
Abstract
Modeling long-range DNA dependencies is crucial for understanding genome structure and function across diverse biological contexts. However, effectively capturing these dependencies, which may span millions of base pairs in tasks such as three-dimensional (3D) chromatin folding prediction, remains a major challenge. A comprehensive benchmark suite for evaluating tasks that rely on long-range dependencies is notably absent. To address this gap, we introduce DNALONGBENCH, a benchmark dataset covering five key genomics tasks with long-range dependencies up to 1 million base pairs: enhancer-target gene interaction, expression quantitative trait loci, 3D genome organization, regulatory sequence activity, and transcription initiation signals. We assess DNALONGBENCH using five methods: a task-specific expert model, a convolutional neural network (CNN)-based model, and three fine-tuned DNA foundation models – HyenaDNA, Caduceus-Ph, and Caduceus-PS. We envision DNALONGBENCH as a standardized resource to enable comprehensive comparisons and rigorous evaluations of emerging DNA sequence-based deep learning models that account for long-range dependencies.
Similar content being viewed by others


Introduction
Genomic DNA sequences are the blueprint of life, guiding the development of cellular complexity. Although protein-coding DNA sequences encode diverse biochemical functions within organisms, most eukaryotic genomes consist predominantly of non-coding sequences interspersed with protein-coding regions. These non-coding sequences contain a variety of regulatory elements, such as promoters, enhancers, non-coding RNAs, and other functional elements, which orchestrate when and where genes are activated or silenced. Over the past two decades, large-scale functional genomics projects, such as ENCODE1, have cataloged extensive collections of putative non-coding regulatory elements in the human genome. However, our understanding of how these elements regulate gene expression remains limited. A key challenge is that genomes are dynamically folded into multi-scale 3D structures within the nucleus, leading to widespread physical DNA-DNA interactions, even between regions located megabases apart2,3,4. Determining which of these interactions are functionally relevant across diverse biological contexts requires significant experimental effort.
The increasing availability of genomic data, such as ChIP-seq5, ATAC-seq6, and Hi-C and its derivatives7, has spurred the development of supervised deep learning methods that show great promise in systematically delineating sequence-to-function relationships. For example, convolutional neural networks (CNNs) and transformer-based methods have proven effective for characterizing regulatory elements8,9,10,11, predicting spatial proximity between genomic loci12,13, and predicting gene expressions from local sequence contexts14. Despite these advances, capturing dependencies across very long distal DNA elements remains a major computational challenge due to both the scarcity of experimental data and the difficulty of modeling long-range sequence dependencies15.
Recently, large language models have revolutionized the field of natural language processing, demonstrating remarkable capabilities across a wide range of applications16,17,18,19. These models leverage self-supervised learning to capture complex patterns from vast amounts of unlabeled text data, followed by fine-tuning for specific tasks. Recognizing structural similarities between DNA sequences and natural language20, several DNA foundation models have emerged21,22,23,24,25,26. However, their utility in addressing meaningful biological questions remains a topic of debate, leaving a critical question unsolved: Could foundation models pre-trained on genomic DNA sequences offer a new paradigm shift in understanding the interactions between regulatory elements and genes? Answering this question requires robust benchmark datasets to evaluate their performance, identify limitations, and guide future improvements. Yet, most existing DNA foundation models have only been evaluated on prediction tasks involving sequences up to a few thousand base pairs, such as regulatory element identification or local gene expression prediction26,27,28,29,30. Their potential for modeling long-range interactions in diverse biological contexts has not been well evaluated.
Benchmark datasets specifically designed to assess the ability of DNA foundation models to capture long-range dependencies remain limited. Most existing benchmarks focus on short-range tasks (spanning thousands of base pairs) and binary classification. To date, BEND27 and the Genomics Long-range Benchmark (LRB)30 are the only two benchmark datasets that include long-range genomic DNA prediction tasks. BEND comprises two long-range tasks: enhancer annotation and gene finding, both of which involve classifying regulatory elements. LRB, adapted from the Enformer14 paper, curated three datasets focused on gene expression prediction and variant effects on expression. However, both are limited in scope: they emphasize regulatory element identification or gene expression prediction while overlooking other critical long-range tasks. For example, neither includes structure-related tasks requiring ultra-long sequences, such as contact map prediction or enhancer-target gene prediction. Furthermore, they lack base-pair-resolution regression tasks for quantitative assays. As a result, a comprehensive benchmark suite covering a broader range of tasks dependent on long-range DNA interactions remains absent.
Here, we introduce DNALONGBENCH, the largest collection to date of biologically meaningful long-range genomic DNA prediction tasks. DNALONGBENCH comprises five different tasks and datasets spanning critical aspects of gene regulation across multiple length scales. A comparison of existing benchmarks with DNALONGBENCH is shown in Table 1. Our contributions are threefold:
-
We introduce DNALONGBENCH, a benchmark for long-range DNA prediction tasks spanning up to 1 million base pairs (bp) across five distinct tasks. To our knowledge, DNALONGBENCH is the most comprehensive benchmark specifically designed for long-range DNA prediction to date.
-
We evaluate DNALONGBENCH using three representative models, demonstrating that while DNA foundation models capture long-range dependencies to some extent, expert models consistently outperform them across all tasks.
-
We show that model performance varies substantially across tasks, highlighting the diverse challenges posed by DNALONGBENCH and revealing differences in task difficulty.
We envision DNALONGBENCH as a valuable resource for evaluating DNA foundation models, with particular emphasis on their ability to model long-range genomic interactions.
Results
Proposed dataset: DNALONGBENCH
The selection of suitable long-range DNA prediction tasks for DNALONGBENCH is crucial to ensure diversity, comprehensiveness, and rigor. To achieve this, we established the following criteria to guide our task selection process.
-
Biological significance: Tasks should be realistic and biologically significant, addressing genomics problems important for understanding genome structure and function.
-
Long-range dependencies: Tasks should require modeling long input contexts spanning hundreds of kilobase pairs or more.
-
Task difficulty: Tasks should pose significant challenges for current models.
-
Task diversity: Tasks should be as diverse as possible, spanning various length scales and including different task types such as classification and regression. This diversity also includes task dimensionality (1D or 2D) and granularity (binned, nucleotide-wide, or sequence-wide).
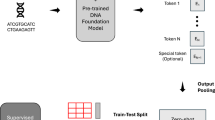
As a result, we selected five long-range DNA prediction tasks, each covering different aspects of important regulatory elements and biological processes within a cell, as illustrated in Fig. 1. An overview of our dataset is presented in Table 2. The input sequences for all tasks are provided in BED format, which lists the genome coordinates of the sequences. This format allows flexible adjustment of the flanking context without requiring reprocessing. The selected tasks are described in detail in “Methods”. Additional details on data processing, data access, and data license are provided in Supplementary Information.

The tasks in DNALONGBENCH include contact map prediction, regulatory sequence activity, enhancer-target gene linking, transcription initiation, and eQTL mapping, covering key layers of chromatin architecture and gene regulation that depend on long-range DNA interactions.
Benchmarking experiments
In this section, we conduct a comprehensive performance comparison by evaluating three distinct types of models: a lightweight CNN, existing expert models that have demonstrated state-of-the-art results, and two types of recent DNA foundation models—HyeynaDNA24 and Caduceus25—distinguished by their support for reverse complement DNA during the training process.
Representative models
We explore the performance of the following three types of models:
-
(1)
CNN: We evaluate the lightweight convolutional neural network31, known for its simplicity and robust performance in various DNA-related tasks. For classification tasks, we trained a three-layer CNN using cross-entropy loss. For contact map prediction, we designed a CNN combining 1D and 2D convolutional layers, trained with mean squared error (MSE) loss. For the regulatory sequence activity and transcription initiation signal prediction tasks, we used CNNs trained with Poisson loss and MSE loss, respectively.
-
(2)
Expert Model: We assess the current state-of-the-art specialized models for each specific long-range DNA prediction task, collectively referred to as the expert model. Specifically, we use:
-
(3)
DNA Foundation Model: We selected three long-range DNA foundation models—HyenaDNA (medium-450k)24 and Caduceus (Ph and PS)25—for evaluation, as they are published works specifically designed for long-range DNA prediction tasks. For the eQTL task, we extracted last-layer hidden representations from both the reference and allele sequences, averaged and concatenated them, and applied a binary classification layer to predict whether the variant was positive. For the remaining tasks, we fed the DNA sequences into the DNA foundation model to obtain feature vectors, then applied linear layers to predict logits at different resolutions.
More detailed model implementations for each task are provided in the Supplementary Information.
Expert models achieve the highest scores on all tasks
We summarize our evaluation results into five tables for each task, respectively, as shown in Tables 3–7. For instance, Table 3 shows the AUROC and AUPR metrics for the enhancer-target gene prediction task, with additional results in Table S1 and Table S2. Table 4 and Table S3 summarize stratum-adjusted correlation coefficient and Pearson correlation for the contact map prediction task for five primary cell lines, with results for four additional cell types shown in Table S4. Figure 2 and Fig. S1 demonstrate examples of contact maps predicted by different methods and ground truth. Table 7 and Table S5 show the AUROC and AUPRC for the eQTL prediction task. In general, we observed that highly parameterized and specialized expert models consistently outperform DNA foundation models. Notably, the advantages of these expert models appear greater in regression tasks such as contact map prediction and transcription initiation signal prediction than in classification task (e.g., enhancer-target gene prediction). For instance, the expert model Puffin achieves an average score of 0.733 on the transcription initiation signal prediction task (TISP), significantly surpassing CNN (0.042), HyenaDNA (0.132), Caduceus-Ph (0.109), and Caduceus-PS (0.108).

The columns show contact maps predicted by HyenaDNA, Caduceus, and Akita model, alongside the ground truth contact map for two genomic regions: a chr6:145,205,248–145,614,848 and b chr3:139,341,824–139,751,424. Colors represent the intensity of contact frequency between paired loci. Pearson correlation coefficient (PCC) and stratum-adjusted correlation coefficient (SCC) metrics are shown beneath each contact map to indicate prediction performance relative to the ground truth. Source data are provided as a Source Data file.
This disparity may stem from the challenge posed by multi-channel regression on long DNA contexts, which makes fine-tuning of DNA foundation models less stable and less capable of capturing sparse real-valued signals. We acknowledge that these expert models are specially designed for their respective tasks, and that some—such as Enformer—have more parameters than HyenaDNA and Caduceus, thereby serving as both strong baselines and potential upper bounds for all tested models. Overall, these observations confirm the Expert Model’s superior ability to capture long-range dependencies, a capability where CNN falls short and DNA foundation models demonstrate reasonable performance in certain tasks.
The contact map prediction presents greater challenges
Unlike the other four tasks, where the Expert Model or DNA foundation models achieve reasonable performance, the contact map prediction task proves significantly more difficult. The highest stratum-adjusted correlation coefficient achieved in this task is 0.233 by the Expert Model (Akita), indicating only a moderate positive correlation. Although contact map prediction is crucial for understanding 3D genome structure, it has received less attention in previous benchmarks, which focused primarily on 1D prediction tasks. This highlights both the difficulty of modeling long-range genomic interactions and the varying levels of complexity across tasks in DNALONGBENCH.
Longer contexts improve model performance
To investigate whether the tasks in our benchmark require long contexts to achieve strong results, we performed ablation studies. This was done by either using varying context lengths or shuffling the central proportion of the input sequence, with results reported in Tables S6-S11. For instance, for the contact map prediction task, we chose Caduceus-Ph for ablation since it showed the highest SCC among the DNA foundation models, and evaluated its performance with input sizes of 409,600, 307,200, and 204,800 bp, corresponding to 200, 150, and 100 bins, respectively. Our results show that model performance increases as context length increases. Similar trends are observed in the other tasks as well, suggesting the model benefits from longer contexts.
Further analysis of DNALONGBENCH evaluations
In this section, we provide further analysis to gain insight into how long-range dependencies are captured in our proposed DNALONGBENCH.
Case Study: Can long-range dependency be captured?
To intuitively demonstrate the presence of extensive long-range dependencies across millions of base pairs and their capture by machine learning methods, we present two examples in Fig. 2 and more examples in Fig. S1. Specifically, in Fig. 2a, b, we visualize the contact maps predicted by HyenaDNA, Caduceus-Ph, and the Expert Model (Akita), alongside the ground truth contact maps for two genomic regions spanning around 400 kb. From these contact maps, we observe the presence of large-scale domains (e.g., blocks in the contact map) and long-range interactions (e.g., off-diagonal dots in the contact map) spanning over 300 kb. Notably, the contact maps predicted by Akita align more closely with the ground truth, confirming its superior ability to capture long-range interactions. In contrast, DNA foundation models show a limited capacity to predict domain structures. This is particularly evident in Fig. 2b, where only Akita accurately predicts the three blocks. These examples highlight DNALONGBENCH’s value in evaluating models for capturing long-range genome structure and function, and provide a foundation for future developments in DNA foundation models.
Base pair-resolution prediction of transcription initiation signal
We visualized the transcription initiation signals predicted by different models for one of the test chromosomes, chromosome 8 (Fig. 3). Predictions from the Expert model Puffin-D closely align with the ground truth, accurately capturing peaks in transcription initiation signal intensity across both large and small genomic regions. In contrast, DNA foundation models tend to underpredict signal intensities or miss certain peaks. In the zoomed-in view (right side of the figure), Puffin-D continues to align well with the ground truth, demonstrating strong performance even at high resolution. By contrast, the DNA foundation models show less precise and broader signals. These findings suggest that base pair-resolution regression tasks remain challenging for current DNA foundation models.

The genomic track on the left displays the ground truth signals (top) alongside predictions from Puffin-D, HyenaDNA, and the two Caduceus models. The X-axis represents genomic coordinates, while the Y-axis indicates signal density. A zoomed-in view of a 1000 bp region centered at the TSS of the gene ZC2HC1A is shown on the right. Source data are provided as a Source Data file.
Discussion
In this paper, we introduce DNALONGBENCH, a benchmark suite comprising five important genomics tasks involving long-range dependencies: enhancer-target gene interaction, eQTL, 3D genome organization, regulatory sequence activity, and transcription initiation signals. We evaluated three baseline methods: a task-specific expert model, a fully supervised CNN-based model, and three fine-tuned DNA foundation models, HyenaDNA, Caduceus-Ph, and Caduceus-PS. The benchmarking results consistently showed that expert models achieved the highest scores across all tasks. Additionally, our analysis revealed that long-range dependencies could be captured across hundreds of thousands of base pairs, underscoring the importance of context length for downstream performance. However, the results also highlight that current DNA foundation models are less effective than expert models in capturing long-range dependencies. It is important to note that each expert model was specifically designed and trained for its respective task. In contrast, DNA foundation models are intended as a “one-to-all” general-purpose solution across diverse applications. Consequently, simple fine-tuning may not be sufficient to outperform these highly specialized expert architectures. There remains substantial room to improve foundation models through novel architectural designs, advanced fine-tuning strategies, and task-specific training objectives. Nevertheless, we believe that DNALONGBENCH will serve as a valuable resource for enabling comprehensive comparisons and rigorous evaluations of emerging DNA sequence-based deep learning models that account for long-range dependencies.
One limitation of this study is the exclusion of transformer-based DNA foundation models, such as DNABERT-1, DNABERT-2, and Nucleotide Transformer, due to the computational challenges posed by training them on long-range tasks. The quadratic cost of the self-attention mechanism renders such tasks infeasible for these models. Exploring strategies to extend the context length of transformer-based models and effectively fine-tune them for long-range tasks remains an important avenue for future research, albeit beyond the scope of this study.
Methods
Benchmark dataset: enhancer-target gene prediction
In eukaryotic cells, enhancers play a key role in gene regulation by forming enhancer-promoter interactions that activate the transcription of target genes, even those located up to several megabases away34. However, the detailed mechanism by which sequence information encodes enhancer–promoter interactions remains poorly understood. Predictive methods that incorporate the entire sequence between enhancers and promoters as input could not only improve prediction performance but also help identify the sequence determinants driving these interactions. To this end, we formulated a task to predict true enhancer–promoter interactions from a list of putative candidates based on the DNA sequence.
We collected experimentally verified enhancer–promoter interactions in K562 cells from three studies32,35,36. Using CRISPRi-mediated perturbation techniques, the authors perturbed thousands of candidate sequences, quantified their effects on gene expression, and identified both positive and negative enhancer-promoter interactions. We filtered this data by retaining enhancer-promoter pair candidates within 450 kb of the gene transcription start site (TSS) and applied additional filtering criteria. Model performance was evaluated using AUROC. We compared models that rely solely on sequence information with the expert model, the Activity-by-Contact (ABC) model32, which incorporates DNase-seq, H3K27ac ChIP-seq data, and a Hi-C matrix to prioritize true enhancer-promoter interactions. It should be noted that the ABC model has inherent advantages over sequence-only models due to its more comprehensive input data types. The primary motivation here is to compare sequence-only models and understand their strengths and limitations.
Benchmark dataset: 3D chromatin contact map prediction
Chromosomes are folded in a well-organized manner within the cell nucleus, affecting various critical cellular functions such as gene transcription and DNA replication37,38. Developing prediction models that connect 1D DNA sequences with 2D contact maps enables the identification of key sequence determinants of 3D chromatin folding, providing valuable insights into the underlying mechanisms of genome organization4,39. We formulated a 3D chromatin contact map prediction task, defined as a 2D regression task to predict pairwise chromatin interactions between every pair of genomic loci within a given context window.
These contact frequencies are expressed as 2D contact maps derived from genomic mapping data such as Hi-C and Micro-C4. We used the processed data from Akita12, which includes chromatin interaction data from five cell lines: HFF, H1-hESC, GM12878, IMR-90, and HCT116. To increase the number of cell types, we curated and processed additional Hi-C data for four cell lines: HAP1, HeLa, HepG2, and K562 from the 4DN data portal40, following the same data processing steps as in the Akita model. Each input sequence, spanning 1 million base pairs (Mbp), is divided into 512 genomic bins at a resolution of 2 kb per bin. For the final prediction, 32 genomic bins are cropped from each side, resulting in a contact map of 448 × 448. Since the contact map is symmetric, predictions are made only for the upper triangular region, with a diagonal offset of 2. The human genome was divided into non-overlapping virtual contigs and randomly assigned to training, validation, and testing sets with an 8:1:1 ratio. The dataset contains 7008 training sequences, 419 validation sequences, and 413 test sequences. Model performance on the held-out test set was evaluated using the stratum-adjusted correlation coefficient (SCC) and the Pearson correlation coefficient (PCC).
Benchmark dataset: regulatory sequence activity prediction
Cell type-specific regulatory activities are encoded by the compositions and interactions of functional DNA segments, such as promoters, enhancers, and insulators, which can regulate genes from distant genomic locations. Predicting functional signals directly from DNA sequences spanning large genomic distances could help identify distal regulatory elements and uncover key sequence features that enable long-range gene regulation. For this task, we compiled human and mouse genomic tracks from the Enformer paper14. The goal of this task is to predict thousands of epigenomic profiles directly from DNA sequence spanning up to 100 kb. We formulated the task as a multitask regression problem aimed at predicting epigenetic and transcriptional signals from long DNA sequences alone.
The dataset includes experimentally determined regulatory activity signal tracks and corresponding DNA sequences from human and mouse genomes. Each input DNA sequence spans 196,608 bp, centered on the TSS of protein-coding genes. Each input sequence consists of a core region and flanking regions. The core sequence is 114,688 bp in length, corresponding to 896 bins at a resolution of 128 bp per bin. The target labels consist of 5313 human tracks and 1643 mouse tracks measuring epigenomic marks. The dataset contains 38,171 human sequences and 33,521 mouse sequences. For the human genome, the data is split into 34,021 training, 2213 validation, and 1937 test sequences. For the mouse genome, the dataset is split into 29,295 training, 2209 validation, and 2017 test sequences. Model performance was evaluated using the Pearson correlation coefficient, calculated by comparing predicted and target signal tracks. Specifically, the Pearson correlation coefficients were computed for each sample across all positions and tracks, and the mean was taken across all samples in the test set.
Benchmark dataset: eQTL prediction
Expression quantitative trait loci (eQTL) are nucleotide variants that affect the expression of one or more genes. Deep learning-based approaches for predicting gene expression from DNA sequences have gained increasing popularity. One practical application of these methods is the identification and interpretation of eQTLs, a traditionally labor-intensive and time-consuming process when relying on genome-wide association studies. We designed an eQTL prediction task to provide an efficient approach for evaluating eQTLs, where the goal is to predict whether a nucleotide variant modulates the expression of a target gene using DNA sequence alone.
We adapted the eQTL dataset used in Enformer14. Positive SNPs were identified using the statistical fine-mapping tool SuSiE41. The original dataset includes positive and matched negative variants across 48 tissues14. For this study, we selected the top nine tissues based on the number of variants. Within these tissues, eQTL-gene pairs were filtered to retain eQTL candidate loci within 450 kb of the gene TSS. Genes with fewer than two positive pairs, two negative pairs, or five combined pairs were excluded. The sequences between variants and promoters were extracted, extending 3 kb downstream of the gene TSS. To reduce bias caused by putative eQTLs within the interval between an eQTL candidate and the gene promoter pair, we masked the sequences of all variants within each variant-promoter pair. The dataset was randomly split into training, validation, and test sets using a stratified sampling approach with an 8:1:1 ratio. To ensure robustness, at least one positive pair and one negative pair were included in both the training and validation sets. Model performance was evaluated using AUROC.
Benchmark dataset: transcription initiation signal prediction
Promoters are specialized DNA sequences at the TSS of genes that support the assembly of transcription machinery and transcription initiation42. Each promoter exhibits a unique profile of transcription initiation signals, which may reflect the mechanisms underlying transcription initiation. Solving the machine learning task of predicting these profiles from promoter sequences provides insights into sequence-based regulation of transcription initiation33. Using long sequences as input and improving the information flow between distal elements could enhance the predictive accuracy of transcription initiation signal prediction. We include a task in DNALONGBENCH aimed at predicting transcription initiation signal profiles from DNA sequences. Specifically, the task predicts transcription initiation signals on both strands for five experimental techniques: FANTOM CAGE, ENCODE CAGE, ENCODE RAMPAGE, GRO-cap, and PRO-cap33. Unlike the regulatory sequence activity prediction task, which predicts sequence coverage at 128 bp genomic bins, this task requires predictions at base-pair resolution, making it significantly more challenging.
We used processed labeled data from the Puffin model33. Predictions were generated for entire test chromosomes (chr8 and chr9) using a sliding window with a step size of 50 kb, with the center 50 kb of each 100 kb prediction being evaluated. Regions within 1 kb of unknown bases or within 25 kb of chromosome ends were excluded. Model performance was evaluated using the Pearson correlation coefficient.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The benchmark datasets have been deposited to Harvard DataVerse under the following DOI link: 1. Regulatory Sequence Activity Prediction Data: https://doi.org/10.7910/DVN/MNUEZR; 2. Transcription Initiation Signal Prediction Data: https://doi.org/10.7910/DVN/VXQKWO; 3. Enhancer-Target Gene Prediction Data: https://doi.org/10.7910/DVN/CTEQXX; 4. 3D Chromatin Contact Map Prediction Data: https://doi.org/10.7910/DVN/AZM25S; 5. Expression Quantitative Trait Loci (eQTL) Prediction Data: https://doi.org/10.7910/DVN/YUP2G5 The enhancer-target gene dataset was obtained from CRISPRi-based screening data from three studies32,35,36. The contact map prediction data were derived from the previous Akita12 paper at https://github.com/calico/basenji/tree/master/manuscripts/akita and four additional in situ Hi-C datasets (4D Nucleome Data Portal accession IDs: 4DNFIWGGYEW2, 4DNFI65WJKMT, 4DNFIQ4G74OW, 4DNFI2R1W3YW). The eQTL and regulatory sequence activity data were obtained from the Basenji43 paper, which was previously used by the Basenji244 and Enformer14 models, available at https://console.cloud.google.com/storage/browser/basenji_barnyard/data. The transcriptional initiation signal prediction data were obtained from Zenodo at 10.5281/zenodo.795497145. Source data are provided with this paper.
Code availability
The source code is available on GitHub at https://github.com/ma-compbio/DNALONGBENCH, under the BSD-3-Clause license. The specific version of the code associated with this publication is archived in Zenodo and is accessible via https://doi.org/10.5281/zenodo.1717956846.
References
ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57 (2012).
Dekker, J. & Misteli, T. Long-range chromatin interactions. Cold Spring Harb. Perspect. Biol. 7, a019356 (2015).
Furlong, E. E. & Levine, M. Developmental enhancers and chromosome topology. Science 361, 1341–1345 (2018).
Zhang, Y. et al. Computational methods for analysing multiscale 3D genome organization. Nat. Rev. Genet. 25, 123–141 (2024).
Furey, T. S. ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nat. Rev. Genet. 13, 840–852 (2012).
Klemm, S. L., Shipony, Z. & Greenleaf, W. J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 20, 207–220 (2019).
Kempfer, R. & Pombo, A. Methods for mapping 3D chromosome architecture. Nat. Rev. Genet. 21, 207–226 (2020).
Zhou, J. & Troyanskaya, O. G. Predicting effects of noncoding variants with deep learning–based sequence model. Nat. Methods 12, 931–934 (2015).
Alipanahi, B., Delong, A., Weirauch, M. T. & Frey, B. J. Predicting the sequence specificities of DNA-and RNA-binding proteins by deep learning. Nat. Biotechnol. 33, 831–838 (2015).
Quang, D. & Xie, X. DanQ: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Res. 44, e107–e107 (2016).
Avsec, Ž. et al. Base-resolution models of transcription-factor binding reveal soft motif syntax. Nat. Genet. 53, 354–366 (2021).
Fudenberg, G., Kelley, D. R. & Pollard, K. S. Predicting 3D genome folding from DNA sequence with akita. Nat. Methods 17, 1111–1117 (2020).
Schwessinger, R. et al. DeepC: predicting 3D genome folding using megabase-scale transfer learning. Nat. Methods 17, 1118–1124 (2020).
Avsec, Ž. et al. Effective gene expression prediction from sequence by integrating long-range interactions. Nat. Methods 18, 1196–1203 (2021).
Karollus, A., Mauermeier, T. & Gagneur, J. Current sequence-based models capture gene expression determinants in promoters but mostly ignore distal enhancers. Genome Biol. 24, 56 (2023).
Devlin, J., Chang, M.-W., Lee, K. & Toutanova, K. Bert: Pre-training of deep bidirectional transformers for language understanding. In Proc. Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, Volume 1 (long and short papers), 4171–4186 (2019).
Wei, J. et al. Emergent abilities of large language models. Transact. Mach. Learn. Res. 2835–8856, https://openreview.net/pdf?id=yzkSU5zdwD (2022).
Achiam, J. et al. GPT-4 technical report. Preprint at https://doi.org/10.48550/arXiv.2303.08774 (2023).
Touvron, H. et al. LLaMA: Open and efficient foundation language models. Preprint at https://doi.org/10.48550/arXiv.2302.13971 (2023).
Tang, Z., Somia, N., Yu, Y. & Koo, P. K. Evaluating the representational power of pre-trained DNA language models for regulatory genomics. Genome Biol. 26, 203 (2025).
Dalla-Torre, H. et al. Nucleotide transformer: building and evaluating robust foundation models for human genomics. Nat. Methods 22, 287–297 (2025).
Nguyen, E. et al. Sequence modeling and design from molecular to genome scale with Evo. Science 386, eado9336 (2024).
Brixi, G. et al. Genome modeling and design across all domains of life with Evo 2. bioRxiv, 2025.02.18.638918, https://doi.org/10.1101/2025.02.18.638918 (2025).
Nguyen, E. et al. HyenaDNA: long-range genomic sequence modeling at single nucleotide resolution. Advances in Neural Information Processing Systems 36 (2024).
Schiff, Y. et al. Caduceus: Bi-directional equivariant long-range DNA sequence modeling. Proc. Mach. Learn. Res. 235, 43632 (2024).
Zhou, Z. et al. DNABERT-2: Efficient foundation model and benchmark for multi-species genome. In Proc. Twelfth International Conference on Learning Representations (2024).
Marin, F. I. et al. BEND: Benchmarking DNA language models on biologically meaningful tasks. In Proc. Twelfth International Conference on Learning Representations (2023).
Grešová, K., Martinek, V., Čechák, D., Šimeček, P. & Alexiou, P. Genomic benchmarks: a collection of datasets for genomic sequence classification. BMC Genom. Data 24, 25 (2023).
Dalla-Torre, H. et al. Nucleotide transformer: building and evaluating robust foundation models for human genomics. Nat. Methods 1–11 (2024).
Kao, C. H. et al. Advancing DNA language models: the genomics long-range benchmark. In Proc. ICLR 2024 Workshop on Machine Learning for Genomics Explorations (2024).
LeCun, Y., Bengio, Y. & Hinton, G. Deep learning. Nature 521, 436–444 (2015).
Fulco, C. P. et al. Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 51, 1664–1669 (2019).
Dudnyk, K., Cai, D., Shi, C., Xu, J. & Zhou, J. Sequence basis of transcription initiation in the human genome. Science 384, eadj0116 (2024).
Schoenfelder, S. & Fraser, P. Long-range enhancer–promoter contacts in gene expression control. Nat. Rev. Genet. 20, 437–455 (2019).
Gasperini, M. et al. A genome-wide framework for mapping gene regulation via cellular genetic screens. Cell 176, 377–390 (2019).
Schraivogel, D. et al. Targeted perturb-seq enables genome-scale genetic screens in single cells. Nat. Methods 17, 629–635 (2020).
Misteli, T. Beyond the sequence: cellular organization of genome function. Cell 128, 787–800 (2007).
Bonev, B. & Cavalli, G. Organization and function of the 3D genome. Nat. Rev. Genet. 17, 661–678 (2016).
Yang, M. & Ma, J. Machine learning methods for exploring sequence determinants of 3D genome organization. J. Mol. Biol. 434, 167666 (2022).
Dekker, J. et al. The 4D Nucleome Project. Nature 549, 219–226 (2017).
Wang, G., Sarkar, A., Carbonetto, P. & Stephens, M. A simple new approach to variable selection in regression, with application to genetic fine mapping. J. R. Stat. Soc. Ser. B: Stat. Methodol. 82, 1273–1300 (2020).
Haberle, V. & Stark, A. Eukaryotic core promoters and the functional basis of transcription initiation. Nat. Rev. Mol. Cell Biol. 19, 621–637 (2018).
Kelley, D. R. et al. Sequential regulatory activity prediction across chromosomes with convolutional neural networks. Genome Res. 28, 739–750 (2018).
Kelley, D. R. Cross-species regulatory sequence activity prediction. PLoS Comput. Biol. 16, e1008050 (2020).
Dudnyk, K., Cai, D., Shi, C., Xu, J. & Zhou, J. Sequence basis of transcription initiation in the human genome. Zenodo https://doi.org/10.5281/zenodo.7954971 (2023).
Cheng, W. et al. DNALONGBENCH: a benchmark suite for long-range DNA prediction tasks. Zenodo https://doi.org/10.5281/zenodo.17179568 (2025).
Acknowledgements
This work was supported, in part, by National Institutes of Health Common Fund 4D Nucleome Program grant UM1HG011593 (J.M.); National Institutes of Health Common Fund Cellular Senescence Network Program grant UH3CA268202 (J.M.); and National Institutes of Health grants R01HG007352 (J.M.), R01HG012303 (J.M.), U24HG012070 (J.M.), and R21DA061481 (J.M.). J.M. was additionally supported by the Ray and Stephanie Lane Professorship, a Guggenheim Fellowship from the John Simon Guggenheim Memorial Foundation, a Google Research Award, and a Single-Cell Biology Data Insights award from the Chan Zuckerberg Initiative. L.L. is supported by an NEC Faculty Research Award and the Neocortex Award from the Pittsburgh Supercomputing Center.
Author information
Authors and Affiliations
Contributions
Conceptualization, W.C., Z.S., Y.Z., L.L., J.M.; Methodology, W.C., Z.S., Y.Z.; Software, W.C., Z.S., Y.Z., S.W., D.W., M.Y.; Investigation, W.C., Z.S., Y.Z., S.W., D.W., M.Y., L.L., J.M.; Writing, W.C, Z.S., Y.Z., J.M.; Funding Acquisition, L.L., J.M.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Bartek Wilczynski, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cheng, W., Song, Z., Zhang, Y. et al. DNALONGBENCH: a benchmark suite for long-range DNA prediction tasks. Nat Commun 16, 10108 (2025). https://doi.org/10.1038/s41467-025-65077-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65077-4
