
Abstract
Eosinophils accumulate in different tissues in allergies, cancer, and infectious diseases, adopting distinct properties. Herein, we profiled murine esophageal eosinophils during allergic inflammation using single-cell sequencing, epigenomic mapping, and flow cytometry. Esophageal eosinophils displayed an altered chromatin accessibility profile compared to bone marrow eosinophils, with 761 epi-transcriptionally regulated genes enriched in inflammation, immunoregulation, bacterial sensing, angiogenesis, migration, and apoptosis. The local environment entrains the unique esophageal eosinophil immunophenotype, as suggested by eosinophil-esophageal epithelial co-cultures, esophageal eosinophil transcriptional similarities regardless of the upstream cytokines driving their esophageal localization, and transcription factor gene editing altering esophageal eosinophilia and the associated eosinophil and global esophageal transcriptomes. Finally, the epigenomic and transcriptomic properties of murine esophageal eosinophils are largely conserved in humans. Thus, our data indicate that tissue specialization of esophageal eosinophils is entrained by local environmental cues that induce genome-wide epigenetic reprogramming and regulated by discrete transcription factors and provide a public, epigenetic database of murine tissue eosinophils.
Similar content being viewed by others
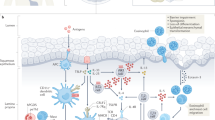

Introduction
Eosinophils are historically regarded as pro-inflammatory leukocytes involved in mediating a variety of diseases, such as asthma; however, mounting evidence suggests that they also contribute to various tissue homeostatic and immune regulatory processes1,2,3,4,5,6. Mature eosinophils develop in the bone marrow under the control of select transcription factors, including GATA1, PU.1, CEBPε and XBP17,8. Although these transcription factors are also involved in the development of other hematopoietic lineages, the specific manner in which they regulate eosinophil development differs, such as employing double palindromic GATA1 binding motifs and interacting with co-factors (e.g., FOG1)7. Though eosinophils are typically considered blood cells, their main function is executed in tissue locations during both homeostasis and disease states. It has recently been appreciated that tissue eosinophils exist as heterogeneous populations of cells with a range of functions4,5,9. Small intestinal eosinophils can be divided into subsets by expression of CD80 and CD274 (PD-L1)5, with active eosinophils expressing both co-stimulatory molecules and demonstrating bactericidal and T cell regulatory activity5, partially regulated by the aryl hydrocarbon receptor (AHR)10. Two pulmonary eosinophil subsets have been identified by differential CD101 expression and cellular morphologies, anatomic localization, and transcriptional signatures at baseline and during inflammation4. More recently, Notch2 signaling was found to contribute to eosinophil survival, function, and intestine-specific adaptations, with no effect in the context of allergic airway inflammation11 – further highlighting tissue-specific drivers of diverse eosinophil phenotypes.
Single-cell RNA sequencing (scRNA-seq) of esophageal tissue has elucidated the importance and functional phenotypes of mast cells12, macrophages and dendritic cells13, and innate lymphoid cells14 in eosinophilic esophagitis (EoE). However, esophageal eosinophils have traditionally been absent from murine single-cell studies, mainly due to their scarcity in the tissue under homeostasis. Furthermore, traditional fluidics‒based single-cell technologies and the enzymatic digestion processes to isolate eosinophils from tissues lead to cellular stress and damage, which result in loss of eosinophil gene transcripts due to their high levels of intracellular RNases. Development of gravity-based single-cell technologies, with their gentler methodologies and increased reaction volumes, have improved identification of eosinophils from enzymatically digested tissue both in humans and mice5,15. Understanding eosinophils in this tissue location is relevant to a number of diseases characterized by variable levels of esophageal eosinophilia, including EoE, gastroesophageal reflux disease (GERD), and esophageal cancer16.
Herein, we investigate whether the external environment controls the phenotype of tissue-dwelling eosinophils via an epigenetic mechanism. Accordingly, we uncovered the epigenetic (chromatin accessibility), transcriptional, and immunophenotypic features of murine eosinophils isolated from the inflamed esophagus. The murine esophageal eosinophils are remarkably similar regardless of the upstream signals leading to the tissue eosinophilia. Furthermore, the transcriptomic and epigenetic programs involved in tissue specialization are conserved across mice and humans. Although there are some similarities between eosinophil tissue subtypes, eosinophils residing in the esophagus are transcriptionally programmed by AP-1 family members in a unique manner compared with eosinophils resident in other tissues, with gene targeting of AP-1 family member ATF3 augmenting eosinophilia in the esophagus but not the lung or peripheral blood. Furthermore, eosinophil-specific depletion of ATF3 impacts both the global esophageal transcriptome and the expression of eosinophil ATF3 target genes predicted within the murine esophageal eosinophil epitranscriptome. Taken together, we have determined that tissue environmental cues epigenetically engrain esophageal eosinophils to adopt an AP-1‒regulated transcriptional profile.
Results
Single-cell RNA sequencing of murine esophageal eosinophils uncovers enrichment for metabolic, immunomodulatory, and extracellular sensing pathways
We subjected the murine esophagus to scRNA-seq utilizing the gravity-based BD Rhapsody platform. Esophageal cells were isolated from CC10-iIL13 and CD2-IL5 transgenic mice, which both have esophageal eosinophilia17,18 (Supplementary Fig. 1A). Clustering analysis revealed 13 major cell lineages present in the murine esophagus that were identified by hallmark gene expression patterns (Fig. 1A, B). The number of genes, unique molecular identifiers (UMIs) and percentage of mitochondrial genes for the different clusters identified by scRNA-seq are shown in Fig. 1C. Only cells that passed the quality control filtering thresholds detailed in the methods section were included in the analysis. A full list of enriched genes for each cellular cluster can be found in Supplementary Data 1A. Eosinophils were a prominent cell type present in the esophageal tissue, accounting for ~25% of the total cells (2613/10,532 single cells, Fig. 1D). Eosinophils were distinguished from other cell populations by expression of Siglecf, Il5ra, and Ccr3 (Fig. 1D).

A UMAP of scRNA-seq esophageal cell populations with population annotations. B Heatmap showing gene expression levels normalized per row of identified hallmark gene expression for esophageal resident cell populations. C Violin plot showing the number of genes, number of UMI’s and percentage mitochondrial genes per cluster in the esophageal scRNA-seq dataset. D PlotProfile showing two esophageal eosinophil populations (Eo1, Eo2) with gene expression of eosinophil marker genes. Plots are colored by the relative expression of gene transcripts. E Radar map showing top gene ontology terms enriched in esophageal eosinophils compared to neutrophils and mast cells, emphasizing metabolic, extracellular sensing, and immunomodulatory gene pathways. F Heatmap showing normalized gene expression values for select genes enriched in the murine esophageal eosinophil scRNA-seq dataset. Gene expression is normalized within each row. G PlotProfile showing expression of eosinophil granule protein genes and early activation marker genes in esophageal eosinophil populations present in the murine esophagus. Plots are colored by the relative expression of gene transcripts. H Left; PlotProfile of C3ar1 gene expression. Right; Quantification of esophageal eosinophil surface expression from CC10-iIL13Tg mice (isotype: n = 3, + C3aR1 antibody: n = 14, 3 independent experiments) with, Bottom; representative histogram compared to isotype control. Scatter plots show the mean with standard deviation error bars. Statistical analysis shows a two-sided paired T test. I Heatmap showing expression (gMFI) of eosinophil markers between tissue locations. Expression is normalized per row. J Bar chart, with representative histograms, showing eosinophil expression of C3aR1 from diverse tissue locations (bone [blue], blood [red], esophagus (esoph. [orange]), lung [pink], duodenum (duo. [light green]), and ileum [dark green]) in CD2-IL5Tg mice (blood, bone and esophagus: n = 8, lung, duodenum and ileum: n = 7, 2 independent experiments). Scatter plots show the mean with standard deviation error bars. Statistical analysis shows one-way ANOVA with Tukey’s multiple comparisons. *** p < 0.0001 compared to blood. ### p < 0.0001 compared to bone marrow. ## p = 0.0076 compared to bone marrow.
Compared with non-eosinophils, esophageal eosinophils were enriched for 1690 unique genes (Supplementary Data 1B [Eo1 eosinophils were enriched for 848 genes, and Eo2 eosinophils were enriched for 1491, totaling 2339 genes; 649 of these were shared by both eosinophil populations, giving a total of 1690 unique genes]), which were enriched for pathways involved in degranulation, biosynthetic, extracellular sensing, immunomodulatory, angiogenic, and apoptosis-related pathways (Fig. 1E and Supplementary Data 1C). Several biosynthetic factors involved in leukotriene and lipoxin biosynthesis (Alox5ap, Alox15, Cyp4f18), prostaglandin biosynthesis (Ptgs2), GTP synthesis (Dennd4a), and lipolysis (Pde3b) were highly expressed by esophageal eosinophils compared to neutrophils and mast cells (Fig. 1F and Supplementary Fig. 1B). In contrast, mast cells were enriched for genes involved in antigen-mediated signaling, B cell receptor signaling, and myeloid development, whereas neutrophils were enriched for TNF signaling and IL-6 production (Supplementary Fig. 1C and Supplementary Data 1C). These data highlight that distinct gene programs are elicited in esophageal eosinophils compared with related cells in the same inflammatory environment.
Key genes enriched in extracellular sensing and immunomodulatory pathways in esophageal eosinophils included co-stimulatory molecules (Cd80, C274), complement- and platelet-related proteins (C3ar1, F5, Ptafr, Pla2g7), extracellular sensing receptors (Antxr2, Ahr, Il1rl1, Nr4a3, Pirb), and angiogenic factors (Thbs1, Olr1, Vegfa) (Fig. 1F and Supplementary Fig. 1B). These data highlight that esophageal eosinophils are likely uniquely poised to respond to the specific environment of the inflamed esophagus through select extracellular sensing and co-stimulatory molecules, likely resulting in inducing degranulation, immunomodulatory, angiogenic, and biosynthetic pathways.
Cluster analysis revealed two populations of eosinophils; the minority were designated Eo1 and represented ~10% of eosinophils, whereas the majority population was designated Eo2 (90%) (Fig. 1D). Differential gene analysis between Eo1 and Eo2 eosinophils revealed 530 genes (0.1 log2 fold change [FC] < 0.05 pAdj [FDR]) that were differently enriched between Eo1 and Eo2 eosinophils (247 genes enriched in Eo1 vs. 256 genes enriched in Eo2) Supplementary Data 1D. Eo1 eosinophils were preferentially enriched for genes involved in the expression of eosinophil granule protein (Prg2, Epx), early activation markers (Itgal, Itgax, Pecam1) and tissue recruitment (Cd34, Adamdec1, Mmp25) transcripts; in contrast, Eo2 eosinophils had relatively low expression of transcripts encoding eosinophil granule proteins (Fig. 1G). These data suggest that the Eo1 subpopulation represents a more immature esophageal eosinophil population. Furthermore, expression of esophageal eosinophil‒enriched genes, including those involved in biosynthesis, extracellular sensing, and immunomodulation, was increased in Eo2 eosinophils compared to Eo1 eosinophils (Fig. 1F and Supplementary Fig. 1B). These data further highlight that esophageal eosinophils can dynamically regulate their transcriptional expression of select key functional genes upon recruitment to the tissue.
Cellular populations, including eosinophils, were similarly present between genotypes, with both CD2-IL5Tg and CC10-iIL13Tg mice contributing to all cell lineages (Supplementary Fig. 1D). Pseudobulk analysis of the eosinophil populations revealed 98% (3585/3650) concordance of genes expressed by eosinophils in the two genotypes, revealing only a limited number—65 genes (< 2% of all genes) —that were differently expressed (0.1 log2FC < 0.05 pAdj [FDR]) between genotypes (Supplementary Fig. 1E and Supplementary Data 1E). Furthermore, a strong correlation between gene expression levels was observed between genotypes (Supplementary Fig. 1F), suggesting that esophageal eosinophil gene expression is dependent on the tissue environment (e.g., esophagus) rather than on distinct processes associated with exposure to IL-5 vs. IL-13, thereby providing a rationale for merging data between genotypes.
Murine esophageal eosinophils are a distinct gastrointestinal eosinophil subset
To determine whether esophageal eosinophils have a unique transcriptome, we compared highly expressed genes between esophageal and other tissue-resident eosinophils5,19. Interestingly, the tissue-specific transcriptomes had many shared and unique genes shown in Table 1. These data indicate that tissue eosinophils have substantial plasticity, adopting shared and unique transcriptomic properties in distinct tissues. Next, we aimed to compare the esophageal eosinophil phenotype with single-cell data derived from murine eosinophils in other tissues. Publicly available murine eosinophil scRNA-seq data5 were derived from an earlier version of the Rhapsody platform, proving difficult to successfully integrate the datasets. Thus, we directly immunophenotyped eosinophils residing in different tissues via marker expression by flow cytometry. Markers were incorporated on the basis of those identified in the scRNA-seq analysis, in addition to others present on tissue-specific populations of eosinophils4,9,10,20. Eosinophils were identified in all tissues using the flow cytometry gating panel in Supplementary Fig. 1A.
We first characterized the esophageal eosinophil immunophenotype in CC10-iIL13Tg mice (Supplementary Fig. 2). Expression of gene transcripts for both C3ar1 and C5ar1 was observed in the esophageal eosinophil scRNA-seq dataset. However, only C3aR1, and not C5aR1, was detectable on the esophageal eosinophil cell surface (Fig. 1H and Supplementary Fig. 2A). Furthermore, surface expression of C3aR1 was only detectable on esophageal eosinophils compared to other tissue cell populations (Supplementary Fig. 2B). Esophageal eosinophils, similar to small-intestine eosinophils5, expressed Cd44, Cd80, Cd274, Il1rl1 and Tlr4 at both the mRNA and protein level (Supplementary Fig. 2A). CD101 and Clec4a4, which have been identified on inflammatory eosinophil populations in the lung and gastrointestinal tract, respectively4,9,20, were not detected either at a transcriptional or proteomic level in esophageal eosinophils (Supplementary Fig. 2A).
Next, to further characterize the immunophenotype of multiple eosinophil tissue populations within the same mouse, we compared eosinophils isolated from discrete tissues of CD2-IL5Tg mice using an extended 17-color panel of antibodies. The transcriptional expression profile of markers included in the extended flow cytometry panel, not shown in Supplementary Fig. 2A, are shown in Supplementary Fig. 3A. Esophageal eosinophils had a unique proteomic profile and were distinct from peripheral blood, bone marrow resident, gastrointestinal, and pulmonary inflammatory eosinophils (Fig. 1I). Despite transcriptional expression of C3ar1 by multiple subsets of tissue eosinophils (Supplementary Fig. 3B), C3aR1 was only expressed by esophageal eosinophils and was absent on blood, bone, pulmonary, duodenal, and ileal eosinophils in CD2-IL5Tg mice (Fig. 1J). Duodenal and ileal eosinophils were exclusively Clec4a4 positive as previously reported9,10; in contrast, esophageal eosinophils were Clec4a4 negative (Fig. 1I and Supplementary Fig. 3C). As in the small intestine, esophageal eosinophils expressed CD274 and CD80; however, expression was lower on eosinophils from the esophagus than from the duodenum or ileum (Fig. 1H and Supplementary Fig. 3D, E). Esophageal, pulmonary, and small intestinal eosinophils all expressed high levels of CD44; however, esophageal eosinophils had higher surface expression of CD44 (Supplementary Fig. 3F). This unique signature demonstrates that esophageal eosinophils are distinct from those found within other tissues on the basis of enrichment of C3aR1 and CD44 and absence of CD101 and Clec4a4. Furthermore, the similarities between the esophageal eosinophil phenotype in CC10-iIL13Tg and CD2-IL5Tg mice agree with the findings from the scRNA-seq, suggesting that tissue location, not inflammatory stimuli, drives the esophageal eosinophil phenotype.
To further examine the unique esophageal eosinophil marker C3aR1, esophageal eosinophils were visualized using the ImageStream21 platform. Eosinophils were identified using the flow cytometry gating panel in Supplementary Fig. 1A without the viability discrimination step, due to limitations with the number of fluorophore combinations possible. ImageStream demonstrated punctate expression of C3aR1 (Supplementary Fig. 3G). Next, C3aR1 + and C3aR1- esophageal eosinophils were sorted from esophageal tissue and visualized by light microscopy. Eosinophils were identified using the gating strategy in Supplementary Fig. 1A; the C3aR1 gate was set on the eosinophil populations using an isotype control (Supplementary Fig. 3H). Visible intracellular vacuoles were increased in C3aR1 + esophageal eosinophils compared with C3aR1- eosinophils (Supplementary Fig. 3I). Furthermore, C3aR1- eosinophils had increased nuclear condensation and blebbing (Supplementary Fig. 3I), indicative of apoptosis.
Murine esophageal eosinophils exhibit a specialized epigenetic program
Having defined the transcriptomic signature of eosinophils in the esophagus, we aimed to elucidate the associated epigenetic landscape of these cells; this has not yet been achieved for tissue eosinophils in any location. Ideally, we would compare the epigenome of esophageal eosinophils to blood eosinophils; however, recovery of blood eosinophils was insufficient, so we focused on bone marrow‒resident eosinophils for our comparisons. Flow cytometric analysis of bone marrow eosinophils revealed two populations of SIGLEC-F+ eosinophils, identified by high or low CCR3 expression (Supplementary Fig. 4A), present in equal proportions in CC10-iIL-13Tg and wild-type mice (Supplementary Fig. 4B, left). CCR3lo bone marrow eosinophils represent an immature eosinophil population and are not a subset of mature eosinophils22. Esophageal eosinophils, detectable only in the IL-13Tg mice, were present as a single population of SIGLEC-F + /CCR3 + cells (Supplementary Fig. 4A, B, right). To examine the mechanism of transcriptional entraining of tissue eosinophils23,24, we profiled the chromatin accessibility landscape of purified populations of the two bone marrow eosinophil groups (CCR3hi and CCR3lo) from CC10-iIL13-Tg animals and compared the epigenome to that of esophageal eosinophils by ATAC-seq. For each cell type, we obtained > 50,000 peaks in both replicates, with Fraction of Reads in Peaks (FRiP) scores > 0.25 and Transcription Start Sites (TSS) enrichment scores > 30 (average of 39.3) (Supplementary Table 1), all of which meet or exceed the recommendations set forth by the ENCODE Project consortium25,26,27,28.
The patterning of the identified accessible chromatin regions was similar between the bone marrow eosinophil and esophageal eosinophil populations, being located predominantly distal to the TSS and identified within intergenic and intronic regions, suggesting putative enhancers; however, proximal accessible regions suggestive of promoters were also identified (Supplementary Fig. 4C). Accessible regions of chromatin between bone marrow eosinophils and esophageal eosinophils revealed strong commonality (81% shared; 67,917/83,302) (Fig. 2B), reflecting programs that likely define the eosinophil lineage. Indeed, eosinophils isolated from both locations were found to be enriched for regions of open chromatin within ± 1 kB from the TSS of key eosinophils markers: Il5ra, Prg2, and Cd63 (Supplementary Fig. 4D). Bone marrow eosinophils (CCR3lo and CCR3hi) were more like each other than esophageal eosinophils, supporting the importance of niche signals in determining the epigenome of eosinophils (Supplementary Fig. 4E, F).
As CCR3hi CC10-iIL13Tg eosinophils represented the most mature bone marrow eosinophil population (most like blood eosinophils)22, we next sought to compare mature (CCR3hi) bone marrow eosinophils to those isolated from the esophagus. There were over 15,000 individual regions of open chromatin that distinguished bone marrow from esophageal eosinophils (18.5% of total peaks using FDR < 0.05, 2 FC) (Fig. 2A, B and Supplementary Data 2A). Of the differentially accessible regions of chromatin, 7844 (51.0%, Supplementary Data 2B) were found to be enriched in esophageal eosinophils, and 7541 (49.0%, Supplementary Data 2C) were enriched in bone marrow CCR3hi eosinophils (Fig. 2B). Differentially regulated open regions of chromatin were assigned to genes by location. The accessible regions of chromatin enriched in esophageal eosinophils annotated to 3854 genes, indicating that some genes were associated with multiple open regions of chromatin. Esophageal eosinophils were enriched for regions of open chromatin surrounding genes involved in tissue specialization (e.g., Ikzf3 and Retnla), whereas bone marrow eosinophils had increased accessibility in the promoters of genes associated with eosinophil development (e.g., Prg3) (Supplementary Fig. 4F). Comparing the transcriptionally and epigenetically enriched genes in esophageal eosinophils revealed 761 common genes, constituting the esophageal eosinophil epitranscriptome.
On the basis of the genes implicated by the esophageal eosinophil epitranscriptome, esophageal eosinophils (red-ringed circle, Fig. 2C) were enriched for pathways involved in inflammation, immunoregulation, bacterial sensing, migration, and apoptosis, whereas bone marrow eosinophils were enriched for pathways associated with cell structural organization, intracellular signaling, and protein phosphorylation (blue-ringed triangle, Fig. 2C). Esophageal eosinophils exhibited distinct epigenetic profiles and differed in their receptors, cytokines, and intracellular molecules, indicating epigenetic regulation of highly specialized tissue functions. Key genes enriched for regions of open chromatin in esophageal eosinophils included extracellular sensing and co-stimulatory molecules (Tlr4, Cd274, Cd80 [Fig. 2D]); complement-related proteins (C3ar1 [Fig. 2D]); angiogenic signals (Thbs1, Vegfa, Olr1); bioactive markers (Retnla [Fig. 2D], Ptgs2, Itgam, Itgax, Cd44); functional receptors including inhibitory and nuclear receptors (Ahr, P2ry10, P2ry14, Nr4a3, Il1rn); oxidative stress signals (Hif1a); and metabolic processes (Pde3b, Denda4a, Pla2g7, Cyp4F18, Entpd1 [CD39], Cdk17). UCSC genome browser tracks for visualizing the entire epigenetic dataset are available at http://genome.ucsc.edu/s/Ledsall/esophageal_epigenome. These data support our hypothesis that eosinophils develop their specialization in situ, emphasizing that this process is not stochastic, but tissue regulated.

A Heatmap showing peak score intensity of differentially bound regions of open chromatin from bone marrow and esophageal eosinophils as determined by ATAC-seq (red box: esophageal eosinophils, dark blue box: mature bone marrow eosinophils, light blue box: immature bone marrow eosinophils). B Volcano plot showing regions with differential chromatin accessibility with FDR < 0.05, Fold Change ≥ 2, as determined by DESeq2. Accessible regions of chromatin are annotated with key genes based on the closest location. Note that the y-axis scale has been log-transformed. C Ranked gene ontology plot displaying enrichment score (Fisher exact test) for key gene pathways expressed as log10(p-value) in esophageal (red-ringed circle) and mature bone marrow (blue-ringed triangle) enriched accessible regions of chromatin. Gene ratio, the proportion of genes in the described gene ontology (GO) term which are enriched in our gene list. D Genome profile view showing chromatin accessibility surrounding differentially regulated regions of the eosinophil epigenome. Genes shown; C3ar1, Cd80, Retnla and Mpo. The histogram shows gene expression levels (data taken from GEO: GSE811352) as mean ± SD; individual markers represent gene expression in individual mice (n = 3). Statistical analysis shows a two-sided Student t test. ESOPH, esophagus; BM, bone marrow.
The murine esophageal eosinophil epigenome has a unique transcription factor repertoire
To elucidate which transcription factors are involved in regulating the esophageal epigenome, we examined DNA sequences within bone marrow and esophageal-enriched regions for enriched transcription factor binding site motifs using the HOMER software package29. Motifs for C/EBP family members were highly enriched in bone marrow eosinophils in comparison to esophageal eosinophils (Fig. 3A). In esophageal eosinophils, motif analysis revealed that the DNA binding sites for AP-1 members (Jun [p = 1E-1236], Fos [p = 1E-1320) and associated transcription factors (Batf3 [p = 1E-1217], Atf3 [p = 1E-1164], Nfe2 [p = 1E-932]) were the most highly enriched motifs (Fig. 3A). Other highly represented motifs included those for Bach1/Bach2 (p = 1E-608), Klf7 (p = 1E-81), Ctcf (p = 1E-221), and Spi1/PU.1 (p = 1E-512) (Fig. 3A). Bulk RNA sequencing of FACS-sorted esophageal eosinophils demonstrated transcriptional enrichment of the AP-1 family transcription factors identified to have enriched presence of binding site motifs within accessible regions of chromatin in esophageal eosinophils (Fig. 3B). These findings highlight a likely important role for AP-1‒related transcription factors in regulating tissue-specific functions in eosinophils in allergic inflammation.

A HOMER motif enrichment p-values (hypergeometric test) for esophageal (red bar) and mature bone marrow (blue bar) enriched ATAC-seq regions, indicating highly represented transcription factor binding site motifs. The DNA sequence of each transcription factor binding motif is shown to the right, with the nucleotide position and probability shown visually. Significance is expressed as -log(P-value). B Gene expression levels for select, enriched AP-1‒like transcription factors in bone marrow (BM) and esophageal FACS-sorted eosinophils from CC10-iIL13Tg mice (data taken from GEO: GSE811352). Data are presented as mean ± SD; individual markers represent gene expression in individual mice (n = 3). Statistical analysis shows a two-sided Student t test. C String-db derived interaction diagram showing predicted interactions of enriched AP-1 complex‒associated transcription factor family members with each other and with other proteins. D, E Upset plot showing top 10 predicted combinations of transcription factor binding sites present in each accessible region of chromatin and their frequency within (D) esophageal eosinophil‒enriched regions and (E) bone marrow eosinophil‒enriched regions. Color key: filled dots (red [esophageal] or blue [bone marrow]) indicate that transcription factor binding sites are co-present within the same accessible region of chromatin, with gray dots indicating that the putative transcription factor binding site is not present within a given ATAC-seq peak. The number on the y-axis indicates the number of peaks with that combination of putative transcription factor binding sites. F Genome profile view of chromatin accessibility surrounding C3ar1 showing the differentially regulated regions of the eosinophil epigenome enriched in esophageal eosinophils with putative transcription factor binding sites predicted by HOMER. G Genome profile view of chromatin accessibility surrounding Il1rl1(ST2/IL33R) showing the differentially regulated regions of the eosinophil epigenome enriched in esophageal eosinophils with putative transcription factor binding sites predicted by HOMER.

A Experimental schema of eosinophil and EPC2 co-culture. Created in BioRender. Felton, J. (2025) https://BioRender.com/hhmye42. B Western blot showing JUN (top) and ATF3 (bottom) expression in CRISPR-transfected EOL-1 cell lines: wild-type EOL-1 (WT) and JUN knock-out (KO), ATF3 KO, and JUN/ATF3 double KO cells, representative of n = 3 experiments. C PCA plot showing bulk RNA sequencing of WT and ATF3-KO cells co-cultured with EPC2 cells for 3 days. D Gene ontology analysis of enriched gene pathways between WT and ATF3-KO co-cultured cells. E C3AR1 gene expression levels in WT, ATF3-KO and JUN-KO cells co-cultured with/without EPC2 cells for 3 days in vitro (WT and ATF3-KO: n = 10, JUN-KO: n = 12, 4 independent experiments). Scatter plots show the mean with standard deviation error bars. Statistical analysis shows two-way ANOVA with Tukey’s multiple comparisons. F Bar chart, with representative histograms showing expression of C3ar1 on WT and transcription factor KO EOL-1 cells co-cultured with EPC2 cells for 3 days in vitro (WT: n = 7, ATF3-KO: n = 8, JUN-KO: n = 9, 3 independent experiments). Scatter plots show the mean with standard deviation error bars. Statistical analysis shows two-way ANOVA with Tukey’s multiple comparisons. For (E) and (F), WT EOL-1 cells (white). WT cells co-cultured with EPC2 cells (gray). ATF3-KO cells (light blue). ATF3-KO cells co-cultured with EPC2 cells (dark blue). JUN-KO cells (light orange). JUN-KO cells co-cultured with EPC2 cells (dark orange).
Examining enriched transcription factors using the String database, a database of known and predicted protein-protein interactions that stem from computational predictions that rely on direct (physical) and indirect (functional) associations between proteins30,31, revealed interactions between bZIP family transcription factors that recognize AP-1‒like motifs (Jun, Fos, Atf3, Batf3, Nfe2, Bach1, Bach2), Ctcf, and Klf7 in esophageal eosinophils (Fig. 3C). Furthermore, application of UpSet32, a data visualization method for comparing intersection counts between multiple datasets, revealed that the five most common predicted transcription factor interaction combinations, accounting for >35% (2760/7844) of peaks in esophageal eosinophils, involved a core of AP-1‒like motifs along with KLF7 (Fig. 3D). In contrast, this core-binding combination accounted for a significantly lower number of regions (< 5%; 357/7541, Fisher’s exact test; p < 0.0001) in the bone marrow eosinophil‒enriched regions (Fig. 3E). It is notable that numerous genes identified as transcriptionally enriched in esophageal eosinophils were found to have open chromatin regions associated with predicted transcription factor binding site motifs for AP-1‒like motifs and Klf7 (Fig. 3F, G). Putative transcription factor binding sites for all enriched genes can be visualized using the manuscript genome browser track.
In vitro modeling of human esophageal environment
As esophageal eosinophils exist in interepithelial locations, we reasoned that local eosinophil-epithelial interactions may govern eosinophil tissue specialization. We purified human eosinophils from the peripheral blood eosinophils at levels that were sufficient to test this hypothesis by examining transcriptomic changes when eosinophils were co-cultured with esophageal epithelial cells (EPC2 cells)33 (Fig. 4A). Out of the 761 genes in the murine esophageal eosinophil epitranscriptome, 317/761 (41.7%, p-value = 5.45 × 10−79 [hypergeometric test]) had significantly altered expression levels when human eosinophils were co-cultured with EPC2 cells for 7 days; select genes are shown in Supplementary Fig. 5A. Gene ontology analysis revealed that these collective 317 genes were enriched in pathways involved in protein phosphorylation; intracellular signaling; regulation of migration; angiogenesis; cytokine, specifically related to response to IL-1 family members; and integrin-mediated signaling (Supplementary Fig. 5B).
Gene pathways enriched in murine esophageal eosinophils are regulated by JUN/ATF3
To elucidate the transcriptional control mechanism involved in acquisition of the esophageal eosinophil epitranscriptome, we examined the consequences of genetically deleting members of the AP-1 complex of transcription factors implicated by predictive algorithms (Fig. 3). Accordingly, ATF3 and/or JUN (AP-1 component) deficiencies were generated using CRISPR/Cas9-mediated knock-out (KO) of ATF3 and JUN in a human eosinophilic cell line (EOL-1) (Fig. 4B). KO of ATF3 and/or JUN caused dysregulation of 42% (317/761) of the human homologs of the murine esophageal eosinophil epitranscriptome. Many of the genes associated with the esophageal eosinophils epitranscriptome were upregulated in ATF3 KO cells. In contrast, loss of JUN either alone or in combination with ATF3 had opposing effects, resulting in mainly a decrease in gene expression (Supplementary Fig. 5C). These data suggest that ATF3 works in combination with JUN as a negative regulator of esophageal eosinophil gene expression.
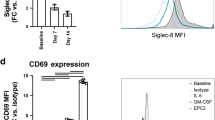
To further test the hypothesis that the external environment entrained eosinophils to acquire tissue specialization, we co-cultured EOL-1 (eosinophilic leukemia) cells with EPC2 (primary esophageal epithelial) cells. Indeed, co-culture was sufficient to induce eosinophil differentiation and the key eosinophil surface markers SIGLEC8, CCR3, and IL-5Rα (Supplementary Fig. 5D). Furthermore, C3AR1 gene expression was observed to be upregulated when EOL-1 cells were co-cultured with EPC2 cells (Supplementary Fig. 5E). We co-cultured ATF3 KO EOL-1 with EPC2 cells and performed bulk RNA sequencing to assess the transcriptional differences (Fig. 4C). A total of 520 genes were differentially regulated between WT and ATF3 KO EOL-1 cells co-cultured with EPC2 cells; these were found to be enriched in pathways associated with immunomodulation (antigen presentation and T cell activation), response to the extracellular environment (response to bacteria and LPS), apoptosis and angiogenesis (Fig. 4D). Indeed, loss of ATF3, but not JUN, reduced both C3AR1 gene expression (Fig. 4E) and C3aR1 surface expression (Fig. 4F) in EOL-1 cells co-cultured with EPC2 cells. C3aR1 surface expression was assessed compared to the isotype control using the gating strategy in Supplementary Fig. 5G. Loss of either transcription factor (TF) had no effect on expression of the canonical eosinophil gene CCR3 (Supplementary Fig. 5F). These data highlight that phenotypic esophageal eosinophil genes are regulated at least in part by the AP-1‒associated transcription factor ATF3.
Next, we generated a mouse model of eosinophil-specific depletion of ATF3 (ATF3-KOeoCre) to determine the impact of loss of ATF3 on esophageal eosinophilia. Esophageal eosinophilia was induced following sensitization and allergen challenge (Fig. 5A). Esophageal eosinophilia was augmented in ATF3-KOeoCre mice compared to WT mice (Fig. 5B). In contrast, genotype had no impact on eosinophil number in either the peripheral blood (Supplementary Fig. 6A) or lung (Supplementary Fig. 6B), suggesting that ATF3 is a tissue-specific transcription factor. Esophageal eosinophil granularity (SSC-A) was decreased in both allergen-treated compared to PBS-treated groups, with a trend for decrease in the ATF3-KOeoCre mice compared to WT mice (Supplementary Fig. 6C). Surface expression of SIGLEC-F was comparable between genotypes and elevated in allergen-treated mice compared to PBS-treated mice (Supplementary Fig. 6D).

A Allergen-induced esophageal eosinophilia model. Calci., calcipotriol; Alt., Alternaria alternata; EtOH, ethanol; iso., isotype control; SigF, SIGLEC-F; DCs, dendritic cells. B Proportion (of CD45 + cells) of esophageal eosinophils (WT PBS: n = 9, ATF3-KOeoCre PBS: n = 10, WT Allergen challenged: n = 15, ATF3-KOeoCre Allergen challenged: n = 16, 3 independent experiments). Scatter plots show the mean with standard deviation error bars. Statistical analysis shows two-way ANOVA with Tukey’s multiple comparisons. C UMAP of scRNA-seq esophageal cell populations with population annotations. D Heatmap showing gene expression levels normalized per row of identified hallmark gene expression for esophageal resident cell populations. E Volcano plot of global pseudobulk analysis (DESeq2; log2FC = 0.1, pdaj < 0.05) between WT allergen-treated and ATF3-KOeoCre allergen-treated esophageal scRNA-seq datasets. F Esophageal eosinophil sub-clustering on WT allergen-treated and ATF3-KOeoCre allergen-treated samples. G Heatmap showing normalized expression of the 44 genes differentially regulated between ATF3-KOeoCre allergen-treated esophageal eosinophils and WT allergen-treated esophageal eosinophils, which overlap with the esophageal eosinophil epitranscriptome. Of these, only one - Crtc3 (annotated with asterix) - was significantly different in PBS- treated WT and ATF3-KOeoCre mice. H Histogram showing esophageal eosinophil expression levels of C3aR1 between genotypes compared to isotype control (blue), WT: black, ATF3-KO: red, with quantification (isotype: n = 3, WT: n = 15, ATF3-KO: n = 16, 3 independent experiments). Data are presented as mean ± SD; individual markers represent an individual mouse. Statistical analysis shows one-way ANOVA with Tukey’s multiple comparisons.
To further determine the impact of eosinophil-specific depletion of ATF3 on the transcriptome of the esophagus and esophageal eosinophils, we performed scRNA-seq on esophageal tissue from PBS- and allergen-treated WT and ATF3-KOeoCre mice. Clustering analysis revealed 14 populations present in the murine esophagus that were identified by hallmark gene expression patterns (Fig. 5C, D). All populations were represented in each experimental group (Supplementary Fig. 6E, F). The number of genes, UMIs and percentage mitochondrial genes for the different clusters identified by scRNA-seq are shown in Supplementary Fig. 6D. Only cells which passed the quality control filtering thresholds detailed in the methods section were included in the analysis. A full list of enriched genes for each cellular cluster can be found in Supplementary Data 3A. Pseudobulk analysis was performed on the whole esophageal scRNA-seq dataset, comparing WT allergen-treated and ATF3-KOeoCre allergen-treated mice and revealing 117 genes globally differently regulated (log2FC = 0.1, pAdj < 0.05) between genotypes (Fig. 5E and Supplementary Data 3B). A total of 23/117 genes were enriched in WT allergen-treated mice and were associated with pathways for B cell signaling, proliferation and activation, lymphocyte differentiation, response to virus and IL-6 production (Supplementary Fig. 6H). In comparison, 94/117 genes were enriched in ATF3-KOeoCre allergen-treated mice, including Cd80, Vegfa, Thbs1, Il4, Hif1a, Trib1, Ccr3 and Ahr, and were associated with angiogenesis, metabolic processes, migration, inflammation, hypoxia, and apoptosis (Supplementary Fig. 6H). These data suggest that loss of ATF3 in eosinophils alone is sufficient to induce major transcriptional changes in the global murine esophagus.
As in the flow cytometry analysis, both the proportion and total number of eosinophils present in the scRNA-seq dataset were enriched in the allergen-treated ATF3-KOeoCre mice (Supplementary Fig. 6I). Genotype had no effect on the proportion or total number of cells within the other 13 clusters in the allergen-treated groups. No differences in the PBS group across genotypes were seen (Fig. 5B and Supplementary Fig. 6I), arguing against a role of ATF3 in the esophagus in the absence of inflammation; this is in line with the known role of ATF3 as an immune regulatory, stress-induced transcription factor34. Thus, to determine the effect of loss of ATF3 on the expression of ATF3 target genes in inflammatory eosinophils, eosinophils from the allergen-treated groups were subjected to sub-setting and re-clustering (Fig. 5F). Differential expression analysis was performed on subset eosinophils using the FindMarkers function (log2FC = 0.1, p-value < 0.05), identifying 483 genes differentially regulated between genotypes (Supplementary Data 3C). A total of 141/483 (29%) genes were enriched in WT eosinophils, whereas 342/483 (71%) were enriched in ATF3-KOeoCre eosinophils. Overlap of the genotype-dependent differentially regulated genes with genes in the esophageal eosinophil epitranscriptome revealed differential regulation of >9% of genes (Fig. 5G). Of these genes, only one, Crtc3 (asterix Fig. 5G), was differentially regulated between PBS-treated WT and ATF3-KOeoCre mice, further highlighting ATF3’s role in inflammatory rather than homeostatic conditions in the esophagus. In line with the results for the scRNA-seq analysis, expression of C3aR1 was increased in ATF3-KO-eoCre eosinophils compared with ATF3 WT eosinophils (Fig. 5H). These data suggest that ATF3 operates as a negative regulator of esophageal eosinophil gene expression and esophageal eosinophil phenotype.
Translational relevance of the murine esophageal eosinophil epitranscriptome in human esophageal eosinophils
To determine the potential significance of the murine esophageal eosinophil phenotype in humans, we compared the murine scRNA-seq dataset to a reanalyzed public scRNA-seq derived from esophageal biopsies obtained from patients with EoE15,35 (Fig. 6A). Differential expression analysis was performed to identify genes significantly enriched in human esophageal eosinophil populations (defined by CLC, SIGLEC8, CCR3, and IL-5Rα) compared to the other cell populations identified within the esophageal biopsies (Supplementary Fig. 7A). Of 898 genes that defined human esophageal eosinophils, 412 (46%, p-value = 3.05 ×10-10 [hypergeometric test]) overlapped with the murine esophageal scRNA-seq transcriptome (Supplementary Fig. 7B). These genes were found to be enriched in pathways key for immune activation, extracellular sensing and complement activation pathways, NFκB signaling, angiogenesis, and immunoregulation (Fig. 6B, C).

A UMAP of scRNA-seq esophageal immune populations (Morgan et al.35, GEO: GSE175930). B Ranked gene ontology plot displaying enrichment score for key pathways expressed as log10(p-value) for the 412 genes transcriptionally enriched in the human esophageal eosinophil scRNA-seq and shared with the murine esophageal transcriptome. C Heatmap showing normalized gene expression levels for select genes of the 412 genes transcriptionally enriched in the human esophageal eosinophil scRNA-seq and shared with the murine esophageal transcriptome. Gene expression levels are normalized within each row. D C3AR1 mRNA expression on eosinophilic esophagitis (EoE) groups from the Eosinophilic Esophagitis Diagnostic Panel (EDP) dataset91 (non-lesional (NL): n = 48, inactive: n = 51, and active EoE: n = 147). Data are presented as median. Statistical analysis shows one-way ANOVA Kruskal-Wallis, with Dunn’s multiple comparison. E C3AR1 and CLC levels correlate; r = 0.4821 p < 0.0001 (linear regression, non-parametric Spearman correlation). F C3AR1 1 mRNA level correlates with eosinophil count; r = 0.4240 p < 0.0001 (linear regression, non-parametric Spearman correlation). G Gating strategy for identifying human esophageal eosinophils obtained from esophageal biopsy (Fig. 6H, I). H Histogram (left) and bar chart (right) showing gMFI of C3aR1 expression on human peripheral blood eosinophils and esophageal eosinophils from 4 individual active EoE biopsies (n = 4). Data are presented as mean ± SD; individual markers represent an individual patient. Statistical analysis shows two-way ANOVA with Tukey’s multiple comparisons. I Percentage of human esophageal eosinophils that are C3aR1 + from active EoE biopsies (n = 4 individuals). Data are presented as mean ± SD; individual markers represent an individual patient. J Representative histogram showing eosinophil intracellular ERK1/2 phosphorylation levels between unstimulated (gray) and hC3a-stimulated (200 ng for 5 min) human esophageal biopsies (red) and peripheral blood (blue). Bar chart showing the fold-change in esophageal eosinophil ERK1/2 expression (gMFI) between unstimulated and hC3a-stimulated samples (n = 4 individuals). Data are presented as mean ± SD; individual markers represent an individual patient. Statistical analysis shows a two-sided unpaired t test.
Of the 412 genes that were transcriptionally enriched in both human and murine esophageal eosinophils, 322 (78%, Supplementary Data 5A) were differentially regulated in the human EoE transcriptome (GSE5864036) at the RNA level, including select genes C3AR1, VEGFA, ALOX5AP, NFKB1, IL1RL1, and DENND4A (Supplementary Fig. 7C). Expression levels of 112 of these genes (35%, Supplementary Data 4B) were found to strongly correlate with esophageal eosinophil levels, including genes involved in extracellular sensing (C3AR1 [Supplementary Fig. 7D], OLR1 [Supplementary Fig. 7D]), angiogenesis and immunoregulation (VEGFA [Supplementary Fig. 7D]), metabolism (DENND4A [Ext. Fig. 7D]), chromatin remodeling, and transcriptional regulation. Of these 112 genes, 72% positively correlated with eosinophil levels and were enriched in gene pathways associated with NFκB signaling, regulation of cell migration and adhesion, apoptosis, and tissue specialization.
C3AR1 mRNA was significantly increased in active EoE in an independent dataset (Fig. 6D). C3AR1 mRNA levels correlated with both peak tissue eosinophil count (r = 0.4240) and CLC level (r = 0.4821) (Fig. 6E, F). Both human esophageal eosinophils and peripheral blood eosinophils, identified using the gating strategy in Fig. 6G and Supplementary Fig. 7E, respectively, were found to express C3aR1 (Fig. 6G, H). The proportion of human esophageal eosinophils that expressed C3aR1 (23.9% ± 8.6% [SD]) was comparable to murine esophageal eosinophils, as demonstrated in Supplementary Fig. 2B (20.9% ± 9.2 [SD]). To determine the functionality of eosinophil-expressed C3aR1, human eosinophils were treated with C3a, and intracellular ERK1/2 phosphorylation was assessed. ERK1 activation has a critical role in regulating eosinophil function, particularly in the context of inflammation and regulating cell fate decisions37. C3a was an activator of human esophageal eosinophils as assessed by induction of intracellular ERK1/2 phosphorylation but had no effect on peripheral blood eosinophils (Fig. 6J).
Lastly, we aimed to determine whether the uniquely established esophageal eosinophil epitranscriptome might be enriched for genes associated with genetic susceptibility to relevant human diseases. We concentrated on the overlapping genes enriched in both murine and human esophageal eosinophils (n = 218) and determined whether they were associated with genetic risk loci in the genome-wide association study (GWAS) catalog. Eosinophil-specific genes were enriched for loci leading to susceptibility for eosinophilia (p = 8.02E-09), allergic disease (p = 5.89E-06), asthma (p = 1.34E-04), and eczema (p = 6.72E-06) (Supplementary Fig. 7F). Taken together, these data demonstrate that esophageal eosinophils have a distinct, tissue-specific epigenome that is enriched for genes whose human homologs are associated with allergic disease risk loci and that correlates with eosinophilia in EoE.
Discussion
Herein, we have uncovered the transcriptional, epigenetic, and proteomic landscape of esophageal eosinophils. Our results demonstrate several major findings including that (1) mature eosinophils residing in a hematopoietic location (the bone marrow) and the esophagus are readily distinguishable by modification of a remarkable number of chromosomal regions (>30,000); (2) parallel RNA- and ATAC-seq has elucidated a combined epigenetic and transcriptomic signature, referred to as the murine esophageal eosinophil epitranscriptome, that is composed of 761 genes enriched in pathways involved in inflammation, immunoregulation, bacterial sensing, angiogenesis, migration, and apoptosis; (3) although there is some shared programming, eosinophils residing in the esophagus are transcriptionally programmed in a unique manner compared with eosinophils residing in other tissues; (4) eosinophil tissue immunophenotypes are unique and are likely entrained by local inflammatory environmental cues rather than pre-determined by stochastic random events, as is evident from co-culture of eosinophils with esophageal epithelial cells and similarities of esophageal eosinophils regardless of the upstream cytokines that drive their tissue localization; (5) the acquisition of tissue specialization at the molecular level is remarkably conserved across species, on the basis of comparing mouse and human scRNA-seq findings; (6) esophageal eosinophils have a distinct, tissue-specific epigenome that is enriched for genes whose human homologs are associated with allergic disease risk loci and eosinophilia in humans; (7) the murine and human conserved transcriptome is enriched in pathways involved in cytokine signaling and production, cellular activation and degranulation, NFκB signaling, angiogenesis, and innate immune function involving Toll-like receptor, IL-1 and bacterial sensing pathways; and (8) the transcription factors involved in tissue specialization are largely distinct from those known to be involved in eosinophil development (e.g., GATA1, PU.1, XBP1). We present evidence for the involvement of a complex of transcription factors containing AP-1 family members in the acquisition of the esophageal phenotype of eosinophils. Furthermore, we highlight the potential role of ATF3 as a negative transcriptional regulator of the esophageal eosinophil phenotype. Collectively, our findings advance the mechanistic understanding of eosinophil plasticity in the context of type 2 immune responses in the esophagus. We demonstrate that the unique tissue specialization of eosinophils is entrained by local inflammatory environmental cues that induce genome-wide epigenetic programming regulated by discrete transcription factors; our data implicate a complex of AP-1 family members in dictating the esophageal program, at least in part (Supplementary Fig. 8). In addition to these findings, we provide a resource to the field, the first public epigenetic database of tissue eosinophils, available at http://genome.ucsc.edu/s/Ledsall/esophageal_epigenome.
Herein, we have employed three different murine models to characterize the inflammatory esophageal eosinophil phenotype in EoE. We recognize that these models have limitations, as they do not fully replicate human disease. The CD2-IL5Tg mouse model is driven by constitutive overexpression of IL-5 under the control of the CD2 promoter active in T cells. This leads to systemic overproduction of IL-5, driving bone marrow eosinopoiesis and the resultant peripheral blood and tissue eosinophilia observed in this model. By contrast, the CC10-iL13Tg mouse model is driven by doxycycline-induced expression of IL-13 by the CC10 promoter in airway epithelial cells. The produced IL-13 accumulates in the esophagus and lungs, leading to tissue eosinophilia. Both CD2-IL5Tg and CC10-iIL13Tg have attributes of human EoE, including esophageal eosinophilia in all layers of the esophagus, albeit at lower levels in the epithelium. These models are also associated with prominent esophageal remodeling, including epithelial hyperplasia, angiogenesis and collagen deposition17,38,39. Furthermore, changes in esophageal transcripts overlap with the human EoE esophageal transcriptome17. In addition, the allergen-induced EoE model demonstrates esophageal eosinophilia, enrichment of eosinophil-associated genes, epithelial hyperplasia, and a transcript profile that overlaps with the human EoE transcriptome40. The observed similarities in the esophageal eosinophil transcriptome and immunophenotype between these distinct murine EoE models emphasize that the eosinophil phenotype is likely dependent on the inflammatory tissue location rather than upstream cytokine exposure, as Th2 cytokine expression levels vary between these models. These data suggest that the inflammatory tissue environment is sufficient to induce the eosinophil phenotype independent of the strength or driver of the inflammatory response. The immunophenotype of esophageal eosinophils are distinct from those isolated from the lung and small intestine on the basis of C3aR1+/CD44high/Clec4a4-/CD101- expression. This combination of markers enables distinguishing esophageal eosinophils from small intestine and lung eosinophils using a minimal number of phenotypic markers. It is interesting to speculate on the important functional consequences of the esophageal eosinophil immunophenotype. CD44 is traditionally considered to be involved in eosinophil recruitment and tissue activation; however, there have been a number of studies in other CD44-expressing cells suggesting that the hyaluronan receptor helps to facilitate immunomodulatory effects by facilitating interactions with T cells41,42,43, regulating inflammatory and fibrotic responses44,45,46,47 and regulating efferocytosis48. Sano et al.49 demonstrated that CD44 expression is higher on pulmonary eosinophils from patients with well-controlled asthma, further suggesting that elevated CD44 maybe be important for effective eosinophil activation and function. The observed increase in C3aR1 expression in esophageal eosinophils may be a consequence of the upregulated expression of C3a present in EoE13,50. As in our scRNA-seq datasets (Supplementary Data 3A), Ding et al.13 also demonstrated that the most prominent C3-expressing population in the esophagus are fibroblasts13. The combined enrichment for both CD44 and C3aR1 on esophageal eosinophils suggests that direct interactions between esophageal eosinophils and fibroblasts may be an operational event in EoE pathogenesis.
In relation to other tissue eosinophil populations, esophageal eosinophils, similar to small intestinal eosinophils5, express the surface receptors TLR4 and ST2 (IL-33 receptor) and the co-stimulatory molecules CD80, CD86, and CD274. Interestingly, a recent elegant study identified NMUR1 + eosinophils as a key regulator of epithelial cell differentiation and barrier protection in the small intestine19. Esophageal eosinophils did not express Nmur1, further highlighting the differential regulation of esophageal and gastrointestinal eosinophils. The distinct immunophenotype of eosinophils between the esophagus, small intestine, and lung emphasizes that eosinophils undergo plastic changes based on the unique microenvironment of each tissue location. The findings presented in this study suggest that eosinophils contribute an immunoregulatory role in the esophagus, specifically through upregulation of gene pathways involved in extracellular sensing, co-stimulation of T cells, angiogenesis, and the production of bioactive molecules (e.g., leukotrienes and lipoxins), supporting that esophageal eosinophils may not exclusively mediate tissue pathology in EoE51,52,53,54,55,56. These observations are consistent with those of others, suggesting that tissue eosinophils are readily able to modulate T cell responses5,57, stimulate anti-bacterial defense57, drive tissue remodeling58,59,60, and aid in energy and metabolic regulation61,62,63.
Using ATAC-seq to assess chromatin accessibility, we demonstrate that eosinophils undergo dynamic chromatin rearrangement when they enter the tissue, although a limitation of our study was that we did not directly examine blood eosinophils but rather bone marrow‒resident eosinophils. Much is known about the generation of mature eosinophils and their development in the bone marrow under the control of select hematopoietic transcription factors, including GATA1 and CEBPε7. The findings presented herein provide evidence that eosinophil tissue-specific processes are regulated by transcription factors distinct from those involved in eosinophil hematopoietic development, extending the recent implication of IKZF364 and KLF365 in this process. Analysis revealed a select group of enriched AP-1 transcription factors, with predicted interactions involving a core complex containing JUN, FOS, ATF3, NFE2, and BATF, along with the non‒AP-1 protein KLF7 (Supplementary Fig. 8). Considering the critical role of AP-1 in mediating the inflammatory response in allergy66, we further investigated the role of these transcription factors in eosinophil gene expression by generating deficiency in ATF3 and/or JUN (components of AP-1) using CRISPR/Cas9-mediated editing in an eosinophilic cell line (EOL-1). ATF3 and JUN were selected because these transcription factors were both transcriptionally and epigenetically enriched in esophageal eosinophils. Loss of ATF3 had the most effect on genes in the esophageal eosinophil signature, resulting in the upregulation of many genes (55%; 233/425) that were associated with biosynthetic, extracellular sensing, angiogenic, and immunomodulatory pathways. To test this hypothesis further, we generated mice with eosinophil-specific deficiency of ATF3; these mice demonstrated increased esophageal eosinophilia and augmented esophageal eosinophil C3aR1 expression in a model of EoE. Furthermore, eosinophil-specific deficiency of ATF3 resulted in changes to both the total global esophageal transcriptome and the transcriptome of esophageal eosinophils themselves, specifically enriched in the expression of esophageal eosinophil epitranscriptome-associated genes. Gilchrist et al. previously demonstrated that ATF3 interacted with two major complexes involved in TLR signaling, AP-1, and NFκB to dampen inflammation and antagonize NFκB-induced responses67. Similar to the findings presented herein, ATF3-global deficient mice developed significantly increased pulmonary eosinophilia, airway hyperresponsiveness, and enhanced chemokine and Th2 cytokine responses in an ovalbumin-induced model of lung inflammation68. Furthermore, in a recent study examining transcriptional regulation in adipose eosinophils, the binding sites for AP-1 family members, including ATF3, were enriched within the promoters of differentially regulated genes in adipose-derived eosinophils compared to blood-derived eosinophils69. These combined findings highlight the role of ATF3 in regulating tissue eosinophil abundance and function.
Collectively, we have demonstrated that eosinophils residing in the bone marrow and the esophagus are readily distinguishable by the marked organization of their chromatin landscape, gene expression, and surface immunophenotype. We have shown that acquiring of tissue specialization at the molecular level is remarkably conserved across species based on the basis of comparing mouse and human scRNA-seq findings. We have shown that esophageal eosinophils have a distinct tissue-specific epigenome that is enriched for genes whose human homologs are associated with allergic disease risk loci and eosinophilia in humans. We have provided molecular evidence that esophageal eosinophils exist as at least two populations; one is most related to the eosinophil in the hematopoietic compartment, and the other is a tissue-differentiated, mature, activated eosinophil enriched for biosynthetic, extracellular sensing, angiogenic and immunomodulatory functions. Eosinophil tissue phenotypes are likely entrained by local environmental cues rather than pre-determined by stochastic random events. Furthermore, the transcription factors involved in tissue specialization are largely distinct from those known to be involved in eosinophil development. We present evidence for the involvement of a complex of transcription factors, including AP-1 family members, in acquiring the esophageal phenotype of eosinophils. It is anticipated that the findings reported herein and the genomic datasets now released to the public will facilitate a better understanding of eosinophils in the context of allergic and esophageal inflammation.
Methods
Mice
Bitransgenic (CC10-iIL13Tg70) mice and appropriate WT:WT co-housed littermates control were subjected to DNA-, RNA-, ATAC- and scRNA-seq and phenotyping experiments. CD2-IL5Tg18 mice were subjected to scRNA-seq and phenotyping experiments. ATF3-KO-eoCre mice were generated by crossing ATF3-Flox mice71 with eoCre72, and the genotype was confirmed by examining for recombination of exon 3. Co-housed littermate controls were used for all experiments. Animals were bred in-house and housed under standard, specific pathogen‒free conditions at 73°F with 36.1% humidity and a 14/10 light-dark cycle and handled under approved protocols of the Institutional Animal Care and Use Committee of Cincinnati Children’s Hospital Medical Center (IACUC 2022-0036 and 2019-0058). Male and female mice aged 6-14 weeks were used for all experiments; male and female mice were housed in separate cages and were randomly assigned to treatment and control groups to ensure an equal balance. To induce esophageal inflammation, bitransgenic mice (CC10-iIL13Tg) were fed a doxycycline-enriched diet for 14 days prior to harvest70,73. Mice were euthanized by intraperitoneal injection of an overdose of sodium pentobarbital (100 mg/kg).
Murine allergen-induced esophageal eosinophilia model
Esophageal eosinophilia was induced in ATF3-KO-eoCre mice and WT littermate controls following sensitization and challenge to Alternaria alternata (Alt.) over 31 days (Fig. 5A). Briefly, mice were sensitized three times on each ear with either 20 mL 100% EtOH followed by 20 mL PBS or 20 mL calcipotriol (4 ng/mL) followed by 20 mL Alt.(600 mg/mL) on days 1, 3 and 5. Following sensitization, mice were challenged with either 50 mL intranasal PBS or Alt. (600 mg/mL) three times per week for 4 weeks, starting 24 h following the last sensitization. Tissue was harvested 24 h following the final intranasal challenge. Esophagus, lung, and blood were collected and processed as below. Experimental sample size was determined by the availability of genetically engineered mice produced through breeding strategies after controlling for cohort age and survival rate in allergen treated group to generate a minimum of three mice per group per experiment.
Preparing single-cell suspensions from tissues
All tissues were collected and processed in media supplemented with serum and inhibitors (RPMI-1640, 20% FBS, 100 U/mL penicillin/streptomycin, Q-VD-OPH [Cayman Chemical; 15260], DNAse1 [Zymo; E1010], and protease inhibitors [Sigma; P8340]). Individual harvest protocols for each tissue are detailed below. All centrifugation steps were performed at 400 g for 5 min at 4 °C unless otherwise stated.
The esophagus was collected, and small pieces (~ 2 mm) of the distal end were saved for both RNA expression and histology. Following collection, the remaining esophagus was washed in EDTA stripping buffer (HBSS, 10% FSC, 5 mM EDTA, 25 mM HEPES) for 10 minutes at 37 °C at 1000 rpm on a heated thermomixer. Stripping buffer was replaced, and samples were incubated for 5 min prior to washing with PBS. Esophagi were dissected longitudinally, placed in media supplemented with 2.4 mg/mL collagenase A (Sigma; COLLA-RO), minced into small pieces, and incubated for 30 minutes at 37 °C at 1000 rpm on a heated thermomixer. Following digestion, the samples were passed through a 19-gauge needle and then passed through a 70-mm filter.
The duodenum was anatomically defined as 6 cm distal to the pylorus, and the ileum was defined as 10 cm proximal to the cecum. Intestinal tissues were dissected longitudinally, washed with PBS to remove fecal matter, and incubated in EDTA for 15 minutes (HBSS, 10% FSC, 5 mM EDTA, 40 mM HEPES) at 37 °C at 1000 rpm on a heated thermomixer. Following an HBSS wash, the tissues were minced into small pieces and incubated in media supplemented with 2.4 mg/mL collagenase A (Sigma; COLLA-RO) for 30 minutes at 37 °C at 1000 rpm on a heated thermomixer. Following digestion, the samples were passed through a 19-gauge needle and through a 70-mm filter.
Lungs were perfused with 10 mL PBS, harvested, and cut into individual lobes prior to mincing and digestion in media supplemented with bovine pancreatic DNAse1 (Sigma; 10104159001) and liberase TL (Sigma; 05401020001) for 45 minutes at 37 °C at 1000 rpm on a heated thermomixer. Lung digests were then passed through a 70-mm cell strainer with syringe plunger before washing with media and lysis of red blood cells using ACK lysis buffer (Gibco; A1049201). Cells were washed in PBS and centrifuged prior to resuspension.
Blood was harvested by post-mortem cardiac puncture and collected in a K2EDTA microtainer blood collection tube (BD; 365974). A total of 80 mL was removed and mixed with 150 mL Alsever’s solution (Sigma; A3551) for flow cytometric processing. Following staining, cells were washed with PBS, and the red blood cells were lysed with FACS lysis buffer (Becton Dickinson [BD]; 349202).
For bone marrow single-cell suspensions, the femur and tibia were harvested and flushed with 10 mL of media with a 21-gauge needle. The suspension was disaggregated with a 19-gauge needle prior to filtering through a 70-mm cell strainer. The sample was centrifuged, and the cell pellet was resuspended in 2 mL ACK red blood cell lysis buffer (Gibco; A1049201). Cells were washed in 20 mL PBS and centrifuged prior to resuspension.
Flow cytometry for quantifying eosinophils in murine peripheral tissues
Cells were stained in PBS at room temperature for 5 min with ZombieNIR (1;1000, Biolegend; 423106) prior to 30 min staining with antibodies against CD45 (BV510, Biolegend: 103138), SIGLEC-F (BV421, BD; 562681), CCR3 (AF647, Biolegend; 144508), C3aR1 (PE, BD; 568276 [isotype, rat Ig2aK; 400507]), FcεR1 (PerCPCy5.5, Biolegend; 134320), and C5aR1 (AF700, R&D; FAB6467N [isotype, rat IgG2b; IC013N]), antibody information in Supplementary Table 2.
Spectral flow cytometry for immunophenotyping of murine eosinophil populations
Cells were stained in PBS at room temperature for 5 minutes with the Live/Dead Blue fixable viability dye (1:1000, Invitrogen; L34961) prior to 30 min staining with a combination of the following antibodies (1:100, unless otherwise stated): CD45 (BV510, Biolegend, 103138), CCR3 (BUV563, BD; 741211), SIGLEC-F (AF647, BD; 562680), CD125 (BV750, BD; 746945), FcεR1 (PerCP-eFlour710, Invitrogen; 46-5898-82), CD44 (BV570, Biolegend; 103037 [isotype, rat IgG2aK; 400641]), CD11b (PerCP, Biolegend; 101229 [isotype, rat IgG2bK; 400629]), CD101 (APC, Invitrogen; 17-1011-82 [isotype, rat IgG2aK; 17-4321-81]), ST2 (BUV737, BD; 749323 [isotype, rat IgG2aK; 612760]), TLR4 (PE-Cy7, Biolegend; 145408 [isotype, rat IgG2aK; 400521]), C3aR1 (BUV661, BD; 753366 [isotype, rat IgG2aK; 612973]), C5aR1 (AF700, R&D; FAB6467N [isotype, rat IgG2b; IC013N]), CD80 (PE/Dazzle594, Biolegend; 104738 [isotype, hamster IgG; 400951]), CD86 (BV711, Biolegend; 137029 [isotype, rat IgG2aK; 400551]), Clec4a4 (AF488, Biolegend; 124910 [isotype, rat IgG2bK; 400625]), AHR (PE, Biolegend; 162703 [isotype, rat IgG2bk; 400636]), and CD274 (BV421, Biolegend; 124315 [isotype, rat IgG2bK; 400639]), antibody information in Supplementary Table 2.
Image stream
Cells were stained for C3aR1 (PE, BD; 568276), FcεR1 (PerCPCy5.5, Biolegend; 134320), SIGLEC-F (BV241, BD; 562681), and CD45 (BV510, Biolegend: 103138) for 30 minutes at room temperature, antibody information in Supplementary Table 2. Samples were fixed with 4% paraformaldehyde (PFA) for 15 minutes. Images were recorded at X60 magnification.
Cell sorting of murine tissues
Samples were stained and sorted in 400 mL PBS with added Q-VD-OPH (Cayman Chemical; 15260), DNAse1 (Zymo; E1010) and protease inhibitors (Sigma; P8340). Eosinophils were FACS-sorted from the esophageal tissue based on viability (1:1000, ZombieNIR, Biolegend, 423106) and expression of CD45 (BV510, Biolegend: 103138), SIGLEC-F (AF647, BD; 562680), and CCR3 (PE, Biolegend; 144506). Following sorting, cells were collected in PBS with added preservatives and processed for ATAC sequencing as below.
Flow cytometry analysis
High-dimensional spectral flow-cytometry analysis was performed on the Cytek Aurora 5 L (Cytek Bioscience). Conventional flow cytometry was performed on BD/FACSCanto. BD FACSDiva software (BD Biosciences) was employed for data acquisition and cell sorting. Flow cytometric data was analyzed using FlowJo software (TreeStar). Positive expression of surface markers was determined compared to the appropriate isotype control.
ATAC sequencing
A total of 6-9 × 104 viable eosinophils were pelleted at 500 g for 5 min at 4 °C prior to resuspension in 50 mL ATAC Resuspension Buffer (ATAC-RSB; 10 mM Tris-HCl, 10 mM NaCl, and 3 mM MgCl2 prepared to a total volume of 50 mL in double-distilled water (ddH2O)) with added 0.1% NP-40, 0.1% Tween-20, and 0.01% Digitonin and were processed for assay for transposase-accessible chromatin with high-throughput sequencing (ATAC sequencing) as previously described64,74,75. Sequencing was performed on Illumina HiSeq 4000, paired-end, and 150 base pairs long (Novogene).
Data processing and functional analysis
ATAC sequencing analysis was performed with the ENCODE ATAC-seq pipeline version 2.0.0 (https://github.com/ENCODE-DCC/atac-seq-pipeline). Briefly, adapter sequences were trimmed, using CutAdapt76, prior to alignment of sequence reads to the mm10 genome using Bowtie 277. Duplicates were removed using Picard. Peaks were called on a per-replicate basis using MACS278 with a p-value cutoff of 0.01. The overlap peak sets generated by the ENCODE pipeline were used for downstream analysis. Read and peak information for ATAC sequencing samples are found in Supplementary Data 2. Presence of ATAC sequencing peaks within 20 kB of expressed eosinophil genes was determined using BEDtools/closest79. Gene ontology (GO) and genome-wide association study (GWAS) analysis was performed using Enrichr80,81. ATAC sequencing tracks were visualized using the Integrative Genomics Viewer (IGV) browser (UC San Diego and the Broad Institute, MIT & Harvard82). ATAC sequencing datasets were analyzed for differential binding using DiffBind/2.16.0 (with default parameter settings)83. Transcription factor binding motif enrichment analyses of identified regulatory elements were performed using the HOMER software package (with default parameter settings)29. Interaction analysis was performed using v12.0 of the full STRING network on the String database30,31 using default settings. UpSet32 analysis was performed to intersect HOMER FindMotif output between esophageal eosinophil enriched transcription factors using the command line tool Intervene84.
Murine bulk RNA sequencing
Bulk RNA sequencing data from purified esophageal and bone marrow eosinophils from CC10-iIL13Tg mice were obtained from publicly available data (GEO accession: GSE81135)2.
Murine esophageal single-cell RNA sequencing single-cell capture and library preparation
Single-cell transcriptome analysis of the murine esophagus was performed using the BD Rhapsody Single-Cell Analysis System (BD, Bioscience). Cells were pooled from 4‒8 mice per sample. Tissue processing was performed as above. Samples were labeled with sample tags (BD Mouse Single-Cell Multiplexing Kit; 633793) according to the manufacturer’s protocol. Briefly, cells were resuspended at 0.8-1.5 × 106/mL in staining buffer and incubated with the respective sample tag for 20 minutes at room temperature. Cells were washed twice before counting and resuspended in 300 mL of sample buffer. Next, cells from up to 4 barcoded samples were pooled for a total of 60,000‒80,000 cells and resuspended in 650 mL BD Sample BD Sample Buffer supplemented with 1:1000 SUPERase in (20 U µL−1, Thermo Fisher Scientific; AM2694) and NxGen Rnase Inhibitor (40 U µl−1; Lucigen; 30281-2), as dictated by the protocol. BD Rhapsody cartridges were super-loaded with 80,000 cells each. Single cells were isolated with the BD Rhapsody Express Single-Cell Analysis System according to the manufacturer’s recommendations (BD Biosciences). The cDNA libraries were prepared using the BD Rhapsody Whole Transcriptome Analysis Amplification Kit (BD Biosciences; 633801) following the BD Rhapsody System mRNA Whole Transcriptome Analysis (WTA) and Sample Tag Library Preparation Protocol (BD Biosciences). The final libraries were quantified using the Qubit dsDNA HS (Thermo Fisher Scientific; Q33230) and the TapeStation D1000 dsDNA HS Kit (Thermo Fisher Scientific; 5067-5584). Sequencing was performed using an S2 flow cell, PE-100bp on the NovaSeq6000, with NovaSeq6000 S2 Reagent Kit v1.5. Reads were calculated using the quantification table provided by BD Rhapsody with targets reads of 40,000 per cell for the WTA libraries and 10,000 per cell for the sample tag libraries. Sequencing was performed in the Cincinnati Children’s Hospital Medical Center (CCHMC) Genomics Sequencing Facility, Cincinnati, Ohio.
Murine esophageal single-cell RNA sequencing data pre-processing and normalization and differential gene expression
The raw scRNA-seq FASTQ files were aligned to the GRCm38 reference genome using the BD Rhapsody WTA Analysis Pipeline v1.12.1 available on the Seven Bridges Genomics platform. The feature-barcode matrices generated from multiple runs were merged and converted to the commonly used Market Exchange (MEX) format. All subsequent data analysis steps were conducted in SciDAP, following the guidelines from Kotliar et al.85. Low-quality cells were removed using the following filtering thresholds: remove doublets, include cells that have ≥ 100 but ≤ 5000 expressed genes, exclude cells with < 500 unique molecular counts and with > 15% of transcripts mapped to mitochondrial genes. Sample integration and merging was performed, and each of the merged samples was normalized with SCTransform. Next, normalized samples were integrated using the 3000 top scored genes. Dimensionality reduction was performed on the integrated dataset using principal component analysis (PCA) with 40 dimensions. For visualization purposes, PCA results were projected with the uniform manifold approximation and projection (UMAP) algorithm using all 40 dimensions. Clustering of dimensionally reduced data was conducted using 40 dimensions and 0.3 clustering resolution. Downstream analysis was conducted in R v.4.2.1 with the package Seurat v.4.3.0. Gene markers for each cluster were identified using the FindAllMarkers function with the default parameters. Top-ranked genes were used to define clusters. Clusters were annotated manually based on marker gene expression. Eosinophils were subclustered by sub-setting populations that expressed Siglecf, Ccr3 and Il5ra. GO analysis for Biological Pathways was performed using Enrichr80,81, and the top unique pathways were selected based on adjusted P-value. Differential gene expression analysis between clusters was performed using the FindMarkers function using the Wilcoxon Rank Sum test. A minimum expression threshold was set using min.pct = 0.1, ensuring that only genes expressed by at least 10% of cells in either group were tested. Differential expression between datasets was performed using Pseudobulk analysis. Raw reads per cell count were summed up per dataset and processed with the DESeq2 R package86. Genes were considered significant with a log2FC of > 0.1 and a pAdj[FDR] < 0.05. Results were visualized using Seurat’s plotting functions, including VlnPlot, FeaturePlot, DimPlot and DotPlot.
Collection of human specimens
All human material was collected under the approved CCHMC IRB protocol 2008-0090. Recruited volunteers provided written informed consent.
Isolating human peripheral blood eosinophils and co-culture with epithelial cells
Eosinophils were isolated from healthy volunteers as described33. An immortalized human esophageal epithelial cell line (EPC2)87, gifted by Dr. Anil Rustgi, was used for co-culture experiments. Prior to use, EPC2 cells were subjected to short tandem repeat (STR) profiling (Genetica Inc.). The STR profile exhibited a 100% match to the reference profile, and the cells were mycoplasma negative. EPC2 cells were seeded at 5 × 105 cells/well of a 24-well plate. For coculture, a 1:1 mixture of eosinophil culture media and KSFM was used. Human eosinophils were added at a concentration of 1 × 106 cells/mL (4 × 105 cells/well), and half of the media was replaced every 48 h for the duration of the experiment (7 days). Following co-culture, eosinophils were isolated from non-adherent cells by CD45 expression. Bulk RNA-sequencing of human eosinophil co-culture experiments was obtained from previously published data33.
Human esophageal RNA sequencing
The EoE transcriptome was derived from differentially expressed genes from bulk RNA sequencing of human esophageal biopsies taken from 6 healthy controls and 10 patients with active EoE; (GEO accession: GSE58640)36. The EDP dataset comprised gene expression values from 147 active EoE, 51 inactive EoE, and 48 normal control (NL; non-lesion) individual bulk esophageal RNA samples88. Human esophageal scRNA-seq data were compiled from 6 biopsies obtained from patients with EoE with > 15 eosinophils per high-power field in the esophageal biopsy from previously published data (GEO accession: GSE175930)15,35. Eosinophils were identified by hallmark gene expression, including CLC, SIGLEC8, CCR3, and IL5RA. Differential expression analysis was performed using the FindMarkers function to identify genes significantly enriched in eosinophil populations compared to the other cell populations identified within the esophageal biopsy, revealing 898 genes significantly enriched in eosinophils.
Stimulating human esophageal biopsies with recombinant human C3a
Human esophageal biopsies were procured from 4 patients with active EoE. Briefly, 3 biopsies per patient were obtained from the distal esophagus and submerged in RPMI 1640 + 10% FCS. Biopsies were processed as above (Preparing single-cell suspensions from tissues). Cells were washed in PBS and centrifuged prior to resuspension and divided into stimulated, unstimulated, and unstained samples. Samples were incubated with human Fc block (BD; 564219) for 5 min prior to staining with CD45 (PerCP, Biolegend; 304026), SIGLEC8 (PE, Biolegend; 347104), FcεR1 (PerCP-eFlour710, Invitrogen; 46-5899-84), and C3aR1 (BV750, BD; 746906 [isotype, mouse IgG2bK; 568006]) for 25 min on ice, followed by Live/Dead Blue fixable viability dye (Invitrogen; L34961) for an additional 5 min. Biopsies were rested at 37 °C for 30 min prior to stimulation with 200 ng/400 mL human recombinant C3a (R&D Systems, 3677-C3) for 5 min at 37 °C. Samples were fixed with 4% PFA for 15 minutes, prior to overnight permeabilization (eBioscience, 00-8333-56). Finally, samples were stained with anti-ERK1/2 (AF647, Cell Signaling, #13148) for 30 min before flow cytometry analysis was performed on the Cytek Aurora 5 L (Cytek Bioscience).
Ribonucleoprotein CRISPR/Cas9 nucleofection of EOL-1 cells
The EOL-1 cell line89 was supplied by the European Collection of Cell Cultures (ECACC; Salisbury, United Kingdom) as catalog number 94042252 and purchased from Sigma Aldrich. Nucleofection of EOL-1 cells was performed using the Neon Transfection System to delete ATF3 and/or JUN (sgRNA information Supplementary Table 3). EOL-1 cells were examined and had > 95% viability, and the media (RPMI + 10% FCS + 1% PSG) was refreshed the day before transfection to remove dead cells.
To anneal CRISPR RNAs (crRNA) and trans-activating CRISPR RNAs (tracrRNA), RNA oligos (200 mM) were resuspended in IDTE buffer as per manufacturers guidelines. Following reconstitution, crRNA and trans-activating tracrRNA were combined with IDTE buffer to a final volume of 10 mL, incubated on a heat block at 95 °C for 5 min, and then removed from heat and allowed to cool to room temperature.
Prior to ribonucleoprotein (RNP) CRISPR/Cas9 nucleofection, media (RPMI + 20% FCS, no antibiotics) was pre-warmed at 37 °C. Cas9-GFP enzyme (62 mM stock) was diluted to a working concentration with resuspension ‘Buffer R’, and the RNP complex was combined with the diluted Cas9 enzyme in a 1:1 ratio and incubated at room temperature for 10‒20 min. While incubating, EOL-1 cells were resuspended in an appropriate volume for nucleofection (3 × 105 cells/well in a 12-well plate) prior to centrifugation and washing in PBS. Following final centrifugation and removal of supernatant, cells were resuspended in 9 µL resuspension ‘Buffer R’ prior to adding of 1 µL RNP complex. A volume of 10 µL of the cell/RNP was aspirated using the Neon Tip with the Neon Pipette. The Neon Tip containing the cell/RNP mixture was inserted into the Pipette Station and electroporated at 1475 V for 20 ms with 2 pulses. Following electroporation, cells were transferred to wells containing pre-warmed media and incubated at 37 °C for 72 h.
For selecting single-cell clones, cells were sorted into 96-well U-bottom plates as single cells using the BDFACSMelody Cell Sorter, selecting for live cells using a viability dye (7-AAD). The single sorted cells were cultured in standard growth media for 2 weeks, with media added after 1 week, and were selected for further screening after colonies were visible to the naked eye.
To confirm gene targeting by western blot and Sanger sequencing, a sample of each colony was transferred to a 96-well PCR plate. The cells were pelleted by centrifugation, the supernatant was removed, and QuickExtract DNA Extract Solution was added to each well. The plate was heated in a thermal cycler at 65 °C for 6 min followed by 98 °C for 2 min. A 30 µL PCR reaction was set up using 5 µL of the QuickExtract DNA and Q5 High-Fidelity DNA Polymerase according to the manufacturer’s instructions. Unpurified PCR products of the correct PCR amplicon size were sequenced by the Australian Genome Research Facility (Sydney) by Sanger sequencing to identify successfully edited clones. Sequencing traces were aligned to the wild-type sequence using SnapGene software (v5.1) to identify clones containing successful modifications. Clones that contained homozygous insertion or deletion mutations that were not a multiple of 3 (e.g., 7-bp deletion) were expanded and screened by western blot to identify clones in which no protein could be detected.
For western blotting of ATF3 or JUN, 50 µg of nuclear protein extracts were denatured by heating in 6X SDS sample buffer at 95 °C for 5 minutes. Samples were loaded onto a 4–12% NuPAGE Bis-Tris Precast Gel in an Invitrogen Novex Mini-Cell Electrophoresis tank in 1X NuPAGE MES SDS Running Buffer for 50 min at 200 V. A volume of 7 µL Amersham ECL Rainbow Marker (full range) was electrophoresed on each gel as a size reference. Proteins were transferred onto a nitrocellulose membrane in transfer buffer (25 mM Tris, 0.2 M glycine and 20% (v/v) methanol) using the Invitrogen Novex Mini-Cell Electrophoresis tank and Blot Module wet-transfer system at 30 V for 60 minutes. The membranes were blocked with 3.5% skim milk (w/v) in TBST (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, and 0.1% Tween-20) twice for 1 hour each, followed by two 5 min washes in TBST. The membranes were incubated overnight at 4 °C with 1:1000 anti-ATF3 antibody (ab207434, Abcam) or 1:1000 anti-JUN antibody (ab40766, Abcam) in 5% skim milk (w/v) in TBST. The next day, membranes were washed 5 times for 5 min each with TBST and then incubated with horseradish peroxidase (HRP)-linked anti-Rabbit IgG secondary antibody (NA934, GE Healthcare) diluted 1:15,000 in TBST for 1 h at room temperature. Membranes were washed 5 times for 5 min each with TBST and then visualized using the ImageQuant LAS 500 imager in the presence of Immobilon Western Chemiluminescent HRP substrate. Blots were quickly washed 5 times with water, stripped for 8 minutes using 0.5 M NaOH, washed again for 5 times with RO water, and re-blocked with skim milk for 2 times of 15 minutes each. The blots were then probed with anti-β-actin antibody (A1978, Sigma) diluted 1:20,000 in TBST for 1 h at room temperature. Following 5 cycles of 5 min TBST washes, blots were incubated with HRP-linked anti-mouse IgG (NA931, GE Healthcare) secondary antibody diluted 1:20,000 in TBST for 1 h at room temperature and then imaged as described above. Antibody information in Supplementary Table 2.
Co-culture of transcription factor KO EOL-1 cells with epithelial cells
EPC2 cells were seeded at 2.5 × 105 cells/well of a 48-well plate. For coculture, a 1:1 mixture of eosinophil culture media and KSFM was used. WT and transcription factor KO EOL-1 cells were added at a concentration of 1 × 106 cells/mL (2.5 × 105 cells/well), and half of the media was replaced every 48 h for the duration of the experiment. Following co-culture, EOL-1 were collected, washed and processed for bulk RNA sequencing. Sequencing was performed (PE-100bp) on the NovaSeq X in the CCHMC Genomics Sequencing Core, Cincinnati, Ohio.
Data analysis
Data were analyzed with GraphPad Prism software (v10; GraphPad Software). A p < 0.05 cutoff was considered statistically significant. The number of replicates, individual experiments and information on statistical tests and post-hoc tests for each dataset are described in the appropriate figure legends. All analysis was conducted in a blinded manner. Samples were randomized and assigned numerical identifiers that did not reflect their experimental group allocation. Group identities were only revealed after completing data analysis.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data are included in the Supplementary Information or available from the authors, as are unique reagents used in this Article. The raw numbers for charts and graphs are available in the Source Data file whenever possible. The next-generation sequencing data have been deposited in NCBI’s Gene Expression Omnibus90 under the accession codes, GSE262178 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi), GSE287703 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi) and GSE287704 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi). A UCSS Genome Browser session containing the ATAC-seq data has been generated and is accessible at http://genome.ucsc.edu/s/Ledsall/esophageal_epigenome. Source data are provided in this paper.
References
Ignacio, A. et al. Small intestinal resident eosinophils maintain gut homeostasis following microbial colonization. Immunity 55, 1250–1267 (2022).
Sugawara, R. et al. Small intestinal eosinophils regulate Th17 cells by producing IL-1 receptor antagonist. J. Exp. Med. 213, 555–567 (2016).
Chu, V. T. et al. Eosinophils promote generation and maintenance of immunoglobulin-A-expressing plasma cells and contribute to gut immune homeostasis. Immunity 40, 582–593 (2014).
Mesnil, C. et al. Lung-resident eosinophils represent a distinct regulatory eosinophil subset. J. Clin. Invest. 126, 3279–3295 (2016).
Gurtner, A. et al. Active eosinophils regulate host defence and immune responses in colitis. Nature 615, 151–157 (2023).
Grisaru-Tal, S. et al. Metastasis-entrained eosinophils enhance lymphocyte-mediated antitumor immunity. Cancer Res. 81, 5555–5571 (2021).
Fulkerson, P. C. Transcription factors in eosinophil development and as therapeutic targets. Front. Med. 4, 115 (2017).
Bettigole, S. E. et al. The transcription factor XBP1 is selectively required for eosinophil differentiation. Nat. Immunol. 16, 829–837 (2015).
Wang, W.-L. et al. The aryl hydrocarbon receptor instructs the immunomodulatory profile of a subset of Clec4a4+ eosinophils unique to the small intestine. Proc. Natl. Acad. Sci. USA 119, e2204557119 (2022).
Diny, N. L. et al. The aryl hydrocarbon receptor contributes to tissue adaptation of intestinal eosinophils in mice. J. Exp. Med. 219, e20210970 (2022).
Schworer, S. A. et al. Notch 2 signaling contributes to intestinal eosinophil adaptations in steady state and tissue burden following oral allergen challenge. J. Leukoc. Biol. 116, 379–391 (2024).
Ben-Baruch Morgenstern, N. et al. Single-cell RNA sequencing of mast cells in eosinophilic esophagitis reveals heterogeneity, local proliferation, and activation that persists in remission. J. Allergy Clin. Immunol. 149, 2062–2077 (2022).
Ding, J. et al. An esophagus cell atlas reveals dynamic rewiring during active eosinophilic esophagitis and remission. Nat. Commun. 15, 3344 (2024).
Lim, M. et al. Esophageal ILC2s mediate abnormal epithelial remodeling in eosinophilic esophagitis via Areg-EGFR signaling. Cell Mol. Immunol. 22, 97–110 (2024).
Ben-Baruch Morgenstern, N. et al. Single-cell RNA-sequencing of human eosinophils in allergic inflammation in the esophagus. J. Allergy Clin. Immunol. 154, 974–987 (2024).
Liacouras, C. A. et al. Eosinophilic esophagitis: updated consensus recommendations for children and adults. J. Allergy Clin. Immunol. 128, 3–20.e6 (2011).
Zuo, L. et al. IL-13 Induces esophageal remodeling and gene expression by an eosinophil-independent, IL-13Rα2–inhibited pathway. J. Immunol. 185, 660–669 (2010).
Dent, L. A., Strath, M., Mellor, A. L. & Sanderson, C. J. Eosinophilia in transgenic mice expressing interleukin 5. J. Exp. Med. 172, 1425–1431 (1990).
Li, Y. et al. Neuromedin U programs eosinophils to promote mucosal immunity of the small intestine. Science 381, 1189–1196 (2023).
Wiese, A. V. et al. C5aR1 activation in mice controls inflammatory eosinophil recruitment and functions in allergic asthma. Allergy 119, 1893–1908 (2023).
Zuba-Surma, E. K., Kucia, M., Abdel-Latif, A., Lillard, J. W. & Ratajczak, M. Z. The ImageStream System: a key step to a new era in imaging. Folia Histochem. Cytobiol. 45, 279–290 (2007).
Voehringer, Rooijen, D., van, N. & Locksley, R. M. Eosinophils develop in distinct stages and are recruited to peripheral sites by alternatively activated macrophages. J. Leukoc. Biol. 81, 1434–1444 (2007).
Mattick, J. S., Amaral, P. P., Dinger, M. E., Mercer, T. R. & Mehler, M. F. RNA regulation of epigenetic processes. BioEssays 31, 51–59 (2009).
Hansen, A. S. & O’Shea, E. K. Encoding four gene expression programs in the activation dynamics of a single transcription factor. Curr. Biol. 26, R269–R271 (2016).
Dunham, I. et al. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Luo, Y. et al. New developments on the Encyclopedia of DNA Elements (ENCODE) data portal. Nucleic Acids Res. 48, D882–D889 (2020).
Kagda, M. S. et al. Data navigation on the ENCODE portal. Preprint at http://arxiv.org/abs/2305.00006 (2023).
Hitz, B. C. et al. The ENCODE uniform analysis pipelines. 2023.04.04.535623 Preprint at https://doi.org/10.1101/2023.04.04.535623 (2023).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Szklarczyk, D. et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 49, D605–D612 (2021).
Szklarczyk, D. et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 51, D638–D646 (2023).
Lex, A., Gehlenborg, N., Strobelt, H., Vuillemot, R. & Pfister, H. UpSet: Visualization ofintersecting sets. IEEE Trans. Vis. Comput. Graph 20, 1983–1992 (2014).
Dunn, J. L. M. et al. Bidirectional crosstalk between eosinophils and esophageal epithelial cells regulates inflammatory and remodeling processes. Mucosal Immunol. 14, 1133–1143 (2021).
Hai, T., Wolfgang, C. D., Marsee, D. K., Allen, A. E. & Sivaprasad, U. ATF3 and Stress responses. Gene Expr. 7, 321–335 (2018).
Morgan, D. M. et al. Clonally expanded, GPR15-expressing pathogenic effector TH2 cells are associated with eosinophilic esophagitis. Sci. Immunol. 6, eabi5586 (2021).
Sherrill, J. D. et al. Analysis and expansion of the eosinophilic esophagitis transcriptome by RNA sequencing. Genes Immun. 15, 361–369 (2014).
Kano, G., Almanan, M., Bochner, B. & Zimmermann, N. Eosinophil cell death determination: role for ERK in the mechanism regulating decision between eosinophil activation, apoptosis or regulated necrosis. Blood 120, 2134 (2012).
Mishra, A., Hogan, S. P., Brandt, E. B. & Rothenberg, M. E. IL-5 Promotes eosinophil trafficking to the rsophagus. J. Immunol. 168, 2464–2469 (2002).
Mishra, A. et al. Esophageal remodeling develops as a consequence of tissue specific IL-5-induced eosinophilia. Gastroenterology 134, 204–214 (2008).
Kellerman, K. et al. Identification of a neuroimmune circuit that regulates allergic inflammation in the esophagus. Preprint at https://doi.org/10.1101/2024.11.16.623883 (2024).
Huet, S. et al. CD44 contributes to T cell activation. J. Immunol. 143, 798–801 (1989).
Katoh, S. Critical involvement of CD44 in T helper type 2 cell-mediated eosinophilic airway inflammation in a mouse model of acute asthma. Front. Immunol. 12, 811600 (2021).
Rothenberg, M. E. CD44–a sticky target for asthma. J. Clin. Invest. 111, 1460–1462 (2003).
Huebener, P. et al. CD44 is critically involved in infarct healing by regulating the inflammatory and fibrotic response. J. Immunol. 180, 2625–2633 (2008).
Henke, C., Bitterman, P., Roongta, U., Ingbar, D. & Polunovsky, V. Induction of fibroblast apoptosis by anti-CD44 antibody: implications for the treatment of fibroproliferative lung disease. Am. J. Pathol. 149, 1639–1650 (1996).
Teder, P. et al. Resolution of lung inflammation by CD44. Science 296, 155–158 (2002).
Svee, K. et al. Acute lung injury fibroblast migration and invasion of a fibrin matrix is mediated by CD44. J. Clin. Invest. 98, 1713–1727 (1996).
Hart, S. P., Dougherty, G. J., Haslett, C. & Dransfield, I. CD44 regulates phagocytosis of apoptotic neutrophil granulocytes, but not apoptotic lymphocytes, by human macrophages. J. Immunol. 159, 919–925 (1997).
Sano, K. et al. CD44 expression on blood eosinophils is a novel marker of bronchial asthma. Int. Arch. Allergy Immunol. 114, 67–71 (1997).
Zhang, S. et al. Effects of C3aR activated mast cells in eosinophilic esophagitis. Preprint athttps://doi.org/10.1101/2025.05.23.655738 (2025).
Doyle, A. D., Masuda, M. Y., Kita, H. & Wright, B. L. Eosinophils in eosinophilic esophagitis: the road to fibrostenosis is paved with good intentions. Front. Immunol. 11, 603295 (2020).
Stein, M. L. et al. Anti-IL-5 (mepolizumab) therapy for eosinophilic esophagitis. J. Allergy Clin. Immunol. 118, 1312–1319 (2006).
Straumann, A. et al. Anti-interleukin-5 antibody treatment (mepolizumab) in active eosinophilic oesophagitis: a randomised, placebo-controlled, double-blind trial. Gut 59, 21–30 (2010).
Assa’ad, A. H. et al. An antibody against IL-5 reduces numbers of esophageal intraepithelial eosinophils in children with eosinophilic esophagitis. Gastroenterology 141, 1593–1604 (2011).
Spergel, J. M. et al. Reslizumab in children and adolescents with eosinophilic esophagitis: results of a double-blind, randomized, placebo-controlled trial. J. Allergy Clin. Immunol. 129, 456–463 (2012).
Conus, S., Straumann, A., Bettler, E. & Simon, H.-U. Mepolizumab does not alter levels of eosinophils, T cells, and mast cells in the duodenal mucosa in eosinophilic esophagitis. J. Allergy Clin. Immunol. 126, 175–177 (2010).
Zhou, J. et al. Eosinophils promote CD8+ T cell memory generation to potentiate anti-bacterial immunity. Signal Transduct. Target Ther. 9, 43 (2024).
Efraim, A. H. N. B., Eliashar, R. & Levi-Schaffer, F. Hypoxia modulates human eosinophil function. Clin. Mol. Allergy. CMA 8, 10 (2010).
Tabary, M., Gheware, A., Peñaloza, H. F. & Lee, J. S. The matricellular protein thrombospondin-1 in lung inflammation and injury. Am. J. Physiol. Cell Physiol. 323, C857–C865 (2022).
Hoshino, M., Nakamura, Y. & Hamid, Q. A. Gene expression of vascular endothelial growth factor and its receptors and angiogenesis in bronchial asthma. J. Allergy Clin. Immunol. 107, 1034–1038 (2001).
Brigger, D. et al. Eosinophils regulate adipose tissue inflammation and sustain physical and immunological fitness in old age. Nat. Metab. 2, 688–702 (2020).
Arita, M. Eosinophil polyunsaturated fatty acid metabolism and its potential control of inflammation and allergy. Allergol. Int. 65, S2–S5 (2016).
Wu, D. et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science 332, 243–247 (2011).
Felton, J. M. et al. Aiolos regulates eosinophil migration into tissues. Mucosal Immunol. 14, 1271–1281 (2021).
Knights, A. J. et al. Eosinophil function in adipose tissue is regulated by Krüppel-like factor 3 (KLF3). Nat. Commun. 11, 2922 (2020).
Escoubet-Lozach, L., Glass, C. K. & Wasserman, S. I. The role of transcription factors in allergic inflammation. J. Allergy Clin. Immunol. 110, 553–564 (2002).
Gilchrist, M. et al. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature 441, 173–178 (2006).
Gilchrist, M. et al. Activating transcription factor 3 is a negative regulator of allergic pulmonary inflammation. J. Exp. Med. 205, 2349–2357 (2008).
Shah, M., Knights, A. J., Vohralik, E. J., Psaila, A. M. & Quinlan, K. G. R. Blood and adipose-resident eosinophils are defined by distinct transcriptional profiles. J. Leukoc. Biol. 113, 191–202 (2023).
Fulkerson, P. C., Fischetti, C. A., Hassman, L. M., Nikolaidis, N. M. & Rothenberg, M. E. Persistent Effects Induced by IL-13 in the Lung. Am. J. Respir. Cell Mol. Biol. 35, 337–346 (2006).
Cheng, Y.-C. et al. Topoisomerase I inhibition and peripheral nerve injury induce DNA breaks and ATF3-associated axon regeneration in sensory neurons. Cell Rep. 36, 109666 (2021).
Doyle, A. D. et al. Homologous recombination into the eosinophil peroxidase locus generates a strain of mice expressing Cre recombinase exclusively in eosinophils. J. Leukoc. Biol. 94, 17–24 (2013).
Ben Baruch-Morgenstern, N. et al. Paired Ig-like Receptor B Inhibits IL-13-Driven Eosinophil Accumulation and Activation in the Esophagus. J. Immunol. 197, 707–714 (2016).
Corces, M. R. et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017).
Felton, J. M. et al. Epigenetic analysis of the chromatin landscape identifies a repertoire of murine eosinophil-specific PU.1-bound enhancers. J. Immunol. 207, 1044–1054 (2021).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12 (2011).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Gaspar, J. M. Improved peak-calling with MACS2. Preprint at https://doi.org/10.1101/496521 (2018).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Chen, E. Y. et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinforma. 14, 128 (2013).
Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Ross-Innes, C. S. et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393 (2012).
Khan, A. & Mathelier, A. Intervene: a tool for intersection and visualization of multiple gene or genomic region sets. BMC Bioinform. 18, 287 (2017).
Kotliar, M., Kartashov, A. & Barski, A. Accelerating single-cell sequencing data analysis with SciDAP: a user-friendly approach. Preprint at https://doi.org/10.1101/2024.02.28.582604 (2024).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Harada, H. et al. Telomerase induces immortalization of human esophageal keratinocytes without p16INK4a inactivation1. Mol. Cancer Res. 1, 729–738 (2003).
Shoda, T. et al. Eosinophilic esophagitis endotype classification by molecular, clinical, and histopathological analyses: a cross sectional study by the Consortium of Eosinophilic Gastrointestinal Disease Researchers (CEGIR). Lancet Gastroenterol. Hepatol. 3, 477–488 (2018).
Saito, H. et al. Establishment and characterization of a new human eosinophilic leukemia cell line. Blood 66, 1233–1240 (1985).
Edgar, R., Domrachev, M. & Lash, A. E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210 (2002).
Shoda, T. et al. Desmoplakin and periplakin genetically and functionally contribute to eosinophilic esophagitis. Nat. Commun. 12, 6795 (2021).
Acknowledgements
This work was supported in part by the National Institutes of Health (NIH) R01 AI045898, R01 AI24355, P30 DK078392 (Genomics Sequencing Core of the Digestive Diseases Research Core Center in Cincinnati); NIH P30AR070549 and CCHMC ARC Award #53632 to M.T.W.; the Campaign Urging Research for Eosinophilic Disease (CURED); the Buckeye Foundation; and the Sunshine Charitable Foundation and its supporters, Denise A. Bunning and David G. Bunning. Supported in part by grant #PNC23-216751 from the American Association for the Study of Liver Diseases Foundation (T.D.T). This work was supported by an Australian National Health and Medical Research Council grant 2020483 (to K.G.R.Q). All flow cytometric data were acquired using equipment maintained by the Research Flow Cytometry Facility in the Division of Rheumatology at Cincinnati Children’s Hospital Medical Center, supported by NIH S10OD025045. We wish to thank Shawna Hottinger for her editorial assistance and Dr. Anil Rustgi for gifting the EPC2 cell line. This manuscript is the result of funding in whole or in part by the National Institutes of Health (NIH). It is subject to the NIH Public Access Policy. Through acceptance of this federal funding, NIH has been given a right to make this manuscript publicly available in PubMed Central upon the Official Date of Publication, as defined by NIH.
Author information
Authors and Affiliations
Contributions
J.M.F. and M.E.R. conceptualized this project and designed the experiments. J.M.F., L.M., N.B.-B.M., M.R., A.M.K., G.A.O., S.Z., B.W. and J.M.C. performed experiments. J.M.F., T.D.T., G.A.O. and H.R. analyzed data. A.M.P., J.L.M.D., T.S. and K.G.R.Q. provided pre-existing datasets. A.M.P. and K.G.R.Q. edited EOL-1 cell lines. J.M.F., L.E.E., M.K., A.B. and M.T.W. conducted bioinformatic analysis. J.M.F., T.D.T., H.R., M.T.W. and M.E.R. wrote the paper. All authors discussed the results and approved the paper before submission.
Corresponding authors
Ethics declarations
Competing interests
J.M.F., L.E.E., T.D.T., L.M., M.K., N.B-B.M., A.M.P., M.R., A.M.K., G.A.O., S.Z., J.L.M.D., B.W., J.M.C., R.H., K.G.R.Q. and M.T.W. have no conflicts of interest to disclose. A.B. is a co-founder of Datirium. M.E.R. is a consultant for Pulm One, Spoon Guru, ClostraBio, Serpin Pharm, Allakos, Celldex, Uniquity Bio, Santa Ana Bio, EnZen Therapeutics, Bristol Myers Squibb, Astra Zeneca, Pfizer, Glaxo Smith Kline, and Regeneron/Sanofi, and Guidepoint and has an equity interest in the first nine listed and royalties from reslizumab (Teva Pharmaceuticals), PEESSv2 (Mapi Research Trust), and UpToDate. M.E.R. is an inventor of patents owned by Cincinnati Children’s Hospital Medical Center. T.S. is a co-inventor of patents owed by Cincinnati Children’s Hospital Medical Center.
Peer review
Peer review information
Nature Communications thanks Christophe Desmet, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Felton, J.M., Edsall, L.E., Troutman, T.D. et al. Epigenetic and transcriptional programming of murine eosinophils in the esophagus. Nat Commun 16, 10454 (2025). https://doi.org/10.1038/s41467-025-65440-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65440-5
