
Abstract
Apicomplexan parasites, including Plasmodium falciparum and Toxoplasma gondii, contain specialized secretory organelles such as micronemes, rhoptries, and dense granules, which are essential for parasite motility, host cell invasion, development, and egress. DedA superfamily proteins are implicated in lipid mobilization, which is a key requirement for organelle biogenesis. Herein, we identify and investigate the vacuole membrane protein 1 (VMP1), a DedA superfamily member, of P. falciparum (PfVMP1) and T. gondii (TgVMP1). PfVMP1 and TgVMP1 are ER-localized lipid scramblases. TgVMP1 depletion adversely affects parasite development, motility, host cell invasion, and egress. These phenotypes are consistent with impaired rhoptry and dense granule biogenesis, and decreased secretion of micronemes and rhoptries in TgVMP1-depleted parasites, indicating a crucial role for TgVMP1 in the biogenesis and function of these organelles. TgVMP1 depletion impairs lipid droplet homeostasis and ER organization, and causes loss of intravacuolar network in the parasitophorous vacuole, which are key for parasite development. Restoration of the ER-localized lipid scramblase by complementing TgVMP1-depleted parasites with PfVMP1 or a homolog as distant as human VMP1 rescues the depleted parasites, indicating their functional conservation and a crucial role for ER-resident lipid scramblase activity in the biogenesis and function of secretory organelles.
Similar content being viewed by others


Introduction
Protozoans of the phylum Apicomplexa are intracellular obligate parasites, which cause diseases in humans, livestock, and wild animals1. Plasmodium and Toxoplasma are the most studied apicomplexans, causing malaria and toxoplasmosis, respectively. The central feature of apicomplexans is the apical complex, consisting of the apical polar ring at the apex of the cell and specialized secretory organelles called rhoptries and micronemes2. Another specialized secretory organelle of apicomplexans is dense granules, which are present throughout the cytoplasm. Microneme and rhoptry secretions are essential for parasite motility, invasion, formation of the parasitophorous vacuole (PV) around the parasite, and egress2,3,4,5,6,7,8,9. Dense granule secretion contributes to the formation of the intravacuolar network (IVN) within the PV, host cell remodeling, nutrient import, and export of various substances across the parasitophorous vacuole membrane (PVM)6,9,10. As these organelles are unique and essential, proteins present in their secretions are being pursued as drug and vaccine targets. The biogenesis of micronemes, rhoptries, and dense granules is far from clear; nonetheless, the ER and Golgi have been shown to be the sites of biogenesis of the vesicles and proteins destined to these organelles11. Lipid scramblases, which move lipids between the membrane leaflets, have been shown to be crucial for various cellular processes, including membrane/organelle biogenesis, lipid distribution, and regulation of ER-organelle contacts12. DedA (downstream (of hisT) E. coli DNA gene A) superfamily proteins are implicated in lipid distribution and organelle biogenesis, and have members in all the life forms13,14. The vacuole membrane protein 1 (VMP1) is the best studied DedA superfamily member, and DedA superfamily proteins have yet to be reported in parasitic protozoans. This prompted us to identify and investigate the DedA superfamily proteins in apicomplexan parasites.
VMP1 was first identified as an overexpressed gene in the rat model of experimental acute pancreatitis15. All the characterized VMP1 homologs are ER-localized multi-pass transmembrane proteins, and share a stretch of amino acids that is called the DedA domain, a characteristic feature of DedA superfamily13,16. VMP1 has roles in a variety of cellular processes, including the formation of autophagosomes, regulation of the ER-organelle contact sites, lipid transport and distribution, secretion of lipoproteins, and formation of viral replication organelles17,18,19,20,21,22,23,24,25,26,27.
A major function of VMP1 is in the formation of autophagosomes at an early stage. Mammalian VMP1 has been shown to recruit the PI3K complex (VPS34/Beclin 1/Vps15/Atg14L) at the phagophore assembly site via interaction with Beclin 1, which then generates PI3P to initiate phagophore formation28,29. The cellular abnormalities observed due to the loss of VMP1 in mammalian cells are also mirrored in VMP1 knock-out unicellular eukaryotes Dictyostelium discoideum and Chlamydomonas reinhardtii, which exhibited fragmented ER, distorted Golgi, impaired endocytosis and exocytosis, damaged mitochondria, and accumulation of ubiquitinated-protein aggregates and lipid droplets (LDs)30,31,32. VMP1 has been shown to activate the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA), which transports cytosolic Ca2+ into ER33, thereby, inducing dissociation of ER-membrane contacts. Consistently, depletion of Homo sapiens VMP1 (HsVMP1) increased the number of ER contacts with autophagosomes, mitochondria, LDs, and endolysosomes33,34.
Mammalian VMP1 and the transmembrane protein 41B (TMEM41B), a DedA superfamily protein as well, have recently been shown to function as lipid scramblases and are implicated in maintaining lipid asymmetry across the ER leaflets and at the ER contact sites with the membranes of other organelles23,35. VMP1 and TMEM41B have been shown to be required for the formation of virus-induced replication organelles in contact with ER, and this function has been attributed to their interaction with viral proteins and mobilization/distribution of lipids at the ER-virus replication organelle site26,27,36,37. VMP1 has been shown to be key for the formation of LDs and secretion of lipoproteins, which likely depend on its scramblase activity24,34.
The pleiotropic functions of VMP1 are plausible based on its scramblase activity and involvement in several ER-associated processes, as ER is the hub of lipid synthesis, organelle biogenesis, and formation of dynamic physical contacts with various organelles and structures38. In this study, we identified a VMP1 homolog in T. gondii (TgVMP1) and P. falciparum (PfVMP1), and investigated whether it has a role in the biogenesis and function of micronemes, rhoptries, and dense granules. We show that TgVMP1 and PfVMP1 are ER-localized lipid scramblases. T. gondii VMP1 is crucial for the biogenesis and function of secretory organelles, maintenance of ER organization, formation of IVN, and LD homeostasis.
Results
Apicomplexan parasites contain DedA superfamily members
The phylum Apicomplexa contains obligate intracellular parasites, including Plasmodium, Toxoplasma, Cryptosporidium, Babesia, Eimeria, Theileria, and Neospora, which cause diseases in humans, livestock and wild animals. To identify the apicomplexan DedA superfamily members, we searched the genome databases of P. falciparum and T. gondii (https://veupathdb.org/veupathdb/app) by BLAST using the amino acid sequences of DedA superfamily proteins of human (HsVMP1), S. cerevisiae (ScTVP38), and Escherichia coli (YohD, YdjZ, YqaA) as queries. The BLAST searches predicted multiple DedA domain-containing proteins in T. gondii (TGME49_244370, TGME49_279370, TGME49_248870, and TGME49_301350) and P. falciparum (PF3D7_1474600, PF3D7_1365700, and PF3D7_0611000). Of these predicted proteins, TGME49_244370 and PF3D7_1474600 showed higher sequence identity (29.3-30.3%) with HsVMP1 than the remaining proteins (10.2–20.1%), and we termed TGME49_244370 and PF3D7_1474600 as the T. gondii VMP1 (TgVMP1) and P. falciparum VMP1 (PfVMP1), respectively. Since VMP1 homologs have crucial roles in several processes, including organelle biogenesis, we focussed on TgVMP1 and PfVMP1. TgVMP1 and PfVMP1 homologs are also present in several apicomplexans, which share 28.1-32.6% sequence identity with HsVMP1, and 34.5-83.3% sequence identity among themselves (Supplementary Table 1). Apicomplexan VMP1 homologs, including TgVMP1 and PfVMP1, contain the characteristic multi-transmembrane domain architecture and DedA domain of the DedA superfamily (Supplementary Fig. 1A). The DedA domain is characterized by [F/Y]XXX[R/K] and GXXX[V/I/L/M]XXXX[F/Y] sequence motifs, and two predicted re-entrant loops (Supplementary Fig. 1B)13. These motifs are YXXXY and GXXXMXXXXF in PfVMP1 and TgVMP1. The Gly residue in the GXXX[V/I/L/M]XXXX[F/Y] motif has been shown to be essential for the function of D. discoideum VMP134, and is also conserved in TgVMP1 and PfVMP1. The AlphaFold structures of PfVMP1 and TgVMP1 also overlapped with that of HsVMP1 (RMSD of 3.736 Å between HsVMP1 and PfVMP1, and 3.362 Å between HsVMP1 and TgVMP1) (Supplementary Fig. 1C)39,40. The conserved DedA superfamily domain architecture and structural similarity of PfVMP1 and TgVMP1 with HsVMP1 indicate that these belong to the DedA superfamily.
PfVMP1 and TgVMP1 localize to ER
Towards characterization of apicomplexan VMP1 proteins, the endogenous PfVMP1 and TgVMP1 coding sequences in P. falciparum D10 and T. gondii RHTIR1 strains were tagged in-frame at their 3’-ends with the coding sequences of GFP and AIDHA, respectively. Cloned recombinant P. falciparum (PfVMP1GFPKI) and T. gondii (RHTIR1-VMP1AID) parasites showed desired modification of the respective target locus (Supplementary Fig. 2). PfVMP1GFPKI parasites showed expression of PfVMP1GFP throughout the asexual erythrocytic stages (Fig. 1A, B), which co-localized with ER-Tracker (Fig. 1C), indicating that PfVMP1 is an ER-resident protein.

A Total lysates of synchronized PfVMP1GFPKI parasites corresponding to the ring (R), early trophozoite (ET), late trophozoite (LT) and schizont (S) stages were processed for western blotting using antibodies to GFP (VMP1) and β-actin (β-Ac) as a loading control. The arrowhead indicates PfVMP1GFP band (∼73 kDa). Protein size markers (M) are in kDa. B The indicated stages of PfVMP1GFPKI parasites were evaluated for localization of PfVMP1GFP by live-cell fluorescence microscopy. The panels are for PfVMP1GFP (VMP1), nucleus (Hoechst), and overlap of these two panels with the bright field panel (Merged). Scale bar = 2 μm. C PfVMP1GFPKI trophozoite stage was evaluated for localization of PfVMP1GFP and ER-Tracker by live-cell fluorescence confocal microscopy. The images show signal for PfVMP1GFP (VMP1), ER-Tracker, nucleus (Hoechst), and overlap of these three images with a bright field image (Merged). The merged image shows co-localization of VMP1 with ER-Tracker (Pearson’s correlation coefficient = 0.643 ± 0.09; n = 30 images). Scale bar = 2 µm. D The total lysates of parental RHTIR1 strain (Par) and RHTIR1-VMP1AID (KI) parasites were processed for western blotting using antibodies to HA (VMP1) and rhoptry protein 1 (ROP1) as a loading control. The arrowhead indicates TgVMP1AIDHA (~76.3 kDa). Protein size markers (M) are in kDa. E The ER reporter (ER-GFP)-expressing RHTIR1-VMP1AID tachyzoites were evaluated for localization of TgVMP1AIDHA and ER-GFP by IFA. The confocal images show signal for TgVMP1AIDHA (VMP1), ER-GFP, nuclear stain (DAPI), and overlap of the three images (Merged). The merged image shows co-localization of TgVMP1AIDHA with ER-GFP (Pearson’s correlation coefficient = 0.68 ± 0.06; n = 43 images). Scale bar = 5 µm. F The Golgi-reporter (GRASP-GFP)-expressing RHTIR1-VMP1AID tachyzoites were evaluated for localization of TgVMP1AIDHA and GRASP-GFP by IFA. The confocal images show signals for TgVMP1AIDHA (VMP1), GRASP-GFP, nuclear stain (DAPI), and overlap of the three images (Merged). The merged image shows partial co-localization of TgVMP1 with GRASP-GFP (Pearson’s correlation coefficient = 0.53 ± 0.11; n = 29 images). Scale bar = 5 µm. Source data are provided as a Source Data file.
RHTIR1-VMP1AID parasites would express the AID receptor TIR1 and TgVMP1 with C-terminal mini-AID-3×HA (TgVMP1AIDHA). mAID or mAID-fusion protein undergoes proteasomal degradation in the presence, but not in the absence, of auxin/indole-3-acetic acid (IAA) in the cells expressing TIR141. RHTIR1-VMP1AID tachyzoites expressed TgVMP1AIDHA (Fig. 1D). To check the subcellular localization of TgVMP1, we designed a GFP-based ER-reporter (ER-GFP) that contained BiP signal sequence at the beginning and ER-retention signal (HDEL) at the end of GFP. We also used the P. falciparum Golgi reassembly stacking protein (GRASP)-GFP as a Golgi-reporter (GRASP-GFP)42,43. We episomally expressed ER-GFP or GRASP-GFP in RHTIR1-VMP1AID parasites, which showed substantial co-localization of ER-GFP with TgVMP1AIDHA (Fig. 1E, F), indicating that TgVMP1 is present in the ER. On the other hand, only a minor proportion of TgVMP1AIDHA co-localized with GRASP-GFP in the region proximal to the ER.
TgVMP1 is essential for parasite development
We used two knockdown approaches for PfVMP1. In the first approach, the endogenous PfVMP1 coding region was tagged in-frame at the 3’-end with cDDHA-glmS ribozyme coding sequence in P. falciparum D10 strain to obtain a trimethoprim- and glucosamine-inducible conditional knockdown line (PfVMP1dKD), which would express PfVMP1cDDHA fusion protein (Supplementary Fig. 3A). In the second approach, the endogenous PfVMP1 coding region was tagged in-frame with mAIDHA-PcDT5U/TIR1-Flag-T2A/HD/PcDT3’U sequence in P. falciparum 3D7 strain to generate an auxin-inducible knockdown line (PfVMP1AID), which would express PfVMP1AIDHA and TIR1-Flag-human DHFR proteins (Supplementary Fig. 3E). PfVMP1dKD and PfVMP1AID parasites showed desired modification of the target loci and expressed PfVMP1cDDHA and PfVMP1AIDHA (Supplementary Fig. 3B, C, F, G), respectively. However, both the parasites neither showed any noticeable depletion of the fusion protein nor any growth defect under knockdown conditions (Supplementary Fig. 3C, D, G, H). PfVMP1AID parasites expressed TIR1 (Supplementary Fig. 3I), which ruled out failure of auxin-inducible knockdown due to its absence. It is possible that the intrinsic nature and ER membrane localization of PfVMP1 rendered it refractory to degradation in case of cDD and glmS systems, as has also been reported for other proteins44,45. The auxin-inducible knockdown was quite effective for TgVMP1 in T. gondii but not for PfVMP1 in P. falciparum, which might be due to differences in the specificity of E3 ligases of P. falciparum and T. gondii for TIR1. Similar to our observation, previous studies showed limited success of the auxin-inducible system in P. falciparum46. Hence, we could not investigate PfVMP1 further in P. falciparum.
On the other hand, RHTIR1-VMP1AID parasites showed efficient depletion of TgVMP1AIDHA in the presence of IAA (+IAA), with a near complete depletion in 3 h, whereas the levels of control proteins (SAG1 and ROP1) remained unchanged (Fig. 2A), confirming IAA-inducible depletion of TgVMP1AIDHA. Consistently, the TgVMP1AIDHA signal, but not the SAG1 signal, was almost absent in the IFA images of +IAA RHTIR1-VMP1AID parasites, whereas both the signals were prominent in -IAA parasites (Fig. 2B).

A The lysates of RHTIR1-VMP1AID parasites cultured with IAA for the indicated durations were evaluated for TgVMP1AIDHA (VMP1), SAG1, and ROP1 proteins by western blotting. The arrowhead indicates TgVMP1AIDHA, and protein size markers (M) are in kDa. B RHTIR1-VMP1AID parasites were cultured without (-IAA) or with (+IAA) IAA, and evaluated for the localization of TgVMP1AIDHA and SAG1 proteins by IFA. The Airyscan images are for SAG1, TgVMP1AIDHA (VMP1), nuclear stain (DAPI), and overlap of all the three images (Merged). Scale bar = 10 µm. C RHTIR1 and RHTIR1-VMP1AID parasites were cultured without (-IAA) or with (+IAA) IAA, stained with Giemsa, and observed for plaques (white areas). D The plaque sizes (Plaque size (mm2)) in C are shown on the y-axis for the indicated parasites on the x-axis. Data are mean ± SD error bar based on 120 plaques from three independent experiments. E RHTIR1 and RHTIR1-VMP1AID tachyzoites were cultured with (+IAA) or without (-IAA) IAA, stained for the PV marker GRA1, and the number of PVs containing different numbers of parasites is shown as a percentage of the total number of PVs (% Vacuoles) on the y-axis for the indicated parasites on the x-axis. Data are mean ± SD error bar based on ≥98 PVs in each of the three independent experiments. The significance of the difference is for PVs containing a single parasite only. F Mice were infected with RHTIR1-VMP1AID parasites or used as uninfected controls. The infected mice were maintained with (Infected, +IAA; n = 10) or without (Infected, -IAA; n = 10) IAA, and control mice were given IAA (Naïve, +IAA; n = 5). The plot shows percent survival of mice (% Survival) on the y-axis over days post-infection (Days-PI) on the x-axis. G Peritoneal exudates of infected +IAA and –IAA mice that succumbed to infection were evaluated for SAG1 and TgVMP1AIDHA. The representative images show bright field (DIC), TgVMP1AIDHA (VMP1), and SAG1 (scale bar = 5 µm). ns: non-significant p-value (>0.05). Source data are provided as a Source Data file.
We next assessed the effect of TgVMP1AIDHA depletion on parasite development. T. gondii tachyzoites invade host cells, replicate every 6–8 h for about 2–3 days to produce 64-128 cells within the PV, and finally egress to infect the neighboring uninfected host cells47. Repeated rounds of lytic cycles cause lysis of infected host cells, leaving clear areas in between the uninfected cells, which are called plaques. RHTIR1-VMP1AID and parental RHTIR1 parasites were cultured in the presence (+IAA) or absence (-IAA) of IAA for 8 days, and assessed for plaques. +IAA or -IAA RHTIR1 parasites and -IAA RHTIR1-VMP1AID parasites formed plaques of similar size, indicating similar growth (Fig. 2C, D). On the other hand, +IAA RHTIR1-VMP1AID parasites did not form plaques (Fig. 2C, D), indicating that TgVMP1 is essential for overall parasite development. To assess intracellular development, RHTIR1 and RHTIR1-VMP1AID tachyzoites were cultured for 24 h with (+IAA) or without (-IAA) IAA, and the number of parasites/PV was counted. The majority of +IAA RHTIR1-VMP1AID parasites did not progress beyond 1 parasite/PV stage, whereas the majority of -IAA RHTIR1-VMP1AID parasites and ±IAA RHTIR1 parasites developed to 4–8 parasites/PV stage (Fig. 2E), indicating an essential role of TgVMP1 in intracellular tachyzoite development.
We next evaluated the effect of TgVMP1 depletion on in vivo development of RHTIR1-VMP1AID tachyzoites. BALB/c mice were infected with RHTIR1-VMP1AID tachyzoites, maintained with (+IAA) or without (-IAA) IAA, and monitored for 21 days. As a control, a group of naïve mice was also maintained with IAA (+IAA). All the infected -IAA mice succumbed to infection by day 10, whereas only 50% of the infected +IAA mice succumbed to infection by day 15 (Fig. 2F). This is in contrast to the complete block of parasite development upon TgVMP1 depletion in plaque assay, which might be due to differences in auxin bioavailability in in vitro and in vivo experimental settings. The peritoneal exudates of dead +IAA mice had few parasites as compared with several parasites in the peritoneal exudates of dead –IAA mice (Fig. 2G). The peritoneal exudates of infected +IAA mice that survived for 21 days did not show parasites. The +IAA naïve mice survived and did not exhibit any visible symptoms, indicating that IAA was not toxic at the concentration used. The delayed death and 50% survival of infected +IAA mice indicate that TgVMP1 is critical for tachyzoite development in vivo.
TgVMP1 is crucial for gliding motility, invasion, and egress
To understand how TgVMP1 depletion affected parasite development, we next evaluated TgVMP1-depleted parasites for gliding motility, invasion, and egress, which are the major steps of the lytic cycle.
The motile and invasive stages of apicomplexan parasites, including P. falciparum and T. gondii, rely on gliding motility to migrate, invade the host cells, egress from the infected cell, and disperse48. Tachyzoites shed surface proteins like SAG1 during gliding, which can be used to track gliding trails. For assessing motility, RHTIR1-VMP1AID parasites were grown with (+IAA) or without (-IAA) IAA, purified, allowed to glide on FBS-coated coverslips, fixed, and stained for SAG1. 64.0% of the -IAA RHTIR1-VMP1AID tachyzoites exhibited gliding motility with an average trail length of 53.0 µm as compared with 21.8% of the +IAA RHTIR1-VMP1AID tachyzoites with an average trail length of 26.4 µm (Fig. 3A–C), indicating a significant defect in gliding motility upon TgVMP1 depletion. Gliding motility is powered by glideosome, an actomyosin motor between the parasite plasma membrane and the inner membrane complex. The plasma membrane protein SAG1 and inner membrane complex protein 1 (IMC1) showed similar localization in +IAA and –IAA RHTIR1-VMP1AID tachyzoites (Supplementary Fig. 4), indicating that these two cellular structures are unaffected in TgVMP1-depleted parasites and unlikely to be responsible for the gliding motility defect. Furthermore, the viability of purified +IAA (82.6% ± 6.8) and –IAA (87.7% ± 4.0) RHTIR1-VMP1AID tachyzoites was comparable, indicating that the decreased motility of +IAA parasites is not associated with parasite viability.

A Purified +IAA and –IAA RHTIR1-VMP1AID tachyzoites were labeled with anti-SAG1 antibodies for gliding trails (scale bar = 5 µm). B The number of parasites showing gliding trails in A is shown as a percentage of the total number of parasites (% Motile) on the y-axis for the indicated parasites on the x-axis. Data are mean ± SD error bar based on ≥100 parasites in each of the three independent experiments. C The length of gliding trails in A is shown as mean Trail length (μm) ± SD error bar on the y-axis for the indicated parasites on the x-axis. Data are based on ≥90 parasites in each of the three independent experiments. D Purified +IAA and –IAA RHTIR1 and RHTIR1-VMP1AID parasites were allowed to invade HFF cells, and labeled with a red fluorophore-conjugated antibody, which would label extracellular parasites (Red). The parasites were permeabilized and labeled again with a green fluorophore-conjugated antibody, which would label both extracellular and invaded parasites (Green). The representative images show invaded or extracellular parasites (scale bar = 5 µm). E The number of invaded parasites in D is shown as a percentage of the total number of parasites (% Invasion) on the y-axis for the indicated parasites on the x-axis. Data are mean ± SD error bar based on ≥100 parasites from each of the three independent experiments. F Intracellular RHTIR1 and RHTIR1-VMP1AID parasites were grown with (+IAA) or without (-IAA) IAA, treated with ionomycin to induce egress, fixed, and stained with antibodies to the PV protein GRA1 and nuclear stain DAPI. The representative images show PVs from which parasites egressed and dispersed or did not egress and remained as clumps (scale bar = 10 µm). G The number of PVs with parasite clumps in F is shown as a percentage of the total number of PVs (% PVs with parasite clumps) on the y-axis for the indicated parasites on the x-axis. Data are mean ± SD error bar based on ≥100 PVs from each of the three independent experiments. ns: non-significant p-value (>0.05). Source data are provided as a Source Data file.
For assessing invasion, purified +IAA and –IAA RHTIR1 and RHTIR1-VMP1AID parasites were allowed to invade HFF cells, fixed, and labeled for extracellular and invaded parasites. The +IAA RHTIR1-VMP1AID parasites showed significantly reduced invasion efficiency as compared with –IAA RHTIR1-VMP1AID parasites and ±IAA RHTIR1 parasites (Fig. 3D, E), indicating that TgVMP1 is crucial for host cell invasion.
For assessing egress, +IAA and –IAA intracellular RHTIR1-VMP1AID and RHTIR1 tachyzoites were treated with the calcium ionophore ionomycin, which has been used to induce rapid egress of intracellular parasites49, and processed for IFA using antibodies to the PV protein GRA1. RHTIR1 tachyzoites showed dispersal irrespective of the IAA treatment. The majority of -IAA RHTIR1-VMP1AID parasites were dispersed away from the PV, whereas +IAA RHTIR1-VMP1AID parasites remained as clumps near the PV (Fig. 3F, G), indicating a key role of TgVMP1 in egress.
TgVMP1 depletion did not affect conoid extrusion
Conoid extrusion, gliding motility, and secretions of micronemes and rhoptries are key sequential events for host cell invasion by T. gondii. Conoid is a hollow cone-like structure of tubulin fibers in the apical complex of T. gondii and certain other apicomplexans50. Conoid extrusion regulates motility, invasion, and egress by activating microneme secretion and actomyosin motor3. Since TgVMP1 depletion decreased gliding motility, invasion, and egress, we compared purified –IAA and +IAA RHTIR1-VMP1AID tachyzoites for conoid extrusion by treating them with the calcium ionophore A23187, which induces conoid extrusion51. The number of parasites showing conoid extrusion was similar in +IAA and –IAA parasites (Supplementary Fig. 5A, B), ruling out a role of TgVMP1 in this key event.
TgVMP1 is crucial for microneme secretion
Decreased motility, host cell invasion, egress, and intracellular development of TgVMP1-depleted parasites prompted us to look into the effect of TgVMP1 depletion on micronemes, rhoptries, and dense granules, which are crucial for these processes in Toxoplasma and Plasmodium2,52. Micronemes are situated at the apical end, and their secretion is required for motility, invasion, and egress3. We compared the localization and secretion of selected microneme proteins in +IAA and –IAA RHTIR1-VMP1AID tachyzoites. For microneme organization, +IAA and -IAA intracellular parasites were processed for IFA and compared for localization of selected microneme proteins (apical membrane antigen 1 (AMA1), microneme protein 2 (MIC2), and microneme protein 3 (MIC3)). All these proteins showed similar predominant apical localization in +IAA and –IAA parasites, ruling out any adverse effect of TgVMP1 depletion on microneme organization (Fig. 4A–D; Supplementary Fig. 6A, B). Transmission electron microscopy (TEM) images confirmed the presence of both apical and lateral micronemes in +IAA and –IAA parasites (Supplementary Fig. 6C), which was also corroborated by MIC2 labeling in immunoelectron microscopy (IEM) images (Supplementary Fig. 7A). Furthermore, the TEM micrographs showed comparable number of apical micronemes in –IAA (9.9 ± 5.9/apical region, n = 22 parasite sections) and +IAA (9.0 ± 4.4/apical region, n = 22 parasite sections) parasites, which together with the IFA data indicate that TgVMP1 depletion did not affect microneme organization. For microneme secretion, purified +IAA and –IAA RHTIR1-VMP1AID parasites were treated with ethanol, which induces microneme secretion that is known as extracellular secreted antigens (ESA)53. We used MIC2 as an ESA marker, which is proteolytically processed by the microneme protein proteases MPP1 and MPP2, and is predominantly present in the shorter form in ESA54. The level of larger MIC2 form was higher in the pellet fraction of +IAA parasites than that of –IAA parasites (Fig. 4E, F). The ESA fraction of +IAA parasites had less of the processed form of MIC2, along with the larger MIC2 form, as compared with the presence of only the processed form of MIC2 in –IAA parasites (Fig. 4E, G), which indicated both impaired microneme secretion and processing of MIC2 upon TgVMP1 depletion.

A, C -IAA and +IAA intracellular RHTIR1-VMP1AID parasites were assessed for microneme organization by labeling with antibodies to AMA1 (A) or MIC2 (C). The representative Airyscan micrographs show localization of AMA1 or MIC2, TgVMP1AIDHA (VMP1), nucleus (DAPI), and overlap of the three images (Merged). Scale bar = 10 µm, and the boxed AMA1 and MIC2 panels are zoomed in (Zoom). B, D The number of parasites with apical localization of AMA1 (in A) or MIC2 (in C) is shown as a percentage of the total number of parasites observed (% Parasites with apical AMA1 or MIC2) on the y-axis for the indicated parasites on the x-axis. Data are mean ± SD error bar based on ≥100 observations from each of the two independent experiments. E Equal number of purified -IAA and +IAA RHTIR1-VMP1AID parasites were induced for microneme secretion. The pellet (Pel) and ESA fractions were assessed for the presence of MIC2 and ROP1 by western blotting. The immunoblot shows the larger form of MIC2 in the pellet fraction of both the parasites, processed form of MIC2 in the ESA fraction of -IAA parasites, and both larger (filled arrowhead) and processed (empty arrowhead) forms of MIC2 in the ESA fraction of +IAA parasites. The protein size markers (M) are in kDa. F The signal intensities of ROP1 and MIC2 in the pellet fractions in E were measured, the signal intensity of MIC2 was normalized with that of ROP1 for the corresponding sample, and plotted as times of the signal intensity of ROP1 (MIC2 (times of ROP1)) on the y-axis for the pellet fractions (Pel) of indicated parasites on the x-axis. G The signal intensities of MIC2 in ESA fractions were measured and plotted as times of the signal intensity of MIC2 in the corresponding pellet fraction (MIC2 (times of Pel)) on the y-axis for the indicated parasites on the x-axis. Data are mean ± SD error bar from four independent experiments. ns: non-significant p-value (>0.05). Source data are provided as a Source Data file.
TgVMP1 is crucial for rhoptry biogenesis and secretion
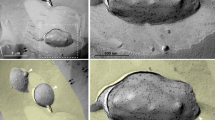
Rhoptries are situated at the apical end, and their secretion is essential for host cell invasion, formation of PV around the parasite inside the host cell, and modulation of host cell responses4,5. We compared intracellular +IAA and –IAA RHTIR1-VMP1AID parasites for rhoptry organization and secretion using ROP1 as a marker. For rhoptry organization, intracellular +IAA and –IAA tachyzoites were processed for IFA, TEM, and IEM. IFA images showed ROP1 localization to characteristic rhoptry-like elongated structures with a bulbous base in the majority (85.3%) of -IAA parasites as compared with only a small number (11.2%) of +IAA parasites (Fig. 5A, B). 59.4% of +IAA parasites also showed ROP1 signal in the PV compared to 10.9% of -IAA parasites (Fig. 5C, D). Similar to the intracellular parasites, purified -IAA parasites also exhibited ROP1 localization to rhoptry-like elongated structures, which was not the case with +IAA parasites (Fig. 5E). In agreement with the IFA data, TEM images showed apical rhoptries in 52.2% of -IAA parasites (n = 23 parasites), whereas only 18.2% of +IAA parasites (n = 22 parasites) had apical rhoptries (Fig. 6A). ROP1 labeling of the similar structures in IEM micrographs indicated ROP1 targeting to these structures (Supplementary Fig. 7B). However, compared to the IFA data, IEM micrographs did not show a clear ROP1 signal in the PV of –IAA and +IAA parasites, which might be due to differences in the two experimental procedures. The loss of rhoptries in TgVMP1-depleted parasites indicates a crucial role for TgVMP1 in rhoptry formation.

+IAA and –IAA RHTIR1-VMP1AID parasites were compared for rhoptry organization. A The representative Airyscan micrographs of intracellular +IAA and –IAA RHTIR1-VMP1AID parasites show rhoptries (ROP1), TgVMP1AIDHA (VMP1), nucleus (DAPI), and overlap of all three images (Merged). Scale bar = 10 µm, and the boxed ROP1 panel is zoomed-in (Zoom). Note that the ROP1 signal is restricted to elongated structures in -IAA parasites, as compared with +IAA parasites. B The number of parasites showing ROP1-labeled elongated structures in A is shown as a percentage of the total number of parasites observed (% Parasites with normal rhoptries) on the y-axis for the indicated parasites on the x-axis. Data are mean ± SD error bar based on ≥100 parasites from each of the three independent experiments. C The representative confocal micrographs show rhoptries (ROP1), bright field (BF), and overlap of the two images (Merged). Scale bar = 10 µm. Note that while the ROP1 signal is predominantly localized to elongated structures within the parasites in -IAA parasites, it is diffuse and also localized to the PVs of +IAA parasites. D The number of PVs with ROP1 signal in C is shown as a percentage of the total number of PVs observed (% ROP1 positive PVs) on the y-axis for the indicated parasites on the x-axis. Data are mean ± SD error bars based on multiple PVs (-IAA, n = 259; +IAA, n = 257) from three independent experiments. E The representative Airyscan confocal micrographs show signal of ROP1, ER-GFP, nuclear stain DAPI, and overlap of the three images (Merged) in purified -IAA and +IAA parasites. Scale bar = 5 µm. Note the ROP1-labeled fragmented structures in +IAA panel as compared with the elongated structure in –IAA panel. Source data are provided as a Source Data file.

A The TEM micrographs show rhoptries (Rh) and other cellular structures in –IAA and +IAA parasites. The labels are for host cell (H), parasite (P), apical end (A), dense granules (DG), micronemes (M), nucleus (N), parasitophorous vacuole (PV), intravacuolar network (IVN), lipid droplet (LD), endoplasmic reticulum (ER), and mitochondria (Mt). Note that –IAA parasites contain multiple club-shaped rhoptries extending from the apical end, which are absent or fewer in +IAA parasites. Scale bar = 1 µm. Two images are shown for each of the –IAA and +IAA parasites, which represent multiple observations from two independent experiments (-IAA: n = 24, +IAA: n = 22). B Cytochalasin D (CytD)-treated purified -IAA and +IAA RHTIR1-VMP1AID parasites were incubated with HFF cells, and labeled with anti-ROP1 antibodies. The representative micrographs show ROP1 signal (ROP1), bright field (DIC), and overlap of the two panels (Merged) for the indicated parasites. Scale bar = 2 µm. The ROP1 signal close to the parasite apical end in the host cell represents e-vacuoles, which are evident in –IAA parasites. On the other hand, the ROP1 signal remained inside the +IAA parasites. C The number of parasites exhibiting e-vacuoles in B was determined, and shown as a percentage of the total number of parasites observed (% E-vacuole positive) on the y-axis for the indicated parasites on the x-axis. Data are mean ± SD error bar based on ≥100 parasites from each of the three independent experiments. Source data are provided as a Source Data file.
For rhoptry secretion, we checked for ROP1-labeled e-vacuoles, which are discharged into the host cell during invasion. E-vacuole discharge can be monitored by treating the parasites with cytochalasin D (CytD) that prevents invasion without affecting e-vacuole secretion. The CytD-treated purified +IAA and –IAA parasites were allowed to invade HFF cells, the cells were processed for IFA using anti-ROP1 antibodies, and scored for ROP1 positive e-vacuoles. ~16% of the +IAA parasites secreted e-vacuoles into the host cell as compared with ~34% of the –IAA parasites (Fig. 6B, C), indicating a crucial role for TgVMP1 in rhoptry secretion.
TgVMP1 is required for dense granule biogenesis and IVN formation
Dense granules are secretory organelles throughout the parasite cytoplasm, which secrete an arsenal of secretory factors, collectively called GRA proteins, into the PV lumen immediately after invasion and during parasite development6,9,10. Proteins secreted by dense granules associate with and contribute to the formation of IVN, which has been suggested to act as a scaffold to position and organize the replicating parasites within the PV9,10. Some GRA proteins have also been shown to be transported to the PVM and host cell to mediate transport across the PVM and modulate host processes, respectively6,52. We evaluated intracellular +IAA and –IAA RHTIR1-VMP1AID parasites for dense granules by IFA using GRA1 and GRA2 as markers, and by TEM. The IFA images showed GRA1 and GRA2 localization to distinct structures in the PV of -IAA parasites, whereas these proteins had diffuse localization in the PV of +IAA parasites (Fig. 7A; Supplementary Fig. 8A). The number of parasites with diffuse GRA1 or GRA2 localization was significantly higher in +IAA parasites (83.6% for GRA1, 79.0% for GRA2) than that in –IAA parasites (9.6% for GRA1, 14.6% for GRA2) (Fig. 7B; Supplementary Fig. 8B). TEM images showed the absence of tubulo-vesicular IVN in the PV of 57.7% +IAA parasites (n = 123 PVs) as compared with 7.8% –IAA parasites (n = 64 PVs) (Fig. 7C), indicating a crucial role for TgVMP1 in IVN formation.

A The representative Airyscan micrographs of intracellular –IAA and +IAA parasites show localization of the dense granule protein GRA2, TgVMP1AIDHA (VMP1), nucleus (DAPI), and overlap of the three images (Merged). Scale bar = 10 µm, and the boxed GRA2 panel is zoomed-in (Zoom). B The number of PVs showing diffuse GRA2 signal in A is shown as a percentage of the total number of PVs (% PVs with diffuse GRA2) on the y-axis for the indicated parasites on the x-axis. Data are mean ± SD error bar based on ≥ 200 PVs from three independent experiments. C The TEM micrographs show tubulo-vesicular IVN in the PVs of –IAA and +IAA parasites, and labels are as described in Fig. 6A. Scale bar = 1 µm, and the boxed area is zoomed-in (Zoom). The images represent multiple observations (-IAA, n = 64; +IAA, n = 123) in two independent experiments. D The mean signal intensity of GRA2 in purified +IAA parasites is shown as times of that in -IAA parasites (Signal intensity (times of –IAA)) on the y-axis for the indicated parasites on the x-axis. Data are mean signal intensity ± SD error bar based on multiple parasites (-IAA, n = 338; +IAA, n = 383) from three independent experiments. E The representative Airyscan micrographs show a merged image of GRA2 and DAPI panels for –IAA and +IAA parasites. Scale bar = 5 µm, and arrowhead indicates perinuclear accumulation of GRA2 in +IAA parasites. F The number of parasites showing perinuclear accumulation of GRA2 in E is shown as a percentage of the total number of parasites (% Perinuclear accumulation) on the y-axis for the indicated parasites on the x-axis. Data are mean ± SD error bar based on multiple parasites (-IAA, n = 338; +IAA, n = 383) from three independent experiments. G The representative Airyscan micrographs show localization of GRA2, ER-GFP, DAPI, and overlap of the three images (Merged) in the indicated parasites. Scale bar = 5 µm. Source data are provided as a Source Data file.
For dense granule biogenesis, purified +IAA and –IAA tachyzoites were processed for IFA and compared for the levels of intracellular GRA1 and GRA2. +IAA parasites showed higher levels of these proteins than –IAA parasites (Fig. 7D; Supplementary Fig. 8C). -IAA RHTIR1-VMP1AID parasites showed distinct GRA1- or GRA2-labeled dense granules in the cytoplasm (Fig. 7E; Supplementary Fig. 8D). On the other hand, in addition to the cytoplasmic GRA1 and GRA2-labeled dense granules, +IAA RHTIR1-VMP1AID parasites also showed perinuclear accumulation of GRA1 and GRA2. The perinuclear accumulation of GRA1 or GRA2 was significantly higher in +IAA parasites than that in –IAA parasites (Fig. 7F; Supplementary Fig. 8E), and both GRA1 and GRA2 co-localized with ER-GFP in +IAA parasites (Fig. 7G; Supplementary Fig. 8F), which indicated a key role of TgVMP1 in dense granule biogenesis. TEM micrographs of both –IAA and +IAA parasites showed dense granules (Figs. 6A and 7C), which was corroborated by IEM showing GRA2 labeling of dense granules in the parasite cytoplasm and IVN in the PV (Supplementary Fig. 7C). The TEM micrographs of +IAA parasites contained less number of cytoplasmic dense granules (1.5/parasite section, n = 153) than those of –IAA parasites (3.2/parasite section, n = 94), which together with the IFA data indicate a crucial role of TgVMP1 in dense granule biogenesis. We next compared dense granule secretion in the ESA of purified –IAA and +IAA parasites by IEM using antibodies to GRA2. The number of GRA2-gold particles was comparable (Supplementary Fig. 7D, E), suggesting that impaired dense granule biogenesis in TgVMP1-depleted parasites did not affect secretion. Nonetheless, the majority of the gold particles were associated with electron-dense material in –IAA parasites as opposed to +IAA parasites (Supplementary Fig. 7D), suggesting a defect in the post-secretion process that might be required for the formation of IVN.
TgVMP1-depletion altered LD homeostasis
The DedA superfamily proteins are proposed to be lipid scramblases; human VMP1 and TMEM41B have been shown to have roles in lipoprotein and lipid transport, LD biogenesis, and lipid distribution22,23,24. This prompted us to evaluate the effect of TgVMP1 depletion on lipid homeostasis using LDs as a marker. LDs contain neutral lipids such as triglycerides and cholesterol esters, are formed in the ER bilayer, and bud off into the cytoplasm55. We stained purified +IAA and –IAA RHTIR1-VMP1AID tachyzoites with Nile red to label LDs56, and observed the parasites by live-cell confocal microscopy. +IAA parasites had a significant reduction in the number and increase in the size of LDs as compared with -IAA parasites (Fig. 8A–C). TEM micrographs confirmed the presence of much larger-sized LDs in +IAA parasites than in -IAA parasites (Fig. 8D), indicating a key role for TgVMP1 in LD homeostasis. Additionally, LDs were closely associated with the ER membrane in +IAA parasites, which was not evident in –IAA parasites. Although we attempted to determine membrane lipid distribution in TgVMP1-depleted parasites using click lipids and fluorophores, we did not succeed due to widespread non-specific labeling of the parasite by click fluorophores. Further optimization of the use of click lipids or alternative approaches is required to determine if TgVMP1-depletion affects membrane lipid distribution.

A The representative confocal micrographs of purified +IAA and –IAA RHTIR1-VMP1AID parasites show Nile red-stained LDs (LD), bright field (BF), and overlap of the two images (Merged). Scale bar = 5 µm. B The number of LDs/parasite in A is shown on the y-axis for the indicated parasites on the x-axis. Data are mean ± SD error bar based on multiple parasites (-IAA, n = 137; +IAA, n = 139) from two independent experiments. C The size of each LD (μm2) in A is shown on the y-axis for the indicated parasites on the x-axis. Data are mean ± SD error bar based on multiple parasites (-IAA, n = 471; +IAA, n = 232) from two independent experiments. D The TEM micrographs of +IAA and -IAA parasites show LDs and other indicated structures as described in Fig. 6A. The boxed area is zoomed-in (Zoom), and shows LDs closely associated with the ER membrane (open arrowheads) in +IAA parasites. Scale bar = 500 nm. The micrographs represent multiple observations (-IAA, n = 119; +IAA, n = 109) from two independent experiments. E The representative confocal images of intracellular -IAA and +IAA RHTIR-VMP1AID::BiP-GFP-HDEL parasites show signal for ER-GFP, nucleus (DAPI), and overlap of the two panels (Merged). Arrowheads indicate perinuclear clustering of ER-GFP, and scale bar = 5 µm. F The number of parasites with perinuclear clustering of ER-GFP in E is shown as a percentage of the total number of parasites observed (% Parasites with perinuclear ER clustering) on the y-axis for the indicated parasites on the x-axis. Data are mean ± SD error bar based on multiple parasites (-IAA, n = 2058; +IAA, n = 1136) from three independent experiments. G The TEM micrographs of –IAA and +IAA parasites show ER and other indicated structures as described in Fig. 6A. The boxed area is zoomed in (Zoom), and scale bar = 1 µm. The micrographs represent multiple observations (-IAA, n = 118; +IAA, n = 108) from two independent experiments. Source data are provided as a Source Data file.
TgVMP1 depletion disrupted ER organization
The microneme, rhoptry, and dense granule proteins have been shown to be synthesized in ER, and vesicles containing these proteins bud off the ER/Golgi regions and follow the endosomal vesicular trafficking pathway to reach/mature into these organelles11. Perinuclear clustering of dense granule proteins GRA1 and GRA2 with ER-GFP in TgVMP1-depleted parasites prompted us to investigate whether TgVMP1 depletion compromised the ER. We compared the localization of ER-GFP in intracellular -IAA and +IAA RHTIR-VMP1AID::BiP-GFP-HDEL parasites by IFA and TEM. –IAA parasites showed uniform ER-GFP localization around the nucleus, which was also extended toward the apical region (Fig. 8E, F). On the other hand, ER-GFP was prominently clustered around the nucleus in a significant number of +IAA parasites (Fig. 8E, F). TEM micrographs showed ER tubules stretched throughout the +IAA parasites, including in close contact with LDs, whereas these were mostly perinuclear in –IAA parasites (Fig. 8G). These data indicate impaired ER organization, which might contribute to various phenotypes observed in TgVMP1-depleted parasites. These data highlight a key role of TgVMP1 in the ER functioning, which could include the maintenance of ER structure, formation of functional subdomains, and communication with other organelles.
TgVMP1 interactome supports its roles in the secretory organelle function and biogenesis
To understand how TgVMP1 depletion might impair the function and biogenesis of secretory organelles, we determined and analyzed the TgVMP1 interactome (Supplementary Fig. 9; Supplementary Table 2). TgVMP1 interactome contained proteins that have been shown to localize to micronemes (apical cap protein AC5 and MIC7), rhoptries (RON3, ROP8, ROP26, and ROP37), and dense granules (phosphatidylserine decarboxylase, cathepsin CPC2, and GRA12). The interactome also contained the proteins associated with gliding motility (PhiL1, GAPM3, GAPM2B, GAPM1A, and GRA12), processing of the microneme and rhoptry proteins (V-type proton ATPase subunit a1), and vesicle transport (sortilin-like receptor, Rab5b, glutathione S-transferase 2, and Rab2). Additionally, the TgVMP1 interactome contained a large number of proteins related to protein biosynthesis, cytoskeleton, maintenance of the parasite shape, and virulence. Among these proteins, the sortilin-like receptor (TgSORTLR) and V-type proton ATPase are specifically relevant in the context of secretory organelles. TgSORTLR is required for trafficking of the microneme and rhoptries proteins, and TgSORTLR knockdown impairs the biogenesis of these organelles57. The V-type proton ATPase is localized to the plasma membrane and endosomal compartments, and is required for the processing of microneme and rhoptry proteins during their transport through the endosomal compartments58. Furthermore, the V-type proton ATPase is also required for activation of the proteases that process microneme and rhoptry proteins. The presence of proteins localized to/associated with micronemes, rhoptries, dense granules and the motor complex in the TgVMP1 interactome suggests a direct or indirect role of TgVMP1 in the functioning and biogenesis of these organelles, which is in agreement with the defects observed in these organelles upon TgVMP1 depletion. The presence of several motility-associated proteins in the TgVMP1 interactome suggests an alternative mechanism by which TgVMP1 could contribute to parasite motility; however, this requires further investigation.
TgVMP1 and PfVMP1 are lipid scramblases, and restoration of the ER-localized scramblase activity rescued TgVMP1-depleted parasites
TgVMP1-depleted parasites showed dense granule accumulation in the ER, impaired LD homeostasis, and disrupted ER organization, suggesting a role for TgVMP1 in ER and ER-organelle contact sites. The ER-localized DedA superfamily proteins HsVMP1 and HsTMEM41B have been shown to have lipid scramblase activity, which has been proposed to be required for lipoprotein and lipid transport, LD biogenesis, lipid distribution, and the formation of viral replication organelles22,23,26,35. TgVMP1, PfVMP1, and HsVMP1 share similar domain architecture and AlphaFold structure, which prompted us to ask whether TgVMP1 and PfVMP1 function as lipid scramblases and restoration of the ER-localized scramblase activity in RHTIR1-VMP1AID parasites could rescue the growth defects.
Lipid scramblases translocate lipids between two membrane leaflets, which can be measured by monitoring the fluorescence signal intensity of fluorescent phospholipid (NBD-PS)-containing liposomes in the presence or absence of a membrane impermeable quencher (dithionite). The fluorescence signal of NBD-PS on the outer liposome leaflet would be quenched by dithionite, causing ~50% decrease in fluorescence intensity (Fig. 9A). On the other hand, translocation of NBD-PS between the outer and inner leaflets of the liposome by a lipid scramblase would result in the »50% decrease in fluorescence intensity (Fig. 9A). Addition of Triton X-100 would permeabilize the liposomes, allowing dithionite to enter and quench the NBD-PS fluorescence completely (Fig. 9A). For lipid scramblase activity, PfVMP1AIDHA and TgVMP1AIDHA were purified from PfVMP1AID and RHTIR1-VMP1AID parasites, respectively (Supplementary Fig. 10A). Purified TgVMP1AIDHA or PfVMP1AIDHA was incorporated into NBD-PS-containing liposomes to obtain TgVMP1AIDHA-liposomes or PfVMP1AIDHA-liposomes. Only NBD-PS-containing liposomes (empty-liposomes) were used as a control. Indeed, TgVMP1-liposomes and PfVMP1-liposomes showed »50% decrease in fluorescence intensity upon addition of dithionite as compared with ~50% decrease in fluorescence intensity of empty-liposomes (Fig. 9B), indicating that TgVMP1 and PfVMP1 are lipid scramblases.

A The schematic shows empty-liposomes and scramblase-liposomes with the indicated constituents. Addition of dithionite, a membrane impermeable quencher, would decrease the fluorescence of NBD-PS in the outer liposome leaflet, causing a ~50% decrease in the fluorescence signal intensity of the empty-liposomes. On the other hand, continuous translocation of NBD-PS by scramblase between outer and inner leaflets of the scramblase-liposomes would cause a »50% decrease in fluorescence signal intensity. The addition of Triton X-100 would make the liposome membrane permeable to dithionite, causing a decrease in fluorescence signal intensity to almost zero. B The plot shows relative fluorescence intensities (Ft/F0) of empty-liposomes, TgVMP1AIDHA-containing liposomes (TgVMP1-liposomes), and PfVMP1AIDHA-containing liposomes (PfVMP1-liposomes) before, after addition of dithionite, and after addition of Triton X-100. Arrows indicate the time points at which dithionite and Triton X-100 were added. Data are the average of three independent scramblase assays. C +IAA and –IAA intracellular RHTIR1-VMP1AID (TgVMP1KD), RHTIR1-VMP1AID/PfVMP1 (PfVMP1COM), and RHTIR1-VMP1AID/HsVMP1 (HsVMP1COM) parasites were stained for the PV marker GRA1. The number of PVs containing different numbers of parasites is shown as a percentage of the total number of PVs (% Vacuoles) on the y-axis for the indicated parasites on the x-axis. Data are mean ± SD error bar based on ≥100 PVs in each of three independent experiments. The p-values are for the PVs containing a single parasite only. D Purified +IAA and -IAA tachyzoites were evaluated for dense granule biogenesis defect using antibodies to GRA2. The Airyscan micrographs show signal for GRA2, nuclear stain (DAPI), and overlap of the two panels (Merged) for the indicated parasites. Scale bar = 5 µm. The arrowheads indicate perinuclear accumulation of GRA2 in +IAA TgVMP1KD, but not in +IAA PfVMP1COM and +IAA HsVMP1COM parasites, indicating restoration of dense granule biogenesis by PfVMP1 and HsVMP1, respectively. E The percentage of parasites showing perinuclear accumulation of GRA2 in D (% Perinuclear accumulation) is shown on the y-axis for the indicated parasites on the x-axis. Data are mean ± SD error bar based on ≥100 parasites in each of the two independent experiments. Source data are provided as a Source Data file.
For restoration of the ER-localized scramblase activity in RHTIR1-VMP1AID parasites, we ectopically expressed Myc-tagged PfVMP1 (PfVMP1Myc) and HsVMP1 (HsVMP1Myc) proteins in RHTIR1-VMP1AID parasites to obtain complemented parasites (RHTIR1-VMP1AID/PfVMP1Myc and RHTIR1-VMP1AID/HsVMP1Myc, respectively). We next compared the complemented parasites with RHTIR1-VMP1AID parasites for overall growth and intracellular development in the presence (+IAA) or absence (-IAA) of IAA. The complemented parasites expressed PfVMP1Myc or HsVMP1Myc (Supplementary Fig. 10B), which co-localized with TgVMP1AIDHA (Supplementary Fig. 10C), indicating that these proteins retain ER localization, even in a heterologous system. As expected, all three parasites formed plaques under –IAA conditions, whereas only complemented parasites, but not RHTIR1-VMP1AID parasites, formed plaques under +IAA conditions (Supplementary Fig. 10D), which indicated restoration of the overall growth by PfVMP1 or HsVMP1 in the absence of TgVMP1. 90.7% of the +IAA RHTIR1-VMP1AID parasites were arrested at the single parasite/PV stage as compared to 27.5% (for HsVMP1) and 56.1% (for PfVMP1) of the +IAA complemented parasites (Fig. 9C), indicating restoration of intracellular development by PfVMP1 or HsVMP1 in the absence of TgVMP1. We next checked if PfVMP1 and HsVMP1 could reverse the perinuclear accumulation of the dense granule protein GRA2 in TgVMP1-depleted parasites. <5.7% of the +IAA complemented parasites showed perinuclear accumulation of GRA2 as compared with 44.7% of the +IAA RHTIR1-VMP1AID parasites (Fig. 9D, E), indicating rescue of the dense granule biogenesis defect. The rescue of growth and dense granule biogenesis defects of TgVMP1-depleted parasites by PfVMP1 and HsVMP1 indicates their functional conservation. This provides a mechanistic proof for a critical role of the ER-localized scramblase activity in the biogenesis and function of apicomplexan secretory organelles.
Discussion
Micronemes, rhoptries, and dense granules are specialized secretory organelles in apicomplexan parasites, and have essential roles in parasite motility, invasion of the host cells, development, and virulence2,3,4,6,52. ER is the site for synthesis of the proteins and biogenesis of the vesicles destined to these organelles, which would require lipid mobilization. Since DedA superfamily proteins are implicated in lipid distribution and organelle biogenesis13,14, we identified and investigated the P. falciparum and T. gondii DedA superfamily protein VMP1 towards understanding its role in the biogenesis and function of secretory organelles. We show that PfVMP1 and TgVMP1 are ER-resident lipid scramblases. TgVMP1 is crucial for the biogenesis of rhoptries and dense granules, and secretion of micronemes and rhoptries (Supplementary Fig. 13). Notably, restoration of the ER-resident scramblase activity by complementing TgVMP1-depleted parasites with PfVMP1 or HsVMP1 alleviated the growth and dense granule biogenesis defects caused by TgVMP1 depletion, indicating their functional conservation and supporting a crucial role of the ER-localized scramblase activity in the biogenesis and function of apicomplexan secretory organelles.
PfVMP1 and TgVMP1 proteins share the DedA superfamily domain organization, are structurally similar to HsVMP1, and localize to the ER, indicating that the predicted PfVMP1 and TgVMP1 proteins belong to the DedA superfamily. Although the ER-retention/retrieval signals are yet to be identified for VMP1 proteins, their C-terminal cytoplasmic tails contain one or more “di-Lys” motif or its variants (Supplementary Fig. 1D), which might mediate ER localization of VMP1 proteins, as has been reported for several ER-membrane proteins59.
TgVMP1 depletion decreased parasite motility, host cell invasion, egress, and intracellular development, indicating a crucial role for it in these processes. TgVMP1 depletion caused decreased microneme secretion, impaired rhoptry formation and secretion, and impaired dense granule formation, which indicated that TgVMP1 is critical for the biogenesis and function of these secretory organelles. As micronemes, rhoptries, and dense granules are essential for the parasite motility, invasion, egress, and intracellular development2,3,4,5,6,7,8,9,10,52, the phenotypic defects of TgVMP1-depleted parasites could be directly attributed to the impaired biogenesis and functions of these secretory organelles.
TgVMP1 depletion did not affect overall microneme organization, but decreased microneme secretion. Microneme secretion is induced by a signaling cascade of cyclic guanosine monophosphate (cGMP), cytosolic Ca2+, and phosphatidic acid60,61. +IAA RHTIR1-VMP1AID parasites showed elevated Ca2+ level as compared with -IAA RHTIR1-VMP1AID parasites (Supplementary Fig. 11), which would enhance microneme secretion. On the contrary, TgVMP1 depletion decreased MIC2 secretion and also impaired MIC2 processing, which has been shown to be crucial for the secretion of MIC2 and several other microneme proteins54,62. This suggests that TgVMP1 depletion adversely affected the MIC2-processing proteases or the processing environment, thereby decreasing microneme secretion. TgVMP1 interactome contained V-type proton ATPase, which causes acidification of endosomal-like compartments wherein microneme and rhoptry proteins are processed during their transport to the destined organelles58, suggesting that TgVMP1 regulates microneme secretion via interaction with V-type proton ATPase.
Rhoptry proteins are synthesized in the ER and transported in secretory vesicles arising from the trans-Golgi network (TGN) to precursor structures that undergo maturation during transport through endosomal-like compartments to form rhoptries4,63. Rhoptry discharge process is not clear, except that microneme secretion is one of the triggers64,65, and decreased microneme secretion in TgVMP1-depleted parasites could be a reason for reduced rhoptry secretion. TgVMP1-depleted parasites had none or fragmented rhoptries, which could be responsible for decreased e-vacuole secretion during invasion. TgVMP1 interactome contained multiple proteins involved in ER-Golgi endosomal transport, including TgSORTLR, which has been shown to be required for the trafficking of microneme and rhoptry proteins57. V-type proton ATPase, which has been reported to be critical for the maturation of microneme and rhoptry proteins, was also present in the TgVMP1 interactome58. In fact, the loss of apical rhoptries and secretion of ROP1 in the PV of TgVMP1-depleted parasites are similar to those observed in parasites with ablation of TgSORTLR, V-type proton ATPase or Vps1157,58,66. It is possible that TgVMP1 contributes to rhoptry biogenesis via interaction with TgSORTLR and V-type proton ATPase, and loss of this interaction in TgVMP1-depleted parasites could have adversely affected rhoptry biogenesis.
Dense granules secrete GRA proteins into the PV, which contribute to the formation of IVN within the PV and host cell remodeling6,9,10. Most of the characterized GRA proteins are synthesized in the ER, transported through the endo-membrane pathway to the TGN, wherein these proteins assemble into aggregates, forming dense granules6. TgVMP1-depleted parasites contained less number of dense granules and also showed accumulation of dense granule proteins in the ER. Impaired ER organization in TgVMP1-depleted parasites might have affected the transport and assembly of dense granule proteins during transport through the ER/Golgi endo-membrane system, thereby causing their accumulation in the ER. Although fewer, cytoplasmic dense granules in TgVMP1-depleted parasites showed normal secretion. While the mechanism by which TgVMP1 contributes to dense granule biogenesis is not clear at present, the presence of several dense granule proteins (phosphatidylserine decarboxylase, cathepsin CPC2, and GRA12) in the TgVMP1 interactome suggests their interaction with TgVMP1, which could facilitate the biogenesis of dense granules. Both control and TgVMP1-depleted parasites secreted comparable GRA2 in the ESA. However, in contrast to TgVMP1-depleted parasites, the majority of the GRA2 was associated with electron-dense material in the ESA of control parasites, which resembled the GRA2-labeled IVN in IEM micrographs and could be membranous structures. This suggests a defect in the form of secreted-GRA2 or post-secretion assembly of GRA2 that might be necessary for IVN formation, and might be a reason for the absence of IVN in the majority of TgVMP1-depleted parasites. Additionally, as IVN formation requires lipids from the host as a primary source and parasite as a secondary source9,67, impaired lipid homeostasis in TgVMP1-depleted parasites could have limited lipid supply to the PV, thereby, adversely affecting IVN formation and parasite growth.
Different effects of TgVMP1 depletion on micronemes, rhoptries, and dense granules could be due to differences in the pathways leading to their biogenesis and secretion3,4,6,50,68,69. It is possible that TgVMP1 directly/indirectly regulates the proteins in the ER/Golgi that are involved at different stages of transport/maturation of the proteins destined to these organelles, which is also supported by the presence of several proteins in TgVMP1 interactome that regulate biogenesis of secretory organelles at different stages. Similar observations have been reported earlier for TgSORTLR and V-type proton ATPase, which are required for trafficking of microneme and rhoptry proteins57,58, but not for dense granule proteins. Similarly, depletion of apical annuli-associated proteins has been shown to have different effects on dense granule secretion and plasma membrane proteins68,69.
Lipid scramblase activity of TgVMP1 and PfVMP1 and rescue of the TgVMP1-depleted parasites by PfVMP1 or a known ER-localized lipid scramblase as distant as HsVMP1 indicated that ER-resident lipid scramblase activity is crucial for parasite development, which likely functions via translocation of lipids in the ER and the precursor vesicles destined to the secretory organelles. The accumulation of enlarged LDs in close association with the ER membrane in TgVMP1-depleted parasites indicates impaired LD homeostasis, which, together with impaired secretory organelle biogenesis, could have contributed to the overall block of parasite growth. LD enlargement occurs due to fusion of LDs when there is a shortage of phosphatidylcholine transport during LD biogenesis in the ER70, suggesting that the loss of lipid scramblase activity could be a reason for enlarged LDs in TgVMP1-depleted parasites. This further indicates that the loss of ER-localized lipid scramblase activity is the major reason for impaired secretory organelle biogenesis, ER organization, LD homeostasis, and IVN formation in TgVMP1-depleted parasites, consequently blocking parasite development. Since TgVMP1 depletion did not affect trafficking of the proteins destined to micronemes, parasite plasma membrane, and IMC, TgVMP1 may contribute to the formation of specific ER subdomains wherefrom precursor rhoptry and dense granule vesicles arise. This proposal is in agreement with the reported role of HsVMP1 in the formation of ER microdomains that make contacts with diverse organelles34.
HsVMP1 recruits the PI3K complex (VPS34/Beclin 1/Vps15/Atg14L) at the phagophore assembly site via interaction with Beclin 1, which is crucial for autophagosome formation28,29. A striking consequence of the ablation of the autophagy pathway in T. gondii and P. falciparum has been shown to be the loss of apicoplast, a relict plastid that produces fatty acid precursors71. We compared TgVMP1-depleted and control parasites for the presence of apicoplast as a marker to assess whether TgVMP1 depletion affected autophagy. The proportion of apicoplast-containing TgVMP1-depleted and control parasites was comparable (Supplementary Fig. 12), suggesting that the observed phenotypes of TgVMP1-depleted parasites are not associated with autophagy-related defects.
Altogether, we demonstrate that TgVMP1 and PfVMP1 are ER-resident lipid scramblases, which are crucial for the biogenesis and function of apicomplexan secretory organelles, most likely via translocation of lipids in the ER wherefrom vesicles destined to the secretory organelles arise (Supplementary Fig. 13). Functional conservation of TgVMP1, PfVMP1, and HsVMP1 hints to a fundamental function of this family of proteins in eukaryotes. Since DedA superfamily proteins are present in all life forms, it would be exciting to investigate whether lipid scramblase activity is a general feature of this superfamily and how it mediates specialized functions depending on the subcellular location.
Methods
Materials
All the routinely used biochemicals were of molecular biology or cell culture grade, and were purchased from Thermo Fisher Scientific, Sigma-Aldrich, MP Biomedicals or Serva unless otherwise stated. Cell culture media and supplements, blasticidin S-hydrochloride, gentamicin, penicillin-streptomycin, and Giemsa stain were from Lonza and Thermo Fisher Scientific. Lipids (POPC, POPS, and NBD-PS) were from Avanti Polar Lipids. Dithionite was from Sigma-Aldrich. All the plasticware for cell culture was procured from standard manufacturers, including Thermo Fisher Scientific, Corning Inc., Nalgene, TPP, and Tarsons. Restriction enzymes and DNA-modifying enzymes were from New England Biolabs and Thermo Fisher Scientific. DNA and plasmid isolation kits were from QIAGEN and MACHERY-NAGEL. PrimeScript 1st strand cDNA synthesis kit was from Takara Bio. ProLong Diamond Antifade Mountant, DAPI, Hoechst, ER-Tracker Red stain (BODIPY TR Glibenclamide), and SuperSignal Chemiluminescent substrates were from Thermo Fisher Scientific. Trimethoprim, N-acetylglucosamine, indole-3-acetic acid (IAA), thapsigargin, xanthine, mycophenolic acid, calcium ionophore A23187, formaldehyde and Nile red were purchased from Merck. Ionomycin was from Tocris Bioscience. 5Ph-IAA was from MedChemExpress. Pyrimethamine was from MP Biomedicals. The protease inhibitor cocktail was from Roche. Glutaraldehyde, formaldehyde, osmium tetroxide, and sodium cacodylate were from Electron Microscopy Sciences. Uranyl acetate was from Lobachemie, and epoxy resin components were from TED Pella. Antibodies were from Thermo Fisher Scientific, Cell Signalling Technology, Santa Cruz Biotechnology or Jackson ImmunoResearch. GFP-Trap and HA-Trap antibodies were from ChromoTek. Pierce protein A/G magnetic beads were from Thermo Fisher Scientific. Human Foreskin Fibroblasts (HFF) were from ATCC, P. falciparum 3D7 and D10 strains, T. gondii RH TIR1-3FLAG strain, and antibodies to T. gondii proteins (AMA1, MIC2, MIC3, ROP1, GRA1, GRA2, and SAG1) were procured from the Biodefense and Emerging Infections Research Resources Repository (BEI Resources). WR99210 was a kind gift from David Jacobus (Jacobus Pharmaceutical, Princeton, U.S.A.). Rabbit anti-IMC1 and anti-ACP polyclonal antibodies were a kind gift from Abhijit S. Deshmukh (NIAB, India) and Dhanasekaran Shanmugam (NCL, India), respectively. Rabbit anti-TIR1 antibody was from MYBIOSOURCE. Gold-conjugated goat-anti-mouse IgG was from Electron Microscopy Sciences. The pTKO-HXGPRT plasmid was a kind gift from Dr Nishit Gupta (BITS, India). pTUB1:YFP-mAID-3HA-DHFR-TS:HXGPRT (Addgene#87259) and pSAG1::Cas9-U6::sgUPRT (Addgene #54467) plasmids were a kind gift from Dr. David Sibley. pCTG-Fluc (originally sourced from Dr. Shobhona Sharma, TIFR, India) and pARL1a-PfGRASP-GFP (originally sourced from Dr. Lilach Sheiner, University of Glasgow, UK) were kind gifts from Dr. Swati Patankar (IIT Bombay, India).
Human blood was collected from healthy volunteers after obtaining informed consent according to the approved protocol of the Institutional Ethics Committee (IEC-103/2023, IEC-38/2015, IEC-38-R1/2015, IEC-38-R2/2015 and IEC-38-R3/2015) of Centre for Cellular and Molecular Biology, India. P. falciparum and T. gondii cultures and related experiments were carried out according to the approved protocols of the Institutional Biosafety Committee of Centre for Cellular and Molecular Biology, India. Animals were housed in cabin-type isolators at room temperature (22-25 °C, 40–70% humidity, and 12/12 h dark/light photoperiod), and all the experiments on animals were performed according to the approved protocols (IAEC 09/2023) of the Institutional Animal Ethics Committees (IAEC) of Center for Cellular and Molecular Biology, India. All the studies were carried out as per the Declaration of Helsinki principles.
Sequence analysis
The amino acid sequences of VMP1 proteins of model organisms (Homo sapiens, ID: Q96GC9; Drosophila melanogaster, ID: Q9W2S1; Caenorhabditis elegans, ID: Q9XWU8; Arabidopsis thaliana, ID: Q5XF36 (KSM1) and F4I8Q7 (KSM2); Chlamydomonas reinhardtii, ID: A0A2K3DNE8; Dictyostelium discoideum, ID: Q54NL4; Danio rerio, ID: Q6NYY9), S. cerevisiae TVP38 (ID: P36164), and selected E. coli DedA superfamily proteins (YohD, ID: P33366; YdjZ, ID: P76221; YqaA, ID: P0ADR0) were obtained from the Uniprot database (https://www.uniprot.org/). The amino acid sequences of HsVMP1, ScTVP38, and E. coli DedA superfamily proteins (YohD, YdjZ, YqaA) were used as queries in the BLAST searches of the eukaryotic pathogen database VEupathDB (https://veupathdb.org/) to identify homologs in selected reference apicomplexan parasites, including T. gondii and P. falciparum (Supplementary Table 1). The amino acid sequences of apicomplexan VMP1 proteins were analyzed for the presence of conserved domains using CD BLAST (ncbi.nlm.nih.gov) or MOTIF (genome.jp/tools/motif/), and sequences were aligned using Clustal Omega72.
Parasite culture
P. falciparum 3D7 and D10 strains were maintained in human RBCs (at 2% hematocrit) in RPMI-1640 medium (supplemented with 2 g/l sodium bicarbonate, 2 g/l glucose, 25 mg/ml gentamicin, 300 mg/l L-glutamine, 100 mM hypoxanthine, 0.5% albumax II) under a mixed gas environment (5% CO2, 5% O2 and 90% N2) at 37 °C73,74,75. The synchrony of P. falciparum culture was maintained by treating the cells with 5% sorbitol when the majority of parasites were at the ring stage76. Genomic DNA was isolated from late trophozoite/schizont stage parasites using the NucleoSpin Tissue kit as recommended by the manufacturer.
Human foreskin fibroblast (HFF) cells were maintained in D10 medium (DMEM supplemented with 10% FBS, 2 mM glutamine, 3.7 g/l sodium bicarbonate, 155 mg/l sodium pyruvate, 1% pen-strep) at 37 °C under 5% CO2 environment. T. gondii RH TIR1-3FLAG strain (RHTIR1)77 was grown in confluent HFF cells, as has been reported previously78. Fully confluent HFF cells infected with RHTIR1 tachyzoites were scraped, and the suspension was passed through a 3 µm filter to remove host debris. The filtrate was centrifuged at 800 × g for 8 min, washed with incomplete DMEM medium (DMEM without antibiotics and FBS), and the purified parasite pellet was resuspended in an appropriate buffer as has been described in the following sections.
Genomic DNA (gDNA) was isolated from purified T. gondii RHTIR1 tachyzoites using the NucleoSpin Tissue kit following the manufacturer’s instructions. For T. gondii cDNA preparation, total RNA was isolated from the purified RHTIR1 T. gondii tachyzoites using the NucleoSpin RNA isolation kit according to the manufacturer’s instructions. 5 μg of the DNA-free total RNA was used to prepare cDNA using PrimeScript 1st strand cDNA synthesis kit as per the manufacturer’s instructions.
Generation of recombinant P. falciparum parasites
The primers and synthetic DNAs used for the generation of transfection constructs are listed in Supplementary Table 3. To knock-in GFP immediately downstream of the 3’-PfVMP1 coding sequence, the 3’-coding sequence of PfVMP1 corresponding to 249 amino acid residues was amplified from P. falciparum 3D7 genomic DNA using PfVMP1-F1/PfVMP1-R1 primers, and cloned into the pGEM-T vector to obtain pGEMT-PfVMP1c plasmid. The pGEMT-PfVMP1c plasmid was digested with ApaI/KpnI to excise the PfVMP1c fragment, which was subcloned into similarly digested pGT-GFP plasmid to obtain pGT-PfVMP1c-GFP. The pGT-PfVMP1c-GFP plasmid was digested with ApaI/XhoI to excise the PfVMP1c-GFP insert, which was subcloned into similarly digested pNC vector to obtain pNC-PfVMP1c-GFP transfection plasmid. pNC and pGT-GFP plasmids have been described earlier79,80.
We constructed a dual knockdown plasmid, which would cause glmS-mediated mRNA degradation and mutant E. coli DHFR (cDD)-mediated protein degradation as has been described previously81. The 3’-coding sequence corresponding to 249 amino acid residues (flank1) of the PfVMP1 coding sequence and 1087 bp of PfVMP1 3’-UTR region (flank2) were amplified from P. falciparum 3D7 genomic DNA using PfVMP1-fl1F/PfVMP1-R1 and PfVMP1-Fl2F/PfVMP1-Fl2R primer pairs, respectively. The flank1 and flank2 PCR products were cloned into the HBPFA18/cDDHA/GlmSAc plasmid at NotI/KpnI and AvrII/KasI sites, respectively, to obtain HB-PfVMP1-cDDHA-GlmSAc transfection plasmid. HBPFA18/cDDHA/GlmSAc plasmid has been described earlier82.
We also employed an auxin-inducible knockdown strategy for PfVMP1. We purchased synthetic XTEN-mAID-2×HA (mAIDHA) and PcDT5U/Tir1-Flag-2A/HD/PcDT3’U (TIR1-HD) DNAs, which were cloned into the HBPFA18/cDDHA/GlmSAc at KpnI/XhoI and AgeI/AvrII sites, respectively, to obtain HB-mAIDHA/TIR1-HD plasmid. The mAID-2×HA and TIR1-Flag-2A/HD coding sequences are codon-optimized for P. falciparum, and the TIR1 used here is F74G mutant of OsTIR183, which is inducible with 5Ph-IAA. The above PfVMP1 flank1 and flank2 regions were cloned into the HB-mAIDHA/TIR1-HD plasmid at NotI/KpnI and AvrII/KasI sites, respectively, to obtain the HB-PfVMP1-mAIDHA/TIR1-HD transfection plasmid.
Ring stage P. falciparum D10 (for PfVMP1c-GFP and HB-PfVMP1-cDDHA-GlmSAc plasmids) or 3D7 (for HB-PfVMP1-mAIDHA/TIR1-HD plasmid) parasites were transfected with 100 µg of plasmid DNA by electroporation, and transfected parasites were selected with blasticidin S hydrochloride (at 2 µg/ml for pNC-PfVMP1c-GFP) or WR99210 (at 1 nM for HB-PfVMP1-cDDHA-GlmSAc and HB-PfVMP1-mAIDHA/TIR1-HD) to obtain resistant parasites as has been previously described84,85. Blasticidin-resistant parasites were cloned by dilution cloning to obtain clonal populations. The genomic DNA of each of the clonal parasite lines was isolated, assessed by PCR for plasmid integration using locus-specific primer sets (5’-integration: PfVMP1-5con/GFPSEQR1; 3’-integration: PcDT5U-R1/PfVMP1-3con; wild-type: PfVMP1-5con/PfVMP1-3con; positive control: PfA8fl2-F1/PfA8fl2-R1). WRR99210 resistant parasites were cloned by dilution cloning to obtain clonal lines, genomic DNA was isolated from clonal lines, and assessed for plasmid integration into the target locus by PCR using locus-specific primer sets (HB-PfVMP1-cDDHA-GlmSAc-transfected parasites (5’-integration: PfVMP1-5con/PvAc-Con-R; 3’-integration: Hrp2-SeqF/PfVMP1-3con2; wild type-specific: PfVMP1-5con/ PfVMP1-3con2; positive control: PfA8-F1/PfA8fl2-R1), HB-PfVMP1-mAIDHA/TIR1-HD-transfected parasites (PfVMP1-5con/PfVMP1-3con2; positive control: PfUCH-F/PfUCH-R). The cloned recombinant parasites with PfVMP1c-GFP (PfVMP1GFPKI), PfVMP1/cDDHA-GlmS (PfVMP1dKD) or PfVMP1-mAIDHA/TIR1-HD (PfVMP1AID) locus were used for various experiments.
Generation of recombinant T. gondii parasites
The transfection cassette containing TgVMP1-mAID-3×HA coding sequence and HXGPRT selection cassette (TgVMP1-mAID-3HA/DHFR-TS:HXGPRT) was amplified from the pTUB1:YFP-mAID-3HA, DHFR-TS:HXGPRT plasmid using TgVMP1-Fkd2/TgVMP1-RP primers86. The TgVMP1-Fkd2 primer contained 50 bp homology sequence corresponding to the immediately upstream of the TgVMP1 stop codon and overlapped with the 5’-mAID-3HA sequence. The TgVMP1-RP primer contained 50 bp homology sequence corresponding to 133 bp downstream of the TgVMP1 stop codon. The primer (SgDNA1) corresponding to the 20-nucleotide protospacer sequence in the 3’ UTR of TgVMP1, along with the SgDNARP primer, were used to amplify pSAG1::Cas9-U6::sgUPRT plasmid87, which generated a PCR product with the target guide RNA sequence and Cas9 expression cassette (pSAG1::Cas9-U6::sgTgVMP1). The PCR product was transformed into DH5α cells and sequenced to ensure the correctness of the guide RNA coding sequence.
T. gondii RHTIR1 tachyzoites were transfected with a mixture of TgVMP1-mAID-3HA/DHFR-TS:HXGPRT transfection cassette and pSAG1::Cas9-U6::s-gTgVMP1 plasmid, as has been described previously41. In brief, fully confluent HFF cells infected with RHTIR1 tachyzoites were scraped, tachyzoites were purified using a 3 µm filter to remove host debris, centrifuged at 800 × g for 8 min, and washed in incomplete DMEM medium (DMEM without antibiotics and FBS). 5 × 106 tachyzoites were mixed with 15 µg each of the transfection cassette and plasmid, the volume was adjusted to 400 µl using incomplete medium, transferred to a 2 mm cuvette, and electroporated (at 1500 V, 10 µF capacitance, ∞ resistance). The electroporated parasites were transferred to confluent HFF cells in T25 flasks, grown in drug-free complete medium for 24 h, followed by in selection medium (complete medium with 25 µg/ml mycophenolic acid and 50 µg/ml xanthine) for 48 h. The intracellular parasites were syringe lysed, and 500 µl of the lysed suspension was added to fully confluent HFF cells in a T25 flask, and grown in the selection medium. Resistant parasites were diluted to achieve 0.5 parasite/well in a 96-well tissue culture plate containing HFF monolayer, and allowed to grow until parasites appeared. These parasites were screened by PCR for the presence of the integration locus using TgVMP1-5con/TgVMP1-3con primer set, along with the TgVP1-5con/TgVP1-3con primer set for the TgVP1 gene as a positive control. Parasites showing the absence of wild-type TgVMP1 and presence of the integration locus were checked for expression of the TgVMP1-mAID-3×HA fusion protein (TgVMP1AIDHA) by western blotting using anti-HA antibodies. Pure clonal recombinant parasites (RHTIR1-VMP1AID) were used for further studies.
Expression of ER and Golgi reporters in RHTIR1-VMP1AID parasites
Tubulin (Tub) promoter region was amplified from RHTIR1 gDNA using Tub F/Tub R primers by PCR, and cloned into pMyc3x.LIC-DHFR plasmid at PacI/EcoRV sites to obtain pTub-DHFR plasmid. The N-terminal signal sequence (MTAAKKLSLFSLAALFCLLSVATLRPVAASD) and C-terminal ER-retention signal (HDEL) of T. gondii chaperonin BiP (TGRH88_051090) were added to the N-terminal and C-terminal, respectively, of GFP by PCR to generate the ER reporter BiP-GFP-HDEL. In brief, the BiP N-terminal signal sequence was amplified from T. gondii gDNA using BipSig-F/BipSig-GFP-Rrec primers. The GFP coding sequence was amplified from the pTKO-HXGPRT plasmid using GFP-Bip-Frec/GFP-HDEL-R primers. The two PCR fragments were recombined by PCR using primers BipSig-F/GFP-HDEL-R to obtain BiP-GFP-HDEL (ER-GFP) coding sequence, which was cloned at EcoRV/AvrII sites in pTub-DHFR plasmid to obtain pTub-ER-GFP plasmid. 15 µg of this plasmid was transfected into RHTIR1-VMP1AID parasites to obtain RHTIR1-VMP1AID::ER-GFP parasites as has been described above in the “Generation of recombinant T. gondii parasites” section.
For the expression of Golgi-reporter PfGRASP-GFP, which has been used as a Golgi-reporter previously42, 15 µg of the pARL1a-PfGRASP-GFP plasmid was transfected into RHTIR1-VMP1AID tachyzoites to obtain RHTIR1-VMP1AID::GRASP-GFP parasites as has been described in the “Generation of recombinant T. gondii parasites” section.
VMP1 expression
Cloned PfVMP1GFPKI parasites, which would express PfVMP1GFP fusion protein, and wild-type P. falciparum D10 parasites were cultured and synchronized as described in the “Parasite culture” section. Synchronized PfVMP1GFPKI parasites were harvested at ring, early trophozoite, late trophozoite, and schizont stages, purified by saponin lysis, and parasite pellets were resuspended in 6× pellet volume of 10 mM Tris (pH 7.4). The suspension was mixed with 1/3rd volume of 4 × SDS-PAGE sample buffer (1× buffer has 50 mM Tris-HCl, 10% glycerol (v/v), 2% SDS, 2.5% β-mercaptoethanol (v/v), 0.05% bromophenol blue (w/v), pH 6.8), heated at 99 °C for 10 min, centrifuged at 20,000 × g for 20 min, and supernatants were processed for western blotting. In brief, the lysates were run in a 12% SDS-PAGE gel, transferred to an Immobilon-P-PVDF membrane, incubated with blocking buffer (TBS-T (10 mM Tris, 150 mM NaCl, 0.1% Tween-20, pH 7.4) with 3% fat-free milk powder), probed using rabbit anti-GFP monoclonal antibodies, followed by goat anti-rabbit-HRP-conjugated secondary antibodies (at 1:20,000 dilution). The membrane was washed, and the signal was developed using SuperSignal chemiluminescent substrate. For loading control, the membrane was stripped off, blocked, incubated with mouse anti-β-actin-HRP-conjugated antibodies (at 1:30,000 dilution), and the signal was developed using SuperSignal chemiluminescent substrate.
RHTIR1 and RHTIR1-VMP1AID parasites were cultured in confluent HFF cells for 24 h post-invasion, as has been described in the “Parasite culture” section. Infected HFF cells were syringe lysed to release parasites, the lysate was passed through a 3 µm filter, and the filtrate containing parasites was centrifuged at 800 × g. The parasite pellet was resuspended in 6× pellet volume of 10 mM Tris (pH 7.4), the suspension was mixed with 1/3rd volume of 4 × SDS-PAGE sample buffer, heated at 99 °C for 10 min, centrifuged at 20,000 × g for 20 min, and supernatants were processed for western blotting as has been described above for PfVMP1GFPKI parasites. The membranes were incubated with rabbit anti-HA monoclonal antibodies (for TgVMP1AIDHA, 1:1000 dilution), followed by goat anti-rabbit-HRP-conjugated secondary antibodies (at 1:20,000 dilution). For loading control, the blot was stripped, blocked, and incubated with mouse anti-ROP1 antibodies (at 1:1000 dilution) for 2 h, followed by HRP-conjugated goat anti-mouse antibodies.
To assess for TgVMP1 depletion, fully confluent HFF cells in T25 flasks were infected with the RHTIR1-VMP1AID parasites and allowed to grow for 24 h. Parasites were subjected to grow in the presence of 500 μM auxin (+IAA) for 0, 15, 30, 60, 120 and 180 min, respectively. Following this, cells were harvested by scraping the parasite-infected HFF cells and subjecting them to centrifugation at 800 × g. The obtained pellets were washed with 1 × PBS and centrifuged again at 800 × g. The resultant pellets were processed for western blotting as described above. TgVMP1AIDHA levels were determined by probing the blots using rabbit anti-HA antibodies, followed by goat anti-rabbit-HRP-conjugated secondary antibodies, as described above. Mouse anti-ROP1 and anti-SAG1 antibodies (at 1:1000 dilution), followed by anti-mouse-HRP antibodies (at 1:20,000 dilution) were used to probe ROP1 and SAG1 as the loading controls.
VMP1 localization
To determine the subcellular localization of PfVMP1GFP, mixed-stage PfVMP1GFPKI parasites were washed with PBS, stained with Hoechst (1 µg/ml in PBS), washed with PBS, immobilized on a poly-L-lysine-coated slide, washed again to remove unattached cells, and covered with a glass coverslip. Live parasites were observed under the 100× objective of Zeiss Axioimager.Z2 fluorescence microscope, images were acquired using AxioVision software, and analyzed using ImageJ software. For co-localization, trophozoite stage PfVMP1GFPKI parasites were grown in complete medium containing the ER-Tracker Red stain (200 nM) for 20 min at 37 °C, Hoechst (1 µg/ml in complete medium) was added to the culture, and the culture was incubated for 10 min. The cells were washed with PBS, immobilized on a poly-L-lysine-coated slide, washed again to remove unattached cells, and then covered with a glass coverslip. Images of live cells were captured using the 100× objective of the Olympus FV300 confocal laser scanning microscope. The images were analyzed for co-localization, and Pearson’s correlation coefficient was determined using the JACoP plugin in Fiji.
For localization of TgVMP1AIDHA, RHTIR1-VMP1AID parasites were grown in HFF cells on coverslips in 12-well plates for 38 h post-invasion without indole-3-acetic acid (IAA), and processed for immunofluorescence assay (IFA). In brief, the culture medium was aspirated, cells were washed with PBS, fixed with formaldehyde (4% in PBS) for 30 min, washed with PBS, permeabilized using 0.1% Triton-X 100 for 30 min, washed with PBS, and blocked with blocking buffer (3% BSA in PBS) for 1 h. The cells were incubated with rabbit anti-HA antibodies (at 1:200 dilution) for 2 h. The coverslips were washed with PBS, incubated with Alexa Fluor 488-conjugated F(ab’)2 donkey anti-rabbit IgG (at 1:1000 dilution) for 1 h at room temperature, incubated with DAPI (10 µg/ml in PBS) for 10 min, washed with PBS, air-dried, and mounted using the ProLongTM diamond antifade mountant. The cells were observed under the 100× objective, and images were acquired on a Zeiss LSM 880 confocal laser scanning microscope. Airyscan mode was used in Zeiss LSM 880 microscope to obtain super-resolution micrographs, images were exported using Zen Black software, and processed using ImageJ software. Maximum intensity projections of z-stack images were obtained using the z-projection of ImageJ software. Pearson’s correlation coefficient was determined using the JACoP plugin of Fiji software.
For co-localization of TgVMP1AIDHA with ER-GFP, RHTIR1-VMP1AID::ER-GFP parasites were grown without IAA, and processed for IFA using mouse anti-HA antibodies (at 1:200 dilution) and rabbit anti-GFP antibodies (at 1:200 dilution), followed by incubation with appropriate secondary antibodies (Alexa Fluor 488-conjugated F(ab’)2 donkey anti-rabbit IgG and Alexa Fluor 647-conjugated F(ab’)2 donkey anti-mouse IgG) as has been described above for TgVMP1. Images were acquired using a Zeiss LSM 880 confocal laser scanning microscope. Airyscan mode was used in Zeiss LSM 880 microscope to obtain super-resolution micrographs, images were exported using Zen Black software, and processed using ImageJ software. Pearson’s correlation coefficient was determined using the JACoP plugin in Fiji. For co-localization of TgVMP1AIDHA with GRASP-GFP, RHTIR1-VMP1AID::GRASP-GFP parasites were grown without IAA, and processed for IFA using mouse anti-HA (at 1:200 dilution) and rabbit anti-GFP antibodies (at 1:200 dilution), followed by incubation with appropriate secondary antibodies (Alexa Fluor 488-conjugated F(ab’)2 donkey anti-rabbit IgG and Alexa Fluor 647-conjugated F(ab’)2 donkey anti-mouse IgG) as has been described above for TgVMP1. The cells were observed under the 100 × objective of Olympus FV3000 confocal laser scanning microscope, images were captured using Fluoview FV31S-SW software, and processed as described above for TgVMP1. Pearson’s correlation coefficient was determined using the JACoP plugin in Fiji.
For TgVMP1 localization under IAA-induced depletion condition, RHTIR1-VMP1AID parasites were grown in HFF cells on coverslips in 12-well plates for 24 h post-invasion, followed by growth for an additional 14 h in the presence of 0.1% ethanol (-IAA) or 500 µM (+IAA) indole-3-acetic acid, and processed for IFA as has been described for TgVMP1. Infected HFF cells were processed for IFA using rabbit anti-HA (at 1:200 dilution) and mouse anti-SAG1 (at 1:500 dilution) antibodies for 2 h, followed by incubation with appropriate secondary antibodies (Alexa Fluor 488-conjugated goat anti-rabbit and Alexa Fluor 647-conjugated goat anti-mouse antibodies) as has been described above for TgVMP1. The cells were observed under the 100× objective, and images were acquired using Airyscan mode on a Zeiss LSM 880 confocal laser scanning microscope, exported using Zen Black software, and processed using ImageJ software. Maximum intensity projections of the z-stacks were obtained using the z-projection of ImageJ software.
To study the effect of TgVMP1 depletion on the inner membrane complex, RHTIR1-VMP1AID parasites were grown with (+IAA) or without (-IAA) indole-3-acetic acid, and processed for IFA using rabbit anti-IMC1 antibodies (at 1:500 dilution) and mouse anti-HA (at 1:200 dilution) antibodies, followed by appropriate secondary antibodies (Alexa Fluor 488-conjugated F(ab’)2 donkey anti-rabbit IgG and Alexa Fluor 647-conjugated F(ab’)2 donkey anti-mouse IgG (1:1000 dilution)) as has been described above for TgVMP1. The cells were observed under the 100× objective of Zeiss LSM 880 confocal laser scanning microscope, images were acquired using Airyscan mode, exported using Zen Black software, and processed using ImageJ software. Maximum intensity projections of the z-stacks were obtained using the z-projection option in ImageJ software.
To study the effect of TgVMP1 depletion on ER, the RHTIR1-VMP1AID::ER-GFP parasites were grown in HFF cells on coverslips in 12-well plates for 24 h post-invasion, and then grown for an additional 14 h with (+IAA) or without (-IAA) IAA. The intracellular or purified parasites were fixed in formaldehyde (4%) and processed for IFA using rabbit anti-GFP (at 1:200 dilution) and mouse anti-HA (at 1:200 dilution) antibodies, followed by appropriate secondary antibodies (Alexa Fluor 488-conjugated goat anti-rabbit or Alexa Fluor 647-conjugated goat anti-mouse antibodies) as has been described above for TgVMP1. The cells were observed under the 100× objective of Olympus FV3000 confocal laser scanning microscope, and images were acquired using Fluoview FV31S-SW software and processed using ImageJ software.
Effect of PfVMP1 depletion on parasite growth
Wild-type P. falciparum D10 and 3D7 strains, PfVMP1dKD, and PfVMP1AID parasites were cultured and synchronized as described in the “Parasite culture” section. PfVMP1dKD and PfVMP1AID parasites were assessed for PfVMP1 depletion and its effect on parasite growth. To check conditional PfVMP1 depletion, ring stage PfVMP1dKD parasites were grown at 8-10% parasitemia under control (+TMP-GlcN), single knockdown (-TMP-GlcN, +TMP+GlcN) or dual knockdown (-TMP+GlcN) conditions for two consecutive cycles (96 h) as has been described in the “Parasite culture” section. Parasites (at 8-10% parasitemia) were harvested at 48 and 96 h time points, and parasite lysates were processed for western blotting using rabbit anti-HA antibodies (1:1000 dilution), followed by HRP-conjugated goat anti-rabbit antibodies (1:20,000 dilution) as has been described in the “VMP1 expression” section. For loading control, the membrane was stripped off and probed with mouse HRP-conjugated anti-β-actin antibodies (at 1:30,000 dilution). To assess the effect of PfVMP1 deletion on parasite growth, synchronized ring stage PfVMP1dKD parasites were grown under control (+TMP-GlcN), single knockdown (-TMP-GlcN; +TMP+GlcN) or dual knockdown (-TMP+GlcN) conditions for 4 consecutive cycles (192 h). The number of parasite-infected RBCs was determined by FACS using a BD Fortessa FACS analyzer, as has been described earlier88, data were presented as % parasitemia over 4 cycles using GraphPad Prism.
Likewise, PfVMP1AID parasites were assessed for 5Ph-IAA-inducible PfVMP1 depletion and the effect of depletion on parasite growth. Briefly, three different clonal lines of the ring stage PfVMP1AID parasites were cultured in the presence or absence of 1 μM 5Ph-IAA for two consecutive erythrocytic cycles, as has been described in the “Parasite culture” section. Parasites (at 8-10% parasitemia) were harvested at 24, 48, 72, and 96 h time points. The parasite lysates were processed for western blotting using rabbit anti-HA antibodies (1:1000 dilution), followed by HRP-conjugated goat anti-rabbit antibodies (1:20,000 dilution) as has been described in the “VMP1 expression” section. The membrane was stripped off and probed for β-actin as a loading control using mouse HRP-conjugated anti-β-actin antibodies (at 1:30,000 dilution). For assessing the effect of PfVMP1 depletion on parasite growth, three different clonal lines of the ring stage PfVMP1AID parasites were cultured in the presence (+A) or absence (-A) of 1 μM 5Ph-IAA for three consecutive erythrocytic cycles as has been described in the “Parasite culture” section. Parasitemia was maintained at 3–4%, Giemsa-stained smears were made at the end of each cycle, observed under the 100× objective of a light microscope, and at least 1000 RBCs were counted to determine the percent parasitemia. Data were presented as % parasitemia over three cycles using the GraphPad Prism.
To check TIR1 expression in PfVMP1AID parasites, wild-type 3D7 and three clonal lines of PfVMP1AID parasites were harvested at mid-trophozoite/schizont stage at 8-10% parasitemia. Parasite lysates were processed for western blotting using rabbit anti-TIR1 antibodies (1:1000 dilution), followed by HRP-conjugated goat anti-rabbit antibodies (1:20,000 dilution) as has been described in the “VMP1 expression” section. The membrane was stripped off and probed for β-actin as a loading control using mouse HRP-conjugated anti-β-actin-antibodies (at 1:30,000 dilution).
Effect of TgVMP1 depletion on parasite growth
To assess the effect of TgVMP1 depletion at the protein level, we compared RHTIR1 and RHTIR1-VMP1AID parasites for the overall development by plaque assay and intracellular development by replication assay, essentially as has been previously described89,90. For plaque assay, fully confluent HFF monolayers in 6-well plates were infected with 500 freshly egressed RHTIR1 or RHTIR1-VMP1AID parasites per well, grown in the presence of 0.1% ethanol (-IAA) or 500 µM IAA (+IAA) for 8 days at 37 °C. The cells were fixed with methanol for 5 min at room temperature, stained with Giemsa for 10 min, and washed with distilled water. The plates were air-dried, observed for plaques, and images were acquired using the ChemiDoc Imaging System (Bio-Rad). The area of each of the 100 plaques from 3 independent experiments was measured using ImageJ software, and presented as mean plaque area (mm2) using the GraphPad Prism software.
For the replication assay, approximately 106 freshly purified RHTIR1 or RHTIR1-VMP1AID parasites were added to fully confluent HFF cells grown on coverslips in 12-well plates. After 3 h of invasion, extracellular parasites were removed by washing with PBS, and infected cells were grown for 24 h in the presence of 0.1% ethanol (-IAA) or 500 µM (+IAA) IAA. The culture media was carefully aspirated, cells were fixed with formaldehyde (4% in PBS), and processed for IFA using mouse anti-GRA1 antibodies (1:500 dilution), followed by Alexa Fluor 647-conjugated F(ab’)2 donkey anti-mouse IgG antibodies and DAPI (10 µg/ml) staining as has been described for TgVMP1 in the “VMP1 localization” section. The number of parasites/PV was determined by observing at least 100 random PVs from 3 independent experiments, and presented as the number of PVs containing different numbers of parasites as a percentage of the total number of PVs.
Effect of TgVMP1 depletion on in vivo development
Female BALB/cJ mice were infected with RHTIR1-VMP1AID parasites to determine the effect of TgVMP1 depletion on in vivo development, as has been described previously91. RHTIR1-VMP1AID parasites were cultured without IAA for 48 h post-infection, and purified as described in the “Parasite culture” section. 8-10 weeks old 20 female BALB/cJ mice were infected intraperitoneally with purified RHTIR1-VMP1AID tachyzoites (200 parasites/mouse), and five age-matched mice were used as uninfected naïve controls. The infected mice were divided into 2 groups (10 mice/group). Mice in one infected group were given 0.2 ml water with 12.5 mg/ml indole-3-acetic acid (+IAA) by oral gavage every day, and also provided drinking water with IAA (0.5 mg/ml) and sucrose (5% (w/v)). Another infected group was given 0.2 ml water only by oral gavage every day and also provided drinking water with (5% sucrose (w/v)). The naïve control group was given 0.2 ml water with 12.5 mg/ml IAA by oral gavage every day and also provided drinking water with IAA (0.5 mg/ml) and sucrose (5% (w/v)). Drinking water was changed every 2–3 days, and mice were monitored for 21 days. Peritoneal exudates of mice were collected on the day of their death and examined for the presence of parasites using mouse anti-SAG1 antibodies (for SAG1) and rabbit anti-HA antibodies (for TgVMP1AIDHA) by IFA as has been described in the “VMP1 localization” section.
Effect of TgVMP1 depletion on gliding motility
To assess the effect of TgVMP1 depletion on gliding motility of tachyzoites, we compared the motility of RHTIR1-VMP1AID parasites cultured with or without IAA as has been previously described92. In brief, RHTIR1-VMP1AID parasites were added to confluent HFF cells in a T75 flask, allowed to invade the cells for 3 h, and extracellular parasites were removed by washing the cells with D10 medium. The cells were grown for 24 h, followed by an additional 14 h in the presence of 0.1% ethanol (-IAA) or 500 µM IAA (+IAA), and parasites were purified as has been described in the “Parasite culture” section. For assessing parasite viability, purified -IAA or +IAA parasites were incubated in PBS with propidium iodide (PI; 5 μg/ml) for 15 min at room temperature, washed with PBS, and immediately observed under the 100 × objective of Zeiss Axioimager.Z2 fluorescence microscope. Images were acquired using AxioVision software, analyzed using ImageJ software, and the number of PI-negative parasites was counted and presented as a percentage of the total number of parasites observed using GraphPad Prism.
2 × 106 purified tachyzoites were resuspended in HBSS, allowed to glide on FBS-coated coverslips in a 12-well plate for 15 min, fixed with formaldehyde (4% in PBS), and processed for IFA using mouse anti-SAG1 antibodies (at 1/500 dilution), followed by Alexa Fluor 488-conjugated F(ab’)2 donkey anti-mouse IgG antibodies as has been described for TgVMP1 in the “VMP1 localization” section. Parasites were imaged for gliding trails under 100× objective of the Axioimager.Z2 using AxioVision software, and analyzed using ImageJ software. At least 100 parasites were scored for the presence of gliding trails in each of the three independent experiments, and the number of parasites showing gliding trails was presented as a percentage of the total number of parasites observed. Motile parasites in each of the three experiments were observed for measuring the gliding trail length, and presented as mean trail length (μm) using GraphPad Prism.
Effect of TgVMP1 depletion on host cell invasion
The invasion efficiency of RHTIR1-VMP1AID parasites was determined according to the previously reported two-color invasion assay93. RHTIR1-VMP1AID parasites were cultured with or without IAA as has been described in the “Effect of TgVMP1 depletion on gliding motility” section. Purified tachyzoites were added to confluent HFF cells grown on coverslips in 12-well plates (MOI 2:1), and incubated for 3 h with 0.1% ethanol (–IAA) or 500 µM IAA (+IAA). The cells were washed with PBS, fixed with formaldehyde (4% in PBS), and processed for IFA as has been described for TgVMP1 in the “VMP1 localization” section. In brief, the cells were first stained for extracellular parasites with mouse anti-SAG1 antibodies, followed by Alexa Fluor 647-conjugated F(ab’)2 donkey anti-mouse IgG antibodies (at 1:1000 dilution). The cells were washed with PBS, permeabilized with 0.1% Triton-X100, washed with PBS, blocked, incubated again with mouse anti-SAG1 antibodies to stain intracellular parasites, followed by Alexa Fluor 488-conjugated F(ab’)2 donkey anti-mouse IgG antibodies. The cells were observed for red/green (failed to invade) and green only (successful invasion) parasites under the 100 × objective of a Zeiss Axioimager Z2 fluorescence microscope, and images were acquired using AxioVision software. The number of red and green parasites was determined, and the number of green parasites was presented as a percentage of the total number of parasites (% Invasion) observed using GraphPad Prism. The experiment was repeated three times, and at least 100 parasites were observed in each experiment.
Effect of TgVMP1 depletion on egress
HFF cells were grown on coverslips in 12-well plates, infected with purified RHTIR1 or RHTIR1-VMP1AID parasites (at 1:1 MOI), and grown for 48 h, followed by an additional 14 h with 0.1% ethanol (-IAA) or 500 µM IAA (+IAA). The cells were washed with PBS, treated with HBSS containing 2 µM ionomycin, a calcium ionophore that induces rapid T. gondii egress94. HBSS was gently removed from the wells, the cells were fixed with 4% formaldehyde (in PBS), processed for IFA using mouse anti-GRA1 antibodies, followed by Alexa Fluor 647-F(ab’)2 donkey anti-mouse IgG and DAPI as described in the “VMP1 localization” section. The cells were observed under 100× objective of the Zeiss Axioimager.Z2 microscope, images were acquired using AxioVision software, and the total number of sites with well-dispersed parasites or clumps of parasites, which mark the PVs from which parasites egressed, was determined by counting at least 100 vacuoles in each of the three independent experiments. The number of PVs with parasite clumps was presented as a percentage of the total number of PVs observed using GraphPad Prism.
Conoid extrusion assay
RHTIR1-VMP1AID parasites were cultured with 0.1% ethanol (-IAA) or 500 µM IAA (+IAA) as has been described in the “Effect of TgVMP1 depletion on gliding motility” section, and evaluated for conoid extrusion using the calcium ionophore A23187 as has been previously described95. Briefly, ~2×106 purified tachyzoites were washed with PBS, resuspended in HBSS, and allowed to settle on coverslips in a 12-well plate for 10 min. The parasites were treated with 2 µM A23187 (in HBSS) for 10 min, fixed with 4% formaldehyde (in PBS), observed under 100× objective of the Axioimager.Z2 microscope, and images were acquired using AxioVision software, and analyzed using ImageJ software. At least 100 parasites were observed for conoid extrusion in each of the three independent experiments, and the number of parasites showing conoid extrusion was presented as a percentage of the total number of parasites observed using GraphPad Prism.
Effect of TgVMP1 depletion on microneme organization and secretion
RHTIR1-VMP1AID parasites were cultured with 0.1% ethanol (-IAA) or 500 µM IAA (+IAA) as has been described in the “Effect of TgVMP1 depletion on gliding motility” section, and assessed for microneme organization and secretion using antibodies to microneme proteins. For microneme organization, parasite-infected cells were fixed with 4% formaldehyde (in PBS), and processed for IFA as has been described in “VMP1 localization” section. The cells were incubated with antibodies to microneme proteins (rabbit anti-AMA1 at 1:500 dilution; mouse anti-MIC2 at 1:500 dilution; mouse anti-MIC3 at 1:500 dilution) and rabbit or mouse anti-HA antibodies (for TgVMPAIDHA at 1:200 dilution), followed by appropriate secondary antibodies (Alexa 488- or Alexa 647-conjugated F(ab’)2 donkey anti-mouse or anti-rabbit IgG) and DAPI (10 µg/ml). The cells were observed under 100× objective of the Zeiss LSM 880 confocal laser scanning microscope, and images were acquired in Airyscan mode using Zen Black software, and processed using ImageJ software. Maximum intensity projections of z-stack images were obtained using the z-projection option of ImageJ software.
Purified tachyzoites were assessed for microneme secretion according to the previously reported method96. Briefly, ~3 × 107 purified tachyzoites were resuspended in 100 µl of DGH medium (DMEM with 2 mM glutamine and 10 mM HEPES), treated with ethanol (1% v/v) at 37 °C for 2 min to induce secretion, incubated on ice to block further secretion, and centrifuged at 1000 × g for 8 min. The supernatant containing the extracellular secreted antigens (ESA) and the pellet were separated, each fraction was mixed with an equal volume of SDS-PAGE sample buffer, and assessed for MIC2 level by western blotting using mouse anti-MIC2 (at 1:2000 dilution) and anti-ROP1 antibodies as has been described in the “VMP1 expression” section. The signal intensities of ROP1 and MIC2 in pellet fractions were measured using ImageJ, the signal intensity of MIC2 was normalized as times of the ROP1 signal intensity for the corresponding parasites, and plotted using GraphPad Prism. The signal intensities of MIC2 in ESA fractions were measured, normalized as times of the MIC2 signal intensity in the corresponding pellet fraction, and plotted using GraphPad Prism. The experiment was repeated four times.
Effect of TgVMP1 depletion on rhoptry organization
RHTIR1-VMP1AID parasites were cultured with 0.1% ethanol (-IAA) or 500 µM IAA (+IAA) as has been described in the “Effect of TgVMP1 depletion on gliding motility” section, and assessed for rhoptry organization by IFA using anti-ROP1 antibodies as has been described in the “VMP1 localization” section. The infected cells were incubated with mouse anti-ROP1 (at 1:500 dilution) and rabbit anti-HA antibodies (for TgVMPAIDHA, at 1:200 dilution), followed by incubation with appropriate secondary antibodies (Alexa Fluor 488-conjugated F(ab’)2 donkey anti-rabbit IgG, Alexa Fluor 647-conjugated F(ab’)2 donkey anti-mouse IgG) and DAPI (10 µg/ml). The cells were observed under the 100× objective of the Zeiss LSM 880 confocal laser scanning microscope, and images were acquired in Airyscan mode using Zen Black software and processed using ImageJ software. Maximum intensity projections of z-stack images were obtained using Z-projection of ImageJ software.
For co-localization of rhoptries with the ER-GFP, RHTIR1-VMP1AID::ER-GFP parasites were cultured with 0.1% ethanol (-IAA) or 500 µM IAA (+IAA) as has been described in the “Effect of TgVMP1 depletion on gliding motility” section, tachyzoites were purified, and processed for IFA as described in the “VMP1 localization” section using mouse anti-ROP1 (at 1:500 dilution) and rabbit anti-GFP antibodies (for ER-GFP, at 1:200 dilution), followed by incubation with appropriate secondary antibodies (Alexa Fluor 488-conjugated F(ab’)2 donkey anti-rabbit IgG, Alexa Fluor 647-conjugated F(ab’)2 donkey anti-mouse IgG) and DAPI (10 µg/ml). The cells were observed under 100× objective of the Zeiss LSM 880 confocal laser scanning microscope, images were acquired in Airyscan mode using Zen Black software, and processed using ImageJ software.
Effect of TgVMP1 depletion on rhoptry secretion
Rhoptries discharge their contents as vacuoles (e-vacuoles) into the host cell upon attachment with the host cell during invasion, which can be monitored using ROP1 as a marker, as has been previously described97. Briefly, RHTIR1-VMP1AID parasites were cultured with 0.1% ethanol (-IAA) or 500 µM IAA (+IAA) as has been described in the “Effect of TgVMP1 depletion on gliding motility” section. 107 purified tachyzoites were treated with 1 µM cytochalasin D (CytD) for 10 min, which prevents invasion, but not rhoptry discharge98. 5 × 106 CytD-treated parasites were added to confluent HFF cells grown on coverslips in 12-well plates, incubated at 37 °C for 15 min, washed with PBS, fixed with formaldehyde (4% in PBS), and processed for IFA using mouse anti-ROP1 antibodies, followed by incubation with Alexa Fluor 647-conjugated F(ab’)2 donkey anti-mouse IgG and DAPI (10 µg/ml) as has been described in the “VMP1 localization” section. The cells were observed under 100× objective of the Zeiss Axioimager. Z2 microscope, and at least 100 parasites were observed for the presence of e-vacuoles. The experiment was repeated three times, and data was presented as the number of e-vacuole-positive parasites as a percentage of the total parasites using GraphPad Prism.
Effect of TgVMP1 depletion on dense granule biogenesis
RHTIR1-VMP1AID parasites were cultured with 0.1% ethanol (-IAA) or 500 µM IAA (+IAA) as has been described in the “Effect of TgVMP1 depletion on gliding motility” section. Intracellular parasites were evaluated for localization of dense granule proteins GRA1 and GRA2 in the PV by IFA, as has been described in the “VMP1 localization” section. In brief, intracellular parasites were incubated with mouse anti-GRA1 (at 1:500 dilution) or mouse anti-GRA2 (at 1:500 dilution) and rabbit anti-HA antibodies (for TgVMPAIDHA, at 1:200 dilution), followed by incubation with appropriate secondary antibodies (Alexa Fluor 488-conjugated F(ab’)2 donkey anti-rabbit IgG and Alexa Fluor 647-conjugated F(ab’)2 donkey anti-mouse IgG) and DAPI (10 µg/ml). The cells were observed for GRA1 and GRA2 localization patterns under 100× objective of the Zeiss LSM 880 confocal laser scanning microscope, images were acquired in Airyscan mode using Zen Black software, and processed using ImageJ software. Maximum intensity projections of z-stack images were obtained using z-projection of ImageJ software. ≥ 50 PVs were examined for the presence of GRA1 and GRA2 localization patterns, and the experiment was repeated three times. The number of PVs with IVN was presented as a percentage of the total number of PVs observed using GraphPad Prism.
To determine biogenesis of dense granules, purified -IAA and +IAA tachyzoites were added on poly-L-lysine-coated slides, and processed for IFA using mouse anti-GRA1 (at 1:500 dilution) or mouse anti-GRA2 (at 1:500 dilution) antibodies, followed by incubation with appropriate secondary antibodies (Alexa Fluor 488-conjugated F(ab’)2 donkey anti-mouse IgG) and DAPI (10 µg/ml). The cells were observed under the 100× objective of the Olympus FV3000 confocal laser scanning microscope, and images were captured using Fluoview FV31S-SW software. For super-resolution images, cells were observed under the 100× objective of the Zeiss LSM 880 confocal laser scanning microscope, and images were acquired in Airyscan mode using Zen Black software. Images were processed using ImageJ software. The fluorescence signal intensity of GRA1 or GRA2 in each tachyzoite was measured using the Multi Measure option of the ROI manager, and calculated using the formula “IntDen-(Area of interest × mean signal intensity of the background)”. The mean signal intensity of +IAA parasites was plotted as a percentage of the mean signal intensity of -IAA parasites (% Signal intensity) based on multiple -IAA (n = 338) and +IAA (n = 383) parasites from three independent experiments. The cells were also analyzed for intraparasitic distribution of GRA1 or GRA2 signal, and the number of parasites with perinuclear accumulation of GRA1 or GRA2 signal was presented as a percentage of the total number of parasites observed using GraphPad Prism.
For co-localization of dense granules with ER-GFP, RHTIR1-VMP1AID::ER-GFP parasites were cultured with 0.1% ethanol (-IAA) or 500 µM IAA (+IAA) as has been described in the “Effect of TgVMP1 depletion on gliding motility” section, tachyzoites were purified, and processed for IFA as described in the “VMP1 localization” section using mouse anti-GRA1 (at 1:500 dilution) or mouse anti-GRA2 (at 1:500 dilution) and rabbit anti-GFP antibodies (for ER-GFP, at 1:200 dilution), followed by incubation with appropriate secondary antibodies (Alexa Fluor 488-conjugated F(ab’)2 donkey anti-rabbit IgG and Alexa Fluor 647-conjugated F(ab’)2 donkey anti-mouse IgG) and DAPI (10 µg/ml). The cells were observed under a 100× objective of the Zeiss LSM 880 confocal laser scanning microscope, images were acquired in Airyscan mode using Zen Black software, and processed using ImageJ software.
Effect of TgVMP1 depletion on lipid droplets
RHTIR1-VMP1AID parasites were cultured with 0.1% ethanol (-IAA) or 500 µM IAA (+IAA) as has been described in the “Effect of TgVMP1 depletion on gliding motility” section, tachyzoites were purified, stained with Nile red (250 ng/ml in complete DMEM) at 37 ⁰C for 30 min, washed with PBS, and observed for fluorescence (λex = 488 nm, λem = 530 nm) under 100× objective of the Zeiss LSM880 confocal laser scanning microscope, images were acquired using Zen Black software, and processed using ImageJ software. The number of lipid droplets (LDs) was determined and presented as the number of LDs/parasite using GraphPad Prism. We also measured the surface area (µm2) of each LD using the analyze particles option of ImageJ software, and presented it as mean LD size (μm2) using GraphPad Prism.
Immunoprecipitation and mass spectrometry
For immunoprecipitation of TgVMP1AIDHA, RHTIR1 and RHTIR1-VMP1AID parasites were cultured without IAA for 48 h post-invasion, as has been described in the “Parasite culture” section. The purified tachyzoite pellets were resuspended in 300 µl of lysis buffer (10 mM Tris, 150 mM NaCl, 5 mM EDTA, 0.1% NP-40, 10% glycerol, 1× protease inhibitor cocktail, pH 7.5), incubated for 30 min on ice with intermittent mixing, sonicated using Bioruptor for 5 min (30 s On/30 s Off at high amplitude), and the lysates were incubated for 30 min in ice. The lysates were centrifuged at 4 °C for 15 min at 20,000 × g, and the supernatants were collected. The pellets were again extracted with 150 µl of lysis buffer as described above, and supernatants were pooled with the respective first supernatant. The supernatant was incubated with 5 µg of rabbit anti-HA antibodies for 12 h at 4 °C, mixed with protein A/G magnetic beads (10 µl slurry/2 mg of protein) for 2 h at 4 °C, and washed with lysis buffer. The beads were washed with lysis buffer, mixed with an equal volume of 2× SDS-PAGE sample buffer, incubated at 99 °C for 10 min, and the supernatant was collected as an eluate. Aliquots of input, flow through, wash, and eluate were processed for western blotting using mouse anti-HA antibodies (1:1000 dilution), followed by goat HRP-conjugated anti-mouse antibodies (1:20,000 dilution), as has been described in the “VMP1 expression” section. The remaining eluate was subjected to mass spectrometry analysis.
The immunoprecipitates were run on a 12% SDS-PAGE gel until the protein size ladder completely entered the gel. The protein-containing region was excised and processed for trypsin digestion and peptide extraction as has been described previously81,82. The peptide solution was run on Q Executive HF (Thermo Scientific) for HCD mode fragmentation and LC-MS/MS analysis. The raw data were imported into the Proteome Discoverer 2.2 software (Thermo Scientific), and proteins were identified by searching against the T. gondii RH-88 Uniprot databases using the SEQUEST HT algorithm. The analysis parameters included trypsin enzyme specificity, a maximum of two missed cleavages, variable modifications, such as carbamidomethylation of cysteine, oxidation of methionine, and deamidation of asparagine/glutamine. Precursor tolerance was set to 5 ppm, and fragmentation tolerance was set to 0.05 Da. Peptide spectral matches (PSM) and peptide identifications grouped into proteins were validated using the Percolator algorithm and filtered to a 1% false discovery rate (FDR). The peptide abundances in RHTIR1 and TgVMP1AIDHA immunoprecipitates from three biological repeats were compared for enrichment analysis using MetaboAnalyst 6.0 (https://www.metaboanalyst.ca/) with a single-factor statistical analysis. Imputation was done by replacing missing values with 1/5th of the minimum positive value of each variable, and normalization was performed based on the sum of the abundances. The abundance value was transformed to Log10, and the auto-scaling option was used for data scaling. Proteins enriched ≥1.5-fold with a p-value < 0.05 in TgVMP1AIDHA immunoprecipitate compared to the RH-TIR1 immunoprecipitate were considered significant interactors of TgVMP1.
Scramblase assay
TgVMP1AIDHA and PfVMP1AIDHA proteins were purified from RHTIR1-VMP1AID and PfVMP1AID parasites, respectively, as described in the “Immunoprecipitation and mass spectrometry” section. Briefly, the parasite lysate supernatants were incubated with HA-Trap magnetic agarose beads (ChromoTek; 25 µl beads for 2 mg/ml protein) for 12 h at 4 °C. The beads were washed (wash buffer: 20 mM Tris, 500 mM NaCl, 0.03% n-dodecyl β-D-maltoside (DDM), pH 7.4), and the bound proteins were eluted in 150 µl elution buffer (25 mM Tris, 150 mM NaCl, pH 7.4) with HA peptide (0.64 mg/ml). The eluate was diluted ~100 fold with storage buffer (20 mM Tris, 150 mM NaCl, 0.03% DDM, 5% glycerol and 1× protease inhibitor cocktail, pH 7.4), concentrated to ~50 µl using Amicon Ultra centrifugal filter (10 kDa cut-off) at 4 °C, flash frozen in liquid nitrogen, and stored at -80 °C until further use. Aliquots of input, flow through, washes, and eluate fractions were checked for the presence of TgVMP1AIDHA or PfVMP1AIDHA proteins by western blotting using rabbit anti-HA antibodies (at 1:1000 dilution), followed by goat HRP-conjugated anti-rabbit antibodies (1:20,000 dilution) as has been described in the “VMP1 expression” section.
Liposomes were prepared as has been reported previously22,99. In brief, POPC, POPS, and NBD-PS were dissolved in chloroform. POPC and POPS were mixed at 9:1 molar ratio along with 0.4 mol% NBD-PS. The lipid mixture was dried in a glass bottle under a nitrogen stream to form a uniform layer, followed by vacuum drying at room temperature for 12 h. The dried lipid layer was rehydrated using rehydration buffer (50 mM HEPES, 100 mM NaCl, pH 7.5) to obtain multilamellar vesicles (MLVs) at a final concentration of 10.5 mM. MLVs were subjected to 10 rounds of freeze-thaw cycles and diluted in rehydration buffer to 5.25 mM. The sample was passed 21 times through a 0.4 µm filter, followed by 11 times through a 0.2 µm filter using a mini extruder (Avanti Polar Lipids) to obtain uniform-sized liposomes. The liposomes were stored at 4 °C until further use.
Scramblase assay was performed as has been described previously22,99. Liposomes (5.25 mM) were destabilized by mixing with DDM (0.36% w/v final), and the mixture was incubated at room temperature for 3 h at 10 rpm on a nutator. The destabilized liposomes were mixed with storage buffer (with 0.36% DDM) or purified proteins (TgVMP1AIDHA or PfVMP1AIDHA in storage buffer with 0.36% DDM) at a 2:1 ratio (v/v) to prepare empty-liposomes or proteoliposomes (TgVMP1-liposomes or PfVMP1-liposomes), respectively. The samples were incubated at room temperature for 1.5 h to allow protein incorporation into the liposomes. DDM was removed by incubating the samples serially with 25 mg Bio-Beads for 1 h at room temperature, 25 mg Bio-Beads for 2 h at room temperature, and 50 mg Bio-Beads for overnight at 4 °C. The liquid phase containing liposomes was separated and diluted to 200 µM using rehydration buffer. 50 µl of the empty or proteoliposome sample was transferred to a 96-well flat-bottom white polystyrene plate, and assessed for fluorescence signal (λex = 470 nm, λem = 530 nm) using the Spectra Max iD3 multi-mode microplate reader (Molecular Devices). The fluorescence signal intensity was measured at every 26 s interval for 208 s, followed by dithionite (10 mM final concentration) for 416 s, and then with Triton X-100 (0.5% (w/v)) for 208 s. Relative fluorescence intensities of empty and proteoliposomes were calculated for each time point, and plotted as (Ft – FTriton)/(F0 – FTriton), where Ft is the fluorescence intensity at the corresponding time point, F0 is the initial fluorescence intensity, and FTriton is the fluorescence intensity after addition of Triton X-100 (at the 208 s time point). The relative fluorescence intensity was plotted on the y-axis over time on the x-axis using GraphPad Prism.
Expression of PfVMP1 and HsVMP1 in RHTIR1-VMP1AID parasites
A synthetic and mammalian-codon optimized PfVMP1 coding sequence (PfVMP1syn) was amplified from the pUC-PfVMP1syn plasmid using PfVMP1-Frec/PfVMP1-R primers. The T. gondii tubulin promoter region was amplified from pCTG-Fluc plasmid using TgTub-F/TgTub-PfVMP1-Rrec primers100. The PfVMP1-Frec and TgTub-PfVMP1-Rrec primers contain overlapping sequences, which allowed recombination of PfVMP1syn and tubulin promoter fragments by PCR using TgTub-F/PfVMP1-R primers. The recombined PCR product was digested with PacI/AvrII and cloned into similarly digested pMyc3 × .LIC-DHFR plasmid to obtain pTub-PfVMP1syn-3 ×Myc-DHFR plasmid. A synthetic and T. gondii-codon-optimized HsVMP1 coding sequence (HsVMP1syn) was cloned into pJET1.2/Blunt vector, excised with EcoRV/AvrII, and subcloned into similarly digested pTub-PfVMP1syn-3 ×Myc-DHFR plasmid to obtain pTub-HsVMP1syn-3 ×Myc-DHFR plasmids. 15 µg of pTub-PfVMP1syn-3 ×Myc-DHFR or pTub-HsVMP1syn-3 ×Myc-DHFR plasmid was transfected into RHTIR1-VMP1AID parasites as has been described in the “Generation of recombinant T. gondii parasites” section. The transfected parasites were selected with 1 µM pyrimethamine without IAA until the resistant parasites emerged. The resistant parasites with pTub-PfVMP1syn-3 ×Myc-DHFR plasmid (RHTIR1-TgVMP1AID::PfVMP1Myc) would express PfVMP1Myc, and the resistant parasites with pTub-HsVMP1syn-3 ×Myc-DHFR plasmid would express HsVMP1Myc. These parasites were cultured with 0.1% ethanol (-IAA) or 500 µM IAA (+IAA). The parasite lysates were evaluated for expression of TgVMP1AIDHA (rabbit anti-HA antibodies), PfVMP1Myc or HsVMP1Myc (mouse monoclonal anti-Myc antibodies at 1:1000 dilution), and SAG1 (mouse anti-SAG1 antibodies) as a loading control by western blotting, as has been described in the “VMP1 expression” section. The RHTIR1-TgVMP1AID::PfVMP1Myc and RHTIR1-TgVMP1AID::HsVMP1Myc parasites were assessed for co-localization of TgVMP1AIDHA with PfVMP1Myc or HsVMP1Myc, respectively, by IFA as has been described in the “VMP1 localization” section. The parasites were incubated with mouse anti-Myc (at 1:200 dilution) and rabbit anti-HA antibodies (for TgVMP1, at 1:200 dilution), followed by appropriate secondary antibodies (Alexa Fluor 488-conjugated F(ab’)2 donkey anti-rabbit IgG and Alexa Fluor 647-conjugated F(ab’)2 donkey anti-mouse IgG) and DAPI (10 µg/ml). The cells were observed under a 100× objective of the Zeiss LSM 880 confocal laser scanning microscope, images were acquired in Airyscan mode using Zen Black software, and processed using ImageJ software. Pearson’s correlation coefficient for co-localization was determined using the JACoP plugin in Fiji.
To study whether PfVMP1Myc or HsVMP1Myc complement the growth of TgVMP1-depleted parasites, the parental RHTIR1-VMP1AID and complemented (RHTIR1-TgVMP1AID::PfVMP1Myc, RHTIR1-VMP1AID::HsVMP1Myc) parasites were grown without (–IAA) or with (+IAA) IAA, and compared for overall growth by plaque assay and intracellular development by replication assay as has been described in the “Effect of TgVMP1 depletion on parasite growth” section. Plaques were observed using the 1× objective at 40× digital zoom of AXIO Zoom.V16 microscope, and images were taken using the Zen Blue software.
To evaluate whether PfVMP1Myc or HsVMP1Myc can rescue the dense granule biogenesis defect, the parental RHTIR1-VMP1AID and complemented (RHTIR1-TgVMP1AID::PfVMP1Myc, RHTIR1-VMP1AID::HsVMP1Myc) parasites were grown without (–IAA) or with (+IAA) IAA, and compared for localization of dense granule protein GRA2 as has been described in the “Effect of TgVMP1 depletion on IVN formation, and dense granule secretion and biogenesis” section.
Intracellular calcium monitoring
RHTIR1-VMP1AID parasites were cultured without (-IAA) or with (+IAA) IAA for 14 h, and tachyzoites were purified as described in the “Parasite culture” section. The parasites were evaluated for intracellular calcium levels using Fluo-4 AM as has been described previously101. The purified parasites were washed with Ringer’s solution (10 mM HEPES, 2 mM CaCl2, 1 mM MgCl2, 4.5 mM KCl, 155 mM NaCl, 10 mM D-glucose, pH 7.4), incubated with 250 nM of Fluo-4 AM for 10 min, washed with and resuspended in Ringer’s solution, transferred to poly-L-lysine-coated glass slides, and observed under the 100× objective of Olympus FV3000 confocal microscope. Images were acquired using Fluoview FV31S-SW software, the fluorescence signal intensity/tachyzoite was measured using the Multi Measure option of the ROI manager of ImageJ software, and calculated using the formula “IntDen-(Area of interest × mean signal intensity of the background)”. Multiple parasites from three independent experiments were observed, and the mean signal intensity/parasite of +IAA parasites was presented as a percentage of the mean signal intensity/parasite of -IAA parasites using GraphPad Prism.
Transmission electron microscopy
RHTIR1-VMP1AID parasites were cultured with 0.1% ethanol (-IAA) or 500 µM IAA (+IAA) as has been described in the “Effect of TgVMP1 depletion on gliding motility” section. Intracellular parasites were processed for transmission electron microscopy (TEM) as has been previously described102. Briefly, parasites were washed with 0.1 M phosphate buffer (PB, pH 7.4) and fixed (PB with 2.5% glutaraldehyde and 2% paraformaldehyde) overnight at 4 °C. The fixed cells were scraped off, washed with PB, incubated in 1% osmium tetroxide (in PB) for 1 h at room temperature, washed twice with distilled water, stained en bloc with 2% aqueous uranyl acetate for 1 h, and washed twice with distilled water. The sample was dehydrated using increasing percentages of ethanol (30%, 70%, 80%, 90%, and 95%) for 10 min each, followed by three treatments with 100% ethanol for 15 min each. The sample was infiltrated with epoxy and ethanol mixtures (1:3 for 2 h, 1:1 for 12 h, and 3:1 for 3 h) at room temperature. This was followed by sequential incubations in 100% epoxy resin for 3 h, 12 h, and 3 h. The cells were transferred to 100 % epoxy and polymerized at 62 ⁰C for 72 h. Ultrathin sections (50–70 nm thickness) were cut using an ultramicrotome (Ultracut UCT; Leica Microsystems) using a glass knife. The sections were transferred onto a 200 mesh copper grid, air-dried, and stained with uranyl acetate and lead citrate for 5 min each. The sections were visualized using a Talos L120C transmission electron microscope operating at 120 kV, and imaged using 4k × 4k Ceta CMOS camera. Images were acquired and processed using Velox software.
Immunoelectron microscopy
RHTIR1-VMP1AID parasites were cultured with 0.1% ethanol (-IAA) or 500 µM IAA (+IAA) as has been described in the “Effect of TgVMP1 depletion on gliding motility” section. Immunoelectron microscopy (IEM) of intracellular parasites was performed essentially as previously described102. Briefly, cells were washed with 0.1 M phosphate buffer (PB), fixed (PB with 4% paraformaldehyde and 0.1% glutaraldehyde) for 15 min at room temperature, followed by fresh fixative for 2 h at 4 °C. The cells were scraped off, washed with PB, dehydrated sequentially using increasing percentage of ethanol (30%, 50%, 70%, 80%, 90%, and 95%) for 10 min each, and then dehydrated twice with absolute ethanol for 15 min each. The cells were incubated twice in ethanol-LR white mixture (1:1) for 1 h each with a change of the mixture. The cells were incubated in 100% LR white for 8 h at 4 °C, followed by incubation in 100% LR white for 2 h at room temperature, and incubated at 55 °C for 72 h to polymerize. Ultrathin sections (50–70 nm) were cut using an ultramicrotome and transferred onto nickel-coated grids. The grids were quenched with 50 mM NH₄Cl (in PB) for 10 min and blocked (2% skimmed milk in PBT (PB with 0.05% Tween-20)) for 1 h at room temperature. The grids were incubated with mouse anti-ROP1, anti-GRA2 or anti-MIC2 antibodies (1:20 dilution in PBT with 3% BSA) for overnight at 4 °C without agitation, washed with PBT, incubated with 6 nm gold-conjugated goat-anti-mouse IgG antibodies (1:20 dilution in PBT with 3% BSA) for 1 h, and washed with PBT. The grids were post-fixed with 2.5% glutaraldehyde for 10 min, washed with distilled water, stained with uranyl acetate, washed again with distilled water, and air-dried. The sections were imaged as described above in the “Transmission electron microscopy” section.
We evaluated GRA2 secretion by extracellular parasites with modification of a previously described method103. Briefly, RHTIR1-VMP1AID parasites were cultured with 0.1% ethanol (-IAA) or 500 µM IAA (+IAA) as has been described in the “Effect of TgVMP1 depletion on gliding motility” section. 5 × 103 purified tachyzoites (in 10 μl DGH medium) were placed onto a glow-discharged carbon-coated 200-mesh nickel grids, and allowed to glide for 15 min at room temperature. Excess solution was gently wicked off, the grids were fixed with 1% paraformaldehyde for 10 min, and probed with anti-mouse-GRA2 primary antibodies (1:20 dilution), followed by 6 nm gold-conjugated anti-mouse IgG (1:20 dilution) as has been described above for ultrathin sections. Gold particles localized in the vicinity of each parasite were counted and presented as the number of particles/1 µm² area of the micrograph.
Effect of TgVMP1 depletion on apicoplast
RHTIR1-VMP1AID parasites were cultured with 0.1% ethanol (-IAA) or 500 µM IAA (+IAA) as has been described in the “Effect of TgVMP1 depletion on gliding motility” section, and assessed for the presence of apicoplast by IFA using antibodies to acyl carrier protein (ACP) as has been described in the “VMP1 localization” section. The infected cells were incubated with rabbit anti-ACP (at 1:500 dilution) and mouse anti-HA (at 1:200 dilution) antibodies, followed by incubation with appropriate secondary antibodies (Alexa Fluor 488-conjugated F(ab’)2 donkey anti-rabbit IgG, Alexa Fluor 647-conjugated F(ab’)2 donkey anti-mouse IgG) and DAPI (10 µg/ml). The cells were observed under the 100× objective of the Olympus FV3000 confocal laser scanning microscope, and images were acquired using Fluoview FV31S-SW software and processed using ImageJ software. Maximum intensity projections of z-stack images were obtained using the Z-projection of the ImageJ software. At least 150 parasites were observed to determine apicoplast/nucleus ratio in each of the three independent experiments, and data were plotted using GraphPad Prism.
Statistical analysis
The significance of the difference between the two groups was determined by using a two-tailed Student’s t-test, and the p-values are mentioned in the respective figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
TgVMP1 IP-MS data has been deposited to PRIDE database with the identifier PXD056007. Source data are provided with this paper.
References
Seeber, F. & Steinfelder, S. Recent advances in understanding apicomplexan parasites. F1000Research 5, https://doi.org/10.12688/f1000research.7924.1 (2016).
Harding, C. R. & Frischknecht, F. The Riveting Cellular Structures of Apicomplexan Parasites. Trends Parasitol. 36, 979–991 (2020).
Dubois, D. J. & Soldati-Favre, D. Biogenesis and secretion of micronemes in Toxoplasma gondii. Cell. Microbiol. 21, e13018 (2019).
Ben Chaabene, R., Lentini, G. & Soldati-Favre, D. Biogenesis and discharge of the rhoptries: Key organelles for entry and hijack of host cells by the Apicomplexa. Mol. Microbiol. 115, 453–465 (2021).
Counihan, N. A., Kalanon, M., Coppel, R. L. & de Koning-Ward, T. F. Plasmodium rhoptry proteins: why order is important. Trends Parasitol. 29, 228–236 (2013).
Griffith, M. B., Pearce, C. S. & Heaslip, A. T. Dense granule biogenesis, secretion, and function in Toxoplasma gondii. J. Eukaryot. Microbiol. 69, e12904 (2022).
Matz, J. M., Beck, J. R. & Blackman, M. J. The parasitophorous vacuole of the blood-stage malaria parasite. Nat. Rev. Microbiol. 18, 379–391 (2020).
Clough, B. & Frickel, E. M. The Toxoplasma Parasitophorous Vacuole: An Evolving Host-Parasite Frontier. Trends Parasitol. 33, 473–488 (2017).
Mercier, C. et al. Biogenesis of nanotubular network in Toxoplasma parasitophorous vacuole induced by parasite proteins. Mol. Biol. Cell 13, 2397–2409 (2002).
Magno, R. C., Lemgruber, L., Vommaro, R. C., De Souza, W. & Attias, M. Intravacuolar network may act as a mechanical support for Toxoplasma gondii inside the parasitophorous vacuole. Microsc. Res. Tech. 67, 45–52 (2005).
Kats, L. M., Cooke, B. M., Coppel, R. L. & Black, C. G. Protein trafficking to apical organelles of malaria parasites - building an invasion machine. Traffic 9, 176–186 (2008).
Wang, Y. & Kinoshita, T. The role of lipid scramblases in regulating lipid distributions at cellular membranes. Biochem. Soc. Trans. 51, 1857–1869 (2023).
Okawa, F. et al. Evolution and insights into the structure and function of the DedA superfamily containing TMEM41B and VMP1. J. Cell Sci. 134, https://doi.org/10.1242/jcs.255877 (2021).
Tábara, L. C., Vincent, O. & Escalante, R. Evidence for an evolutionary relationship between Vmp1 and bacterial DedA proteins. Int. J. Dev. Biol. 63, 67–71 (2019).
Dusetti, N. J. et al. Cloning and Expression of the Rat Vacuole Membrane Protein 1 (VMP1), a New Gene Activated in Pancreas with Acute Pancreatitis, Which Promotes Vacuole Formation. Biochem. Biophys. Res. Commun. 290, 641–649 (2002).
Hama, Y., Morishita, H. & Mizushima, N. Regulation of ER-derived membrane dynamics by the DedA domain-containing proteins VMP1 and TMEM41B. EMBO Rep. 23, e53894 (2022).
Ropolo, A. et al. The Pancreatitis-induced Vacuole Membrane Protein 1 Triggers Autophagy in Mammalian Cells*. J. Biol. Chem. 282, 37124–37133 (2007).
Tian, Y. et al. C. elegans Screen Identifies Autophagy Genes Specific to Multicellular Organisms. Cell 141, 1042–1055 (2010).
Itakura, E. & Mizushima, N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 6, 764–776 (2010).
Kishi-Itakura, C., Koyama-Honda, I., Itakura, E. & Mizushima, N. Ultrastructural analysis of autophagosome organization using mammalian autophagy-deficient cells. J. Cell Sci. 127, 4089–4102 (2014).
Tábara, L. C. et al. Vacuole membrane protein 1 marks endoplasmic reticulum subdomains enriched in phospholipid synthesizing enzymes and is required for phosphoinositide distribution. Traffic 19, 624–638 (2018).
Li, Y. E. et al. TMEM41B and VMP1 are scramblases and regulate the distribution of cholesterol and phosphatidylserine. J. Cell Biol. 220, https://doi.org/10.1083/jcb.202103105 (2021).
Ghanbarpour, A., Valverde, D. P., Melia, T. J. & Reinisch, K. M. A model for a partnership of lipid transfer proteins and scramblases in membrane expansion and organelle biogenesis. Proc. Natl Acad. Sci. USA 118, https://doi.org/10.1073/pnas.2101562118 (2021).
Morishita, H. et al. A critical role of VMP1 in lipoprotein secretion. eLife 8, e48834 (2019).
Hoffmann, H. H. et al. TMEM41B Is a Pan-flavivirus Host Factor. Cell 184, 133–148.e120 (2021).
Ji, M. et al. VMP1 and TMEM41B are essential for DMV formation during β-coronavirus infection. J. Cell Biol. 221, https://doi.org/10.1083/jcb.202112081 (2022).
Ji, M., Li, M., Sun, L., Deng, H. & Zhao, Y. G. DMV biogenesis during β-coronavirus infection requires autophagy proteins VMP1 and TMEM41B. Autophagy 19, 737–738 (2023).
Molejon, M. I., Ropolo, A. & Vaccaro, M. I. VMP1 is a new player in the regulation of the autophagy-specific phosphatidylinositol 3-kinase complex activation. Autophagy 9, 933–935 (2013).
Molejon, M. I., Ropolo, A., Re, A. L., Boggio, V. & Vaccaro, M. I. The VMP1-Beclin 1 interaction regulates autophagy induction. Sci. Rep. 3, 1055 (2013).
Calvo-Garrido, J., Carilla-Latorre, S., Lázaro-Diéguez, F., Egea, G. & Escalante, R. Vacuole membrane protein 1 is an endoplasmic reticulum protein required for organelle biogenesis, protein secretion, and development. Mol. Biol. Cell 19, 3442–3453 (2008).
Calvo-Garrido, J. & Escalante, R. Autophagy dysfunction and ubiquitin-positive protein aggregates in Dictyostelium cells lacking Vmp1. Autophagy 6, 100–109 (2010).
Tenenboim, H., Smirnova, J., Willmitzer, L., Steup, M. & Brotman, Y. VMP1-deficient Chlamydomonas exhibits severely aberrant cell morphology and disrupted cytokinesis. BMC Plant Biol. 14, 121 (2014).
Zhao, Y. G. et al. The ER-localized transmembrane protein EPG-3/VMP1 regulates SERCA activity to control ER-isolation membrane contacts for autophagosome formation. Mol. cell 67, 974–989. e976 (2017).
Tábara, L.-C. & Escalante, R. VMP1 establishes ER-microdomains that regulate membrane contact sites and autophagy. PLoS ONE 11, e0166499 (2016).
Huang, D. et al. TMEM41B acts as an ER scramblase required for lipoprotein biogenesis and lipid homeostasis. Cell Metab. 33, 1655–1670.e1658 (2021).
Trimarco, J. D. et al. TMEM41B is a host factor required for the replication of diverse coronaviruses including SARS-CoV-2. PLoS Pathog. 17, e1009599 (2021).
Yousefi, M. et al. TMEM41B and VMP1 modulate cellular lipid and energy metabolism for facilitating dengue virus infection. PLoS Pathog. 18, e1010763 (2022).
Phillips, M. J. & Voeltz, G. K. Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell Biol. 17, 69–82 (2016).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Varadi, M. et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444 (2022).
Brown, K. M., Long, S. & Sibley, L. D. Conditional Knockdown of Proteins Using Auxin-inducible Degron (AID) Fusions in Toxoplasma gondii. Bio Protoc. 8, https://doi.org/10.21769/BioProtoc.2728 (2018).
Struck, N. S. et al. Re-defining the Golgi complex in Plasmodium falciparum using the novel Golgi marker PfGRASP. J. Cell Sci. 118, 5603–5613 (2005).
Sangaré, L. O. et al. Unconventional endosome-like compartment and retromer complex in Toxoplasma gondii govern parasite integrity and host infection. Nat. Commun. 7, 11191 (2016).
de Koning-Ward, T. F., Gilson, P. R. & Crabb, B. S. Advances in molecular genetic systems in malaria. Nat. Rev. Microbiol. 13, 373–387 (2015).
Wichers, J. S. et al. PMRT1, a Plasmodium-Specific Parasite Plasma Membrane Transporter, Is Essential for Asexual and Sexual Blood Stage Development. mBio 13, e0062322 (2022).
Kreidenweiss, A., Hopkins, A. V. & Mordmüller, B. 2A and the auxin-based degron system facilitate control of protein levels in Plasmodium falciparum. PLoS One 8, e78661 (2013).
Black, M. W. & Boothroyd, J. C. Lytic cycle of Toxoplasma gondii. Microbiol. Mol. Biol. Rev. 64, 607–623 (2000).
Frénal, K., Dubremetz, J.-F., Lebrun, M. & Soldati-Favre, D. Gliding motility powers invasion and egress in Apicomplexa. Nat. Rev. Microbiol. 15, 645–660 (2017).
Endo, T., Sethi, K. K. & Piekarski, G. Toxoplasma gondii: calcium ionophore A23187-mediated exit of trophozoites from infected murine macrophages. Exp. Parasitol. 53, 179–188 (1982).
Dos Santos Pacheco, N., Tosetti, N., Koreny, L., Waller, R. F. & Soldati-Favre, D. Evolution, Composition, Assembly, and Function of the Conoid in Apicomplexa. Trends Parasitol. 36, 688–704 (2020).
Del Carmen, M. G., Mondragón, M., González, S. & Mondragón, R. Induction and regulation of conoid extrusion in Toxoplasma gondii. Cell. Microbiol. 11, 967–982 (2009).
Mercier, C. & Cesbron-Delauw, M.-F. Toxoplasma secretory granules: one population or more? Trends Parasitol. 31, 60–71 (2015).
Carruthers, V. B., Moreno, S. N. & Sibley, L. D. Ethanol and acetaldehyde elevate intracellular [Ca2+] and stimulate microneme discharge in Toxoplasma gondii. Biochem. J. 342, 379–386 (1999).
Carruthers, V. B., Sherman, G. D. & Sibley, L. D. The Toxoplasma Adhesive Protein MIC2 Is Proteolytically Processed at Multiple Sites by Two Parasite-derived Proteases. J. Biol. Chem. 275, 14346–14353 (2000).
Gao, M., Huang, X., Song, B.-L. & Yang, H. The biogenesis of lipid droplets: Lipids take center stage. Prog. Lipid Res. 75, 100989 (2019).
Greenspan, P., Mayer, E. P. & Fowler, S. D. Nile red: a selective fluorescent stain for intracellular lipid droplets. J. Cell Biol. 100, 965–973 (1985).
Sloves, P.-J. et al. Toxoplasma Sortilin-like receptor regulates protein transport and is essential for apical secretory organelle biogenesis and host infection. Cell Host Microbe 11, 515–527 (2012).
Stasic, A. J. et al. The Toxoplasma vacuolar H+-ATPase regulates intracellular pH and impacts the maturation of essential secretory proteins. Cell Rep. 27, 2132–2146.e2137 (2019).
Teasdale, R. D. & Jackson, M. R. Signal-mediated sorting of membrane proteins between the endoplasmic reticulum and the golgi apparatus. Annu. Rev. Cell Dev. Biol. 12, 27–54 (1996).
Bullen, H. E., Bisio, H. & Soldati-Favre, D. The triumvirate of signaling molecules controlling Toxoplasma microneme exocytosis: Cyclic GMP, calcium, and phosphatidic acid. PLoS Pathog. 15, e1007670 (2019).
Bullen, H. E. et al. Phosphatidic acid-mediated signaling regulates microneme secretion in Toxoplasma. Cell Host Microbe 19, 349–360 (2016).
Dowse, T. & Soldati, D. Host cell invasion by the apicomplexans: the significance of microneme protein proteolysis. Curr. Opin. Microbiol. 7, 388–396 (2004).
Nishi, M., Hu, K., Murray, J. M. & Roos, D. S. Organellar dynamics during the cell cycle of Toxoplasma gondii. J. Cell Sci. 121, 1559–1568 (2008).
Kessler, H. et al. Microneme protein 8 – a new essential invasion factor in Toxoplasma gondii. J. Cell Sci. 121, 947–956 (2008).
Valleau, D. et al. A conserved complex of microneme proteins mediates rhoptry discharge in Toxoplasma. EMBO J. 42, e113155 (2023).
Morlon-Guyot, J., Pastore, S., Berry, L., Lebrun, M. & Daher, W. Toxoplasma gondii Vps11, a subunit of HOPS and CORVET tethering complexes, is essential for the biogenesis of secretory organelles. Cell. Microbiol. 17, 1157–1178 (2015).
Caffaro, C. E. & Boothroyd, J. C. Evidence for host cells as the major contributor of lipids in the intravacuolar network of Toxoplasma-infected cells. Eukaryot. cell 10, 1095–1099 (2011).
Chelaghma, S. et al. Apical annuli are specialised sites of post-invasion secretion of dense granules in Toxoplasma. Elife 13, https://doi.org/10.7554/eLife.94201 (2024).
Fu, J. et al. An unconventional SNARE complex mediates exocytosis at the plasma membrane and vesicular fusion at the apical annuli in Toxoplasma gondii. PLoS Pathog. 19, e1011288 (2023).
Jackson, C. L. Lipid droplet biogenesis. Curr. Opin. Cell Biol. 59, 88–96 (2019).
Chen, X., Suo, X., Zhu, G. & Shen, B. The apicoplast biogenesis and metabolism: current progress and questions. Trends Parasitol. 40, 1144–1158 (2024).
Madeira, F. et al. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 50, W276–w279 (2022).
Trager, W. & Jensen, J. B. Human malaria parasites in continuous culture. Science 193, 673–675 (1976).
Binh, V. Q., Luty, A. J. & Kremsner, P. G. Differential effects of human serum and cells on the growth of Plasmodium falciparum adapted to serum-free in vitro culture conditions. Am. J. Trop. Med. Hyg. 57, 594–600 (1997).
Cranmer, S. L., Magowan, C., Liang, J., Coppel, R. L. & Cooke, B. M. An alternative to serum for cultivation of Plasmodium falciparum in vitro. Trans. R. Soc. Trop. Med. Hyg. 91, 363–365 (1997).
Lambros, C. & Vanderberg, J. P. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 65, 418–420 (1979).
Brown, K. M., Long, S. & Sibley, L. D. Conditional knockdown of proteins using auxin-inducible degron (AID) fusions in Toxoplasma gondii. Bio. Protoc. 8, e2728–e2728 (2018).
Khan, A. & Grigg, M. E. Toxoplasma gondii: Laboratory Maintenance and Growth. Curr. Protoc. Microbiol. 44, 20c.21.21–20c.21.17 (2017).
Sijwali, P. S. & Rosenthal, P. J. Functional evaluation of Plasmodium export signals in Plasmodium berghei suggests multiple modes of protein export. PLoS One 5, e10227 (2010).
Bhattacharjee, M. et al. Characterization of Plasmodium falciparum NEDD8 and identification of cullins as its substrates. Sci. Rep. 10, 20220 (2020).
Tanneru, N. et al. Plasmodium DDI1 is a potential therapeutic target and important chromatin-associated protein. Int. J. Parasitol. 53, 157–175 (2023).
Sudhakar, R., Das, D., Thanumalayan, S., Gorde, S. & Sijwali, P. S. Plasmodium falciparum Atg18 localizes to the food vacuole via interaction with the multi-drug resistance protein 1 and phosphatidylinositol 3-phosphate. Biochem. J. 478, 1705–1732 (2021).
Yesbolatova, A. et al. The auxin-inducible degron 2 technology provides sharp degradation control in yeast, mammalian cells, and mice. Nat. Commun. 11, 5701 (2020).
Govindarajalu, G., Rizvi, Z., Kumar, D. & Sijwali, P. S. Lyse-Reseal Erythrocytes for Transfection of Plasmodium falciparum. Sci. Rep. 9, 19952 (2019).
Fidock, D. A. & Wellems, T. E. Transformation with human dihydrofolate reductase renders malaria parasites insensitive to WR99210 but does not affect the intrinsic activity of proguanil. Proc. Natl. Acad. Sci. USA 94, 10931–10936 (1997).
Brown, K. M., Long, S. & Sibley, L. D. Plasma membrane association by N-acylation governs PKG function in Toxoplasma gondii. mBio 8, https://doi.org/10.1128/mBio.00375-17 (2017).
Shen, B., Brown, K. M., Lee, T. D. & Sibley, L. D. Efficient gene disruption in diverse strains of Toxoplasma gondii using CRISPR/CAS9. mBio 5, e01114–e01114 (2014).
Rizvi, Z. et al. Plasmodium falciparum contains functional SCF and CRL4 ubiquitin E3 ligases, and CRL4 is critical for cell division and membrane integrity. PLoS Pathog. 20, e1012045 (2024).
Kong-Hap, M. A. et al. Regulation of ATG8 membrane association by ATG4 in the parasitic protist Toxoplasma gondii. Autophagy 9, 1334–1348 (2013).
Ye, S. et al. Micronemal protein 13 contributes to the optimal growth of Toxoplasma gondii under stress conditions. Parasitol. Res. 118, 935–944 (2019).
Brown, K. M. & Sibley, L. D. Essential cGMP Signaling in Toxoplasma is initiated by a hybrid P-type ATPase-guanylate cyclase. Cell Host Microbe 24, 804–816.e806 (2018).
Håkansson, S., Morisaki, H., Heuser, J. & Sibley, L. D. Time-lapse video microscopy of gliding motility in Toxoplasma gondii reveals a novel, biphasic mechanism of cell locomotion. Mol. Biol. Cell 10, 3539–3547 (1999).
Wetzel, D. M., Chen, L. A., Ruiz, F. A., Moreno, S. N. J. & Sibley, L. D. Calcium-mediated protein secretion potentiates motility in Toxoplasma gondii. J. Cell Sci. 117, 5739–5748 (2004).
Vella, S. A. et al. The role of potassium and host calcium signaling in Toxoplasma gondii egress. Cell Calcium 94, 102337 (2021).
Long, S. et al. Calmodulin-like proteins localized to the conoid regulate motility and cell invasion by Toxoplasma gondii. PLoS Pathog. 13, e1006379 (2017).
Teo, C. F., Zhou, X. W., Bogyo, M. & Carruthers, V. B. Cysteine protease inhibitors block Toxoplasma gondii microneme secretion and cell invasion. Antimicrob. Agents Chemother. 51, 679–688 (2007).
Suarez, C., Lodoen, M. B. & Lebrun, M. Assessing rhoptry secretion in T. gondii. Methods Mol. Biol. 2071, 143–155 (2020).
Håkansson, S., Charron, A. J. & Sibley, L. D. Toxoplasma evacuoles: a two-step process of secretion and fusion forms the parasitophorous vacuole. EMBO J. 20, 3132–3144 (2001).
Wu, L., Liu, L., Xu, B., Huang, D. & Chen, X.-W. In vitro and in vivo assay of the ER lipid scramblase TMEM41B. STAR Protoc. 3, 101333 (2022).
Walunj, S. B. et al. High-throughput screening to identify inhibitors of Plasmodium falciparum importin alpha. Cells 11, https://doi.org/10.3390/cells11071201 (2022).
Nagamune, K., Beatty, W. L. & Sibley, L. D. Artemisinin induces calcium-dependent protein secretion in the protozoan parasite Toxoplasma gondii. Eukaryot. Cell 6, 2147–2156 (2007).
Koiri, D. et al. Real-time visualization reveals Mycobacterium tuberculosis ESAT-6 disrupts phagosome-like compartment via fibril-mediated vesiculation. Cell Rep. 44, 115328 (2025).
Stewart, M. J. & Vanderberg, J. P. Electron microscopic analysis of circumsporozoite protein trail formation by gliding malaria sporozoites. J. Protozool. 39, 663–671 (1992).
Acknowledgements
The authors are thankful to the staff of the Advanced Microscopy and Imaging Facility, Proteomics Facility, Electron Microscopy Facility, and Animal House Facility of the Centre for Cellular and Molecular Biology for their assistance in imaging, mass spectrometry, EM imaging, and animal-related experiments, respectively. The authors thank Mr. Harikrishna Adicherla for training and assistance with TEM. This study was supported by funds from the Council of Scientific and Industrial Research, India (MLP0148) and the Department of Biotechnology, India (BT/PR53849/BMS2/156/10/2024 (CN 20169)) to P.S.S. G.S.R., K.S., and M.K.B. are the recipients of PhD fellowships from the Council of Scientific and Industrial Research, India. S.M.G. is a recipient of a PhD fellowship from the University Grants Commission, India. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
G.S.R. and P.S.S. conceived the study, designed the experiments, interpreted the data, and wrote the manuscript. G.S.R. performed the majority of experiments. S.M.G. performed egress assays, in vivo mouse experiments, and mass spectrometry. K.S. maintained and provided HFF cells for infection, carried out invasion and microneme secretion assays, and assisted in confocal imaging. M.K.B. constructed ER-GFP and HsVMP1 plasmids and assisted in IFA. A.S.D. assisted in establishing the T. gondii culture and provided technical guidance for the construction of plasmids and the transfection of Toxoplasma. P.S.S. supervised the entire study and obtained funds.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Stanislas Tomavo, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Reddy, G.S., Gorde, S.M., Saxena, K. et al. An ER-resident lipid scramblase is crucial for biogenesis and function of apicomplexan parasite secretory organelles. Nat Commun 16, 11463 (2025). https://doi.org/10.1038/s41467-025-66303-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66303-9
