
Abstract
Broadly neutralizing antibodies (bnAbs) prevent HIV infection but face administration and cost challenges. We present single-chain mRNA-encoded bnAbs that improve heavy-light chain assembly and enable three enhancement approaches: co-expression, isotype selection, and engineered nanobodies. We observe enhanced in vitro neutralization through co-expression of PGT121 and VRC07 (targeting V3-glycan and CD4-binding site, respectively) and by replacing IgG constant heavy chain (IgG-CH) with IgA-CH or incorporating an IgM tailpiece into IgG-CH. While IgG versions fail to neutralize several SHIV/HIV strains, co-expressing PGT121 and VRC07 as IgM-like multimers restores neutralizing capability (IC50 < 100 ng/mL) and substantially improves breadth. Vaginal explants from rhesus macaques, treated with intravaginal aerosolized mRNAs, show robust protection against ex vivo challenges with multiple SHIV strains when PGT121 and VRC07 are co-expressed as IgM-like multimers. Our data present novel drug compositions for intravaginal mRNA delivery to prevent HIV acquisition and advance antibody-design strategies that enhance breadth and potency of existing bnAbs.
Similar content being viewed by others

Introduction
Human immunodeficiency virus (HIV) remains a significant global health challenge, with approximately 39 million people living with HIV in 2022 and 1.3 million new infections reported in the same year1. Despite advances in treatment, a definitive cure for HIV infection remains elusive, necessitating lifelong administration of antiretroviral drugs to suppress viral replication. This emphasizes the critical need for efficacious preventative strategies to suppress the spread of HIV. Pre-exposure prophylaxis (PrEP) and post-exposure prophylaxis regimens, which involve daily oral administration of antiretrovirals such as tenofovir and emtricitabine, or more recent long-acting formulations, have demonstrated efficacy in preventing HIV infection, particularly among men who have sex with men (MSM)2,3,4. However, their effectiveness in heterosexual women is less certain, primarily due to challenges in adherence to PrEP and significantly lower retention of antiretrovirals in vaginal tissue compared to rectal tissue5. Moreover, tenofovir-based PrEP has been associated with renal and bone toxicity, raising concerns about its long-term safety profile6. Broadly neutralizing antibodies (bnAbs) have emerged as a promising alternative due to their ability to target multiple strains of HIV, with numerous clinical trials demonstrating that bnAbs, such as PGT121 and VRC07, can provide robust protection against HIV7,8. Although systemically administered bnAbs have shown promising antiviral properties, their practical use is limited by the large doses required to achieve protective antibody concentrations in genital secretions and tissues7,9, the high manufacturing costs of purified antibodies10, and the potential development of anti-drug antibodies11. Direct vaginal application of bnAbs was assessed as a potential on-demand approach, but it showed rapid elimination and poor stability of bnAbs in acidic human cervicovaginal secretions12. These limitations highlight the need for alternative formulations and delivery methods that can improve adherence and achieve protective drug concentrations in relevant tissues while minimizing adverse effects and costs.
The field of messenger RNA (mRNA) has made remarkable progress over the past decade, establishing itself as a promising approach for encoding and producing a wide range of proteins13,14,15,16. This progress has led to the development of in vitro transcribed mRNA-based antibody approaches, allowing antibody expression via mRNA delivery in target cells or tissues16. mRNA-encoded antibodies offer several advantages, including low immunogenicity achieved through RNA modifications17 and in situ protein production, as well as potential cost-effectiveness compared to traditional purified antibody manufacturing processes18. Due to the excellent permeability and absorption of compounds through the vaginal mucosa, the vaginal route of administration has been investigated as an alternative to systemic delivery, with numerous advantages, such as lower dosage requirements, ease of application, and reduced adverse effects19,20. Our previous study on intravaginal aerosol delivery of mRNA-encoded bnAb has demonstrated its potential, suggesting that mRNA technology, coupled with intravaginal delivery, could address many limitations associated with current HIV prevention strategies21, but breadth and potency still required optimization.
Here, we introduce mRNA-based approaches that enhance the breadth and potency of existing bnAbs through single-chain antibody design with improved heavy-light chain assembly, exploring three enhancement strategies: antibody co-expression, isotype selection, and nanobody engineering. We confirm expression by cell staining and Western blotting and assess function using binding kinetic measurements and in vitro neutralization assays against diverse SHIV/HIV strains, demonstrating that these strategic modifications can overcome neutralization resistance observed with conventional IgG formats. Notably, isotype and valency markedly affect the neutralizing potency of defined bnAbs against specific viral isolates, with findings corroborated in ex vivo viral challenges of vaginal explants from rhesus macaques following intravaginal mRNA dosing. We demonstrate that single-chain mRNA-encoded modified bnAbs provide a more effective and user-controlled HIV prevention strategy for women while offering advantages over traditional protein-based prophylaxis. This approach emphasizes the need to revisit antibody design strategies to further optimize clinically ready antibodies and can be extended to prevent other sexually transmitted infections, establishing a versatile platform for mucosal immunity.
Results
Single-chain mRNA-encoded IgGs are expressed with enhanced assembly compared to conventional separate heavy and light chain mRNA system
Several studies have demonstrated the expression of IgGs from mRNAs by using two mRNAs encoding the heavy and light chains, respectively. However, the optimal assembly ratio of heavy and light chains varies for each IgG, typically ranging from 1:1 to 4:121,22,23,24,25 (Fig. 1a). To optimize the heavy and light chain assembly, we designed single-chain mRNAs by connecting the heavy and light chains with 12 repeats of Gly-Gly-Gly-Gly-Ser (G4S) linker26 (Fig. 1b). Lindsay et al. previously reported dual-chain mRNA-encoded PGT121 in two formats: secreted and glycosylphosphatidylinositol (GPI)-anchored IgGs, demonstrating more durable antibody expression with the GPI-anchored IgG in sheep21. Building on this work, we developed a single-chain version of the GPI-anchored PGT121 IgG (SC-PGT121 aIgG). While SC-PGT121 aIgG is expected to persist longer on the cell membrane, it was revealed that its molecular weight and neutralizing capability remained comparable to its secreted format (SC-PGT121 sIgG) (Supplementary Fig. 1).

a Schematic of antibody expression using the conventional dual-chain approach with separate heavy chain (HC) and light chain (LC) mRNAs. The IgG HC mRNA encodes the variable (VH) and constant (CH) domains of the heavy chain, while the IgG LC mRNA encodes the variable (VL) and constant (CL) domains of the light chain. Both mRNAs are delivered into the cytoplasm and translated into HC and LC proteins, respectively. The HC and LC are assembled in the endoplasmic reticulum, transported to the cell membrane, and secreted outside the cells. b Schematic of single-chain (SC) mRNA-based antibody expression. The LC and HC mRNAs are linked into a single mRNA strand using 12 repeats of Gly-Gly-Gly-Gly-Ser (G4S) linker ((G4S)12). The SC-IgG mRNA is translated into a fully assembled HC-LC complex. c Representative immunostaining images of aIgG expression in A549 cells. Glycosylphosphatidylinositol (GPI)-anchored PGT121s (PGT121 aIgG) expressed from dual- or single-chain mRNAs localize on the cell membrane (AF-488, green). The nuclei were stained with DAPI (blue). The Scale bar represents 20 µm. d Quantification of PGT121 aIgG expression in Expi293F cells from dual- and single-chain mRNAs. A total of 75 µg of each mRNA was used for transfection in 75 × 106 Expi293F cells. Data from n = 3 independent experiments are shown as individual values and their mean. e Capillary electrophoresis-based Western blot (JESS) analysis of PGT121 aIgG expression from dual- or single-chain mRNAs produced in Expi293F cells. The antibodies were detected using HRP-conjugated anti-human IgG antibody, with chemiluminescence signals normalized to the highest value. f JESS analysis of single-chain mRNA-expressed PGT121 aIgG under reducing conditions. g Combinational treatment of multiple antibodies. In the dual-chain mRNA system, partial or complete heavy-light mismatched antibodies (highlighted in red and purple) can form, whereas the SC mRNA system produces antibodies with correctly paired heavy and light chains. h TZM-bl neutralization assays against tier 2 SHIV strains from clades A, B, and C. The 50% inhibitory concentrations (IC50) are summarized in the table. All neutralization assays were performed in duplicates and repeated at least twice in independent assays (*: IC50 values from literature). i Correlation of IC50 values between mRNA-encoded and conventional bnAbs from in vitro neutralization assays. Statistical comparison by Pearson correlation: r(10) = 0.6921, p = 0.0126 (two-tailed), 95% CI [0.1960, 0.9061]. a, b, g Created in BioRender. Santangelo, P. (2025) https://BioRender.com/fowauum.
To compare the novel single-chain mRNA system with the conventional dual-chain system, we transfected A549 cells with each mRNA. Staining the expressed IgGs using anti-human secondary antibodies revealed similar expression on the cell membrane (Fig. 1c). Additionally, we produced both IgGs in the Expi293F expression system for further characterization. As the expressed IgGs were bound to the cell surface by GPI anchors, we treated the cells with phosphatidylinositol-specific phospholipase C (PI-PLC), which cleaves the GPI-anchor and releases the anchored IgGs from the membrane, allowing for collection from supernatant fluids. The expression levels of PGT121 aIgGs from dual- and single-chain mRNAs were measured using ELISA, revealing 1.5× higher IgG production by the single-chain system (Fig. 1d). We then performed capillary-based Western blot (JESS) to visualize the expressed IgGs and determine their molecular weights. In both single- and dual-chain mRNA-expressed PGT121 aIgGs, fully assembled IgGs with major bands around 180 kDa were confirmed (Fig. 1e). However, in the dual mRNA system, an additional band was detected at around 150 kDa, suggesting the presence of heavy-chain-only IgGs, which lost their pairing light chain. In the single-chain system, a minor band was observed at approximately 260 kDa. To investigate this, we treated the single-chain aIgG with 40 mM dithiothreitol to reduce disulfide bonds between single-chain monomers, resulting in a single 90 kDa band (Fig. 1f). This suggests that the 260 kDa band represents a single-chain trimer or 1.5 IgG, potentially increasing the number of binding sites. Overall, these results indicate that the single-chain mRNA system is a reliable alternative to the conventional dual-chain system, offering improved heavy and light chain assembly.
The single-chain mRNA format offers additional benefits for combinational treatment of bnAbs, a strategy frequently seen in several clinical trials7,8. Unlike the traditional dual-chain system, which can and will produce at least one arm of the antibody with mismatched heavy and light chain pairings, the single-chain system ensures the expression of antibodies with correctly paired heavy and light chains (Fig. 1g). Considering this, we designed six single-chain mRNAs encoding potent bnAbs (PGT12127, VRC0728,29,30, 10E8.431, iMab32,33, CAP25634, PGDM14008,35) in a GPI-anchored IgG format. The six aIgGs were expressed in A549 cells and isolated for Western blot analysis, showing IgG bands at around 180 kDa (Supplementary Fig. 2a, b). Their neutralizing capabilities were evaluated in in vitro TZM-bl neutralization assays against three tier 2 SHIV strains representing different clades. While PGDM1400 and CAP256 did not neutralize the tested SHIV strains, VRC07 exhibited the most efficient neutralizing capability (Fig. 1h and Supplementary Fig. 2c). For comparison, conventional recombinant bnAbs were also tested in parallel and showed neutralization trends consistent with their mRNA-encoded counterparts (Fig. 1h, i and Supplementary Fig. 2d). Based on these results, we selected PGT121 and VRC07 for a co-expression approach, targeting the V3-glycan36 and CD4 binding site29 of the HIV envelope (Env), respectively.
Enhanced neutralizing capability through multimerization of mRNA-encoded antibodies
Among the five isotypes of mammalian immunoglobulins (IgA, IgD, IgE, IgG, and IgM), only IgA and IgM can multimerize, providing higher avidity37, and these isotypes generally present better stability in mucosal environments38. We hypothesized that the potency of mRNA-encoded antibodies could be enhanced by increasing avidity through antibody multimerization. Without any modifications to the antibody binding sites (variable regions), we designed: (1) IgA-like PGT121 (PGT121 IgA) by replacing the IgG constant heavy chain (IgG-CH) with the IgA-CH, and (2) IgM-like PGT121 (PGT121 IgM-tail) by incorporating an IgM tailpiece sequence (PTLYNVSLVMSDTAGTCY) at the end of IgG-CH39 (Fig. 2a).

a Schematics of single-chain mRNA-based antibody modifications. A GPI-anchor sequence (purple) was inserted into the secreted IgG (sIgG, Supplementary Fig. 1) to produce GPI-anchored IgG (aIgG). Dimeric IgAs were made by replacing the IgG constant heavy (IgG-CH) domain with the IgA-CH (green), and their dimerization was facilitated by co-delivery of secreted J-chain (sJC) mRNA. A GPI-anchored J-chain (aJC) was designed by incorporating the GPI-anchor sequence into the sJC mRNA, enabling IgAs to attach to the cell membrane. The IgM-tail version was created by inserting IgM tailpiece sequence (red) at the end of IgG-CH domain, forming pentameric IgM-like antibodies. Created in BioRender. Santangelo, P. (2025) https://BioRender.com/yuy35vv. b Representative immunostaining images of sIgA and IgM-tail PGT121 expression in A549 cells. Dual- or single-chain mRNA-encoded IgAs and IgM-tails expressed in A549 cells were visualized with anti-human IgA or IgG antibodies with AF-488 (green), and the nuclei were stained with DAPI. The scale bar represents 20 µm. c Quantification of PGT121 sIgA and IgM-tail expressions in Expi293F cells from dual- and single-chain mRNAs. A total of 75 µg of each mRNA was used for transfection in 75 × 106 Expi293F cells. Data from n = 3 independent experiments are shown as individual values and their mean. d JESS Western blot analysis of PGT121 sIgA and IgM-tail. The peaks at approximately 170 kDa represent monomeric antibodies, while the peaks around 300 kDa indicate dimeric antibodies. e Traditional Western blot of mRNA-encoded PGT121 IgM-tail antibody. f TZM-bl neutralization assays. The neutralization efficacy of three different isotype antibodies (aIgG, sIgA, and IgM-tail) expressed from dual- or single-chain mRNAs was evaluated against tier 2 SHIV strains from clades A, B, and C, and their 50% inhibitory concentrations (IC50) are summarized in the table. All neutralization assays were performed in duplicates and repeated at least twice in independent assays.
To facilitate multimerization, we also designed mRNAs encoding the joining (J) chain, which plays a critical role in multimeric IgA and IgM formation. We optimized the ratio of the mRNA-encoded J-chain to mRNA-encoded antibodies (Ab: J-chain = 1 : 0.15) to maximize multimeric IgA formation and minimize monomeric IgA formation (Supplementary Fig. 3). The J-chain mRNA was designed in two forms: a secreted J-chain (sJC) for secreted IgA (SC-sIgA) and a GPI-anchored J-chain (aJC) for membrane-bound IgA (SC-aIgA). For IgM-tail, only sJC was used to support antibody multimerization in this study.
We compared the single-chain and conventional dual-chain systems for both IgA and IgM-tail formats, showing that both antibodies were expressed in A549 cells without observable differences (Fig. 2b). Expression levels of PGT121 sIgAs and IgM-tails from dual- and single-chain mRNAs were compared, demonstrating comparable levels of sIgAs and higher IgM-tail production in the single-chain system (Fig. 2c). Notably, increased formation of multimers of PGT121 sIgA and IgM-tail were detected in the single-chain system, with higher ratios of multimers (around 300 kDa) to monomers (around 180 kDa) (Fig. 2d). Given that JESS Western blots can only detect proteins up to 440 kDa, we conducted traditional Western blots to visualize multimers with higher molecular weights, such as pentamers and hexamers, which are naturally formed in IgM. Accordingly, the mRNA-encoded PGT121 IgM-tail clearly produced pentameric and hexameric populations (Fig. 2e).
We then compared the neutralizing capabilities of mRNA-expressed PGT121s from the single-chain and dual heavy/light chains platforms against three tier 2 SHIV strains from different clades, demonstrating no marked differences between both expression systems (Fig. 2f). However, we found a 2-log difference in the half-maximal inhibitory concentration (IC50) between PGT121 aIgG (~ 440 ng/mL) and PGT121 IgM-tail (~ 3 ng/mL) against SHIV-C109 (clade C), and a log difference between PGT121 aIgG and PGT121 sIgA (~ 30 ng/mL). These results imply that multimerization via isotype modifications can enhance the neutralizing ability of existing bnAbs.
Co-expressed, isotype-modified PGT121 and VRC07 exhibit enhanced potency and breadth compared to their individual IgG forms
Considering the advantages of the single-chain system and isotype modifications, we co-expressed PGT121 and VRC07 as different isotypes. Single-chain GPI-anchored PGT121 and VRC07 IgG (SC-PGT121 + SC-VRC07 aIgG) was produced by delivering each mRNA at a 1:1 ratio (Fig. 3a). Additionally, single-chain PGT121 and VRC07 secreted or GPI-anchored IgAs (SC-PGT121 + SC-VRC07 sIgA or aIgA, respectively) and IgM-tailed single-chain PGT121 and VRC07 (SC-PGT121 + SC-VRC07 IgM-tail) were produced using three mRNAs: PGT121 mRNA, VRC07 mRNA, and J-chain mRNA with or without the GPI-anchor sequence, at a 1:1:0.3 ratio. Antibody expression was confirmed in A549 cells without notable differences compared to individually expressed PGT121 and VRC07 (Fig. 3b). The antibodies were co-expressed in Expi293F cells and visualized by JESS Western blots, showing molecular weights consistent with the individual ones: around 180 kDa for aIgGs and approximately 180 and 320 kDa for monomeric and dimeric sIgAs and IgM-tails (Fig. 3c).

a Schematic of antibody co-expression. Two SC-mRNAs encoding different antibodies were co-transfected, resulting in the expression of a combined antibody (SC-aIgG #1 + 2) as well as the individual antibodies (SC-aIgG #1 and SC-aIgG #2) separately. Created in BioRender. Santangelo, P. (2025) https://BioRender.com/v81ivzo. b Representative immunostaining images of mRNA-expressed antibodies in A549 cells. The expression of PGT121, VRC07, and PGT121 + VRC07 (combinational treatment) with four different isotypes (anchored IgG, secreted IgA, anchored IgA, and IgM-tail) was visualized after staining with AF488-conjugated anti-human IgA or IgG antibodies. The scale bar represents 20 µm. c JESS Western blot analysis of SC-aIgG, SC-sIgA, and SC-IgM-tail variants of PGT121, VRC07, and PGT121 + VRC07. d, e TZM-bl neutralization assays against five SHIV and six HIV strains from different clades. f Summary of 50% inhibitory concentrations (IC50). All neutralization assays were performed in duplicate and independently repeated at least twice.
Next, we evaluated the neutralizing capability of nine antibodies produced from transduced Expi293F cells, representing three isotypes (SC-aIgG, SC-sIgA, and SC-IgM-tail) for two different bnAbs or their combination (PGT121, VRC07, and PGT121 + VRC07), against five SHIV and six HIV-1 strains (Fig. 3d–f). All antibodies demonstrated potent neutralization (IC50 < 100 ng/mL) against six strains: SHIV-KNH1144p2 (clade A), SHIV-162p3 (clade B), SHIV-1157ipd3N4 (clade C), SHIV-C109 (clade C), HIV-1 JRFL (clade B), and HIV-1 93/MW/965 (clade C). However, SC-PGT121 aIgG poorly neutralized SHIV-C109 (clade C) with an IC50 of 598.7 ng/mL, but its potency was dramatically enhanced upon co-expression with VRC07 aIgG (IC50 = 5.9 ng/mL) and when expressed in sIgA and IgM-tail formats, reducing IC50 values to 17.7 ng/mL and 1.0 ng/mL, respectively. All aIgG variants failed to neutralize SHIV-AE16 (clade AE), HIV-1 92/RW/024 (clade A), HIV-1 93/RW/002 (clade A), HIV-1 94/UG/118 (clade D), and HIV-1 RV217 (clade AE), with IC50 values exceeding 5000 ng/mL. In contrast, while sIgA formats showed partial neutralization against HIV-1 92/RW/024 (clade A), HIV-1 94/UG/118 (clade D), and HIV-1 RV217 (clade AE), co-expressed IgM-tail variants demonstrated markedly enhanced potency, achieving IC50 values of 8.9, 47.3, and 9.9 ng/mL, respectively. Although less potent against certain strains, co-expressed IgM-tails still recovered measurable neutralization activity against SHIV-AE16 (clade AE) and HIV-1 93/RW/002 (clade A), with IC50 values of 118.2 ng/mL and 388.6 ng/mL, respectively. These results demonstrate that antibody potency and breadth can be substantially improved through co-expression strategies and isotype-driven multimerization.
To assess the binding affinity of these antibodies to HIV envelope (Env) trimers, we conducted bio-layer interferometry (BLI), which measures real-time biomolecular interactions40. Four synthetic HIV Env gp140 proteins, including UG37 (clade A), SF162 (clade B), JRFL (clade B), and C1086 (clade C), were immobilized onto biosensors. For each mRNA-encoded antibody, we measured association rate constant (ka) and dissociation rate constant (kd), from which equilibrium dissociation constant (KD) values were calculated, where lower KD values indicate stronger binding affinity. Association/dissociation curves revealed that SC-IgM-tail antibodies exhibited higher binding signals, with this trend being particularly notable in the PGT121 + VRC07 co-expressed group (Fig. 4a). To compare binding affinity across groups, Ka and Kd values were plotted on iso-affinity kinetic plots, with diagonal lines representing KD values (Fig. 4b). Most PGT121 antibodies showed KD values between 10 and 100 nM, while VRC07 antibodies demonstrated stronger affinity, with KD values between 1 and 10 nM. Co-expression of PGT121 and VRC07 resulted in clustered KD values around 10 nM, indicating that VRC07’s strong binding affinity could complement PGT121 when co-expressed.

a Association and dissociation curves showing the interactions between HIV Env proteins and antibodies at three different antibody concentrations. b Two-dimensional iso-affinity kinetic plots of mRNA-encoded PGT121, VRC07, and their combination (PGT121 + VRC07). The table summarizes the equilibrium dissociation constants (KD), where lower values indicate higher binding affinity, along with the association (Ka) and dissociation constants (Kd). Diagonal dashed lines represent KD values. Reported KD values are the average of three experiments using an independent antibody dilution series.
Repeating VHH domains via nanobody engineering enhances potency
Heavy chain-only antibodies from llama and their variable regions (VHH), also known as nanobodies, are potential alternatives to conventional mammalian immunoglobulins due to their high binding affinity and smaller sizes. In particular, J3-VHH has demonstrated broad and potent neutralization against various HIV-1 strains41. Building on this, we designed mRNAs encoding GPI-anchored J3-VHH (aJ3) derivatives with the following modifications: (1) addition of 1×, 2×, and 4× repeated VHH domains and (2) addition of an IgG constant heavy chain fragment (Fc) to VHHs (Fig. 5a). To confirm expression of the six resulting engineered VHH nanobodies, A549 cells were transfected with each mRNA and stained with anti-VHH and anti-human IgG secondary antibodies. aJ3s with or without Fc were similarly expressed on the cell surface regardless of the number of repeated domains. Decreased Fc signal was observed for aJ3-4×-Fc, likely due to steric hindrance and potentially decreased translation rate of the longer mRNA42 (Fig. 5b). JESS Western blot results confirmed increased molecular weights proportionate to the number of VHH repeats: 33 kDa for aJ3, 42 kDa for aJ3-2×, and 60 kDa for aJ3-4× (Fig. 5c). The aJ3s-Fcs showed two populations of monomeric and dimeric heavy chain-only antibodies: 51 and 103 kDa for aJ3-Fc, 57 and 123 kDa for aJ3-2×-Fc, and 73 and 151 kDa for aJ3-4×-Fc.

a Schematic of GPI-anchored J3-VHH constructs with repeated domains (1×, 2×, and 4×) and J3-VHH-Fc constructs. VHH domains are linked by two repeats of G4S linker ((G4S)2), and the IgG CH2 and CH3 with the hinge region are attached to VHH to form J3-VHH-Fc. Created in BioRender. Santangelo, P. (2025) https://BioRender.com/z117hh5. b Expression of J3-VHH derivatives in A549 cells. Each J3 nanobody was visualized using anti-VHH-AF488 (green) and anti-human IgG-AF647 (white). The scale bar represents 20 µm. c JESS Western blot analysis of aJ3, aJ3-2×, aJ3-4× (without Fc) and aJ3-Fc, aJ3-2×-Fc, aJ3-4×-Fc (with Fc). d BLI binding kinetic assays of J3-VHH derivatives. Association and dissociation curves showing the biomolecular interactions between HIV Env proteins and nanobodies are presented in the graphs, with three different concentrations of each nanobody. The equilibrium dissociation constants (KD) are summarized in the corresponding table. e TZM-bl neutralization assays of J3-VHH derivatives against SHIV/HIV. IC50s are summarized in the corresponding table. All neutralization assays were performed in duplicates and repeated at least twice in independent assays.
To test our hypothesis that increasing the number of VHH domains would improve affinity, we conducted BLI binding kinetic assays for the six VHH derivatives under the same conditions used to test the co-expressed, isotype-modified PGT121 and VRC07. In most cases, the binding affinity was proportionate to the number of VHH repeats, particularly within the VHH-Fc groups (Fig. 5d and Supplementary Fig. 4). An exception was represented by GPI-anchored J3s without Fc, where aJ3’s binding affinity was stronger than aJ3-2×. This may be due to its low molecular weight and rapid diffusion, and lower probability of steric hindrance, which facilitates ligand-nanobody interaction. Additionally, the results showed that incorporating Fc increased binding affinity by inducing the formation of two populations of nanobodies, monomeric and dimeric, with 4 and 8 VHH domains, respectively, in the case of aJ3-4×-Fc.
In TZM-bl neutralization assays, the six VHH variants displayed robust neutralizing capabilities against SHIV-KNH1144p2 (clades A), SHIV-162p3 (clade B), SHIV-1157ipd3N4, and SHIV-C109 (clade C) but were ineffective against SHIV-AE16 (clade AE), HIV-1-RV217 (Fig. 5e). Like the BLI results, their neutralizing capabilities increased with the number of VHH domains and with the addition of Fc. For instance, aJ3-Fc neutralized SHIV-KNH1144p2 (clade A) with an IC50 of 125.3 ng/mL, but aJ3-2×-Fc and aJ3-4×-Fc showed enhanced neutralizing capabilities against the same virus with IC50 of 25.4 ng/mL and 3.7 ng/mL, respectively. aJ3 also showed exceptional neutralization, likely due to its small size. Overall, these results suggest that the potency of nanobodies can be enhanced by increasing the number of VHH domains and incorporating Fc.
mRNA-transduced macaque vaginal explants are protected from SHIV infection
Before conducting an in vivo challenge, we performed ex vivo tissue infection studies. To confirm local protein expression in vaginal mucosa of rhesus macaques, we first administered 1 mg of GPI-anchored nanoluciferase (aNluc)-encoding mRNA intravaginally using an aerosolizer, as previously described21. Whole reproductive organs were collected 24 h post-administration, and luminescent signal was visualized using an in vivo imaging system (IVIS) upon its substrate application. Robust and localized expression was detected across the vaginal surface, while no luminescence signal was observed in the upper reproductive tracts (ovaries and uterus) or adjacent iliac lymph nodes, confirming spatial restriction of expression to the lower tract (Supplementary Fig. 5a).
To assess pharmacokinetics of antibody expression, SC-PGT121 aIgG mRNA was administered intravaginally, and vaginal secretions were collected over time. IgG levels in vaginal secretions increased on day 3, indicating that the expressed aIgGs remained in the tissue until day 1 before gradually being released (Supplementary Fig. 5b). Based on these observations, we selected 24 h post-administration as the optimal time point for vaginal biopsy collection in the subsequent ex vivo challenge study.
For the ex vivo challenge study, mRNAs were delivered intravaginally, and vaginal pinch biopsies were collected 24 h post-treatment. Biopsies were infected ex vivo with SHIV strains from different clades and incubated in tissue culture media. Excess virus was washed out after 2 h, and the culture media were collected and replaced on days 4, 7, and 10 to measure the levels of viral protein p27, serving as an indicator of SHIV replication in the tissues (Fig. 6a).

a mRNAs encoding each antibody were delivered intravaginally into rhesus macaques using an aerosolizer (n = 2 per group). Cervicovaginal tissue biopsies were collected 24 h post-administration and subsequently infected with different SHIV strains. Untreated explants were served as a negative control. Tissue culture media were collected on days 4, 7, and 10, and the viral replication in the biopsies was determined using SIV p27 antigen capture assays. Created in BioRender. Santangelo, P. (2025) https://BioRender.com/u70z502. b Efficacy of tissue protection via IgG co-expression and J3-VHH-Fc against SHIV strains from clade A, B, and C. The mRNA dose was 1 mg, except for group 4, which received 3 mg. (dpi: days post-infection) c Efficacy of tissue protection via SC-PGT121 with various isotypes against SHIVs from clades B and C. All groups received 1 mg doses. d Efficacy of co-expressed, isotype-modified SC-PGT121 and VRC07 in protecting tissues against SHIVs from clades A, B, C, and AE. All groups received 1 mg doses. The lower detection limit of the assay is 31.25 pg/mL (dashed vertical line). The p27 values shown for each collection represent the amount of viral replication which occurred between the successive collections.
We first tested protection upon co-expressing GPI-anchored PGT121 and VRC07 IgGs, and Fc-incorporated aJ3-2× (aJ3-2×-Fc) (Fig. 6b). Vaginal biopsies from both control macaques showed robust viral production after infection with all three SHIV strains tested. Biopsies from macaques administered mRNA encoding SC-PGT121 aIgG showed lower yet measurable viral production than controls. However, complete protection was achieved upon either co-expression of aIgG (SC-PGT121 + VRC07 aIgG) or expression of aJ3-2×-Fc, regardless of doses tested (1 or 3 mg). These results demonstrated that the combination of PGT121 and VRC07 and incorporation of Fc in the nanobody efficiently protected vaginal tissues ex vivo. We then tested protection by different isotypes, including PGT121 aIgG, sIgA, aIgA, and IgM-tail (Fig. 6c). Incomplete protection was observed with PGT121 aIgG and PGT121 sIgA, while aIgA and IgM-tail successfully protected the tissues from SHIV-162p3 (clade B) and SHIV-C109 (clade C). Lastly, the effects of co-expressed, isotype-modified antibodies were tested against four different SHIV strains (Fig. 6d). Compared to SC-PGT121 aIgG, SC-VRC07 aIgG conferred complete protection against SHIV strains from clades A, B, and C, suggesting that protection observed with SC-PGT121 + VRC07 aIgG in Fig. 6b was primarily attributable to VRC07. However, SC-VRC07 aIgG failed to protect against SHIV-AE16, aligning with in vitro neutralization data shown in Fig. 3f. Although co-expressed SC-aIgG was not tested against SHIV-AE16, in vitro data suggest limited expected ex vivo efficacy. While co-expressed SC-sIgA showed lower protection against SHIV strains from clades A, B, and C compared to co-expressed aIgGs, it provided partial protection against SHIV-AE16. Most importantly, the combination of mRNA-encoded PGT121 and VRC07 as an IgM-like multimer achieved near-complete protection against all the strains tested.
Intravaginal mRNA delivery induces minimal systemic or local toxicity in female rhesus macaques
To evaluate the safety of mRNA-encoded antibodies delivered intravaginally, we first assessed systemic responses in female rhesus macaques treated with SC-PGT121 + VRC07 IgM-tail, SC-PGT121 + VRC07 aIgA, or a negative control. Blood samples were collected at baseline and 24 h post-administration, and changes in clinical parameters were analyzed. Blood chemistry results showed no significant alterations in liver enzymes, renal markers, or other systemic indicators, with exception of a modest increase in alanine aminotransferase (ALT) and total protein in the aIgA group (Fig. 7a and Supplementary Fig. 6a). A similar ALT elevation was also observed in the untreated control group, while the IgM-tail group showed no increase. These data suggest that observed ALT elevations are unlikely to be treatment-related and may instead reflect transient, non-specific stress responses, such as those caused by anesthesia or handling43. Importantly, ALT levels in all groups mostly remained within normal physiological range.

a–c Blood samples were collected at baseline (0 h) and 24 h post-administration from three groups (n = 6 per group): negative control, SC-PGT121 + VRC07 IgM-tail, and SC-PGT121 + VRC07 aIgA. Heatmaps show log2 fold changes between pre- and post-treatment values. a Blood chemistry analysis (see Supplementary Fig. 6a for detailed data). b Peripheral blood mononuclear cell (PBMC) profiling by flow cytometry (see Supplementary Fig. 6b). c Cytokine and chemokine levels measured by multiplexed immunoassays (see Supplementary Fig. 10a). d Representative H&E-stained sections of female reproductive tissues from untreated (negative control) and mRNA-treated animals. The scale bar represents 100 µm. Additional images are shown in Supplementary Fig. 10b.
Similarly, the effects of delivered mRNA on peripheral blood mononuclear cells (PBMCs) were evaluated. Most immune cell populations showed no significant alterations in cell number, proliferation, or activation. In the IgM-tail group, a slight increase in proliferation of non-classical monocytes and a slight decrease in proliferation of plasmacytoid DCs (pDCs) were observed in the heatmap, but these changes were not statistically significant (Fig. 7b and Supplementary Fig. 6b). Furthermore, multiplexed cytokine/chemokine analysis demonstrated no evidence of systemic inflammation in any groups (Fig. 7c and Supplementary Fig. 10a).
To evaluate local tissue responses, reproductive tract tissues (vaginal, cervical, and uterine) were collected from two female macaques, one treated with 1 mg of aNluc mRNA and one untreated control, at 24 h post-administration. Hematoxylin and eosin (H&E) staining was performed to evaluate histopathological changes (Fig. 7d and Supplementary Fig. 10b). A board-certified veterinary pathologist scored the tissues based on epithelial integrity, leukocyte infiltration, edema, and vascular congestion. No histological abnormalities were observed in the tissues treated. Morphological features were comparable to the control group, with no evidence of inflammation, necrosis, epithelial or stromal injury, atypia, or degenerative changes.
Taken together, these findings indicate that intravaginal mRNA delivery is well tolerated, without notable signs of systemic toxicity, immune activation, or local tissue damage at 24 h post-treatment. These results support the safety of localized mRNA administration and its potential for clinical application in mucosal immunoprophylaxis.
mRNA-encoded antibodies are effectively expressed and retain functionality in two different species via alternative delivery methods
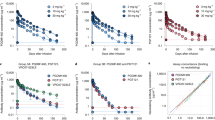
We evaluated potency enhancement through co-expression and isotype modifications using an alternative delivery method and animal model. mRNAs encoding SC-PGT121 and SC-VRC07 in their sIgA or IgM-tail forms were formulated into lipid nanoparticles (LNPs) and administered intravenously to mice in two doses (Fig. 8a). Serum samples were collected and quantified, with concentrations of 19.2 µg/mL for sIgA and 82.8 µg/mL for IgM-tail (Fig. 8b). Their neutralizing capabilities were then evaluated using TZM-bl neutralization assays against SHIV and HIV strains from different clades (Fig. 8c). Consistent with our previous results, IgM-tail groups demonstrated substantially enhanced neutralizing ability against all the viruses tested compared to the IgA group, particularly rescuing potency against HIV-1-RV217 (clade AE). These results suggest that mRNA-encoded antibodies can be effectively expressed in different species while maintaining their functional properties. Additionally, this suggests that systemic mRNA delivery via LNPs offers a potential complementary approach to localized intravaginal delivery, potentially enhancing overall neutralizing effect for HIV prevention.

a Experimental design. For SC-PGT121 + VRC07 IgA or IgM-tail formulations, LNPs were diluted in saline at a 4:4:2 ratio. The LNP mixtures were delivered two times on days 0 and 2 via retro-orbital (RO) injection at 2 mg/kg dose (n = 5 per group). Serum samples were collected on day 3. Created in BioRender. Santangelo, P. (2025) https://BioRender.com/hj0jyqn. b Quantification of IgA and IgM-tail antibodies by ELISA. Data are presented as mean ± SD. c TZM-bl neutralization assays of mice serum samples against SHIV/HIV. Data are presented as mean ± SD. The 50% inhibitory concentrations (IC50) are summarized in the corresponding table. All neutralization assays were performed in duplicates and repeated at least twice in independent assays.
Discussion
Despite rapid progress in long-duration antiretroviral therapy (ART) formulations for PrEP, the need for a sustained drug regimen has not gained uniform approval among various populations at risk, especially those at low risk, and/or necessitating only transient on-demand protection. Thus, alternative strategies, such as local administration of a preventative at the portal of infection, remain desirable. HIV bnAbs have been shown to prevent infections of non-human primates with SHIV, yet the route of administration of such a drug would require a medical environment, and the cost of bnAb production remains markedly high44.
This study demonstrates the feasibility of single-chain mRNA-encoded antibodies, exhibiting comparable expression profiles to conventional dual heavy and light chain mRNA systems but with improved heavy and light chain assembly. This simplified production approach offers numerous advantages, including consistent antibody expression in complex combinations and potential cost reductions. In addition, while improper stoichiometry between heavy and light chains in the dual-chain system may lead to excess free light chains, raising potential concerns about light chain deposition disease, the single-chain system mitigates this risk45,46. Previous studies by Narayanan et al. have developed single-chain variable fragment (scFv)-Fc constructs47, and Stadler et al. have created bispecific Fab-(scFv)2 constructs48, both excluding the constant heavy 1 (CH1) domain. In contrast, our single-chain mRNA system includes entire antibody sequences with the CH1 domain, which may play a crucial role in controlling the binding affinity of the variable region49,50.
Several clinical trials have demonstrated that delivering multiple bnAbs is effective for HIV prevention and treatment, as it can increase coverage against high HIV-1 diversity and arising bnAbs-resistant variants7,8,51. While enhancing breadth, co-expressing PGT121 and VRC07 maintains potency levels comparable to those observed with each bnAb expressed individually. The mRNA expression platform simplifies this and avoids costly antibody purification processes. While here we enhanced breadth and potency by combining potent bnAbs targeting different sites in HIV Env, this single-chain mRNA-encoded antibody approach could be multiplexed, with inclusion of immuno-contraceptives and antibodies against other sexually transmitted infections in a single dose. In addition to the mRNA platform’s versatility, we further explored modifications to enhance potency and breadth by designing IgA forms of bnAbs and incorporating an IgM-tail, aiming to improve antibody functionality within mucosal environment.
Abundance and active transport of IgA and IgM in mucosal immunity, compared to IgG, can be attributed to their specific structural and functional properties37. Mucosal IgA primarily exists as a dimer with a J-chain, making it resistant to proteolytic degradation in harsh mucosal environment52. IgM exists as a pentamer, providing high avidity for antigens and effectively agglutinating pathogens38. Leveraging these characteristics for mucosal delivery, we designed IgA forms of bnAbs and incorporated an IgM tailpiece to IgG, while keeping variable regions unchanged. These modifications remarkably enhanced neutralizing ability against various SHIV/HIV strains, particularly enabling neutralization of SHIV-C109, SHIV-AE16, HIV-1 92/RW/024, HIV-1 93/RW/002, HIV-1 94/UG/118, and HIV-1-RV217, which were not neutralized by the same mRNA-expressed bnAbs in their IgG forms. These findings are critical as they demonstrate a remarkable increase in the breadth of these antibodies, crucial for combating highly mutating viruses. While underlying mechanisms are not fully understood, several explanations have been suggested for the difference in antibody affinity and specificity with identical variable regions but different constant regions. Recent studies have shown that IgM provides more effective viral protection than IgG, attributed to increased avidity and epitope-dependent steric hindrance53,54. Another possible explanation is that modification of constant regions can affect electrostatic and hydrophobic interactions at variable regions, making the antigen-binding site more flexible or rigid, with subtle conformational changes55,56. Our study demonstrated potency enhancement with mRNA-expressed IgM-tail antibodies by adding IgM tailpiece sequence to IgG. These findings underscore the critical role of constant region modifications in optimizing antibody efficacy.
Similarly, nanobody engineering results suggest that increasing the number of VHH domains can enhance potency (Fig. 5e). The exception of monomeric aJ3 is probably due to its smaller size than the other five VHH derivatives, which likely allows it to access the Env trimers with less steric hindrance and more rapid diffusion. This property suggests the potential for broader applications when co-expressed with other antibodies. Although VHH modifications improved binding affinity and neutralizing capability, they did not confer the ability to neutralize SHIV/HIV strains from clade AEs, in contrast to the enhanced potency observed with the PGT121 and VRC07 in IgM-tail format. This is probably due to differences in spatial arrangement of binding domains. In multimerized sIgA and IgM-tail formats, flexible configuration may enable simultaneous engagement of multiple Env proteins on a single virion or even crosslink multiple virions, potentially resulting in viral aggregation. In contrast, VHH domains multimerized in a linear string may be sterically constrained, limiting such multivalent interactions. The observation that potency enhancement was more pronounced with Fc-fused VHHs further supports this interpretation. Furthermore, incorporating an Fc could offer several benefits beyond doubling VHH domains. Apart from directly neutralizing viruses, antibodies can also activate immune effectors via their Fc domains, interacting with Fc receptor-expressing immune cells and the complement system57,58. Additionally, successful incorporation of an Fc can open opportunities for isotype modifications, such as IgA or IgM-tailed form of VHHs, expecting similar enhancements seen with PGT121 and VRC07 IgA and IgM-tail constructs. Overall, our findings demonstrate feasibility of mRNA-encoded nanobody system and its design strategies.
Previously, our group has shown successful expression of functional IgGs in the female reproductive tracts (FRT) of sheep and rhesus macaques. We achieved this through aerosolized intravaginal delivery of conventional dual heavy and light chain mRNAs, and assessed their neutralizing abilities using ex vivo explant SHIV challenge21. The ex vivo method provides a rigorous assessment as it directly exposes the epithelium, removing natural physical barriers. Building on this work, we have further demonstrated enhanced potency in rhesus macaques using co-expressed, isotype-modified bnAbs based on the single-chain mRNA system. It is worth noting that our ex vivo challenges utilized a small number of animals per group (n = 2). However, despite this limitation, the results indicated high sensitivity to viral infection in non-protected tissues, with all control animal biopsies productively infected by all SHIV strains tested. Groups with one of two animals protected could be interpreted as having some level of effectiveness, though with insufficient potency. Despite these limitations, co-expressed IgM-tail group showed robust protection against all tested viruses, indicating potential efficacy for in vivo challenges.
With intravaginal administration of SC-PGT121 aIgG mRNA, increased PGT121 IgG levels were detected in day 3 vaginal secretions, suggesting delayed release from local tissue expression. While this supports mucosal antibody expression, we acknowledge limitations in our pharmacokinetic (PK) assessment. Specifically, vaginal swab sampling method only captures antibodies present in luminal fluids, whereas a potentially significant portion may remain within the tissue, where protective activity is most relevant. Furthermore, unlike conventional PK studies using blood samples, mucosal PK assessment is complicated by biological factors, such as menstrual cycle-dependent fluid variability, mucus accumulation, natural secretion, and menstrual shedding. These variables can affect both the amount of fluid recovered and assay accuracy. Given these challenges, we recognize the need for a more refined and standardized approach to characterize PK in the female reproductive tract. In addition, our findings suggest that intravaginal delivery of mRNA-encoded antibodies is well tolerated in female rhesus macaques. All of the data support the safety of localized mRNA delivery to the FRT and provide a strong foundation for future studies involving mucosal mRNA-based antibody approach.
While we demonstrated localized protection in the FRT of rhesus macaques, we also administered LNPs containing mRNAs encoding for SC-PGT121 and SC-VRC07 sIgA or IgM-tail intravenously to mice, confirming that the IgM-tail group exhibited remarkably enhanced neutralizing ability compared to the IgA group, consistent with our results in NHPs. This study suggests that our mRNA-encoded antibodies are effectively expressed and retain their functional properties regardless of the host tissue in which they are expressed. Moreover, this approach offers an additional mode for HIV protection, potentially synergizing the neutralizing effect with localized intravaginal delivery.
In conclusion, this study demonstrates how the single-chain mRNA-encoded antibody system has the potential to enhance breadth and potency of current bnAbs, highlighting the need to reevaluate current antibody discovery approaches. Our findings also indicate new potential drug formulations for intravaginal mRNA delivery to protect women from HIV infection, as well as possible applications in preventing other sexually transmitted infections. Future research will focus on durability studies, further optimization of delivery methods, and in vivo challenge studies to validate the effectiveness of our approach to prevent HIV acquisition. This, in turn, will contribute to the development of more efficient and versatile mRNA-based HIV prophylactics.
Methods
Ethics approval
All the experiments using rhesus macaques were performed under ethical guidance and upon approval from the University of Louisiana Institutional Animal Care and Use Committee (IACUC protocol 2020-8803-09). All the mouse studies were approved by the Emory University Institutional Animal Care and Use Committee (IACUC protocol PROTO202300033).
Animals
Sexually mature female rhesus macaques (Macaca mulatta), aged six to fourteen years, were used for the ex vivo SHIV challenge studies. Animals were obtained from the New Iberia Research Center Specific Pathogen-Free (SPF) breeding cohorts and housed individually in barrier facilities under BSL2+ containment at the New Iberia Research Center. The animals were cared for in compliance with the Guide for the Care and Use of Laboratory Animals, and all experiments were approved by the University of Louisiana IACUC. They were fed a diet of monkey chow (Teklad), supplemented daily with fresh fruits or vegetables, and water ad libitum. All procedures were performed under anesthesia with Telazol and/or ketamine. The vaginal mucosa of animals was examined by a veterinarian before and after procedures, and animals in their menstrual period were excluded from this study. Animals were humanely euthanized via intravenous barbiturate administration, consistent with American Veterinary Medical Association recommendations.
Six to eight-week-old C57BL/6J female mice (Mus musculus) (Jackson Laboratory, stock #: 000664) were used in the LNP study in accordance with the approved IACUC protocol. Mice were maintained in a SPF barrier facility with controlled environmental conditions (20–24 °C, 30–70% humidity, 12:12 light/dark cycle). Experimental and control groups were housed separately but within the same facility. The animals were acclimatized for a week before any procedures and were fed ad libitum. The LNP injection and blood sample collection were carried out under sedation by isoflurane inhalation. Following blood collection by cardiac puncture, animals were euthanized by cervical dislocation.
Cell culture
A549 cells were sourced from the American Type Culture Collection (ATCC, CCL-185) for the cell staining experiments to confirm the expression of mRNA-encoded antibodies. The cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Gibco, 11995) supplemented with 10% fetal bovine serum (FBS, Cytiva, SH30071.03) and 1% penicillin-streptomycin (Corning, 30-002-CI). Passaging was performed when cells reached 90% confluence, and the cells were maintained at 37 °C with 5% CO2.
Expi293F cells, purchased from Thermo Fisher Scientific (A14528), were used for antibody production and were cultured in Expi293 expression media (Thermo Fisher Scientific, A1435101) using an orbital shaker at 125 rpm. Sub-culturing was performed according to the manufacturer’s protocol, and the cells were maintained at 37 °C with 8% CO2.
SHIV/HIV stocks
The SHIV-KNH1144p2 (clade A), SHIV-162p3 (clade B), SHIV-1157ipd3N4 (clade C) and SHIV-C109p5 (clade C)59 viruses were propagated at New Iberia Research Center (NIRC) by infecting ConA (1 µg/mL)-stimulated rhesus macaque PBMCs and culturing infected cells with complete media supplemented with 50 U/mL rMamuIL-2 (Resource for nonhuman primate Immune Reagents, NIRC) for 21 days after stimulation. Viral supernatants were tested for the amount of virus (SIV p27) by ELISA using the SIV p27 antigen capture assay kit (ABL, 5436). Supernatants with p27 values >100 ng were pooled, sterile filtered, and stored separately in 1 mL aliquots in liquid nitrogen. The SHIV-AE16 (clade AE) virus stock was kindly provided by Dr. Dan Barouch’s laboratory60, SHIV-KNH1144 by Dr. Ruth Ruprecht’s laboratory61, and SHIV-C109 by Dr. Cecilia Cheng-Mayer’s laboratory62. The HIV-1-RV217 (clade AE, 40100) viral stocks were obtained from the National Institutes of Health (NIH) AIDS Research and Reference Reagent Program. Those SHIV or HIV-1 stocks were titrated on TZM-bl reporter cells for 50% tissue culture infective dose (TCID50) by luminescence [relative luciferase unit (RLU)] and on vaginal explants from untreated naïve macaques. The same virus stocks were used throughout the study.
In vitro transcription of mRNA
Plasmid DNAs encoding different antibodies were synthesized by GenScript based on the mRNA sequences provided in Supplementary Data 1. mRNA synthesis was performed as previously described63,64. First, constructs were in vitro transcribed (IVT) using the Hiscribe T7 kit (New England Biolabs, E2040S), incorporating N1-methylpseudouridine (Crystal Chem, 13946). Resulting RNA was enzymatically capped with a Cap-1 structure using 2′-O-methyltransferase (Aldevron, 9130), guanylyl transferase (Aldevron, 9131), and polyadenylated. Finally, the IVT mRNA was treated with Antarctic Phosphatase (New England Biolabs, M0289L). Purity was determined by capillary electrophoresis (Agilent, DNF-471). All mRNA concentrations were greater than 5 mg/mL.
In vitro mRNA transfection and antibody collection
For A549 cell transfection, the cells were prepared in 24-well glass-bottom plates with 80% confluency. The mRNAs and Lipofectamine MessengerMAX reagent (Thermo Fisher Scientific, LMRNA015) were diluted in Opti-MEM medium (Gibco, 31985070) separately. The mRNAs (1 µg total) were mixed with 1.5 µL of the reagent, and the resulting mRNA-MessengerMAX complex was added to the cells. The transfected cells were incubated for 24 h prior to cell staining.
For Expi293F cells, 7.5 × 107 cells were prepared in a 125 mL flask with 30 mL of Expi293 expression medium. A total of 75 µg of mRNAs and 112.5 µL of Lipofectamine MessengerMAX reagent were diluted in Opti-MEM medium separately and then mixed. The mRNA-MessengerMax complex was added to the cells, with incubation for 48 h. To collect the secreted antibodies, the cells were centrifuged at 1000 × g for 10 min, and the supernatant was collected and concentrated with Amicon Ultra-15 Centrifugal Filters (Millipore Sigma, UFC9050). To collect the GPI-anchored antibodies, the PI-PLC (Millipore Sigma, P5542) solution was prepared and used to release the surface-bound antibodies, according to the method described in Vanover et al.65. The transfected cells were centrifuged at 300 × g for 5 min, and the cell supernatant was discarded to remove unanchored proteins. The cell pellet was then washed with 1 × PBS and centrifuged again. After discarding the supernatant, the PLC solution (2.5 U/mL) was added to the cells and incubated for 1 h at 37 °C on the orbital shaker. The cell suspension was then centrifuged at 1000 × g for 10 min, and the supernatant was collected. The collected solution was concentrated with Amicon Ultra-15 Centrifugal Filters. The antibody concentrations were quantified by human IgG or IgA ELISA kits (Abcam, ab195215, ab196263) according to the manufacturer’s instructions. Data from n = 3 independent experiments are shown as individual values and their mean.
Immunofluorescence
mRNA-transfected A549 cells were washed with 1 × PBS and fixed using 4% paraformaldehyde (Electron Microscopy Sciences, 157-8). To detect membrane-bound antibodies (aIgGs and aIgAs), the fixed cells were blocked with 5% BSA (Gibco, 15260037). For the detection of secreted antibodies (sIgAs and IgM-tails), the cells were permeabilized using 0.1% Triton X-100 (Millipore Sigma, X100) for 5 min at RT, followed by blocking with 5% BSA. Alexa Fluor 488 AffiniPure Donkey Anti-Human IgG (H + L) (Jackson ImmunoResearch Laboratories, 709-545-149), Goat Anti-Human IgA alpha chain (DyLight 488, pre-adsorbed) (abcam, ab98553), and MonoRab Rabbit Anti-Camelid VHH Cocktail [iFluor 488] (GenScript, A02021) were used to capture the mRNA-encoded IgGs, IgAs, and J3 nanobody derivatives, respectively. The nuclei were stained with DAPI (Sigma-Aldrich, D9542). After staining, ProLong Gold Antifade Mountant (Invitrogen, P10144) was applied, and cover glasses were placed over the cells. Andor BC43 microscope system (Oxford Instruments) was used to image the cells, and the images were analyzed with Imaris Image Analysis Software (v10.1.0). This was performed at least twice, and representative images were presented in the figure.
Capillary electrophoresis-based Western blots (JESS)
The JESS automated Western blot system (Bio-Techne, 004-650) was used to analyze mRNA-expressed antibodies according to the manufacturer’s instructions. Capillaries with size ranges of 12–230 kDa or 66–440 kDa were selected based on the size of the analytes (Bio-Techne, SM-W001, SM-W005). Samples were diluted and mixed with 5× fluorescent master mix. To avoid reducing the antibodies, DDT solution was omitted from the master mix. The samples were denatured by heating at 95 °C for 5 min and then kept on ice until loading into the cartridge. For IgG detection, an anti-human secondary-HRP (Bio-Techne, 043-491-2) was used, and for IgAs, goat anti-human IgA alpha chain (HRP) (abcam, ab97215) was used as a secondary antibody without using primary antibodies. For VHH detection, MonoRab Rabbit Anti-Camelid VHH Cocktail (GenScript, A02014) and anti-rabbit secondary HRP antibody (Bio-Techne, 042-206) were used as primary and secondary antibodies, respectively.
Western blots for IgM-tail antibody
The IgM-like multimers larger than 440 kDa were characterized by Western blots as described by Karola Vorauer-Uhl et al.66. Samples were diluted with NuPAGE LDS sample buffer (Invitrogen, NP0007) and loaded onto NativePAGE 3–12% Bis-Tris gels (Invitrogen, BN1001BOX). The samples were run with Nupage Tris-Acetate SDS running buffer (Invitrogen, LA0041) at 200 V for 2 h. The gels were briefly washed and transferred into 0.45 µm PVDF membranes (Thermo Fisher Scientific, 88518) at 20 V for 1 h. The membrane was blocked with 5% non-fat dry milk (Santa Cruz Biotechnology, sc-2324) in TBST at RT for 2 h. For detection, a rabbit anti-human IgG antibody (Abcam, ab109489) was applied with a 1/1000 dilution and incubated for 2 h at RT. As a secondary antibody, goat anti-rabbit IgG-HRP (Abcam, ab6721) was used with a 1/1000 dilution and incubated at RT for 1 h. Radiance Q chemiluminescent substrate (Azure Biosystems, AC2101) was treated on the membrane and waited for 1 min at RT. The membrane was imaged by Western blot imaging systems c400 (Azure Biosystems).
Biomolecular interaction analysis using Octet Biolayer Interferometry (BLI)
Binding kinetics of antibodies and nanobodies were performed with HIV-1 Env proteins (obtained from NIH AIDS Reagent Program) including clade A: UG37 gp140 (Cat. #12063), clade B: SF162 gp140 Trimer recombination Protein (Cat. #12026), and JRFL gp140 recombinant protein (B.JRFL gp140CF; Cat. #12573), and clade C: C.1086 gp140C (Cat. #12581). Briefly, HIV-1 Env ligands were biotinylated using EZ-Link Sulfo-NHS-LC-Biotinylation kit (Thermo Fisher Scientific, 21435). The proteins were incubated with biotin for 2 h on ice. Residual biotin was removed using a desalting column and buffer exchanged with PBS.
The analyte-ligand binding kinetics assay was run on the BLItz platform (Fortebio) using the advanced kinetics module with a default shaker speed of 2200 rpm at 25 °C. The run parameters for each assay are mentioned below. Prior to running the assay, the Octet Streptavidin (SA) Biosensors (Sartorius, 18-5020) were hydrated for 10 min in kinetics buffer (0.1% BSA in 1× PBS buffer). The same buffer was used to dilute the ligand and analyte (twofold dilution series). The hydrated probe was then loaded with specific concentrations of biotinylated ligand (UG37: 80 µg/mL, SF162: 10 µg/mL, JRFL: 40 µg/mL, C1086: 10 µg/mL) for 5 min. Following the loading step, association with the analyte was monitored for 2 min. The tips were then moved to the tube containing buffer to monitor dissociation for 2 min. The association and dissociation curves were fitted with a 1:1 binding model using BLItz Pro software (Fortebio), which provided the association and dissociation rate constants (ka, kd) and the equilibrium dissociation constant (KD). Reported KD values are the average of 3 experiments using independent antibody dilution series.
TZM-bl neutralization assays
The neutralization activity of antibody against multiple SHIV strains (clades A, B, C, or AE) was measured using a standard protocol of luciferase-based HIV-1 neutralization assay in TZM-bl cells (Dr. Montefiori laboratory, Duke University). Briefly, equal volume (50 µL) of fivefold serial diluted antibody and titrated SHIV virus (105 RLU) were pre-incubated for 1 h at 37 °C. Freshly trypsinized TZM-bl cells (10,000 cells in 100 µL of 10% FBS-supplemented DMEM media containing 15 µg/mL DEAE-dextran) were added to each well and cultured for 48 h at 37 °C. At 48 h, the cells were lysed, and luminescence (RLU) was measured using a BioTek Synergy HT microplate reader. The background luminescence (RLU) from cell control wells was subtracted from the luminescence for each experimental well. The neutralization curves and 50% inhibitory concentration (IC50) were calculated and generated using GraphPad Prism (v10.3.1) software. All neutralization assays were performed with duplicate wells and repeated at least twice in independent assays.
mRNA delivery in rhesus macaques
The mRNA samples were prepared by diluting them into 3.33 mg/mL in nuclease-free H2O. The diluted mRNA solutions were loaded onto 1 mL high-pressure syringes (Medline, DYNJSYRPC01W). The MADgic Laryngo-Tracheal Mucosal Atomizer (Teleflex, MAD720) was connected to the syringe. Rhesus macaques were sedated with Telazol/ketamine and positioned in ventral recumbency. The cervicovaginal mucosa was visualized with a speculum to check up, and mucus on the cervicovaginal wall was cleared away by gently swabbing with cotton tips. The delivery was performed as previously described21. Two doses of 150 µL mRNA samples were sprayed on each animal: one onto the cervical os at a 1 cm distance and the other onto the vaginal surface at a 2–3 cm distance from the cervix. All animals received 1 mg of mRNAs in 300 µL in total. All physical exams, mRNA doses, administration times, and other notes were recorded.
Luminescence imaging of local protein expression in rhesus macaques
A total of 1 mg of mRNA encoding GPI-anchored nanoluciferase (aNluc) was administered intravaginally to two female rhesus macaques. After 24 h, animals were euthanized, and FRTs and iliac lymph nodes were collected. The FRTs were bisected longitudinally along the coronal plane to expose the luminal surface. Tissues were incubated in Nano-glo Luciferase Assay Substrate (Promega, N1150) diluted 1:50 in 1× PBS for 5 min. Luminescence signals were captured using an IVIS (Perkin Elmer) and analyzed using Living Image software (v4.8.2, Revvity).
Macaque vaginal secretion collection
Vaginal secretions were collected using Weck-cel swabs (Beaver-Visitec International, 0008685) pre-soaked with 50 µL of PBS. Under sedation, swabs were placed into the vaginal lumen for 5 min, then transferred to QIAshredder columns (Qiagen, 79656) and centrifuged at 20,000 × g for 10 min. Subsequently, 200 µL of PBS containing 1× Halt protease inhibitor cocktail (Thermo Scientific, 78429) was added to each swab, followed by incubation on ice for 15 min. After additional centrifugation, the recovered secretions were collected and stored at −80 °C until analysis.
Quantification of mRNA-expressed antibody in macaque vaginal secretions
SOSIP BG505.664 Env proteins67 (provided from Albert Cupo’s laboratory) were diluted to 10 µg/mL in 1 × PBS, coated onto high-binding 96-well plates (Corning, 9018), and incubated O/N at 4 °C. Plates were washed, and vaginal secretion samples, alongside SC-PGT121 aIgG standards produced in Expi293F cells, were added and incubated for 1 h at RT. After washing, goat anti-human IgG-HRP (1:5000; Abcam, ab6858) was added and incubated for 1 h. Following a final wash, TMB substrate solution (Thermo Scientific, 34018) was added and incubated for 20 min. Reactions were stopped using stop solution (Thermo Scientific, N600), and absorbance values were measured using a microplate reader (BioTek). Antibody concentrations were calculated by interpolation from a standard curve using GraphPad Prism 10 (v10.3.1).
Macaque ex vivo explant SHIV challenges
Vaginal tissue explants (similar size of 2–3 mm in diameter) were collected 24 h post-administration in testing groups of rhesus macaques or from untreated macaques. Each vaginal explant was placed individually in a well of a 24-well plate and challenged ex vivo with 2.0 × 104 TCID50 of SHIV-KNH1144p2 (clade A), 5.5 × 104 TCID50 of SHIV-162p3 (clade B), 4.8 × 104 TCID50 of SHIV-C109p5 (clade C), and 1.4 × 104 TCID50 of SHIV-AE16 (clade AE) for 2 h at 37 °C. The untreated explants served as negative controls. After 2 h of infection, explants were thoroughly washed out with PBS, and then placed on presoaked Surgifoam rafts (Ethicon, 1969) in individual wells of 24-well plates containing 500 µL of explant culture media (RPMI 1640, 10% FBS, 1% antibiotic-antimycotic). On days 4, 7, and 10, supernatants from the cultures were harvested and replaced with fresh explant culture media. The extent of viral replication in tissue explants was determined by measuring SIV p27 antigen concentrations in supernatants using the SIV p27 antigen capture assay kit (ABL, 5436) according to the manufacturer’s instructions.
Systemic assessment of mRNA toxicity
EDTA plasmas were collected before and 24 h after intravaginal mRNA administration. Plasma samples were aliquoted and stored at −80 °C until analysis. Blood chemistry was conducted using the Superchem panel, performed by the Emory National Primate Research Center. Multiplex cytokine and chemokine profiling was conducted by the Emory Multiplexed Immunoassay Core using U-PLEX and V-PLEX custom non-human primate (NHP) biomarker assay kits (Meso Scale Diagnostics).
Immunophenotyping by flow cytometry
Immunophenotyping of T cells, B cells, NK cells, monocytes, and dendritic cells (DCs) was conducted by flow cytometry using cryopreserved peripheral blood mononuclear cells (PBMCs) isolated from whole blood on day 0 (pre-mRNA dosing) and day 1 (post-24 h dosing). One million cryopreserved PBMCs were thawed in complete RPMI medium supplemented with 10% FBS and rested overnight at 37 °C before staining. The next day, PBMCs were first stained with live/dead viability dye (Invitrogen, L3224) to exclude dead cells. The cells were washed with PBS supplemented with 2% FBS and surface-stained at 4 °C for 30 min with the following antibodies: anti-CD3 Alexa 700 (BD Biosciences, 557917), anti-CD4-FITC (BD Biosciences, 550628), anti-CD8-BV650 (BioLegend, 301042), anti-CD20-APC-Cy7 (BD Biosciences, 335812), anti-NKG2a (CD159a)-APC (Beckman Coulter, A60797), anti-CD14-PE (BioLegend, 301806), anti-CD16-BV711 (BioLegend, 302044), anti-CD123-PerCP-Cy5.5 (BD Biosciences, 558714), anti-CD11c-PE-Cy7 (BioLegend, 301608), anti-BDCA-1 (CD1c)-Pacific Blue (BioLegend, 331508), anti-HLA-DR-PE-CF594 (BD Biosciences, 562304), and anti-CD86-BV605 (BioLegend, 305430). For intracellular staining, the cells were first fixed and permeabilized with Cytofix/Cytoperm (BD Biosciences, 554714) for 30 min, washed with 1× Perm/Wash, followed by staining with anti-Ki-67-BV786 (BD Biosciences, 563756). Stained cells were fixed in 1% formaldehyde and acquired on a BD FACSAria Fusion cell sorter. Flow cytometry data were analyzed using FlowJo (v10.7.1). For each cell population, a singlet, followed by live cells were first gated. For T cells: CD3+ cells were gated and divided into CD4+ and CD8+ T cells. For B cells: CD20+ CD14− cells were gated. For NK cells, CD16+ NKG2a+ cells were gated from CD8+ cells. Monocytes were gated from CD3− CD8− CD20− HLA-DR+ cells and further divided into three subsets: classical CD14+ CD16− monocytes, intermediate CD14+ CD16+ monocytes, and non-classical CD14− CD16+ monocytes. DCs were gated from CD3− CD8− CD16− HLA-DR+ cells and further divided into two subsets: plasmacytoid DC (pDC), identified as CD123+ CD11c−, and myeloid DC (mDC), identified as CD123− CD11c− BDCA-1+. In addition, each gated population was further analyzed for the proliferation (Ki67+) and activation (HLA-DR+ or CD86+), respectively.
Histologic evaluation of female reproductive tissues
Histopathological assessment was conducted to evaluate potential local changes in the female reproductive tract (FRT) by intravaginal aNluc mRNA administration. Female reproductive tissue samples were collected from two rhesus macaques (one control, one treated) and fixed in 10% neutral buffered formalin (VWR, 10790-714). Tissue blocks approximately 1 cm3 in size were prepared from the vagina, cervix, and uterus, and processed by the Emory Cancer Tissue and Pathology core. Sections were embedded in paraffin, sectioned, stained with H&E, and mounted for analysis. Histological examination was done by a board-certified veterinary pathologist. Evaluation criteria included epithelial architecture, glandular organization, stromal integrity, vascular morphology, and presence or absence of inflammatory infiltrates.
LNP formulation
The mRNAs were diluted in 10 mM citrate buffer (pH 3) to create the aqueous phases. To prepare the organic phases, cKK-E12 (Organix, O-8744), cholesterol (Sigma, C8667), C14-PEG 2000-PE (Avanti Polar Lipids, 880150P), and DOPE (Avanti Polar Lipids, 850725 P) were added to 100% ethanol at a ratio of 35:46.5:2.5:16. A mass ratio of 20 (lipid/mRNA) was used for all in vivo formulations. The two phases were mixed using a NanoAssemblr benchtop device containing a microfluidic cartridge (Precision NanoSystems, NIN0061) at an aqueous to organic flow rate ratio of 3:1 at 12 mL/min. The LNPs were next diluted 6× in 10 mM Tris buffer and concentrated using 30 kDa MWCO centrifugal filters (Millipore Sigma, UFC903096), followed by a 7× diafiltration with 6% (w/v) sucrose. After sterile filtering, particle size was determined by dynamic light scattering on a Zetasizer Pro (Red) (Malvern Panalytical). A RiboGreen assay (Invitrogen, R11490) was performed to calculate the encapsulation percentage and concentration of mRNA cargos. After size and concentration were characterized, LNP solutions were diluted with 6% sucrose to the desired final concentration, aliquoted, and stored at −80 °C until use.
LNP delivery to mice
The LNPs encapsulating mRNAs of SC-PGT121 IgA, SC-VRC07 IgA, and J chain were mixed at a 4:4:2 ratio and diluted to 0.4 mg/mL in 0.9% Sodium Chloride Injection (Hospira, 00409-4888-10) to prepare the dose for IgA. Likewise, the LNP samples for IgM-tail antibodies were prepared by mixing the mRNAs encoding SC-PGT121 IgM-tail, SC-VRC07 IgM-tail, and J chain at the same ratio and concentration. The LNP mixtures were loaded onto a 1-mL syringe, and a 27-G needle was connected to the syringe. C57BL/6J mice were anesthetized by isoflurane inhalation before each retro-orbital (RO) injection. In the first injection (day 0), 40 µg in 100 µL (2 mg/kg) was delivered to the left side of the eye, and the same dose was delivered to the alternate side of the eye two days after the first dose (day 2). On day 3, mice blood was collected from cardiac puncture and centrifuged at 2000 × g for 10 min. The serum was collected and stored at −80 °C until TZM-bl neutralization assays. The concentrations of mRNA-expressed antibodies in mouse serum were determined by human IgG or IgA ELISA kits (Abcam, ab195215, ab196263) according to the manufacturer’s protocols. Data are presented as mean ± SD.
Statistics and reproducibility
Experimental data were analyzed and plotted using GraphPad Prism 10 (v10.3.1). Statistical analyses included two-way ANOVA with Šidák’s multiple comparisons and Pearson correlation analysis. Error bars represent standard deviation (SD). Sample sizes (n) and the number of independent biological replicates are specified for each experiment in the figure legends. Representative images and data are from experiments that were independently repeated at least three times with consistent results.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data are included in the Supplementary Information or available from the authors, as are unique reagents used in this article. The raw numbers for charts and graphs are available in the Source Data file whenever possible. Source data are provided with this paper.
References
Global HIV & AIDS statistics — Fact sheet. https://www.unaids.org/en/resources/fact-sheet.
Baeten, J. M. et al. Antiretroviral prophylaxis for HIV prevention in heterosexual men and women. N. Engl. J. Med. 367, 399–410 (2012).
Grant, R. M. et al. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N. Engl. J. Med. 363, 2587–2599 (2010).
Rogers, B. G. et al. Perspectives on long-acting formulations of pre-exposure prophylaxis (PrEP) among men who have sex with men who are non-adherent to daily oral PrEP in the United States. BMC Public Health 23, 1643 (2023).
Nuttall, J. et al. Pharmacokinetics of Tenofovir following Intravaginal and Intrarectal Administration of Tenofovir Gel to Rhesus Macaques. Antimicrob. Agents Chemother. 56, 103–109 (2012).
Fioroti, C. E. A. et al. Tenofovir-induced renal and bone toxicity: report of two cases and literature review. Rev. Inst. Med. Trop. Sao Paulo 64, e10 (2022).
Mahomed, S., Garrett, N., Baxter, C., Abdool Karim, Q. & Abdool Karim, S. S. Clinical trials of broadly neutralizing monoclonal antibodies for human immunodeficiency virus prevention: a review. J. Infect. Dis. 223, 370–380 (2020).
Julg, B. et al. Safety and antiviral activity of triple combination broadly neutralizing monoclonal antibody therapy against HIV-1: a phase 1 clinical trial. Nat. Med. 28, 1288–1296 (2022).
Brandenberg, O. F. et al. Predicting HIV-1 transmission and antibody neutralization efficacy in vivo from stoichiometric parameters. PLoS Pathog. 13, e1006313 (2017).
Murphy, C., Devine, T. & O’Kennedy, R. Technology advancements in antibody purification. Antib. Technol. J. 6, 17–32 (2016).
van Brummelen, E. M. J., Ros, W., Wolbink, G., Beijnen, J. H. & Schellens, J. H. M. Antidrug antibody formation in oncology: clinical relevance and challenges. Oncologist 21, 1260–1268 (2016).
Morris, G. C. et al. MABGEL 1: first phase 1 trial of the anti-HIV-1 monoclonal antibodies 2F5, 4E10 and 2G12 as a vaginal microbicide. PLoS ONE 9, e116153 (2014).
Qin, S. et al. mRNA-based therapeutics: powerful and versatile tools to combat diseases. Signal Transduct. Target. Ther. 7, 1–35 (2022).
Parhiz, H., Atochina-Vasserman, E. N. & Weissman, D. mRNA-based therapeutics: looking beyond COVID-19 vaccines. Lancet 403, 1192–1204 (2024).
Van Hoecke, L. & Roose, K. How mRNA therapeutics are entering the monoclonal antibody field. J. Transl. Med. 17, 54 (2019).
Deal, C. E., Carfi, A. & Plante, O. J. Advancements in mRNA encoded antibodies for passive immunotherapy. Vaccines 9, 108 (2021).
Alameh, M.-G. & Weissman, D. Chapter 7 - Nucleoside modifications of in vitro transcribed mRNA to reduce immunogenicity and improve translation of prophylactic and therapeutic antigens. In RNA Therapeutics (eds Giangrande, P. H., de Franciscis, V. & Rossi, J. J) 141–169 (Academic Press, 2022).
Kwon, S., Kwon, M., Im, S., Lee, K. & Lee, H. mRNA vaccines: the most recent clinical applications of synthetic mRNA. Arch. Pharm. Res. 45, 245–262 (2022).
Salatin, S. et al. Chapter 1 - Introduction: an overview of the non-parenteral delivery of nanomedicine. In Theory and Applications of Nonparenteral Nanomedicines (eds Kesharwani, P., Taurin, S. & Greish, K.) 1–25 (Academic Press, 2021).
Wang, X. et al. Vaginal drug delivery approaches for localized management of cervical cancer. Adv. Drug Deliv. Rev. 174, 114–126 (2021).
Lindsay, K. E. et al. Aerosol delivery of synthetic mRNA to vaginal mucosa leads to durable expression of broadly neutralizing antibodies against HIV. Mol. Ther. J. Am. Soc. Gene Ther. 28, 805–819 (2020).
Thran, M. et al. mRNA mediates passive vaccination against infectious agents, toxins, and tumors. EMBO Mol. Med. 9, 1434–1447 (2017).
Rybakova, Y. et al. mRNA delivery for therapeutic anti-HER2 antibody expression in vivo. Mol. Ther. 27, 1415–1423 (2019).
Tiwari, P. M. et al. Engineered mRNA-expressed antibodies prevent respiratory syncytial virus infection. Nat. Commun. 9, 3999 (2018).
Pardi, N. et al. Administration of nucleoside-modified mRNA encoding broadly neutralizing antibody protects humanized mice from HIV-1 challenge. Nat. Commun. 8, 14630 (2017).
Koerber, J. T., Hornsby, M. J. & Wells, J. A. An improved single-chain fab platform for efficient display and recombinant expression. J. Mol. Biol. 427, 576–586 (2015).
Walker, L. M. et al. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 477, 466–470 (2011).
Gaudinski, M. R. et al. Safety and pharmacokinetics of broadly neutralising human monoclonal antibody VRC07-523LS in healthy adults: a phase 1 dose-escalation clinical trial. Lancet HIV 6, e667–e679 (2019).
Rudicell, R. S. et al. Enhanced potency of a broadly neutralizing HIV-1 antibody in vitro improves protection against lentiviral infection in vivo. J. Virol. 88, 12669–12682 (2014).
Wu, X. et al. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 329, 856–861 (2010).
Kwon, Y. D. et al. Optimization of the solubility of HIV-1-neutralizing antibody 10E8 through somatic variation and structure-based design. J. Virol. 90, 5899–5914 (2016).
Huang, Y. et al. Engineered bispecific antibodies with exquisite HIV-1-neutralizing activity. Cell 165, 1621–1631 (2016).
Burkly, L. C. et al. Inhibition of HIV infection by a novel CD4 domain 2-specific monoclonal antibody. Dissecting the basis for its inhibitory effect on HIV-induced cell fusion. J. Immunol. 149, 1779–1787 (1992).
Doria-Rose, N. A. et al. New member of the V1V2-directed CAP256-VRC26 lineage that shows increased breadth and exceptional potency. J. Virol. 90, 76–91 (2016).
Sok, D. et al. Recombinant HIV envelope trimer selects for quaternary-dependent antibodies targeting the trimer apex. Proc. Natl. Acad. Sci. USA 111, 17624–17629 (2014).
Barnes, C. O. et al. Structural characterization of a highly-potent V3-glycan broadly neutralizing antibody bound to natively-glycosylated HIV-1 envelope. Nat. Commun. 9, 1251 (2018).
Matsumoto, M. L. Molecular mechanisms of multimeric assembly of IgM and IgA. Annu. Rev. Immunol. 40, 221–247 (2022).
Michaud, E., Mastrandrea, C., Rochereau, N. & Paul, S. Human secretory IgM: an elusive player in mucosal immunity. Trends Immunol. 41, 141–156 (2020).
Sørensen, V., Sundvold, V., Michaelsen, T. E. & Sandlie, I. Polymerization of IgA and IgM: roles of Cys309/Cys414 and the secretory tailpiece. J. Immunol. 162, 3448–3455 (1999).
Jug, A., Bratkovič, T. & Ilaš, J. Biolayer interferometry and its applications in drug discovery and development. TrAC Trends Anal. Chem. 176, 117741 (2024).
McCoy, L. E. et al. Potent and broad neutralization of HIV-1 by a llama antibody elicited by immunization. J. Exp. Med. 209, 1091–1103 (2012).
Valleriani, A., Zhang, G., Nagar, A., Ignatova, Z. & Lipowsky, R. Length-dependent translation of messenger RNA by ribosomes. Phys. Rev. E 83, 042903 (2011).
Lugo-Roman, L. A., Rico, P. J., Sturdivant, R., Burks, R. & Settle, T. L. Effects of serial anesthesia using ketamine or ketamine/medetomidine on hematology and serum biochemistry values in rhesus macaques (Macaca mulatta). J. Med. Primatol. 39, 41–49 (2010).
Moog, C. et al. Protective effect of vaginal application of neutralizing and nonneutralizing inhibitory antibodies against vaginal SHIV challenge in macaques. Mucosal Immunol. 7, 46–56 (2014).
Sayed, R. H. et al. Natural history and outcome of light chain deposition disease. Blood 126, 2805–2810 (2015).
Light chain deposition disease: pathogenesis, clinical characteristics and treatment strategies. Ann. Hematol. https://link.springer.com/article/10.1007/s00277-024-05911-9.
Narayanan, E. et al. Rational design and in vivo characterization of mRNA-encoded broadly neutralizing antibody combinations against HIV-1. Antibodies 11, 67 (2022).
Stadler, C. R. et al. Elimination of large tumors in mice by mRNA-encoded bispecific antibodies. Nat. Med. 23, 815–817 (2017).
Tudor, D. et al. The immunoglobulin CH1 constant region modulates antigen binding affinity and functional activities of the broadly neutralizing 2F5 HIV specific antibody. Retrovirology 7, P23 (2010).
Torres, M., May, R., Scharff, M. D. & Casadevall, A. Variable-region-identical antibodies differing in isotype demonstrate differences in fine specificity and idiotype. J. Immunol. 174, 2132–2142 (2005).
Sobieszczyk, M. E. et al. Safety, tolerability, pharmacokinetics, and immunological activity of dual- and triple-combinations of anti-HIV monoclonal antibodies PGT121, PGDM1400, 10-1074, and VRC07-523LS administered intravenously to HIV-uninfected adults: results of a phase 1 randomized trial. Lancet HIV 10, e653–e662 (2023).
Li, Y., Jin, L. & Chen, T. The effects of secretory IgA in the mucosal immune system. BioMed. Res. Int. 2020, 2032057 (2020).
Ku, Z. et al. Nasal delivery of an IgM offers broad protection from SARS-CoV-2 variants. Nature 595, 718–723 (2021).
Law, E. C. Y. et al. IgM antibodies can access cryptic antigens denied to IgG: hypothesis on novel binding mechanism. Front. Immunol. 10, 1820 (2019).
Torres, M. & Casadevall, A. The immunoglobulin constant region contributes to affinity and specificity. Trends Immunol. 29, 91–97 (2008).
Chen, Q., Menon, R., Calder, L. J., Tolar, P. & Rosenthal, P. B. Cryomicroscopy reveals the structural basis for a flexible hinge motion in the immunoglobulin M pentamer. Nat. Commun. 13, 6314 (2022).
Zhang, A. et al. Beyond neutralization: Fc-dependent antibody effector functions in SARS-CoV-2 infection. Nat. Rev. Immunol. 23, 381–396 (2023).
Goldberg, B. S. & Ackerman, M. E. Antibody-mediated complement activation in pathology and protection. Immunol. Cell Biol. 98, 305–317 (2020).
Bose, D. et al. SHIV-C109p5 NHP induces rapid disease progression in elderly macaques with extensive GI viral replication. J. Virol. 98, e0165223 (2024).
Tartaglia, L. J. et al. Production of mucosally transmissible SHIV challenge stocks from HIV-1 circulating recombinant form 01_AE env sequences. PLoS Pathog. 12, e1005431 (2016).
Zhou, M. et al. Adaptation of an R5 simian-human immunodeficiency virus encoding an HIV clade A envelope with or without ablation of adaptive host immunity: differential selection of viral mutants. J. Virol. 93, e02267-18 (2019).
Ren, W. et al. Generation of lineage-related, mucosally transmissible subtype C R5 simian-human immunodeficiency viruses capable of AIDS development, induction of neurological disease, and coreceptor switching in rhesus macaques. J. Virol. 87, 6137–6149 (2013).
Beyersdorf, J. P. et al. Robust, durable gene activation in vivo via mRNA-encoded activators. ACS Nano 16, 5660–5671 (2022).
Blanchard, E. L. et al. Treatment of influenza and SARS-CoV-2 infections via mRNA-encoded Cas13a in rodents. Nat. Biotechnol. 39, 717–726 (2021).
Vanover, D. et al. Nebulized mRNA-encoded antibodies protect hamsters from SARS-CoV-2 infection. Adv. Sci. 9, 2202771 (2022).
Vorauer-Uhl, K., Wallner, J., Lhota, G., Katinger, H. & Kunert, R. IgM characterization directly performed in crude culture supernatants by a new simple electrophoretic method. J. Immunol. Methods 359, 21–27 (2010).
Sanders, R. W. et al. A next-generation cleaved, soluble HIV-1 Env trimer, BG505 SOSIP.664 gp140, expresses multiple epitopes for broadly neutralizing but not non-neutralizing antibodies. PLoS Pathog. 9, e1003618 (2013).
Acknowledgements
This study was supported by NIH grant 1R01AI155007 and 5R01AI139288 to P Santangelo and F Villinger. We would like to thank the staff of the New Iberia Research Center for their care of the animals and their help in conducting the studies. The authors also wish to thank the AIDS Reagent Program for the supply of HIV gp140 proteins, Dr. Barouch, Ruprecht, and Cheng-Mayer for the procurement of SHIV isolates. Figures 1a, b, g, 2a, 3a, 5a, 6a, 8a, and Supplementary Fig. 1a were created in part using BioRender.com.
Author information
Authors and Affiliations
Contributions
J.Y.J., D.V., C.Z., F.V., and P.J.S. designed the experiments and interpreted the data. D.V., C.Z., F.V., and P.J.S. supervised the project. J.Y.J., Y.J., and J.H. performed in vitro characterizations and mouse studies. P.X. conducted in vitro neutralization, ex vivo explant SHIV challenges and analyzed the data. S.P.J. performed biolayer interferometry and analyzed the data. H.P. synthesized the mRNAs, and L.E.S. formulated the LNPs. D.B. produced SHIV. J.Y.J., P.X., S.P.J., D.V., D.B., and F.V. performed macaque studies. J.Y.J., F.V., and P.J.S. wrote the manuscript. All authors reviewed and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Mariangela Cavarelli and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Joo, J.Y., Xiao, P., John, S.P. et al. Intravaginal delivery of mRNA-encoded antibodies with enhanced breadth and potency for SHIV/HIV protection. Nat Commun 16, 10463 (2025). https://doi.org/10.1038/s41467-025-65456-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65456-x
