
Abstract
All-solid-state batteries face critical mechanical challenges, particularly when employing Li-metal, Si-based, or initially anode-free configurations, due to substantial volume changes of the negative electrode within rigid cell architectures during cycling. Such fluctuations generate considerable mechanical stress, which can lead to both bulk and interfacial degradation, posing serious risks to long-term stability. This problem is further amplified in stacked cell configurations, where mechanical integrity is critical. To address this, we investigate metal–organic frameworks as low-strain negative electrodes integrated with the argyrodite Li6PS5Cl0.5Br0.5 solid electrolyte in sulfide-based all-solid-state batteries. Notably, the Co-based metal–organic framework containing a thiophenedicarboxylic acid linker exhibits high structural reversibility, retaining 82% of its capacity with only 1.04% volume change after 700 cycles at 30 °C (1.5 mA cm−2). Operando pressure and displacement analysis further confirm negligible mechanical strain during cell operation. Moreover, a pouch-type full cell maintains stable performance for 50 cycles at 30 °C under a low stack pressure of 5 MPa (0.5 mA cm−2), demonstrating the promise of metal–organic frameworks as low-strain electrodes for durable all-solid-state battery architectures.
Similar content being viewed by others


Introduction
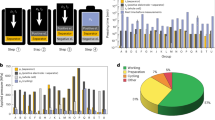
Sulfide-based all-solid-state batteries (ASSBs) have emerged as a promising alternative to traditional liquid electrolyte (LE)-based lithium-ion batteries (LIBs) due to their high energy density and enhanced safety1,2,3,4,5,6. Despite these advantages, the commercialization of ASSBs is hindered by several challenges, particularly, finding negative electrode materials that exhibit high specific capacity, chemical compatibility with sulfide solid electrolytes (SEs), and mechanical stability against volume changes during lithiation/delithiation7,8,9,10. Graphite (Gr), the standard negative electrode material in LIBs, is limited in sulfide-based ASSBs by its relatively low specific capacity (372 mAh g−1) and low interfacial stability with sulfide SEs11,12. While silicon (Si) and Li-metal offer significantly higher specific capacities13,14, they suffer from severe volume expansion, interfacial degradation as well as dendrite formation during cycling15,16,17. Initially, anode-free ASSBs concept incorporating Ag-C interlayer has shown potential18,19, yet they still face problems associated with large volume changes from Li plating/stripping and irreversible Ag dissolution20,21. These mechanical issues, often masked in lab-scale pellet cells, can become increasingly significant at the module or pack level. Therefore, to enable the practical realization of ASSBs, it is crucial to develop new negative electrode materials that offer high specific capacity while maintaining minimal volumetric changes for long-term cycling (Fig. 1).

Schematic representation of the exacerbating effect of mechanical stress generated by the negative electrode in large-format cells. Panels show a comparison of volume changes and strain produced by various negative electrode materials during the lithiation and delithiation processes.
Metal–organic frameworks (MOFs), present a potential solution to these challenges. MOFs, a class of hybrid materials composed of metal ions and organic ligands, have been extensively explored for energy storage applications owing to their mechanical stability, adjustable pore structure, and high specific capacity22,23. Their high degree of customizability allows precise tailoring of pore size, shape, and chemical composition by altering the metal ions and organic ligands used in their synthesis24,25,26,27,28,29,30. These features make MOF materials an ideal choice for low-strain negative electrodes tailored for various applications. For instance, in a study by Chen et al., a ZIF-8 MOF-derived carbon matrix was utilized as a 3D host for Si nanoparticles (SiNPs). This significantly reduced strain compared to conventional SiNP-based electrodes, effectively mitigating the volume changes during cycling in LIBs31. Similarly, Zhang et al. demonstrated that Sn-MOF-derived Sn3(PO4)2 nanocrystal confined in an interlinked carbon network functioned effectively as a negative electrode in both potassium-ion batteries (PIBs) and sodium-ion batteries (SIBs), exhibiting high strain tolerance during potassiation/sodiation and maintaining stable cycling performance32. Despite these advantages, the application of MOFs and their derivatives has largely been limited to LE-based battery systems. Particularly, the direct use of pure MOFs as low-strain materials in sulfide-based ASSBs, along with comprehensive electrochemo-mechanical analysis during the lithiation/delithiation process, remains underexplored. Therefore, detailed electrochemical and structural investigations are imperative to fully harness their potential and establish design protocols for high-performance MOF electrodes in ASSBs.
In this work, we present a systematic screening of various 3d transition metal ions to identify the most promising MOF candidates for negative electrode applications in sulfide-based ASSBs. For these tests, we use two representative organic ligands, heterocyclic thiophenedicarboxylic acid (TPDC) and homocyclic benzenedicarboxylic acid (BDC), as model systems to elucidate the interplay between metal ions and organic ligands on the electrochemical performance of MOF in sulfide-based ASSBs. Our findings reveal that the Co-TPDC-MOF, featuring a thiophene group, exhibits better cyclability and a more reversible discharge/charge process compared to its benzene-based counterpart. We attribute this to the electronegative sulfur atom in the thiophene group facilitates a reversible charging/discharging process by providing favorable Li-ion migration pathways and minimizing Li-ion trapping within its organic matrix. To verify this, we investigate the lithiation/delithiation mechanisms of these MOF through a combination of X-ray absorption spectroscopy (XAS), X-ray photoelectron spectroscopy (XPS), Fourier transform infrared spectroscopy (FT-IR), and density functional theory (DFT) calculations. Furthermore, we perform a comparative mechanical analysis against conventional negative electrode materials (Li, Si, and Gr) via operando electrochemical pressiometry (OEP) and operando displacement measurement (ODM) to examine the near strain-free properties of the reported MOF electrodes during the cell operation. This is further validated by cross-sectional focused ion beam-scanning electron microscope (FIB-SEM), which reveals negligible volume change during the lithiation/delithiation process. Finally, using full cells, comprising the Co-TPDC-MOF, argyrodite Li6PS5Cl0.5Br0.5 (LPSClBr) SE, and LiNbO3-coated NCM811, we assess their electrochemical performance under practical conditions such as high loading capability, operation at 30 °C, and low stack pressure. These combined mechanical strain and mechanistic studies demonstrate the origin of their low mechanical strain properties and establish the groundwork for the practical application of MOF negative electrodes in sulfide-based ASSB systems.
Results
High-throughput screening of 3d transition metal ion-based MOF negative electrodes with TPDC and BDC organic ligand combinations
To investigate the influence of metal centers and organic ligands on the electrochemical performance of MOF electrodes in sulfide-based ASSBs, we conducted a high-throughput material screening of different 3d metal ions combined with TPDC and BDC organic ligands. These MOFs were synthesized via an ultrasonic-assisted method, using tetraethylammonium (TEA) as a deprotonating agent (Fig. 2a). Electrochemical testing over the first 5 cycles was performed with Li-metal as the counter electrode at the current density of 0.3 mA cm−2. Initial coulombic efficiency (ICE) and initial capacity retention (ICR) were established as performance metrics to identify the most viable candidates. In the ICE vs. 1st charge capacity plot, both TPDC- and BDC-based 3d metal ion-MOFs demonstrated a similar linear trend, with Co- and Ni-based MOFs exhibiting the highest ICEs and charge capacity (Fig. 2b, c). Notably, the ICR vs. 5th charge capacity plots revealed that Co-TPDC-MOF and Co-BDC-MOF showed better capacity retention compared to other candidates within their respective groups, indicating their highly reversible charge/discharge behavior (Fig. 2d, e). These findings are further supported by the voltage profiles and compiled electrochemical performance data of various 3d metal ion-MOFs with TPDC and BDC ligands (Supplementary Fig. 1a, b and Supplementary Tables 1–2). Additionally, other heterocyclic and homocyclic ligands-based MOFs were also tested using Co as metal ion, but they exhibited comparatively weaker performance in terms of ICE and ICR compared to Co-TPDC-MOF and Co-BDC-MOF (Supplementary Fig. 2 and Supplementary Table 3).

a Schematic representation of the MOF synthesis process incorporating various 3d metal ions (Co, Ni, Mn, Fe, Cu, and Zn) with TPDC and BDC ligands. Initial coulombic efficiency (ICE) of 3d metal ion-MOFs with TPDC (b) and BDC (c) ligands plotted against the 1st charge capacity in half-cells. Initial capacity retention (ICR) of 3d metal ion-MOFs with TPDC (d) and BDC (e) ligands plotted against the 5th charge capacity in half-cells. All samples were cycled at a current density of 0.3 mA cm−2.
The structural characterization of Co-TPDC-MOF and Co-BDC-MOF
Both Co-TPDC-MOF and Co-BDC-MOF were thus selected as model systems to further investigate the difference in their redox mechanism and electrochemical properties. Figure 3a, b shows the synthesis process and SEM image of Co-TPDC-MOF, revealing individual particles with a randomly arranged trapezoidal shape and a smooth nanosheet structure. Co-BDC-MOF also exhibits a similar polygonal-shaped nanosheet structure with a more densely packed morphology (Supplementary Fig. 3a, b). The energy dispersive X-ray spectroscopy (EDS) elemental mapping of both samples (Fig. 3c and Supplementary Fig. 3c) shows the homogeneous distribution of cobalt metal ions and organic ligand components, i.e., Co, C, O, and S (for Co-TPDC-MOF) throughout the sample surface. The nanosheet architecture of both MOFs offers several advantages, including intimate interfacial contact between active materials and the sulfide SE, short ion diffusion length to the inner bulk electroactive sites, and abundant open metal sites exposure at the surface. These features are anticipated to stimulate the redox activity of the metal center, thereby facilitating Li-ion and charge transport kinetics33,34,35.

a Schematic representation of the synthesis process of Co-TPDC-MOF. SEM image (b) and EDS elemental mapping (c) of Co-TPDC-MOF, showing the uniform distribution of Co, O, C, and S elements. d XPD pattern of Co-TPDC-MOF. e FT-IR spectra of the TPDC ligand and Co-TPDC-MOF. f High-resolution Co 2p XPS spectra of Co-TPDC-MOF. g Nitrogen adsorption and desorption isotherms and the corresponding mesoporous analysis of Co-TPDC-MOF. h Crystal structure of Co-TPDC-MOF along the a-axis, visualized with VESTA54.
The crystal structure of the prepared samples was examined using powder X-ray diffraction (XRD). The XRD patterns of Co-TPDC-MOF and Co-BDC-MOF (Fig. 3d and Supplementary Fig. 4a) align with previously reported MOF electrodes employing the same metal cation and organic ligand36,37. The samples were further analyzed using FT-IR spectroscopy with their corresponding TPDC and BDC ligands to assess the coordination between metal cation and organic ligand during synthesis. As illustrated in Fig. 3e, the FT-IR spectrum shows that the characteristic peaks of TPDC ligand at 1659 (νC=O), 1523, 1414 (νring), 1267 (νC-O) vanish and shift to the dicarboxylate peaks of Co-TPDC-MOF at 1573 and 1363 cm−1, corresponding to νas(COOCo) and νs(COOCo), respectively. A similar transition is observed for the BDC ligand and Co-BDC-MOF, confirming the successful transformation of cobalt metal ions and organic ligands into a coordinated structure (Supplementary Fig. 4b). High resolution Co 2p XPS analysis was also conducted to probe the electronic state of the central metal atoms (Fig. 3f and Supplementary Fig. 4c). For both Co-TPDC-MOF and Co-BDC-MOF, the analysis reveals two characteristic symmetric peaks corresponding to Co 2p1/2 and Co 2p3/2, with an observed binding energy difference of 16.1 eV between the two peaks indicating that the element Co primarily exists in the form of Co2+. For the organic part, the C1s XPS spectrum of Co-TPDC-MOF comprises three peaks at 284.8, 286.4, and 288.7 eV, which can be assigned to C=C/C–C, C–O, and C=O, respectively. The O1s XPS spectrum shows complementary results, consisting of two peaks at 532.2 and 533.6 eV, which can be assigned to O–C=O and O–H (Supplementary Fig. 5a, b). Likewise, a similar carbon and oxygen configuration is observed for Co-BDC-MOF, further proving the successful completion of coordination reaction (Supplementary Fig. 6a, b). Additionally, the microstructure and micropore distribution of the MOF samples were examined through the Brunauer−Emmett−Teller (BET) surface area measurement for N2 adsorption. Co-TPDC-MOF displayed a calculated surface area of 167.69 m2 g−1 (Fig. 3g) whereas Co-BDC-MOF had a surface area of 135.62 m2 g−1 (Supplementary Fig. 4d). Notably, Co-BDC-MOF presents a relatively broader micropore distribution, which could enhance Li-ion accessibility and storage capacity by offering more available sites for ion adsorption. However, difference in pore connectivity, local framework dynamics, and coordination environments may also influence the redox kinetics and diffusion behavior, as reflected in the differing electrochemical performance between the two MOFs. Furthermore, the crystal structure models were constructed based on the experimental results. Co-TPDC-MOF exhibits asymmetric tetradentate (κ1–κ1)‒ (κ1–κ1)‒μ4 coordination with alternating direction of sulfur atom above and below plane, resulting a corrugated sheet structure (Fig. 3h)38,39,40. While the Co-BDC-MOF exhibits planar structure, containing two types of octahedrally coordinated metallic sites (denoted as Co1 and Co2) wherein each Co1 is connected to four µ3‒OH and two O-carboxylate atoms and each Co2 is connected to two µ3‒OH and four O-carboxylate atoms (Supplementary Fig. 4e)41,42.
The electrochemical characterization of the Co-TPDC-MOF and Co-BDC-MOF negative electrodes in the sulfide-based ASSBs
Co-TPDC-MOF and Co-BDC-MOF electrodes were prepared by incorporating a SE, serving as a Li-ion conduction channel in an optimized ratio of 45 wt% (Supplementary Fig. 7). XRD analysis confirmed the presence of crystalline MOF and SE phases without additional peaks, indicating a purely physical mixture of the two components (Supplementary Fig. 8). All half-cell tests were performed with Li-metal as the counter electrode (Fig. 4a). To investigate the redox behavior of the two MOFs, cyclic voltammetry (CV) measurements were conducted within a potential window of 0.01–3.0 V (vs. Li+/Li) at a scan rate of 0.1 mV s−1 under the temperature of 30 °C. As shown in Fig. 4b, the CV curve of Co-TPDC-MOF in the initial cathodic scan exhibits two reduction peaks at 0.7 and 1.1 V that differ from subsequent scans. This behavior can be ascribed to the stepwise Li-ion insertion process, involving gradual interactions with carboxylate groups and aromatic rings. Meanwhile, the anodic scan reveals multiple peaks in the range of 1.0–1.9 V, arising from the Li-ion extraction process, but the peak at 1.0 V vanishes in the subsequent cycles with the emergence of a new anodic peak at 2.6 V. This behavior can be attributed to the restructuring of the MOF frameworks to form reversible passage of Li-ions for the following charging/discharging process. Importantly, all peaks remain consistent after the first cycle, signifying a reversible and stable redox activity. On the contrary, the CV profile of Co-BDC-MOF (Fig. 4c) during the first cathodic and anodic scans shows two redox peaks in the ranges of 0.8–1.1 V and 0.9–2.1 V, respectively, ascribed to the insertion/extraction of Li-ions within the metal centers and organic ligands. Additionally, a new anodic peak above 2.5 V appears in the subsequent anodic scans of Co-BDC-MOF, albeit less pronounced compared to that of Co-TPDC-MOF. In particular, the reduction peaks of Co-BDC-MOF remain unstable after the first cycle, indicating irreversible and unsteady Li storage behavior. Figure 4d, e depict the galvanostatic discharge/charge profiles of the Li-metal half cells with MOF electrodes, which were cycled within a potential range of 0.01–3.0 V at a current density of 0.3 mA cm−2 for the first 5 cycles and at 1.0 mA cm−2 for the subsequent cycles. As shown in Fig. 4d, Co-TPDC-MOF delivers an initial discharge capacity of 1597 mAh g−1 and a reversible charge capacity of 1047 mAh g−1, resulting in an ICE of 65.5%. This relatively small ICE, compared to conventional negative electrodes such as Gr or Si, can be attributed to the combined effects of SEI layer formation and partial Li-ion trapping at active sites. Notably, Co-TPDC-MOF exhibits highly reversible performance with negligible capacity fading during the first 5 cycles and throughout subsequent cycling at higher current density. In contrast, Co-BDC-MOF delivers higher initial discharge and reversible charge capacities of 1833 and 1182 mAh g−1, respectively, corresponding to an ICE of 64.5% (Fig. 4e). However, it suffers significant capacity fading, losing nearly 35% of its initial reversible capacity by the 5th cycle and exhibiting a pronounced capacity drop when transitioning to high current density conditions. To understand this distinction, electrochemical impedance spectroscopy (EIS) measurement of the cycled MOF electrodes was conducted. The Nyquist plots (Fig. 4f and Supplementary Table 4) reveal that Co-TPDC-MOF has a much lower charge transfer resistance (Rct) compared to Co-BDC-MOF after cycling (123.64 vs. 400.05 Ω), suggesting a more facile formation of a stable SEI layer, accompanied by better Li-ion transfer kinetics. Rate capability tests were also conducted at current densities, ranging from 0.3 to 1.0 mA cm−2 (Fig. 4g). Even at the high current density of 1.0 mA cm−2, Co-TPDC-MOF retained a higher capacity than Co-BDC-MOF (696 vs. 606 mAh g−1), demonstrating its enhanced rate performance. Furthermore, a long-term cycling test was performed at 1.0 mA cm−2 after precycling at 0.3 mA cm−2 (Fig. 4h). Co-TPDC-MOF outperformed Co-BDC-MOF in both reversible capacity (710 vs. 479 mAh g−1) and capacity retention (82.4 vs. 70.3%) after 200 cycles. These results suggest that Co-TPDC-MOF exhibits improved reversibility and cyclability, verifying its highly reversible Li-ion storage capability and effective ionic/electronic transport kinetics.

a Schematic illustration of the fabricated sulfide-based ASSB with Li-metal as a counter electrode. CV profiles of Co-TPDC-MOF (b) and Co-BDC-MOF (c) half-cells. Discharge/charge voltage profiles of Co-TPDC-MOF (d) and Co-BDC-MOF (e) at current densities of 1.0 mA cm−2, following 5 precycles at 0.3 mA cm−2 at 30 °C. f EIS spectra of half-cells with MOF electrodes. g Rate capability at varying current densities ranging from 0.3 to 1.0 mA cm-2 at 30 °C. h Long-term cycling performance at a current density of 1.0 mA cm-2 at 30 °C.
To assess the electrochemical reaction kinetics of the two MOFs, CV measurements were carried out over a potential range of 0.01–3 V at varying scan rates, ranging from 0.1 to 1.0 mV s−1 (Supplementary Fig. 9a, b). The redox peak currents (i) undergo continuous shift trends with the increasing scan rate (v). This correlation between the peak current (i) and scan rate (v) can be expressed using the equation below:
Here, b serves as a parameter that characterizes the charge storage kinetics of Li-ion and it can be determined from the slope of the log (i) vs. log (v) plot. In general, when the b value is close to 0.5, the Li-ion storage mechanism follows a diffusion-controlled behavior, while b value close to 1 suggests capacitive-controlled behavior. The computed b values for the primary reduction peaks are 0.833 and 0.671 for Co-TPDC-MOF and Co-BDC-MOF, respectively, indicating a highly capacitive-controlled process for Co-TPDC-MOF compared to Co-BDC-MOF (Supplementary Fig. 9c). While, the b values for the primary oxidation peaks of Co-TPDC-MOF and Co-BDC-MOF are 0.584 and 0.603, respectively, suggesting similar diffusion-dominated Li storage kinetics (Supplementary Fig. 9d). Furthermore, the diffusivity of Li-ions within the active material was evaluated using the galvanostatic intermittent titration technique (GITT) to provide detailed insight into the kinetic factors. The calculated Li-ion diffusion coefficient (DLi) is presented as a function of lithiation/delithiation states (Supplementary Figs. 10, 11 and Supplementary Table 5). Notably, during the first cycle, Co-TPDC-MOF displayed slightly higher diffusivity during lithiation than delithiation. However, in the second cycle, a distinct shift was observed, delithiation diffusivity exceeded that of lithiation, suggesting a transformation in the material’s internal transport characteristics. This evolution in transport behavior highlights a structural activation process occurring during the initial cycle. The first lithiation induces irreversible morphological rearrangements that lead to a more disordered framework in subsequent cycles. As a result, ionic transport becomes increasingly facilitated, particularly during delithiation. Especially, Co-TPDC-MOF undergoes a more pronounced activation process in early cycles compared to Co-BDC-MOF, resulting in improved Li transport pathways and redox kinetics in subsequent cycles.
Investigation of the lithiation/delithiation mechanism of Co-TPDC-MOF and Co-BDC-MOF negative electrodes
To gain a deeper understanding of the Li-ion storage mechanism in Co-MOFs, DFT calculations were conducted43,44 (Supplementary Data 1). The Co-MOFs primarily reduced Co2+ rather than organic ligand when Li-ions are inserted into the MOFs. In the case of Co-TPDC-MOF, besides a small region above the Fermi level that can accept electrons corresponding to the organic ligand, it was observed that cations are dominantly utilized as the redox center (Fig. 5a). Similarly, in Co-BDC-MOF, there are unoccupied states that can accept electrons through cation redox. Concurrently, Li-ions are preferentially stored at electrostatically favorable sites within the Co-MOFs, which are negatively charged. There are two distinctive sites in Co-MOFs: oxygen atoms of CoO6 and organic ligands (Fig. 5b, c). Among them, at the initial stage of lithiation, Li ions preferentially coordinate with the oxygen atoms of the CoO6 units in both Co-TPDC-MOF and Co-BDC-MOF, exhibiting relatively high equilibrium potentials (~1.1 V vs. Li+/Li), as shown in Fig. 5d and Supplementary Table 6. In Co-TPDC-MOF, a broader distribution of calculated potentials is observed compared to Co-BDC-MOF, reflecting the greater structural heterogeneity of the ligand environment. Moreover, some CoO6 sites in Co-TPDC-MOF exhibit lower potentials than those in Co-BDC-MOF, indicating weaker stabilization of Li ions and implying their more facile extraction during delithiation. In contrast, the organic ligand sites correspond to significantly lower potentials (~0.7 V vs. Li+/Li), consistent with the second reduction peak in the CV profiles, suggesting the presence of two distinct Li insertion sites in Co-MOFs. The lower potential response is likely attributable to local structural distortion and partial framework collapse occurring despite the nominally similar coordination environments (Supplementary Fig. 12). This interpretation is further supported by the observation that, after the second cycle, the pristine framework no longer retains its original integrity indicating that the high-potential (~1.1 V vs. Li+/Li) redox centers associated with CoO6 coordination are lost. As a result, Li insertion/extraction becomes reversibly confined to the lower-potential sites, consistent with the stable cycling behavior observed in subsequent electrochemical profiles.

a Density of states of pristine Co-TPDC-MOF (left panel) and Co-BDC-MOF (right panel). The Fermi level is set to 0 eV. Front and side views of probable Li adsorption site of Co-TPDC-MOF and Co-BDC-MOF at b CoO6 and c organic ligand site, visualized with VESTA54. Blue, green, gray, red, yellow, and white balls represent Co, Li, C, O, S, and H atoms, respectively. d Calculated equilibrium potential of Co-TPDC-MOF and Co-BDC-MOF at CoO6 and organic ligand sites. e Ex situ Co K-edge XANES spectra of Co-TPDC-MOF and Co-BDC-MOF at pristine, fully lithiated (0.01 V vs. Li+/Li) and fully delithiated (3.0 V vs. Li+/Li) states after cycling at 0.3 mA cm-2 at 30 °C. f Corresponding FT-EXAFS spectra in R space for selected states of Co-TPDC-MOF and Co-BDC-MOF. g Volumetric change of lithiated bulk Co-TPDC-MOF and Co-BDC-MOF obtained from DFT calculations. Note that the shaded area represents the range of standard deviation for each point.
Then, the lithiation/delithiation mechanism of Co-TPDC-MOF and Co-BDC-MOF was explored by first analyzing the changes in the local electronic environments and valence states of metallic centers via the ex situ XAS analysis. Figure 5e, presents the Co K-edge X-ray absorption near edge structure (XANES) spectra of each MOF at three different states, i.e., pristine, fully lithiated (0.01 V), and fully delithiated (3.0 V) during the first cycle. The results show that the XANES spectra for both MOFs at pristine states closely align with CoO, confirming an average valence state of metal ion center as 2+. However, in the subsequent fully lithiated/delithiated states, the disparity in the degree of absorption energy shift between the two MOFs becomes evident. As illustrated in the half-height edge energy (E0.5) plots depicted in the insets of Fig. 5e, the XANES spectrum of fully lithiated Co-TPDC-MOF nearly converges with the standard Co foil, indicating a near-complete conversion to Co0. In contrast, the E0.5 shift of fully lithiated Co-BDC-MOF stops slightly at a higher energy than the standard Co foil, suggesting a partial conversion to 0 valence states. In the fully delithiated state, the XANES spectra of both MOFs return to higher absorption energy, indicating a progressive recovery of Co2+. Notably, the E0.5 of the fully delithiated Co-TPDC-MOF aligns with its initial state and CoO, signifying the re-conversion to 2+ state. On the contrary, the fully delithiated Co-BDC-MOF does not fully return to its initial state, suggesting an incomplete recovery of metal ion coordination. Corroborating this findings, ex situ Co 2p XPS results for Co-TPDC-MOF demonstrate a reversible behavior with the loss of satellite peaks (characteristic features of Co 2p XPS spectrum for Co metal) in the fully lithiated state, along with a recovery to the original binding energy position in the fully delithated state (Supplementary Fig. 13).
In addition, the extended X-ray absorption fine structure (EXAFS) in R space for the selected lithiation/delithiation states was examined to obtain more insights into the structural change of metallic center. Figure 5f shows FT of the EXAFS results during the first cycle. The EXAFS spectra for both pristine MOFs exhibit a strong peak at 1.53 Å, associated with Co–O interaction, alongside two additional peaks at 2.61 and 3.49 Å, corresponding to CoOx species (0 <x < 1). At the fully lithiated state, a new strong peak appears at ~2.17 Å, associated with Co–Co interactions, accompanied by weaker CoOx bonds. The Co–Co peak intensity is notably higher in Co-TPDC-MOF than in Co-BDC-MOF, indicating more extensive dissociation of Co–O bonds during Li-ion insertion. In the following fully delithated state, the Co–Co peak diminishes with the recovery of Co–O peak. However, the CoOx peaks do not fully return to their original state, likely due to significant disordering of CoOx species. This is consistent with the ex situ XRD analysis of delithiated Co-TPDC-MOF, which shows partial loss of the long-range order (Supplementary Fig. 14). Interestingly, the recovery of the Co–Co peak in the fully delithiated state of Co-TPDC-MOF is more pronounced compared to Co-BDC-MOF, signifying enhanced reversibility and redox activity in Co-TPDC-MOF. This greater reversibility of Co-TPDC-MOF was further examined through DFT calculations. The lattice parameters of the lithiated Co-MOFs were obtained from fully relaxed crystal structures at varying degrees of Li-ion insertion. As illustrated in Fig. 5g, Co-BDC-MOF exhibits significant volumetric expansion with increasing state of charge (SOC), particularly along the b and c axis aligned with the spacing between organic ligands (Supplementary Table 7). This anisotropic expansion suggests elongation of the ligand framework and destabilization of the coordination environment during lithiation. In contrast, Co-TPDC-MOF maintains a nearly zero-strain profile up to 2.0 e⁻/Co without undergoing abrupt structural collapse. Although partial amorphization may eventually occur at higher Li content, the onset of structural disorder in Co-TPDC-MOF requires deeper lithiation compared to Co-BDC-MOF, indicating a delayed and more gradual degradation pathway. This resilience reflects the structural buffering capability of the TPDC ligand. Collectively, these results underscore Co-TPDC-MOF’s intrinsic ability to accommodate Li-ions while maintaining structural integrity over a broad range of lithiation, contributing to its electrochemical reversibility. In contrast, the pronounced and early-onset lattice strain in Co-BDC-MOF leads to irreversible framework collapse and rapid capacity fading. Furthermore, the elastic properties of the two MOFs were evaluated using the Hill approximation (Supplementary Table 8). The calculated bulk and Young’s modulus of Co-TPDC-MOF are significantly lower than those of Co-BDC-MOF, indicating greater mechanical flexibility and tolerance to tensile or compressive stress. These properties underscore Co-TPDC-MOF’s advantage in maintaining structural integrity and ensuring prolonged electrochemical performance.
To study the local electronic environments around organic ligands of two MOF electrodes during electrochemical process, the combination of ex situ XPS and FT-IR analysis was conducted under pristine, fully lithiated (0.01 V), and fully delithiated states (3.0 V) (Fig. 6a, e). As shown in the C 1s XPS spectra (Fig. 6b, f), pristine MOFs exhibit three major deconvolution peaks, corresponding to C=O, C–O, and C=C/C–C bond. At the fully lithiated state, new peaks corresponding to CO–Li and C–Li emerge for both MOF electrodes at ~289.0 eV and ~284.5 eV, respectively, which can be ascribed to the Li-ion interaction at the aromatic ring of organic ligands. The O 1s spectra corroborate this result, showing an increased intensity of the C–O peak relative to the C = O peak (Fig. 6c, g). Meanwhile, an additional interaction at the ligand site of Co-TPDC-MOF could be observed from the S 2p spectrum where a peak shift in S 2p3/2 and S 2p1/2 pair, associated with C–S–C group occur during the lithiation process. This behavior can be attributed to the interaction of Li-ion with the sulfur atom of the thiophene backbone or at the interspace between two sulfur atoms of neighboring ligand species (Supplementary Fig. 15). Upon delithiation, the C 1s spectra of both Co-MOFs return to an original state, but C–Li peak remains, indicating irreversible Li-ion incorporation into the organic ligands. Furthermore, the Li 1s spectra at the fully delithiated states for both electrodes show no significant signal below 53 eV and plasmon-loss features around 60 eV, confirming the absence of Li-metal plating at the interface45. Spectral deconvolution in the lithiated state reveals the presence of SEI-related species, such as Li2S/Li2O, C-Li, and LiCl. Importantly, residual SEI components remain even after delithiation, indicating partial Li-ion trapping within electrochemically inactive SEI phases. Nevertheless, Co-TPDC-MOF displays noticeably lower overall peak intensities than Co-BDC-MOF after delithiation, suggesting a more favorable SEI layer formation and reduced irreversible Li consumption within its framework (Supplementary Fig. 16).

a First cycle voltage profile of Co-TPDC-MOF. Corresponding ex situ XPS C 1s (b) and O 1s (c) spectra and ex situ FT-IR spectra (d) of Co-TPDC-MOF at pristine, fully lithiated (0.01 V vs. Li+/Li) and fully delithiated (3.0 V vs. Li+/Li) states after cycling at 0.3 mA cm−2 at 30 °C. e First cycle voltage profile of Co-BDC-MOF. Ex situ XPS C 1s (f) and O 1s (g) spectra and ex situ FT-IR spectra (h) of Co-BDC-MOF under the same electrochemical conditions, sampled at pristine, fully lithiated (0.01 V vs. Li+/Li) and fully delithiated (3.0 V vs. Li+/Li) states.
The ex situ FT-IR analysis for the selected lithiation/delithiation states was carried out to further verify the change in the chemical structure of organic moieties. As presented in Fig. 6d, h, the FT-IR spectra of both MOF electrodes reveal changes in the asymmetric (va(COO−)) and symmetric (vs(COO−)) vibrations within the 1650–1450 cm−1 range, associated with carbonyl compounds. Additionally, the peak change of the remaining species, including C–H vibration during lithiation/delithiation, further confirms the existence of organic redox reaction occurring at the aromatic rings. Moreover, the FT-IR spectrum of fully delithiated Co-TPDC-MOF almost regains its original shape, demonstrating the reversible redox activity of the thiophene backbone.
Operando pressure and displacement variation analysis of Co-TPDC-MOF ASSB full cell
To evaluate the pressure induced by MOF electrodes during cycling in an ASSB full cell configuration (incorporating a LiNbO3-coated NCM811), we conducted operando electrochemical pressiometry (OEP) following established protocols46,47. The schematic of the OEP setup is depicted in Fig. 7a. For comparison, conventional negative electrodes such as Li-metal and μSi were tested under identical conditions. All full cells were cycled at 0.3 mA cm−2 (30 °C) for 10 cycles during the measurement. The moderate stack pressure of 35 MPa was applied to ensure consistent interfacial contact and ionic conductivity across all test configuration (Supplementary Fig. 17 and Supplementary Table 9). To decouple positive electrode induced-strain, a reference cell employing a zero-strain LTO negative electrode paired with NCM811 was used. As shown in Supplementary Fig. 18, the LTO full cell exhibited a reversible negative pressure change (−0.20 MPa) throughout the cycles, attributed to the contraction of NCM811 upon full delithiation and expansion upon full lithiation. This baseline enabled calculation of the negative electrode-induced pressure (ΔPt) in other full cells: Li-metal (3.29 MPa), μSi (1.48 MPa), Co-BDC-MOF (0.53 MPa), and Co-TPDC-MOF (0.57 MPa) (Fig. 7b and Supplementary Fig. 18). To account for the difference in specific capacity, we normalized the pressure change with respect to the specific capacity (denoted as (ΔP)Q), allowing fair comparison of intrinsic strain generation across electrodes. Figure 7c presents the capacity normalized pressure change as a function of cycle number. The ASSB full cells with Co-TPDC-MOF and Co-BDC-MOF exhibited lower average (ΔP)Q values of 3.34 and 3.36 MPa g (A h)−1, respectively, compared to Li-metal (15.86 MPa g (A h)−1) and μSi (8.97 MPa g (A h)−1). These results were even lower than that of the Gr (4.70 MPa g (A h)−1), highlighting the advantageous volume stability and strain-buffering capabilities of MOF electrodes (Supplementary Fig. 19). Furthermore, we implemented a new analysis called operando displacement measurement (ODM) using a custom-built thickness monitoring setup. Unlike conventional fixed-gap jigs, the system utilizes a spring-loaded pressure jig to maintain constant stack pressure (35 MPa) while allowing for precision real-time detection of vertical cell displacement using a high-resolution digital indicator (0.0005 mm) (Supplementary Fig. 20). Similarly, the LTO||NCM811 reference cell was used to isolate the mechanical response of the positive electrode. The cell exhibited a reversible thickness contraction (ΔZ) of ~−0.5 μm at 100 SOC, which returns to baseline at 100 DOD. While variations below ±0.5 μm fall beneath the resolution threshold of the indicator, the results consistently capture the periodic and directional displacement pattern. Using this baseline, the net negative electrode-specific thickness variation (ΔZt) was calculated as 1.5 μm for Co-TPDC-MOF and 6.5 μm for Li-metal. These results were corroborated by cross-section FIB-SEM analysis of Co-TPDC-MOF electrodes at pristine, fully lithiated, and delithiated states. As shown in Supplementary Fig. 21, the electrode thickness increased from 36.13 μm (pristine) to 37.59 μm (fully lithiated), corresponding to a 4.04% increase (+1.46 μm). This closely aligns with the ODM result of Co-TPDC-MOF, which recorded a thickness change of ~1.5 μm. The small discrepancy may arise from ex situ nature of sample preparation, or from mechanical variables such as spring tension in the ODM setup. Nevertheless, both measurements consistently indicate a similarly low degree of expansion. Upon full delithiation, the thickness decreased to 36.28 μm, indicating an overall thickness change of 0.42% from the pristine state. In addition, the FIB-SEM images reveal clear morphological changes in electrode porosity. In the fully lithiated state, the electrode appeared denser with reduced visible porosity. Upon full delithiation, the porous structure partially recovered, suggesting a minor degree of irreversible densification, but overall, the data supports the conclusion of high mechanical reversibility of Co-TPDC-MOF during cycling.

a Schematic representation of the OEP setup for a sulfide-based ASSB full cell with NCM811 as the positive electrode. b Pressure changes (ΔP) in the NCM811 ASSB full cell using Li-metal, µSi, Co-BDC-MOF and Co-TPDC-MOF negative electrodes as a function of cycle number, measured during cycling at 0.3 mA cm−2 at 30 °C. The dataset labels are indicated directly in the panel. c Corresponding capacity-normalized pressure change (ΔP)Q as a function of cycle number. d Voltage profile of the NCM811 positive electrode and Co-TPDC-MOF negative electrode as a function of areal capacity. e Rate capability of the full cell with the Co-TPDC-MOF negative electrode at 30 °C. f Long-term cycling performance of the Co-TPDC-MOF|LPSClBr|NCM811 ASSB full cell at a current density of 1.5 mA cm−2 at 30 °C. g Cross-sectional SEM images showing volume changes in the Co-TPDC-MOF negative electrode within the ASSB full cell after 700 cycles. h Long-term cycling performance of a high-loading NCM811 ASSB full cell with the Co-TPDC-MOF negative electrode at a current density of 2.0 mA cm−2 at 30 °C. i Cycling performance of the Co-TPDC-MOF-based ASSB pouch-type full cell operated under a low stack pressure (5 MPa) at a current density of 0.5 mA cm−2 at 30 °C.
The long-term cyclability of the full cell with an N/P ratio of 1.1 was evaluated after the rate capability test at a current density of 1.5 mA cm−2 (Fig. 7d–f). Notably, the full cell demonstrated stable cycling performance, achieving 82% capacity retention after 700 cycles. Simultaneous pressure change measurement over the entire cycling period revealed an average ΔP value of 0.10 MPa, signifying minimal volumetric strain during the charge/discharge process. Ex situ cross-sectional SEM analysis of the cycled Co-TPDC-MOF confirmed only negligible thickness changes, with only a 0.49% increase after the initial cycle and 1.04% after 700 cycles, underscoring the mechanically robust, near-zero-strain characteristics of Co-TPDC-MOF (Fig. 7g and Supplementary Fig. 22). Even when paired with a high-loading LiNbO3-coated NCM811 (31.2 mg cm−2), the full cell maintained stable cyclability even at a higher current density of 2.0 mA cm−2. This performance was extended to low stack pressure conditions, with the full cell maintaining stable cycling under 10 MPa stack pressure. The performance improved at elevated temperatures, attributed to the enhanced ionic conductivity of the solid electrolyte (Supplementary Fig. 23 and Supplementary Table 10).
To further validate the practicality of the Co-TPDC-MOF electrode, a pouch-type ASSB full cell was assembled using dry processed LiNbO3-coated NCM811 and LPSClBr separator, followed by compaction via cold isostatic pressing (CIP) at 500 MPa. Notably, the pouch cell delivered a reversible capacity of 2.51 mAh cm−2 at 0.2 mA cm−2 and 1.55 mAh cm−2 at 0.5 mA cm−2, with negligible capacity fading over 50 cycles under a low stack pressure condition of only 5 MPa (30 °C) (Fig. 7i and Supplementary Fig. 24). This corresponds to a high specific energy of ~232 Wh kg−1 based on positive electrode active mass, and 101 Wh kg−1 at the cell level, highlighting the mechanical stability and practical feasibility of Co-TPDC-MOF electrodes in scalable ASSB configurations (Supplementary Tables 11, 12).
Discussion
A comparative investigation of two cobalt-based MOFs, featuring distinct organic ligands (heterocyclic TPDC and a homocyclic BDC), reveals that Co-TPDC-MOF outperforms Co-BDC-MOF in both reversibility and rate capability. This performance is attributed to the favorable metal ion coordination and enhanced π-conjugation provided by the TPDC ligand, which facilitates efficient Li-ion insertion/extraction within the molecular frameworks. Systematic ex situ analyses and DFT calculations further unveiled the advantages of the thiophene backbone in Co-TPDC-MOF, which provides multiplanar ionic pathways. The sulfur atoms in the TPDC ligand matrix promote accelerated Li-ion migration into the inner aromatic ring, creating more efficient Li-ion binding sites compared to the BDC ligand.
Notably, both MOFs have shown to be highly capable of accommodating volume changes during the discharge/charge process, with significantly lower strain generation compared to conventional negative electrode materials such as Li-metal and μSi. As a proof of concept, an NCM811 ASSB full cell incorporating the Co-TPDC-MOF as the negative electrode achieved a capacity retention of 82% (1.5 mA cm−2), with an average pressure variation of just 0.10 MPa and a minimal volume change of 1.04% after 700 cycles. Furthermore, the low-strain property of the Co-TPDC-MOF enabled stable operation of pouch-type ASSB full cell under low stack pressure conditions (5 MPa), reinforcing its practical applicability in sulfide-based ASSBs. These findings provide valuable insights into the fundamental factors governing the electrochemical properties of MOFs and offer critical guidance for the design and optimization of MOF-based negative electrodes for sulfide-based ASSBs.
Methods
Synthesis of Co-TPDC-MOF and Co-BDC-MOF
All chemicals and solvents used in this study were of analytical grade and utilized without any further purification. Initially, a mixture of 20 mL of N,N-Dimethylformamide (DMF, Sigma-Aldrich, 99.8%), 20 mL of deionized water, and 20 mL of methanol (Sigma-Aldrich, ≥99.9%) was prepared in a 100 mL vial. After thorough mixing, the solution was divided into two equal volumes. In one solution, 3.6 mmol of CoCl2·6H2O (Sigma-Aldrich, ACS reagent, ≥98%) was added, while in the other, 8 mmol of NaOH (Sigma-Aldrich, ≥98%) and 4 mmol of 2,5-thiophenedicarboxylic acid (TPDC, Sigma-Aldrich, ≥99%) were introduced. Upon complete dissolution, the solution containing CoCl2·6H2O was slowly added dropwise into the ligand solution. Then, a rapid injection of 0.8 mL of Triethylamine (TEA, Sigma-Aldrich, ≥99.5%) into the mixed solution followed. After stirring for 5 min, the solution underwent continuous ultrasonication for 6 h (40 kHz) within an airtight environment. Finally, the resulting Co-TPDC-MOF was collected via centrifugation, washed with ethanol three times, and dried under vacuum at 110 °C for 12 h. The synthesis of Co-BDC-MOF followed the same procedure, except 4 mmol of TPDC was replaced with 4 mmol of 1,4-benzenedicarboxylic acid (BDC, Sigma-Aldrich, ≥98%). The synthesis of other 3d metal ion MOFs was carried out using the same protocol, with CoCl2·6H2O replaced by equivalent amounts of NiCl2·6H2O (Sigma-Aldrich, ≥98%), MnCl2·4H2O (Sigma-Aldrich, ≥98%), FeCl2·6H2O (Sigma-Aldrich, ≥98%), CuCl2·2H2O (Sigma-Aldrich, ≥98%), or ZnCl2·2H2O (Sigma-Aldrich, ≥98%).
Material characterization
Powder X-ray diffraction (PXRD) measurement of the samples was performed using a Rigaku Ultima IV X-ray diffractometer with Cu Kα (λ = 1.5406 Å) radiation at a low scanning speed of 0.1° per min and step size of 0.02°. X-ray photoelectron spectroscopy (XPS) analyses were conducted using a K-Alpha system (Thermo VG, U.K.) with monochromatic Al Kα X-ray to analyze the chemical composition of sample surface. Fourier transform infrared spectroscopy (FT-IR) spectra were collected via a Bruker Vertex 70 FT-IR spectrometer. N2 sorption isotherms were obtained at 77 K with a Quantachrome Autosorb IQ, and the specific surface area and the corresponding pore size distribution were calculated through Brunauer-Emmett-Teller (BET) and Barrett-Joyner-Halenda (BJH) methods. Surface and cross-sectional morphologies of the electrode samples were analyzed by SEM (7610F-Plus, JEOL Ltd.) at an accelerating voltage of 15 kV with an energy cut-off of 0.5 eV. Elemental mapping was carried out using energy-dispersive X-ray spectroscopy (EDS). Cross-sectional images were obtained from crosscuts using an argon ion beam (Cross Section Polisher (CP) IB-19510, JEOL). For ex situ analyses, samples were harvested in an argon-filled glovebox and transferred in airtight containers to prevent exposure to ambient air. Ex situ Co K-edge X-ray absorption spectra of electrode samples at different lithiation/delithiation states were collected in both transmission and fluorescence mode at beamline BL1D KIST-PAL of Pohang Light Source (PLS-II), using a Si (111) double-crystal monochromator in the 3.0 GeV storage ring, with a ring current of 252.12 mA. A linear pre-edge background was fitted and subtracted, and spectra was normalized by scaling the post-edge region to a unit edge step. Energy calibration was performed using the first-derivative maximum of a simultaneously measured Co foil reference. The normalized spectra were then background-subtracted and Fourier transformed into R space by standard procedures in ATHENA (Demeter package).
Computational details
Density functional theory (DFT) calculations were conducted using the Vienna ab initio simulation package (VASP)48, incorporating the effects of spin-polarization. We employed the Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation49. To precisely address the electron-electron interactions, we used the Dudarev approach for the Hubbard model50, assigning a Hubbard-U parameter of 3.32 eV to the 3d electrons of Co element51. Additionally, to account for the van der Waals (vdW) dispersion energy corrections, we integrated the DFT-D3 method into our calculations52. We set a plane-wave cutoff energy at 520 eV and arranged the k-point grids to maintain an actual spacing of 0.04 Å−1. Notably, density of states was estimated by the increased k-point grids with an actual spacing of 0.02 Å−1. The structural optimization was carried out until the forces between atoms reached a threshold below 0.02 eV/Å. The initial crystal structures of pristine Co-TPDC-MOF and Co-BDC-MOF were adopted from reported structures in the literatures33,38, chosen based on close agreement with our experimental XRD data. To reflect the actual composition of our synthesized materials, the metal centers in the selected reference structures were substituted with cobalt (Co). These modified structures were then used as the starting point for geometric optimization. To obtain the model system of the lithiated Co-MOFs, we identified the Li adsorption site by estimating the local minima of the electrostatic potential in both the pristine and lithiated Co-MOFs. Initially, we conducted a full relaxation of all probable Li sites, allowing freedom of lattice parameters, with one Li atom per unit cell. Subsequently, the model system exhibiting the lowest energy was selected as the starting system for the preparation of lithiated Co-MOFs for subsequent Li adsorption. The maximum quantity of Li atoms incorporated into the lithiated Co-MOFs was determined based on the full capacity of Co2+/Co0 redox. The calculated equilibrium potential of lithiated MOF (V) was calculated as follows,
E(Lix1MOF) and E(Lix2MOF) are the DFT calculated energies of lithiated Co-MOF with different lithium contents or pristine Co-MOF (x2 = 0). It is noted that E(Li) was defined as the total energy per atom of DFT-optimized body-centered cubic (bcc) lithium metal53.
Cell assembly and electrochemical characterizations
Both Co-TPDC-MOF and Co-BDC-MOF electrodes used in this study consist of the synthesized MOF active material, Li6PS5Cl0.5Br0.5 (LPSClBr, supplied by Hyundai Motor Company), carbon nanofiber (CNF, Sigma-Aldrich, ≥98%), and nitrile butadiene rubber (NBR, supplied by Hyundai Motor Company) binder in a composition of 45/50/4/1. These components were mixed in dibromomethane (DBM, Sigma-Aldrich, ≥99%) solvent using a Thinky mixer (Thinky USA) to create a slurry mixture (~70 wt% solid content). The prepared slurry was cast onto Ni foil (10 μm thickness, Alfa Aesar, 99.9%) using a doctor blade and then vacuum dried at 60 °C for 2 h. The prepared electrodes were punched into discs with diameters of 12.9 mm, and the mass loading of the active materials was 2.0–2.5 mg cm–2. To fabricate a half-cell, 150 mg of LPSClBr powder was pressed in a polyether ether ketone (PEEK) die (12.9 mm in diameter) under a pressure of 100 MPa. Then the punched electrode was placed on one side of the SE pellet and pressed together at a pressure of 430 MPa. A punched Li-metal foil (12.9 mm in diameter with 200 μm thickness, Honjo Metal, Japan) was inserted on the other side of the pellet and used as a counter electrode. Finally, a stack pressure of 35 MPa was applied to the cell, maintained by tightening the screws of a stainless-steel framework. For the positive electrodes, LiNbO3-coated NCM811, LPSClBr, and Super C65 carbon (TIMCAL) with a mass ratio of 70/30/3 were utilized. The mass loading of the active material was 16.3 mg cm–2. The full cell was fabricated by placing the electrodes on opposite sides of the SE pellet and pressed at 430 MPa, with the stack pressure of 35 MPa. The pouch-type ASSB full cell was assembled by stacking a dry-processed LiNbO3-coated NCM811 positive electrode (AM/SE/CA/B—66/31/3/0.5) with an areal loading of 24.0 mg cm−2, an LPSClBr separator film (0.1 wt% PTFE, 130 μm thick), and a Co-TPDC-MOF negative electrode (casted on Ni foil) with an areal loading of 6.0 mg cm−2, followed by compaction via CIP at 500 MPa for 3 min. The N/P ratio was 1.1. During electrochemical testing, a constant stack pressure of 5 MPa was maintained using a custom-built, spring-loaded uniaxial pressure jig. The entire cell assembly was carried out in an argon-filled glove box. The electrochemical performance and GITT measurements were assessed using a battery cycler (WBCS 3000, WonATech) in a chamber set at 30 ± 1 °C, under the constant current-constant voltage (CC–CV) procedure. The data were recorded at a sampling rate of 1 Hz. The current density conditions for all half/full cell tests are specified in the legends of individual figures. Rate capability tests were conducted at symmetric discharge/charge rates, and to verify capacity recovery, the tests concluded with a return to 0.3 mA cm–2. The reproducibility of each electrochemical measurement was verified by evaluating two cells simultaneously. CV and EIS measurements were performed using an electrochemical workstation (ZIVE MP1, WonATech). For CV, the scan rate of 0.2 mV s−1 was used. EIS was conducted in potentiostatic mode with a 10 mV AC signal over a frequency range of 10−2–105 Hz, with 10 data points collected per frequency decade. Prior to measurements, the cells were stabilized for 1 h at open-circuit voltage to ensure quasi-stationary conditions. For operando electrochemical pressiometry (OEP), a pressure sensor with a resolution of 0.1 kg (load cell, BONGSHIN) was placed in the pressurized cell, as depicted in Fig. 7a. The pressure changes during the charge/discharge process were monitored using in-house software. The capacity-normalized pressure change difference (ΔP)Q, was calculated by the following equation: (ΔP)Q = (ΔP)/Qdischarge where Qdischarge is the discharge capacity at a corresponding cycle. The μSi and Gr used for comparison in OEP analysis were fabricated using the same electrode composition as the MOF electrodes, with active material mass loading of 9.30 and 1.24 mg cm–2, respectively. For operando displacement measurement (ODM), a digital indicator (Mitutoyo Corp) was integrated with a spring-loaded jig to monitor real-time thickness change of the full cell during cycling.
Data availability
All data are available in the main text or the supplementary information. Source data are provided with this paper.
References
Wang, S. et al. Lithium argyrodite as solid electrolyte and cathode precursor for solid-state batteries with long cycle life. Adv. Energy Mater. 11, 2101370 (2021).
Zhang, Q. et al. Sulfide-based solid-state electrolytes: synthesis, stability, and potential for all-solid-state batteries. Adv. Mater. 31, 1901131 (2019).
Wang, C. et al. All-solid-state lithium batteries enabled by sulfide electrolytes: from fundamental research to practical engineering design. Energy Environ. Sci. 14, 2577–2619 (2021).
Zhang, J. et al. Challenges and strategies of low-pressure all-solid-state batteries. Adv. Mater. 37, 2413499 (2024).
Zhong, Y. et al. Protecting lithium metal anodes in solid-state batteries. Electrochem. Energy Rev. 7, 30 (2024).
Kim, K. H. et al. Near-strain-free anode architecture enabled by interfacial diffusion creep for initial-anode-free quasi-solid-state batteries. Nat. Commun. 15, 3586 (2024).
Janek, J. & Zeier, W. G. Challenges in speeding up solid-state battery development. Nat. Energy 8, 230–240 (2023).
Oh, P. et al. Development of high-energy anodes for all-solid-state lithium batteries based on sulfide electrolytes. Angew. Chem. Int. Ed. 61, e202201249 (2022).
Lim, H.-D. et al. A review of challenges and issues concerning interfaces for all-solid-state batteries. Energy Storage Mater. 25, 224–250 (2020).
Kerman, K., Luntz, A., Viswanathan, V., Chiang, Y.-M. & Chen, Z. Review—practical challenges hindering the development of solid state Li ion batteries. J. Electrochem. Soc. 164, A1731–A1744 (2017).
Höltschi, L. et al. Performance-limiting factors of graphite in sulfide-based all-solid-state lithium-ion batteries. Electrochim. Acta 389, 138735 (2021).
Davis, A. L. et al. Rate Limitations in composite solid-state battery electrodes: revealing heterogeneity with operando microscopy. ACS Energy Lett. 6, 2993–3003 (2021).
Tan, D. H. S. et al. Carbon-free high-loading silicon anodes enabled by sulfide solid electrolytes. Science 373, 1494–1499 (2021).
Cao, D. et al. Processing strategies to improve cell-level energy density of metal sulfide electrolyte-based all-solid-state Li metal batteries and beyond. ACS Energy Lett. 5, 3468–3489 (2020).
Huo, H. et al. Chemo-mechanical failure mechanisms of the silicon anode in solid-state batteries. Nat. Mater. 23, 543–551 (2024).
Tong, Z., Wang, S., Liao, Y. K., Hu, S. F. & Liu, R. Interface Between solid-state electrolytes and Li-metal anodes: issues, materials, and processing routes. ACS Appl. Mater. Interfaces 12, 47181–47196 (2020).
Zhan, X. et al. Challenges and opportunities towards silicon-based all-solid-state batteries. Energy Storage Mater. 61, 102875 (2023).
Lee, Y.-G. et al. High-energy long-cycling all-solid-state lithium metal batteries enabled by silver–carbon composite anodes. Nat. Energy 5, 299–308 (2020).
Kim, J.-S. et al. Surface engineering of inorganic solid-state electrolytes via interlayers strategy for developing long-cycling quasi-all-solid-state lithium batteries. Nat. Commun. 14, 782 (2023).
Han, S. Y. et al. Stress evolution during cycling of alloy-anode solid-state batteries. Joule 5, 2450–2465 (2021).
Wang, J.-C., Wang, P.-F. & Yi, T. Challenges and optimization strategies at the interface between sulfide solid electrolyte and lithium anode. Energy Storage Mater. 62, 102958 (2023).
Wang, L. et al. Metal–organic frameworks for energy storage: batteries and supercapacitors. Coord. Chem. Rev. 307, 361–381 (2016).
Lee, H. H. et al. Coordination polymers for high-capacity Li-ion batteries: metal-dependent solid-state reversibility. ACS Appl. Mater. Interfaces 10, 22110–22118 (2018).
Li, S. et al. Design principles and direct applications of cobalt-based metal-organic frameworks for electrochemical energy storage. Coord. Chem. Rev. 438, 213872 (2021).
Chulgi, N. H., Crom, A. B., Feldblyum, J. I. & Lukatskaya, M. R. Metal-organic frameworks for fast electrochemical energy storage: mechanisms and opportunities. Chem 9, 798–822 (2023).
Ngue, C.-M., Baskoro, F., Wong, H. Q., Yen, H. & Leung, M.-F. Co- and Ni-based electroactive metal–organic frameworks for stable lithium storage: electrochemical and charge-storage behavior in response to different metal centers. Cryst. Growth Des. 22, 5872–5882 (2022).
Li, C., Shen, J., Wu, K. & Yang, N. Metal centers and organic ligands determine electrochemistry of metal–organic frameworks. Small 18, 2106607 (2022).
Ye, Z., Jiang, Y., Li, L., Wu, F. & Chen, R. Rational design of MOF-based materials for next-generation rechargeable batteries. Nano Micro Lett. 13, 203 (2021).
Wu, H. B. & Lou, X. W. (David). Metal-organic frameworks and their derived materials for electrochemical energy storage and conversion: promises and challenges. Sci. Adv. 3, eaap9252 (2017).
Wang, L., Zhu, Y., Du, C., Ma, X. & Cao, C. Advances and challenges in metal–organic framework derived porous materials for batteries and electrocatalysis. J. Mater. Chem. A 8, 24895–24919 (2020).
Chen, B. et al. Zero-Strain high-capacity silicon/carbon anode enabled by a MOF-derived space-confined single-atom catalytic strategy for lithium-ion batteries. Adv. Mater. 34, 2200894 (2022).
Jiang, H. et al. Stress-dispersed superstructure of Sn3(PO4)2@PC derived from programmable assembly of metal–organic framework as long-life potassium/sodium-ion batteries anodes. Adv. Sci. 10, 2206587 (2023).
Zhao, S. et al. Ultrathin metal–organic framework nanosheets for electrocatalytic oxygen evolution. Nat. Energy 1, 16184 (2016).
Xiao, X. et al. Ultrathin two-dimensional nanosheet metal-organic frameworks with high-density ligand active sites for advanced lithium-ion capacitors. Nano Energy 103, 107797 (2022).
Song, E. et al. Two-dimensional bimetallic cobalt-copper metal organic framework for improved desalination performance of capacitive deionization. J. Ind. Eng. Chem. 123, 116–124 (2023).
Ning, Y., Lou, X., Li, C., Hu, X. & Hu, B. Ultrathin cobalt-based metal-organic framework nanosheets with both metal and ligand redox activities for superior lithium storage. Chem. Eur. J. 23, 15984–15990 (2017).
Li, C. et al. Ultrathin manganese-based metal–organic framework nanosheets: low-cost and energy-dense lithium storage anodes with the coexistence of metal and ligand redox activities. ACS Appl. Mater. Interfaces 9, 29829–29838 (2017).
Wang, C.-P. et al. Self-optimized metal–organic framework electrocatalysts with structural stability and high current tolerance for water oxidation. ACS Catal. 11, 7132–7143 (2021).
Demessence, A., Rogez, G., Welter, R. & Rabu, P. Structure and magnetic properties of a new cobalt (II) thiophenedicarboxylate coordination polymer showing unprecedented coordination. Inorg. Chem. 46, 3423–3425 (2007).
Eddaoudi, M. et al. Geometric requirements and examples of important structures in the assembly of square building blocks. PNAS 99, 4900–4904 (2002).
Mesbah, A. et al. From hydrated Ni3(OH)2(C8H4O4)2(H2O)4 to anhydrous Ni2(OH)2(C8H4O4): impact of structural transformations on magnetic properties. Inorg. Chem. 53, 872–881 (2014).
Sibille, R. et al. Magnetic measurements and neutron diffraction study of the layered hybrid compounds Mn(C8H4O4)(H2O)2 and Mn2(OH)2(C8H4O4). J. Solid State Chem. 186, 134–141 (2012).
Gaber, S. et al. MOFite: a high-density lithiophilic and scalable metal–organic framework anode for rechargeable lithium-ion battery. Angew. Chem. Int. Ed. 63, e202409256 (2024).
Chu, J. et al. Electronic band structure engineering of π-d conjugated metal-organic framework for sodium organic batteries. Nat. Commun. 16, 3549 (2025).
Alt, C. D. et al. Quantifying multiphase SEI growth in sulfide solid electrolytes. Joule 8, 2755–2776 (2024).
Lim, H. et al. Operando electrochemical pressiometry probing interfacial evolution of electrodeposited thin lithium metal anodes for all-solid-state batteries. Energy Storage Mater. 50, 543–553 (2022).
Jun, S. et al. Operando differential electrochemical pressiometry for probing electrochemo-mechanics in all-solid-state batteries. Adv. Funct. Mater. 30, 2002535 (2020).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J. & Sutton, A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA+U study. Phys. Rev. B 57, 1505–1509 (1998).
Ong, S. P. et al. Python Materials Genomics (pymatgen): a robust, open-source python library for materials analysis. Comput. Mater. Sci. 68, 314–319 (2013).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, S. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Urban, A., Seo, D.-H. & Ceder, G. Computational understanding of Li-ion batteries. npj Comput. Mater. 2, 16002 (2016).
Momma, K. & Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44, 1272–1276 (2011).
Acknowledgements
This work was mainly supported by Hyundai Motor Company. J.H.Park acknowledges the support by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (RS-2024-00354154,RS-2023-00302697).
Author information
Authors and Affiliations
Contributions
M.R., S.O.P. conceptualized and designed the overall experiment, conducted data analysis, and wrote the manuscript. M.R. performed all electrochemical experiments and conducted ex situ spectroscopy and operando pressure/displacement measurements. S.L. and H.L. carried out MOF synthesis and morphological characterizations. J.H.L., G.L., K.Y.B., J.Y.K., and Y.K. contributed to the analysis and assisted in interpreting experimental results. S.O.P. performed the density functional theory calculations on the Li-ion storage mechanism of MOF electrodes. J.H.P., K.K., and S.S. provided supervision for the research. All the authors engaged in discussions regarding the results presented in the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Bingwen Hu and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ryu, M., Park, SO., Lim, S. et al. Low-strain metal–organic framework negative electrode for stable all-solid-state batteries. Nat Commun 16, 9722 (2025). https://doi.org/10.1038/s41467-025-64711-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-64711-5
