
Abstract
Bone homeostasis mainly depends on the equilibrium of osteoclasts and osteoblasts, overactivated osteoclasts play a pivotal role in the progression of osteoporosis. Here, we revealed that Pla2g7 (phospholipase A2 group VII) was positively correlated with bone resorption in clinic. By single-cell RNA-seq data analysis, Pla2g7 was found highly enriched in osteoclasts along the developmental trajectory, which promoted osteoclast differentiation. Inhibition of Pla2g7 by Darapladib impaired both human and mice osteoclast differentiation, meanwhile, Pla2g7-deficient mice showed higher bone mass and restored the ovariectomy-induced bone loss. Mechanistically, we identified that Alox12 (arachidonate 12-lipoxygenase) mediated-arachidonic acid metabolism is a key determinant in Pla2g7 enhanced osteoclast differentiation. Its metabolite 12-HETE (12-hydroxyeicosatetraenoic acid) activated Gpr31 to regulate osteoclast formation via p38 MAPK pathway and mitochondrial energy metabolism. Collectively, our study uncovers an Alox12/12-HETE/Gpr31 axis that regulates Pla2g7-induced osteoclast differentiation, and provides a new insight for osteoporosis treatment.
Similar content being viewed by others


Introduction
Bone homeostasis is a dynamic equilibrium, which mainly depends on osteoclast-mediated bone resorption and osteoblast-mediated bone formation1. Osteoporosis is a bone homeostasis disorder characterized by a loss of bone mineral density (BMD) and deterioration of bone structure, increasing the risk of fragile fracture2. Osteoblast originates from the mesenchymal stem cells (MSCs) of bone-marrow, and promotes the formation of new bone and the mineralization of bone matrix3. Osteoclasts arise from hematopoietic precursors via a two-step process: M-CSF promotes their differentiation into macrophages, which are then induced by RANKL to become osteoclasts4. However, overactivated osteoclasts cause a series of clinical diseases, like osteoporosis, Paget’s disease of bone, osteogenesis imperfecta and periodontal disease5. Therefore, investigating the regulatory mechanism of osteoclast differentiation and discovering a novel therapy method to prevent disorder are needed.
Phospholipases A2 (PLA2s) are a diverse group of enzymes that release phospholipid-related production and initiate arachidonic acid cascade reaction via hydrolyzing fatty acids at sn-2 position of phospholipids6. PLA2s are largely divided into cytosolic PLA2s (cPLA2s), Ca2+-independent PLA2s (iPLA2s), secretory PLA2s (sPLA2s), and lipoprotein-associated PLA2s (Lp-PLA2s)7. Pla2g7, which encodes lipoprotein-associated phospholipase A2, also known as platelet-activating factor acetylhydrolase (PAF-AH), is a secretory enzyme mainly produced by myeloid cells and exerts a significant role in a spectrum of metabolic and inflammatory disorders, including atherosclerosis, diabetes, and autoimmune diseases8,9,10. However, the detailed function of Pla2g7 and underlying mechanism in regulating bone homeostasis are unknown.
Arachidonic acid plays a pivotal role in osteoclast differentiation through its downstream metabolites, such as prostaglandin E (PGE) and leukotriene B4 (LTB4), which are well-characterized mediators involved in this process11,12. As a key enzyme in the lipoxygenase pathway of arachidonic acid metabolism, Alox12 regulates the production of these bioactive lipids and has been implicated in pathological conditions including atherosclerosis, thrombosis, and hepatic ischemia-reperfusion injury13,14,15. 12-HETE is a crucial metabolite that is converted from arachidonic acid by Alox1216. Notably, previous studies have already focused on its positive regulation on phosphorylation of NF-κB subunit p65, which is tightly linked with the modulation of osteoclasts17. As is well known, G-protein-coupled receptors (Gprs) could be receptors for lipid metabolites as reported previously, including Gpr40, Gpr120, Gpr31, Gpr75 that serve as receptors of HETEs18,19,20,21. More importantly, Gpr31 has been widely recognized as the high-affinity receptor responsible for 12-HETE-elicited signaling15,21. The 12-HETE/ Gpr31 axis triggers various downstream cascades, such as MAPK and NF-κB pathways, which have been reported to be involved in the regulation of thrombosis and tissue injury16,22. Nevertheless, whether 12-HETE acts on bone homeostasis via Gpr31 remains unclear.
In this study, Pla2g7 was found to be positively associated with bone resorption in both human and mice. Pla2g7 knockout markedly increased bone mass and restored the ovariectomy-induced bone loss in vivo. The Alox12/12-HETE/Gpr31 axis, as downstream of Pla2g7 regulation, modulated osteoclast via mitochondrial energy metabolism and p38 MAPK pathway. Furthermore, Darapladib is a promising method for clinical osteoporosis treatment by impeding osteoclast formation.
Results
Pla2g7 is associated with bone homeostasis in mice and humans
To access the potential role of Pla2g7 in bone homeostasis, we analyzed its expression in human bone tissue using single-cell RNA-seq data (GSM4423510, GSE169396) (Fig. 1A), and found that Pla2g7 is specifically expressed in mono-macrophages, but not in BMSC or other immune cells (Fig. 1B). Moreover, no obvious expression of Pla2g7 was detected during the differentiation from osteoblast to osteocyte (GSE154718) (Fig. 1C and Supplementary Fig. 1A). We further analyzed the dynamic expression profile of Pla2g7 during developmental trajectory from mono-macrophages to osteoclasts (GSE147174), which showed that Pla2g7 expression is continuously enhanced in pre-osteoclasts and osteoclasts (Fig. 1D, E and Supplementary Fig. 1B, C). Both gene and protein levels of Pla2g7 were confirmed to be upregulated with a time-dependent manner in mice primary BMMs (bone marrow monocytes) treated with RANKL (Fig. 1F, G). Meanwhile, immunohistochemistry staining showed that an elevated of Pla2g7 in femoral of ovariectomized (OVX) or metatarsal bone of K/BxN administrated mice (Fig. 1H, I), and immunofluorescence staining revealed that Pla2g7 was highly expressed in Nfatc1+ osteoclasts in above models (Supplementary Fig. 1D–F).

A Uniform manifold approximation and projection (UMAP) plot showing the main cell types of the single-cell RNA sequencing dataset and the enrichment of Pla2g7 in the main cell types. B The expression of Pla2g7 in the main cell types of bone tissue from human. C The expression of Pla2g7 during the differentiation from osteoblast to osteocyte. D, E The expression of Pla2g7 in pre-osteoclasts and osteoclasts with the RANKL-induced time gradient. F, G The gene and protein expression of Pla2g7, Nfatc1 and c-Fos in response to RANKL-induced time gradient, and the densitometric analysis of Western blot bands. For RNA analysis, n = 3 biologically independent wells per group, for Western blot analysis, n = 3 biologically independent cell samples per group. H Immunohistochemistry staining images of femur in OVX model and metatarsus in K/BxN model. Cells labeled in red are Pla2g7-positive. (Scale bar, OVX models, 200 μm. K/BxN models, 100 μm). I Quantification of immunohistochemistry staining in Pla2g7 positive cells. n = 6 biologically independent mice per group. J Elisa detection of Pla2g7 concentration in normal patients (NP) and osteoporosis patients (OP). NP, n = 75. OP, n = 75. Each n represents one independent patient. K Pla2g7 concentration in Sham and OVX groups. n = 6 biologically independent mice per group. Data were showed as mean ± SD. Two-sided Student’s t test (I–K) was employed to assess the difference between the two groups. One-way ANOVA (F, G) was utilized for multiple comparisons test. Significance: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001, ns non-significance.
We then collected serum samples from 150 donors, including normal bone mass and osteoporosis (Supplementary Tables 2 and 3). Compared with the normal bone mass group (NP), the serum level of PLA2G7 was significantly elevated in osteoporosis patients (OP), and PLA2G7 was identified to be positively correlated with CTX-1 but not PINP (Fig. 1J and Supplementary Fig. 1G, H). Similarly, Pla2g7 was positively associated with CTX-I and negatively associated with PINP in mice with OVX operation (Fig. 1K and Supplementary Fig. 1I, J). In conclusion, these results indicated that the highly association of Pla2g7 with bone homeostasis, especially bone resorption.
Osteoclast-specific ablation of Pla2g7 displays higher bone mass in mice
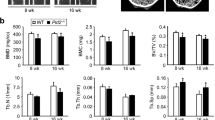
To further ascertain the role of Pla2g7 in bone resorption, we constructed 12-week-old Pla2g7 conditional knockout (cKO) mice by crossing Pla2g7f/f mice with mice expressing Lysozyme 2 or Cathepsin K promotor-Cre (LysM-Cre or CTSK-Cre). Using immunofluorescence staining and fluorescence intensity analysis to confirm the knockdown efficiency at the tissue level (Supplementary Fig. 2A–D). In micro-CT analysis, compared with WT (wild type) littermates, both Pla2g7LysM and Pla2g7CTSK female mice exhibited higher trabecular bone parameters, like bone volume/tissue volume (BV/TV), trabecular number (Tb.N) and trabecular thickness (Tb.Th) but decreased trabecular separation (Tb.Sp) (Fig. 2A–C). In contrast, osteoclast-specific ablation of Pla2g7 did not effect on cortical bone thickness (Ct.Th) (Supplementary Fig. 2E, F). Similarly, the results of bone mass in the male mice were consistent with those in female mice (Supplementary Fig. 2G–K). Notably, the effect of Pla2g7 deficiency on female bone mass was more significantly than that on the male. A decreased presence of osteoclasts was observed in the distal femur of Pla2g7LysM and Pla2g7CTSK mice through HE and TRAP staining (Fig. 2D, F and Supplementary Fig. 2L, M), and the histomorphometry analysis showed decreased ratios of osteoclast number to bone tissue area (Oc.N/BS) and osteoclast area to bone tissue area (Oc.S/BS) (Fig. 2E, G and Supplementary Fig. 2N, O). The serum levels of the CTX-I were decreased and the PINP had no significant change in Pla2g7 cKO mice (Fig. 2H, I). Notably, no significant differences were observed in body length or hindlimb long bone length in mice, suggesting that the epiphyseal cartilage was not affected and longitudinal bone growth remained intact (Fig. 2J and Supplementary Fig. 2P).

A The diagrams of bone mass in Pla2g7 f/f, Pla2g7 LysM and Pla2g7 CTSK female mice (scale bar, 1 mm). B, C Trabecular-related parameters in Pla2g7 f/f, Pla2g7 LysM and Pla2g7 CTSK female mice, including BV/TV, Tb.Th, Tb.N and Tb.Sp. n = 6 biologically independent mice per group. D–G HE and TRAP staining of distal of femur, and the histomorphometric parameters of osteoclasts in each group, including Oc.N/BS and Oc.S/BS. The bottom image enlarges the black box labeled region of the middle image. n = 6 biologically independent mice per group. (scale bar, normal images, 200 μm, enlarged images, 100 μm). H, I The expression of serum CTX and PINP using Elisa assays. n = 6 biologically independent mice per group. J The body and hindlimbs length in Pla2g7 f/f, Pla2g7 LysM and Pla2g7 CTSK female mice. K, L The rescued-condition of Pla2g7 conditional knockout in OVX-induced bone loss. OVX is performed at the stage of 12 weeks and the model lasts for 8 weeks (scale bar, 1 mm). M, N Morphological parameters of trabecular bone in each group. n = 6 biologically independent mice per group. Data were showed as mean ± SD. Two-sided Student’s t test (B, C, E, G, H, I) was employed to assess the difference between the two groups. Two-way ANOVA (M, N) was utilized for multiple comparisons test. Significance: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001, ns non-significance.
Then, an OVX mice model was employed to further unveil the role of Pla2g7 in pathological osteoporosis. We found that the deficiency of Pla2g7 overtly rescued OVX-induced bone mass loss (Fig. 2K, L), including increasing BV/TV, Tb.Th, Tb.N and decreasing Tb.Sp. (Fig. 2M, N). Accordingly, HE and TRAP staining displayed a decreased osteoclast formation in Pla2g7 conditional knockdown group, and histomorphometry analysis demonstrated a reduced N.Oc/BS and Oc.S/BS (Supplementary Fig. 2Q–T). However, the cortical bone loss and morphological parameters did not show significant changes (Supplementary Fig. 2U, V). Taken together, these data suggested that the loss of Pla2g7 increased bone mass via suppressing the osteoclast-mediated bone resorption in vivo.
Darapladib suppresses osteoclast differentiation as a Pla2g7 specific inhibitor
To clarify the role of Pla2g7 in osteoclast differentiation, we isolated primary BMMs from Pla2g7LysM mice and its WT littermates. Knockout efficiency was confirmed by RT-qPCR and immunoblotting analysis (Supplementary Fig. 3A, B). The osteoclast differentiation was inhibited appreciably as indicated by TRAP and F-actin staining (Fig. 3A, B), meanwhile, the expression of osteoclast specific markers and bone resorption activity were reduced in Pla2g7LysM group (Fig. 3C, D and Supplementary Fig. 3C–E). Darapladib, as the chemical structure shown in Fig. 3E, is the most advanced Pla2g7 inhibitor and has undergone Phase II and Phase III clinical trials for treatment of Alzheimer’s disease, diabetic retinopathy, and atherosclerosis23,24,25,26. The osteoclast differentiation was potently suppressed by Darapladib treatment with a concentration-dependent manner as indicated by TRAP and F-actin staining (Fig. 3F, G). Similarly, the osteoclast markers Nfatc1, c-Fos and Ctsk were reduced after Darapladib supplementation (Fig. 3H–J). In the hydroxyapatite resorption assay, less bone erosion areas were observed after Darapladib treatment (Fig. 3K, L).

A TRAP and F-actin staining in BMMs derived from Pla2g7 f/f and Pla2g7 LysM mice following osteoclast induction (scale bar, normal, 500 μm, enlarged, 200 μm). B Quantification of TRAP positive MNCs and average osteoclast size. n = 6 biologically independent wells per group. C, D Relative gene and protein expression of osteoclast markers, and the densitometric analysis of Western blot bands. RNA, n = 3 biologically independent wells per group. WB, n = 3 biologically independent cell samples per group. E The molecular structure diagram of Darapladib. F, G TRAP and F-actin staining and quantification of osteoclasts stimulated with RANKL in the absence or presence of Darapladib in a concentration-dependent manner (0, 100, 200, and 400 nM), with Darapladib administered throughout the entire induction period. n = 6 biologically independent wells per group. (scale bar, normal, 500 μm, enlarged, 200 μm). H, I The gene and protein expression of osteoclast markers. n = 3 biologically independent wells per group. J The immunoblotting analysis in osteoclasts induced by RANKL with or without Darapladib at different time points. K, L Hydroxyapatite resorption assay and statistical analysis of BMMs incubated with RANKL with or without Darapaldib (400 nmol/L). n = 3 biologically independent cell samples per group. (scale bar, normal, 200 μm). M TRAP and F-actin staining images of osteoclasts stimulated by RANKL in the absence or presence of recombinant Pla2g7 protein (50, 100, or 200 ng/mL) for 5 days, with Pla2g7 administered throughout the entire induction period (scale bar, normal, 500 μm, enlarged, 200 μm). N, O Relative gene and protein expression of osteoclast-specific markers. n = 3 biologically independent wells per group. P Western blot analysis of BMMs incubated with RANKL in the absence or presence of Pla2g7 at various time points. Data were showed as mean ± SD. Two-sided Student’s t test (B–D, L) was employed to assess the difference between the two groups. One-way ANOVA (G) and two-way ANOVA (H, N) were utilized for multiple comparisons test. Significance: *p < 0.05; **p< 0.01; ***p < 0.001; ****p < 0.0001, ns non-significance.
Then, we used exogenous recombinant protein to comprehensively determine the effect of Pla2g7 on osteoclast differentiation. Firstly, we found that recombinant Pla2g7 protein significantly promoted osteoclast differentiation (Fig. 3M and Supplementary Fig. 3F), and the expression of osteoclast markers were markedly increased (Fig. 3N–P). Also, there were more bone plate erosion areas than the group without administration of recombinant Pla2g7 protein (Supplementary Fig. 3G, H).
To well elucidate the role of Pla2g7 in bone homeostasis, we next investigated whether Darapladib or Pla2g7 recombinant protein have an effect on osteoblast differentiation. ALP (Alkaline phosphatase) and alizarin red S staining revealed that both of them had no effects on osteoblast activity (Supplementary Fig. 3I, L), and no obvious alteration of osteoblast differentiation markers at the levels of mRNA and protein (Supplementary Fig. 3J, K, M, N). These results indicated that Darapladib and exogenous Pla2g7 protein exert a strong effect on osteoclast differentiation but no effect on osteoblast differentiation.
Darapladib inhibits osteoclast differentiation via regulating arachidonic acid metabolism
Phospholipase A2 is the key rate-limiting enzyme mainly involved in the fatty-acid metabolism, various inflammatory responses, signal transduction and hemodynamics27,28,29,30. Thus, lipidomics was performed to explore the underlying mechanism of Pla2g7 regulating osteoclast differentiation (Fig. 4A). Principal-component analysis (PCA) suggested marked lipidomic profile between the two groups (Supplementary Fig. 4A). Using the screening criteria of fold change >1.5 and p < 0.05, we identified 401 differentially secondary metabolites, including 135 upregulated and 266 downregulated metabolites by Darapladib treatment (Fig. 4B). Then, we separately screened 5 upregulated and downregulated metabolites with the most significant differences (Supplementary Fig. 4B). Interestingly, half of the ten significant metabolites were closely associated with the arachidonic acid metabolism (Fig. 4C). Correspondingly, FA 20:4, exactly the chemical formula of arachidonic acid, was decreased significantly after Darapladib administration (Fig. 4D). Kyoto encyclopedia of Genes and Genomes (KEGG) analysis on the all differentially secondary metabolites showed that the lipid metabolism is the most significant enriched pathway in KEGG Main Class (Supplementary Fig. 4C). Subsequently, we also found the arachidonic acid metabolism is highly enriched in Lipid metabolism (Supplementary Fig. 4D). Therefore, we speculated Darapladib may inhibit osteoclast differentiation through regulation of arachidonic acid metabolism.

A The design of lipid metabolomics assay, in which Darapladib treatment was conducted concurrently with RANKL induction for 3 days until osteoclasts emerged. n = 5 biologically independent cell samples per group. B Volcano plot indicating differentially expressed genes in BMMs incubated with RANKL in the absence or presence of Darapladib (400 nmol/L). C Network analysis of the top 5 most significantly upregulated and downregulated secondary metabolites according to the integrated analysis of Fold change and P value. D Heatmap of differentially expressed secondary metabolites. FFA free fatty acid, SM sphingomyelin, LPC lysophosphatidylcholine, LPI lysophosphatidylinositol, LPE lysophosphatidylethanolamine, PC phosphatidylcholine, PS phosphatidylserine, PI phosphatidylinositol, PG phosphatidylglycerol, PE phosphatidylethanolamine. n = 5 biologically independent cell samples per group. E A schematic diagram of the RNA-sequencing of BMMs from WT mice with or without Darapladib, following 3 days of RANKL induction until osteoclasts emerged. n = 3 biologically independent cell samples per group. F Volcano plot showing 1036 upregulated and 1979 downregulated genes for RNA-seq data. G A heatmap of osteoclast-specific genes between RANKL and RANKL treated by Darapladib groups. n = 3 biologically independent cell samples per group. H The metabolism class in KEGG analysis of RNA-seq data. I, J The concentration of arachidonic acid in Darapladib, recombinant Pla2g7 and Pla2g7 cKO groups. n = 4 biologically independent mice per group. K–M The expression of ferroptosis-related proteins in Darapladib, recombinant Pla2g7 and Pla2g7 cKO groups, and these densitometric analysis of Western blot bands. n = 3 biologically independent cell samples per group. Data were showed as mean ± SD. Two-sided Student’s t test (I, J and K–M) was employed to assess the difference between the two groups. Significance: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001, ns non-significance.
To firmly determine the arachidonic acid metabolism as the major regulation pathway, we conducted RNA sequencing (RNA-seq) on osteoclasts with or without Darapladib treatments (Fig. 4E). We identified 1036 upregulated and 1979 downregulated differentially expressed genes (DEGs) (Fig. 4F). Among the total DEGs, osteoclast-associated genes were decreased in Darapladib treatment group (Fig. 4G). Importantly, arachidonic acid metabolism also exhibited a selective enrichment within the metabolic pathways classified by KEGG (Fig. 4H). Based on the above analysis, we re-confirmed that arachidonic acid concentration was decreased in Darapladib treatment, Pla2g7LysM and Pla2g7CTSK group (Fig. 4I, J). Pla2g7 was found to resist ferroptosis by remodeling lipid metabolism24,31, while ferroptosis itself exerts complex regulatory effects on osteoclast, with its inhibition potentially enhancing osteoclast activity32. However, in our study, Darapladib or Pla2g7 knockout inhibited ferroptosis pathway in osteoclast, whereas exogenous supplementation of Pla2g7 enhanced it. These findings suggest that Pla2g7 regulates osteoclast differentiation through a mechanism independent of ferroptosis (Fig. 4K–M). Collectively, these results revealed that arachidonic acid metabolism is the core downstream pathway of Pla2g7 regulating osteoclast differentiation according to the multi-omics.
Pla2g7 regulates osteoclast differentiation through Alox12/12-HETE signaling axis
As it is well known, the formation of arachidonic acid metabolites, including prostaglandin, oxylipins, leukotrienes and hydroxyeicosatetraenoic acid, is catalyzed mainly by cyclooxygenases (COX), lipoxygenases (LOX) and cytochrome P450 enzymes (CYP450) (Supplementary Fig. 4E)27. Therefore, we screened these pivotal enzymes from the levels of mRNA and protein27,33,34, and surprisingly found that the expression changes of Alox12 was the most significant after supplementation of Darapladib or Pla2g7 knockout (Fig. 5A–D). Similarly, it was also been observed as the most significant upregulated enzyme after recombinant Pla2g7 protein treatment (Supplementary Fig. 4F–H). These findings were confirmed by immunofluorescence staining, supporting the regulatory effect of Pla2g7 on Alox12 expression (Fig. 5E, F and Supplementary Fig. 4I, J). To verify the effect of Pla2g7 inhibition on Alox12 expression in vivo, we conducted an OVX-induced osteoporosis mice model with Darapladib treatment. Immunofluorescence staining demonstrated osteoclast differentiation and Alox12 expression were decreased in Darapladib treatment group by double staining of Nfatc1 with Alox12 (Fig. 5G, H), as well as the Pla2g7 conditional knockout group (Supplementary Fig. 4K-L). Consistently, the protein expression of Alox12 peaked at the early edge of osteoclast differentiation (Supplementary Fig. 4M). Furthermore, exogenous supplementation of arachidonic acid effectively restored Alox12 expression, suggesting that the observed downregulation of Alox12 is predominantly mediated by the diminished availability of Pla2g7-derived arachidonic acid, rather than being a direct effect of Pla2g7 protein itself (Supplementary Fig. 4N, O). Therefore, we speculated that Pla2g7 regulated osteoclast differentiation through Alox12-mediated arachidonic acid metabolic pathway.

A–D The expression of enzymes associated with the arachidonic acid metabolism pathway in osteoclasts treated with the inhibitor or ablation of Pla2g7 according to immunoblotting and relative RNA analysis. n = 3 biologically independent wells per group associated with cyclooxygenase (COX) and cytochrome P450 (CYP) pathways and n = 4 per group associated with lipoxygenase (LOX) pathways. E, F The immunofluorescence images of Alox12 in osteoclasts treated with Darapladib and quantification of fluorescence intensity. n = 3 biologically independent wells per group (scale bar, 10 μm). G, H The expression of Alox12 and Nfatc1 in mice treated with drug and statistical analysis of fluorescence intensity. n = 6 biologically independent mice per group (scale bar, 100 μm). I TRAP staining indicating Alox12 inhibitor treatment on Pla2g7-induced osteoclasts differentiation. (scale bar, 200 μm). J–L ML355 treatments (200 nM) on Pla2g7-induced osteoclasts in immunoblotting and RT-qPCR. n = 3 per group. n = 3 biologically independent wells per group. M The concentration of 12-HETE in RANKL or RANKL and Darapladib treatments were assessed following a 3-day induction period, coinciding with osteoclast formation. n = 4 biologically independent mice per group. N, O Darapladib or 12-HETE treatments (100 ng/mL) on RANKL-induced BMMs in TRAP staining and quantification of TRAP positive MNCs and average osteoclast size in each group. n = 6 biologically independent wells per group. P, Q Relative mRNA and protein expression in osteoclasts treated with Darapladib or 12-HETE, and the densitometric analysis of Western blot bands. For RNA analysis, n = 3 biologically independent wells per group. For Western blot analysis, n = 3 biologically independent cell samples per group. R Immunoblotting showing these markers in osteoclasts incubated with or without 12-HETE. Data were showed as mean ± SD. Two-sided Student’s t test (B, D, F, H, M) was employed to assess the difference between the two groups. Two-way ANOVA (K, O–Q) was utilized for multiple comparisons test. Significance: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001, ns non-significance.
To this end, we used ML355 as a selective inhibitor of Alox12 and found that ML355 treatment significantly suppressed Pla2g7-enhanced osteoclast formation from TRAP staining (Fig. 5I), as well as the western blot and RT-qPCR detection (Fig. 5J, K). A similar result was obtained in RANKL-induced time gradient, ML355 inhibited Pla2g7-enhanced osteoclast differentiation compared to the control group (Fig. 5L). To further elucidate the relationship between other key enzymes involved in arachidonic acid metabolism and Pla2g7-mediated osteoclast differentiation, we selected a panel of selective inhibitors, including Zileuton (Alox5), PD146176 (Alox15), Aspirin (Cox1), Celecoxib (Cox2), TP0472993 (Cyp450 4A11/4F2), Sulfaphenazole (Cyp450 2C9), and LKY-047 (Cyp450 2J2), to systematically assess the involvement of them in osteoclast differentiation. We found that both aspirin and celecoxib, as cyclooxygenase (COX) inhibitors, only partially suppressed the enhancement of osteoclast differentiation induced by Pla2g7 (Supplementary Fig. 4P, Q). In contrast, zileuton slightly promoted osteoclast differentiation, suggesting a potential negative regulatory role of Alox5 in this context (Supplementary Fig. 4R). Furthermore, we assessed the effects of PD146176 and several cytochrome P450 (CYP450) inhibitors, including those targeting Cyp2C9, Cyp2J2, Cyp4F2, and Cyp4A11 (Supplementary Fig. 4S–V). None of these agents showed significant effects on osteoclast differentiation, indicating that these pathways may not play a dominant role in Pla2g7-mediated osteoclast regulation.
Alox12 is a lipoxygenase to converse arachidonic acid to 12-HETE. Subsequently, we investigated the role of 12-HETE in osteoclasts. Firstly, a remarkable decreased of 12-HETE was observed after Darapladib administration (Fig. 5M). Supplementation of 12-HETE promoted osteoclast differentiation (Supplementary Fig. 5A), moreover, partially rescued osteoclastogenesis which was impeded by Darapladib treatment or Pla2g7 knockout (Fig. 5N, Q and Supplementary Fig. 5B–E). In RANKL-induced time ingredient assay, 12-HETE also rescued osteoclast-specific genes expression compared to control group (Fig. 5R and Supplementary Fig. 5F). In conclusion, Alox12/12-HETE signaling axis was involved in Pla2g7-regulated osteoclast differentiation.
Pla2g7/12-HETE/Gpr31 axis mediates osteoclast differentiation via regulating p38 MAPK and mitochondrial energy metabolism
G-protein-coupled receptor 31 (Gpr31) has been reported to be receptor for 12-HETE21, and Gpr31 knockdown affects 12-HETE-induced activation of MAPK signaling15,35. Therefore, we proceeded to elucidate the effect of Gpr31 on osteoclast differentiation. Firstly, we used small interfering RNA (siRNA) to silence the expression of Gpr31, and found that osteoclast markers were significantly downregulated with Gpr31 knockdown (Fig. 6A, B). TRAP staining demonstrated siGpr31 downregulated osteoclast differentiation (Fig. 6C). We also found Gpr31 knockdown dramatically suppressed osteoclast markers expression with a time-dependent manner (Fig. 6D). In addition, knockdown of Gpr31 obviously inhibited 12-HETE-induced osteoclastogenesis via TRAP staining and RT-qPCR and immunoblotting detection of osteoclast markers (Supplementary Fig. 5G–J). Similarly, the Pla2g7-promoted osteoclastogenesis was suppressed by siGpr31, as demonstrated by TRAP staining and detection of osteoclast markers (Supplementary Fig. 5K–N).

A, B Knockdown efficiency of Gpr31 with siRNA from the level of gene and protein. n = 3 biologically independent wells per group. C TRAP staining to demonstrated the knockdown efficiency of Gpr31. D Immunoblotting showing deceased osteoclast markers expression in Gpr31 siRNA knockdown compared to control group during osteoclast formation. E KEGG analysis according to the differentially expressed genes of RNA-seq. F GSEA of oxidative phosphorylation and MAPK signaling pathway between RANKL and RANKL with Darapladib treatment. G, H The expression of MAPK signaling pathway in RANKL-induce osteoclast formation treated with drug or Gpr31 siRNA knockdown in Western blot. I–L OCR (left) and ECAR (right) of RANKL-induced BMMs with or without Darapladib, and statistical analysis of related parameters including Maximal respiration, ATP-linked respiration and Maximal ECAR. n = 3 biologically independent wells per group. M–P OCR (left) and ECAR (right) of BMMs incubated with Gpr31 siRNA compared with control group, and quantification of parameters Maximal respiration and Maximal ECAR. n = 3 biologically independent wells per group. (Q, R) The expression of mitochondrial respiratory chain, electron transport chain and osteoclast markers in WT BMMs treated with Darapladib or Gpr31 siRNA compared with control group. S The densitometric analysis of Western blot bands. n = 3 biologically independent cell samples per group. T, U Immunoblotting showing the expression of mitochondrial dynamics and osteoclast markers in WT BMMs treated with Darapladib or Gpr31 siRNA compared with control group. V The densitometric analysis of Western blot bands. n = 3 biologically independent cell samples per group. W–Z The flow diagram of MitoSOX and quantification of fluorescence intensity of probe. n = 3 biologically independent wells per group. Data were showed as mean ± SD. Two-sided Student’s t test (S, V, Z) was employed to assess the difference between the two groups. One-way ANOVA (A, X) and two-way ANOVA (J, L, N, P) were utilized for multiple comparisons test. Significance: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001, ns non-significance.
To gain a better understanding of how Pla2g7/12-HETE/Gpr31 axis regulates osteoclastogenesis, we reviewed the results of transcriptome analysis. Remarkably, MAPK signaling pathway and oxidative phosphorylation were highly enriched after Darapladib administration (Fig. 6E), and both of them were suppressed by the inhibitor according to the results of GSEA (Fig. 6F). Particularly, p38 MAPK signaling was suppressed by Pla2g7 inhibition or deficiency (Fig. 6G and Supplementary Fig. 6A), as well as Gpr31 knockdown but increased by Pla2g7 stimulation (Fig. 6H and Supplementary Fig. 6B). The p38 MAPK represents a master signaling pathway that is implicated in several cellular functions such as cell proliferation, cell cycle, and mitochondrial functions, especially involved in mitochondrial biogenesis and oxidative phosphorylation, via the phosphorylation of a panel of substrates36. Therefore, we further determined the transformation of mitochondrial energy metabolism. Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) analysis indicated that both of the Darapladib treatment and Pla2g7-deficient significantly decreased RANKL-induced excessive ATP-linked respiration, maximal respiration (Fig. 6I, J and Supplementary Fig. 6C, D) and maximal ECAR capacity (Fig. 6K, L and Supplementary Fig. 6E, F). A similar result was observed in Gpr31 knockdown group (Fig. 6M–P). In contrast, Pla2g7 stimulation markedly increased the parameters of mitochondrial energy metabolism (Supplementary Fig. 6G–J).
Meanwhile, we assessed the activity of mitochondrial electron transport chain and mitochondrial respiratory chain, including Cyc1, Uqcrc2 and Ndufs6. We found that these proteins were upregulated after recombinant Pla2g7 protein supplementation but downregulated after Pla2g7 inhibition or knockout treatment, whose result was same as Gpr31 knockdown (Fig. 6Q–S and Supplementary Fig. 6K–M). Then, we detected mitochondrial dynamics by analyzing fission proteins Drp1 and fusion proteins Mfn1, Mfn2 and Opa1. Decreased mitochondrial fission and increased mitochondrial fusion were found in Pla2g7 inhibition or deficiency group (Fig. 6T and Supplementary Fig. 6N, O) and in Gpr31 knockdown group (Fig. 6U, V), which followed by reduced ROS and mtROS (mitochondrial reactive oxygen species) production (Fig. 6W–Z and Supplementary Fig. 6Q, R, 6U–X). Notably, the role of Pla2g7 supplementary on mitochondrial dynamics and mtROS was opposite to the above results (Supplementary Fig. 6P, S, T).
To further validate whether p38 MAPK as a downstream effector of Gpr31 signaling, we employed Adezmapimod as a specific inhibitor and Anisomycin as an activator of p38 MAPK. We intended to examine the role of p38 in regulating mitochondrial function more comprehensively. A series of experiments, including OCR (Supplementary Fig. 7A–D), oxidative phosphorylation and electron transport chain activity (Supplementary Fig. 7E, F), mitochondrial dynamics (Supplementary Fig. 7G, H), and mitochondrial reactive oxygen species (ROS) production (Supplementary Fig. 7I–L), were performed. All of the aforementioned mitochondria-related experimental results support that p38 MAPK can serve as a downstream signaling pathway of the Pla2g7/Gpr31 signaling axis.
Therefore, these results suggested that Pla2g7/12-HETE/Gpr31 axis mediated osteoclast differentiation by regulating p38 MAPK signaling and mitochondrial energy metabolism.
Darapladib ameliorates various pathological bone loss in mice models
Darapladib has been shown as a potent therapy of atherosclerosis, inflammatory and some lipid-related diseases37,38. Therefore, we constructed high-fat diets (HFD) mice models to explore whether Darapladib treatment restored the bone mass loss of HFD mice (Fig. 7A). Interestingly, we found Darapladib definitely reduced the body weight and liver weight of HFD mice, and decreased the lipid-related indexes including TG, TCHO and LDL-C (Supplementary Fig. 8A, B). The lipid formation of livers was also downregulated by Darapladib via HE staining and lipid droplets analysis (Supplementary Fig. 8C, D). Besides, Darapladib rescued HFD-induced bone mass loss (Fig. 7B), including improving BV/TV, Tb.N and Tb.Th (Fig. 7C). HE and TRAP staining revealed that Darapladib obviously decreased HFD-induced osteoclast formation (Fig. 7D, E). However, cortical bone mass and parameter was not altered (Fig. 7F, G). Moreover, osteocalcin (OCN) immunofluorescence assay suggested that Darapladib had no effect on osteoblast in cortical bone (Supplementary Fig. 8E, F). Therefore, Darapladib restore HFD-induced bone mass loss.

A The schematic diagram of high fat diets induced bone loss model. B Micro CT images of trabecular in CD or HFD treated with or without Darapladib groups. CD Chow diets, HFD high fat diets (scale bar, 1 mm). C Analysis of bone mass parameters in the three groups. n = 6 biologically independent mice per group. D, E Staining and statistical analysis of osteoclasts presence. n = 6 biologically independent mice per group. CD group served as control, HFD group represents high-fat diet model, and HFD + Darapladib group represents treatment with Darapladib on HFD background (scale bar, normal, 200 μm. enlarged, 100 μm). F, G Micro CT images and statistical analysis of cortical thickness. n = 6 biologically independent mice per group. (scale bar, 1 mm). H The schematic diagram of the ovariectomy experiment. I, J The images and bone mass analysis of trabecular in female mice divided into sham, OVX, OVX supplemented with Darapladib groups. n = 6 biologically independent mice per group (scale bar, 1 mm). K, L The expression of osteoclasts in two staining assays and statistical analysis. n = 6 biologically independent mice per group (scale bar, normal, 200 μm, enlarged, 100 μm). M, N The diagram of cortical bone and morphological parameters of WT, OVX and Darapladib-treated OVX mice. n = 6 biologically independent mice per group (scale bar, 1 mm). Data were showed as mean ± SD. Two-way ANOVA (C, E, G, J, L, N) was utilized for multiple comparisons test. Significance: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001, ns non-significance.
To demonstrated the effect of Darapladib comprehensively, we constructed OVX model, and continuous administration of Darapladib throughout the whole process (Fig. 7H). Consistently, Darapladib significantly ameliorated the OVX-induced osteoporosis, as evidenced by increased BV/TV, Tb.N and Tb.Th (Fig. 7I, J). HE and TRAP staining also revealed that Darapladib effectively reduced the osteoclastogenesis, which was embodied in the analysis of Oc.S/BS and Oc.N/BS (Fig. 7K, L). The cortical bone analysis and OCN immunofluorescence indicated that no significant differences were observed between the two group with or without Darapladib administrations (Fig. 7M, N and Supplementary Fig. 8G, H).
Lastly, we investigated whether Darapladib could restore bone mass loss induced by acute inflammation. The mice were injected with LPS under the cranium and administrated with 20 mg/kg Darapladib for 2 weeks (Supplementary Fig. 8I). Similarly, Darapladib treatment significantly decreased the osteolysis caused by LPS and displayed less osteoclast as evidenced by HE and TRAP staining (Supplementary Fig. 8J–L). In conclusion, Darapladib effectively suppressed bone loss in various pathological models.
PLA2G7 as a potential therapeutic target for human osteoporosis
To further investigate the potential clinical usage of Darapladib for bone loss diseases, we examined its effect on human osteoclastogenesis and osteoblastogenesis. Firstly, we collected parts of cancellous bone from 16 osteoporosis patients and 16 normal bone mass patients.
Consistently, immunohistochemical analysis revealed a marked increase in the number of PLA2G7-positive cells in osteoporosis groups (Fig. 8A, B). Furthermore, immunofluorescence staining demonstrated that PLA2G7-positive multinucleated osteoclasts were highly expressed in OP groups (Fig. 8C, D). Protein and mRNA detection verified that the expression levels of PLA2G7 in osteoporosis patients were higher than the control group (Fig. 8E, F). Next, we collected peripheral blood and bone marrow fluid to attain hPBMCs (human peripheral blood mononuclear cells) and hBMSCs (human mesenchymal stem cells) from healthy donors. We conducted osteoclast induction on hPBMCs by human RANKL treatment and found PLA2G7 was elevated in response to time-dependent manner (Fig. 8G). To further define the role of PLA2G7 on human osteoclast differentiation, we treated hPBMCs with Darapladib (400 nM) or recombinant Pla2g7 protein (200 ng/mL) separately. TRAP staining displayed that Darapladib effectively inhibited human osteoclast differentiation while PLA2G7 potently increased the differentiation capacity (Fig. 8H, K). These results were also demonstrated as indicated by Western blot and RT-qPCR assay (Fig. 8I, J, L, M).

A, B Immunohistochemistry staining and statistical analysis of Pla2g7 in normal patients (NP) and osteoporosis patients (OP). n = 16 biologically independent patients per group (scale bar, normal, 200 μm. enlarged, 25 μm). C, D The immunofluorescence images of Pla2g7 and NFATc1 in NP and OP (scale bar, 25 μm), and the statistical analysis of Pla2g7 +NFATc1 + cells. n = 6 biologically independent patients per group. E, F Relative gene expression of osteoclasts in NP and OP, and protein expression with densitometric analysis of Western blot bands. RNA, n = 6 biologically independent wells per group. WB, n = 3 biologically independent cell samples per group. G The expression of Pla2g7 and osteoclast markers during human osteoclast differentiation. H TRAP staining indicating Darapladib treatments (400 nM) on RANKL-induced osteoclast differentiation. (scale bar, 100 μm). I, J The expression of protein and gene in osteoclast differentiation stimulated with RANKL or RANKL and Darapladib, and the densitometric analysis of Western blot bands. RNA, n = 3 biologically independent wells per group. WB, n = 3 biologically independent cell samples per group. K TRAP staining indicating Pla2g7 treatments (200 ng/mL) on RANKL-induced osteoclast differentiation. (scale bar, 100 μm). L, M Immunoblotting and RT-qPCR showing increased osteoclast markers expression in Pla2g7 treatment group compared to control group, and the densitometric analysis of Western blot bands. RNA, n = 3 biologically independent wells per group. WB, n = 3 biologically independent cell samples per group. N, O ALP, ARS and protein expression in human mesenchymal stem cells (MSCs) incubated with standard induction mode with a dose-dependent manner of Darapladib (scale bar, 4 mm). P Osteoblast-related staining indicating the differentiation and mineralization of human mesenchymal stem cells treated with various concentration of Pla2g7. (scale bar, 4 mm). Q Immunoblotting showing the expression of osteoblast markers in human MSCs incubated with dexamethasone, ascorbic acid, β-glycerophosphate and Pla2g7 with different concentration Data were showed as mean ± SD. Two-sided Student’s t test (B, D, E, F, I, J, L, M) was employed to assess the difference between the two groups. Significance: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001, ns non-significance.
Besides, we continued to explore the role of Darapladib and PLA2G7 on human mesenchymal stem cell differentiation, ALP and ARS staining indicated that both Darapladib and recombinant PLA2G7 protein had no effects on osteoblast activity (Fig. 8N, P), which also was proved by the immunoblot analysis (Fig. 8O, Q). In summary, PLA2G7 not only regulated osteoclast induction in mice but also exerted a similar effect on human, which revealed Darapladib may be a potential treatment for osteoporosis in clinical practice.
Discussion
Osteoporosis is a common age-related disease, particularly prevalent among postmenopausal women, where hormonal changes contribute to its development39. As the population ages, it is increasingly becoming a burden on families and society, with rising demands for healthcare and social support40. However, bisphosphonate therapy, as a traditional approach, is often limited by poor patient adherence and severe adverse effects, like gastrointestinal distress and osteonecrosis of the jaw41. Therefore, it is imperative to discover novel therapeutic targets or identify effective and practical methods.
In this study, we firstly uncovered that Pla2g7 was a novel target for osteoporosis treatment. We found Pla2g7 was high expression in osteoporosis patients and was positively correlated with bone resorption marker CTX-I. Compared with wild-type littermates, Pla2g7-deficient mice displayed high bone mass and could evidently restore OVX-induced bone mass loss. Darapladib, an anti-atherosclerotic drug, has been rigorously tested in Phase III clinical trials, demonstrating its safety in mice and human. Darapladib treatment not only suppressed lipid accumulation in HFD group but also ameliorated the bone loss caused by high-fat diets. Because of its capacity of inhibiting osteoclastogenesis, Darapladib also rescued bone loss resulting from these pathological mice models. Meanwhile, Darapladib wielded an influence on bone health by suppressing osteoclast differentiation but had no effect on osteoblasts in human. Therefore, Darapladib has the potential to be a promising method for osteoporosis, with Pla2g7 serving as a novel therapeutic target.
Arachidonic acid metabolism was revealed as the critical downstream pathway according to the integrated analysis of lipidomics and transcriptomics. Arachidonic acid pathway is a complex metabolic network, which mainly contains cyclooxygenase, lipoxygenase and cytochrome P450 pathway27,42. A wide array of bioactive lipid mediators was generated by these pathways to impose a profound influence on inflammation, pain and immune responses43,44,45. Consequently, a variety of classic drugs targeting these mediators have been developed, such as Aspirin and Ibuprofen that suppressing Cox1 enzyme to decrease prostaglandins production46. In our study, we discovered that Alox12 exhibited the most pronounced differentiation expression among the enzymes associated with arachidonic acid pathway. ML355, known as an inhibitor of Alox1247, effectively suppressed osteoclast differentiation promoted by Pla2g7. 12-HETE is generated by the action of Alox12 on arachidonic acid. Thus, 12-HETE is naturally considered as a Pla2g7-regulated metabolite. Not only did 12-HETE independently promoted osteoclast function and activity, but it also counteracted the inhibition of osteoclast differentiation caused by Darapladib. Collectively, Alox12/12-HETE axis acted as a crucial role in Pla2g7-mediated osteoclast differentiation.
Gpr31 has been identified as a high-affinity receptor for 12-HETE, however, its potential mechanism in osteoclast formation is largely unknown. Osteoclast formation and function require a large amount ATP provided by mitochondrial through OXPHOS process48. Mitochondrial is involved in the dynamic regulation of calcium ion to affect bone resorption processes of osteoclasts, and produces reactive oxygen which promotes osteoclast formation and regulates inflammation reaction as signaling molecule49,50,51. Therefore, mitochondrial metabolism is closely associated with osteoclastogenesis. Here, we uncovered that Gpr31 knockdown suppressed mitochondrial energy production and MAPK cascades to impede the induction of osteoclasts. In conclusion, the Alox12/12-HETE/Gpr31 axis modulated by Pla2g7, represented a novel pathway that was the first discovered in osteoclast.
However, our study also has some limitations. Firstly, we identified the high expression of PLA2G7 in osteoporosis via testing the serum of 150 patients, but we unable to collect more sufficient cancellous bone to demonstrated PLA2G7 expression in tissue levels. Additionally, the role of Gpr31 on osteoclastogenesis only has been demonstrated in vitro, it remains to be explored whether Gpr31 also wields an influence on osteoclasts in vivo. Thirdly, although arachidonic acid metabolism was identified as a key pathway by integrating transcriptomic and metabolomic analyses, this approach may overlook other biologically relevant metabolic processes that do not reach statistical significance. In addition, other polyunsaturated fatty acid (PUFA) metabolic pathways—such as linoleic acid and α-linolenic acid metabolism—have been implicated in inflammatory responses and bone remodeling52, and their potential roles in osteoclast regulation warrant further in-depth investigation.
Methods
Reagents
Fetal bovine serum (FBS) and alpha-MEM were sourced from Gibco. Recombinant mouse M-CSF (Cat#CB34, Novoprotein; Cat#AM10003, Amizona Scientific) and RANKL (Cat#462-TEC, R&D System), as well as recombinant human M-CSF (Cat#11682-HNCH, SinoBiological) and RANKL (Cat#11792-HNAH, SinoBiological). ML355 (Cat#HY-12341) and Darapladib (Cat#HY-10521) were purchased from MedChemExpress. Pla2g7 (Cat#C624, Novoprotein). 12-HETE (Cat#GC40429, Gibco).
Mice
Pla2g7f/f mice constructed by CRISPR-Cas9 technology were obtained from GemPharmatech. LysM-Cre and CTSK-Cre mice were provided by Cyagen Biosciences.
Mice were housed under specific pathogen-free (SPF) conditions with a 12-h light/dark cycle, temperature maintained at 22–24 °C, and humidity at 50–60%. They were fed standard chow (Cat#P1101F, SLACOM, China) with the following composition: 20% protein, 4% fat, 5% fiber, 8% ash, and 1% calcium. The chow was provided by the Experimental Animal Center of Zhejiang University School of Medicine. In our investigation, WT littermates of the same sex mice served as control groups and compared with Pla2g7LysM and Pla2g7CTSK mice. All strains, including Pla2g7f/f, LysM-Cre and CTSK-Cre mice, were maintained on a C57BL/6 genetic background.
Serums and tissue specimens from patients
Peripheral blood samples were collected and centrifuged at 1600g to obtain serum (Centrifuge model, Cat#75003181, ThermoFisher scientific, USA), which were then processed using a human monocyte isolation kit to obtain PBMCs. (Cat#P9011, Solarbio, China). In addition, surgically excised cancellous bone specimens from both NP and OP were collected. Given that osteoporosis predominantly affects postmenopausal women, the known relationship between age and bone mass, and the inter-individual heterogeneity in bone mass, the study was not sufficiently powered to detect sex- or gender-specific differences.
Osteoclast differentiation, TRAP staining and bone resorptive assay
Bone marrow-derived macrophages (BMMs) were obtained from the femurs and tibias of 6-week-old C57BL/6 J mice, and cultured in alpha-MEM containing 10% FBS, 1% penicillin-streptomycin, 25 ng/mL M-CSF under a humidified atmosphere with 95% air and 5% CO2 at 37 °C. Then, osteoclasts differentiation was induced from BMMs, which were stimulated with 25 ng/mL M-CSF and 50 ng/mL RANKL for 5 days.
Human peripheral blood mononuclear cells (hPBMCs) were isolated using Ficoll separation liquid and cultured in alpha-MEM with 10% FBS and 25 ng/ml human M-CSF. To differentiate hPBMCs into osteoclasts, the cells were treated with 25 ng/ml human M-CSF and 50 ng/ml human RANKL for 9 days.
Osteoclasts originated from BMMs and PBMCs were washed with PBS, fixed with 4% paraformaldehyde for 15 min and stained with TRAP solution (Cat#387A-1 KT, Sigma-Aldrich, USA) for 1 h at 37 °C. After staining, TRAP-positive osteoclasts were counted under a microscope and average multinucleated osteoclasts size were measured via image J software.
The bone resorption assay was performed onto the hydroxyapatite-coated plate (Cat#3989, Corning Inc, USA) in triplicate. The BMMs were induced for 5 days then were rinsed with 10% sodium hypochlorite solution. The resorption pits were taken under a microscope and quantified using Image J software.
Osteoblast differentiation, alkaline phosphatase and alizarin red staining
Craniums were isolated from 3-day-old C57BL/6 J neonatal mice and subjected to enzymatic digestion with Collagenase II for 16 h. Osteoblasts derived from Craniums were harvested from filtered medium and cultured in DMEM containing 10% FBS and 1% penicillin-streptomycin.
Human mesenchymal stem cells (hMSCs) were isolated from marrow fluid and purified using Ficoll separation liquid. The cells were cultured in commercial special medium (Cat#HUXMA-90011, OriCell, China) and maintained in cell culture incubator.
Osteoblast differentiation was induced in DMEM supplemented with 5 µM ascorbic acid (Cat#A4403, Sigma-Aldrich, USA), 10 mM β-glycerophosphate (Cat#G9422, Sigma, USA) and 10 nM dexamethasone (Cat#D4902, Sigma-Aldrich, USA). After 7 days induction, alkaline phosphatase (ALP) staining was performed according to the protocol of the kit (Cat#MA0197, MeilunBio, China), followed by alizarin red S (ARS) staining (Cat#A5533, Sigma, USA) on day 21 to assess mineralization. The culture medium was refreshed every 2 days and the cells were incubated in the condition as described above throughout the induction process.
RNA extraction and quantitative PCR
Cells were seeded (2 × 105 per well) in 12-well plates. Total RNA was isolated using TRIzol reagents (Cat#CW0581, CWBIO, China) as indicated as the instruction of manufacturer, and mRNA reverse transcription was performed using the HiFiScript cDNA Synthesis Kit (Cat#CW2569, CWBIO, China). The RT-qPCR amplification reactions were carried out using cDNA, primers and Hieff qPCR SYBR Green Master Mix (Cat#11201ES08, Yeasen, China). The relative mRNA expression levels were normalized to GAPDH levels. The primer sequences were presented in Supplementary Table 4. (The primer was purchased from TSINGKE Biosciences).
Western blot
The proteins were extracted from adherent cells using RIPA lysate mixed with 1% proteinase inhibitor, 1% phosphotransferase inhibitor and 100 mM phenylmethanesulfonyl fluoride, followed by sonicating and centrifuging at 13,800g for 15 min to collect the supernatant that were heated at 100 °C for 10 min (Centrifuge model, Cat#75003424, ThermoFisher scientific, USA). SDS–polyacrylamide gel electrophoresis was performed as previously described53. Protein bands were exposed with an enhanced chemiluminescence (ECL) solution (Cat# FD8020, Fdbio science, China), and analyzed using the FUSION FX Bio Imaging System (VILBER, Paris, France). The antibodies for western blot were as follows: OPA1 (Abcam, ab42364, 1:1000), RUNX2 (Cell Signaling Technology, 12556, 1:1000), Erk (Cell Signaling Technology, 5013, 1:1000), P-P38 (Cell Signaling Technology, 4511, 1:1000), Pla2g7 (Proteintech, 15526-1-AP, 1:1000), CYP2J2 (Proteintech, 13562-1-AP, 1:1000), CYP2C9 (Proteintech, 16355-1-AP, 1:1000), CYP4A11 (Proteintech, 11688-1-AP, 1:1000), P-Erk (Proteintech, 28733-1-AP, 1:1000), JNK (Proteintech, 51151-1-AP, 1:1000), P-JNK (Proteintech, 80024-1-RR, 1:1000), P38 (Proteintech, 14064-1-AP, 1:1000), GAPDH (Proteintech, 60004-1-Ig, 1:1000), OCN (Proteintech, 23418-1-AP, 1:1000), DRP1(Proteintech, 12957-1-AP, 1:1000), NDUFS6(Proteintech, 14417-1-AP, 1:1000), NFATc1 (Santa Cruz, sc-7294, 1:1000), ALOX5 (Santa Cruz, sc-136195, 1:1000), ALOX15 (Santa Cruz, sc-133085, 1:100), MFN1 (Santa Cruz, sc-166644, 1:1000), MFN2 (Santa Cruz, sc-100560, 1:1000), CYC1 (Santa Cruz, sc-514435, 1:1000), CTSK (Abclonal, A5871, 1:1000), ALOX12 (Abclonal, A14703, 1:1000), Gpr31 (HUABIO, ER1910-06, 1:1000) and UQCRC2 (HUABIO, ER1803-08, 1:1000), CYP4F2(Affinity, AF9051, 1:1000).
Immunofluorescence staining
For immunofluorescence staining assays, BMMs were plated (1 × 105 cells/well) on coverslips in a 24-well plate. The cells were cultured as described above. After washing with phosphate-buffered saline (PBS), the cells were fixed with 4% paraformaldehyde (Cat#G1101, Servicebio, China) for 15–20 min. Permeabilization was achieved with 0.1% Triton X-100 for 15 minutes at room temperature, followed by blocking with QuickBlock immunostaining blocking solution (Cat#P0260, Beyotime, China) for one hour at room temperature. The blocking solution was washed off, the primary antibody was added and incubated overnight at 4 °C. The plates were then washed three times for 10 min each, before being incubated with a fluorescently labeled secondary antibody (Cat#4412S, Cell Signaling Technology; Cat#4409S, Cell Signaling Technology), DAPI and F-actin at room temperature for 1 h. The coverslips were using an anti-quenching agent (Cat#G1401-5ML, Servicebio, China), and fluorescence images were captured using Leica TCS sp8 confocal microscope and analyzed with ImageJ software.
For tissue sections, deparaffinization was conducted followed by antigen retrieval with sodium citrate buffer overnight at 55 °C. After cooling the sections to room temperature gradually, primary antibody was added and incubated overnight at 4 °C after blocking with QuickBlock immunostaining blocking solution for 1 h at room temperature. Subsequently, the slides were washed three times with TBST, followed by incubation with a fluorescently labeled secondary antibody and DAPI for one hour at room temperature. The slides were sealed with an anti-quenching agent before fluorescence scanning using digital slide scanner (KF-FL-120, KFBIO, China).
Immunohistochemistry staining
Tissue sections were processed for antigen retrieval. Following this, the sections were blocked with bovine serum albumin (BSA) for 1 h, and then incubated with an anti-Pla2g7 primary antibody overnight at 4 °C. After washing the sections three times with PBS, they were incubated with the secondary antibody (Cat#pv9001, ZsBio, China). The sections were washed again three times with PBS and stained with a DAB (diaminobenzidine) solution (Cat#G1212-200T, ServiceBio, China) for 5–10 min at room temperature. Images were captured using a light microscope (Nikon, Japan).
Transfection of Gpr31 siRNA
SiRNAs targeting Gpr31 were obtained from RiboBio and the sequences were listed in Supplementary Table 5. BMMs were cultured in plates and transfected with SiRNA using LipofectamineTM 3000 Transfection Reagent (Cat#2078159, Invitrogen, USA). After transfection for 24 h, the medium was refreshed without siRNA and the cells was incubated at the above condition for 24 h, followed by conducting relevant treatments associated with our study. The efficiency of the knockdown was assessed by WB, qPCR and TRAP staining.
Micro-CT scanning
The skull, femur and tibia of mice were collected and then scanned using a SkyScan1275 system (Bruker, USA) at X-ray settings of 60 μA and 50 kV with a resolution of 9 μm. To further analyze trabecular bone, 150 slices was selected to assess qualitatively and quantitatively from the growth plate. Cortical bone analysis was performed using 150 slices taken from the region within the range of 300 slices distal to the growth plate. A constant threshold of 65–255 was applied to the analysis of trabecular bone and cortical bone.
Enzyme-linked immunosorbent assay (ELISA)
Blood samples were collected from the orbital venous plexus of mice, and serums were harvested using centrifugating under 4 °C at 1600 g for 15 min (Centrifuge model, Cat#75003181, ThermoFisher scientific, USA). The serums from patients and cell supernatant without FBS were collect after centrifugation at 1600g for 15 min (Centrifuge model, Cat#75003181, ThermoFisher scientific, USA). We used ELISA assay kit of mouse CTX (Cat#CEA665Mu, Cloud-Clone Corp, China) and PINP (Cat#SEA957Mu, Cloud-Clone Corp, China) to detect the serum levels of bone resorption and bone formation. Pla2g7 (Cat#CSB-E08321m, CUSABIO, China) in mice serum was also detected and was unveiled to be correlated with two markers. For serums from patients, Pla2g7 (Cat#E-EL-H2286, Elabscience, China), CTX (Cat#CSB-E11224h, CUSABIO, China) and PINP (Cat#CSB-E11226h, CUSABIO, China) were tested utilizing human ELISA assay kit and the relevance was analyzed using Pearson correlation coefficient.
To further investigate the secondary metabolites expression in mice serum or cell supernatant, we utilized ELISA assay kit to detect the expression of these metabolites, including arachidonic acid and 12-HETE (Cat#EM2143, FineTest, China; Cat#EU3131, FineTest, China). These ELISA assays were used according to the protocol of these manufacturers.
In silico analysis of single-cell RNA sequencing data sets
To validate the specific expression pattern of PLA2G7, we used three independent sets of published single-cell RNA sequencing data sets for analysis. We used data of bone tissue from human beings, including osteoporosis patients (GEO: GSM4423510)54 and healthy controls (GEO: GSE169396)55, osteoblasts and osteocytes from Mus musculus (GEO: GSE154718)56, and differentiation trajectory of osteoclasts (GEO: GSE147174)57. Data analysis was conducted utilizing the Seurat package (version 5.1.0)58 in R (version 4.3.1). Filtering parameters for low-quality cells were adjusted based on the characteristics of each dataset. Normalization and identification of variable features in the single-cell gene expression data were achieved using the ‘LogNormalize’ and ‘vst’ methods. Clustering and differential expression analyses were subsequently performed using default parameters. After merging each dataset, we used the method of Harmony59 or CCA60 in the function of IntegrateLayers to remove the batch effect between each dataset. Cell clusters were visualized using UMAP. Details of the specific parameters and integration method can be found in Supplementary Table 1. Cell groups were identified based on markers previously reported in the original study. Wilcoxon rank-sum test was applied to assess inter-group differences of gene expression.
RNA sequencing and Lipid metabolomics assay
For RNA sequencing (n = 3), BMMs were incubated (1 × 106 cells/well) on 6-well plates with. Cells with or without Darapladib were obtained after 5 days of M-CSF and RANKL culture. For Lipid metabolomics assay (n = 3), BMMs were seeded (1 × 106 cells/dish) in 6-cm dishes. Cells were treated as described above and harvested, followed by freezing rapidly in liquid nitrogen. LC-MS/MS was utilized for Lipid metabolomics assay. Both assays were conducted by LC-Bio (Hangzhou, China). The raw transcriptomic data have been deposited in the Sequence Read Archive (SRA) (PRJNA1344675), and the raw metabolomic data have been deposited in the MetaboLights repository (MTBLS13141).
Flow cytometry
BMMs were seeded onto 24-well plates and subjected to different treatments based on experimental objectives. After culture and intervention, the cells were immersed in diluted fluorescent probes, including DCFH-DA (Cat#S0033S, Beyotime, China) and MitoSOX (Cat#40778ES50, Yeasen, China). The cell samples were obtained according to the instructions of manufacturers. Then, gating strategies for all flow cytometry experiments were based on FSC and SSC parameters to define the main cell populations. Macrophages and osteoclasts were distinguished from dead cells based on differences in size and internal complexity, with clear clusters observed in FSC-A versus SSC-A and FSC-H versus SSC-H. The larger clusters corresponding to macrophages and osteoclasts were selected, and their fluorescence intensity was subsequently measured using the Flow Cytometer (CytoFLEX LX, Beckman). Finally, the images and statistical data were analyzed with FlowJo v10.8 software.
Extracellular flux assay
Seahorse assay and ATP calculations were performed following the manufacturer’s protocols61,62. BMMs were seeded to Seahorse XF96 plates (Cat#102978-100, Agilent, USA), and treated differently according to experimental assignments. After finishing treatments, we utilized the Seahorse XF96 Extracellular Flux Analyzer (Agilent, USA) to measure OCR and ECAR of cultured BMMs. Cellular bioenergetics were evaluated using the Seahorse XF Cell Mito-Stress Test Kit (Cat#103260-100, Agilent, USA) and Seahorse XF Cell Glycolytic Stress Test Kit (Cat#103020-100, Agilent, USA) according to the protocol of manufacturers. Data were normalized to number of cells.
HFD model
Six-week-old male C57BL/6 mice were divided into three groups, which were controlled diet (CD), high-fat-diet (60% fat) with PBS group and high-fat-diet with Darapladib group respectively. After adapting to the diet, PBS or Darapladib (20 mg/kg) were injected intraperitoneally every other day for 3 months to assess the therapeutic effect. Both CD and HFD diets were provided by the Experimental Animal Center of Zhejiang University School Medicine. After 3 months of model establishment, their serums were harvested to detect the TG, TCHO, LDL-C conditions, livers were obtained to assess the size and number of lipid droplets, femur and tibias were collected to analyze bone mass and bone histomorphometry.
OVX-induced osteoporosis model
The osteoporosis model was established by performing bilateral ovariectomy on female mice aged 12 weeks under anesthesia, followed by standard postoperative care, to induce estrogen-deficiency–related bone loss63. WT mice were randomly divided into three groups, sham, OVX with or without Darapladib injection groups respectively. For conditional knockout mice, 12-week-old female C57BL/6 Pla2g7f/f and Pla2g7 conditional knockout (cKO) mice were respectively divided into two groups with or without treatment of bilateral ovariectomy surgery after being anesthetized. These serums, femur and tibias were obtained to analyze bone mass and bone histomorphometry after 8 weeks of model establishment.
LPS-induced inflammation model
The model was performed using 12-week-old male C57BL/6 mice. The mice were randomly divided into three groups: sham, LPS-induced with or without Darapladib treatment groups. The mice were subcutaneously injected with 25 mg/kg LPS after being anesthetized (Cat#L2630, Sigma-Aldrich, USA). After surgery, PBS or Darapladib was injected intraperitoneally every other day for 2 weeks. The craniums were collected to analyze bone mass and bone histomorphometry after 2 weeks of model establishment.
Statistical analysis
The data was presented as mean ± S.D. Statistical analysis was performed using Graph Pad Prism 9.0 (Graph Pad Software, San Diego, CA, USA) before confirming that these data follow a normal distribution. Results were analyzed using two-sided Student’s t test, one-way ANOVA, two-way ANOVA or Pearson correlation coefficient. Two-sided Student’s t test was utilized to compare means between two groups, one-way ANOVA and two-way ANOVA were employed for multiple comparisons test, Pearson correlation coefficient was used to confirm the degree of correlation between the two groups. Significance was determined at p < 0.05. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Ethical statement
All the animal studies and procedures were approved by the Ethics Committee of Zhejiang University School Medicine (Approval NO: 30787). All participants provided written informed consent and voluntarily participated in the clinical study without receiving any compensation. The clinical study was approved by the Ethical Review Board of Sir Run Run Shaw Hospital (Approval NO: 2024-2686-01).
The study design and conduct involving human participants were carried out in full compliance with all applicable regulations and guidelines governing human research, and were conducted in accordance with the ethical principles of the Declaration of Helsinki. The clinical study protocol was reviewed and authorized by the Ethical Review Board of Sir Run Run Shaw Hospital, Zhejiang University School of Medicine (Approval NO: 2024-2686-01).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data necessary to support the conclusions of this study are included in the main text and/or the Source Data files. The metabolomics dataset generated in this study is available in the MetaboLights repository under accession code MTBLS13141. The RNA sequencing data have been submitted to the Sequence Read Archive (SRA) under BioProject accession code PRJNA1344675. Additional processed data supporting the findings of this study are available from the corresponding author upon reasonable request. Source data are provided with this paper.
References
Weitzmann, M. N. & Ofotokun, I. Physiological and pathophysiological bone turnover—role of the immune system. Nat. Rev. Endocrinol. 12, 518–532 (2016).
Reid, I. R. & McClung, M. R. Osteopenia: a key target for fracture prevention. Lancet Diab. Endocrinol. 12, 856–864 (2024).
Marie, P. J. Targeting integrins to promote bone formation and repair. Nat. Rev. Endocrinol. 9, 288–295 (2013).
Veis, D. J. & O’Brien, C. A. Osteoclasts, master sculptors of bone. Annu. Rev. Pathol. 18, 257–281 (2023).
Hascoet, E. et al. New insights into inflammatory osteoclast precursors as therapeutic targets for rheumatoid arthritis and periodontitis. Bone Res. 11, 26 (2023).
Gao, X. et al. Lyso-PAF, a biologically inactive phospholipid, contributes to RAF1 activation. Mol. Cell 82, 1992–2005 e1999 (2022).
Peng, Z., Chang, Y., Fan, J., Ji, W. & Su, C. Phospholipase A2 superfamily in cancer. Cancer Lett. 497, 165–177 (2021).
Ferguson, J. F. et al. Translational studies of lipoprotein-associated phospholipase A(2) in inflammation and atherosclerosis. J. Am. Coll. Cardiol. 59, 764–772 (2012).
Spadaro, O. et al. Caloric restriction in humans reveals immunometabolic regulators of health span. Science 375, 671–677 (2022).
Candels, L. S., Becker, S. & Trautwein, C. PLA2G7: a new player in shaping energy metabolism and lifespan. Signal Transduct. Target. Ther. 7, 195 (2022).
Garcia, C. et al. Leukotriene B4 stimulates osteoclastic bone resorption both in vitro and in vivo. J. Bone Miner. Res. https://doi.org/10.1002/jbmr.5650111105 (1996).
Fuller, K.& Chambers, T. J. Effect of arachidonic acid metabolites on bone resorption by isolated rat osteoclasts. J. Bone Miner. Res. https://doi.org/10.1002/jbmr.5650040212 (1989).
Tourdot, B. E. & Holinstat, M. Targeting 12-Lipoxygenase as a potential novel antiplatelet therapy. Trends Pharm. Sci. 38, 1006–1015 (2017).
Powell, W. S. & Rokach, J. Biosynthesis, biological effects, and receptors of hydroxyeicosatetraenoic acids (HETEs) and oxoeicosatetraenoic acids (oxo-ETEs) derived from arachidonic acid. Biochim. Biophys. Acta 1851, 340–355 (2015).
Zhang, X. J. et al. An ALOX12-12-HETE-GPR31 signaling axis is a key mediator of hepatic ischemia-reperfusion injury. Nat. Med. 24, 73–83 (2018).
Zhang, X. J. et al. Pharmacological inhibition of arachidonate 12-lipoxygenase ameliorates myocardial ischemia-reperfusion injury in multiple species. Cell Metab. 33, 2059–2075.e2010 (2021).
Sharma, I., Liao, Y., Zheng, X. & Kanwar, Y. S. Modulation of gentamicin-induced acute kidney injury by myo-inositol oxygenase via the ROS/ALOX-12/12-HETE/GPR31 signaling pathway. JCI Insight https://doi.org/10.1172/jci.insight.155487 (2022).
Milligan, G., Alvarez-Curto, E., Hudson, B. D., Prihandoko, R. & Tobin, A. B. FFA4/GPR120: pharmacology and therapeutic opportunities. Trends Pharm. Sci. 38, 809–821 (2017).
Lu, J. et al. Structural basis for the cooperative allosteric activation of the free fatty acid receptor GPR40. Nat. Struct. Mol. Biol. 24, 570–577 (2017).
Garcia, V. et al. 20-HETE signals through G-protein-coupled receptor GPR75 (G(q)) to affect vascular function and trigger hypertension. Circ. Res. 120, 1776–1788 (2017).
Guo, Y. et al. Identification of the orphan G protein-coupled receptor GPR31 as a receptor for 12-(S)-hydroxyeicosatetraenoic acid. J. Biol. Chem. 286, 33832–33840 (2011).
Zhang, Q., Lenardo, M. J. & Baltimore, D. 30 Years of NF-kappaB: a blossoming of relevance to human pathobiology. Cell 168, 37–57 (2017).
Morigny, P. et al. Association of circulating PLA2G7 levels with cancer cachexia and assessment of darapladib as a therapy. J. Cachexia Sarcopenia Muscle 12, 1333–1351 (2021).
Oh, M. et al. The lipoprotein-associated phospholipase A2 inhibitor Darapladib sensitises cancer cells to ferroptosis by remodelling lipid metabolism. Nat. Commun. 14, 5728 (2023).
Berman, J. P., Farkouh, M. E. & Rosenson, R. S. Emerging anti-inflammatory drugs for atherosclerosis. Expert Opin. Emerg. Drugs 18, 193–205 (2013).
Serruys, P. W. et al. Effects of the direct lipoprotein-associated phospholipase A(2) inhibitor darapladib on human coronary atherosclerotic plaque. Circulation 118, 1172–1182 (2008).
Wang, B. et al. Metabolism pathways of arachidonic acids: mechanisms and potential therapeutic targets. Signal Transduct. Target. Ther. 6, 94 (2021).
Smani, T. et al. Role of Ca2+-independent phospholipase A2 and store-operated pathway in urocortin-induced vasodilatation of rat coronary artery. Circ. Res. 101, 1194–1203 (2007).
Jiang, W. et al. PGE2 activates EP4 in subchondral bone osteoclasts to regulate osteoarthritis. Bone Res. 10, 27 (2022).
Birts, C. N., Barton, C. H. & Wilton, D. C. Catalytic and non-catalytic functions of human IIA phospholipase A2. Trends Biochem. Sci. 35, 28–35 (2010).
Mihee, O. et al. The lipoprotein-associated phospholipase A2 inhibitor Darapladib sensitises cancer cells to ferroptosis by remodelling lipid metabolism. Nat. Commun. https://doi.org/10.1038/s41467-023-41462-9 (2023).
Peng, D. et al. Mitochondria from osteolineage cells regulate myeloid cell-mediated bone resorption. Nat. Commun. https://doi.org/10.1038/s41467-024-49159-3 (2024).
Chrisnasari, R., Hennebelle, M., Vincken, J. P., van Berkel, W. J. H. & Ewing, T. A. Bacterial lipoxygenases: Biochemical characteristics, molecular structure and potential applications. Biotechnol. Adv. 61, 108046 (2022).
Schebb, N. H. et al. Formation, signaling and occurrence of specialized pro-resolving lipid mediators-what is the evidence so far?. Front. Pharm. 13, 838782 (2022).
King, L. A., Toledo, A. H., Rivera-Chavez, F. A. & Toledo-Pereyra, L. H. Role of p38 and JNK in liver ischemia and reperfusion. J. Hepatobiliary Pancreat. Surg. 16, 763–770 (2009).
Sarg, N. H. et al. The interplay of p38 MAPK signaling and mitochondrial metabolism, a dynamic target in cancer and pathological contexts. Biochem. Pharm. 225, 116307 (2024).
Huang, F., Wang, K. & Shen, J. Lipoprotein-associated phospholipase A2: The story continues. Med. Res. Rev. 40, 79–134 (2020).
Lv, S. L. et al. Lp-PLA2 inhibition prevents Ang II-induced cardiac inflammation and fibrosis by blocking macrophage NLRP3 inflammasome activation. Acta Pharm. Sin. 42, 2016–2032 (2021).
Neer, R. M. et al. Effect of parathyroid hormone (1-34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N. Engl. J. Med. 344, 1434–1441 (2001).
Dong, Y. et al. Global, regional, and national burden of low bone mineral density from 1990 to 2019: results from the Global Burden of Disease Study 2019. Front. Endocrinol. (Lausanne) 13, 870905 (2022).
Ayers, C. et al. Effectiveness and safety of treatments to prevent fractures in people with low bone mass or primary osteoporosis: a living systematic review and network meta-analysis for the American College of Physicians. Ann. Intern. Med. 176, 182–195 (2023).
Yarla, N. S. et al. Targeting arachidonic acid pathway by natural products for cancer prevention and therapy. Semin. Cancer Biol. 40-41, 48–81 (2016).
Back, M., Yurdagul, A. Jr., Tabas, I., Oorni, K. & Kovanen, P. T. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat. Rev. Cardiol. 16, 389–406 (2019).
Baral, P., Udit, S. & Chiu, I. M. Pain and immunity: implications for host defence. Nat. Rev. Immunol. 19, 433–447 (2019).
Wojcik, P., Gegotek, A., Zarkovic, N. & Skrzydlewska, E. Oxidative stress and lipid mediators modulate immune cell functions in autoimmune diseases. Int. J. Mol. Sci. https://doi.org/10.3390/ijms22020723 (2021).
Renda, G. et al. Celecoxib, ibuprofen, and the antiplatelet effect of aspirin in patients with osteoarthritis and ischemic heart disease. Clin. Pharm. Ther. 80, 264–274 (2006).
Guo, S. et al. Baicalein alleviates cisplatin-induced acute kidney injury by inhibiting ALOX12-dependent ferroptosis. Phytomedicine 130, 155757 (2024).
Song, C. et al. Sexual dimorphism of osteoclast reliance on mitochondrial oxidation of energy substrates in the mouse. JCI Insight. https://doi.org/10.1172/jci.insight.174293 (2023).
Park-Min, K. H. Metabolic reprogramming in osteoclasts. Semin. Immunopathol. 41, 565–572 (2019).
Bushinsky, D. A. & Krieger, N. S. Effects of acid on bone. Kidney Int. 101, 1160–1170 (2022).
Agidigbi, T. S. & Kim, C. Reactive oxygen species in osteoclast differentiation and possible pharmaceutical targets of ROS-mediated osteoclast diseases. Int. J. Mol. Sci. https://doi.org/10.3390/ijms20143576 (2019).
Hongda, L. et al. Efficacy and mechanism of the Ermiao San series of formulas for rheumatoid arthritis based on Chinmedomics strategy. Phytomedicine https://doi.org/10.1016/j.phymed.2024.155903 (2024).
Xie, Z. et al. A novel diterpenoid suppresses osteoclastogenesis and promotes osteogenesis by inhibiting Ifrd1-mediated and IkappaBalpha-mediated p65 nuclear translocation. J. Bone Min. Res. 33, 667–678 (2018).
Wang, Z. et al. Single-cell RNA sequencing deconvolutes the in vivo heterogeneity of human bone marrow-derived mesenchymal stem cells. Int. J. Biol. Sci. 17, 4192–4206 (2021).
Qiu, X. et al. Single-cell RNA sequencing of human femoral head in vivo. Aging (Albany NY) 13, 15595–15619 (2021).
Wang, J. S. et al. Control of osteocyte dendrite formation by Sp7 and its target gene osteocrin. Nat. Commun. 12, 6271 (2021).
Tsukasaki, M. et al. Stepwise cell fate decision pathways during osteoclastogenesis at single-cell resolution. Nat. Metab. 2, 1382–1390 (2020).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e3529 (2021).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Mookerjee, S. A., Gerencser, A. A., Nicholls, D. G. & Brand, M. D. Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J. Biol. Chem. 292, 7189–7207 (2017).
Ma, E. H. et al. Metabolic profiling using stable isotope tracing reveals distinct patterns of glucose utilization by physiologically activated CD8(+) T cells. Immunity 51, 856–870.e855 (2019).
Yu, B. et al. Addendum: Wnt4 signaling prevents skeletal aging and inflammation by inhibiting nuclear factor-kappaB. Nat. Med. 21, 1101 (2015).
Acknowledgements
This investigation was supported by the National Natural Science Foundation of China (Grant No. 82472472 to JCe, Grant No. 82402873 to ZF, Grant No. 82272520 to JCe, Grant No. 82272521 to FZ) and Zhejiang Provincial Natural Science Foundation of China (Grant No. LZ23H060002 to FZ). We thank Chao Bi and Xiaoli Hong from the Core Facilities, Zhejiang University School of Medicine for their technical support. The graphical abstract was generated at FigDraw.com.
Author information
Authors and Affiliations
Contributions
J.J., Z.Z. and J.G. administrated the project, collected clinical samples, analyzed experimental data and wrote the original draft. Q.Z., Z.X. and Z.F. provided methodology and validated the experimental results. S.T., S.H. and J.Ch. performed the mice experiments. J.W., H.Z. and J.Ce. provided datasets and organized these data. S.L., F.Z. and J.Ce. conceived the project, provide fund support and edited the paper. Authors with similar initials are distinguished as J.Ch. (Jiao Cheng), J.Ce. (Jian Chen).
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Eun-Woo Lee and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jin, J., Zheng, Z., Gu, J. et al. Pla2g7 regulates bone homeostasis via Alox12/12-HETE/Gpr31 signaling axis. Nat Commun 16, 11449 (2025). https://doi.org/10.1038/s41467-025-66285-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66285-8
