
Abstract
Traditional phenotypic drug discovery platforms suffer from poor scalability and/or a lack of mechanistic understanding of discovered probes. We address this by creating Endo-GeneScreen (EGS), a high-throughput platform that identifies small molecules that regulate endogenous protein levels encoded by a preselected target gene within disease-modeling contexts. Two initial screens identify >40 validated small molecules that boost endogenous neuronal Syngap1 levels, a gene that causes a neurodevelopmental disorder when haploinsufficient. EGS assays also accelerate preclinical development of drug candidates and facilitate mode-of-action deconvolution studies of orphaned probes. SR-1815 represents a fully validated proof-of-concept candidate from the platform. It is a previously unknown drug-like small molecule multikinase inhibitor that regulates splicing of Syngap1 transcripts. It restores SynGAP protein abundance to wildtype levels and mitigates major cellular consequences of Syngap1 loss-of-function. Thus, the EGS platform promotes identification and development of small molecules that alter the abundance of disease-linked proteins in a translationally-relevant context.
Similar content being viewed by others

Introduction
High-throughput screening (HTS) for phenotypes has re-emerged as a powerful approach in drug discovery and functional genomics1,2,3. Unlike target-based strategies that begin with a predefined molecular target, phenotypic screening starts with an observable biological outcome (phenotype) and then identifies compounds or genetic perturbations that produce the desired effect. This forward pharmacology approach allows for the unbiased discovery of bioactive agents, often capturing complex, system-level responses that target-centric methods might overlook. As a result, phenotypic strategies have been credited with the discovery of many first-in-class drug candidates that regulate essential cell signaling pathways implicated in disease4,5,6.
Phenotypic assays that measure expression of proteins encoded by critical genes required for cellular health promotes advancements in drug development and discovery biology7. By measuring changes in a protein’s native expression level, it is possible to identify drug-like probes that increase (up-regulate)8 or decrease (down-regulate)9 their abundance, thereby expanding utility to discover both activators and inhibitors of disease-relevant pathways. Performing assays in native cellular models further improves translational relevance, as hits are identified within a context that closely mirrors relevant pathobiology. By embracing this unbiased, context-rich strategy, one can reveal previously unknown regulatory pathways and therapeutic strategies that would likely remain hidden in target-centric approaches. Traditionally, there has been a significant trade-off between biological appropriateness and scalability (e.g., how many agents can be screened); as the context becomes more relevant, fewer compounds can be screened because the physiologically relevant environment is typically more complex. Moreover, phenotypic screening in general suffers from a lack of target-based information (e.g., orphaned probes) with respect to how a cellular phenotype is altered in response to the newly discovered agent. Together, these factors have limited the application of phenotypic screening to disease biology and drug development, particularly for disorders of the brain.
We reasoned that recent advances in biotechnology can be leveraged to develop phenotypic screening platforms that increase the scale of screening campaigns without sacrificing the appropriateness of the biological context. Additional advances in chemical biology and chemo-proteomics have also improved the success rate of molecular target deconvolution of phenotypic probes10. Here, we demonstrate the integration of advances in endogenous protein detection within native cells11, lab automation, and the recent widespread availability of massive, commercially available libraries of drug-like bioactive molecules to create the Endo-GeneScreen (EGS) platform (Fig. 1). This flexible HTS-enabled screening platform identifies small molecules capable of regulating endogenous protein expression encoded from any preselected target gene within biologically appropriate cellular contexts. We focus on small molecule screening because these agents remain the gold standard therapeutic for most disease indications12, and their chemical diversity can reveal previously unknown druggable pockets in nucleic acids and proteins13, a process that facilitates therapeutic discovery.

a A relevant screening library is selected based on the cellular context of interest (e.g. a CNS-targeted library for brain cells; only relevant if drug-development is a future goal). b Mouse models are developed; FLAG-HiBIT tag is inserted into gene of interest. This tag could be inserted into a transgene if protein lowering in a cellular context is the goal. Inclusion of a cKO mouse model is a must if protein upregulation is desired within a gene loss-of-function cellular context. c DLR assay workflow showing how endogenous proteins are detected within a specific cellular context; screening plates required for 100k compound screen for screening within cortical mouse neurons. d After completion of the screen, data are visualized on a per-plate basis and hit detection and filtering algorithms are implemented to identify compounds of interest. e Preliminary hits are run through a biological validation workflow that focuses on replication and repeatability, ultimately relying on protein detection using KO-validated antibodies (if available). f Probes that are validated at the protein level within the cellular context of interest are then tested in a series of low-throughput phenotypic assays that ideally are known to be modified by altered expression of the protein of interest. g Validated probes that also regulate cell-context-specific phenotypes are then explored at the chemical level by obtaining ~100 closely related analogues. This determines the extent to which a chemical series with promise can be modified by medicinal chemistry.
As a proof-of-concept, we demonstrate how each component of the platform is used to identify, validate, develop, and define the mode-of-action of drug-like small molecules that upregulate expression of the target gene, SYNGAP1/Syngap1 (HUMAN/Mouse; Mouse from now on). Syngap1 was selected for multiple reasons. First, de novo mutations in this gene that lead to haploinsufficiency are one of the most common genetic causes of sporadic neurodevelopmental disorders associated with intellectual disability, autism, and epilepsy14,15,16,17,18. Thus, among many potential uses, this platform can be used to screen for boosters of autosomal dominant (AutD) haploinsufficiency genes. These genes cause disease through de novo mutations that lead to reduced functional protein expression19,20. As such, small molecule boosters would, in principle, address the root cause of these genetic conditions. Second, this gene principally functions in differentiated cortical excitatory neurons21,22,23, a notoriously difficult cellular system to work with at scale24. Third, excellent in vitro and in vivo models for Syngap1 haploinsufficiency have been developed and extensively validated25,26,27. These models, if combined with a probe discovery platform, would form an integrated ecosystem that can be used to first screen for probes that upregulate SynGAP protein in an appropriate context (e.g., cortical neurons) and then eventually validate the effectiveness of these probes in the same system. Here, a Syngap1-focused variation of the EGS platform led to the discovery of SR-1815, a drug-like small molecule that restores low SynGAP expression and mitigates synaptic and neuronal hyperexcitability, three major cellular consequences of Syngap1 haploinsufficiency. Moreover, we demonstrate how EGS workflows accelerate preclinical drug development by jump-starting medicinal chemistry-based improvement of identified compounds. Finally, EGS assays can be combined with emerging chemical biology and molecular genetic approaches to reveal the mode- and mechanism(s)-of-action of previously uncharacterized phenotypic probes. Indeed, in a parallel study, we report that SR-1815 is a drug-like kinase inhibitor that boosts SynGAP levels through alternative splicing linked to activity at certain kinase targets (Douglas et al., BioRxiv, 2025; https://www.biorxiv.org/content/10.1101/2025.03.13.643041v1)28. This related study highlights an additional feature of the EGS platform. It provides an opportunity to discover unconventional biological probes that regulate disease-associated gene expression through cumulative efficacy across multiple molecular targets and cellular pathways [e.g., polypharmacology29,30]. Therefore, de-orphaning probes discovered through EGS can advance disease-associated cell biology in addition to promoting precision drug discovery.
Results
Development of EGS scalable assays for tracking endogenous target protein expression within disease-appropriate cellular contexts
EGS is built upon a series of scalable phenotypic assays that report relative changes in endogenous protein encoded by a pre-selected target gene within a biologically relevant cellular context. The foundational principle of the platform concept was to develop scalable endogenous protein expression assays within relevant disease-modeling cellular contexts, such as 2D/3D cultures from primary cells derived from animal models or induced cellular models derived from patient iPSCs (Fig. 1). When engineering the scalable assays, we set several design parameters: 1) flexible targeting—any gene of interest can be targeted with the assays; 2) relevant cellular context—assay must be carried out in disease-modeling cellular systems; 3) sufficient scalability—HTS-capable to enable screening of expansive chemical libraries of drug-like molecules. After several iterations, we settled on a design utilizing genetic engineering to enable an HTS-compatible Dual Luciferase Reporter (DLR) screening assay performed in primary cells from mice expressing two distinct luminescence-based reporters (Fig. 2a). Primary cells are extracted from the organ system causally linked to the disorder, such as the cortex for the Syngap1 version of EGS21,23. Moreover, a conditional allele was required to both enable induction of Syngap1 haploinsufficiency and to facilitate a streamlined breeding strategy (Fig. 2a, g). The first luminescence signal was envisioned as a readout of endogenous target protein through activation of a split luciferase based on an engineered variant of Nano-luciferase11. The second luminescence signal, generated by a firefly luciferase transgene (fLUC) expressed from a ubiquitous promoter, was envisioned to report global changes in protein expression and/or cellular toxicity in response to library compounds added to screening plates harboring primary cells. Inducible Syngap1 haploinsufficiency, to enable disease modeling, would be achieved through a conditional Syngap1 allele with LoxP sites flanking essential exons. Three distinct strains of mice were required to achieve the noted design goals, and once obtained, they would be crossed to yield offspring expressing the engineered genetic components (Fig. 2a, g).

a Concept of required engineered mouse line to enable endogenous SynGAP protein measurements within an HTS-like workflow. Neurons from the mouse line are cultured in 384-well plates are then treated with library compounds for 2–4 days before execution of the DLR assay. b Schematic of the design and location of the HiBIT tag to be inserted into the mouse Syngap1 gene. c Endogenous SynGAP-HiBIT fusion protein activates nBIT luciferase activity. d Immunoblots from PND14 mice heterozygous for the inserted HiBIT tag. Samples were probed with Pan-SynGAP and FLAG antibodies. Samples were also probed with a detection kit that enables a direct luminescent readout of proteins that contain a HiBIT tag. e Quantification of endogenous SynGAP protein from immunoblots containing Syngap1+/+ (blue) and Syngap1+/HiBIT (purple) mice (n = 6 per genotype). Syngap1+/HiBIT lysate was serially diluted and probed for correlation of SynGAP levels detected using a Pan-SynGAP antibody or LgBIT. Simple linear regression was performed, Pan-SynGAP p = <0.001, and nBIT p = <0.001, n = 2 per concentration. f Syngap1+/HiBIT mice were crossed to each other. Syngap1+/HiBIT and Syngap1HiBIT/HiBIT mice were cultured in 384-well plates and assayed at DIV14. g Breeding strategy to yield mouse offspring to feed the Syngap1 version of EGS. h Neuronal plating density was varied to measure how the two DLR signals reflect changes in the amount of total protein in the well. n = 128 for densities 5000–15,000 cells/well and n = 64 for 20,000 cells/well. Data are presented as mean values +/- SEM. i Amitriptyline, a known neurotoxic agent in primary cultured neurons, induces a dose-dependent reduction in both luciferase signals (fLUC: black, nBIT: orange) indicating that a coordinated drop in both signals reflect neuronal toxicity (n = 32 per dose). Data are presented as mean values +/- SD (e, i). p-value for main effects and interaction are indicated as n.s.: p > 0.05, detailed statistics and source data are provided as a Source Data file. Created in BioRender. Samowitz, P. (2025) https://BioRender.com/cnld7d3.
The conditional Syngap1 knockout mouse31 and a transgenic mouse ubiquitously expressing fLUC (see methods) were already constructed, validated, and available commercially. To detect endogenous SynGAP protein in mouse cells/tissues, a mouse knock-in strain that expressed a HiBIT ligand11 in-frame within protein made from the murine Syngap1 gene was created (Figs. 2b; S1A-B). For targeting, we selected an exon included in all major isoforms that codes for an unstructured portion of the SynGAP protein with no known functional domains. When the HiBIT tag binds to an inactive and purified fragment of Nano-luciferase, called LargeBIT, it reactivates dormant luciferase activity (Fig. 2C). The reconstituted and catalytically active HiBIT-LargeBIT complex is referred to as NanoBIT luciferase (nBIT from now on). Photons generated from nBIT activity produced from an endogenous SynGAP-HiBIT fusion protein were hypothesized to report proportional changes in endogenous SynGAP protein induced by test agents within primary cultured cortical neurons. To validate this, we first confirmed that the genetic insertion within the Syngap1 coding region did not alter Mendelian ratios of offspring. We also confirmed that the insertion did not disrupt endogenous full-length SynGAP protein expression levels in mouse brain (Fig. 2d, e). Insertion of the tag accurately reported relative changes in SynGAP expression within a diluted sample of cortical tissue extract. Moreover, detection of endogenous SynGAP protein through addition of LargeBIT on a traditional immunoblot predicted the levels of endogenous protein on par with antisera. We next confirmed that primary cultured neurons prepared from the Syngap1-HiBIT knock-in mouse enabled detection of relative changes to endogenous SynGAP protein within HTS-compatible 384-well screening plates (Fig. 2f). To do this, neurons from either heterozygous or homozygous Syngap1-HiBIT knock-in mice were added to separate wells of the same assay plate. nBIT signal from homozygous knock-in neurons were on average ~2-fold higher than signals obtained from heterozygous neurons (Fig. 2f). Signals derived from wildtype mice yielded ~60 relative luciferase units (RLUs), resulting in a very high signal-to-noise ratio of >10e3 for detecting endogenous HiBIT-tagged SynGAP protein within primary cells or tissue. Moreover, signals derived from assay wells with either heterozygous or homozygous neurons resulted in completely non-overlapping populations (Fig. 2f), indicative of low inherent assay variance. These properties suggested that this signal would be suitable to identify small molecules that regulate physiological changes in endogenous SynGAP protein within primary cortical neurons.
Primary neurons extracted from postnatal day (PND) 0 mice (Fig. 2g) were then cultured in assay plates to determine to what extent the non-specific fLUC signal reports global changes in protein expression and/or cellular toxicity. Indeed, we found that incrementally raising neuronal plating density drove a proportional increase in fLUC signal, confirming that this signal approximates global changes in total protein (and plating density) within assay wells (Fig. 2h). This signal also reliably reported neuronal toxicity induced by amitriptyline (Fig. 2i), a known neurotoxic agent32. As a result, the non-specific fLUC signal serves as a counter-screen to eliminate test agents that globally induce changes in protein expression, while also reliably reporting cellular toxicity. The former is important for rejecting test agents that fail to stimulate enrichment of the target (SynGAP) protein, while the latter is critical for directing downstream medicinal chemistry approaches aimed at improving the drug-like properties of probes that advance through a lead optimization program. Finally, we confirmed that AAV-driven Cre expression within neurons derived from this cross induced a dose-dependent decrease in SynGAP protein (Fig. S1C). This confirmed our ability to induce Syngap1 haploinsufficiency in neurons expressing the two luciferase reporters, a requirement for modeling the Syngap1 genetic disorder in a dish.
We next optimized the assay for HTS-level scalability so that large chemical libraries of drug-like probes could be screened. Internal in silico modeling suggested that a strategic selection of at least 100 K distinct compounds would be sufficient to sample diverse chemical space within a > 2 M compound collection of drug-like lead molecules available through various commercial sources (see methods). To screen at least 100 K compounds (in duplicate) using the primary neuron DLR assay, ~624 assay plates would be required (Fig. S2A-B). To achieve this desired level of scalability, ~52 individual litters of mice would be needed to produce cultured cortical neurons at scale. With an average output of two batches of assay plates per week, a screen would take ~6 months to complete. Scalability is largely dictated by the availability of mouse primary neurons. When using an optimized breeding strategy (Fig. 2g), we previously demonstrated that two batches of neurons per week (up to 32 plates; Fig. 1) is possible using one full-time-equivalent laboratory position24. However, to minimize rates of false positive/negative data points, the assay must exhibit reliability and reproducibility, especially when performed in an iterative screening environment spanning months. Indeed, it was unclear at the time to what extent primary cortical cultures from postnatal mouse brains would exhibit the required scalability and reliability to sustain a screening platform carried out over an extended timeframe. To quantify culture reliability, plate-level quality control (QC) metrics, such as raw luciferase values from each independent signal and the coefficient of variance (CV) of these signals in negative control wells ( < 0.1% DMSO), were used as “pass/fail” flags for individual compound screening plates within and between culture batches (Fig. 3a). In pilot studies, plates within a screening “batch” shared pooled primary neurons obtained from a single mouse litter, and as expected, QC metrics were highly similar between plates within the same batch (Fig. 3b). This experiment revealed that the CV of raw luciferase values from individual plates remained within the acceptable range ( < 15%) even when culture density varied over a 1.5-fold range. This suggested that culture batches produced over a long timeframe within the same screening project would demonstrate the requisite reliability (e.g. few failed batches) and thus could sustain a screening platform. Consistent with this prediction, post-hoc analysis of several hundred neuronal culture screening plates prepared over ~1 year were found to be highly consistent, with only 4% of plates being failed (CV > 15%: Fig. 3c). When plates were failed, post hoc analysis of these simple QC metrics enabled a quick diagnosis, and most fails were traced back to an issue with one or more channels within the robotic liquid dispenser (e.g., clogging).

a Schematic of data hierarchy generated from the proposed iterative high-throughput screen. b Luminescent values and coefficient of variance (CV) for fLUC (black) and nBIT (orange) signals across two batches of neurons plated at 15,000 cells per well (Batch 1) or 10,000 cells per well (Batch 2), n = 36 per plate. c Raw luminescence values (top) and CV (bottom) for negative control wells in Screen 1 ( ~ 59,840 compounds) and Screen 2 ( ~ 47,360 compounds). n = 204 and n = 148 plates with 96 or 72 samples per compound plate for Screen 1 and 2, respectively. Data are presented as mean values +/- SEM (b, c). Source data are provided as a Source Data file. Created in BioRender. Samowitz, P. (2025) https://BioRender.com/cnld7d3.
EGS assays enable HTS-style screening of chemical libraries required for Lead Identification
To identify compounds that upregulate SynGAP protein expression within haploinsufficient neurons, we performed two independent screens, which together comprised >100 K distinct drug-like small molecules. Two independent screens were carried out because we identified important principles in the first screen, which led to modest modifications to the screening workflow and assay parameters, which were implemented in Screen 2. For example, at the time of implementing Screen 1, there were no known small molecules that raised SynGAP protein in our culture model system. Therefore, assay plates in Screen 1 did not contain a positive control (Fig. S2A). Instead, an extra row of negative controls was included. Additionally, the final compound concentration was varied so that a post hoc analysis of data could be used to determine how concentration impacted assay performance. In Screen 1, the concentration of library compounds ranged from ~3.125–12.5 μM in ~0.03125–0.125% DMSO. Moreover, compounds were pinned into primary culture assay wells and allowed to incubate for 48 h– a notably conservative approach for inducing steady-state changes in protein abundance. The DLR assays were always carried out on day in vitro (DIV) 14. All compounds were screened in duplicate (N = 2 screen).
To detect hits from Screen 1, an outlier algorithm was developed to identify compounds that selectively increased or decreased the nBIT signal (Fig. 4a–c)-the proxy measure for SynGAP protein expression in neurons. Before this could be implemented, the median fLUC and nBIT signal of the negative control wells (DMSO only) from each plate were first used as a normalization value for every individual well on each plate, including all compound wells. This enabled a z-score to be calculated, with the normalized fLUC signal on the X-axis and the nBIT signal on the Y-axis (Fig. 4a). When visualized this way, both the negative control wells, as well as most compound wells, clustered near the origin. The large number of compounds interspersed with the negative controls was indicative of most compounds lacking activity in the assay, something that was expected in screens of this type, with hit rates often below 1%. However, on each plate, some wells exhibited signals that significantly deviated from the origin, which was indicative of biological activity in the assay. For example, compounds often appeared in the lower left quadrant, which was an indication of cellular toxicity. We also commonly observed a fraction of compounds that significantly increased both signals, suggesting a global increase in protein. Because the goal was to identify compounds that selectively up-regulated endogenous SynGAP expression within haploinsufficient neurons, we used the normalized data to identify Hits, which were defined by setting 3x standard deviation (SD) threshold lines (orange vertical and horizontal lines) above and below the average of the normalized negative control wells for both fLUC and nBIT signals. Compounds showing activity above 3x SD for the nBIT signal and within 3x SD of fLUC were considered preliminary hits. We next filtered out compounds showing unacceptably high variability between replicate data points. This was done by calculating the CV of each initial compound hit, and compounds showing <10% variability across both fLUC and nBIT reads were retained on the final Hit List for each compound plate (Fig. 4a—blue data points). All accepted data from each compound plate data was then collapsed onto a single 2D-scatter plot (Fig. 4b, c). Visualizing the complete screening data set revealed the relative position of the final Hits (blue points), the inactive compounds (grey points), and the negative controls (green points). Using this approach, Screen 1 yielded 127 compounds of interest across 60 K compounds assayed (0.21% Hit rate).

a One plate of screening data from Screen 1 visualized on a 2D scatter plot—nBIT (SynGAP) signal vs. fLUC (toxicity/selectivity) signal. Threshold lines set at 3X standard deviation of the mean for negative control wells. Red shaded section above corresponds up-regulator hit zone and blue shaded section below corresponds down-regulator hit zone. Gray dots appearing in hit zone did not meet established variability criteria. b All data from Screen 1 visualized on 2D scatter plot. c Zoomed in view of Fig. 3b to visualize hit compounds from Screen 1 (127 compounds = 0.21% hit rate). d One plate of screening data from Screen 2 with adjusted threshold lines. e All data from Screen 2 visualized on 2D scatter plot. f) Zoomed in view of Fig. 3d to visualize hit compounds from Screen 2 (171 compounds = 0.36% hit rate). Source data are provided as a Source Data file.
We next screened a distinct library of ~48,000 compounds that were pre-selected based on chemical features known to be conducive to developing neuroactive small molecule therapeutics. For this second screen (e.g., Screen 2—Fig. 4d–f), several workflow improvements were implemented, which were inspired by experience gained from Screen 1. Consequential modifications included a single screening concentration ( ~ 3.125 μM), doubling the compound incubation time to 96 h (DIV10-14), and incorporation of a positive control on all screening plates (Fig. S2A). The positive control was discovered during Screen 1 (Fig. 4b—red dot). Including this positive control on screening plates led to an improved computational approach for identifying compounds with significant activity in the assay (Fig. S3A–D). By quantifying the pattern/spread of positive control, negative control, and inactive compound data points across all screening plates, a clear positive correlation between fLUC and nBIT signals in each of these three populations was identified. This relationship functioned as a type of “loading control” for each well, which allowed us to estimate the relative difference in the amount of neuronal material from well-to-well, and was instrumental in developing an improved hit detection algorithm (denoted by sloped lines; Fig. 4d). The preliminary Hits were next filtered for obvious false positives by assigning a non-repeatedness value. This is a single numerical value that considers both the embedded positive relationship between fLUC and nBIT signals and the continuity of the duplicate data points afforded by the N = 2 screen design (see “methods”; Fig. S3E, F). A value that approaches “1” reflects concordant luciferase values for each replicate of a given test compound. Thus, higher values increase confidence that the compound is a true “hit”. Implementation of a hit detection algorithm and filtering for non-repeatedness revealed 171 additional Hits in Screen 2 (0.36% hit rate) (blue dots; Fig. 4e, f). Several Hits met, and in some cases exceeded, the performance of the positive control. This, combined with a similar hit rate, suggested that the somewhat narrower chemical space within CNS-focused library did not negatively impact overall screening performance, which may be explained by the improved assay workflows implemented in Screen 2.
EGS assays support biological validation of preliminary screening Hits
An essential component of the EGS platform is a workflow dedicated to validating the most promising compounds of interest identified in HTS-style screens (Figs. 1, 5a). The goal of this workflow was to develop confidence that a preliminary Hit obtained in the original screen reliably upregulates endogenous steady-state SynGAP protein abundance within Syngap1 haploinsufficient neurons. SR-1815 (Fig. 4b, c, red dot) was chosen as a proof-of-concept molecule to demonstrate the effectiveness of the Hit Validation workflow because it was the very first molecule of interest to emerge from Screen 1. Searches of the literature (Scifinder, PubMed) were conducted for SR-1815 analogs and identified no known chemistry, biology, or pharmacology related to this scaffold. They were not represented in the public domain other than as analogs in a screening collection.

a Validation workflow encompassing luciferase-based assays, counter-screens, and protein validation assays. b, c SR-1815 in DLR or nBIT counterscreen 8-point dose-response assay. n = 12 per dose for DLR and n = 10 per dose for cell free assay, fLUC (black) and nBIT (orange). d Total protein and SynGAP protein expression in a single dose Dot Blot experiment with SR-1815 (1.5 µM) for 14 days (e, f) Dot blot dose response of SR-1815 treated for 7 days utilizing a SynGAP antibody that recognizes either all isoforms (e) or only the α2 C-terminal isoform (f); n = 28 per dose. g SynGAP protein expression from Pan-SynGAP antibody for WT rat neurons in a Dot Blot experiment after 7 days treatment (DIV 7- 14); n = 28 per dose. Total protein (black) and SynGAP (orange) expression (d–g). h Western blot with samples extracted from primary neuron cultures treated for 14 days with DMSO or SR-1815 (1.5 μM) derived from either Syngap1+/+ (blue) and Syngap1+/- (red). i Representative images from an immunocytochemistry experiment in Syngap1+/- primary cultures treated with either DMSO (v) or SR-1815 (1.5 µM) (d) 14 days and labeled with SynGAP (red), PSD95 (green), or SV2A (blue) antisera. Somatic signals (square) and quantified synaptic signals (rectangle) are represented, right. j Left, Representative image of dendritic branch illustrating structures where multiple signals co-localize (e.g., synapses). SynGAP expression was localized to dendrites as evidenced by non-overlapping linear signal with the synaptic markers. Right, Quantification of SynGAP intensity (as measured by pixel intensity) across neuronal compartments. Dendritic signal was measured as peak intensity from a line series. Somatic and synapse signal intensity was measured as average pixel intensity from thresholded area within square and rectangle, respectively (i). Data are presented as mean values +/- SEM (b–g, i, j) or +/- SD (h). p-value for main effects and interaction are indicated as n.s.: p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, detailed statistics and source data are provided as a Source Data file. Source data are provided as a Source Data file. Created in BioRender. Samowitz, P. (2025) https://BioRender.com/cnld7d3.
The probe validation workflow was comprised of up to five additional levels of validation beyond the initial screening data (Fig. 5a). The first steps in the validation pipeline were designed to determine to what extent a compound regulates the nBIT signal in a dose-dependent manner. SR-1815 demonstrated dose-response activity from freshly sourced compound powder (Fig. 5b). To rule out a direct effect of the small molecule on nBIT enzymatic activity, we utilized a cell-free counter-screen, where LargeBIT, a purified Halo-HiBIT fusion protein, and the compound of interest were added together with the assay reagents. In this cell-free assay, SR-1815 did not increase nBIT activity at any concentration tested (Fig. 5c), suggesting that it may regulate SynGAP protein expression.
Given these results, we hypothesized that the compound increases SynGAP steady-state protein abundance in neurons. Evidence supporting this hypothesis necessitated the development of a scalable orthogonal assay that directly measures SynGAP protein. A scalable Dot Blot protein assay was developed that combined a 384-array pin tool, a nitrocellulose membrane, a label that reports total protein, and a series of knock-out validated SynGAP antibodies (Fig. S4A). After neuronal lysis directly within the 384-well plate, samples were incubated to fluorescently label lysine residues as a measure of total protein using derivatizer, 3-(2-Furoyl)quinoline-2-Carboxaldehyde, and activator, Mandelonitrile. Lysate from each well was pinned onto a nitrocellulose membrane. After optimization, this technique reliably reported accurate levels of total protein in each sample on the membrane (Fig. S4A). The membrane was also exposed to a monoclonal antibody that detects a motif expressed in all SynGAP protein isoforms (e.g., pan-SynGAP). After washing and secondary antibody exposure, the membrane was imaged for total protein and incubated with chemiluminescent substrate. This resulted in a strong signal that was linear across protein concentrations that spanned an order of magnitude (Fig. S4A). The Dot Blot assay was then tested to determine how well it measures changes in steady-state SynGAP abundance in primary cortical neurons. To do this, we utilized an already validated Syngap1 conditional rescue mouse line31,33, which was engineered to express an artificial exon containing a stop codon and an exogenous poly-A sequence within the mouse Syngap1 gene. This exon is efficiently spliced into Syngap1 transcripts as evidenced by disruption to SynGAP protein expression. However, this exon is flanked by LoxP sites, and therefore expression of Cre recombinase re-activates SynGAP protein expression due to excision of the artificial exon (Fig. S4B). It was previously shown that mice heterozygous for this targeted allele have roughly half SynGAP protein expressed in neurons relative to wildtype littermates, while homozygous mice nominally express SynGAP protein31. This Dot Blot technique accurately reported the known expression changes of SynGAP in neurons derived from this mouse line, with nominal expression of SynGAP protein in homozygous neurons and roughly half the normal levels of protein expressed in the heterozygous neuronal population (Fig. S4C). Importantly, the Dot Blot was also able to detect the Cre-dependent re-expression of SynGAP protein in both homozygous and heterozygous neurons (Fig. S4C). Furthermore, the Dot Blot assay was validated using antisera that detects three of the four major SynGAP C-terminal isoforms. Importantly, these isoform-specific antibodies were themselves validated using the Syngap1 KO mouse line (Fig. S4D). These reagents are important because individual C-terminal isoforms have unique spatial/temporal expression profiles and distinct biological functions in vivo34,35. Finally, we developed custom software that automated analysis of Dot Blots (Figs. S5A-D, S6A-B), which dramatically increased the scalability of SynGAP protein detection.
Using the now-validated Dot Blot assay, we found that SR-1815 could double endogenous SynGAP levels in heterozygous neurons compared to DMSO controls (Fig. 5d). Moreover, the compound stimulated a dose-dependent increase in SynGAP protein abundance as measured by both a pan-SynGAP antibody that detects all isoforms (Fig. 5e), and an antibody that recognizes only the α2 isoform (Fig. 5f). Given that the anti-α2 signal matched the anti-pan-SynGAP signal within the same samples, this result indicated that SR-1815 stimulates relatively equal expression of all SynGAP C-terminal isoforms34. The compound also stimulated SynGAP expression in neurons derived from typically developing wildtype rats (Fig. 5g). This demonstrated that SR-1815 efficacy is not limited to mouse neurons, and the compound is effective in both a typically developing genetic background and in a background of Syngap1 haploinsufficiency. Pairing optimized assay conditions with a traditional Western blot technique, SR-1815 rescued SynGAP protein levels in heterozygous KO neurons (Fig. 5h).
To ascertain how subcellular pools of SynGAP change in response to SR-1815 treatment, we performed immunofluorescence paired with confocal microscopy in Syngap1 haploinsufficient neurons. An integrated measure of isolated synaptic SynGAP signal (e.g., thresholded area x pixel intensity) was upregulated ~4-fold by SR-1815 relative to vehicle (Fig. 5i), while somatic SynGAP signal was unchanged (Fig. 5j), indicating that SR-1815 induced an outsized effect on synaptic SynGAP. The average intensity of synaptic SynGAP structures was also modestly, but significantly, increased by the compound. Further, we observed a ~ 3.7-fold change in the average number of thresholded pixels in SR-1815 treated neurons compared to vehicle (U = 115, vehicle, 2285 + /−231; drug, 8540 + /−485; (SEM); p = <10e-6). These results indicated that the 4-fold increase in synaptic SynGAP signal was due largely to a greater thresholded synapse area, with a modest increase in SynGAP molecules per synapse. SR-1815 could regulate a change in synapse area through increasing synapse number, synapse size, or both. SV2A, a presynaptic marker, was much less affected ( ~ 1.3-fold change) by the compound compared to the SynGAP (Fig. 5i). This suggested that the major effect of the drug was on postsynaptic structural changes rather than synapse number. The size of the postsynaptic density (PSD) is correlated with dendritic spine size/volume36. If SynGAP levels are increased in part through a change in the size of the PSD and/or dendritic spines, then this would predict a similar change in isolated PSD95 synaptic signal. Consistent with this, a ~ 3-fold increase in integrated PSD-95 signal was observed in response to SR-1815 (Fig. 5i). Dendritic SynGAP signal was also modestly, but significantly, increased by the drug (Fig. 5i). This may be due to either increased extrusion of SynGAP molecules out of spines and into dendrites37, or through an increase in abundance of SynGAP isoforms known to be relatively enriched in dendrites35. Importantly, our confocal imaging studies are consistent with the findings of Douglas et al.28, which found that SR-1815 regulated transcriptional dynamics and splicing events overrepresented in genes that encode postsynaptic proteins. Moreover, using immunoblotting, this study also reported several postsynaptic proteins, including SynGAP, PSD95, and GluN1 were substantially increased by SR-1815, while SV2A abundance was slightly reduced.
We next asked if probes discovered using EGS mitigate functional phenotypes that are caused by reduced target protein expression. Syngap1 haploinsufficiency in mice has been shown to increase the frequency and amplitude of miniature excitatory postsynaptic events (mEPSCs) in primary cultures22,38, which is a measure of excitatory synapse strength. Therefore, we designed an experiment that enabled an assessment of how excitatory synapse strength was impacted by SR-1815 in both wildtype and Syngap1 haploinsufficient neurons. We observed a strong interaction between drug and genotype for both mEPSC frequency (Interaction: (1,64) = 9.168, p = 0.0035) and amplitude (Interaction: F (1, 64) = 7.344, p = 0.0086), demonstrating that these effects of the compound were genotype-dependent (Fig. 6a). Post-hoc comparisons revealed that both mEPSC frequency and amplitude were significantly increased in neurons derived from Syngap1 heterozygous animals (Frequency: DF = 64, p = 0.015, Amplitude: DF = 64, p = 0.017), a result that agreed with past studies. The strong interaction was driven by the opposing effect of the compound on mEPSC measures within each genotype. The compound tended to increase excitatory synapse strength in wildtype neurons but tended to decrease it in haploinsufficient neurons. These opposing trends are what drove the strong interaction in the statistical model. Thus, while the effect of the treatment fell short of post hoc significance within each genotype, the strength of the interaction overall provides strong evidence that the compound has genotype-specific bidirectional effects on excitatory synapse function (e.g., increases synapse strength in wildtype neurons; decreases synapse strength in haploinsufficiency neurons). This finding is consistent with our data demonstrating that SR-1815 induces a change in SynGAP abundance within the synaptic compartment (Fig. 5i). Moreover, this finding is therapeutically relevant because SYNGAP1/Syngap1 haploinsufficiency causes neural hyperexcitability and seizures in humans and rodents21. Furthermore, reducing excitatory synapse strength through AMPA receptor inhibition improves cognition-linked brain rhythms in Syngap1 heterozygous mice39.

a) Representative traces from mEPSC recordings in Syngap1+/+ (blue) and Syngap1+/- (red) neurons treated with vehicle or SR-1815 (left). Plot showing mEPSC frequency for the four different conditions (middle) and mEPSC amplitudes for the four different conditions (right) n = 17 for Syngap1+/+: vehicle; n = 16 for Syngap1+/+: SR-1815; n = 17 for Syngap1+/-: vehicle; n = 18 for Syngap1+/-: SR-1815. Error bars represent SEM. b Syngap1 + /+ and Syngap1 + /- neurons transduced with AAV9 vectors expressing Flex-gCAMP8f and Cre (to control labeling). Neurons were treated with DMSO or 1.5 μM SR-1815 for 14 days. Calcium imaging performed on DIV14. Plot showing the spiking frequency (spikes per second) for the four different conditions. For DMSO control 12 fields were imaged for Syngap1+/+ Syngap1+/- and for SR-1815 respectively 14 and 17 fields for Syngap1+/+ Syngap1+/- were imaged from at least 4 wells per conditions. The total number of segmented neurons were DMSO Syngap1+/+, n = 1693; Syngap1+/-, n = 2047; SR-1815 Syngap1+/+, n = 1803 and Syngap1+/-, n = 1253. Dots with error bar represent mean and SEM, individual neuron value (small dot) and well average (large dot) are plotted, within the violin plot median, 25th and 75th percentile. p-value for main effects and interaction are indicated as n.s.: p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, detailed statistics and source data are provided as a Source Data file.
The observed compound-induced genotype-specific effects on excitatory synapse strength suggested that SR-1815 may also regulate neuronal activity in a genotype-specific manner. To test this, GCaMP8 dynamics were measured across thousands of individual neurons treated with vehicle or SR-1815 from cortical cultures derived from each genotype (Fig. 6b). Overall, the effect of SR-1815 within each genotype was consistent with results obtained from measurements of excitatory synapse strength. For example, this analysis detected a main effect of genotype (F(1,6792) = 29.4, p < 0.001), and a posthoc comparison confirmed that vehicle-treated heterozygous neurons had increased activity compared to vehicle-treated wildtype neurons (mean diff = −0.0065, 95% CI [−0.0096, −0.0034], p < 0.001). Moreover, there was a main effect of treatment (F(1,6792) = 33.8, p < 0.001), indicating that the SR-1815 significantly regulated spike rates in neurons from both genotypes. However, a significant interaction was detected between genotype and treatment (F(1,6792) = 200.9, p < 0.001), demonstrating that the compound regulated activity in a genotype-specific manner. Indeed, SR-1815 significantly increased activity in wildtype (WT) neurons (WT-DMSO vs. WT-drug, mean diff= 0.0069, 95% CI [0.0037, 0.0101], p < 0.001), while it substantially decreased it in heterozygous knockout (Het) neurons (Het-DMSO vs. Het-drug, mean diff = −0.0189, 95% CI [−0.0223, −0.0155], p < 0.001). Thus, the compound drove normally hyperactive heterozygous KO neurons to activity levels at or below that of wildtype neurons. Given the potential significance of this result, we repeated this experiment in heterozygous KO neurons, but this time using multiple doses. Importantly, a dose-dependent decrease in neuronal activity by SR-1815 was observed in this additional experiment (Fig. S7), demonstrating that adjusting the dose of the compound can tune hyperactive heterozygous KO neurons to levels approximating the wildtype state. Taken together, EGS can identify drug-like probes that raise endogenous expression of the targeted gene, and this probe can counteract the functional consequences of genetic haploinsufficiency within a disease modeling cellular context. Furthermore, the genotype-specific effects of SR-1815 highlight the importance of working in the appropriate cellular contexts when exploring the function of phenotypic probes.
With the hit validation pipeline pressure-tested through extensive characterization of SR-1815, we next used the first few stages of this pipeline to evaluate the most promising hits from Screen 2 (CNS-focused library; Fig. 4f). Seventy-two (72) compounds from this initial pool were selected for follow-up testing based on efficacy, non-repeatedness, and chemical diversity. Because of the extensive number of preliminary hits, highly similar compounds could be excluded, allowing us to enrich the pool with lead candidates that were as chemically diverse as possible. From freshly sourced powder, 40 compounds exhibited significant activity in the SynGAP DLR assay (Fig. S8A). Sixteen (16) compounds exhibited at least a two-fold change in nBIT signal, which suggests they may have the potential to rescue SynGAP protein abundance in haploinsufficient neurons. Many candidates demonstrated dose-response activity beginning in the low-mid nanomolar range (Fig. S8B). Eight (8) of the most promising compounds were next tested in the Dot Blot protein assay, which was also executed in Syngap1 haploinsufficient neurons (Fig. S8C). Critically, each of these selected candidates exhibited a dose-dependent change in SynGAP protein abundance that was predicted by the nBIT signal (Fig. S8B-C). In total, the Syngap1 EGS campaign yielded at least 42 potential lead molecules suitable for entering preclinical drug development workflows. This includes 40 candidates from Screen 2, as well as multiple promising and validated lead compounds discovered during Screen 1, including SR-1815. These initial compounds are advancing through the early stages of preclinical drug development, while the subsequent compounds from Screen 2 are undergoing more advanced biological (e.g., mechanistic) validation and determination of initial drug-like properties.
EGS assays facilitate preclinical drug development
EGS yields drug-like probes that intersect with AutD gene biology and function. Therefore, it is critical to demonstrate that the platform can support preclinical drug development workflows. An initial first step in the drug development pipeline is to optimize the lead candidate scaffold through synthetic chemistry. As a first step in this process, medicinal chemists often perform a shotgun approach, where substitutions are made throughout the different motifs within the compound to identify the positions on the molecule that can tolerate significant modifications.
We developed an optimized workflow that can jumpstart preclinical development by bypassing this initial random shotgun approach to scaffold modification. Instead, our approach is to identify and then purchase small quantities of close analogs of a lead molecule that exist within the extensive small molecule collections held by various commercial partners. DataWarrior40 is an open-source cheminformatics tool that can interface with the full compound collection of various commercial entities that source screening collections for drug discovery. This tool is used to identify compounds within the full collection that are chemically similar to a lead originally identified from the screening library (Fig. 7a). To begin the SR-1815 development process, we identified 200 structurally-related compounds within the full collection. Next, these compounds were sorted based on structural diversity. This ensured that none of the structural motifs within the lead compound were overweighted in the identification process. Then, 100 compounds that exhibited the broadest structural diversity across the pool of 200 selected compounds were sourced. This process yielded one hundred SR-1815 analogs that resembled first-order derivatives of the lead (Fig. 7b), many of which would have been made by a medicinal chemist in a traditional “shotgun” approach. The selected compounds were sourced from the vendor, usually delivered within three weeks, and then tested for dose-response activity in the Syngap1 DLR assay (Fig. 7c).

a Top, Screen capture from DataWarrior exploration of SR-1815-like compounds within the full collection of small molecules. Bottom, Process for selecting 100 structurally similar SR-1815-like compounds. b Visualization of relative structural diversity among identified SR-1815-like compounds. Colored regions reflect different core motifs within SR-1815. c Relationship between molecular structure and SynGAP assay activity from 104 SR-1815-related compounds. The 104 compounds are comprised of 100 close analogs of SR-1815 (a), 3 related compounds in the original screening library (e.g, SR-1821; grey circle), and SR-1815 (reference compound). d Identification of clearly modifiable R-groups derived from structure-activity analysis of SR-1815 analogs shown in (c). Source data are provided as a Source Data file.
Three of the compounds flagged in the cheminformatic profiling were present in the original screening library—SR-1819, SR-1821, SR-1823—and each analog retained activity in the SynGAP DLR assay. One of these compounds, SR-1821, was flagged as a Hit in Screen 1, which supports the validity of this profiling approach. Moreover, the retained activity from these three analogs indicated that the 5-position of the pyrazole (R2) was amenable to substitution (Fig. 7d), allowing for cyclic alkanes and ethers as well as linear and branched alkanes. Analyzing DLR data from the other 100 related compounds revealed additional areas of the compound that could sustain substitutions (Fig. 7b, c). The pyrazole ring was difficult to replace with many group substitutions rendering the molecule completely inactive. However, some substituted imidazole derivatives did retain activity, indicating that a free NH group was not required. 1-N-alkyl substitution of the urea was tolerated with a range of groups from ethyl and cyclopropyl to cyclopentyl affording active molecules (R1). It is not yet clear if the 3-NH-urea is required for activity or the cyclic urea itself because the molecules available for purchase to test these positions were not available and will need to be synthesized. The C5 amide appears to be important for activity as well, but modifications including the reverse amide, amines, ethers and cyclic versions were not available for purchase. This strategy will be addressed during traditional SAR studies. In summary, the “SAR by purchase” approach greatly facilitated the development plan for how and where substitutions can be made to potentially optimize potency and efficacy within identified phenotypic probes. The ability to identify and then quickly receive 100 analogs of the original library molecule greatly accelerated the SAR program and validated SR-1815 as a viable preclinical lead candidate. Finally, this experiment demonstrated that EGS assays are sufficiently scalable and reliable to direct an SAR preclinical development program.
Discussion
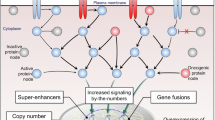
EGS is a significant technological advance because it provides a platform to identify drug-like small molecules that regulate endogenous expression of proteins that directly regulate cellular states, such as the shift from health to disease, in a translationally-relevant context. Our findings suggest that these small molecules may advance drug development for genetic disorders defined by altered protein abundance, such as AutD haploinsufficiency disorders. Moreover, because small molecules are discovered in EGS through their ability to alter endogenous levels of a pre-selected target protein in the disease context, they likely act through modulation of cell signaling pathways that regulate gene expression, protein synthesis, and/or protein stability/degradation (Fig. 8a). Due to the phenotypic nature of the platform, the small molecules identified in our screens act through unknown, potentially complex mechanisms and are, by definition, biologically active. Because these probes regulate disease-modifying protein abundance, identifying their mechanism-of-action (MOA) also offers opportunities for deeper insights into disease-associated cellular biology. This may benefit biological discovery for the intended disease indication, or perhaps even seemingly unrelated diseases. For these reasons, EGS was developed from the outset to not only seed preclinical drug candidates, but to facilitate MOA deconvolution of orphaned probes (Fig. 8b).

a Entry point for platform is to choose a gene and then screen chemical or biological agents using HTS-compatible assays that read-out endogenous protein expression of the gene of interest within disease-modeling state. The test agents could increase protein expression in a variety of ways, with the expected outcome of boosting protein resulting in a switch from “disease” to “healthy” cellular state. b (a, left box) Molecules suitable for further study enter the development pipeline workflows. A compound travels through two distinct sub-paths: (b, top right box) the probe undergoes pre-clinical development toward eventual IND application; (c, bottom right box) probe enters phenotype-to-mechanism studies aimed at identifying its molecular targets and how it regulates signaling to switch the state from “disease” and toward “health”. Created in BioRender. Samowitz, P. (2025) https://BioRender.com/cnld7d3.
The promise of previously undescribed phenotypic probes for biological discovery and drug development are strongly supported by both the initial discovery of SR-1815 (this study) and our parallel efforts to de-orphan it28. This current study illustrates an important proof-of-concept discovery. It demonstrates that small molecules identified through EGS have the potential to counteract both the root cause of genetic haploinsufficiency disorders (e.g., deficient protein expression) and to resolve phenotypes caused by their low abundance (e.g., SynGAP variants). At present, and to our knowledge, SR-1815 is the only known small molecule capable of boosting SynGAP abundance in haploinsufficient neurons to wildtype levels, while also mitigating functional cellular consequences of Syngap1 haploinsufficiency, such as tempering increased excitatory synapse function and neuronal hyperexcitability. Therefore, SR-1815, as well as the ~40 other validated SynGAP boosting small molecules, are currently undergoing initial preclinical evaluation for suitability to advance into later stages of drug development. Moreover, as described in our companion study28, we identified the molecular targets of SR-1815 and elucidated its mode/mechanism-of-action, including regulation of a splicing event known to regulate SynGAP protein abundance. Some of its kinase targets are implicated in cancer biology and SR-1815 exhibited potent activity in cancer cell lines known to be sensitive to these molecular targets. These results demonstrate an additional value of de-orphaning hits from the EGS platform, as mechanistic insights inspired by neuronal assays can reveal drug targets with therapeutic potential beyond the brain.
The small molecule probes revealed by this platform offer unique translational opportunities. The current state of the art in treating haploinsufficiency disorders is to develop targeted antisense oligonucleotides (ASOs) that disinhibit mRNA-dependent gene suppression mechanisms or to utilize viral vectors that either edit or replace a dysfunctional gene copy41. While these approaches have shown successes for some genetic disorders, they do not always successfully translate, nor are they necessarily the best treatment modality for every patient. For example, ASOs are not brain penetrant and, therefore, must be given intrathecally for CNS disorders, which introduces discomfort and risk for the patient, as well as high costs42. Further, ASOs have half-lives of weeks, and viral-based clinical approaches lack effective shut-off mechanisms, resulting in challenges related to tuning the effectiveness of these therapeutic approaches and managing side effects. Small molecules remain the gold standard therapeutic agent for treating neurological disorders because they can be improved through chemistry to achieve brain penetrance and optimized for more desirable routes of delivery (e.g. oral). Moreover, the level of small molecules in the body can be easily adjusted through dose titration. This is an important issue for treating genetic haploinsufficiency disorders because overexpression of the target gene/protein above levels seen in healthy cells can cause toxicity. Indeed, it is well established that genes that cause AutD disease require tightly controlled expression levels to maintain cellular health43. Too little or too much expression of these powerful genes can, on their own, cause disease states. Because overexpression is a concern, a once-per-day oral medication that regulates the target is easier to control and, therefore, could be better tolerated by patients. As a result, when a choice exists among small molecule (PO), gene replacement therapy (IV/SC), and intrathecal ASO injection for the same genetic disease, small molecules may be viewed as the superior first-line treatment option44. At the very least, small molecule development provides an additional parallel approach for treating genetic loss-of-function disorders, and they may also be useful as a combination approach with ASOs or gene therapy to boost overall efficacy by targeting complementary mechanisms.
Selection of the cellular context is an important factor for the success of phenotypic screening. In the Syngap1 version of EGS, we chose neurons over a more traditional scalable heterologous cell line for a clear reason: SynGAP protein is enriched in neurons and dysfunction of these cells is linked to disease states. The enrichment of endogenous SynGAP in brain, and in particular neurons26,34, is clear evidence that its expression is regulated in a cell-specific manner. Therefore, there is a higher probability of translational success, as well as discovery of previously unknown biology, when neurons are used as the screening material. Going a step further, genotype (e.g., wildtype versus Syngap1 haploinsufficiency) may also be important when screening for expression boosters. In support of this, SR-1815 has distinct functions in wildtype versus Syngap1 haploinsufficient neurons, with the latter existing in an altered cellular state due to reduced abundance of this critical protein. Moreover, we chose mouse primary cortical neurons from Syngap1 haploinsufficient mice rather than patient-derived human neurons for several reasons. First, primary cortical neurons have been, and continue to be, the gold standard in vitro discovery model for understanding neuronal cell biology, especially the processes related to synapse biology and how synapse biology regulates network activity45,46. Second, there is overwhelming evidence supporting the fundamental function of SynGAP expression in cortex and in cortical neurons. Patients diagnosed with SYNGAP1-related neurodevelopmental disorders express irregular cortical EEG rhythms16,17 and altered cortical sensory processing47, which when combined with reports of disrupted higher cognitive functions14, support the role of this protein in regulation of cortical neuron function. Syngap1 haploinsufficient mice and rats model domains of brain dysfunction observed in human SYNGAP1 patients48,49. In mouse models, regulation of Syngap1 expression selectively within cortical neurons is both necessary and sufficient to modulate disease-associated phenotypes21,23, including higher cognitive functions and seizure susceptibility caused by neural hyperexcitability. Cultured primary cortical neurons derived from Syngap1 haploinsufficient mice express analogous phenotypes, including synaptic22,38 (Fig. 5i) and cellular50 (Fig. 5j) hyperfunction. Indeed, these key phenotypes were ameliorated by SR-1815. Third, human excitatory neurons derived from patient iPSCs are viewed as substandard relative to primary rodent neurons for replicating aspects of disease-linked biology related to synapse function, neural plasticity, and the emergent phenotypes that arise from these complicated processes45. Fourth, the scalability of iPSC-induced neurons is severely limited relative to mouse primary neurons because the former develop much more slowly. It can take at least one month to achieve reliable synapse function in induced human neurons, and to achieve functional synapses, they must be co-cultured with astroglia, which further impacts economies of scale. For certain phenotypes, iPSC-derived human neurons may be the more appropriate choice, such as regulation of neurite outgrowth51. However, for complicated phenotypes that integrate cellular processes linking synapse function and plasticity with network dynamics, primary rodent neurons can often be the superior cellular context.
EGS is not limited to identification of small molecule boosters linked to genetic loss-of-function disorders. Rather, the platform was developed to be modular and flexible, where the FLAG-HiBIT tag can be inserted into any gene of interest using standard gene editing approaches. It is straightforward to breed any new HiBIT-tagged mouse line with the existing fLUC-expressing line, leading to an in-mouse DLR assay for any target protein of interest. Similar strategies could be applied to endogenous proteins expressed in induced cells derived from patient iPSCs. As a result, DLR assays within models characterized by either low or high/toxic proteins can be used to perform endogenous screening within relevant cellular contexts. This may be particularly useful for neuropsychiatric disorders. For example, the MECP2 gene causes Rett Syndrome when it is expressed at low levels, yet causes a distinct brain disorder when this same gene is overexpressed through gene duplication, and excellent models exist for each disorder52,53. A single EGS platform screen for endogenous regulators of MECP2 protein expression in wildtype cortical neurons would, in theory, identify both up- and down-regulating compounds to test in models for both disorders. In support of this, we have identified several candidate SynGAP down-regulators using our EGS approaches.
Although the EGS platform represents an advance for discovering small molecules that regulate endogenous protein abundance in disease-relevant contexts, several limitations should be considered. Because the screen is phenotypic, early compounds, such as SR-1815, likely act through multiple mechanisms, and their full molecular targets and signaling effects remain to be defined. In the case of a transcriptional and splicing regulator, broad changes are expected at this early stage and will require ongoing medicinal chemistry to improve selectivity and safety. In addition, while primary cortical neurons provide a biologically relevant discovery context, relevant phenotypes induced by small molecules may not fully generalize to intact biological in vivo systems. The current lead compound has poor brain exposure and a short half-life due to active efflux by P-glycoprotein, limiting in vivo evaluation until optimized analogs are available. Finally, because excessive stimulation of increased protein abundance could be harmful, careful titration and safety testing will be essential as these compounds advance. These limitations define the current scope of the platform and point to clear next steps for improving selectivity, pharmacokinetics, and translational potential for SR-1815, as well as the several dozen additional lead compounds that apparently up-regulate SynGAP abundance in cultured neurons.
Methods
Reagents table
All essential reagents required for EGS platform assays, as well as related information on how to use these reagents, can be found in Supplemental Table 1.
Mice and rats
Both males and females (M/F) were used in all experiments. The design and maintenance of the constitutive Syngap1 KO and two conditional Syngap1 lines have been described previously31 and are available at Jackson Labs (germline knockout line = #008890; conditional knockout line = #029303; conditional rescue line = #029304). The firefly luciferase mouse is also available from Jackson Labs (008450). Syngap1-HiBIT knock-in strain was created by standard CRISPR methods in collaboration with the Salk Institute Transgenesis Core facility. Briefly, mouse blastocysts were injected with repair template (Fig. S1). Chimeras were identified and bred to germline confirm transmission. F1 mice from one of the chimeras was bred to C57/BL6J and the line was crossed with new C57/BL6j mice from JAX for three generations before crossing to the other two strains noted in Fig. 1g. Timed pregnant Sprague-Dawley rats (E16.5) were purchased from Charles River Laboratories. Upon arrival, animals were housed in a temperature- and humidity-controlled facility with a 12 h light/dark cycle and ad libitum access to food and water. On postnatal day 0 (P0), litters were collected, and male and female pups were used for culture experiments as described below. All procedures were conducted in accordance with institutional animal care guidelines and approved protocols.
Primary cell culture protocol for HTS screening
Forebrains from mice with desired genotype (Fig. 1g) were dissected from post-natal day 0 (PND0) mouse pups to isolate primary cortical neurons in dissection media (culture grade H2O (Fisher Scientific: SH3052902), 10% 10x HBSS without Ca2+ and Mg2+ (Invitrogen: 14185052), 2% HEPES (Invitrogen: 15630080), 1% pyruvate (Invitrogen: 11360070), 1% Glucose solution (Thermo Fisher: A2494001), and 0.02% Gentamicin (Invitrogen: 15710064). The cortices were placed in a digestion solution containing dissection media and 20 active units/mL of papain (Worthington: LS003124) for 30 min at 37 °C. Tissues were washed and triturated in plating medium consisting of Neurobasal (Invitrogen: 21103049) containing 5% heat inactivated FBS, (Invitrogen: 10082139), 2% Glutamax-I, (Invitrogen: 35050061), and 0.02% Gentamicin (Invitrogen: 15710064). Cells were then centrifuged for 5 min at 800 g and resuspended in plating medium at 300 µL per brain. Cell suspension was then diluted into Feeding medium consisting of Neurobasal-A (Invitrogen: 10888022), 2% Glutamax-I, and 0.02% Gentamicin, 2% B-27 supplement (Invitrogen: 17504044), 10 µM 5-fluoro-2′-deoxyuridine (FUDR) (Sigma:F0503) to suppress the proliferation of glia, and 1 × 10e4—3x10e4 viral particles/cell of pENN.AAV.hSyn.Cre.WPRE.hGH (AAV9) (Addgene: 105553-AAV9; single-use aliquots) to induce haploinsufficiency (Fig. S1C). Using a BioTek EL406 microplate washer dispenser (Agilent Technologies), cells were dispensed into 384-well plates pre-coated with poly-D-lysine (PDL) (Aurora ABE2-01200B-PDL) at 10,000 cells in 80 µL/ well and placed in 37 °C incubator. A solution of 1% agarose was placed in the evaporation border wells prior to plating to minimize edge effects. At 7 days in vitro (DIV7), 50% of the conditioned media was replaced with fresh feeding media, and cultures were maintained undisturbed until assayed (usually DIV14).
HTS-compatible dual-luciferase reporter (DLR) assay
Neuronal culture plates were assayed (usually at DIV 14) using the Promega Nano-Glo® HiBiT Dual-Luciferase® Reporter Assay System (DLR) (Promega: N1620). Frozen reagents ONE-Glo™ EX Luciferase assay buffer and NanoDLR™ Stop & Glo® Buffer were thawed overnight at 4 °C. ONE-Glo™ EX Luciferase Assay Substrate was then resuspended in ONE-Glo™ EX Luciferase assay buffer. All buffers were then equilibrated to room temperature before use. LargeBIT protein was diluted 1:100 into ONE-Glo™ EX Luciferase Assay Reagent and NanoDLR™ Stop & Glo® Substrate was diluted 1:100 into NanoDLR™ Stop & Glo® Buffer. 60 µL of culture media from each well of the assay plates was removed using a BioTeK ELx405 (Agilent Technologies) for screen 1 anda BioTek EL406 for screen 2. 10 µL of ONE-Glo™ EX Luciferase Assay Reagent with LgBIT protein was added to each well of the 384-well plate using a BioTeK ELx405 for screen 1 and a Certus Flex liquid dispenser (Trajan Scientific and Medical) for screen 2. Plates were then shaken at 1500 rpm for 10 min. The plates were then measured for firefly luciferase (fLUC) luminescence using an EnVision plate reader (Perkin Elmer) for screen 1 and a CLARIOstar Plus Microplate Reader (BMG Labtech) for screen 2. 10 µL of NanoDLR™ Stop & Glo® Reagent was added to each well of the 384-well plate and shaken at 1500 rpm for 10 min. The plates were then measured for NanoBiT (nBIT) luminescence on the same reader.
Compound administration
Primary screening
Library compounds were administered to neuronal cultured plates on DIV 12 (screen 1) using at 100 nL 384-array pintool (V&P Scientific) or DIV 10 (screen 2) using a 25nL 384-array pin tool (V&P Scientific). The final concentration of the compounds was 3.125 µM or 12.5 µM for screen 1 and 3.125 µM for screen 2 in 80 µL of culture medium. The pin tool was cleaned between each plate by sonicating in water, submerging in DMSO, isopropanol, and methanol, and then dried using house air.
Validation pipeline (Fig. 4)
Neuronal cultures were generated as described above with one modification—feeding occurred every 3–4 days. On feeding days, compounds were readministered using two methods: 50% media exchange with subsequent pinning or by diluting compounds into feeding media and using a Certus Flex dispenser (Trajan Scientific and Medical) to administer media and compound. Pinning compounds requires 100X more compound than feeding, thus the method was chosen based on experimental needs and compound availability.
Dot blot assay
A lysis buffer was prepared containing 2% SDS, 2 mM TCEP, 10% ethylene glycol, 50 mM Sodium Borate, 500 µM 3-(2-Furoyl)quinoline-2-Carboxaldehyde (FQ) (VWR: 102987-910), and 500 µM Mandelonitrile (Sigma:116025) dissolved into water. Using the BioTek EL406, 384-well assay plates containing cortical neurons were washed with 60 µL of PBS three times and then liquid was completely removed using centrifugation. 20 µL of lysis buffer was added to each well using the BioTek EL406 and then shaken at 800 rpm for 10 min. The plate was then heated at 75 °C using an Envirogenie Incubator (Scientific Industries) for 20 min and then cooled at room temperature for 30 min. The plate was centrifuged at 3000 g for 3 min and shaken for 2 min at 400 rpm. Using a pintool array with 384 channels (100nL/channel; V&P Scientific) lysate was pinned on to a 0.2 µm pore size nitrocellulose membrane (Sigma: GE10600004), dried for 1 min, and placed into a container with 1X TBS-T. The membrane was then blocked with 1% BSA-TBS-T for 1 h Antibodies (see below) were then diluted into 1% BSA-TBS-T at appropriate concentration and incubated overnight at 4 °C on a platform rocker. The membranes were washed 3X with TBS-T for 10 min and then incubated with HRP-conjugated secondary antibodies (see below). The membranes were washed 3X with TBS-T and once with TBS. Membranes were then imaged for total protein using BioRad ChemiDoc imaging system using the Stain Free Blot setting. 15 mL of SuperSignal West Pico Plus Chemiluminescent substrate (Thermo Fisher:34580) was added to the membrane and incubated for 2 h on a platform rocker, followed by addition of 2 mL SuperSignal West Femto Maximum Sensitivity substrate (Thermo Fisher: 34096), and incubated for 5 min, and imaged for chemiluminescence.
SDS-PAGE and immunoblotting
Primary neuronal cultures were prepared as described above (validation protocol) from Syngap1 heterozygous cRescue mice and plated within PDL coated 24-well plates (Corning:353847) at 250,000 cells / well. FUDR was added at DIV 3 instead of at plating. Cultures were maintained by 50% media exchanges every 3–4 days. On DIV 14, plates were washed with PBS twice and proteins were extracted by sonication in a buffer consisting of 2% SDS, 50 mM Sodium Borate, 1X Halt Protease and Phosphatase inhibitors. Sample protein concentrations were measured using Pierce BCA Protein Assay Kit (Thermo Fisher: 23225) and adjusted to normalize protein content. 10 µg of protein per sample was loaded and separated by SDS-PAGE on 10% Criterion TGX Stain-Free gels (BioRad: 5678035) and then transferred to low fluorescence PVDF membranes (45 µm pore size) (BioRad: 1620262) with the Trans-Blot Turbo System (BioRad). Membranes were imaged for total protein using BioRad ChemiDoc imaging system, blocked with 1% BSA-TBS-T for 1 hour, and then probed with primary antibodies at 4 °C overnight. Membranes were washed 3X with TBS-T, incubated with secondary antibodies, washed, and imaged for chemiluminescence. Direct nBIT luciferase signal was also measured using the Nano-Glo® HiBiT Blotting System (Promega: N2410). Following chemiluminescence detection, 30% H2O2 was added to the membrane to quench the chemiluminescence and washed 3X with TBS-T. LgBiT protein was added in Nano-Glo Blotting buffer, incubated overnight at 4 °C, and Nano-Glo® Luciferase assay substrate was diluted 500-fold into the Nano-Glo blotting buffer. The membrane was incubated for 5 min and then imaged for chemiluminescence.
Immunocytochemistry (ICC)
Primary neuronal cultures were generally prepared as described above from constitutive heterozygous Syngap1 KO mice and plated into 96-well plates (Aurora: ABN2-10201F) at 25,000 cells / well. FUDR was added at DIV 3. Cultures were maintained by 50% media exchanges and treated with vehicle (DMSO) or SR-1815 (1.5μM [final]) on DIV 0, 3, 7, and 10. On DIV 14, plates were fixed with 4% PFA and 4% sucrose in PBS pH 7.4 for 10 min at room temperature. Plates were washed 3X with PBS and blocked in 2% BSA and 0.1% Triton X in PBS for 1 hour at room temperature. Primary antibodies pan-SynGAP, PSD95, and SV2A were diluted (1:500) into blocking solution and incubated overnight at 4 °C. Plates were washed with PBS 3X and secondary antibodies anti-rabbit Alexa Fluor (AF)−568, anti-mouse AF-488, and anti-guinea pig AF-647 were diluted (1:1000) in blocking solution and incubated for 1 hat room temperature. Plates were washed 3X with PBS and incubated with Prolong Gold antifade reagent with DAPI (Thermo Fisher: P36931) for 24 h 4 °C. Images were taken using Molecular Devices HCS.ai high content imager at 60X magnification water immersion with a confocal 50/500 µm pinhole spinning disk. Each channel was imaged with 500 ms exposure. Images were taken for 9 fields of view (FOVs) across 6 wells for each condition. Using the DAPI signal, 2 FOVs were selected from each well to ensure equivalent numbers of somas (e.g., to correct for variation in neuronal density across surface of culture wells) for signal quantification. Synaptic expression was measured by thresholding within 4 sub-fields of view per image (without a soma) to restrict analysis to the brightest punctate objects. The integrated density/intensity of all thresholded objects was calculated along with the thresholded area and the average intensity of the thresholded pixels. Somatic SynGAP was measured by placing regions of interest on each soma and measuring the average pixel intensity. To quantify dendritic SynGAP, background subtraction was performed across all images and line series were placed on regions of a suspected dendrite where SynGAP expression was non-overlapping with PSD95 and SV2A. The maximum pixel intensity was measured for each line scan for each dendrite. Experimenters were blinded to conditions during analysis.
Primary Antibodies
Pan-SynGAP antibody (1:1000 for Immnoblotting, 1:500 for ICC)—Cell Signaling #5539
SynGAP-α2 antibody (1:1000)—Cell Signaling #56927
SynGAP-α1 antibody (50 ng/mL)—Cell Signaling Test sample (VSP-137655); Rabbit mAb #34124
SynGAP-β antibody (50 ng/mL)—Cell Signaling Test sample (VSP-143511); Rabbit mAb #28580
ANTI-FLAG M2 antibody (1:1000)—Sigma #F1804
PSD95 antibody (1:500)- Thermo Fisher MA1-045
SV2A—Synaptic Systems 119004
Secondary Antibodies:
Anti-mouse IgG HRP Conjugate (1:2500)—Promega W402B
Anti-rabbit IgG HRP Conjugate (1:2500)—Promega W401B
Alexa Fluor 488 goat anti-mouse IgG—Thermo Fisher A11029
Alexa Fluor 568 goat anti-rabbit IgG—Thermo Fisher A11036
Alexa Fluor 647goat anti-guinea pig IgG—Thermo Fisher A21450
Cell-free nBIT (counter-screen) assay
A counter screening assay was developed to determine to what extent small molecules directly regulated nBIT activity. Starting with the Nano-Glo® HiBIT Extracellular Detection System (Promega: N2420), a HiBIT control protein (Promega: N3010) was diluted to 100 pM in buffer containing 0.1% BSA-PBS and 10 µL was dispensed into black opaque 384-well assay plates (Greiner: 781900). Compounds were pinned into the plate and shaken at 1500 rpm for 1 minute. 10 µL of Nano-Glo® HiBIT Extracellular buffer with 1:50 Nano-Glo® HiBIT Extracellular substrate and 1:100 LargeBIT protein was dispensed into the plate, shaken for 1 minute, centrifuged at 100 g for 1 minute, and incubated for 10 min. The plate was then measured for luminescence using the CLARIOstar Plus plate reader.
Screening libraries
Both custom designed and pre-selected screening libraries were obtained from Enamine (Ukraine) and ChemBridge (San Diego). Libraries were delivered in 384-well plates and each well contained ~30uL of compound (10 mM). Compounds were absent in columns 1, 2, 23, 24 to accommodate controls. Assay plates used in the HTS-style screen were pinned directly from the library compound plates. Controls were pinned separately.
Electrophysiology and mEPSC analysis
Primary neuronal cultures were prepared as described above from wildtype and constitutive heterozygous Syngap1 null mice and plated onto PDL coated glass coverslips (Neuvitro: GG-12-pdl) in 24-well plates at 250,000 cells / well. FUDR was added at DIV 3 instead of at plating. Cultures were treated with vehicle (DMSO) or SR-1815 (1.5 µM) on DIV 0, 3, 7, and 10 using a 50% media exchange. Whole-cell patch clamp recordings were conducted from forebrain cultures between DIV 13-15. Putative excitatory neurons were visually identified using infrared DIC optics. Recordings were made using boroscilicate glass pipettes (3-6 MΩ; 0.86 mm inner diameter; 1.5 mm outer diameter; Harvard Apparatus) made using a P-97 pipette puller (Sutter Instruments). All signals were amplified using Multiclamp 700B amplifier (Molecular Devices), filtered at 2.4 KHz, digitized at 10 KHz and stored on a personal computer for off-line analysis. Analogue to digital conversion was performed using a Digidata 1400 A system (Molecular Devices). Data acquisition and analysis was performed using pClamp 10.2/11.2 software (Molecular Devices), along with Minianalysis software (Synaptosoft) for semi-automated mEPSC event detection. To isolate AMPA-mediated mEPSCs, cultures were continuously perfused with artificial cerebrospinal fluid (aCSF), composed of (in mM): 150 NaCl, 3.1 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 10 Glucose, 0.05 D-2-amino-5-phosphonovalerate, 0.001 tetrodotoxin and 0.1 picrotoxin. Osmolarity was adjusted to 305-310 mOsm and pH adjusted to 7.3-7.4. The internal solution consisted of (in mM): 135 Cs-methanesulfonate, 10 CsCl, 10 HEPES, 5 EGTA 2 MgCl2, 4 Mg-ATP, and 0.1 Na-GTP. The internal solution was adjusted to pH 7.3 and to 290-295 mOsm. Following the establishment of whole-cell configuration, passive membrane properties were monitored throughout the experiment. Cells with access resistance > 30 MΩ or unstable ( > 20 % change) were discarded from analysis. A minimum of 100 and a maximum of 200 events were collected from each neuron, while 2-4 neurons were patched per culture. The amplitude threshold for event detection was set to √RMS x3 (typically ~4 pA). Data acquisition and analysis were performed by the same experimenter blinded to both treatment and genotype. Data from each group was averaged and statistical significance was determined using 2-way ANOVA with Fisher’s LSD posthoc test. Data are expressed as mean ± SEM.
Calcium imaging and analysis
Imaging
Primary neuronal cultures were prepared as described above and plated into PDL coated 384-well plates at 15,000 cells / well. FUDR was added at DIV 3 instead of at plating. Cultures were maintained by 50% media exchanges and treated with vehicle (DMSO) or SR-1815 on DIV 0, 3, 7, and 10. On DIV 3 cultures were transduced with AAV9 vectors expressing pGP-AAV-syn-FLEX-jGCaMP8f-WPPRE (AAV9) (Addgene: 162379-AAV9) (MOI = 300,000 vp/cell) and pENN.AAV.hSyn.Cre.WPRE.hGH (AAV9) (Addgene: 105553-AAV9) (MOI = 10,000 vp/cell). On DIV 14, imaging was performed on an InCell Analyzer 6000. Image series were acquired through a 20x Objective (Nikon 20X/0.45, Plan Fluor, ELWD, Corr Collar 0-2.0, CFI/60) at a rate of ~10 Hz using a 478 nM laser for sensor excitation. Neuronal activity was analyzed from time-lapse fluorescence microscopy images. Images were first segmented using the Cellpose deep learning model to identify individual neurons. The average fluorescence intensity within each segmented region was extracted over time. These signals were then detrended and normalized to obtain ΔF/F values. Spikes were detected in the ΔF/F traces by setting a threshold based on the baseline noise level. Spike frequency was calculated for each neuron. The results were aggregated across all analyzed images, combining spike metrics with segmentation properties and experimental metadata (e.g., drug treatment, GCaMP type).
Hit identification for screen 1
Each compound plate was tested in duplicate on two plates containing cultured neurons at DIV 12. At DIV 14, the DLR assay was performed and yielded two fLUC and two nBIT reads per compound plate tested. To identify hits from each compound plate, the median fLUC and nBIT values of the negative controls (DMSO only) were calculated for each plate. This value was used to normalize each plate and then each duplicate read was averaged for both fLUC and nBIT. Using the averaged normalized data, the standard deviation and mean of the negative controls was used to calculate a z-score for each data point. The data was plotted on a 2D scatter plot with the z-score of the normalized data for fLUC on the x-axis and the z-score of the normalized data for nBIT on the y-axis. To identify hit compounds, vertical threshold lines were drawn at 3 X standard deviation (SD) left and right for fLUC and horizontal lines above and below for nBIT (z-score = ± 3). Compounds with fLUC values within 3X SD and nBIT values above 3X SD were identified as potential upregulators, whereas those with nBIT values below 3X SD were considered down-regulators. To ensure reproducibility, variability filtering was applied. The coefficient of variation (CV) was calculated for each compound’s fLUC and nBIT replicate reads. Only compounds with <10% variability for both fLUC and nBIT were retained as final hits. 2D scatter plots were color coded for each category: negative control (green), compound field (gray), and hits (blue).
Development of hit detection algorithm for screen 2
All experiments are performed in duplicate. This yields two fLUC (f1, f2) and two nBIT (b1, b2) reads per experiment. The 1st, 2nd, 23th, and 24th datapoints from rows “A”, “B”, “O”, and “P” are excluded from the analysis as these are often affected by edge effects. First, a logarithmic transformation is applied to all raw luminescence intensities (log2f1, log2f2, log2b1, log2b2).
Second, a Mahalanobis-distance54 based filter is applied to the negative control data, allowing us to correctly estimate the distance of each negative control datapoint from the center of mass of the distribution in a direction dependent manner, to remove obvious outliers.
The Mahalanobis-distance \({{DM}}_{i}\) is calculated as follows for all the \(n\) observations in the negative control:
where \({\vec{x}}_{i}\) is a column vector representing the i th observation:
\(\vec{\mu }\) is a column vector representing the mean of the negative control:
S-1 is the inverse of the covariance matrix, and T denotes transposition. Any datapoint with a Mahalanobis distance larger than 3 is considered an outlier.
Next, data reduction is done by calculating the mean of the two reads for all datapoints:
Where \({F}_{j}\) and \({B}_{j}\) are the reduced fLUC and nBIT signals of the j th observation, respectively.
The reduced data is then normalized by subtracting the mean of the outlier-filtered negative control group from each datapoint:
Where \({F}_{j}^{,}\) and \({B}_{j}\) are the normalized fLUC and nBIT signals of the j th observation, \({F}_{k}\) and \({B}_{k}\) are the reduced firefly and nanoBIT signals of the kth observation among m total observations in the outlier filtered negative control group, respectively.
This transformation shifts the coordinates of the center of the negative control ellipsoid to (0,0) in a two-dimensional double-logarithmic scatter plot (Fig. S3A).
The effect size for each data point is calculated as the difference of the observed and expected nBIT signals (Fig. S3B). The observed signal is simply the normalized nBIT signal (as defined above), while the expected nBIT signal equals the normalized firefly signal:
This relationship is the consequence of the linear correlation between cell density and protein levels in a sample. Any relatively small change in the cell density that does not result in a change of the “fundamental” biological processes (e.g. protein expression levels) in the sample must be followed by a similar change in both the fLUC and nBIT signals. Interestingly, one can observe this correlation in positive and negative control data (Fig. S3C), even if these controls are not designed to detect this phenomenon. In the controls from screening, only the random experimental variability (e.g. dispensing of a cell suspension) results in different cell densities.
Due to random experimental variability and the above-described relationship, compounds with the same effect size are expected to be scattered along lines with a slope = 1 in double logarithmic scatter plots (Fig. S3A-B). To identify hits in a screening experiment, threshold lines with a slope of 1 are required (Fig. S3A-B). Compounds showing significant firefly effects relative to the negative control (e.g. due to toxicity, induction of non-specific protein expression changes, inhibition, activation or stabilization of firefly luciferase55) should not be considered as hits. To identify such compounds, two vertical threshold lines are used at 0 ± 3 x standard deviation of the negative control (Fig. S3A-B, vertical orange lines).
Last, the non-repeatedness defined as (log2f2—log2f1)—(log2b2—log2b1) is calculated for all datapoints and reflects how much the two parallel experiments do not replicate each other. If the non-repeatedness value is above 2 x standard deviation then the compound is not selected as a hit. (Fig. S3F).
Automated Dot Blot analysis
An Image J macro was developed to automate the Dot Blot image analysis process. The analysis involves fitting a grid to the image representing the microplate from which the samples were transferred to the membrane (Fig. S5A-D). The distance between the center of any two immediate neighbors in this array (in pixel units) is theoretically constant and represents the distance of neighboring wells in the plate. In order to find dots representing the real samples, shape-based sample identification is performed by running a sequence of operations. First, edge detection is performed and the resulting image is transformed into a binary (black and white) image (Fig. S5B). This is followed by local maxima identification, which gives a set of experimental coordinates representing the center of samples. To remove high intensity artifacts, the flood-fill tool is used followed by selecting the whole object (region of interest), which includes the area surrounded by the circular edge and the edge itself. Real dots on the membrane give selections with very similar area and almost perfectly circular shape. Artifacts, however, typically give selections with much smaller area and irregular shapes (Fig. S5B). Therefore, the identified objects are filtered by size and median width, median height and median circumference are calculated. These statistics are used as shape-based filters to remove any remaining artifacts from the list.
Next, with a set of coordinates representing real samples only, the optimal distance between the closest neighbors is determined (in pixel units) by generating a histogram from the calculated Euclidian distance between the center of each spot and the center of all other identified spots in the image using the following formula:
Where (x1, y1) and (x2, y2) are the coordinates of the 2 spots in pixel units (Fig. S5C). A theoretical distance distribution is also calculated (Fig. S5C) by assuming unit distance ( = 1) between the closest neighbors in an idealized grid representing the plate. The optimal (experimental) distance “d” is determined by fitting the theoretical histogram to the experimental histogram. This is done by multiplying the peak positions in the theoretical histogram by a variable “d” and optimizing the value of “d” such that the peak positions in the theoretical histogram overlap with the peak positions in the experimental histogram. The position of the peaks is compared by defining a zone for each peak in the theoretical distance histogram such that the boundaries of the zone fall half-way in between the neighboring peaks. The sum of the distances of each theoretical peak and the experimental peaks within its zone are calculated. The global minimum of this penalty function gives the optimal distance (d) in pixel units.
Next, the orientation of the membrane within the image needs to be determined. Each sample has a maximum of four closest neighbors allowing a displacement vector to be defined pointing to each of these neighbors with coordinates (x2-x1, y2-y1), where x2-x1 and y2-y1 are the displacement components along the “x” and “y” axes, respectively, (x1, y1) are the coordinates of the sample, and (x2, y2) are the coordinates of the neighbor in pixel units (Fig. S5D). Plotting these vectors for all samples yields four distinct populations around the origin. The distance between the center of each of these populations and the origin is expected to be equal to the optimal distance between the closest neighbors (see previous step). Relative to any arbitrarily chosen one of the four vectors pointing to the center of these populations, the other three are rotated by 90°, 180° and 270°. Therefore, for each datapoint, we calculate all possible rotations (0°, 90°, 180° and 270°) and chose the one with the angle closest to zero in all cases. The mean of all the angles represented by the datapoints yields the optimized orientation (α).
Next, a relationship between the coordinates of samples (in pixel units) and coordinates in an idealized plate (row, column) needs to be determined (indexing). Image coordinates are transformed by a combination of a translation, a rotation by α, and a division with the optimal distance (d) determined above:
Where xi’ and yi’ are the transformed coordinates, xi and yi are the image coordinates of the ith sample, a and b are the image coordinates of one randomly selected sample, and Round() denotes rounding to the nearest integer. The transformed coordinates are then translated to set the smallest xi’ and yi’values to (0,0).
The indices xi’ and yi’ are used to generate a set of idealized coordinates by using the optimal distance and reversing the transformations. The sum of Euclidian distances between each sample and the corresponding location in the idealized grid is calculated and used as a penalty function in coordinate refinement. A series of translations is performed along the x and y axes until the global minima of the penalty function (a 2D surface) is found (Fig. S6A). At this point, the image coordinates of sample “A01” (column = 1, row =1) define the optimal location of the grid in the original image (xA01, yA01; see Fig. S5A).
Next, missing samples are identified in the list of indices, and the above defined transformations are performed in reverse order to obtain the corresponding coordinates of the grid.
In the last step, sample integration is done with local background correction (Fig. S6B). Pixels around each grid point in the unmodified image are selected within a circle covering the entire spot to calculate a “raw” integral. This raw integral needs to be corrected for background, which can be estimated from the integral of the immediate surroundings of the circle. We selected pixels in a larger circle such that the center of both circles is located at the coordinates defined by the grid point, and the area of the large circle is exactly 2 times the area. This gives a radius of rL = r*√2. The integral within the small circle is a sum of the signal (S) and the background (B): I1 = S + B. The integral within the large circle contains the signal (S) and twice the background (B): I2 = S + 2B. Thus, the background corrected signal can be calculated as S = 2I1—I2. Although the radius can be easily optimized by testing all possible r values between 1 pixel and d/(2*√2) pixels we typically chose r = d/(2*√2) to avoid high variability due to partially lost signal in areas of local membrane distortions and to minimize the effect of overestimated background due to high intensity local artifacts.
Statistics and reproducibility
Data analyses were conducted in GraphPad Prism (version 9.4.1, GraphPad Software) or custom Python scripts (version 3.9). No statistical method was used to predetermine sample size. The study design and sample sizes were chosen to align with the principles of high-throughput screening and validation workflows, allowing for the screening of large chemical libraries and the subsequent testing of promising compounds. No data were excluded from the analyses, except for the removal of edge-affected data points from rows in 384-well plates. Experiments were not randomized, and the investigators were blinded to allocation during experiments and outcome assessment. Data are presented as mean ± SEM or ± SD unless otherwise noted. Statistical analyses were conducted using methods such as simple linear regression, Mann-Whitney U tests, one-way ANOVA with Tukey’s post-hoc tests, and two-way ANOVA with Fisher’s LSD post-hoc tests, as detailed in the methods and figure legends. For hit detection in Screen 1, a simple “outlier-detection” algorithm was used, while Screen 2 employed an improved algorithm (see Fig. S3). Mahalanobis-distance-based filtering was used to remove outliers from the negative control in Screen 2, while false positive hits were identified based on their “non-repeatedness” values (see Fig. S3). All experiments were performed in duplicate to ensure reproducibility. For calcium imaging, neuronal segmentation was performed using the Cellpose deep learning model, and spike frequency was calculated based on a threshold set above the baseline noise level. A custom ImageJ macro was developed for automated analysis of Dot Blots, including shape-based filtering of artifacts and local background correction to ensure accurate quantification of protein.
Ethics statement
All mouse procedures were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals, and all methods were authorized by the Scripps/UF Scripps Biomedical Research Institutional Animal Care and Use Committee and the Institutional Biosafety Committee.
Data availability
The data generated in this study are provided in the Supplementary Information/Source Data file. Source data are provided with this paper.
Code availability
Custom code was written in Python for data analysis. Codes which are not already available in public repository (see Methods) are available from: https://github.com/RumbaughLab/Samowitz_etal_NatCom_2025 and used without restriction under GPL-3.0 license.
References
Johannessen, C. M., Clemons, P. A. & Wagner, B. K. Integrating phenotypic small-molecule profiling and human genetics: the next phase in drug discovery. Trends Genet. 31, 16–23 (2015).
Moffat, J. G., Vincent, F., Lee, J. A., Eder, J. & Prunotto, M. Opportunities and challenges in phenotypic drug discovery: an industry perspective. Nat. Rev. Drug Discov. 16, 531–543 (2017).
Shalem, O., Sanjana, N. E. & Zhang, F. High-throughput functional genomics using CRISPR-Cas9. Nat. Rev. Genet. 16, 299–311 (2015).
Palacino, J. et al. SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat. Chem. Biol. 11, 511–517 (2015).
Vincent, F. et al. Developing predictive assays: the phenotypic screening “rule of 3”. Sci. Transl. Med. 7, 293ps15 (2015).
Swinney, D. C. & Anthony, J. How were new medicines discovered?. Nat. Rev. Drug Discov. 10, 507–519 (2011).
Vincent, F. et al. Phenotypic drug discovery: recent successes, lessons learned and new directions. Nat. Rev. Drug Discov. 21, 899–914 (2022).
Ratni, H., Scalco, R. S. & Stephan, A. H. Risdiplam, the first approved small molecule splicing modifier drug as a blueprint for future transformative medicines. ACS Med. Chem. Lett. 12, 874–877 (2021).
Zhong, G., Chang, X., Xie, W. & Zhou, X. Targeted protein degradation: advances in drug discovery and clinical practice. Signal Transduct. Target Ther. 9, 308 (2024).
Jörg, M. & Madden, K. S. The right tools for the job: the central role for next generation chemical probes and chemistry-based target deconvolution methods in phenotypic drug discovery. RSC Med. Chem. 12, 646–665 (2021).
Dixon, A. S. et al. NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chem. Biol. 11, 400–408 (2016).
Kostic, M. & Jones, L. H. Critical assessment of targeted protein degradation as a research tool and pharmacological modality. Trends Pharm. Sci. 41, 305–317 (2020).
Meyer, S. M. et al. Small molecule recognition of disease-relevant RNA structures. Chem. Soc. Rev. 49, 7167–7199 (2020).
Hamdan, F. F. et al. Synapse to Disease G. Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N. Engl. J. Med. 360, 599–605 (2009).
Carvill, G. L. et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat. Genet. 45, 825–830 (2013).
Jimenez-Gomez, A. et al. Phenotypic characterization of individuals with SYNGAP1 pathogenic variants reveals a potential correlation between posterior dominant rhythm and developmental progression. J. Neurodev. Disord. 11, 18 (2019).
Vlaskamp, D. R. M. et al. SYNGAP1 encephalopathy: a distinctive generalized developmental and epileptic encephalopathy. Neurology 92, e96–e107 (2019).
Satterstrom, F. K. et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180, 568–584.e23 (2020).
Bartha, I., di Iulio, J., Venter, J. C. & Telenti, A. Human gene essentiality. Nat. Rev. Genet. 19, 51–62 (2018).
Johnson, A. F., Nguyen, H. T. & Veitia, R. A. Causes and effects of haploinsufficiency. Biol. Rev. Camb. Philos. Soc. 94, 1774–1785 (2019).
Ozkan, E. D. et al. Reduced cognition in Syngap1 mutants is caused by isolated damage within developing forebrain excitatory neurons. Neuron 82, 1317–1333 (2014).
Rumbaugh, G., Adams, J. P., Kim, J. H. & Huganir, R. L. SynGAP regulates synaptic strength and mitogen-activated protein kinases in cultured neurons. Proc. Natl. Acad. Sci. USA 103, 4344–4351 (2006).
Vaissiere, T. et al. Syngap1 promotes cognitive function through regulation of cortical sensorimotor dynamics. Nat. Commun. 16, 812 (2025).
Spicer, T. P. et al. Improved scalability of neuron-based phenotypic screening assays for therapeutic discovery in neuropsychiatric disorders. Mol. Neuropsychiatry 3, 141–150 (2018).
Kilinc M. et al. Species-conserved SYNGAP1 phenotypes associated with neurodevelopmental disorders. Mol. Cell Neurosci. 2018. PubMed PMID: 29580901.
Gamache, T. R., Araki, Y. & Huganir, R. L. Twenty years of SynGAP research: from synapses to cognition. J. Neurosci. 40, 1596 (2020).
Vermaercke, B. et al. SYNGAP1 deficiency disrupts synaptic neoteny in xenotransplanted human cortical neurons in vivo. Neuron 112, 3058–68.e8 (2024).
Douglas C. J. et al. Mesoscale proximity labeling to study macro changes to chromatin occupancy. bioRxiv. 2025:2025.03.13.643041.
Ryszkiewicz, P., Malinowska, B. & Schlicker, E. Polypharmacology: promises and new drugs in 2022. Pharm. Rep. 75, 755–770 (2023).
Kampasis, D., Uliassi, E. & Bolognesi, M. L. Polypharmacology approaches for brain disorders aimed to enhance brain permeability and circadian clock targeting. Adv. Drug Deliv. Rev. 223, 115621 (2025).
Clement, J. P. et al. Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell 151, 709–723 (2012).
Binder G., Rappold G. A. SHOX Deficiency Disorders. In: Adam M. P., Feldman J., Mirzaa G. M., Pagon R. A., Wallace S. E., Amemiya A., editors. GeneReviews(®). Seattle (WA): University of Washington, Seattle Copyright © 1993-2025, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved. (1993).
Creson T. K. et al. Re-expression of SynGAP protein in adulthood improves translatable measures of brain function and behavior. Elife. 2019;8 Epub 2019/04/27. PubMed PMID: 31025938; PMCID: PMC6504227.
Kilinc M. et al. Endogenous Syngap1 alpha splice forms promote cognitive function and seizure protection. Elife. 11, e75707 (2022).
Gou, G. et al. SynGAP splice variants display heterogeneous spatio-temporal expression and subcellular distribution in the developing mammalian brain. J. Neurochem. 154, 618–634 (2020).
Arellano J. I., Benavides-Piccione R., DeFelipe J., Yuste R. Ultrastructure of dendritic spines: correlation between synaptic and spine morphologies. Front. Neurosci. 2007; Volume 1-2007. https://doi.org/10.3389/neuro.01.1.1.010.2007.
Araki, Y., Zeng, M., Zhang, M. & Huganir, R. L. Rapid dispersion of SynGAP from synaptic spines triggers AMPA receptor insertion and spine enlargement during LTP. Neuron 85, 173–189 (2015).
Wang, C. C., Held, R. G. & Hall, B. J. SynGAP regulates protein synthesis and homeostatic synaptic plasticity in developing cortical networks. PLoS One 8, e83941 (2013).
Sullivan, B. J. et al. Low-dose perampanel rescues cortical gamma dysregulation associated with parvalbumin interneuron GluA2 upregulation in epileptic syngap1(+/-) Mice. Biol. Psychiatry 87, 829–842 (2020).
Sander, T., Freyss, J., von Korff, M. & Rufener, C. DataWarrior: an open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 55, 460–473 (2015).
Carvill, G. L., Matheny, T., Hesselberth, J. & Demarest, S. Haploinsufficiency, dominant negative, and gain-of-function mechanisms in epilepsy: matching therapeutic approach to the pathophysiology. Neurotherapeutics 18, 1500–1514 (2021).
Hammond S. M. et al. Antibody-oligonucleotide conjugate achieves CNS delivery in animal models for spinal muscular atrophy. JCI Insight. 2022;7 Epub 20221222. PubMed PMID: 36346674; PMCID: PMC7614086.
Lombardi, L. M., Baker, S. A. & Zoghbi, H. Y. MECP2 disorders: from the clinic to mice and back. J. Clin. Invest. 125, 2914–2923 (2015).
Waldrop, M. A. Clinical decision making around commercial use of gene and genetic therapies for spinal muscular atrophy. Neurotherapeutics 21, e00437 (2024).
Mateos-Aparicio P., Bello S. A., Rodríguez-Moreno A. Challenges in Physiological Phenotyping of hiPSC-Derived Neurons: From 2D Cultures to 3D Brain Organoids. Front. Cell Dev. Biol. 8. https://doi.org/10.3389/fcell.2020.00797 (2020).
Verstraelen P. et al. Image-based profiling of synaptic connectivity in primary neuronal cell culture. Front. Neurosci. 12. https://doi.org/10.3389/fnins.2018.00389 (2018).
Côté, V. et al. Differential auditory brain response abnormalities in two intellectual disability conditions: SYNGAP1 mutations and Down syndrome. Clin. Neurophysiol. 132, 1802–1812 (2021).
Kilinc, M. et al. Species-conserved SYNGAP1 phenotypes associated with neurodevelopmental disorders. Mol. Cell Neurosci. 91, 140–150 (2018).
Katsanevaki D. et al. Key roles of C2/GAP domains in SYNGAP1 -related pathophysiology. Cell Rep. 43. https://doi.org/10.1016/j.celrep.2024.114733 (2024).
Gao, Y. et al. Proximity analysis of native proteomes reveals phenotypic modifiers in a mouse model of autism and related neurodevelopmental conditions. Nat. Commun. 15, 6801 (2024).
Sridharan, B. et al. A simple procedure for creating scalable phenotypic screening assays in human neurons. Sci. Rep. 9, 9000 (2019).
Guy, J., Gan, J., Selfridge, J., Cobb, S. & Bird, A. Reversal of neurological defects in a mouse model of Rett syndrome. Science 315, 1143–1147 (2007).
Sztainberg, Y. et al. Reversal of phenotypes in MECP2 duplication mice using genetic rescue or antisense oligonucleotides. Nature 528, 123–126 (2015).
Mahalanobis, P. C. Reprint of: Mahalanobis, P.C. (1936) “On the Generalised Distance in Statistics.”. Sankhya A. 80, 1–7 (2018).
Auld, D. S., Thorne, N., Nguyen, D. T. & Inglese, J. A specific mechanism for nonspecific activation in reporter-gene assays. ACS Chem. Biol. 3, 463–470 (2008).
Acknowledgements
We are grateful to the entities that provided sponsored research funding that supported the development of this platform (National Institute for Mental Health—U01MH136567 and R01MH113648; Praxis Precision Medicines—UFAS-A00002). A custom chemical library was curated by Andy Jennings and shared with the Rumbaugh Laboratory by Praxis Precision Medicines. Jordan Hirschfeld of Cell Signaling Technology (CST) developed and shared SynGAP isoform-specific antibodies (see methods). Lina Deluca handled all compound management activities.
Author information
Authors and Affiliations
Contributions
P.S. and L.R. performed experiments, designed experiments, analyzed data, co-wrote the manuscript, and edited the manuscript. T.V., S.D.M. performed experiments, designed experiments, analyzed data, and edited the manuscript. C.R., R.M., M.K., A.E., J.S., R.H. and V.F-V performed experiments. T.P.S., L.S., T.K., and C.A.M. secured funding, designed experiments, interpreted data, and edited the manuscript. G.R. conceived the study, secured funding, developed tools, designed experiments, interpreted data, co-wrote the manuscript, and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Mootaz Salman and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Samowitz, P., Radnai, L., Vaissiere, T. et al. The Endo-GeneScreen platform identifies drug-like probes that regulate endogenous protein levels within physiological contexts. Nat Commun 16, 10970 (2025). https://doi.org/10.1038/s41467-025-65971-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-65971-x
