
Abstract
Plants have evolved an extensive repertoire of specialized metabolites to adapt to complex environmental changes. Here, we identify two paralogous dirigent proteins (DPs) in cotton that serve as gatekeepers of extracellular terpenoid phytoalexin production in green organs, directing the transition of hemigossypol away from gossypol synthesis toward a hydroxylation pathway that leads to the biosynthesis of highly toxic hemigossypolone and heliocides. Under oxidative conditions, these proteins function synergistically with aldo-keto reductases to catalyze the hydroxylation of hemigossypol, followed by spontaneous oxidation that yields hemigossypolone, revealing a noncanonical role for aldo-keto reductases in extracellular terpenoid metabolism. Notably, mutants lacking these dirigent proteins produce gossypol but are devoid of hemigossypolone and heliocides in green organs exhibit heightened susceptibility to multiple biotic stresses, underscoring the enhanced protective role of these metabolites. This study describes a DPs-mediated mechanism of extracellular hydroxylation and highlights the potential ecological advantages of redirecting specialized metabolism extracellularly for enhanced defense against varying types of pathogens and herbivores.
Similar content being viewed by others

Introduction
Plants employ a multifaceted defense system against pests and pathogens, characterized by an integrated network of physical barriers, pattern-recognition receptors, defensive proteins, and bioactive specialized metabolites1,2,3. These metabolic pathways are often unique to specific species, exhibiting organ- and cell-specificity and distinct regulatory profiles. This specialization equips plants to effectively respond to fluctuating environmental conditions and various biotic stressors caused by pathogens and herbivores1,4,5. Of these metabolites, terpenoids represent the specialized metabolites with the highest structural diversity in plants6.
A fundamental aspect of plant defense is metabolic redirection, wherein different tissues and organs synthesize related yet distinct metabolites to counter specific antagonists. In some Solanaceae species, for instance, leaves produce steroidal saponins while berries generate steroidal glycoalkaloids7. Despite sharing a common precursor, these metabolites are differentially synthesized and deployed to target distinct threats. This organ-specific metabolite production underscores the precision with which plants have evolved to optimize their defenses, tailoring responses to the ecological challenges posed by various pests and pathogens.
Within the genus Gossypium, a diverse array of terpenoid phytoalexins characterized by the δ-cadinene skeleton is synthesized, including the sesquiterpenoid hemigossypol (1), the sesquiterpenoid quinone hemigossypolone (3), the disesquiterpenoid gossypol (4), and the heterodimers of monoterpenes and 3 known as heliocides (5a ~ 5d) (Fig. 1a). These specialized metabolites play critical roles in plant defense, exhibiting significant bioactivity against various pathogens and herbivores8,9,10,11. Notably, the spatial distribution of these compounds within plant organs is highly specialized. 4 accumulates predominantly in non-green organs, such as the subepidermal layers of roots, where it offers robust defense against non-specialist pathogens like Verticillium dahliae12. Additionally, in the secretory glandular cells (SGCs) of seeds, 4 functions as a deterrent against herbivorous mammals by providing spermatogenesis-inhibiting effects8,13,14,15. In contrast, 3 and heterocyclic 5a ~ 5d are primarily localized in the SGCs of chlorophyll-rich organs, including leaves and stems (Fig. 1b)11,16 This organ-specific accumulation pattern is reminiscent of other classes of specialized metabolites, such as flavonoids, steroids, and alkaloids, which similarly exhibit distinct localization and activity profiles across different plant organs7,17,18,19. Therefore, such a spatial distribution raises the possibility of a regulatory framework orchestrating localized defense responses. While previous studies have elucidated the accumulation of 3 and 5a ~ 5d in the extracellular space20, the reactions that enable these complex chemical transformations in the apoplastic environment remain unclear.

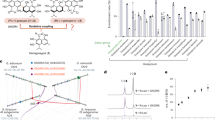
a Proposed biosynthetic pathways of 3, 4, and 5a ~ 5d in cotton. FPP from the MVA pathway is converted into 1 through CDN, P450s, dioxygenases, etc. 1 can either undergo homocoupling to form 4 or be further hydroxylated to produce 3. Previous studies suggest that 3 may originate from deoxyhemigossypol84,85,86,87. 3 and MEP-derived monoterpenes undergo a Diels-Alder reaction to yield 5a ~ 5d. Solid lines, well-characterized steps; Dashed lines, putative steps. b Heatmap of relative abundance of compounds 3 and 4 across various cotton organs. Ratios of 3 to 4 were calculated and log2-transformed. Color gradient, relative abundance (red: 3-dominant; yellow: 4-dominant). Detailed quantitative data in Supplementary Data 1. c Correlation network of 3 levels and the transcript abundance of glandular-specific genes across eight cotton organs. Only genes with P ≤ 0.05 (two-tailed) are included. Edge thickness, correlation magnitude; Node size, -log10(P_value) value; Blue node, compound 3; Red node, GhDP1_A1 and GhDP1_A2; Orange node, known terpenoid biosynthesis genes; Gray node, other significantly correlated but uncharacterized genes. Transcript data analyzed with edgeR; correlations and P values calculated using rcorr in R (Detailed in Supplementary Data 2). d Heatmap illustrating 3 accumulation patterns and organ-specific transcript abundance of GhDP1_A1, GhDP1_A2, and other 4 biosynthetic genes. The FPKM values were normalized by log2 transformation and then used to generate the heatmap. e Dot plots depicting the expression profiles of GhDP1_A1 and GhDP1_A2 across various cell types in cotton leaves. Dot color, scaled expression; Dot size, proportion of cells within each cluster that express the corresponding gene. f GUS staining analysis of wild-type, pGhDP1_A1::GUS and pGhDP1_A2::GUS cotton leaves. Scale bars, 50 μm. g Confocal microscopy images of wild-type and transgenic cotton leaves expressing pGhDP1_A1::GhDP1_A1-GFP and pGhDP1_A2::GhDP1_A2-GFP. Scale bar, 30 μm. For f–g the experiments were independently repeated three times with similar results. h Phylogenetic analysis of AtDIRs alongside functionally characterized dirigent proteins. Functionally characterized proteins are marked with yellow asterisks. Source data are provided as a Source Data file.
In cotton, the biosynthesis of 1 requires the coordinated activities of cytochrome P450 monooxygenases, specialized glyoxalases, dioxygenases, and dehydrogenases, which successively modify the δ-cadinene skeleton produced by terpene synthases from farnesyl pyrophosphate (FPP) produced via the mevalonate (MVA) pathway8,11,21,22 (Fig. 1a). Following its synthesis, 1 can be converted into 4 through the action of laccases and dirigent proteins (DPs) in the extracellular space, where the DPs precisely dictate the axial chirality of 48,23 (Fig. 1a). DPs, as a unique family of extracellular glycoproteins, are pivotal in the stereochemical regulation of biomolecular coupling reactions24. Although they lack intrinsic catalytic activity, these proteins exert significant influence over the stereochemical outcomes of enzymatic reactions, playing an essential role in the biosynthesis of compounds necessitating radical-radical coupling, such as lignans and lignin25,26,27,28; however, their broader functional roles remain underexplored.
Here, we report a distinct extracellular hydroxylation mechanism mediated by DPs and aldo-keto reductases (AKRs) that redirects terpenoid synthesis in SGCs of green organs of cotton. In the presence of oxidants, DPs interact with AKRs to mediate the hydroxylation of 1, yielding the intermediate 5-hydroxy-hemigossypol (2), which is prone to spontaneous oxidation to form 3. This mechanism redirects terpenoid metabolism away from 4 biosynthesis toward the production of 3 and heterocyclic 5a ~ 5d. Intriguingly, DP-knockout plants, which accumulate 4 exclusively while being devoid of 3 and 5a ~ 5d, show increased susceptibility to a range of biotic stresses, including both pathogens and insect predators, underscoring the potential selective advantage conferred by these other terpenoid phytoalexins. These findings broaden our understanding of the functional repertoire of DPs and provide valuable insight into the complexity of plant specialized metabolism even outside the cell.
Results
Identification of GhDP1_A1/A2 using coexpression analysis
The distinct accumulation patterns of 3 and 4 in cotton organs prompted us to investigate the genetic basis of 3 biosynthesis. Through correlation analysis of 3 content across eight organ types—including root, stem, leaf, petal, stamen, pistil, calycle, and ovule—we identified two subgenome-A biased tandemly arrayed DP genes, GhDP1_A1 (Gh_A01G2132) and GhDP1_A2 (Gh_A01G2133), both exhibiting strong positive correlations with 3 accumulation [correlation coefficient (r) ≥ 0.98, Fig. 1b–d and Supplementary Data 1–3]. Although their expression profiles differ markedly from those of genes encoding enzymes responsible for the monomer synthesis in the 4 biosynthetic pathway (Fig. 1d), they are absent in glandless cotton plants, which are devoid of 1, 3, 4, and 5a ~ 5d, consistent with other genes involved in 4 biosynthesis (Supplementary Fig. 1). These genes, GhDP1_A1 and GhDP1_A2, exhibit a remarkable sequence identity of approximately 93% at both the nucleotide and amino acid level (Supplementary Fig. 2a, b). Notably, their D-subgenome homologs, GhDP1_D1 (Gh_D09G0156) and GhDP1_D2 (Gh_D09G0157), exhibit negligible or undetectable expression across all organs in upland cotton (Supplementary Fig. 2c), suggesting potential loss of biological function.
Our previous single-cell RNA sequencing (scRNA-seq) results20, along with promoter-driven GUS and GFP analyses, revealed that the expression of GhDP1_A1 and GhDP1_A2 is confined to SGCs (Fig. 1e–g), which are responsible for the biosynthesis of 4-related specialized metabolites in cotton aboveground organs20. Computational predictions indicate that numerous DPs possess N-terminal signal peptides that are responsible for directing them to the apoplast29, the extracellular space within plant tissues. Both GhDP1_A1 and GhDP1_A2 proteins harbor these signal peptides (Supplementary Fig. 3), suggesting their targeting to the extracellular space, which is further corroborated by immunogold labeling of GhDP1 proteins and the subcellular localization of GFP-tagged GhDP1_A1 (Supplementary Fig. 4). Altogether, these findings substantiate the localization of GhDP1_A1 and GhDP1_A2 within the extracellular cavity of pigment glands, where 3, 4, and allied terpenoids accumulate20, suggesting their central role in the extracellular biosynthesis of terpenoids.
Phylogenetic analysis indicates that GhDP1_A1 and GhDP1_A2 belong to DIR-b/d subfamily (Fig. 1h)30,31,32, which is consistent with the previously characterized DPs (GhDIR5 and GhDIR6) involved in determining the axial chirality of 48. However, GhDP1_A1 and GhDP1_A2 share less than 50% amino acid similarity with the (+)- and (–)−4-forming DPs (Supplementary Fig. 5), suggesting that GhDP1 proteins have diverged functionally from 4 axial chirality-controlling DPs in cotton.
GhDP1_A1/A2 redirect terpenoid metabolism in cotton
To investigate the function of GhDP1_A1 and GhDP1_A2, we employed virus-induced gene silencing (VIGS) technology to attenuate their expression. Due to the high nucleotide homology between the GhDP1_A1 and GhDP1_A2 transcripts, the silencing construct was unable to selectively target either transcript, resulting in the downregulation of both GhDP1_A1 and GhDP1_A2 (Supplementary Fig. 6a, b). This gene silencing led to a marked reduction of more than 50% in the levels of 3 and 5a ~ 5d, alongside a 5- to 22-fold increase in 4 accumulation in green organs (Supplementary Fig. 6c, d).
To further determine the involvement of GhDP1_A1 and GhDP1_A2 in the biosynthesis of 3 and 5a ~ 5d, we employed CRISPR-Cas9 to simultaneously delete both genes in Gossypium hirsutum. Due to the high sequence similarity between GhDP1_A1 and GhDP1_A2, two single-guide RNAs (sgRNAs) were designed to target sequences within the single exon of both genes (Fig. 2a). CRISPR-Cas9 allowed for the efficient knockout of both genes in a single editing event (Fig. 2a and Supplementary Fig. 7a). Editing of both GhDP1_A1 and GhDP1_A2 did not result in any observable effects on plant growth and development, as assessed by visual inspection (Supplementary Fig. 7b). However, analysis of metabolite profiles by liquid chromatography-mass spectrometry (LC-MS) revealed a complete loss of 3 and 5a ~ 5d, the major specialized metabolites in wild-type (WT) leaves, stems, pistils, and calyces, in the edited lines (Fig. 2b). In contrast, these mutant lines exhibited a significant increase in the accumulation of 4, with levels rising 4- to 17-fold across various organs (Fig. 2b). Collectively, these findings establish GhDP1 proteins as essential for 3 and 5a ~ 5d biosynthesis, directing 1 from 4 towards 3 and 5a ~ 5d biosynthesis in green organs.

a Schematic diagram of the GhDP1_A1 (left) and GhDP1_A2 (right) gene structures. Pink rectangles denote the positions of single-guide RNA (sgRNA) target sites within the coding regions. The sequences of the sgRNA target sites are presented for both wild-type (WT) plants and the corresponding mutant alleles. b Statistical analysis of the content levels of 3, 4, and 5a ~ 5d in various organs of GhDP1-knockout plants (CR-Ghdp1 #1/2), in comparison to wild-type (WT) plants (mean ± s.d., n = 3 biological replicates). P values were determined by a two-tailed unpaired t-test. Source data are provided as a Source Data file.
GhDP1-mediated hydroxylation reprograms terpenoid biosynthesis by channeling hemigossypol to hemigossypolone
To explore the biochemical function of GhDP1 in the biosynthesis of 3, we employed the apoplastic fluid (APF) wash method, a widely used technique for isolating and analyzing secreted proteins from plant tissues, particularly in Nicotiana benthamiana33,34. APFs, the extracellular fluids collected from the apoplast, contain secreted proteins, including laccases and other oxidases, which catalyze oxidation reactions in vitro33,34. This method is particularly useful for functional analysis of secretory proteins without the need for further purification33,34.
We firstly used APFs from G. hirsutum leaves to investigate the biochemical role of GhDP1_A1 and GhDP1_A2. The APFs catalyzed the conversion of 1 to 4 in vitro (Fig. 3a and Supplementary Fig. 8). Upon the addition of recombinant GhDP1_A1 or GhDP1_A2, both expressed and purified from Sf9 insect cells (Supplementary Fig. 9a), the reaction mixture produced 3 and a previously unidentified compound with a molecular mass 16 Da higher than 1, tentatively identified as 5-hydroxy-hemigossypol (2), along with a decrease in 4 levels (Fig. 3a–c and Supplementary Fig. 8). These results demonstrate that, in the presence of APFs, both GhDP1_A1 and GhDP1_A2 mediate the hydroxylation of 1 to form 2, which is subsequently converted to 3. As a control, no product was detected when 1 was incubated with GhDP1_A1 or GhDP1_A2 alone in the buffer without APFs (Fig. 3a and Supplementary Fig. 8), confirming that GhDP1 proteins require the presence of factors in the APF for their activity. Time-course analysis employing APFs as a catalytic system revealed a transient accumulation of 2, and a steady increase in 3 levels with increasing concentrations of GhDP1 proteins (Fig. 3d and Supplementary Fig. 9b, c). These findings suggest that 2 serves as a key intermediate on the pathway to 3.

a LC-MS chromatograms demonstrating the formation of 2 and 3 through recombinant GhDP1_A1, GhDP1_A2 and apoplastic fluid. Gh_APF, apoplastic fluid derived from G. hirsutum leaves. Ultraviolet detection for HPLC was conducted at a wavelength of 257 nm. b MS/MS fragmentation spectrum of 2. c MS/MS fragmentation spectrum of 3. d Time-course analysis of the GhDP1_A1-mediated reaction using APF as a catalyst. Comparative chromatograms depict the temporal changes in substrate (1) and the resulting products, including 2, 3, and 4, within the reaction system. e LC-MS chromatograms illustrating the formation of 2 and 3 in apoplastic fluid from N. tabacum plants stably overexpressing GhDP1_A1 and GhDP1_A2. Nt_GhDP1_A1_APF and Nt_GhDP1_A2_APF represent apoplastic fluids from N. tabacum leaves overexpressing GhDP1_A1 and GhDP1_A2, respectively. Nt_EV_APF, apoplastic fluids from wide type N. tabacum leaves. Control groups (no protein or substrate 1) are shown in Supplementary Fig. 11a. The extracted ion chromatograms (EICs) show the following metabolites: m/z 261.1121 for 1, m/z 275.0914 for 3, m/z 277.1067 for 2, and m/z 519.2013 for 4 [M + H+] in positive mode. f EICs of the products obtained from the oxidation of 1 catalyzed by GhDP1_A1 and RvLac. The EICs show the following metabolites: m/z 261.1121 for 1, m/z 277.1067 for 2, m/z 275.0914 for 3 and m/z 519.2013 for 4 [M + H+] in positive mode. Control groups (no protein or substrate 1) are shown in Supplementary Fig. 12a. g Quantification of compound contents in RvLac-catalyzed reactions of 1, with and without purified GhDP1_A1. Substrate and product levels in the reaction without adding GhDP1_A1 was set to 1. Data represent the mean of three biological replicates (mean ± s.d., n = 3). P values were determined by a two-tailed unpaired t-test. Source data are provided as a Source Data file.
To further confirm these results, we extracted APFs from N. benthamiana leaves transiently expressing GhDP1_A1. These APFs catalyzed the conversion of 1 to 2 and then to 3, whereas no 3 was produced in the absence of GhDP1_A1 (Supplementary Fig. 10). Similarly, APFs from Nicotiana tabacum leaves stably expressing either GhDP1_A1 or GhDP1_A2 catalyzed the conversion of 1 to 3 via 2 efficiently, with a marked reduction in 4 production, while APF from control plants exclusively yielded 4 (Fig. 3e and Supplementary Fig. 11). These observations support the functional equivalence of the paralogous proteins GhDP1_A1 and GhDP1_A2. Together, these results indicate that GhDP1 proteins are essential in modulating terpenoid metabolism by blocking 4 production and enabling 3 biosynthesis.
GhDP1 directs laccase-catalyzed hydroxylation rather than homocoupling
Previous studies have demonstrated that the canonical substrates of DPs, phenolic radicals, are generated by laccases or alternative organic oxidants, which are subsequently stereoselectively dimerized into lignans or 4 with the aid of DPs25,35. To evaluate the potential catalytic activity of GhDP1_A1 in oxidant-mediated reaction, we assessed recombinant GhDP1_A1 in conjunction with 1 and a laccase from Rhus verniciflua (RvLac), an enzyme renowned for its multi-electron oxidation capabilities36. Consistent with previous findings, products of laccase alone lack selectivity8,37. In our experiments, laccase alone catalyzed the dimerization of 1 into 4 and the formation of 2 and 3, although their production was very low (Fig. 3f), similar to reports of laccase activity in lignin biosynthesis, where products are generated in the presence of oxidants (H2O2), and DPs substantially augmented specific product yield26. Likewise, our previous findings on 4 axial chirality-controlling DPs indicated that while laccase alone could convert 1 to 4 at minimal levels, the presence of chirality-controlling DPs increased yield significantly8. In accordance with these observations, the addition of recombinant GhDP1_A1 in laccase enzymatic assays resulted in approximately a threefold increase in the total 2 yield and a fourfold increase in 3 yield, while 4 was barely detectable (Fig. 3f, g and Supplementary Fig. 12). These findings highlighted that GhDP1 proteins mediate the chemical reaction in a specific direction, organizing otherwise chaotic products.
We further isolated and purified 2 and used it as a substrate to evaluate the spontaneity of its dehydrogenation process to 3. In a separate experiment, we discovered that 2 dissolved in methanol at low temperature (4 °C) resulted in its gradual conversion to 3, which is relatively more stable at an even lower temperature (−40 °C) (Supplementary Fig. 13a). At 30 °C, 2 quickly and spontaneously produces 3 in mildly acidic buffer (pH 5.8) (Supplementary Fig. 13b). These results showed that 2 can undergo spontaneous dehydrogenation to form 3 in a non-enzymatic manner. The inherent instability and rapid conversion of 2 have thus far precluded its unambiguous structural characterization by nuclear magnetic resonance (NMR), despite our attempts to isolate sufficient quantities for definitive analysis. Therefore, although the mass difference (+16 Da) is consistent with a hydroxylated derivative of 1, the precise position of hydroxylation in 1 remains to be conclusively determined.
Interaction of GhDP1 with aldo-keto reductase in hemigossypolone biosynthesis
Despite the extensive study of DPs, the specific enzyme partners with which they interact remain poorly characterized. To identify potential interacting partners of GhDP1_A1, we utilized anti-GFP magnetic beads to perform immunoprecipitation- mass spectrometry (IP-MS) analysis of 35S::GhDP1_A1-GFP transgenic and wild-type cotton plants. This approach identified approximately 2800 potential interacting proteins, which were specifically detected in the 35S::GhDP1_A1-GFP samples and absent in the wild-type cotton plant samples (Supplementary Data 4). To refine our candidate list, we cross-referenced the identified proteins with our previously published RNA-seq dataset, focusing on genes that are significantly downregulated in glandless cotton cultivars, which are characterized by disrupted biosynthesis of terpenoid specialized metabolites38. This intersection yielded a list of 66 candidate genes, among which a previously uncharacterized aldo-keto reductase, designated GhAKR13D2_A3 (Gh_A08G1908), exhibited the highest correlation coefficient with GhDP1_A1 expression (r = 0.95) (Fig. 4a, b and Supplementary Data 5). The interaction between GhDP1_A1 and GhAKR13D2_A3 was substantiated via a split luciferase complementation (SLC) assay (Fig. 4c). The emergence of yellow fluorescence upon co-expression of GhDP1_A1-cYFP and GhAKR13D2_A3-nYFP in tobacco leaves provided further corroboration of this interaction (Fig. 4d and Supplementary Fig. 14a). A His pull-down assay confirmed the physical binding of GhDP1_A1-His with GhAKR13D2_A3-FLAG and in a co-immunoprecipitation assay, GhAKR13D2_A3-YFP was co-immunoprecipitated with GhDP1_A1-FLAG (Fig. 4e, f). Altogether, these results provided the first evidence of a DP-AKR interaction.

a Venn diagram illustrating the intersection of genes identified through IP-MS targeting GhDP1_A1 and those down-regulated in differential gene expression analyses contrasting glandless cotton (GL) and glandular cotton (G) leaves, excluding GhDP1_A1 itself. b Correlation network showing Pearson’s correlation coefficients between GhDP1_A1 expression and 25 interacting genes, which are significantly down-regulated in GL compared with G leaves. Correlations were calculated across various cotton organs with P ≤ 0.05 using rcorr in R. c SLC assay in tobacco leaves demonstrates the GhDP1_A1-GhAKR13D2_A3 interaction, with a blue-red gradient depicting interaction strength. d BiFC assay confirm the interaction of GhDP1_A1 with GhAKR13D2_A3 in N. benthamiana leaves. Scale bar, 25 μm. e In vitro pull-down assays illustrate the interaction between GhDP1_A1-His and GhAKR13D2_A3-FLAG, using YFP-His as a negative control. f Co-IP assay of GhAKR13D2_A3 with GhDP1_A1 in N. benthamiana. YFP alone served as a negative control. The enriched YFP monomer observed in the GhAKR13D2_A3-YFP IP sample is likely due to partial dissociation or degradation of the fusion protein during extraction or IP procedures. For c–f the experiments were independently repeated twice with similar results. g Dot plot illustrating the expression of GhAKR13D2_A3 across various cell types in cotton leaves. h Correlation plot depicting the co-expression coefficients of gene expression profiles of five GhAKR13D2 genes with 3 and GhDP1_A1. Higher correlation coefficients are represented by darker blue hues. i The transcript expression profiles of GhAKR13D2_A3 among different cotton organs. RNA-seq datasets for various cotton organs were retrieved from the NCBI Sequence Read Archive (SRA) under project accession number PRJNA248163. j Comparative content analysis of terpenoid specialized metabolites in leaves from control plants (TRV:00) and GhAKR13D2-VIGS (TRV:GhAKR13D2) cotton plants. Box plots show the distribution of values with the centre line representing the median, the bounds of the box representing the 25th (Q1) and 75th (Q3) percentiles, and whiskers extending to the minimum and maximum values. Each point represents a biological replicate (n = 4 samples). P values were determined by a two-tailed unpaired t-test. Source data are provided as a Source Data file.
AKRs are ubiquitous across diverse species, participating in various redox reactions with broad substrate specificity and utilizing NAD(P)(H) as cofactors39. In G. hirsutum, the GhAKR13D2 gene family comprises five members, consisting of three tandemly duplicated genes in the A-subgenome (GhAKR13D2_A1 ~ A3) and two in the D-subgenome (GhAKR13D2_D1 ~ D2). ScRNA-seq analysis reveals that although the five AKR genes are not exclusively expressed in SGCs, their expression levels are markedly higher in SGCs compared to other cell types (Fig. 4g and Supplementary Fig. 14b). Notably, experiments utilizing the native promoter of GhAKR13D2 to drive the expression of a GhAKR13D2_A3–GFP fusion protein demonstrated that GhAKR13D2_A3 is actively expressed within cells (Supplementary Fig. 14c). Remarkably, GhAKR13D2_A3 exhibits the strongest correlation with both GhDP1_A1 expression and 3 levels across various organs (Fig. 4h and Supplementary Data 6). Organ-specific expression profiling indicates elevated expression of GhAKR13D2_A3 in photosynthetically active green organs such as leaves and stems, where 3 and 5a ~ 5d accumulate, while expression is minimal in non-photosynthetic organs including ovules and roots (Fig. 4i). Although the computational predictions did not identify any secretory signal peptides (Supplementary Fig. 15a), apoplastic protein extraction followed by mass spectrometry analysis showed the presence of GhAKR13D2_A3 in the extracellular space (Supplementary Fig. 15b and Supplementary Data 7). Furthermore, immunogold labeling revealed the presence of GhAKR13D2_A3 within glandular cavities (Supplementary Fig. 15c). To further elucidate its extracellular distribution, we co-expressed GhAKR13D2_A3 fused to mCherry with the plasma membrane marker AtPIP2A-YFP in N. benthamiana leaf epidermal cells, and stained the cell wall using Fluorescent Brightener 28 (Calcofluor White M2R, FB28). Confocal imaging revealed that GhAKR13D2_A3-mCherry partially co-localized with AtPIP2A-YFP at the plasma membrane, while also exhibiting overlap with the FB28 signal along the cell wall. This dual localization pattern indicates that GhAKR13D2_A3 can be targeted to both the plasma membrane and the apoplastic space (Supplementary Fig. 15d), implicating it in extracellular biochemical processes. Although AKRs have conventionally been categorized as intracellular enzymes40, our findings reveal their potential for extracellular localization, suggesting a promising cooperative role with DPs.
GhAKR13D2_A3 plays a critical role in the biosynthesis of hemigossypolone and heliocides
To investigate the role of GhAKR13D2_A3 in planta, we performed a VIGS assay targeting GhAKR13D2, quantitative real-time PCR (qPCR) analysis confirmed a significant downregulation of GhAKR13D2_A3 expression (Supplementary Fig. 16a). Given the potential for VIGS to simultaneously reduce the expression of multiple GhAKR13D2 genes due to their high sequence similarity (Supplementary Fig. 16b), we performed transcriptome sequencing on leaves from VIGS-silenced and control plants to distinguish between these genes accurately. This analysis revealed that the expression levels of all five GhAKR13D2 genes were down-regulated in VIGS-silenced plants (Supplementary Fig. 16c). The VIGS assay resulted in a significant reduction in the levels of 3 and 5a ~ 5d compared to control plants. Specifically, the level of 3 decreased by approximately eightfold, while the levels of 5a ~ 5d were reduced by 3-6 fold (Fig. 4j).
To further investigate the role of GhAKR13D2_A3, we generated CRISPR- Cas9-mediated knockout mutants using two sgRNAs specifically designed to target GhAKR13D2_A3, with minimal theoretical potential to affect the other four paralogous GhAKR13D2 genes (Supplementary Fig. 16d). Sequence analysis of T2 generation plants confirmed successful gene editing, resulting in two knockout lines: CR-Ghakr13d2_a3#1 and CR-Ghakr13d2_a3#2 (Supplementary Fig. 16d). In CR-Ghakr13d2_a3#1, both target sites induced frameshift mutations, whereas in CR-Ghakr13d2_a3#2, the first target site caused a 5-bp deletion, and the second site led to a 1-bp insertion (Supplementary Fig. 16d). Subsequent DNA sequencing validated the specificity of the gene editing events to GhAKR13D2_A3, with no detectable alterations in the sequences of homologous genes, GhAKR13D2_A1/A2/D1/D2 (Supplementary Fig. 16e), reinforcing the precision of our CRISPR-Cas9 methodology. Metabolic profiling of these CRISPR lines revealed results consistent with those observed in the VIGS assay, demonstrating significantly reduced levels of 3 and 5a ~ 5d compared to wild-type plants (Supplementary Fig. 16f and Supplementary Data 8). These findings demonstrate that GhAKR13D2_A3 and GhDP1 are functionally interdependent during the biosynthesis of 3 and 5a ~ 5d.
Enhanced hydroxylation efficiency via GhDP1-AKRs cooperation with oxidants
To further explore the structural basis underlying the interaction between GhDP1 and GhAKR13D2, we employed AlphaFold3 (AF3) to predict their three-dimensional structures41. Molecular docking analyses of the GhDP1_A1 and GhAKR13D2_A3 complex revealed that 1 preferentially binds within the active site of GhDP1_A1 (Fig. 5a), rather than the larger pocket of GhAKR13D2, which typically accommodates NAD(P)(H). Notably, the hydroxyl group at C3 of 1 forms two hydrogen bonds with the R145 residue of GhDP1, while the hydroxyl group at C8 establishes a hydrogen bond with Y102. C7, the site of 1 dimerization, is encapsulated within the protein complex, a configuration that may hinder the homocoupling of 1 required for 4 biosynthesis. Conversely, C5 is exposed (Fig. 5a), potentially facilitating access for laccases or AKRs to exert their functions.

a Structural model of the GhDP1–GhAKR13D2 heterodimeric complex associated with hemigossypol (1). The structure was predicted by AlphaFold3, with GhDP1_A1 shown in green and GhAKR13D2_A3 in purple. 1 (yellow) is modeled in the active site of GhDP1_A1, positioned in close proximity to two key residues, R145 and Y102. The lower panel highlights the predicted hydrogen bonding interactions between 1 and the catalytic residues, with distances (in Å) indicated in red. Gray numbers indicate the positions of carbon atoms in 1. b Characterization of dirigent activities in vitro using apoplastic fluids from N. benthamiana leaves transiently expressing GhDP1_A1, GhDP1_A1 mutants, and control (empty vector), with 1 as the substrate. Extracted ion chromatograms (EICs) at m/z 277.1067 and m/z 519.2013 indicate 3 and 4, respectively. c, d Functional characterization of GhAKR13D2_A3 and GhDP1_A1 in the presence of Cu2+ by in vitro assays. Chromatographic peaks corresponding to 2 (c) and 3 (d) were detected at EICs m/z 277.1067 and m/z 275.0914, respectively, in positive mode. e LC-MS detection of hydroxylation products of 1 by GhDP1_A1, GhAKR13D2_A3 and homologous proteins of GhAKR13D2_A3 in vitro, in the presence of Cu²⁺. The hydroxylation product 2 was detected by LC-MS with an EIC at m/z 277.1067. The homologous proteins of GhAKR13D2_A3 were derived from Escherichia coli (NP_414953.2), Saccharomyces cerevisiae (NP_014068.1), Chlamydomonas reinhardtii (Cre11.g467622.t1.1), Physcomitrella patens (Pp3c6_770V3.1), Selaginella moellendorffii (270369), A. thaliana (AT1G60690.1), Solanum lycopersicum (Solyc01g097390.2.1), Drosophila melanogaster (NP_996068.1), Danio rerio (XP_021329604.1), Mus musculus (XP_006501119.1), and Homo sapiens (NP_751892.1). The right panel shows quantitative comparisons of hydroxylation product (2) levels, with and without AKRs. Data are presented as mean ± s.d. (n = 3 independent experiments); significance was determined by unpaired two-tailed t-tests. The product level in the group without AKR protein was set to 1. f Phylogenetic analysis and amino acid sequence alignment of GhAKR13D2 proteins across different kingdoms including microorganisms, animals and plants. Conserved catalytic sites are marked by red asterisks, while black asterisks denote NADP(H) binding sites. Source data are provided as a Source Data file.
To confirm the functional significance of residues in proximity to 1, we generated site-directed mutants of GhDP1 and assessed their activity using APFs extracted from N. benthamiana leaves transiently expressing the corresponding constructs. Enzymatic assays revealed that the R145A and Y102A substitutions completely abrogated activity, as evidenced by the absence of product 3 formation (Fig. 5b). In silico stability predictions suggested that the overall protein fold of GhDP1_A1 was not significantly destabilized by these two site-directed mutations (ΔΔG < 5 kcal/mol for R145A and Y102A, Supplementary Data 9), supporting the notion that the abolished activity is due to the loss of critical catalytic interactions rather than global misfolding. Additionally, the S140A, V177A, and I167A mutants of GhDP1_A1 retained the capacity to produce the hydroxylated product 2, as well as the subsequent product 3 in similar assays (Supplementary Fig. 17). These findings highlight the essential roles of R145 and Y102 and demonstrate the effectiveness of structure-guided mutagenesis in elucidating the functional mechanisms of DPs.
We next incubated recombinant GhDP1_A1 with GhAKR13D2_A3 expressed in Escherichia coli and using 1 as the substrate. The addition of standard AKR cofactors, NAD(P)(H), did not yield detectable enzymatic activity (Supplementary Fig. 18). Given our previous findings that laccases, through their high oxidative potential, can generate 2 and 3 independently of DPs in vitro, we sought alternative oxidants. According to the previous studies on the role of DPs in lignin biosynthesis, we evaluated weaker oxidants, specifically oxidative Cu²⁺ (or Fe³⁺) ions, which have been shown to induce hydroxylation in laccase-DPs systems (Fig. 3f). Notably, neither GhDP1_A1 nor GhAKR13D2_A3 alone facilitated significant production of 2 or 3 in the presence of these oxidants (Fig. 5c, d and Supplementary Fig. 19). However, when both proteins were present, a substantial increase in 2 yield was observed, along with a notable accumulation of 3 (Fig. 5c, d and Supplementary Fig. 19). This synergistic effect suggests that the hydroxylation of 1 is controlled by GhAKR13D2 and GhDP1 working together under oxidative conditions.
The promiscuity exhibited by ancestral enzymes is thought to precede the emergence of evolved enzymatic activities42. The identification of GhAKR13D2_A3 as a partner for DPs prompted us to investigate its homologs across multiple species. Among these species, only species of cotton synthesize 3 or 4. Nevertheless, these AKRs can partner effectively with GhDP1_A1 to catalyze the hydroxylation of 1 to 2 in the presence of Cu²⁺ ions (Fig. 5e and Supplementary Fig. 20). These findings suggest that, during the evolution of the 3 pathway, AKRs have been selected that can interact with DPs, thereby enabling complex extracellular hydroxylation reactions.
Sequence alignment revealed that GhAKR13D2 members contain conserved residues essential for NADP(H) binding and substrate interaction (Fig. 5f)39,43,44. Biacore analysis further confirmed a high-affinity interaction between GhAKR13D2_A3 and NADP(H) (Supplementary Fig. 21). Enzymatic assays demonstrated GhAKR13D2_A3 has the typical AKR activity, such as the NADPH-dependent reduction of 2-nitrobenzaldehyde to 2-nitrobenzyl alcohol45 (Supplementary Fig. 22). However, during the 1 oxidation reaction mediated by GhAKR13D2_A3 and GhDP1_A1, the addition of NADPH inhibited the conversion of 1 to 2 and 3 (Supplementary Fig. 23), suggesting that the oxidation reaction may proceed through a mechanism that is incompatible with the reductive conditions introduced by NADPH.
Hemigossypolone biosynthetic DPs and AKRs among Gossypium species
The cotton genus exhibits remarkable genomic diversity from global radiation, with evidence indicating that polyploidization between Gossypium arboreum (A2 genome)-like and Gossypium raimondii (D5 genome)-like species has given rise to at least seven allotetraploid AD-genome species (2n = 4x = 52) [(AD)1 to (AD)7 genomes]46.
To trace the genomic inheritance related to specialized metabolism across species, we conducted a syntenic analysis utilizing the genomes of Durio zibethinus, Theobroma cacao, and selected Gossypium species from the order Malvales, with Arabidopsis thaliana serving as an outgroup reference. Our analysis revealed that orthologues of both GhDP1 and GhAKR13D2 are present in the genomes of Gossypium species but are conspicuously absent in D. zibethinus, T. cacao, and A. thaliana (Fig. 6). Notably, G. raimondii possesses orthologues of GhAKR13D2 but lacks orthologues of GhDP1, which may account for the absence of 3 in this species16. Collectively, the evolutionary trajectories of GhDP1 and GhAKR13D2 have endowed cotton plants with the ability to synthesize complex natural compounds, including 3 and heterocyclic 5a ~ 5d.

Syntenic analysis of genomic loci encoding GhDP1 (left panel) and GhAKR13D2 (right panel) across species within the Gossypieae tribe, durian, and cacao from the Malvales order, with A. thaliana serving as an outgroup. Syntenic relationships between species are indicated by gray ribbons, while loci corresponding to GhDP1 and GhAKR13D2 are highlighted in purple.
Extracellularly hydroxylation-derived hemigossypolone and heliocides display selective advantages against insects and pathogens
The GhDP1-knockout mutant plants, characterized by the exclusive accumulation of 4 and the absence of 3 and heterocyclic 5a ~ 5d (Fig. 2b), provide a unique system to investigate the role of terpenoid metabolic synthesis in biotic resistance. Given that the biosynthesis of plant natural products significantly enhances the Darwinian fitness of plants47, we sought to elucidate the impact of these metabolic changes on the biotic resistance of cotton.
Helicoverpa armigera and Spodoptera frugiperda are among the most destructive chewing herbivores in agricultural systems48,49. In our analysis, GhDP1-knockout mutants exhibited significantly reduced resistance to these pests. Larvae feeding on GhDP1-deficient plants gained 2.2 times more weight for H. armigera and 1.9 times more for S. frugiperda compared to those feeding on wild-type controls (Fig. 7a, b). These results highlight the critical role of 3 and 5a ~ 5d in mediating herbivore deterrence.

a, b Comparative growth analysis of H. armigera (a) and S. frugiperda (b) larvae fed on leaves from wide type (WT) and GhDP1-knockout (CR-Ghdp1) cotton plants (n = 15 individuals). c, f Disease symptoms of different cotton genotypes upon infection by F. oxysporum (c) or R. solani (f). Scale bar, 5 cm. d, g Index analyses of disease grades on cotton leaves. The proportion of plants exhibiting different levels of disease severity is shown. Disease indexes were assessed for 35 plants inoculated with F. oxysporum (d) and 33 plants inoculated with R. solani (g). e, h Relative fungal biomass in infected plants. For e and h FoActin/GhUBQ7 and Rs18S/GhUBQ7 ratios in WT were set to 1, respectively (n = 6 samples). i Morphological comparison of representative leaves from WT and CR-GhDP1 cotton plants after five days of inoculation with B. cinerea. Scale bars, 1 cm. j, k Quantitative assessment of the lesion area induced by B. cinerea (j) (n = 5 samples) and relative biomass of B. cinerea in the inoculated cotton leaves (k) conducted five days post-inoculation (n = 4 samples). l Biosynthetic pathways of major terpenoid specialized metabolites optimized for cotton defense. In non-green organs, such as late-stage ovules, 1 undergoes oxidative homocoupling at the C7 position mediated by laccases and DPs to form 4, conferring reproductive toxicity to mammals. In green organs such as stems and leaves, 1 selective oxidatized at the C5-position by GhDP1, GhAKR13D2 and oxidants, generating specific radical intermediates that lead to the formation of 2, followed by spontaneous oxidation to produce 3. The substantial accumulation of 3 and its derivatives (5a ~ 5d), along with a minor presence of 4, provides significantly enhanced toxicity towards herbivores and pathogens, compared to 4 alone in green organs. This GhDP1-mediated metabolic redirection underscores cotton’s strategically optimized defensive mechanism. For a, b, e, h, j, k, mean ± s.d., P values were determined by a two-tailed unpaired t-test. Source data are provided as a Source Data file.
To further investigate the role of GhDP1 in pathogen resistance, we challenged both WT and CR-Ghdp1 cotton plants with two soil-borne fungal pathogens: Fusarium oxysporum, a vascular wilt-causing ascomycete50, and Rhizoctonia solani, a necrotrophic basidiomycete responsible for root and stem blight51. Disease severity was quantified using a standardized disease grading system, revealing that GhDP1-deficient plants exhibited significantly exacerbated disease symptoms in both pathosystems (Fig. 7c, d, f, g). Consistently, qPCR-based quantification of fungal biomass demonstrated a substantial increase in F. oxysporum and R. solani colonization in CR-Ghdp1 plants compared to WT (Fig. 7e, h), corroborating the observed susceptibility. In parallel, we extended these findings to foliar infections using Botrytis cinerea, a necrotrophic fungus with an exceptionally broad host range and global agronomic significance52,53. Upon leaf inoculation, CR-Ghdp1 plants developed substantially larger necrotic lesions than WT counterparts (Fig. 7i, j). qPCR quantification of fungal DNA revealed a ~ 7-fold increase in B. cinerea biomass in GhDP1-knockout leaves (Fig. 7k), further supporting a critical role for GhDP1 in broad-spectrum pathogen resistance. These results collectively demonstrate that GhDP1 contributes positively to cotton resistance against both vascular and non-vascular fungal pathogens.
Overall, these findings demonstrate that GhDP1 and its homologs, which have evolved specifically within the cotton lineage, are pivotal in enhancing resistance to insect herbivores and pathogens by redirecting terpenoid synthesis towards 3 and heterocyclic 5a ~ 5d. The resultant accumulation of a range of specialized metabolites—characterized notably by elevated levels of 3 and heterocyclic 5a ~ 5d, alongside reduced levels of 4—in the green organs of cotton, signifies an optimized defensive mechanism that supersedes reliance on 4 alone (Fig. 7l). Given that 3 and 5a ~ 5d are cytotoxic, their extracellular biosynthesis and sequestration likely serve to mitigate potential toxicity within plant cells, thereby providing a defensive advantage. These findings exemplify a sophisticated evolutionary optimization of plant specialized metabolism, enabling cotton to mount robust, compartmentalized, and chemically diversified defenses.
Discussion
In this study, we have uncovered a specific branch of terpenoid metabolism that acts extracellularly in the green organs of cotton and elucidated its underlying genetic and biochemical mechanisms. GhDP1, in conjunction with AKR, mediates the oxidative redirection from the 4 pathway toward the biosynthesis of 3 and 5a ~ 5d. This metabolic shift appears to enhance defense capabilities against a broad range of biotic stresses, including herbivorous insects such as H. armigera and S. frugiperda, the airborne fungal pathogen B. cinerea, and soil-borne fungal pathogens such as F. oxysporum and R. solani. Meanwhile, cotton seeds exclusively biosynthesize 4, potentially serving as a chemical deterrent against seed-predating non-ruminant mammals (Fig. 7l). This research underscores the role of divergence in metabolic pathways in augmenting plant defense mechanisms.
Interestingly, although laccase-rich APF from G. hirsutum leaves was capable of converting 1 into gossypol 4, it failed to support the hydroxylation reaction leading to 2 and 3 in the absence of externally supplied GhDP1_A1 or GhDP1_A2. We hypothesize that GhDP1_A1/_A2 may be tightly associated with membrane-bound receptor proteins, limiting their dissociation during standard APF extraction protocols. This is reminiscent of the rice small secreted dirigent protein OsSSP1, which binds to plasma membrane receptors OsSSR1 and OsBAK1 in the apoplast to trigger immune responses during Magnaporthe oryzae infection54. Such interactions could similarly prevent the release of GhDP1 proteins during vacuum infiltration and centrifugation. Consistent with this, proteomic analysis of APF further corroborated these findings, identifying abundant laccases but no GhDP1 peptides (Supplementary Data 7). These observations explain why exogenous supplementation of recombinant GhDP1 was necessary to reconstitute extracellular hydroxylation activity in vitro and highlight the challenges associated with recovering compartmentalized proteins from complex plant tissues.
Hydroxylation of aromatic compounds, the oxygen transfer reaction that introduces the hydroxyl group (-OH) into aromatic rings via substituting functional groups or hydrogen atoms, is viewed as one of the most challenging fields in modern synthesis55. Enzymes such as cytochrome P450 monooxygenases, dioxygenases, significantly streamline this process through facilitating selective oxygenation of organic molecules under benign conditions56. However, the reactions carried out by P450 enzymes rely on NAD(P)H57, which is energy-consuming and requires a carefully designed catalytic environment. Here, we have discovered a mechanism of extracellular hydroxylation reactions distinct from the classical NADPH-dependent P450-catalyzed hydroxylation reactions. Our study reveals that the hydroxylation of 1 occurs extracellularly, facilitated without NADPH, through the synergistic actions of GhDP1 and GhAKR13D2 under oxidative conditions. This mechanism would be well-suited to hydroxylation using white biochemical reactions and contrasts sharply with the traditional multi-electron transfer process involving cytochrome P450 systems that utilize an oxo-ferryl heme radical cation complex [heme(FeIV=O)]·+58. We propose a putative mechanism wherein 1 is attacked by hydroxyl radicals generated by laccases or metal ions (Fig. 7l), following a mechanism akin to previously documented radical-mediated aryl hydroxylation59. Subsequently, 2 spontaneously oxidizes to its benzoquinone form, 3 (Fig. 7l). The participation of GhDP1 is crucial, as its absence impedes the extracellular hydroxylation reaction, underscoring the expanded role of DPs in the biosynthesis of plant specialized metabolites, where they predominantly contribute to bimolecular coupling reactions and determine the chirality of phenolic products60,61,62,63,64,65.
Although DPs are well documented to cooperate with laccases in the oxidative dimerization of extracellular specialized metabolites65, our study elucidates a previously unknown mechanism in which the dirigent protein GhDP1 not only acts as a regulator of extracellular hydroxylation reactions but also recruits an AKR to synergistically mediate this reaction. The hydroxylation mechanism catalyzed by the GhDP1-AKR complex fundamentally differs from the classical DP-laccase system: the catalytic reaction shifts from dimerization coupling to hydroxylation and proceeds independently of the typical AKR cofactor NADPH, occurring extracellularly under oxidative conditions via a radical-mediated process. Moreover, AKR family members are identified as auxiliary catalytic partners of dirigent proteins, significantly expanding the functional scope of DPs from controlling product stereochemistry to modulating metabolic flux direction. This discovery provides valuable insights into how plants precisely regulate extracellular specialized metabolism and defense mechanisms, opening avenues for research into dirigent protein function and plant metabolic regulation.
Moreover, GhAKR13D2 appears to provide ancillary support for this hydroxylation process. Its widespread subcellular localization suggests that it retains primordial AKR family functions, as evidenced by its interaction with the cofactors NADP(H), a hallmark cofactor of AKRs. Notably, unlike GhDP1-knockout lines, GhAKR13D2-VIGS plants and GhAKR13D2_A3-knockout lines exhibit reduced 4 levels compared to control plants, suggesting a multifaceted role for GhAKR13D2 in cotton (Fig. 4j and Supplementary Fig. 16f). This implies its participation not only in the biosynthesis of 3 but also potentially in the production of 4 or in the recycling of enzymatic cofactors, given the broad utilization of NADP(H) by numerous enzymes during 4 biosynthesis. These findings expand our understanding of AKRs beyond their conventional functions, highlighting their critical contributions to oxidation reactions and opening avenues for chemical synthesis and synthetic biology.
The discovery of DPs as regulatory valves in redirecting synthesis represents an instructive paradigm in plant biochemistry, elucidating how plants precisely control the production of specialized metabolites through the regulation of extracellular metabolic flow. This insight advances our understanding of plant defense mechanisms, broadening the research scope concerning selective pressures and evolutionary trajectories that shape metabolic complexity. Moreover, our study offers promising avenues for agricultural innovation. By modulating DPs and their associated pathways, it may possible to fine-tune the production of protective metabolites both in vivo and in vitro using white biochemistry.
Methods
Plant materials and growth conditions
Experiments were conducted using upland cotton G. hirsutum cv. R15, its transgenic derivatives, and the glandless mutant G. hirsutum cv. CCRI12gl. These plants were cultivated under controlled environmental conditions in a greenhouse environment, maintained at 28 °C with a 16-hour light and 8-hour dark photoperiod. Additionally, field trials were performed at two distinct locations: Songjiang (Shanghai) and Sanya (Hainan Province), to evaluate growth under natural conditions. N. benthamiana and N. tabacum were grown under greenhouse conditions in Shanghai. N. benthamiana was maintained at 22 °C, whereas N. tabacum was cultivated at 28 °C, both under a consistent 16-hour light and 8-hour dark photoperiod.
Virus-induced gene silencing (VIGS) and metabolite analysis
Virus-induced gene silencing was employed to investigate the functional roles of specific target genes. DNA fragments, ranging from 300 to 500 bp, corresponding to target gene sequences, were amplified via polymerase chain reaction (PCR) using gene-specific primers detailed in Supplementary Data 10. These amplicons were subsequently cloned into the pTRV2 vector. The recombinant pTRV2 constructs were then introduced into Agrobacterium tumefaciens strain GV3101. The transformed cultures were grown overnight in Luria-Bertani (LB) medium at 28 °C. Following cultivation, the bacterial cells were collected by centrifugation, resuspended in infiltration medium containing 10 mM MgCl2, 10 mM 2-morpholinoethanesulphonic acid, and 150 mM acetosyringone, with the pH adjusted to 5.8. The suspension was standardized to an optical density at 600 nm (OD600) of approximately 0.8 to ensure consistency in infiltration efficiency. For the infiltration procedure, cotyledons from 10-day-old cotton seedlings were treated with a 1:1 mixture of A. tumefaciens carrying the pTRV2 vectors and helper pTRV1 vectors. Two weeks post-infiltration, young second true leaves were harvested for metabolite analysis. Compounds including gossypol (4), hemigossypolone (3) and heliocides (5a ~ 5d) were isolated and subjected to characterization via high-performance liquid chromatography (HPLC), following established protocols11. Briefly, samples were extracted using an acetonitrile/water/phosphoric acid (80:20:0.1, v/v/v) solution and analyzed on an Agilent 1260 Infinity II LC System equipped with a Thermo Syncronis C18 column (150 × 4.6 mm, 5 μm), with detection at 272 nm. The mobile phase, consisting of EtOH: MeOH: IPA: ACN: H2O: EtOAc: DMF: H3PO4 = 16.7: 4.6: 12.1: 20.2: 37.4: 3.8: 5.1: 0.1, was delivered at 1 mL min−1 for 60 min with the column maintained at 40 °C. Compound identification and quantification were based on retention time matching against standards using the Agilent Chemstation Revision B.04.03.
RNA-Seq analysis
Total RNA was isolated with the RNAprep Pure Plant Plus Kit (DP441, TIANGEN). RNA quantification and purity assessment were conducted on a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific), while RNA integrity was evaluated using the RNA Nano 6000 Assay Kit on an Agilent Bioanalyzer 2100 system (Agilent Technologies). For library construction, 1 µg of total RNA per sample was used as starting material with the Hieff NGS Ultima Dual-mode mRNA Library Prep Kit for Illumina (Yeasen Biotechnology). Poly(A)+ mRNA was enriched using magnetic beads coated with oligo(dT). First-strand and second-strand cDNA were sequentially synthesized. The resulting cDNA was end-repaired, adenylated at the 3′ ends, and ligated to NEBNext adaptors. Purification of the library was carried out with the AMPure XP system (Beckman Coulter). USER Enzyme (NEB) was applied to the adaptor-ligated cDNA, followed by incubation at 37 °C for 15 min and 95 °C for 5 min. Amplification was performed using Phusion High-Fidelity DNA polymerase with universal and index primers. The final PCR products were purified (AMPure XP system), and library quality was verified using the Agilent Bioanalyzer 2100. Sequencing was conducted on an Illumina NovaSeq platform. Raw data were processed via the BMKCloud online platform (www.biocloud.net). Reads were aligned to the reference genome using Hisat2, and transcriptome assembly was performed with StringTie in Reference Annotation Based Transcript (RABT) mode to identify both known and novel transcripts66,67. Gene expression levels were quantified based on fragments per kilobase per million mapped fragments. Differential expression analysis was carried out with DESeq268, with genes showing an adjusted P-value < 0.01 and |Fold Change | ≥ 2 considered differentially expressed.
RNA extraction and gene expression analysis
Total RNA was extracted from plant organ using the RNAprep Pure Plant Plus Kit (DP441-H, TIANGEN) according to the manufacturer’s protocol. The isolated RNA was subsequently subjected to reverse transcription using the EasyScript One-Step Genomic DNA Removal and cDNA Synthesis SuperMix Kit (TransGen, AE311-03), ensuring efficient removal of genomic DNA and synthesis of complementary DNA (cDNA) for downstream analyses. qPCR was performed using the SYBR Green Pre-mix Pro Taq HS qPCR Kit (AG11701, AG) to quantify gene expression levels. The housekeeping gene, GhHIS3 (Gh_D03G0370), was employed as a normalization control69. The relative expression levels were calculated using standard methods, thus allowing for the assessment of differential gene expression across various experimental conditions.
Gene editing and cotton transformation
To achieve CRISPR/Cas9-mediated knockout of GhDP1_A1, GhDP1_A2, and GhAKR13D2_A3, we first analyzed the genomic DNA sequences of these targets using an online CRISPR design toolkit (https://www.crispr.hzau.edu.cn/CRISPR2). This process uncovered two common efficient target sites within the single exon of GhDP1_A1 and GhDP1_A2: TCAGTGGTGAAAACCCCAGT and GCTGATTTCGCATTTACGAG, and two specific and effective target sites in different exons of GhAKR13D2_A3: TTAAGCATGCATTCGAAAGA and TTACTGCCGTACAAATCGAG. These sequences were embedded within the pYLCRISPR/Cas9 vector. Through the use of A. tumefaciens strain EHA105, we then transformed these constructs into G. hirsutum cv. R15 to generate gene-knockout plants. Following the sgRNA efficiency checks, stable transformations were conducted. We screened for GhDP1_A1, GhDP1_A2, and GhAKR13D2_A3 knockout lines by isolating genomic DNA from both wild-type and T2 generation gene-editing cotton lines. This isolation was achieved using a DNA extraction kit (DP3112, Bioteke) following the manufacturer’s protocols. The targeted genomic regions were amplified through genotyping with primers flanking the sgRNA target sequences (forward primer: ATGGAGAAGGAACACAATATAC and reverse primer: TTAGTAATGGTACAAAGTAACATTG) for CR-Ghdp1 plants, and primers flanking the sgRNA target sequences (forward primer: AAAGCTGGGATTTGGGTG and reverse primer: TCTTCCTCAACATCACGAGT) for CR-Ghakr13d2_a3 plants. Then the PCR products were cloned into a TA-cloning vector (TransGen, CB501-2) for further analysis. We used Sanger sequencing to validate the presence and accuracy of the CRISPR-induced gene edits.
Transient and stable expression of dirigent proteins in Nicotiana species
For transient expression assays, the coding sequences of dirigent protein genes were amplified from a cotton cDNA library via PCR and cloned into the pEAQ vector to facilitate expression studies. These recombinant plasmids were then introduced into A. tumefaciens strain GV3101. The transformed cultures were incubated overnight at 28 °C in LB medium to achieve optimal bacterial growth. Following cultivation, bacterial cells were harvested by centrifugation and resuspended in an infiltration medium containing 10 mM MgCl2, 10 mM 2-morpholinoethanesulphonic acid, and 150 mM acetosyringone, with the pH adjusted to 5.8. The cell suspension was standardized to an OD600 of approximately 0.8. This A. tumefaciens suspension was then used to infiltrate leaves of four- to six-week-old N. benthamiana plants using a syringe infiltration technique. Two to three days post-infiltration, the plants expressing the target dirigent proteins were harvested for further analyses.
For the stable transformation of N. tabacum, sterilized “Thanxi” leaf segments (0.5–1.0 cm) were immersed in the resuspended A. tumefaciens culture containing the target vectors. These leaf sections were subsequently placed on Murashige and Skoog medium (MSB2) for two days, followed by transfer to MSB2 supplemented with 150 mg/L kanamycin and 500 mg/L ticarcillin for three to four weeks under culture room conditions maintained at 26 °C with diffused light. Explant tissues were then moved to half-strength Murashige and Skoog (1/2 MS0) medium supplemented with 150 mg/L kanamycin and 300 mg/L ticarcillin for an additional two to three weeks, during which antibiotic resistance was indicated by the formation of green calli with regenerating shoots. The regenerating shoots or callus pieces were then transferred to 1/2 MS0 medium containing 200 mg/L kanamycin and 150 mg/L ticarcillin until root formation was observed. Carefully remove the agar from the roots of the regenerated seedlings and transfer them into the soil. After stable growth, collect young leaves for further research.
Extraction of apoplastic fluids (APFs)
To extract plant APFs, an infiltration-centrifugation protocol was employed with minor modifications based on previously reported methods70. Leaf samples were initially submerged in a beaker filled with Phosphate Buffer Saline (PBS), pH 7.4, ensuring complete coverage by the solution. A rotary evaporator was used to apply a vacuum to the beaker, facilitating the removal of air bubbles from the leaf. The vacuum was then gradually released to atmospheric pressure. This step was reiterated until the leaves were entirely infiltrated, as confirmed by the absence of air spaces within the tissues. After infiltration, the leaf surface was carefully dried and placed into a 20 mL syringe body, substituting the usual plunger setup. This syringe assembly was then positioned within a 50 mL centrifuge tube. A two-step centrifugation process was carried out, initially at 750 g for 20 minutes at 4 °C, followed by a subsequent spin at 13,400 × g for another 20 minutes at the same temperature. The APFs were collected at the base of the centrifuge tubes and were deemed ready for subsequent analysis without necessitating any further processing.
Protein expression and purification
For eukaryotic expression, signal peptide truncation of GhDP1_A1 and GhDP1A2 were cloned into pFast-Bac GP67 vector and expressed in the Sf9 insect cell (invitrogen)/baculovirus expression system26. Insect cell cultures were incubated with Ni Sepharose excel and protease inhibitors at 4 °C for 1 h, and then collected by gravity column. Protein was washed with buffer (20 mM NaAc (pH 4.5), 150 mM NaCl, 20 mM imidazole) and eluted from the column using buffer (20 mM NaAc (pH 4.5), 150 mM NaCl, 300 mM imidazole). A Centriprep Centrifugal Filter (4302, Millipore) was used to concentrate and transfer the protein to buffer (20 mM NaAc (pH 4.5), 150 mM NaCl, 5% glycerol). Further purified GhDP1_A1 and GhDP1_A2 by affinity chromatography TALON columns. The protein concentration was determined using the Bradford assay.
GhAKR13D2_A3 was expressed in E. coli strains BL21(DE3). The coding sequence of GhAKR13D2_A3 was first cloned into the pET32a vector and transformed into E. coli. Transformants were then selected on LB medium containing 100 µg/mL ampicillin. The cells were inoculated into 500 mL LB medium containing 100 µg/mL ampicillin and induced by 250 µM IPTG when the cells reached an OD600 of approximately 0.6 at 18 °C for 20 hours. The protein was purified using wash buffer (50mM Tris-HCl, pH 7.5, 300mM NaCl, 20mM imidazole) and elution buffer (50mM Tris-HCl, pH 7.5, 300mM NaCl, 200mM imidazole). Then the proteins were desalted and the buffer was converted to 50 mM Tris-HCl pH 7.5, 300 mM NaCl buffer using PD-10 Desalting Columns (Cytiva).
Dirigent assays
For dirigent activity assays utilizing APFs as catalysts, GhDP1_A1 and GhDP1_A2 proteins expressed in insect cell cultures were assayed in 200 μL APFs of G. hirsutum leaves containing 100 μM hemigossypol (1). Controls were performed either without 1, without GhDP1, or without APF. For the GhDP1_A1 and GhDP1_A2 proteins expressed in N. benthamiana and N. tabacum, 100 μM hemigossypol (1) was incubated with 200 μL of APF that either expressed GhDP1_A1, GhDP1_A2 or the empty vector (EV).
For GhDP1_A1 and laccase activity assays, the phenolic hydroxylation of 1 by the GhDP1_A1 protein and RvLac was carried out in 500 μL of a solution containing 25 mM NaAC buffer pH 4.5, 100 μM CuCl2, 100 μM substrate with 2 μg RvLac and 5 μg GhDP1_A1 protein. Controls were executed either without 1, without GhDP1_A1, without RvLac, or without both GhDP1_A1 and RvLac. The above reaction products and substrates were extracted with ethyl acetate, then evaporated to dryness using a vacuum concentrator. The sample sizes for each analysis are indicated in the figure legends. The residue was subsequently resuspended in 60 μL of acetonitrile and analyzed by Agilent 1260 Infinity II LC System or Q Exactive quadrupole orbitrap high-resolution mass spectrometry coupled with a Dionex Ultimate 3000 RSLC (HPG) ultra-performance liquid chromatography (UPLC-Q-Orbitrap-HRMS) system (Thermo Fisher Scientific) using the Thermo C18 analytical column (150 × 4.6 mm, 5 μm). The mobile phase consisted of solvent A (water with 0.1% formic acid) and solvent B (acetonitrile) at a flow rate of 1 mL/min, with the following 12-minute gradient and detection at 257 nm and 272 nm: 0-3 min, 20-70% B; 3-5 min, 70-80% B; 5-7 min, 80-84% B; 7-8 min, 84-100% B; 8-10 min, 100% B; 10-11 min, 100-20% B; 11-12 min, 20% B. A HESI ionization source was employed to collect the MS data in positive-ion mode. Key parameters of mass spectrometer were as follows: mass range: 100-1000 m/z; ionization voltage: +3.8 kV/–3.0 kV; sheath gas pressure: 35 arbitrary units; auxiliary gas: 10 arbitrary units; auxiliary gas heater temperature: 350 °C; capillary temperature: 320 °C. Mass spectra and retention time matching with standards were used to identify detected compounds. Agilent Chemstation Revision B.04.03 and Thermo Scientific Xcalibur Version 4.2.47 were used for data analysis.
Conjoint enzymatic activity assay of GhDP1_A1 and GhAKR13D2_A3
To analyze the combined catalytic activity of GhDP1_A1 and GhAKR13D2_A3, the reaction mixture (500 µL) contained 25 mM sodium acetate buffer (pH 4.5), 100 μM CuCl₂ or FeCl₃, and 100 μM hemigossypol (1). Subsequently, 5 μg of GhDP1_A1 and 5 μg of GhAKR13D2_A3 were added to the mixture. Control experiments with individual components removed in turn were set. The reaction products were detected using the method as described previously.
Immunogold labeling and TEM observation
Referring to previously reported methods71, the young leaves of cotton seedlings were fixed in 4% (v/v) glutaraldehyde (EM Grade) in 0.1 M PBS (pH 7.4). After being washed three times with PBS, they were post-fix in 2% (w/v) osmium tetroxide in PBS for 1 h, then rinsed thoroughly with Milli-Q water. The samples were then dehydrated through a graded ethanol series, followed by infiltration with acrylic ‘LR White’ resin for several days. Each section was embedded in gelatin capsules with fresh resin and polymerized at 65 °C. The sections were cut to 80 nm thickness and collected on formvar-coated gold grids.
For immunogold labeling and TEM, treat ultrathin sections with 10% H2O2 for 10 min, followed by three washes with distilled water. Block with 1% bovine serum albumin in PBS for 1 hour. Incubate the sections with anti-GhDP1 or anti-GhAKR13D2 monoclonal antibody (1:500 dilution, customized by Abmart) at 37 °C for 2 hours. Wash sections five times with 0.01% Tween 20 in PBS, then incubate with goat anti-mouse IgG (1:30 dilution, Abcam, ab27241) for 1 hour. Rinse with PBS and distilled water. Stain with aqueous uranyl acetate (2% w/v) for 3 min and lead citrate for 5 min, followed by rinsing with distilled water and air drying. Finally, observe the sections under a transmission electron microscope (H7650, Hitachi) at 80 kV accelerating voltage.
Subcellular localization assays
The coding sequences of GhDP1_A1 and GhDP1_A2 were fused with GFP and placed under the control of upstream of the 2 kb promoter of GhDP1_A1 and GhDP1_A2 or the 35S promoter, respectively, and cloned into the pCAMBIA2301 vector. Similarly, GhAKR13D2_A3 and its upstream 2 kb promoter were cloned into the pCAMBIA2301 vector. The above constructs were used for cotton transformation. For transient expression analysis in N. benthamiana, the coding sequence of GhAKR13D2_A3 was fused in-frame to the C-terminus of the mCherry fluorescent protein and introduced into N. benthamiana leaf epidermal cells via Agrobacterium-mediated infiltration. The plasma membrane marker AtPIP2A fused to YFP was co-expressed as a localization reference. Two to three days post-infiltration, leaves were excised and subjected to confocal laser scanning microscopy. The fluorescent dye Fluorescent Brightener 28 (Calcofluor White M2R, FB28) was used at a final concentration of 0.1% (w/v) in water for the cell wall visualization. Colocalization of GhAKR13D2_A3-mCherry signals with AtPIP2A-YFP and FB28 staining was assessed under both non-plasmolyzed and plasmolyzed conditions. For plasmolysis treatment, leaf discs were incubated in 0.8 M mannitol for 20 − 30 minutes before imaging to distinguish the plasma membrane from the apoplastic space.
All fluorescence signals were observed by a Leica TSC SP8 STED 3× confocal microscope. Pictures were analyzed by ImageJ.
Phylogenetic analysis
The full-length amino acid sequences of dirigent proteins or aldo-keto reductases were aligned and trimmed by MUSCLE v5.1 (https://drive5.com/muscle5) and trimAI (http://trimal.cgenomics.org)72,73. Then, a phylogenetic tree was constructed using IQ-TREE v2.2.0 (http://www.iqtree.org) with the maximum likelihood (ML) method74. The bootstrap test method was set to 1000. The polygenetic tree was visualized by iTOL (https://itol.embl.de)75.
Molecular docking and in silico protein stability analysis
AlphaFold3 was applied for protein homology models of GhDP1_A1 and GhAKR13D2_A341. The molecular docking analysis was performed by AutoDock Vina, v1.1.276. The docking parameters are as follows: center_x = −6.862, center_y = −14.904, center_z = 3.977, size_x = 40, size_y = 40, size_z = 50, exhaustiveness = 8. The effects of site-directed mutagenesis on GhDP1_A1 stability were systematically evaluated using three computational approaches: Rosetta’s ddg_monomer, FoldX 5.0, and ThermoMPNN77,78,79.
Western blotting
Protein samples were isolated using a 4-20% ExpressPlus PAGE Gel (GenScript), and subsequently transferred onto an Immobilon-P PVDF 0.45 µm membrane (Merck). First antibodies applied comprised of 5% skim milk (BD) anti-His (1:5000 dilution, Smart-Lifesciences, SLAB2801), anti-GFP (1:5000 dilution, Smart-Lifesciences, SLAB3001), anti-FLAG (1:5000 dilution, Smart-Lifesciences, SLAB0101), or a custom-made mouse monoclonal antibody against GhDP1 produced by Abmart (1:1000). For secondary antibody, horseradish peroxidase (HRP)-labeled goat anti-mouse (1:10,000 dilution, ZB-2305, ZSGB-BIO) in 1×TBST was used. The blotting signals were identified by applying SuperSignal West Femto High Sensitivity Substrate (Thermo Scientific) to the PVDF membrane. Finally, signal detection was performed using Tanon 5200 SF (Tanon).
Split luciferase complementation (SLC) assay
The coding sequences of GhDP1_A1 and GhAKR13D2_A3 were cloned into pJW771 (nLUC) and pJW772 (cLUC) vectors under the control of the CaMV 35S promoter, respectively. The resulting constructs, along with the corresponding empty vector controls, were introduced into A. tumefaciens strain GV3101. The transformed strains were cultured, harvested, and resuspended in infiltration buffer (10 mM MgCl₂, 10 mM MES, 150 µM acetosyringone) to an OD600 of ~1.0 and incubated at room temperature for 2–3 h. Equal volumes of the nLUC and cLUC strains were mixed at a 1:1 ratio and infiltrated into N. benthamiana leaves. After 48 hours, 1 mM D-luciferin (potassium salt; APExBIO) was applied to the infiltrated areas. Luminescence was detected and imaged using a Tanon 5200 SF imaging system to assess protein–protein interaction.
Pull-down assay
GhDP1_A1-His and GhAKR13D2_A3-FLAG proteins were co-expressed in E. coli (BL21) using the pETDuet vector, with YFP-His and GhAKR13D2_A3-FLAG co-expression as the control group. Ni-NTA agarose resin (Cytiva) was used to capture the His-tagged proteins. The Ni-NTA agarose resin was then washed three times with wash buffer (50 mM Tris-HCl pH 7.5, 100 mM NaCl, 20 mM imidazole) to remove non-specifically bound proteins. The precipitates were released by boiling in SDS sample buffer at 100 °C for 10 minutes and detected by Western blot analysis using anti-FLAG (Smart-Lifesciences, SLAB0101) and anti-His (Smart-Lifesciences, SLAB2801) antibodies.
Bimolecular fluorescence complementation (BiFC) assay
pMSH21-mCherry-cYFP, pMSH21-mCherry-nYFP, pMSH21-GhDP1_A1-cYFP and pMSH22-GhAKR13D2_A3-nYFP vectors were constructed and subsequently transformed into A. tumefaciens strain GV3101. Then, the bacterial cultures were expanded and the bacteria were collected. The collected bacteria were suspended until the OD600 was approximately 1.0 and then left at room temperature for 2-3 hours. Equal volumes of pMSH21-GhDP1_A1-cYFP and pMSH22-nYFP, pMSH22-GhAKR13D2_A3-nYFP and pMSH22-cYFP, pMSH21-GhDP1_A1-cYFP and pMSH22-mCherry-nYFP, pMSH22-GhAKR13D2_A3-nYFP and pMSH22-mCherry-cYFP, pMSH22-mCherry-nYFP and pMSH22-mCherry-cYFP were mixed and injected into tobacco leaves. The mCherry protein was used as an unrelated protein control. After 48 h fluorescence was observed using a Leica TSC SP8 STED 3× confocal microscope.
Co-immunoprecipitation (Co-IP)
The expression vectors pCAMBIA1300-GhAKR13D2_A3-YFP, pCAMBIA1300-YFP (empty vector control), and pEAQ-GhDP1_A1-FLAG were used to transiently co-express GhAKR13D2_A3-YFP or YFP and GhDP1_A1-FLAG in N. benthamiana leaves. Proteins were subsequently extracted from the leaves using IP lysis buffer consisting of 50 mM Tris-HCl, 150 mM NaCl, 20% glycerol, 0.5% NP-40, and 1×PIC (TransGen). After centrifugation, supernatant was incubated with anti-GFP beads (Smart-Lifesciences, SM038001) for 2 h at 4 °C, followed by four times washes with the extraction buffer. Western blot analysis was performed using anti-GFP (Smart-Lifesciences, SLAB3001) and anti-FLAG antibodies (Smart-Lifesciences, SLAB0101) to identify proteins that interacted and were tagged with GFP and FLAG.
Biacore assay
GhAKR13D2_A3 purified protein was dissolved with deionized and prepared into 1 mg/mL protein stock solution. The stock solution was diluted to 30 μg/mL with four different acetate buffers (pH 4.0, pH 4.5, pH 5.0, pH 5.5) prior to injection. The response values of proteins under different conditions were detected by the Biacore preenrichment system, and the optimal coupling condition was pH 4.0.
The coupling of purified proteins to the CM5 chip was achieved by activating the carboxyl groups on the surface of the CM5 chip via EDC/NHS, followed by the condensation of carboxylic acids and amines to bond the proteins to the CM5 chip, and finally blocking the unbound carboxyl groups with ethanolamine.
The small molecular compounds were dissolved in PBS and diluted into 64 μmol/L, and the compound concentration was diluted at a half ratio. The concentration range was 0.0625 to 32 μmol/L, and the mobile phase was a PBS solution. The response value of the molecular compounds flowing over the protein surface was analyzed by the Biacore system. The initial concentration of the stock solution was 125 nmol/L, and the buffer system was a phosphate buffer system without DMSO, ranging from 0.488 nmol/L to 62.5 nmol/L, and other parameters were the same as those of small molecular compounds.
A kinetic curve was then drawn based on the dose-effect relationship between the response value and the concentration of the candidate compound. The binding specificity of the mobile phase molecules to the target protein was determined through the curve fitting, and the dissociation constant (KD) value was calculated.
Insect feeding test
The third instar larvae of H. armigera and S. frugiperda were purchased from Keyun Biology and cultured in an incubator at 28 °C with 70% relative humidity, under a 16-hour light / 8-hour dark. G. hirsutum cv. R15 (WT) or GhDP1-knockout (CR-Ghdp1) cotton leaves were used as fodder. Each larva was fed in a separate container, fresh plants were changed daily, and the body weight gain was recorded after three (H. armigera) or five (S. frugiperda) days of feeding.
Botrytis cinerea inoculation assay
Botrytis cinerea strain B05.10 was cultured on potato dextrose agar (PDA) medium for three days. Colonies with a diameter of 5 mm were then transferred to leaves of G. hirsutum cv. R15 (WT) or GhDP1-knockout (CR-Ghdp1) cotton plants for pathogen inoculation. After five days, the lesion area on the leaves infected with B. cinerea was measured using ImageJ software. For reference, wild-type leaves treated with a blank medium (MOCK) served as an additional control. The biomass of the B. cinerea was quantified by measuring the relative abundance of fungal Actin (BcActin) DNA to cotton GhUBQ7 DNA using qPCR.
Fusarium oxysporum and Rhizoctonia solani inoculation assay
Fusarium oxysporum f. sp. lycopersici strain 4287 and Rhizoctonia solani isolate RH-9 were initially cultured on potato dextrose agar (PDA) at 28 °C for 3 days. Subsequently, mycelia were transferred to Czapek-Dox liquid medium and incubated at 28 °C with shaking at 150 rpm for 3 days to promote spore production. Spores were harvested by suspending the cultures in sterile water, and concentrations were adjusted to 106 conidia/mL using a hemocytometer. Seedlings (2–3 weeks old) were inoculated using the root-drenching method, followed by application of the spore suspension to the soil. Plants were grown under controlled conditions (28 °C, 85% relative humidity, 16-hour light/8-hour dark). Disease grades were assessed 7–10 days post-inoculation using a five-point scale: 0 = no visible wilting or yellowing symptoms; 1 = one or two cotyledons wilted or dropped; 2-3 = one or two true leaves wilted or dropped; 4 = all leaves dropped or plant died. Disease grades were calculated from three independent biological replicate experiments. Relative fungal biomass was measured by DNA-based qPCR using the threshold cycle value (CT) of F. oxysporum Actin (FoActin) DNA or R. solani 18S rRNA (Rs18S) DNA against the CT of cotton genomic GhUBQ7 (GenBank: DQ116441)80,81. The data were normalized to cotton GhUBQ7 (GenBank: DQ116441) expression. The ratio of fungal DNA to cotton DNA in WT plants was set to 1. Statistical significance was evaluated using Student’s t-test. The plants were photographed and subjected to disease index analyses 7-10 days post-inoculation.
Synteny analysis
The genomes and complete protein sequences of the species utilized in the phylogenetic study were uploaded to MCscan (Python version), a tool from the JCVI utility libraries (https://github.com/tanghaibao/jcvi)82. Gene pair comparisons were conducted using LAST83. Following bidirectional sequence alignment, removal of potential tandem duplications and low-scoring hits, the anchors derived from the LAST outputs were organized into syntenic blocks. Microsynteny diagrams were produced using the synteny functions with their default parameters.
Immunoprecipitation-mass spectrometry (IP-MS)
Cotton plants (transgenic 35S::GhDP1_A1-GFP and wild type) were grown for three weeks. Leaf organs were immediately frozen in liquid nitrogen, then insoluble polyvinylpolypyrrolidone (PVPP) was added at a ratio of 5:1 (m/m) and thoroughly ground. Proteins were extracted using the extraction buffer (150 mM Tris-HCl pH 7.5, 150 mM NaCl, 10% glycerol, 10 mM EDTA, 0.5% Triton X-100, 20 mM NaF, 100 mM DTT, and the protease inhibitor cocktail (Roche)) by sonication at 4 °C. The homogenate was filtered through a 0.45 µm filter. Wild-type cotton plants were used as negative controls for downstream analyses.
Anti-GFP Magarose Beads (Smart-Lifesciences, SM038001), pre-equilibrated with TBS (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5 mM EDTA), were incubated with the protein extract at 4 °C for 2 hours. Following incubation, the beads were washed three times quickly and three times slowly with TBST (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.5% Triton X-100). The proteins were eluted using the denaturing buffer (2% SDS, 100 mM Tris-HCl pH 8.0, 10 mM TCEP, 50 mM CAA), and the collected eluate was denatured at 95 °C for 10 minutes for further analysis.
Samples were sequentially washed with 8 M urea and 50 mM ammonium bicarbonate using filter-aided sample preparation (FASP) with Vivacon 500 (10,000 MWCO, SARTORIUS, VN01H01). Each sample was digested overnight using 1 μg of trypsin (Promega). Desalting was performed using Pierce™ C18 Spin Tips & Columns (Thermo Fisher, 89870). The eluted peptides were dried by centrifugation at 10 °C and dissolved in an appropriate volume of 0.1% formic acid and then subjected liquid chromatography mass spectrometry (LC-MS) detection.
Mass spectrometry experiments were conducted using a timsTOF Pro2 instrument, which was integrated with a nanoElute liquid chromatography system (Bruker Daltonics). Peptide samples were reconstituted in 0.1% formic acid (FA), and 200 ng of each peptide was subjected to separation via a reversed-phase analytical column (25 cm × 75 μm i.d., Ionopticks). The separation employed a 60-minute gradient (buffer A: 0.1% FA; buffer B: 0.1% FA in acetonitrile), starting at 2% buffer B and increasing to 22% over 45 minutes, followed by a rapid increase to 37% over 5 minutes, a jump to 80% for 5 minutes, and then held at this concentration. The flow rate was set at 300 nL/min, with the column maintained at a temperature of 50 °C. The timsTOF Pro2 was operated in DDA-PASEF mode, configured with a capillary voltage of 1500 V, a dry gas flow rate of 3 L/min, and a drying temperature of 180 °C. During PASEF MS/MS acquisition, collision energy was linearly ramped from 59 eV at a mobility value of 1/K0 = 1.6 V·s/cm² down to 20 eV at 1/K0 = 0.6 V·s/cm².
The mass spectrometry data were processed using Paser version 2023, with a search conducted against the NBI_Gossypium_hirsutum_v1.1.pep.fas.fa (https://www.cottongen.org). The initial search parameters included a precursor mass tolerance of 10 ppm, employing Trypsin as the enzymatic cleavage agent, allowing for a maximum of two missed cleavages, and a fragment ion mass tolerance of 0.02 Da. Carbamidomethylation of cysteine (C) at 57.02 Da was set as a fixed modification, while methionine oxidation (M) at 15.99 Da was considered a variable modification during the database search. A global false discovery rate (FDR) threshold of 0.01 was established for both peptide and protein identification. For the decoy database, a mutated strategy similar to random amino acid sequence shuffling was used, with sequences varying from a minimum of two amino acids to up to half the length of the original peptide. Spectronaut 19 performed automatic calibration and local normalization of the data, and the average peak area of the top three peptides with an FDR below 1.0% was utilized for protein group quantification.
Proteome of cotton apoplastic fluid (APF)
Upland cotton G. hirsutum cv. R15 APF was extracted as follows, then the BBP buffer (100 mM Tris-Base (pH 8.8), 100 mM EDTA, 50 mM Borax, 50 mM ascorbic acid, 1% Triton X-100, 10 mM TCEP, 30%(m/v) sucrose, 50 mM tetraborate, the protease inhibitor 1 mM cocktail (Roche) and 1 mM PMSF was added at a volume ratio of 1:4 (v/v). Subsequently, phenol was added to the mixture at a ratio of 1:1 for protein extraction. After centrifugation, the upper phase was collected and transferred to a solution of 0.1 M ammonium acetate (dissolved in methanol) and incubated at −20 °C overnight for protein precipitation. The precipitated proteins were washed with 90% acetone and subsequently dissolved in 8 M urea. The samples underwent reduction, alkylation, and trypsin digestion. After digestion, desalting was performed using Pierce™ C18 Spin Tips & Columns (Thermo Fisher, 89870). The samples were then dried by centrifugation and dissolved in 0.1% formic acid for instrumental analysis. LC-MS detection, sequence database searching, and data analysis were performed as previously described.
Data analysis and statistics
Compound structures were drawn by ChemDraw 19.0. Enzyme assays or analyses of chemicals were conducted by Origin 2022. Quantification and statistics were conducted using GraphPad Prism 8.2.1. All data are presented as mean ± s.d. and statistical analysis was conducted using a two-tailed unpaired Student’s t-test unless otherwise specified. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The authors declare that all relevant data supporting the findings of this study are available within the article and its Supplementary Information. The RNA-seq data generated in this study have been deposited in the NCBI SRA database under accession number PRJNA1210977, corresponding to the transcriptome of GhAKR13D2 VIGS experiment. Public RNA-seq datasets used for comparative analyses are available under accession numbers PRJNA248163 (gene expression profiles across different organs of upland cotton, https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA248163) and PRJNA493958 (transcriptomes of glanded and glandless cotton varieties, https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA493958). Sequence data and gene IDs can be found in the Cottongen database (https://www.cottongen.org). The single-cell transcriptomic data used in this study can be found at the National Center for Biotechnology Information using the accession number GEO: GSE243419. The mass spectrometry proteomics raw data from the IP-MS and apoplast proteome experiments have been deposited in the ProteomeXchange Consortium under the accession numbers PXD062712 and PXD062792, respectively. In summary, all datasets generated and analyzed in this study have been deposited in publicly accessible repositories, and the relevant accession numbers and links are provided above. Source data are provided with this paper.
References
Dixon, R. A. Natural products and plant disease resistance. Nature 411, 843–847 (2001).
Jones, J. D. G. & Dangl, J. L. The plant immune system. Nature 444, 323–329 (2006).
van Loon, L. C., Rep, M. & Pieterse, C. M. Significance of inducible defense-related proteins in infected plants. Annu Rev. Phytopathol. 44, 135–162 (2006).
Gershenzon, J. & Dudareva, N. The function of terpene natural products in the natural world. Nat. Chem. Biol. 3, 408–414 (2007).
Pichersky, E. & Lewinsohn, E. Convergent evolution in plant specialized metabolism. Annu Rev. Plant Biol. 62, 549–566 (2011).
Croteau, R., Kutchan, T. M. & Lewis, N. G. Natural products (secondary metabolites). Biochem. Mol. Biol. Plants 24, 1250–1319 (2000).
Boccia, M. et al. A scaffold protein manages the biosynthesis of steroidal defense metabolites in plants. Science 386, 1366–1372 (2024).
Lin, J. L. et al. Dirigent gene editing of gossypol enantiomers for toxicity-depleted cotton seeds. Nat. Plants 9, 605–615 (2023).
Fryxell, P. A. A redefinition of the tribe Gossypieae. Bot. Gaz. 129, 296–308 (1968).
Liu, Y. et al. Structure, properties of gossypol and its derivatives-from physiological activities to drug discovery and drug design. Nat. Prod. Rep. 39, 1282–1304 (2022).
Tian, X. et al. Characterization of gossypol biosynthetic pathway. Proc. Natl Acad. Sci. USA 115, E5410-e5418 (2018).
Gao, W. et al. Proteomic and virus-induced gene silencing (VIGS) analyses reveal that gossypol, brassinosteroids, and jasmonic acid contribute to the resistance of cotton to Verticillium dahliae. Mol. Cell. Proteom.: MCP 12, 3690–3703 (2013).
de Peyster, A. & Wang, Y. Y. Gossypol–proposed contraceptive for men passes the Ames test. N. Engl. J. Med. 301, 275–276 (1979).
Coutinho, E. M., Melo, J. F., Barbosa, I. & Segal, S. J. Antispermatogenic action of gossypol in men. Fertil. Steril. 42, 424–430 (1984).
Pösö, H. et al. Gossypol, a powerful inhibitor of human spermatozoal metabolism. Lancet 1, 885–886 (1980).
Bell, A. A. et al. Sesquiterpenoid aldehyde quinones and derivatives in pigment glands of Gossypium. Phytochemistry 17, 1297–1305 (1978).
Shen, N. et al. Plant flavonoids: Classification, distribution, biosynthesis, and antioxidant activity. Food Chem. 383, 132531 (2022).
Wei, G. et al. Biosyntheses characterization of alkaloids and flavonoids in Sophora flavescens by combining metabolome and transcriptome. Sci. Rep. 11, 7388 (2021).
Eljounaidi, K. et al. Variation of terpene alkaloids in Daphniphyllum macropodum across plants and tissues. N. Phytol. 243, 299–313 (2024).
Lin, J.-L. et al. Single-cell RNA sequencing reveals a hierarchical transcriptional regulatory network of terpenoid biosynthesis in cotton secretory glandular cells. Mol. Plant 16, 1990–2003 (2023).
Huang, J. Q. et al. Aromatization of natural products by a specialized detoxification enzyme. Nat. Chem. Biol. 16, 250–256 (2020).
Chen, X. Y. et al. Cloning and heterologous expression of a second (+)-delta-cadinene synthase from Gossypium arboreum. J. Nat. Prod. 59, 944–951 (1996).
Effenberger, I. et al. Dirigent proteins from cotton (Gossypium sp.) for the atropselective synthesis of gossypol. Angew. Chem. Int Ed. Engl. 54, 14660–14663 (2015).
Pickel, B. & Schaller, A. Dirigent proteins: molecular characteristics and potential biotechnological applications. Appl Microbiol Biotechnol. 97, 8427–8438 (2013).
Davin, L. B. et al. Stereoselective bimolecular phenoxy radical coupling by an auxiliary (dirigent) protein without an active center. Science 275, 362–366 (1997).
Gao, Y. Q. et al. A dirigent protein complex directs lignin polymerization and assembly of the root diffusion barrier. Science 382, 464–471 (2023).
Kim, S. S. & Sattely, E. S. Dirigent proteins guide asymmetric heterocoupling for the synthesis of complex natural product analogues. J. Am. Chem. Soc. 143, 5011–5021 (2021).
Wang, Y. et al. A dirigent family protein confers variation of Casparian strip thickness and salt tolerance in maize. Nat. Commun. 13, 2222 (2022).
Teufel, F. et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 40, 1023–1025 (2022).
Ralph, S. et al. Dirigent proteins in conifer defense: gene discovery, phylogeny, and differential wound- and insect-induced expression of a family of DIR and DIR-like genes in spruce (Picea spp.). Plant Mol. Biol. 60, 21–40 (2006).
Ralph, S. G., Jancsik, S. & Bohlmann, J. Dirigent proteins in conifer defense II: Extended gene discovery, phylogeny, and constitutive and stress-induced gene expression in spruce (Picea spp.). Phytochemistry 68, 1975–1991 (2007).
Meng, Q. et al. Pterocarpan synthase (PTS) structures suggest a common quinone methide-stabilizing function in dirigent proteins and proteins with dirigent-like domains. J. Biol. Chem. 295, 11584–11601 (2020).
Gasper, R. et al. Dirigent protein mode of action revealed by the crystal structure of AtDIR6. Plant Physiol. 172, 2165–2175 (2016).
Pen, J. et al. Production of active Bacillus licheniformis Alpha-Amylase in tobacco and its application in starch liquefaction. Biotechnology 10, 292–296 (1992).
Wallace, G. & Fry, S. C. Action of diverse peroxidases and laccases on six cell wall-related phenolic compounds. Phytochemistry 52, 769–773 (1999).
Andréasson, L.-E., Brändén, R. & Reinhammar, B. Kinetic studies of Rhus vernicifera laccase: Evidence for multi-electron transfer and an oxygen intermediate in the reoxidation reaction. Biochim Biophys. Acta 438, 370–379 (1976).
Llevot, A. et al. Selective laccase-catalyzed dimerization of phenolic compounds derived from lignin: Towards original symmetrical bio-based (bis) aromatic monomers. J. Mol. Catal. B Enzym 125, 34–41 (2016).
Wu, W.-K. et al. Regulation of glandular size and phytoalexin biosynthesis by a negative feedback loop in cotton. Adv. Sci. 11, 2403059 (2024).
Penning, T. M. The aldo-keto reductases (AKRs): Overview. Chem. Biol. Interact. 234, 236–246 (2015).
Guo, R. et al. A novel aldo-keto reductase gene, OsAKR1, from rice confers higher tolerance to cadmium stress in rice by an in vivo reactive aldehyde detoxification. J. Hazard Mater. 470, 134212 (2024).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Khersonsky, O. & Tawfik, D. S. Enzyme promiscuity: a mechanistic and evolutionary perspective. Annu Rev. Biochem 79, 471–505 (2010).
Penning, T. M., Wangtrakuldee, P. & Auchus, R. J. Structural and functional biology of aldo-keto reductase steroid-transforming enzymes. Endocr. Rev. 40, 447–475 (2018).
Mindnich, R. D. & Penning, T. M. Aldo-keto reductase (AKR) superfamily: Genomics and annotation. Hum. Genomics 3, 362 (2009).
Salabei, J. K. et al. Functional expression of novel human and murine AKR1B genes. Chem. Biol. Interact. 191, 177–184 (2011).
Huang, G. et al. Recent advances and future perspectives in cotton research. Annu Rev. Plant Biol. 72, 437–462 (2021).
Anarat-Cappillino, G. & Sattely, E. S. The chemical logic of plant natural product biosynthesis. Curr. Opin. Plant Biol. 19, 51–58 (2014).
Jones, C. M. et al. Movement ecology of pest Helicoverpa: Implications for ongoing spread. Annu Rev. Entomol. 64, 277–295 (2019).
Tay, W. T. et al. Spodoptera frugiperda: ecology, evolution, and management options of an invasive species. Annu Rev. Entomol. 68, 299–317 (2023).
Gordon, T. R. & Martyn, R. D. The evolutionary biology of Fusarium oxysporum. Annu Rev. Phytopathol. 35, 111–128 (1997).
Cubeta, M. A. & Vilgalys, R. Population biology of the Rhizoctonia solani complex. Phytopathology 87, 480–484 (1997).
Staats, M., van Baarlen, P. & van Kan, J. A. Molecular phylogeny of the plant pathogenic genus Botrytis and the evolution of host specificity. Mol. Biol. Evol. 22, 333–346 (2005).
Williamson, B. et al. Botrytis cinerea: the cause of grey mould disease. Mol. Plant Pathol. 8, 561–580 (2007).
Zhao, T. et al. Recognition of the inducible, secretory small protein OsSSP1 by the membrane receptor OsSSR1 and the co-receptor OsBAK1 confers rice resistance to the blast fungus. Mol. Plant 17, 807–823 (2024).
Ullrich, R. & Hofrichter, M. Enzymatic hydroxylation of aromatic compounds. Cell Mol. Life Sci. 64, 271–293 (2007).
Alcalde, M. et al. Environmental biocatalysis: from remediation with enzymes to novel green processes. Trends Biotechnol. 24, 281–287 (2006).
Werck-Reichhart, D. & Feyereisen, R. Cytochromes P450: a success story. Genome Biol. 1, 3001 (2000). reviews3003.
Guengerich, F. P. Mechanisms of cytochrome P450-catalyzed oxidations. ACS Catal. 8, 10964–10976 (2018).
Mahdi, J. G. Biosynthesis and metabolism of β-d-salicin: A novel molecule that exerts biological function in humans and plants. Biotechnol. Rep. 4, 73–79 (2014).
Kim, M. K. et al. The western red cedar (Thuja plicata) 8-8’ DIRIGENT family displays diverse expression patterns and conserved monolignol coupling specificity. Plant Mol. Biol. 49, 199–214 (2002).
Pickel, B. et al. An enantiocomplementary dirigent protein for the enantioselective laccase-catalyzed oxidative coupling of phenols. Angew. Chem. Int Ed. Engl. 49, 202–204 (2010).
Kim, K. W. et al. Opposite stereoselectivities of dirigent proteins in Arabidopsis and schizandra species. J. Biol. Chem. 287, 33957–33972 (2012).
Dalisay, D. S. et al. Dirigent protein-mediated lignan and cyanogenic glucoside formation in flax seed: integrated omics and MALDI mass spectrometry imaging. J. Nat. Prod. 78, 1231–1242 (2015).
Yonekura-Sakakibara, K. et al. Seed-coat protective neolignans are produced by the dirigent protein AtDP1 and the laccase AtLAC5 in Arabidopsis. Plant Cell 33, 129–152 (2021).
Paniagua, C. et al. Dirigent proteins in plants: modulating cell wall metabolism during abiotic and biotic stress exposure. J. Exp. Bot. 68, 3287–3301 (2017).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. methods 12, 357–360 (2015).
Pertea, M. et al. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667 (2016).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Shan, C.-M. et al. Control of cotton fibre elongation by a homeodomain transcription factor GhHOX3. Nat. Commun. 5, 5519 (2014).
O'Leary, B. M. et al. The infiltration-centrifugation technique for extraction of apoplastic fluid from plant leaves using Phaseolus vulgaris as an example. J. Vis. Exp. 94, 52113 (2014).
Burton, R. A. et al. Cellulose synthase-like CslF genes mediate the synthesis of cell wall (1,3;1,4)-beta-D-glucans. Science 311, 1940–1942 (2006).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Nguyen, L. T. et al. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–w296 (2021).
Trott, O. & Olson, A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput Chem. 31, 455–461 (2010).
Kellogg, E. H., Leaver-Fay, A. & Baker, D. Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins 79, 830–838 (2011).
Delgado, J. et al. FoldX 5.0: working with RNA, small molecules and a new graphical interface. Bioinformatics 35, 4168–4169 (2019).
Dieckhaus, H. et al. Transfer learning to leverage larger datasets for improved prediction of protein stability changes. Proc. Natl Acad. Sci. USA 121, e2314853121 (2024).
Redkar, A. et al. Marchantia polymorpha model reveals conserved infection mechanisms in the vascular wilt fungal pathogen Fusarium oxysporum. N. Phytol. 234, 227–241 (2022).
Wang, A. et al. Identification of rice (Oryza sativa L.) genes involved in sheath blight resistance via a genome-wide association study. Plant Biotechnol. J. 19, 1553–1566 (2021).
Tang, H. et al. Synteny and collinearity in plant genomes. Science 320, 486–488 (2008).
Kiełbasa, S. M. et al. Adaptive seeds tame genomic sequence comparison. Genome Res. 21, 487–493 (2011).
Opitz, S., Kunert, G. & Gershenzon, J. Increased terpenoid accumulation in cotton (Gossypium hirsutum) foliage is a general wound response. J. Chem. Ecol. 34, 508–522 (2008).
Park, S.-H. et al. Chemical defense responses of upland cotton, Gossypium hirsutum L. to physical wounding. Plant Direct 3, e00141 (2019).
Scheffler, J. A. Evaluating protective terpenoid aldehyde compounds in cotton (Gossypium hirsutum L.) roots. Am. J. Plant Sci. 7, 1086–1097 (2016).
Wagner, T. A. et al. RNAi construct of a cytochrome P450 gene CYP82D109 blocks an early step in the biosynthesis of hemigossypolone and gossypol in transgenic cotton plants. Phytochemistry 115, 59–69 (2015).
Acknowledgements
The authors thank W.L. Hu, X.Y. Xu, L.Y. Jing, Z.H. Wang, W.R. Liu, and Z.Q. Wang for their assistance with HPLC-MS analyses, and also thanks M.L. Ma for assisting with protein expression. The work was supported by grants from National Key R&D Program of China 2022YFD1400800, the National Natural Science Foundation of China (32388201, 32300330, 32422007), the Talent Plan of Shanghai Branch, Chinese Academy of Sciences (CASSHB-QNPD-2023-015) and the Foundation of Youth Innovation Promotion Association of the Chinese Academy of Sciences to J.-Q.H. (2022273), and also by the Yunnan Revitalization Talent Support Program “Top Team” Project (202305AT350001) to X-Y.C. Moreover, the study was also supported by the Taishan Scholars Program (No. tsqn202312303 to Y.L. and No.tsqn202306323 to B.X.) and the Shanghai Rising-Star Project (No. 24QA2710300 to J.-Q.H. and No. 23QA1410100 to Y.L.), the Shanghai Agricultural Science and Technology Innovation Program (Grant No.T2024204) to J.-Q.H, the National Key Research and Development Program of China (No. 2022YFC2804100 to B.X.), the Shandong Laboratory Program (SYS202205 to B.X.), the Shanghai Pujiang Program (23PJ1415300 to B.X.) and the Winall Hi-tech Seed Co., Ltd. (GMLM2023).
Author information
Authors and Affiliations
Contributions
Two authors share the initials J.-X.L.; they are differentiated as J.-X.L.1 and J.-X.L.2 in the statement below. J.-Q.H., J.-L.L. and X-Y.C. conceived the idea and designed this study; J.-L.L., J.-Q.H., W.-K.W. and G.-B.N. conducted the experiments; J.-X.L.1 performed protein modeling; X.F., Y.-G.S., M.-M.W., Q.-Y.Z., X.-X.G., J.-F.H., L.-Y.M., L.-J.W., J.-X.L.2, S.-S.W., B.X., Y.G., Y.L., and D.W. discussed results and provided advice; J.-L.L., W.-K.W., and G.-B.N. wrote the manuscript; J.-Q.H., X.-Y.C., and C.M. reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Adam Jozwiak, Reuben Peters and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lin, JL., Wu, WK., Nie, GB. et al. A dirigent protein redirects extracellular terpenoid metabolism for defense against biotic challenges. Nat Commun 16, 9270 (2025). https://doi.org/10.1038/s41467-025-64323-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-64323-z
