb-I bonds under photothermal stimulation. On the contrary, the films containing TAPB, TAPOB, and TPTA showed gradually increased activation energies of ion migration (Eaion =403.3 meV, Eaion = 452.9 meV and Eaion = 563.3 meV, respectively), which can be explained by the improved coordination effect of Sn-I binding strength. It thus reduces the thermal vibration strength, alleviates stress accumulation, increases the energy barrier of ion migration, and finally effectively inhibits the defect expansion and improves the stability of the lattice structure. These findings demonstrate the key role of bond strength regulation and stress regulation in controlling ion migration and maintaining the stability of perovskite materials.
Time-of-flight secondary ion mass spectrometry (TOF-SIMS) was used to further analyze the iodide migration in Sn-Pb perovskite under long-term light exposure55, the spatial distribution of iodine ions in control and TPTA-incorporated devices after 40 h of light exposure, as shown in Fig. 2h, i. In the control films, a high concentration of iodide ions was detected in the Ag electrode, indicative of severe ion migration, compromising charge extraction efficiency and deteriorating long-term device stability. In contrast, TPTA-incorporated films exhibited significantly lower iodide ion signal intensity, which can be attributed to the increased migration barrier provided by the molecular additive.
For a quantitative analysis of iodide migration and iodine release under prolonged light exposure, perovskite films illuminated for 10, 20, and 30 h were immersed in toluene, and UV-Vis absorption spectra were recorded (Supplementary Fig. 10) to monitor iodine-related peaks. Two characteristic absorption peaks were observed. The peak at ~ 500 nm corresponds to molecular iodine (I2), likely formed through oxidation of I- at the film surface, while the peak at 300–350 nm is associated with iodide ions or iodine-solvent complexes dissolved in toluene56,57. In the control film, the intensities of both peaks increased significantly with extended illumination time, reflecting extensive iodide ion migration to the surface. Further oxidation of iodide ions to molecular iodine contributes to the ~ 500 nm peak, while others dissolve directly into toluene, resulting in the 300–350 nm absorption. This pronounced iodine release, driven by lattice destabilization, Sn-I bond cleavage, and defect formation, highlights the vulnerability of the perovskite layer to light-induced stress. In comparison, films contained with TAPB, TAPOB, and TPTA showed a clear suppression of iodine release, with a much smaller increase in both peak intensities. Notably, the TPTA-incorporated film exhibited the smallest change, even after 30 h of illumination, suggesting that TPTA significantly strengthens Sn-I bonds, suppresses thermal vibrations, mitigates local stress through reversible deformation, increases the migration barrier of I-, and effectively blocks multi-field coupled degradation pathways.
The oxidation process of Sn2+ to Sn4+ mediated by I2 was further analyzed by X-ray photoelectron spectroscopy (XPS), and the relative contents of Sn2+ and Sn4+ in the films before and after 20 and 40 h of illumination were summarized (Supplementary Fig. 11 and Supplementary Table 1). After illumination, the proportion of Sn4+ in the control film increased significantly from 14.9% to 29.5% and 43.2%, indicating severe oxidation. This oxidation disrupts the Sn-I bond network and introduces deep-level defect states. In contrast, the Sn4+ content in the TAPB- and TAPOB-incorporated films increased from 10.1% to 23.0% and 30.5%, and from 7.7% to 16.6% and 28.7%, respectively, indicating moderate suppression of Sn2+ oxidation. Remarkably, the TPTA-incorporated film exhibited the lowest Sn4+ increase, rising only from 4.1% to 8.1% and 17.9% after 20 and 40 hours of illumination, respectively. These results demonstrate the excellent stabilizing capability of TPTA, which enhances Sn-I bond strength, limits I2-mediated oxidation, and maintains structural integrity under prolonged illumination, consistent with the reduced I2 signal observed in the UV-Vis analysis. To further examine whether TPTA inhibits Sn²⁺ oxidation in the precursor solution under ambient conditions, we analyzed the Sn-Pb perovskite precursor solutions (with and without TPTA) exposed to air for different periods of time, as shown in Supplementary Fig. 12. It can be observed that after the perovskite solution (the control group) was exposed to air for 5 minutes, the solution changed from clear to turbid orange-red, indicating that I- and Sn2+ in the perovskite solution had been oxidized. However, the perovskite solution with TPTA added showed only a weak oxidation effect after 5 minutes, with a slight color change on the surface. This indicates that the addition of the additive TPTA effectively inhibited the oxidation of the perovskite solution.
The surface potential distribution and its correlation with electronic properties were further examined using Kelvin probe force microscopy (KPFM)24,58 (Supplementary Fig. 13). Before illumination, the control film exhibited a broad potential distribution centered at ~ 50 mV, indicating substantial inhomogeneity and high surface defect density. In contrast, the film containing TPTA displayed a narrower and more uniform potential distribution centered at 85 mV, indicating enhanced surface uniformity. The more positive surface potential suggests improved n-type character, facilitating surface electron transport and charge extraction. After 40 h of illumination, the control film showed the appearance of high work function regions, indicative of degradation products such as PbI2, likely formed due to photoinduced iodide migration and structural instability. A film formed by spin-coating a precursor containing TPTA, on the other hand, maintained a uniform surface potential distribution with minimal variation, confirming suppressed decomposition.
The enhanced n-type character and VOC can be further attributed to improved energy level alignment induced by TPTA. Ultraviolet photoelectron spectroscopy (UPS) reveals that the valence band maximum (VBM) of the perovskite shifts upward from – 5.32 eV to – 4.97 eV upon TPTA treatment, accompanied by a corresponding upward shift in the conduction band minimum (CBM) (As shown in Supplementary Fig. 14). This results in reduced energetic offsets with both the hole and electron transport layers, promoting more balanced carrier extraction and suppressed interfacial recombination. The improved band alignment is attributed to the strong dipole moment and electron-donating character of TPTA, which modulate the interfacial surface potential and band bending. Overall, these interfacial energetic modifications, in combination with trap state passivation, synergistically contribute to the observed increase in VOC.
The enhanced n-type behavior observed in the film containing TPTA can be attributed to the passivation of undercoordinated metal ions and halide vacancies by triamine groups, modulated through dipole tuning. This result aligns with DFT calculations, as shown in Supplementary Fig. 1, which illustrates the adsorption energies of TAPB, TAPOB, and TPTA on defect-free Sn-Pb perovskite surfaces. The more negative adsorption energy of TPTA suggests a stronger interaction between TPTA and the perovskite surface. This binding is attributed to the introduction of the P = S group in TPTA, which regulates the electron cloud distribution around the polyamine group, facilitates stable anchoring in the lattice, and enhances defect passivation capability. As shown in Fig. 2j, TAPB-, TAPOB-, and TPTA-incorporated films exhibit increased defect formation energies relative to the control film, indicating that TPTA effectively strengthens of the Sn-I bonding, prevents bond elongation and fracture, stabilizes the lattice structure, inhibits the migration of I-, and effectively reduces defect formation14.
Scanning electron microscopy (SEM) images (Fig. 3a, b and Supplementary Fig. 15) reveal distinct morphological differences59,60 between the films containing TAPB, TAPOB, and TPTA and the control film, both before and after extended illumination. Prior to illumination, all three additive-incorporated films displayed significantly enlarged grains compared to the control, with average grain sizes of 710.5, 790.4, and 910.8 nm, respectively (Supplementary Fig. 16). This increment in grain size reflects enhanced crystallinity and reduced grain boundary defects, both of which are critical for suppressing ion migration, reducing nonradiative recombination, and facilitating charge transport. In stark contrast, residual PbI2 at grain boundaries in the control film would induce accelerated degradation. After 20 hours of illumination, the control film showed significant surface roughness and degradation at the grain boundaries, with more PbI2 precipitates. After 40 h, PbI2 accumulation increased further, accompanied by collapse and degradation of the crystal lattice. In contrast, films containing additives, especially TPTA, retained excellent morphological stability. Notably, although strong additive–metal coordination in some cases hinders crystal growth, the observed grain enlargement in TPTA-incorporated films can be attributed to the kinetic modulation effect of TPTA rather than facet blocking. TPTA selectively coordinates with Sn²⁺ in the precursor solution, forming intermediate complexes that delay premature nucleation of Sn-rich domains and thereby suppress vertical compositional gradients of Sn-rich and Pb-rich components. This retards early nucleation and lowers nucleation density, enabling extended lateral crystal growth and promoting uniform grain morphology. Importantly, the coordination strength toward Pb²⁺ remains weak, avoiding inhibition of crystal facet development during the growth stage. This kinetic regulation mechanism4,6, coupled with optimized additive concentration, results in enlarged and compositionally homogeneous grains, as confirmed by SEM, GIWAXS, and TOF-SIMS analyses. Moreover, the TPTA-incorporated film maintained intact grain structure, with a smooth, dense surface morphology, free of voids or amorphization, even after 40 h of aging.

Scanning electron microscopy (SEM) images of the perovskite film without illumination and after illumination for 40 h, pristine perovskite film (a)and TPTA perovskite film(b). c, d Detailed analysis of the (100) diffraction peak of GIWAXS patterns of control and TPTA-treated Sn-Pb perovskite films at different grazing incidence angles (α = 0.1°−0.8°). Transient photocurrent (TPC) decay curves (e). Transient photovoltage (TPV) decay curves (f). g Trap density (Nt) values for control, TAPB-, TAPOB- and TPTA-incorporated Sn-Pb perovskite films determined using space-charge-limited current (SCLC) analysis. The statistical PCE of Sn-Pb PSCs with different treatments (h). J-V curves (i) of the best performing devices under control and modified with TAPB, TAPOB and TPTA. j The EQE spectra of control devices and target devices. k MPP tracking of control devices and devices with TPTA under 1 sun illumination in the N2 environment. l Long-term stability test of control devices and devices with TPTA Sn–Pb PSCs stored in an N2-filled glovebox.
X-ray diffraction (XRD) was used to analyze the effect of additives on film formation (Supplementary Fig. 17a). After treatment with TAPB, TAPOB, and TPTA, the intensity of the (100) diffraction peak increased, and the full-width at half-maximum (FWHM) significantly decreased (Supplementary Fig. 17b), indicating enhanced crystallinity. In addition, a peak at 12.7° corresponding to PbI2 was observed in the control film, but was absent in the additive-treated films46. This is consistent with the PbI2 aggregation seen in the SEM images and confirms that the additives inhibit PbI2 formation.
A key factor influencing crystallization in Sn-Pb perovskites is the distribution of Sn and Pb within the film61. TOF-SIMS depth profiling of the control film (Supplementary Fig. 18) reveals that Sn is concentrated near the top surface, while Pb is more abundant in the bottom region, resulting in a pronounced compositional gradient. This inhomogeneous distribution stems from the differences in solubility and crystallization kinetics between Sn- and Pb-based perovskites, inducing lattice strain and structural distortion that promote photodegradation and reduce device stability. In contrast, the TPTA-incorporated film exhibits a uniform Sn and Pb distribution throughout the film. This homogeneity is attributed to the strong coordination between amine groups in TPTA and Sn2+, which stabilizes Sn intermediates and lowers the nucleation barrier. By selectively retarding Sn crystallization, the growth kinetics of the Sn phase become better matched to that of the Pb phase, mitigating the phase segregation observed in the control film. This uniform composition helps reduce lattice strain and ensures consistent electronic properties across the entire film.
Grazing-incidence wide-angle X-ray scattering (GIWAXS) analysis was further conducted to gain insights into the crystallographic properties of the (100) plane62 (Fig. 3c, d and Supplementary Fig. 19). In the control film, the (100) peak exhibited a broad FWHM across different incidence angles, indicating poor crystallinity and significant lattice strain and structural inhomogeneity along the vertical direction. As the grazing incidence angle (α) increases, the X-ray penetrates deeper into the film, enabling depth-resolved structural analysis. In the control sample, the (100) diffraction peak exhibits a gradual shift toward lower angles with increasing α, indicative of increased d-spacing near the surface4. This behavior arises from a vertical compositional gradient, with Sn enrichment in the upper layers and Pb accumulation at the bottom, as confirmed by TOF-SIMS depth profiles (Supplementary Fig. 16). Although the ionic radius of Sn²⁺ (1.18 Å) is slightly smaller than that of Pb²⁺ (1.19 Å), the Sn-rich regions display local lattice expansion due to the more polarizable and covalent nature of Sn–I bonds, attributed to the higher chemical activity of the Sn²⁺ 5 s² lone pair. This lattice relaxation leads to a depth-dependent increase in d-spacing. In contrast, TPTA-incorporated films show negligible peak shift across different α values, indicating suppressed vertical segregation and a compositionally uniform lattice structure.
The photoluminescence (PL) spectra of the control, TAPB-, TAPOB-, and TPTA-incorporated perovskite films are presented in Supplementary Fig. 20. Among the four samples, the TPTA-treated film exhibited the highest luminescence intensity, followed by the TAPOB- and TAPB-incorporated films, with the control film showing the weakest emission. This trend reflects the superior defect passivation and suppressed nonradiative recombination in TPTA-incorporated films, in agreement with the increased defect formation energy and improved structural uniformity discussed earlier. In addition, a noticeable blue-shift in the PL peak of the film containing TPTA further confirms the suppression of defect states. To complement these findings, time-resolved photoluminescence (TRPL) measurements were performed (Supplementary Fig. 21), and fitting parameters are summarized in Supplementary Table 2. The film containing TPTA exhibited the longest carrier lifetime of 12.52 us, significantly higher than those of the TAPOB-incorporated (11.88 us), TAPB-incorporated (10.06 us), and control films (1.49 us), reflecting a substantial reduction in nonradiative recombination. To evaluate interfacial recombination between the perovskite and C60 layers, photoluminescence quantum yield (PLQY) tests were performed (Supplementary Fig. 22). The results showed a gradual reduction in interfacial recombination loss with the addition of TAPB, TAPOB, and TPTA. Notably, the film containing TPTA exhibited the lowest recombination loss of only 0.0031%, compared to 0.0128% for the control, indicating improved photoluminescence energy conversion efficiency and minimized nonradiative losses at the interface.
Performance and stability of NBG single-junction PSCs
Charge extraction kinetics and carrier lifetimes under operational conditions were investigated via transient photovoltage (TPV) and transient photocurrent (TPC) measurements. TPC analysis (Fig. 3e and Supplementary Fig. 23a) showed that the TPTA-incorporated device had the fastest charge extraction rate with a decay time of 8.37 us, outperforming the TAPOB-incorporated (10.56 us), TAPB-incorporated (10.67 us), and control devices (12.37 us). Faster decay suggests more efficient charge extraction from the perovskite layer to the adjacent transport layer, which could be attributed to the improved grain morphology and superior interface quality imparted by TPTA. These structural enhancements reduce the energy barrier for charge transfer and minimize carrier accumulation at grain boundaries. TPV measurements (Fig. 3f and Supplementary Fig. 21b) highlight the role of defect passivation and lattice stabilization in suppressing carrier recombination. The device fabricated using the perovskite solution with TPTA showed the longest carrier lifetime (6.95 us), exceeding those of TAPOB-incorporated (6.67 us), TAPB-incorporated (5.28 us), and control (3.43 us) devices. A longer TPV lifetime suggests a lower recombination rate and reduced trap state density.
Trap density (Nt) in perovskite films was quantified using space charge-limited current (SCLC) measurements on hole-only and electron-only devices15 (Supplementary Fig. 24 and Fig. 3g). The trap-filled limit voltage (VTFL) and corresponding Nt values are summarized in Supplementary Tables 3, 4. The VTFL values for electron-only devices decreased from 0.84 V (control) to 0.80 V, 0.77 V, and 0.74 V for TAPB-, TAPOB-, and TPTA-incorporated films, respectively. In hole-only devices, VTFL values dropped from 0.75 V (control) to 0.66 V, 0.61 V, and 0.54 V, respectively. Correspondingly, Nt values in control films were 1.16 × 1017 cm−3 (hole type) and 1.30 × 1017 cm−3 (electron type), which decreased to 1.02 × 1017 cm−3 and 1.24 × 1017 cm−3 for TAPB-incorporated, 9.44 × 1016 cm−3 and 1.19 × 1017 cm−3 for TAPOB-incorporated, 8.36 × 1016 cm−3 and 1.15 × 1017 cm−3 for TPTA-incorporated devices. These results confirm that TPTA significantly reduces trap densities, thereby suppressing iodide ion migration and nonradiative recombination.
The built-in potential (Vbi) in PSCs was extracted via Mott-Schottky (MS) analysis (Supplementary Fig. 25). The control device had the lowest Vbi of 0.46 V. In comparison, TAPB- and TAPOB-incorporated devices showed increased values of 0.54 V and 0.62 V, respectively, while the TPTA-incorporated device exhibited the highest Vbi of 0.69 V, suggesting enhanced internal electric field strength and improved interface energy alignment, consistent with TPV and TPC results.
Based on the improved film quality achieved through additive incorporation, single-junction Sn-Pb PSCs were fabricated using an inverted p-i-n architecture: ITO/PEDOT:PSS/NBG perovskite/C60/BCP/Ag. The optimal concentrations for TAPB, TAPOB, and TPTA were determined to be 1.0 mg/mL, 1.1 mg/mL, and 1.0 mg/mL, respectively (Supplementary Fig. 26). The photovoltaic parameters (VOC, JSC, FF, and PCE) are illustrated in Supplementary Table 5, and the corresponding J-V characteristics are shown in Fig. 3i. TPTA-treated devices exhibited significant performance improvements. The champion device achieved a VOC of 0.88 V (vs. 0.83 V for control), attributed to suppressed nonradiative recombination. The JSC increased from 32.2 to 32.6 mA/cm2 due to enhanced charge extraction. EQE spectra confirmed the JSC value (Fig. 3j). Together, these enhancements led to a champion PCE of 23.4%, compared to 20.8% for the control. Furthermore, the average PCE across 50 devices improved from 20.2% (control) to 22.7% (TPTA-incorporated) (Fig. 3h). In a steady power output (SPO) test over 200 s, the devices fabricated by spin-coating a precursor containing TPTA maintained a stable PCE of 23.2% (Supplementary Fig. 27). Under continuous 1-sun MPP tracking, the TPTA device retained 94.9% of its initial efficiency after 950 h, whereas the control dropped to 61.3% after 290 h (Fig. 3k). By reinforcing the Sn-I bond, suppressing thermal vibrations, and relieving local stress through reversible deformation, the P = S and -NH2 functional groups form a multipoint “molecular spring” that increases I- migration barriers and blocks multiple degradation pathways. These combined effects enable TPTA devices to achieve high performance and long-term operational stability. In shelf storage tests, encapsulated TPTA devices retained 90.0% of initial PCE after 3000 h, while control devices declined to 65.1% after 1600 h (Fig. 3l).
Performance and stability of 2 T PTSCs
The optimized NBG perovskite was further employed as the bottom sub-cell in perovskite tandem solar cells (PTSCs). The monolithic 2-terminal (2 T) device utilized a 1.78 eV WBG perovskite top sub-cell and a structure of ITO/NiOx/Me-4PACz/WBG PSC/C60/SnO2/Au/PEDOT:PSS/NBG perovskite/C60/BCP/Ag (Supplementary Fig. 28). Cross-sectional SEM images confirmed successful integration of the two sub-cell layers (Fig. 4a). The J-V curve of the WBG PSC (Supplementary Fig. 29) yielded a PCE of 19.9%. Detailed device parameters are provided in Supplementary Table 6. The champion PTSC achieved a PCE of 29.6%, with VOC = 2.17 V, JSC = 16.4 mA/cm2, and FF = 83.2% (Fig. 4b and Supplementary Table 7). Meanwhile, a reputable third-party organization gives certified PCE of 28.9% (Supplementary Fig. 30). A comparative summary of recent all-perovskite tandem devices is provided in Supplementary Table 8, demonstrating the competitiveness of our device in both efficiency and long-term stability. The corresponding tandem cell also exhibits a stabilized PCE of up to 29.5% (Supplementary Fig. 31). EQE spectra of both WBG and NBG sub-cells (Supplementary Fig. 32) matched the integrated JSC values, confirming the accuracy of the tandem design. Device reproducibility was verified by fabricating 50 PTSCs (Fig. 4c), all showing consistent performance. The TPTA additive also significantly enhanced tandem device stability. Under MPP tracking, encapsulated TPTA-based PTSCs retained 93.4% of their initial PCE after 900 h of continuous operation, while control devices declined to 66.6% after 230 h (Fig. 4d). Long-term storage in a nitrogen-filled glovebox showed that the PCE of the control device dropped to 65.0% after 1200 h, whereas the TPTA device retained 90.1% of its original PCE after 2900 h (Fig. 4e). These results demonstrate the critical role of TPTA in improving lattice stability and mitigating performance losses, enabling efficient and durable perovskite tandem solar cells.

a Cross-section SEM image of PTSCs with TPTA modification. b J-V curves of the Champion PTSCs. c Histogram of PCEs for 50 PTSCs. d MPP tracking of control device and device with TPTA under 1 sun illumination in the N2 environment. e Long-term stability test of control device and device with TPTA PTSCs stored in a N2-filled glovebox.
Discussion
This study proposes an effective molecular design strategy to address key challenges in Sn-Pb NBG perovskites, including lattice instability, strain heterogeneity, and halide migration. Among the additives, TPTA with a high dipole moment and multi-site anchoring effect demonstrated the highest efficacy in strengthening Sn-I bonds, suppressing lattice vibrations, alleviating local stress, and stabilizing the Sn-Pb perovskite lattice. By decoupling the photothermal-mechanical feedback loop and interrupting the multi-scale degradation cascade, TPTA promotes a uniform and robust lattice structure that remains stable under operating conditions. As a result, optimized NBG PSCs achieved a PCE of 23.4% and retained 94.9% efficiency after 950 hours of continuous operation. When integrated into a 2 T PTSC configuration, a PCE of 29.6% (certified PCE of 28.9%) was achieved, with 93.4% retained after 900 hours. This work provides a compelling solution for breaking the self-amplifying photothermal-mechanical degradation cycle and represents a promising approach to achieving high structural and operational stability in Sn-Pb perovskite-based devices, paving the way for commercial deployment of next-generation photovoltaic technologies.
Methods
Materials
All materials were utilized as received without any further purification. FAI, 99.99%, methylammonium chloride (MACl 99.99%), PbI2 (99.9%), lead chloride (PbCl2 99.9%), SnI2 (99.9%), Nickel oxide (NiOx) and patterned ITO substrates were purchased from Advanced Election Technology CO, Ltd. Methylammonium iodide (MAI), cesium iodide (CsI), Pb bromide (PbBr2), 1,3-propane-diammonium iodide (PDAI2) and ethanediamine dihydroiodide (EDAI2) were supplied from Xi’an Polymer Light Technology Corporation. Isopropanol (IPA, 99.9%), chlorobenzene (CB, 99.9%), diethyl ether (DE), N, N-dimethyl formamide (DMF, 99.8%), and dimethyl sulfoxide (DMSO, 99.7%) were obtained from Beijing J&K Scientific Ltd. TAPB (97%), TAPOB (97%) and TPTA (97%) were procured from Aladdin. Sn (II) fluorine (SnF2,99%) and ammonium thiocyanate (NH4SCN, 99.9%) and Pb (II) thiocyanate (Pb (SCN)2, 99.9%) were purchased from Sigma-Aldrich. PEDOT: PSS (CLEVIOS P VP AI 4083) was purchased from Heraeus.
NBG FA0.6MA0.3Cs0.1Pb0.5Sn0.5I3 perovskite solution Preparation
To prepare the 2.0 M perovskite precursor solution, dissolve the following amounts of each compound in 1 mL of a mixed solvent of DMF and DMSO (3:1, v/v): 459.54 mg of PbI₂, 372.79 mg of SnI₂, 206.46 mg of FAI, 95.27 mg of MAI, 51.91 mg of CsI, 15.65 mg of SnF₂, and 2.86 mg of NH₄SCN. Stir the mixture thoroughly until all components are completely dissolved. Finally, filter the solution through a 0.22 μm polytetrafluoroethylene (PTFE) membrane to remove any undissolved particles before using it to prepare perovskite films.
WBG FA0.8Cs0.2PbI1.8Br1.2 perovskite solution Preparation
Dissolve 212.06 mg of PbI₂, 271.58 mg of PbBr₂, 165.1 mg of FAI, 6.67 mg of PbCl2, 62.4 mg of CsI, 1.62 mg of MACl, and 4.8 mg of Pb(SCN)₂ in a 1 mL mixed solvent of DMF and DMSO, with a volume ratio of 4:1 (DMF:DMSO).
NBG Sn-Pb PSCs fabrication
The patterned ITO substrates were first ultrasonically cleaned sequentially with detergent, deionized water, acetone, and isopropanol, with each step lasting 30 min. The substrates were then dried using N₂ gas and treated with plasma for 5 min. Subsequently, the cleaned ITO substrates were spin-coated with a PEDOT: PSS solution at 4000 rpm for 30 s, followed by annealing at 150 °C for 15 min in air. The films were then transferred to an N₂-filled glovebox for further processing. The NBG precursor solution was spin-coated onto the substrates using a two-step process: an initial spin at 1000 rpm for 10 s, followed by a second spin at 4000 rpm for 30 s. During the second spin, CB as the antisolvent was dripped onto the film at 30 s. The coated films were then annealed at 100 °C for 10 min inside the glovebox. Subsequently, the EDAI2 layer precursor solution (1.0 mg mL⁻¹ in IPA) was spin-coated at 4000 rpm for 20 s, followed by annealing at 100 °C for 1 min. Finally, 25 nm of C₆₀, 6 nm of BCP, and 100 nm of Ag were deposited sequentially via thermal evaporation.
All-perovskite tandem solar cell fabrication
NiOx nanocrystal layers (10 mg mL⁻¹ in H₂O) were spin-coated onto ITO substrates at 1500 rpm for 30 seconds and subsequently annealed at 150 °C for 30 min in air. After cooling, the substrates were promptly transferred to an N₂-filled glovebox. Next, self-assembled monolayers of Me-4PACz (0.5 mg mL⁻¹ in ethanol) were spin-coated onto the ITO substrates at 3000 rpm for 30 s, followed by heating at 100 °C for 10 min. The WBG perovskite films were then deposited using a two-step spin-coating procedure: an initial spin at 5000 rpm for 2 s and a second spin at 4000 rpm for 60 s. During the second step, diethyl ether (DE) was dropped onto the spinning substrate at 25 s. The substrates were then transferred onto a hotplate and annealed at 100 °C for 10 min. After the substrates were cooled, post-treatments with PDAI2 were conducted via spin-coating a solution of 2 mg/ml PDAI2 in IPA at 4000 rpm for 30 s, followed by annealing at 100 °C for 5 min. After cooling to room temperature, the substrates were transferred to an evaporation system for the deposition of a 25 nm C₆₀ layer. ALD SnO ₂ layers with a thickness of 20 nm were then deposited onto the WBG perovskite films, followed by the deposition of a 1.0 nm Au layer via thermal evaporation. PEDOT: PSS layers were subsequently spin-coated on top of the front cells at 4000 rpm for 30 s and annealed at 120 °C for 20 min in air. The films were then transferred to an N₂-filled glovebox for further processing. The NBG precursor solution was spin-coated onto the substrates using a two-step process: an initial spin at 1000 rpm for 10 s, followed by a second spin at 4000 rpm for 30 s. During the second spin, CB as the antisolvent was dripped onto the film at 30 seconds. The coated films were then annealed at 100 °C for 10 min inside the glovebox. Subsequently, the EDAI2 layer precursor solution (1.0 mg mL⁻¹ in IPA) was s pin-coated at 4000 rpm for 20 s, followed by annealing at 100 °C for 1 min. Finally, 25 nm of C₆₀, 6 nm of BCP, and 100 nm of Ag were deposited sequentially via thermal evaporation.
DFT calculations
The DFT calculations in this research are performed using the Vienna ab initio simulation software (VASP). To simulate electron exchange-related interactions, the Perdew–Burke–Ernzerhof (PBE)63 functional is utilized, and the projector augmented wave (PAW)64 approach is used for electron–ion–nucleus interactions. In order to handle van der Waals interactions in perovskites, we employ the Grimme DFT-D3 approach with Becke-Johnson damping65. Geometry optimization is carried out with the Γ-centered 2 × 2 × 1 Monkhorst−Pack k-point mesh and the 400 eV plane wave energy cutoff. The geometric structure is regarded as convergent when the energy difference between all ions is smaller than − 10−5eV. The models employed in all calculations were optimized structurally using VASP. The perovskite surface model was selected for the calculations of passivation molecule adsorption and defect formation energy. We established a defect model based on the optimized perovskite surface model and adsorbed TPTA on its surface to simulate the effect of adsorbed organic ions on surface defects.
The defect formation energy is given as,
where \(E({X}^{q})\) is the total energy of the defect system with charge q, E(bulk) is the total energy of the pristine system, and n and μ are the number and the chemical potentials of the species added to or subtracted from the perfect system in order to form the defect. The last term is the energy associated with the exchange of charges with the electrons’ reservoir (the Fermi level of the system E_F), referenced to the valence band maximum (E_VBM) of the pristine system, followed by a correction for the electrostatic potential ∆V induced by the defect66.
Device characterization
The performance characterization of the devices was conducted using a Newport Oriel Sol3A 450 W solar simulator under AM 1.5 G illumination. This setup was used to measure J–V curves and SPO, as well as to characterize conductivity and perform SCLC analysis under dark conditions. The tested solar cell area was 0.116 cm², with an aperture shading mask applied to define an effective area of 0.08 cm². The EQE spectra of the devices were measured using a solar cell quantum efficiency test system (Elli Technology, Taiwan). EQE measurements were performed by applying external voltage/current sources to the PSCs using a REPS measurement instrument (Enlitech). The MS analyses were conducted with a Chenhua CHI760E electrochemical workstation. The EIS measurements were conducted under AM 1.5 G light illumination, using a CHI604E electrochemical workstation. The frequency range was set from 1 MHz to 38.3 Hz (logarithmically distributed), with an AC perturbation amplitude of 10 mV. Operational stability tests for the NBG and tandem solar cells were carried out under MPP conditions in an N₂ environment, RH30-40% and room temperature, illuminated by an AM 1.5 xenon lamp (100 mW /cm², without UV filter). TOF-SIMS analysis was performed using a dual-beam technique. Primary ion bombardment was conducted with Bismuth (Bi3+) ions at an energy of 30 keV and a current of 45 nA, while secondary ion detection utilized Cesium (Cs+) ions at an energy of 1 keV and a current of 80 nA. The secondary ion beam was aligned at 45° to the primary ion path. In addition, a flood gun was employed to neutralize surface charges, ensuring high mass resolution and accurate ion yield.
Film characterization
The FTIR spectroscopy was detected by FTIR-8400S (Shimadzu). XPS was measured by Kratos axial super dald. For the calibration of XPS data, the instrument was calibrated using the binding energy positions of Au 4f7/2, Ag 3d5/2, and Cu 2p3/2 spectral peaks, respectively. The calibration error of the instrument is G0.10 eV, and the binding energy error during the test is within 0.1 eV. SEM images were obtained by scanning electron microscope (verios G4 UC) (Rimono Scientific Company, USA) at 2 kV. The IDPC-TEM images were captured using Titan-G2 (60–300) and JEOL ARM-300 spherical aberration-corrected transmission electron microscopes. The steady PL spectra were recorded on the Horiba Jobin Yvon flfluorolog-3 spectrofluorometer system, and PL mapping was measured by HORIBA HR Evolution. TRPL was analyzed by the FLS 980 fluorescence spectrometer equipped with a 532 nm excitation. The KPFM images were captured by Dimension ICON SPM (Dimension Icon, German). Chi 660e electrochemical measurement workstation (Chengdu equipment company, Shanghai, China) was used for SCLC analysis under dark conditions. GIXRD were characterized using a Rigaku SmartLab five-axis X-ray diffractometer at 45 kV and 200 mA, equipped with Cu Ka radiation (l = 1.54050 A ̊), parallel beam optics and a secondary graphite monochromator. GIWAXS measurements were measured by the XEUSS SAXS/WAXS equipment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data generated in this study are provided in the Supplementary Information/Source Data file. Source data are provided in this paper.
References
Queisser, H. J. Detailed balance limit for solar cell efficiency. Mater. Sci. Eng.: B 159–160, 322–328 (2009).
Pan, Y. et al. Surface chemical polishing and passivation minimize non-radiative recombination for all-perovskite tandem solar cells. Nat. Commun. 15, 7335 (2024).
Lim, J., Park, N.-G., Seok, S. I. & Saliba, M. All-perovskite tandem solar cells: from fundamentals to technological progress. Energy Environ. Sci. 17, 4390–4425 (2024).
Zhang, Y. et al. Synchronized crystallization in tin-lead perovskite solar cells. Nat. Commun. 15, 6887 (2024).
Li, C. et al. Diamine chelates for increased stability in mixed Sn–Pb and all-perovskite tandem solar cells. Nat. Energy 9, 1388–1396 (2024).
Zhou, J. et al. Mixed tin-lead perovskites with balanced crystallization and oxidation barrier for all-perovskite tandem solar cells. Nat. Commun. 15, 2324 (2024).
Tong, J. et al. Carrier control in Sn–Pb perovskites via 2D cation engineering for all-perovskite tandem solar cells with improved efficiency and stability. Nat. Energy 7, 642–651 (2022).
Hu, S. et al. Optimized carrier extraction at interfaces for 23.6% efficient tin–lead perovskite solar cells. Energy Environ. Sci. 15, 2096–2107 (2022).
Duan, C. et al. Scalable fabrication of wide-bandgap perovskites using green solvents for tandem solar cells. Nat. Energy 10, 318–328 (2025).
Zhu, H. et al. Low-dimensional Sn-based perovskites: Evolution and future prospects of solar cells. Chem 8, 2939–2960 (2022).
Kamaraki, C. et al. Charting the irreversible degradation modes of low bandgap Pb-Sn perovskite compositions for de-risking practical industrial development. Adv. Energy Mater. 14, 2302916 (2024).
Tsai, H. et al. Light-induced lattice expansion leads to high-efficiency perovskite solar cells. Science 360, 67–70 (2018).
Ge, Y. et al. Suppressing wide-angle light loss and non-radiative recombination for efficient perovskite solar cells. Nat. Photonics 19, 170–177 (2025).
Bai, Y. et al. Decoupling light- and oxygen-induced degradation mechanisms of Sn–Pb perovskites in all perovskite tandem solar cells. Energy Environ. Sci. 17, 8557–8569 (2024).
Lim, V. J.-Y. et al. Air-degradation mechanisms in mixed lead-tin halide perovskites forsolar cells. Adv. Energy Mater. 13, 2200847 (2023).
Yan, W. et al. Hot-carrier cooling regulation for nixed Sn-Pb perovskite solar cells. Adv. Mater. 36, 2312170 (2024).
Tan, S. et al. Sustainable thermal regulation improves stability and efficiency in all-perovskite tandem solar cells. Nat. Commun. 15, 4136 (2024).
Zeng, P. et al. Control of hot carrier relaxation in CsPbBr3 nanocrystals using damping ligands. Angew. Chem. 134, e202111443 (2022).
Shen, Y. et al. Strain regulation retards natural operation decay of perovskite solar cells. Nature 635, 882–889 (2024).
Zhang, L. et al. Manipulating local lattice distortion for spectrally stable and efficient mixed-halide blue perovskite LEDs. Angew. Chem. Int. Ed. 62, e202302184 (2023).
Dey, K. et al. Substitution of lead with tin suppresses ionic transport in halide perovskite optoelectronics. Energy Environ. Sci. 17, 760–769 (2024).
Lanzetta, L. et al. Degradation mechanism of hybrid tin-based perovskite solar cells and the critical role of tin (IV) iodide. Nat. Commun. 12, 2853 (2021).
Ricciarelli, D., Meggiolaro, D., Ambrosio, F. & De Angelis, F. Instability of tin iodide perovskites: bulk p-doping versus surface tin oxidation. ACS Energy Lett. 5, 2787–2795 (2020).
Sun, Q. et al. Surface charge transfer doping of narrow-bandgap Sn–Pb perovskites for high-performance tandem solar cells. Energy Environ. Sci. 17, 2512–2520 (2024).
Zhang, Z. et al. Revealing superoxide-induced degradation in lead-free tin perovskite solar cells. Energy Environ. Sci. 15, 5274–5283 (2022).
Tong, J. et al. Carrier lifetimes of >1 μs in Sn-Pb perovskites enable efficient all-perovskite tandem solar cells. Science 364, 475–479 (2019).
Rahimnejad, S., Kovalenko, A., Forés, S. M., Aranda, C. & Guerrero, A. Coordination chemistry dictates the structural defects in lead halide perovskites. ChemPhysChem 17, 2795–2798 (2016).
Yuan, H. et al. Component distribution regulation in Sn-Pb perovskite solar cells through selective molecular interaction. Adv. Mater. 35, 2303674 (2023).
Tai, Q. et al. Antioxidant grain passivation for air-stable tin-based perovskite solar cells. Angew. Chem. Int. Ed. 58, 806–810 (2019).
Liu, H. et al. Modulated crystallization and reduced VOC deficit of mixed lead–tin perovskite solar cells with antioxidant caffeic acid. ACS Energy Lett. 6, 2907–2916 (2021).
Yang, X. et al. Understanding and manipulating the crystallization of Sn–Pb perovskites for efficient all-perovskite tandem solar cells. Nat. Photon. 19, 426–433 (2025).
Yeom, K.-W., Lee, D.-K. & Park, N.-G. Hard and soft acid and base (HSAB) engineering for efficient and stable Sn-Pb perovskite solar cells. Adv. Energy Mater. 12, 2202496 (2022).
Yu, D. et al. Electron-withdrawing organic ligand for high-efficiency all-perovskite tandem solar cells. Nat. Energy 9, 298–307 (2024).
Chen, X. et al. Supramolecular Aza crown ether modulator for efficient and stable perovskite solar cells. Adv. Funct. Mater. 34, 2311527 (2024).
Hu, M. et al. Surface Sn(IV) hydrolysis improves inorganic Sn–Pb perovskite solar cells. ACS Energy Lett. 8, 1035–1041 (2023).
Su, H. et al. Modulation on electrostatic potential of passivator for highly efficient and stable perovskite solar cells. Adv. Funct. Mater. 33, 2213123 (2023).
Guo, X. et al. Synergetic surface charge transfer doping and passivation toward high efficient and stable perovskite solar cells. iScience 24, 102276 (2021).
Zhao, W. et al. A special additive enables all cations and anions passivation for stable perovskite solar cells with efficiency over 23%. Nano Micro Lett. 13, 169 (2021).
Liu, B. et al. Imidazole derivative assisted crystallization for high-efficiency mixed Sn–Pb perovskite solar cells. Adv. Funct. Mater. 34, 2310828 (2024).
Hao, F., Stoumpos, C. C., Cao, D. H., Chang, R. P. H. & Kanatzidis, M. G. Lead-free solid-state organic–inorganic halide perovskite solar cells. Nat. Photon 8, 489–494 (2014).
Hong, H. et al. Two-dimensional lead halide perovskite lateral homojunctions enabled by phase pinning. Nat. Commun. 15, 3164 (2024).
Zhang, Y. et al. Efficient tin perovskite solar cells via suppressing autoxidation in inert atmosphere. Small 20, 2306115 (2024).
Heitz, R., Mukhametzhanov, I., Stier, O., Madhukar, A. & Bimberg, D. Enhanced polar exciton-LO-phonon interaction in quantum dots. Phys. Rev. Lett. 83, 4654–4657 (1999).
Wei, Q. et al. Effect of zinc-doping on the reduction of the hot-carrier cooling rate in halide perovskites. Angew. Chem. Int. Ed. 60, 10957–10963 (2021).
Tian, R. et al. Nucleation regulation and mesoscopic dielectric screening in α-FAPbI3. Adv. Mater. 36, 2309998 (2024).
Luo, J. et al. Improved carrier management via a multifunctional modifier for high-quality low-bandgap Sn–Pb perovskites and efficient all-perovskite tandem solar cells. Adv. Mater. 35, 2300352 (2023).
Shi, P. et al. Strain regulates the photovoltaic performance of thick-film perovskites. Nat. Commun. 15, 2579 (2024).
Chen, C.-H. et al. Full-dimensional grain boundary stress release for flexible perovskite indoor photovoltaics. Adv. Mater. 34, 2200320 (2022).
Hutter, E. M. et al. Thermodynamic stabilization of mixed-halide perovskites against phase segregation. Cell Rep. Phys. Sci. 1, 100120 (2020).
Islam, J. & Hossain, A. K. M. A. Semiconducting to metallic transition with outstanding optoelectronic properties of CsSnCl3 perovskite under pressure. Sci. Rep. 10, 14391 (2020).
Shen, B. et al. A single-molecule van der Waals compass. Nature 592, 541–544 (2021).
Shen, B. et al. Atomic imaging of zeolite-confined single molecules by electron microscopy. Nature 607, 703–707 (2022).
Liu, S. et al. Triple-junction solar cells with cyanate in ultrawide-bandgap perovskites. Nature 628, 306–312 (2024).
Song, Z. et al. Universal approach for managing iodine migration in inverted single-junction and tandem perovskite solar cells. Adv. Mater. 37, 2410779 (2025).
Yang, M. et al. Sn-Pb perovskite with strong light and oxygen stability for all-perovskite tandem solar cells. Adv. Mater. 37, 2415627 (2025).
Wang, J. et al. Enhancing photostability of Sn-Pb perovskite solar cells by an alkylammonium pseudo-halogen additive. Adv. Energy Mater. 13, 2204115 (2023).
Zhang, Y. et al. Improved fatigue behaviour of perovskite solar cells with an interfacial starch–polyiodide buffer layer. Nat. Photonics 17, 1066–1073 (2023).
Lu, X. et al. Dynamic reversible oxidation-reduction of iodide ions for operationally stable perovskite solar cells under ISOS-L-3 protocol. Adv. Mater. 36, 2400852 (2024).
Zhou, S. et al. Aspartate all-in-one doping strategy enables efficient all-perovskite tandems. Nature 624, 69–73 (2023).
Liu, F. et al. Is excess PbI2 beneficial for perovskite solar cell performance?. Adv. Energy Mater. 6, 1502206 (2016).
Cao, J. et al. High-performance tin–lead mixed-perovskite solar cells with vertical compositional gradient. Adv. Mater. 34, 2107729 (2022).
Li, M. et al. Acceleration of radiative recombination for efficient perovskite LEDs. Nature 630, 631–635 (2024).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Xie, H. et al. Decoupling the effects of defects on efficiency and stability through phosphonates in stable halide perovskite solar cells. Joule 5, 1246–1266 (2021).
Acknowledgements
This work was financially supported by the National Key Research and Development Program (2024YFF1401100), the National Science Fund for Distinguished Young Scholars (21925506), the National Natural Science Foundation of China (22439004, U21A20331, 81903743, 22279151, and 22275004), and Zhejiang Province “Leading Goose” Plan (2024C01091). Z.G. acknowledges the funding support from the National Science Fund for Distinguished Young Scholars (21925506), the National Key Research and Development Program (2024YFF1401100), the National Natural Science Foundation of China (22439004, U21A20331, 81903743, 22279151, and 22275004); C.L. acknowledges the funding support from the National Natural Science Foundation of China (22279151), and Zhejiang Province “Leading Goose” Plan (2024C01091).
Author information
Authors and Affiliations
Contributions
H.P., Y.B. and K.S. contributed equally to this work. C.L. conceived the idea and guided the work. H.P. and Y.B. fabricated the NBG subcell for device performance. H.P. fabricated the WBG subcell for device performance. R.T. and M.Y. carried out crystal analysis. Y.W. helped with the XPS measurement and data analysis. J.W. and Y.M. carried out GIWAXS measurement and data analysis. J.G. calculated the electrostatic potential. K.S. calculated DFT simulation calculations. X.L. and S.Z. characterized the KPFM test. Z.S. carried out conductivity and associated data analysis. H.P., C.L. and Z.G. wrote and revised the paper. All the authors contributed to the discussion of the results and the final paper preparation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Hong Zhang, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pan, H., Bai, Y., Sun, K. et al. Decoupling photothermal-mechanical degradation through lattice-stabilizing networks in Sn–Pb perovskites and all-perovskite tandem solar cells. Nat Commun 16, 9221 (2025). https://doi.org/10.1038/s41467-025-64274-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-64274-5
