
Abstract
Tools to edit DNA methylation in a targeted manner are vital for establishing causal relationships between DNA methylation and its function, as well as for plant breeding and gene therapy. Here, by constructing dCas9 fusions to a panel of effectors and cofactors, we develop a range of highly effective tools for editing DNA methylation in Arabidopsis, including five tools for DNA methylation and six tools for DNA demethylation. Our tools show a diversity of performance features in terms of specificity and efficiency, offering either the capacity to edit DNA methylation in a target-specific manner or the ability to edit DNA methylation genome-wide due to potent off-target effect. Importantly, DNA methylation edited by these tools is inherited in the absence of transgene. These versatile tools pave the way for diverse applications of DNA methylation editing in not only research but also epigenetic breeding of crops.
Similar content being viewed by others


Introduction
DNA methylation at the C-5 position of cytosine residues is an evolutionarily conserved epigenetic modification that plays critical roles in transposon silencing and gene expression regulation1,2,3,4,5,6,7. Aberrant DNA methylation in humans is associated with multiple diseases, including cancer and neurological disorders8,9. In plants, DNA methylation is a key determinant of development and influences numerous agronomic traits10,11,12,13,14,15,16,17. Thus, the molecular tools that enable efficient editing of DNA methylation in a targeted manner are important in basic research, gene therapy, and plant epigenetic breeding18,19,20,21,22.
Benefiting from in vitro manipulation at the cellular level and the ability to enrich the positive cells via sorting, targeted DNA de/methylation editing tools, especially CRISPR/dCas9-based system, where pre-designed guide RNAs loaded into dCas9 direct DNA de/methylation effectors to specific genomic loci, have been established in mammalian systems23,24,25,26,27,28,29. However, developing corresponding tools for plants has been challenging, as they must be applied at the organismal level and generate transgenerationally heritable edits. Despite this, a few tools have been developed. Targeted DNA demethylation in plants has been achieved in plants using two approaches: fusing artificial zinc finger (ZF) with human demethylase TET1 (TEN-ELEVEN TRANSLOCATION METHYLCYTOSINE DIOXYGENASE 1) catalytic domain (TET1cd) and using a CRISPR/dCas9 Suntag system that recruits multiple TET1cd-fused anti-SunTag antibodies30,31. Targeted DNA methylation has been achieved using the CRISPR/dCas9 SunTag system with Nicotiana tabacum DOMAINS REARRANGED METHYLTRANSFERASE (DRM) catalytic domain (NtDRMcd) or bacterial methyltransferase MQ132,33. Additionally, genome-wide DNA methylation can be induced using ZF-MQ1 fusions34. Random genome-wide demethylation has been achieved through overexpression of TET1cd35.
Here, we present a CRISPR/dCas9-based fusion protein strategy for editing DNA methylation in the plant genome. In this approach, a panel of effector proteins (de/methylases) is fused at the N terminus of dCas9 to edit DNA methylation, while a group of cofactor proteins (proteins involved in epigenetic regulation) is fused to the C terminus of dCas9 to improve the efficacy of these effectors. Our results demonstrate that combining effectors with cofactors is an effective strategy for DNA methylation editing, leading to the development of five tools for DNA methylation and six tools for DNA demethylation. These tools exhibit a range of performance in terms of specificity and efficiency. In addition to the ability to edit DNA methylation in a targeted manner, our tools are also capable of globally methylating or demethylating DNA across the genome through potent off-target effect. Successful DNA methylation editing in regions with different epigenetic landscapes highlights the broad applicability of our tools. Importantly, the DNA methylation edited by our tools is stably and efficiently inherited in the absence of the transgene, making them suitable for practical applications. These tools will enable the investigation of the causal relationship between DNA methylation and its diverse functions, the generation of epialleles with desirable traits, the screening of regulatory elements encoded by DNA methylation, and the implementation of plant epi-breeding.
Results
A dCas9-based single fusion protein to edit DNA methylation
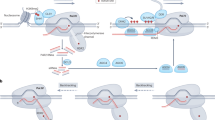
To achieve targeted editing of DNA methylation while minimizing off-target risks associated with effector proteins, we designed a series of CRISPR-based fusion proteins composed of an effector, a cofactor, and catalytically inactive S. pyogenes dCas9 (Fig. 1a). The effectors were positioned at the N terminus of dCas9 to allow its optimal access to cytosine for edit DNA methylation36 (Fig. 1a). For targeted DNA methylation, we chose the mammalian DNA (CYTOSINE-5)-METHYLTRANSFERASE 3 alpha and 3-like (DNMT3A-3L) catalytic domain, Arabidopsis thaliana DNA methyltransferase (DRM2), Nicotiana tabacum DNA methyltransferase catalytic domain (NtDRMcd), and the bacterial DNA methyltransferase variant (MQ1Q147L, hereafter referred to as MQ1v) as effectors (Fig. 1a). In regard to targeted DNA demethylation, Arabidopsis thaliana DNA demethylase ROS1 (REPRESSOR OF SILENCING 1) and its catalytic domain (ROS1cd), as well as human DNA demethylase catalytic domain (TET1cd) were chosen as effectors (Fig. 1a). Meanwhile, we placed at the C terminus of dCas9 epigenetic cofactors that are hypothesized to enhance the efficacy of de/methylation effector (Fig. 1a). The transcriptional repressor (human KRAB (KRÜPPEL-ASSOCIATED BOX) and Arabidopsis thaliana SRDX (SUPERMAN REPRESSION DOMAIN X)) and Arabidopsis thaliana KYP (KRYPTONITE), which encodes histone 3 lysine 9 methyltransferase, were chosen as the cofactors for methylation effectors (Fig. 1a). In parallel, we selected a protein to deposit active histone modification (human histone acetyltransferase catalytic domain p300) and a protein to remove repressive histone modification H3K9me2 (Arabidopsis thaliana H3K9me2 demethylase IBM1 (INCREASE IN BONSAI METHYLATION 1) and its catalytic domain IBM1cd as the cofactors for demethylation effectors (Fig. 1a). Additionally, two promoters (pUBQ1 (Ubiquitin 1) and pRPS5A (Ribosomal protein 5a)) were employed to drive the expression of these dCas9-based DNA methylation editor proteins37 (Fig. 1a).

a Design of dCas9 fusion proteins for editing DNA methylation. Effector and cofactor proteins were fused to the N and C terminus of dCas9, respectively. The pUBQ1 and pRPS5A promoters were chosen to drive the expression of the designed fusion proteins. b Schematic of targeting DNA methylation at the FWA promoter. This methylation-dependent visual phenotype can be used as a readout for successful targeted DNA methylation. c fwa plants and representative early-flowering T1 plants expressing MQ1v-dCas9-KRAB (M22) to illustrate differences in flowering-time. d Bar graph showing FWA expression levels in representative early-flowering T1 plants expressing the indicated tools and control plants. The controls and T1 plants were grown side by side. Error bars are defined as the standard deviation (SD) of three technical replicates; data are presented as mean ± s.d.. e Dot plot of rosette leaf count at flowering-time of T1 plants transformed with indicated tools in fwa, compared to fwa controls. The rosette leaf count reflects the time required for flowering. “-gRNA” indicates the transformed vector without the gRNA expression cassette (the same below). pU-dCas9 indicates that the pUBQ1 promoter drive the expression of single dCas9 protein (the same below); pR-dCas9 indicates that the pRPS5A promoter drive the expression of single dCas9 protein (the same below). Each dot represents the rosette leaf number of an individual plant (the same below). The dashed line corresponds to the leaf number threshold (21) below which plants are considered early-flowering relative to fwa plants. The code names referring to the tools are listed below. The numerator in the fraction at the top indicates the number of early-flowering plants relative to fwa plants, while the denominator represents the total number of plants observed (the same below). The controls and T1 plants were grown side by side. f Screenshot of DNA methylation over FWA promoter in the indicated T1 plants. The grey box highlights the gRNA-targeted site at the FWA promoter (the same below). Red bars indicate the positions of the gRNAs (the same below). The controls and T1 plants were grown side by side. g Genome-wide CG methylation levels in the indicated T1 and fwa plants. Source data are provided as a Source data file.
Targeted DNA methylation at the FWA promoter
To thoroughly test the ability of our designed fusion proteins to methylate DNA in targeted regions of the plant genome, we utilized four DNA methyltransferases (effectors), three cofactors, and two promoters to construct editing tools through permutations and combinations, resulting in a total of 38 tools for targeted DNA methylation (Supplementary Fig. 1; M1 to M38). We first investigated the performance of these tools through methylation of the FWA promoter (Fig. 1b). FWA (Flowering Wageningen) is normally silenced by DNA methylation over its promoter in the vegetative tissue of wild-type (WT) plants and the loss of DNA methylation leads to ectopic expression of FWA, causing a late-flowering phenotype and generating heritable fwa epialleles38. This methylation-dependent visual phenotype can be used as a readout to reflect the status of DNA methylation at the FWA promoter30,31,32,33 (Fig. 1b).
The 38 tools for targeted DNA methylation, which included guide RNAs (gRNA) binding the FWA promoter, along with control vectors, were individually transformed into fwa epiallele plants. It should be pointed out that fwa epiallele plants were derived from the MET1 heterozygous mutant, and thus their genome is CG-hypomethylated. We monitored the flowering time of T1 transformants and observed that several tools driven by the RPS5A promoter (M6, M12, M14, M16, M22, and M30) triggered an early-flowering phenotype compared to both untransformed fwa plants and those transformed with control vectors (Fig. 1e), whereas the tools driven by the UBQ promoter did not result in early-flowering phenotype (Fig. 1c, e), indicating the superior performance of RPS5A promoter in targeted DNA methylation over that of UBQ1 promoter. Of these tools that successfully induced early-flowering phenotype in T1 plants, MQ1v-dCas9(M14) and MQ1v-dCas9-KRAB(M22) most strongly triggered early-flowering phenotype, followed sequentially by DNMT3A-3L-dCas9-KRAB(M16), MQ1v-dCas9-SRDX(M30), NtDRMcd-dCas9(M12), and dCas9-KYP(M6) (Fig. 1e). These results suggest that the efficiencies (as defined by the proportion of T1 plants exhibiting an early-flowering phenotype) of MQ1v-dCas9(M14) and MQ1v-dCas9-KRAB(M22) in targeting DNA methylation are significantly higher than those of the DNMT3A-3L-dCas9-KRAB(M16), MQ1v-dCas9-SRDX(M30), NtDRMcd-dCas9(M12), and dCas9-KYP(M6) (Fig. 1e).
To examine if the observed early-flowering phenotype was due to down-regulation or silencing of FWA expression by targeted DNA methylation in its promoter, we conducted the RT-qPCR and whole-genome bisulfite sequencing (WGBS) on fwa plants, control plants, and two representative early-flowering T1 plants (M6, M12, M14, M16, M22, and M30). As expected, FWA expression was sharply decreased or silenced in all representative early-flowering T1 plants relative to fwa and control plants (Fig. 1d). We then analyzed DNA methylation levels at the FWA promoter and found that dCas9-KYP(M6), NtDRMcd-dCas9(M12), MQ1v-dCas9(M14), MQ1v-dCas9-KRAB(M22), and MQ1v-dCas9-SRDX(M30) successfully induced DNA methylation at the FWA promoter in representative early-flowering T1 plants (Fig. 1f), indicating that the observed early-flowering phenotype resulted from our tools silencing FWA via methylating its promoter. Interestingly, DNMT3A-3L-dCas9-KRAB(M16) did not methylate the FWA promoter, while FWA expression was dramatically decreased in two corresponding representative early-flowering T1 plants (Fig. 1d, f), suggesting that DNMT3A-3L may not function effectively in the plant system, and the decrease in FWA expression was likely caused by the transcriptional repression domain KRAB in M16.
Furthermore, we found that: (1) MQ1v-dCas9(M14) exclusively methylated the CG sites at the FWA promoter, consistent with previous reports26,33 (Supplementary Fig. 2); (2) MQ1v-dCas9-KRAB(M22) and MQ1v-dCas9-SRDX(M30) can trigger both CG and non-CG methylation at the FWA promoter and the level of non-CG methylation in M30-edited T1 plants was higher than that in M22-edited T1 plants (Supplementary Fig. 2), indicating that cofactor SRDX (and to a lesser extent in KRAB) at the C terminus of dCas9 can expand the capacity of MQ1v to target non-CG methylation; (3) the extent of CG methylation induced by NtDRMcd-dCas9(M12) is lower compared to other tools (Supplementary Fig. 2), consistent with the previously reported preference of NtDRM for non-CG methylation39; (4) dCas9-KYP(M6) also could induce both CG and non-CG methylation at the FWA promoter (Supplementary Fig. 2), in absence of a fused DNA methyltransferase, suggesting that M6 may indirectly induce DNA methylation at the FWA promoter via KYP-mediated methylation of histone H3K9, which recruits endogenous DNA methylation machinery.
Characterizing the specificity of tools for methylating the FWA promoter
To assess the specificity of these tools, we observed the DNA methylation levels in the region flanking the targeted site (Fig. 1f) and conducted a genome-wide DNA methylation analysis (Supplementary Fig. 3). dCas9-KYP(M6)- and NtDRMcd-dCas9(M12)-edited T1 plants showed comparable DNA methylation level to the fwa plants both in regions flanking the FWA promoter and genome-wide (Fig. 1f and Supplementary Fig. 3), indicating their high specificity. In contrast, CG sites in the flanking region of the targeted site were methylated by MQ1v-dCas9(M14), to a lesser extent by MQ1v-dCas9-KRAB(M22), and MQ1v-dCas9-SRDX(M30)-editied T1 plants showed the lowest level of methylation at these CG sites (Fig. 1f). Additionally, genome-wide CG methylation levels in MQ1v-dCas9(M14)- and MQ1v-dCas9-KRAB(M22)-, but not the MQ1v-dCas9-SRDX(M30)-edited T1 plants are greatly higher than those in fwa plants (Fig. 1g and Supplementary Fig. 4). The number of DMRs is consistent with the genome-wide DNA methylation analysis (Supplementary Fig. 5a). The hyper-DMRs in M6- and M12-edited T1 plants show enrichment in the pericentromeric regions, whereas those in M14-, M22-, and M39-edited T1 plants are depleted in these regions (Supplementary Fig. 6). The hypo-DMRs in edited T1 plants tend to be located in the pericentromeric regions (Supplementary Fig. 6). Most of hyper-DMRs in edited T1 plants belong to MET1-targeted regions (Supplementary Fig. 5b). The hypo-DMRs in edited T1 plants are enriched in Class I RdDM-targeted regions (Supplementary Fig. 5b). Hence, we conclude that dCas9-KYP(M6) and NtDRMcd-dCas9(M12) are highly specific DNA methylation tools, followed by MQ1v-dCas9-SRDX(M30) with moderate specificity, whereas MQ1v-dCas9(M14), due to its high efficacy but lack in specificity, can be used to methylate CG sites throughout the genome via its potent off-target effect.
MQ1v-dCas9(M14), MQ1v-dCas9-KRAB(M22), and MQ1v-dCas9-SRDX(M30) all contain the same effector MQ1v, differing only in the cofactors at the C terminus of dCas9 (Supplementary Fig. 1). The dramatic difference of their specificity suggests that the cofactor SRDX enhances the specificity of MQ1v-dCas9 for targeting DNA methylation. To rule out the possibility that the improved specificity of MQ1v-dCas9-SRDX(M30) was due to the presence of BFP (blue fluorescent protein; see the construct in Supplementary Fig. 1), we directly fused BFP and SRDX, respectively, to the C terminus of MQ1v-dCas9 (Supplementary Fig. 1; M39 and M40), and measured DNA methylation level of their transformants at the target site and genome-wide. Both MQ1v-dCas9-BFP(M39) and MQ1v-dCas9-cSRDX(M40) induced DNA methylation over the FWA promoter in representative early-flowering T1 plants, and the resulting DNA methylation silenced the FWA expression (Fig. 1d, f). The DNA methylation pattern at the FWA promoter and genome-wide in MQ1v-dCas9-cSRDX(M40)-edited T1 plants but not in MQ1v-dCas9-BFP(M39)-edited T1 plants are consistent with those in MQ1v-dCas9-SRDX(M30)-edited T1 plants (Fig. 1f, g), confirming the role of cofactor SRDX in improving the specificity of MQ1v-dCas9 for targeting DNA methylation. To investigate the relationship between the specificity of the tools and the levels of the corresponding dCas9-fusion proteins, we compared protein levels in plants carrying the tools with varying specificity. For tools containing different effectors, the specificity or lack of it does not correlate with the protein levels of the encoded editors, as exemplified by NtDRMcd-dCas9(M12), which exhibits the highest specificity but has similar protein levels to MQ1v-dCas9(M14), which shows the lowest specificity (Fig. 1f, g and Supplementary Fig. 7). For tools using MQ1v as the effector, although specificity appears to be negatively correlated with protein levels among representative MQ1v-dCas9(M14)-, MQ1v-dCas9-KRAB(M22)-, and MQ1v-dCas9-SRDX(M30)-edited plants, it cannot be conclusively stated that cofactor SRDX or KRAB improves specificity by altering fusion protein levels because the protein level of MQ1v-dCas9-cSRDX(M40) is comparable to those of MQ1v-dCas9-BFP(M39) and MQ1v-dCas9(M14) (Fig. 1f, g and Supplementary Fig. 7). Of note, we discovered that the specificity of these tools is inversely correlated with their efficiency in methylating DNA at targeted site, as exemplified by dCas9-KYP(M6) and NtDRMcd-dCas9(M12), which show the highest specificity but lowest efficiency; MQ1v-dCas9(M14) and MQ1v-dCas9-KRAB(M22), which exhibit the lowest specificity but highest efficiency; and MQ1v-dCas9-SRDX(M30), which displays moderate specificity and efficiency (Fig. 1e-g and Supplementary Fig. 4).
Targeted DNA methylation at the FWA promoter is heritable
To evaluate the stability and heritability of our tools-mediated DNA methylation at the FWA promoter, we analyzed the progeny (T2 plants) of these representative early-flowering T1 plants. Allelic segregation during selfing allows the loss of transgene, creating transgene-free individuals whose change in methylation at the target site must be inherited from the previous generation. 100% of the T2 plants derived from T1 plants where the FWA promoter was methylated by corresponding tools (except for M12-#1) displayed early-flowering phenotype (Fig. 2a and Supplementary Fig. 8b). As a comparison, all T2 plants derived from DNMT3A-3L-dCas9-KRAB(M16)-edited T1 plants flowered late (Supplementary Fig. 8a), further supporting that the DNMT3A-3L was unable to methylate the FWA promoter.

a Dot plot of rosette leaf count at flowering-time of indicated T2 plants in the fwa background. Red dots indicate the corresponding T2 plants lacking the transgene (the same below); black dots indicate the corresponding T2 plants containing the transgene (the same below); gray dots represent untransformed plants in the indicated background (the same below). The dashed line corresponds to the leaf number threshold (21) below which plants are considered early-flowering relative to fwa plants. The code names referring to the tools are listed below. The controls and T2 plants were grown side by side. b Bar graph showing FWA expression level in the indicated T2 and control plants. “+” indicates T2 plants containing the transgene (the same below); “-” indicates T2 plants without the transgene (the same below). The controls and T2 plants were grown side by side. Error bars are defined as the standard deviation (SD) of three technical replicates; data are presented as mean ± s.d.. c Screenshot of DNA methylation over FWA promoter in the indicated T2 plants. Please note that the control WGBS data for fwa-1 and fwa-2 used here are identical to those used in Fig. 1f. Source data are provided as a Source data file.
We then performed RT-qPCR and WGBS on one T2 plant containing the transgene (+) and two T2 plants without transgene (-), along with control plants. Consistent with the early-flowering phenotype, we detected repression of FWA expression and high levels of DNA methylation at the FWA promoter in all T2 plants compared to the fwa or control plants (Fig. 2b, c). Moreover, the extent of FWA repression and the DNA methylation levels of its promoter were similar between T2 with (+) and without (-) transgene (Fig. 2b, c). Taken together, we conclude that our tools-induced DNA methylation at the FWA promoter was well inherited in the absence of transgene. T2 plants lacking the transgene, derived from MQ1v-dCas9(M14)-edited T1 plants, retain their partial genome-wide ectopic CG methylation (Supplementary Figs. 9 and 10).
Targeted DNA methylation at a distal enhancer of FT
To assess the generality of our DNA methylation systems, we used these tools to methylate Block C, a distal enhancer of FT (Flowering locus T), whose ectopic DNA methylation delays flowering in WT plants40 (Fig. 3a). In line with expectation, all tools that were effective on the FWA promoter caused a late-flowering phenotype in T1 plants compared to WT and control plants (Fig. 3b, c), suggesting that our tools also can methylate the FT enhancer Block C. In contrast to the FWA promoter, where dCas9-KYP(M6) had the lowest capacity to trigger early-flowering (Fig. 1e), the efficiency of dCas9-KYP(M6) in inducing late-flowering in T1 plants via editing the Block C was the highest among the tools tested. The efficiencies (as defined by the proportion of T1 plants exhibiting a delayed-flowering phenotype) of NtDRMcd-dCas9(M12), MQ1v-dCas9(M14), MQ1v-dCas9-KRAB(M22), and MQ1v-dCas9-SRDX(M30) in inducing change in flowering time remain consistent between the FWA promoter and the FT enhancer Block C (Figs. 1e and 3b).

a Schematic of targeting DNA methylation at the FT enhancer Block C. DNA methylation at FT enhancer Block C induces a late-flowering phenotype. b Dot plot of rosette leaf count at flowering-time of T1 plants transformed with the indicated tools, compared to control plants. The dashed line corresponds to the leaf number threshold (15) above which plants are considered delayed flowering relative to WT plants. The controls and T1 plants were grown side by side. c WT plants and representative late-flowering T1 plants expressing MQ1v-dCas9 (M14) to illustrate differences in flowering time. d Screenshot of DNA methylation at the FT enhancer Block C in the indicated control, T1, and T2 plants. ATAC-seq (Assay for Transposase-Accessible Chromatin with high-throughput sequencing) shows the position of enhancer Block C. The controls and T1 plants were grown side by side. e Dot plot of rosette leaf count at flowering-time of the indicated T2 plants. The dashed line corresponds to the leaf number threshold (15) above which plants are considered delayed flowering relative to WT plants. The controls and T2 plants were grown side by side. Source data are provided as a Source data file.
To confirm that the late-flowering phenotype was induced by the DNA methylation at the FT enhancer Block C, we performed WGBS on two representative T1 plants for each tested tool. Similar to the results with the FWA promoter, we observed that: (1) NtDRMcd-dCas9(M12) specifically methylated the targeted site without altering global DNA methylation (Fig. 3d and Supplementary Fig. 11); (2) CG sites not only in the targeted site but also in its flanking region were methylated by MQ1v-dCas9(M14), and to a less extent by MQ1v-dCas9-KRAB(M22) and MQ1v-dCas9-SRDX(M30) (Fig. 3d and Supplementary Fig. 12); (3) MQ1v-dCas9(M14), but not MQ1v-dCas9-KRAB(M22) and MQ1v-dCas9-SRDX(M30) can significantly increase genome-wide CG methylation level (Supplementary Fig. 13). The number of DMRs is consistent with the genome-wide DNA methylation analysis (Supplementary Fig. 14a). The hyper-DMRs in M12-edited T1 plants show enrichment in certain pericentromeric regions, whereas those in M14-edited T1 plants are depleted across all pericentromeric regions (Supplementary Fig. 15). Both hyper- and hypo-DMRs in M12-, M22-, and M30-edited T1 plants are enriched in Class I RdDM-targeted regions, whereas only the hypo-DMRs in M14-edited T1 plants show enrichment in these regions (Supplementary Fig. 14b).
Unexpectedly, dCas9-KYP(M6) did not trigger DNA methylation at the FT enhancer Block C in the two representative T1 plants (Fig. 3d), suggesting that the late-flowering phenotype in these plants may be attributed to H3K9me2 induced by KYP.
We found that MQ1v-dCas9(M14) induced stronger change in genome-wide CG methylation levels when targeting the FWA promoter than when targeting the FT enhancer Block C (Fig. 1g and Supplementary Figs. 4 and 13). This is reasonable since MQ1v-dCas9(M14) was transformed into a CG-hypomethylated genome (fwa epiallele plants) to methylate FWA promoter, whereas a WT genome with normal CG methylation was used to methylate FT enhancer Block C. In fwa epiallele plants, numerous unmethylated CG sites could be methylated by MQ1v-dCas9(M14), leading to a significant increase in genome-wide CG methylation levels compared to fwa plants. In contrast, only a limited number of unmethylated CG sites are available for methylation by MQ1v-dCas9(M14) when the FT enhancer Block C was targeted in the WT background, resulting in a mild increase in genome-wide CG methylation levels in corresponding T1 plants compared to WT plants (Supplementary Fig. 13). To confirm this hypothesis, we removed all methylated CG sites (ratio of mCG > 50%) in the WT background, and then compared the remaining CG methylation levels between MQ1v-dCas9(M14)-edited T1 plants and control plants (Supplementary Fig. 16). This result supports our idea, as reflected by the fact that the increase in genome-wide CG methylation in MQ1v-dCas9(M14)-edited T1 plants when targeting the FWA promoter is comparable to that in MQ1v-dCas9(M14)-edited T1 plants when targeting the FT enhancer Block C after removal of all methylated CG sites in the WT background (Supplementary Fig. 16). Together, these findings indicate that the performances of NtDRMcd-dCas9(M12), MQ1v-dCas9(M14), MQ1v-dCas9-KRAB(M22), and MQ1v-dCas9-SRDX(M30) are similar at both sites tested.
We next examined whether our tools-induced DNA methylation at the FT enhancer Block C was heritable. We analyzed the progeny of T1 plants with DNA methylation at the FT enhancer Block C and observed that a portion of the T2 plants exhibited a late-flowering phenotype, regardless of whether they carried transgene (Fig. 3e and Supplementary Fig. 17). Additionally, the DNA methylation levels of the FT enhancer Block C in T2 plants with the transgene (+) are similar to those in T2 plants without transgene (-) (Fig. 3d). Similar to the FWA results, the DNA methylation induced by our tools at the FT enhancer Block C was heritable in the absence of transgene. Genome-wide DNA methylation levels in these T2 plants are comparable to those in WT plants (Supplementary Figs. 18 and 19).
In summary, we developed five DNA methylation tools in plants: dCas9-KYP(M6) and NtDRMcd-dCas9(M12) are highly specific tools that only methylate the targeted region; MQ1v-dCas9-SRDX(M30) shows moderate specificity and efficiency; MQ1v-dCas9-KRAB(M22) and MQ1v-dCas9(M14) displays low specificity but high efficiency. Their induced DNA methylation at the target site was stably inherited in the absence of the transgene. Furthermore, we discovered that the cofactor SRDX can significantly improve the specificity of MQ1v-dCas9, albeit at the cost of decreased efficiency.
Targeted DNA demethylation at the FWA promoter
By combining three effectors, three cofactors, and two promoters, we created 30 tools for targeted DNA demethylation (Fig. 1a and Supplementary Fig. 20; D1 to D30). To test if these tools can be used in plants for targeted demethylation, the DNA methylation at the FWA promoter was selected as the first testing site (Fig. 4a). Loss of DNA methylation at the FWA promoter leads to the activation of FWA and a late-flowering phenotype (Fig. 4a). The 30 designed tools with gRNAs targeting the FWA promoter were individually transformed into WT plants and the flowering time of their T1 transformants were examined. A fraction of T1 plants showed delayed flowering, with a slightly higher proportion of late-flowering in those carrying RPS5A promoter-driven tools than in those carrying UBQ1 promoter-driven tools (Fig. 4b, d). Consequently, we focused on RPS5A promoter-driven tools, including ROS1-dCas9(D10), TET1cd-dCas9(D12), TET1cd-dCas9-p300(D18), TET1cd-dCas9-IBM1cd(D24), and TET1cd-dCas9-IBM1(D30), for further analysis. As expected, the DNA methylation level at the FWA promoter was significantly decreased in these T1 plants, which negatively correlates with the extent of FWA activation (Fig. 4c, e).

a Schematic of targeting demethylation at the FWA promoter. b WT plants and representative late-flowering T1 plants expressing TET1cd-dCas9 (D12) to illustrate differences in flowering time. c Bar graph showing FWA expression level relative to ACT2 in representative late-flowering T1 plants expressing the indicated tools and control plants. The controls and T1 plants were grown side by side. Error bars are defined as the SD of three technical replicates; data are presented as mean ± s.d.. d Dot plot of rosette leaf count at flowering-time of T1 plants transformed with the indicated tools, compared to control plants. The dashed line corresponds to the leaf number threshold (15) above which plants are considered delayed flowering relative to WT plants. The controls and T1 plants were grown side by side. e Screenshot of DNA methylation over FWA promoter in the indicated T1 plants. The controls and T1 plants were grown side by side. f Genome-wide CG methylation levels in the indicated T1 and WT plants. Source data are provided as a Source data file.
A more detailed examination of DNA methylation at the FWA promoter in these representative late-flowering T1 plants revealed that ROS1-dCas9(D10) partially demethylated the FWA promoter, TET1cd-dCas9-p300(D18) substantially reduced its DNA methylation, while TET1cd-dCas9(D12), TET1cd-dCas9-IBM1cd(D24), and TET1cd-dCas9-IBM1(D30) completely removed DNA methylation at the FWA promoter, resulting in varying degrees of demethylation at the 3’ terminus of the FWA gene (Fig. 4e). These results indicate that in our system, TET1cd outperforms ROS1 in the efficacy of DNA demethylation, whereas ROS1 provides better specificity.
Characterizing the specificity of tools for demethylating the FWA promoter
To further assess the specificity of our tools, we analyzed the WGBS data from the representative late-flowering T1 plants. We found that the genome-wide DNA methylation levels in ROS1-dCas9(D10)- and TET1cd-dCas9-p300(D18)-edited T1 plants are similar to those in WT plants (Supplementary Fig. 21); TET1cd-dCas9(D12) and TET1cd-dCas9-IBM1cd(D24) resulted in a slight decrease in genome-wide CG methylation (Fig. 4f and Supplementary Fig. 22); TET1cd-dCas9-IBM1(D30) can greatly reduce global CG methylation levels (Fig. 4f). These results indicate that ROS1-dCas9(D10) and TET1cd-dCas9-p300(D18) are highly specific demethylation editors, while TET1cd-dCas9(D12) and TET1cd-dCas9-IBM1cd(D24) are less specific ones with minor genome-wide effects, and that TET1cd-dCas9-IBM1(D30) is a tool with high efficiency (as defined by the proportion of T1 plants exhibiting a delayed-flowering phenotype) but lowest specificity. The number of DMRs is consistent with the genome-wide DNA methylation analysis (Supplementary Fig. 23a). The hypo-DMRs in D10-, D12-, D18-, D24-, and D30-edited T1 plants are depleted in the pericentromeric regions, and most correspond to MET1-targeted regions (Supplementary Figs. 23b and 24). To investigate whether there is a correlation between the specificity of demethylation editors and their protein levels, we examined the protein levels in plants carrying the respective editors (D10, D12, D18, D24, and D30). Among these, only TET1cd-dCas9(D12), which encodes a protein much smaller than 300 kDa, showed a detectable band at the expected size (Supplementary Fig. 25). There was no detectable protein for the other editors, all of which encode proteins larger than 300 kDa, consistent with the fact that detecting large fusion proteins remains technically challenging (Supplementary Fig. 25). As a result, we cannot determine whether the specificity of these demethylation editors is associated with their protein levels. Meanwhile, these findings suggest that the cofactor IBM1 and p300 can enhance the efficiency and specificity of TET1cd-dCas9 for DNA demethylation, respectively.
Targeted DNA demethylation at the FWA promoter is heritable
To test the heritability of our tools-mediated demethylation at the FWA promoter, we detected the flowering time of the progeny of these representative late-flowering T1 plants, and found that many T2 plants with the transgene (+) or without it (−) displayed a late-flowering phenotype (Fig. 5a and Supplementary Fig. 26). The proportion of late-flowering plants in T2 is depended on the degree of hypomethylation at the FWA promoter in the corresponding T1 plants, as reflected by the observation that only a few of the T2 plants of the partially demethylated T1 plants (D10) at the FWA promoter showed late-flowering (Fig. 5a and Supplementary Fig. 26). Furthermore, complete loss of DNA methylation at the FWA promoter was observed in transgene-free T2 plants (Fig. 5c, Supplementary Fig. 27 and 28). Accordingly, the expression level of FWA was comparable between T2 plants containing the transgene (+) and those lacking it (−) (Fig. 5b).

a Dot plot of rosette leaf count at flowering-time of indicated T2 plants. The dashed line corresponds to the leaf number threshold (15) above which plants are considered delayed flowering relative to WT plants. The controls and T2 plants were grown side by side. b Bar graph showing FWA expression level relative to ACT2 in the indicated T2 and control plants. The controls and T2 plants were grown side by side. Error bars are defined as the SD of three technical replicates; data are presented as mean ± s.d.. c Screenshot of DNA methylation over FWA promoter in the indicated T2 plants. Please note that the control WGBS data for WT-1 and WT-2 used here are identical to those used in Fig. 4e. Source data are provided as a Source data file.
ROS1-dCas9(D10) induced a near-complete loss of DNA methylation at the FWA promoter in T2 plants (Fig. 5c), although the corresponding T1 plants were only partially demethylated at the FWA promoter (Fig. 4e), indicating that ROS1 is capable of completely demethylation at the targeted site, but may require the continuous presence of the transgene over generations. A transgene-free T2 plant generated from ROS1-dCas9(D10)-edited T1 plant showed half of DNA methylation and FWA expression levels compared to WT plants (Fig. 5b, c), suggesting that the DNA methylation at the FWA promoter is heterozygous, with one unmethylated allele inherited from corresponding T1 plant. Overall, we conclude that our tools-mediated DNA demethylation was effectively inherited, even in the absence of transgene.
Targeted demethylation at the SDC promoter
We evaluated the general applicability of our DNA demethylation tools via targeted demethylation at an additional promoter, the SDC (Suppressor of drm1 drm2 cmt3) promoter. In WT plants, the SDC promoter is silenced by DNA methylation, and loss of this methylation activates SDC expression, leading to a curled leaf phenotype41 (Fig. 6a). We transformed the 30 tools containing gRNAs targeting the SDC promoter into WT plants and monitored the T1 transformants for the curled leaf phenotype. Unexpectedly, no curled leaf phenotype was observed in the T1 plants (Fig. 6d). To investigate why no tool induced the curled leaf phenotype in the T1 plants, we examined whether the SDC promoter is targeted by the RNA-directed DNA methylation (RdDM) pathway. Analysis of our published ChIP-seq data revealed high enrichment of Pol IV (a core component of the RdDM pathway) occupancy at the SDC promoter (Supplementary Fig. 29)42. Hence, we inferred that the RdDM pathway counteracts the activity of our tools in targeted demethylation of the SDC promoter, leading to insufficient demethylation below the requirement to induce curled leaf phenotype. To test this hypothesis, we transformed our tools into the nrpe1 mutant, which abolishes the RdDM pathway. Indeed, we observed the curled leaf phenotype in T1 plants transformed with TET1cd-dCas9(D12), TET1cd-dCas9-p300(D18), TET1cd-dCas9-IBM1cd(D24), or TET1cd-dCas9-IBM1(D30) in the nrpe1 background (Fig. 6b, d). Among the tools that induced the curled leaf phenotype, TET1cd-dCas9-IBM1(D30) triggered the highest proportion of curled leaf plants in the T1 generation (Fig. 6d), followed by TET1cd-dCas9(D12) (Fig. 6d). Both TET1cd-dCas9-p300(D18) and TET1cd-dCas9-IBM1cd(D24) resulted in very few curled leaf plants in the T1 generation. These results suggest that, as observed in demethylation of the FWA promoter, the cofactor IBM1 can improve the ability of TET1cd-dCas9, whereas cofactors p300 and IBM1cd reduce its effectiveness in DNA demethylation at the SDC promoter.

a Schematic of targeting demethylation at the SDC promoter. The loss of DNA methylation at the SDC promoter causes a curled leaf phenotype. b nrpe1 plants and representative curled-leaf T1 plants expressing TET1cd-dCas9-IBM1(D30) in the nrpe1 background to illustrate differences in leaf morphology. c Bar graph showing SDC expression level in the indicated T1, T2, and control plants. Error bars are defined as the standard deviation (SD) of three technical replicates; data are presented as mean ± s.d.. The controls and T1/T2 plants were grown side by side. d The proportion of curled-leaf plants among the indicated T1 and T2 plants in the nrpe1 and WT background. #1 to #12 represent the T2 plants generated from different T1 plants. The controls and T1 plants were grown side by side. e Screenshot of DNA methylation over SDC promoter in the indicated plants in the nrpe1 and WT background. The controls and T2 plants were grown side by side. Please note that the control WGBS data for WT-1 and WT-2 used here are identical to those used in Fig. 4e. f Genome-wide CG methylation levels in the indicated T1, T2, nrpe1, and WT plants. Source data are provided as a Source data file.
To verify that the curled leaf phenotype is a consequence of the activation of SDC expression following demethylation at its promoter, we conducted RT-qPCR and WGBS on representative curled leaf T1 plants. All these T1 plants showed activation of SDC relative to control plants (Fig. 6c). Varying degrees of loss of DNA methylation at the SDC promoter were observed in these T1 plants (Fig. 6e), among which TET1cd-dCas9(D12) and TET1cd-dCas9-IBM1(D30) completely demethylated the SDC promoter, whereas TET1cd-dCas9-p300(D18) and TET1cd-dCas9-IBM1cd(D24) partially demethylated the SDC promoter, with TET1cd-dCas9-IBM1cd(D24) having a larger effect than TET1cd-dCas9-p300(D18) (Fig. 6e). Overall, the performance of these tools in DNA demethylation at the SDC promoter is consistent with the results observed at the FWA promoter (Figs. 4e and 6e).
The cumulative demethylation effect of ROS1-dCas9(D10) at the FWA promoter across generations (Figs. 4e and 5c) prompted us to ask whether the above constructs could similarly demethylate the SDC promoter over generations and induce a curled leaf phenotype in the WT background. We checked the leaf phenotypes in T2 plants edited with two tools (D12 and D30) in the WT background. Both tools were chosen because they showed high efficacy in the nrpe1 background. We observed the curled leaf phenotype in one T2 line (1/12) edited with TET1cd-dCas9(D12) (Supplementary Fig. 30a) and in seven T2 lines (7/12) edited with TET1cd-dCas9-IBM1(D30) in the WT background (Fig. 6d, upper right), suggesting that TET1cd-dCas9-IBM1(D30) outperforms TET1cd-dCas9(D12) in demethylating the SDC promoter in the WT background. This result confirms that the cofactor IBM1 enhances the efficacy of TET1cd-dCas9. Activation of SDC expression and substantial or complete loss of DNA methylation at its promoter were observed in the curled leaf T2 plants in the WT background (Fig. 6c, e and Supplementary Fig. 30 b, c). Taken together, these results demonstrate that our tools can effectively demethylate DNA in a region targeted by the RdDM pathway in the WT background, although the effect can be seen only in a later generation.
Building on the above idea, we then tested whether ROS1-based tools (D10 and D28) can demethylate the SDC promoter in T2 plants in nrpe1 background (Fig. 6d). While no curled-leaf plants appeared among the ROS1-dCas9(D10)-edited T2 populations in the nrpe1 background (Supplementary Fig. 30d), six ROS1-dCas9-IBM1(D28)-edited T2 populations displayed this phenotype (Fig. 6d, lower right). Demethylation of the SDC promoter and activation of gene expression were observed in these curled leaf T2 plants (Fig. 6c, e), indicating that the ROS1-based tool, ROS1-dCas9-IBM1(D28), can demethylate the SDC promoter.
Characterising the specificity of tools for demethylating the SDC promoter
To assess the specificity of our tools for targeted demethylation at the SDC promoter, we observed the DNA methylation levels in the region flanking the targeted site (Fig. 6e and Supplementary Fig. 30c) and conducted genome-wide DNA methylation analysis (Supplementary Figs. 31 and 32). TET1cd-dCas9-p300(D18) specifically and partially demethylated the SDC promoter (Fig. 6e); TET1cd-dCas9(D12) and TET1cd-dCas9-IBM1(D30) induced the complete or larger loss of DNA methylation at both the targeted site and the 3’ terminus of SDC gene (Fig. 6e and Supplementary Fig. 30c); TET1cd-dCas9-IBM1cd(D24) and ROS1-dCas9-IBM1(D28) induced partial loss of DNA methylation at both the SDC promoter and the 3’ terminus of SDC gene (Fig. 6e). Consistent with these results, genome-wide DNA methylation levels in TET1cd-dCas9-p300(D18)-edited T1 plants in the nrpe1 background are similar to those in nrpe1 plants (Fig. 6f and Supplementary Figs. 31a and 32a); TET1cd-dCas9(D12)-edited T1 plants exhibited genome-wide CG methylation levels comparable to those of the control in the nrpe1 background, whereas a pronounced decrease was observed in the edited T2 plants in the WT background (Fig. 6f and Supplementary Fig. 32b); TET1cd-dCas9-IBM1(D30) triggered a slight decrease in genome-wide CG methylation in the edited T1 plants in the nrpe1 background, and a great decrease in the edited T2 plant in the WT background (Fig. 6f and Supplementary Fig. 32a); TET1cd-dCas9-IBM1cd(D24) could induce a slight decrease in genome-wide CG methylation in the edited T1 plant in the nrpe1 background (Supplementary Fig. 32a); ROS1-dCas9-IBM1(D28) caused a slight decrease in genome-wide CG methylation in the edited T2 plants in the nrpe1 background (Supplementary Fig. 32c), whereas a significant decrease in genome-wide CHG methylation was observed in these plants (Supplementary Fig. 33c). The number of DMRs is consistent with the genome-wide DNA methylation analysis (Supplementary Fig. 34a). The hypo-DMRs in D30-edited T1 plants are depleted in the pericentromeric regions, and most correspond to MET1-targeted regions (Supplementary Figs. 34b and 35). Both hyper- and hypo-DMRs in D12-, D18-, and D24-edited T1 plants are enriched in Class I RdDM-targeted regions (Supplementary Fig. 34b).
In summary, these results indicate that TET1cd-dCas9-p300(D18) is a demethylation editor with high specificity; TET1cd-dCas9(D12) and TET1cd-dCas9-IBM1cd(D24) show low specificity; TET1cd-dCas9-IBM1(D30) has a strong capacity to demethylate CG sites at the genome-wide level, although not all edited plants exhibited this extensive loss of CG methylation; ROS1-dCas9-IBM1(D28) appears to inducing the loss of genome-wide CHG methylation. Since we were unable to detect the protein for most of the demethylation editors, likely due to their very large molecular weight (greater than 300 kDa, Supplementary Fig. 25), we cannot exclude the possibility that differences in editor specificity are due to variations in protein levels caused by differences in transgene insertion sites.
Targeted DNA methylation at the SDC promoter is heritable
We then investigated whether our tools-induced demethylation at the SDC promoter was heritable. We analyzed the progeny of these tools-edited T1 plants in the nrpe1 background and found that many T2 plants showed a curled leaf phenotype, regardless of whether they contained transgene (Fig. 7a and Supplementary Fig. 36). Both T2 plants containing (+) and lacking (-) transgene showed activation of SDC expression and loss of DNA methylation at its promoter in the nrpe1 background (Fig. 7b, c). Similar results were found in the edited T2 plants in the WT background (Fig. 6c, e and Supplementary Fig. 30b, c). Thus, the demethylation mediated by these tools was efficiently inherited in the absence of the transgene.

a Dot plots showing the leaf phenotype of the indicated T2 plants. “C” and “N” indicate the curled and normal leaf phenotype, respectively. The controls and T2 plants were grown side by side. b Bar graph showing SDC expression level in the indicated T2 and control plants. Error bars are defined as the standard deviation (SD) of three technical replicates; data are presented as mean ± s.d.. The controls and T2 plants were grown side by side. c Screenshot of DNA methylation over SDC promoter in the indicated T2 plants. Please note that the control WGBS data for nrpe1-1 and nrpe1-2 used here are identical to those used in Fig. 6e. Source data are provided as a Source data file.
Noticeably, TET1cd-dCas9-IBM1(D30) induced genome-wide CG hypomethylation, and ROS1-dCas9-IBM1(D28) caused genome-wide CHG hypomethylation were retained in T2 plants in the WT background whose transgenes were segregated out (Supplementary Figs. 32d and 33c). In contrast, in the nrpe1 background, TET1cd-dCas9-IBM1(D30)-edited T2 plants lacking transgene did not retain the genome-wide CG hypomethylation generated in T1 plants (Supplementary Figs. 37 and 38).
In summary, we developed six demethylation tools in plants, including ROS1-dCas9(D10), TET1cd-dCas9(D12), TET1cd-dCas9-p300(D18), TET1cd-dCas9-IBM1cd(D24), ROS1-dCas9-IBM1(D28), and TET1cd-dCas9-IBM1(D30). In terms of specificity, ROS1-dCas9(D10) and TET1cd-dCas9-p300(D18) occupy the first place; TET1cd-dCas9(D12) and TET1cd-dCas9-IBM1cd(D24) come second; ROS1-dCas9-IBM1(D28) and TET1cd-dCas9-IBM1(D30) are the last.
Oppositely, regarding efficiency, TET1cd-dCas9-IBM1(D30) ranks first; TET1cd-dCas9(D12) comes in second; ROS1-dCas9(D10), TET1cd-dCas9-p300(D18), TET1cd-dCas9-IBM1cd(D24), and ROS1-dCas9-IBM1(D28) occupy the lowest positions. Demethylation induced by our tools at the targeted site was inheritable in the absence of transgene.
Discussion
In this study, we adopted the CRISPR/dCas9 system by combining effectors with cofactors to achieve editing DNA methylation in a targeted manner and at the genome-wide level, developing five tools for DNA methylation and six tools for DNA demethylation. Editing DNA methylation in regions with varied genomic features highlights the generality of our tools for editing DNA methylation and their characteristic performance in terms of specificity and efficiency. Importantly, DNA methylation changes induced by these tools were stably inherited in the absence of transgene, a crucial feature for their application in plant breeding.
We demonstrated that selecting appropriate cofactors can significantly improve the specificity and efficiency of editing tools, as evidenced by: (1) SRDX, a 12-amino acid peptide, improves the specificity of MQ1v-dCas9 for targeted DNA methylation; (2) IBM1 and p300 enhance the efficiency and specificity of TET1cd-dCas9 for DNA demethylation, respectively, although the underlying mechanism remains unknown. The co-factor SRDX improves the specificity of MQ1v but reduces its efficiency, while the co-factor IBM1 enhances the efficiency of TET1cd but increases off-target effects. Identifying co-factors that can improve both specificity and efficiency—or enhance one without compromising the other—represents an important direction for future research. The choice of the promoter driving dCas9 expression is crucial for the effectiveness of DNA methylation editing tools37,43,44, as reflected by that tools driven by the RPS5A promoter efficiently methylate DNA, whereas those driven by the UBQ1 promoter are less effective. We described plant demethylase ROS1-based targeted demethylation tools in plants45, showing that ROS1-dCas9(D10) exhibits high specificity, although their efficiency is significantly lower than that of human demethylase TET1cd-based tools. Our strategy may inspire the development of more efficient and/or specific DNA methylation editing tools in mammalian systems.
Taken together, the diverse tools described here make customizable DNA methylation editing possible and open avenues for functional research on DNA methylation and plant epi-breeding. The guidelines for applying our tools for editing DNA methylation are as follows: (1) if a precise change in DNA methylation at a targeted site is necessary, such as in studies of locus-specific effects of DNA methylation on gene expression and plant breeding, one can use dCas9-KYP(M6) and NtDRMcd-dCas9(M12) for targeted DNA methylation, and ROS1-dCas9(D10) and TET1cd-dCas9-p300(D18) for targeted DNA demethylation, although these tools may have low efficiency; (2) if minor off-target risks are acceptable when editing DNA methylation at a site of interest, MQ1v-dCas9-SRDX(M30) is recommended for targeted DNA methylation, while TET1cd-dCas9(D12) and TET1cd-dCas9-IBM1cd(D24) can be chosen for targeted DNA demethylation. These tools offer moderate specificity and efficiency. In general, genome-wide off-target is not retained in T2 plants that lack these editing tools; (3) if broad off-target risks are permissible or high efficiency is prioritized, MQ1v-dCas9-KRAB(M22) is suitable for this situation because it displays high efficiency but low specificity; (4) if the aim is to introduce changes in DNA methylation across the genome, such as creating epialleles, screening for DNA methylation-controlled traits, or investigating the functions of DNA methylation, MQ1v-dCas9(M14) and TET1cd-dCas9-IBM1(D30) can be used for inducing global DNA methylation and demethylation, respectively, through their potent off-target effects.
Methods
Plant materials and growth conditions
All plants were grown under long-day conditions (16 h light/8 h dark). For seedling growth, Arabidopsis seeds were plated on 1/2 Murashige and Skoog (MS) medium with 0.6% agar and 1.5% sucrose and stratified for 7 days at 4 °C in darkness before being transferred to the growth chamber (16 h light/8 h dark, 22 °C). For experiments with adult plants, seedlings were transplanted into soil in the growth chamber. All plants used in this study are in the Columbia-0 (Col-0) background. We used two primer pairs to genotype the T2 population to identify plants with or without the transgene. Each primer pair consists of one specific primer targeting the gRNAs and one primer targeting the vector backbone. Primer sequences are listed in Supplementary Data 1.
Plasmid design and construction
All constructs were cloned into the pCAMBIA1300 binary vector46. Individual modules of our system presented in this work were either amplified from the Arabidopsis thaliana genome or synthesized by the company (Jie Li Biology). The DNMT3A-3L, NtDRMcd, MQ1v, TET1cd, KRAB, SRDX, and p300 sequences were obtained from previously published papers26,28,29,32,47,48. DRM2, ROS1, ROS1cd, KYP, IBM1, IBM1cd, pUBQ1, and pRPS5A sequences were obtained from the Arabidopsis thaliana genome. 1 and 2x SV40-type NLS were placed at the N terminus and C terminus of dCas9, respectively (Supplementary Figs. 1 and 20). The effectors and cofactors were fused to the N terminus and C terminus of dCas9, respectively (Supplementary Figs. 1 and 20). The XTEN80 linker49 was used to link the effectors to dCas9 at the N terminus, whereas the XTEN16 linker50,51 was used to link the cofactors to dCas9 at the C terminus (Supplementary Figs. 1 and 20). All functional plasmids have been deposited to Addgene for use by the scientific community (Plasmid numbers: 239470; 239477; 239478; 239479; 239480, dCas9-KYP (M6); 239481, NtDRMcd-dCas9 (M12); 239482, MQ1v-dCas9 (M14); 239483, MQ1v-dCas9-KRAB (M22); 239484, MQ1v-dCas9-SRDX (M30); 239485, MQ1v-dCas9-BFP (M39); 239486, MQ1v-dCas9-cSRDX(M40); 239487, ROS1-dCas9 (D10); 239488, TET1cd-dCas9 (D12); 239489, TET1cd-dCas9-p300(D18); 239490, TET1cd-dCas9-IBM1cd(D24); 239491, ROS1-dCas9-IBM1 (D28); 239492, TET1cd-dCas9-IBM1(D30)). The construct sequences are listed in Supplementary Data 2.
gRNAs were placed downstream and upstream of the fusion protein expression cassette (Supplementary Fig. 39), respectively. U6, U3b, or 7SL drove the expression of gRNA. Six gRNAs were designed to edit DNA methylation at the FWA promoter; four gRNAs were used to edit DNA methylation at the FT enhancer Block C; and six gRNAs were selected to edit DNA methylation at the SDC promoter. The gRNA sequences are listed in Supplementary Data 1.
Flowering time measurement
The flowering time is measured by counting the number of rosette leaves, and each dot in the corresponding figures represents the rosette leaf number of individual plants. In fwa plants that are used for methylation of the FWA promoter, flowering typically occurs at the 24–36 leaf stage in our experiment conditions. Therefore, in the fwa background, plants are considered early-flowering relative to fwa plants if they flower with fewer than 21 leaves. In WT plants that are used for methylation of the FT enhancer and demethylation of the FWA promoter, flowering normally occurs at the 9–11 leaf stage in our experiment conditions. Accordingly, we define delayed flowering relative to WT plants when plants flower with more than 15 leaves.
RT-qPCR
Total RNA was subjected to reverse transcription using the TransScript One-Step gDNA Removal and cDNA Synthesis SuperMix kit (TransGen Biotech). The resulting cDNA was used as a template in a PCR reaction with Green Premix Ex Taq (Tli RNaseH Plus) (TaKaRa). All reactions were carried out on a CFX96TM Real-Time System (Bio-Rad). The constitutively expressed ACT2 was used as an endogenous control for normalization. Primer sequences are listed in Supplementary Data 1.
Immunoblotting
After 14 days of growth on 1/2 MS medium supplemented with 50 mg/L hygromycin, 20 seedlings (total weight: W mg) were ground into a fine powder in liquid nitrogen. Total proteins were extracted using 2 × W μL of protein extraction buffer (50 mM HEPES, pH 7.5; 150 mM NaCl; 1 mM EDTA; 1% Triton X-100; 0.1% sodium deoxycholate; 1% SDS; and protease inhibitor cocktail). The supernatant was then boiled with SDS loading buffer. Thirty μL of protein extract was separated by 7.5% SDS-PAGE. Proteins were transferred to a PVDF membrane and immunoblotted with an anti-Cas9 antibody (Active Motif, Cat# 61957; 1:5000 dilution). An anti-actin antibody (Sangon Biotech, Cat# D110007; 1:5000 dilution) was used as the loading control.
Whole-genome bisulfite sequencing and analysis
Genomic DNA was extracted from leaves using DNeasy Plant Mini Kit (Qiagen) and sheared to 200 bp with Covaris S220 (Covaris). Libraries were then prepared using Ultra II DNA Library Prep Kit (NEB) in conjunction with EpiTect Plus Bisulfite Kit (Qiagen) following the manufacturer’s instructions.
For data analysis, the raw paired-end reads were processed using a custom pipeline. Adapters and low-quality bases were trimmed using fastp with default parameters, and the clean reads were aligned to the TAIR10 genome using BSMAP with parameters “-v 2 -p 24 -S 1”, allowing for up to two mismatches. The methratio.py script extracted methylation ratios with parameters “-d -o -r -z -p”. The output was further processed to generate BedGraph and BigWig files for different methylation contexts (CG, CHG, CHH, and all) using awk and bedGraphToBigWig.
Two fwa epialleles with different DNA methylation landscapes were used to generate transformants for targeted DNA methylation at the FWA promoter. Consequently, a comparison of genome-wide DNA methylation levels between edited and fwa plants was conducted within the same epigenotype, using metaplot analysis.
Identification of differentially methylated regions
Differentially methylated regions (DMRs) were identified using a 200 bp sliding window with a 50 bp step size across the genome. Within each window, DNA methylation levels between edited plant samples and wild-type samples were compared using Fisher’s exact test. The resulting p-values were adjusted using the Benjamini–Hochberg method to control the false discovery rate (FDR). Differentially methylated cytosines (DMCs) were defined as sites with a methylation fold change of at least 3 and an absolute methylation difference of ≥ 8%. A genomic region was designated as a DMR if it satisfied all three of the following criteria: (1) FDR ≤ 0.05, (2) an overall methylation fold change ≥ 3, and (3) including at least 8 DMCs. Finally, DMRs located within 100 bp of one another were merged.
Class I RdDM-targeted regions are hypo-DMRs in nrpd1 relative to WT (data from GSE8380252); Class II RdDM-targeted regions are hypo-DMRs in ros1 nrpd1 compared with ros1, but not in nrpd1 compared with WT (data from GSE8380252); MET1-targeted regions are hypo-DMRs in met1 relative to WT (data from GSE3990153); CMT3-targeted regions are hypo-DMRs in cmt3 relative to WT (data from GSE3990153); CMT2-targeted regions are hypo-DMRs in cmt2 relative to WT (data from GSE16587742); DRDD-targeted regions are hyper-DMRs in drdd relative to WT (data from GSE16491654).
ATAC-seq and analysis
ATAC-seq that indicates the position of the FT enhancer Block C was performed following a published protocol55. The rosette leaves of 5-week-old WT plants were collected for nuclei extraction. For data analysis, raw paired-end reads were trimmed using Trim Galore with the “--nextera” and “--paired” parameters, and quality control was conducted using FastQC. Trimmed reads were aligned to the TAIR10 reference genome using Bowtie2, and the aligned reads were converted to BAM format using Samtools. The BAM files were sorted and indexed using Samtools, and duplicates were marked and removed with Picard’s “MarkDuplicates” function. Reads with a mapping quality score above 30 were filtered using Samtools, and mitochondrial and chloroplast DNA reads were removed using AWK and Samtools. The filtered BAM files were converted to BedGraph format using bedtools and subsequently to BigWig format using the bedGraphToBigWig tool for efficient visualization.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All high-throughput sequencing data generated in this study have been deposited in the Gene Expression Omnibus (GEO) with accession codes GSE282160 and GSE282162. Source data are provided with this paper.
References
Goll, M. G. & Bestor, T. H. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 74, 481–514 (2005).
Feng, S. et al. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. USA 107, 8689–8694 (2010).
Law, J. A. & Jacobsen, S. E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 11, 204–220 (2010).
Heard, E. & Martienssen, R. A. Transgenerational epigenetic inheritance: myths and mechanisms. Cell 157, 95–109 (2014).
Zhong, X. Comparative epigenomics: a powerful tool to understand the evolution of DNA methylation. N. Phytol. 210, 76–80 (2016).
Zhang, H., Lang, Z. & Zhu, J. K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 19, 489–506 (2018).
Schmitz, R. J., Lewis, Z. A. & Goll, M. G. DNA methylation: shared and divergent features across eukaryotes. Trends Genet. 35, 818–827 (2019).
Robertson, K. D. DNA methylation and human disease. Nat. Rev. Genet. 6, 597–610 (2005).
Brena, R. M. & Costello, J. F. Genome-epigenome interactions in cancer. Hum. Mol. Genet. 16, R96–R105 (2007).
He, L. et al. DNA methylation-free Arabidopsis reveals crucial roles of DNA methylation in regulating gene expression and development. Nat. Commun. 13, 1335 (2022).
Deng, Y. et al. Epigenetic regulation of antagonistic receptors confers rice blast resistance with yield balance. Science 355, 962 (2017).
Manning, K. et al. A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 38, 948–952 (2006).
Ong-Abdullah, M. et al. Loss of Karma transposon methylation underlies the mantled somaclonal variant of oil palm. Nature 525, 533–537 (2015).
Quadrana, L. et al. Natural occurring epialleles determine vitamin E accumulation in tomato fruits. Nat. Commun. 5, 3027 (2014).
Tang, S. et al. Targeted DNA demethylation produces heritable epialleles in rice. Sci. China Life Sci. https://doi.org/10.1007/s11427-021-1974-7 (2021).
Brzycki Newton, C., Young, E. M. & Roberts, S. C. Targeted control of supporting pathways in paclitaxel biosynthesis with CRISPR-guided methylation. Front. Bioeng. Biotechnol. 11, 1272811 (2023).
Veley, K. M. et al. Improving cassava bacterial blight resistance by editing the epigenome. Nat. Commun. 14, 85 (2023).
Gallego-Bartolome, J. DNA methylation in plants: mechanisms and tools for targeted manipulation. N. Phytol. 227, 38–44 (2020).
Gardiner, J., Ghoshal, B., Wang, M. & Jacobsen, S. E. CRISPR-Cas-mediated transcriptional control and epi-mutagenesis. Plant Physiol. 188, 1811–1824 (2022).
Kawakatsu, T. & Ecker, J. R. Diversity and dynamics of DNA methylation: epigenomic resources and tools for crop breeding. Breed. Sci. 69, 191–204 (2019).
Mercé, C., Bayer, P. E., Tay Fernandez, C., Batley, J. & Edwards, D. Induced methylation in plants as a crop improvement tool: progress and perspectives. Agronomy 10, https://doi.org/10.3390/agronomy10101484 (2020).
Li, L., Zhang, D., Zhang, Z. & Zhang, B. CRISPR/Cas: a powerful tool for designing and improving oil crops. Trends Biotechnol. https://doi.org/10.1016/j.tibtech.2024.09.007 (2024).
Nakamura, M., Gao, Y., Dominguez, A. A. & Qi, L. S. CRISPR technologies for precise epigenome editing. Nat. Cell Biol. 23, 11–22 (2021).
Lei, Y., Huang, Y. H. & Goodell, M. A. DNA methylation and de-methylation using hybrid site-targeting proteins. Genome Biol. 19, 187 (2018).
Morita, S. et al. Targeted DNA demethylation in vivo using dCas9-peptide repeat and scFv-TET1 catalytic domain fusions. Nat. Biotechnol. https://doi.org/10.1038/nbt.3658 (2016).
Lei, Y. et al. Targeted DNA methylation in vivo using an engineered dCas9-MQ1 fusion protein. Nat. Commun. 8, 16026, (2017).
Pflueger, C. et al. A modular dCas9-SunTag DNMT3A epigenome editing system overcomes pervasive off-target activity of direct fusion dCas9-DNMT3A constructs. Genome Res. 28, 1193–1206 (2018).
Nuñez, J. K. et al. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell 184, 2503–2519.e2517 (2021).
Liu, X. S. et al. Editing DNA methylation in the mammalian genome. Cell 167, 233–247.e217 (2016).
Gallego-Bartolome, J. et al. Targeted DNA demethylation of the Arabidopsis genome using the human TET1 catalytic domain. Proc. Natl. Acad. Sci. USA 115, E2125–E2134 (2018).
Ghoshal, B. et al. A viral guide RNA delivery system for CRISPR-based transcriptional activation and heritable targeted DNA demethylation in Arabidopsis thaliana. PLoS Genet. 16, e1008983 (2020).
Papikian, A., Liu, W., Gallego-Bartolome, J. & Jacobsen, S. E. Site-specific manipulation of Arabidopsis loci using CRISPR-Cas9 SunTag systems. Nat. Commun. 10, 729 (2019).
Ghoshal, B., Picard, C. L., Vong, B., Feng, S. & Jacobsen, S. E. CRISPR-based targeting of DNA methylation in Arabidopsis thaliana by a bacterial CG-specific DNA methyltransferase. Proc. Natl. Acad. Sci. USA 118, e2125016118 (2021).
Liu, W. et al. Ectopic targeting of CG DNA methylation in Arabidopsis with the bacterial SssI methyltransferase. Nat. Commun. 12, 3130 (2021).
Ji, L. et al. TET-mediated epimutagenesis of the Arabidopsis thaliana methylome. Nat. Commun. 9, 895 (2018).
Zhang, Z. M. et al. Structural basis for DNMT3A-mediated de novo DNA methylation. Nature 554, 387–391 (2018).
Gardiner, J., Zhao, J. M., Chaffin, K. & Jacobsen, S. E. Promoter and terminator optimization for DNA methylation targeting in Arabidopsis. Epigenomes 4, https://doi.org/10.3390/epigenomes4020009 (2020).
Soppe, W. J. et al. The late flowering phenotype of fwa mutants is caused by gain-of-function epigenetic alleles of a homeodomain gene. Mol. Cell 6, 791–802 (2000).
Wada, Y., Ohya, H., Yamaguchi, Y., Koizumi, N. & Sano, H. Preferential de novo methylation of cytosine residues in non-CpG sequences by a domains rearranged DNA methyltransferase from tobacco plants. J. Biol. Chem. 278, 42386–42393 (2003).
Zicola, J., Liu, L., Tanzler, P. & Turck, F. Targeted DNA methylation represses two enhancers of FLOWERING LOCUS T in Arabidopsis thaliana. Nat. Plants 5, 300–307 (2019).
Henderson, I. R. & Jacobsen, S. E. Tandem repeats upstream of the Arabidopsis endogene SDC recruit non-CG DNA methylation and initiate siRNA spreading. Genes Dev. 22, 1597–1606 (2008).
He, L. et al. Pathway conversion enables a double-lock mechanism to maintain DNA methylation and genome stability. Proc. Natl. Acad. Sci. USA 118, e2107320118 (2021).
Tsutsui, H. & Higashiyama, T. pKAMA-ITACHI vectors for highly efficient CRISPR/Cas9-mediated gene knockout in Arabidopsis thaliana. Plant Cell Physiol. 58, 46–56 (2017).
Kang, B. C. et al. Precision genome engineering through adenine base editing in plants. Nat. Plants 4, 427–431 (2018).
Parrilla-Doblas, J. T., Ariza, R. R. & Roldan-Arjona, T. Targeted DNA demethylation in human cells by fusion of a plant 5-methylcytosine DNA glycosylase to a sequence-specific DNA binding domain. Epigenetics 12, 296–303 (2017).
Zhang, Z. et al. A multiplex CRISPR/Cas9 platform for fast and efficient editing of multiple genes in Arabidopsis. Plant Cell Rep. 35, 1519–1533 (2016).
Hiratsu, K., Matsui, K., Koyama, T. & Ohme-Takagi, M. Dominant repression of target genes by chimeric repressors that include the EAR motif, a repression domain, in Arabidopsis. Plant J. 34, 733–739 (2003).
Hilton, I. B. et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 33, 510–517 (2015).
Schellenberger, V. et al. A recombinant polypeptide extends the in vivo half-life of peptides and proteins in a tunable manner. Nat. Biotechnol. 27, 1186–1190 (2009).
Lu, Y. & Zhu, J. K. Precise editing of a target base in the rice genome using a modified CRISPR/Cas9 system. Mol. Plant. 10, 523–525 (2017).
Zong, Y. et al. Precise base editing in rice, wheat and maize with a Cas9- cytidine deaminase fusion. Nat. Biotechnol. https://doi.org/10.1038/nbt.3811 (2017).
Tang, K., Lang, Z., Zhang, H. & Zhu, J. K. The DNA demethylase ROS1 targets genomic regions with distinct chromatin modifications. Nat. Plants 2, 16169 (2016).
Stroud, H., Greenberg, M. V., Feng, S., Bernatavichute, Y. V. & Jacobsen, S. E. Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 152, 352–364 (2013).
Zeng, W. et al. Roles of DEMETER in regulating DNA methylation in vegetative tissues and pathogen resistance. J. Integr. Plant Biol. 63, 691–706 (2021).
Wang, F. X. et al. Protocol for assaying chromatin accessibility using ATAC-seq in plants. STAR Protoc. 2, 100289 (2021).
Acknowledgements
We thank Detlef Weigel and Rosa Lozano-Durán for the discussion and comments on the manuscript. This work was supported by the Chinese Academy of Sciences and the National Natural Science Foundation of China (32188102 to J.-K.Z., 32100458 to L.H.).
Author information
Authors and Affiliations
Contributions
L.H. and J.-K.Z. conceived the project. Y.Y. (Yao Yao) performed the majority of the experiments. Y.Y. (Yin You) performed a few experiments. X.W. and Y.M. carried out bioinformatics analyses. L.H., Y.Y. (Yao Yao), and Z.L. analyzed the data. L.H., W.Y., and J.-K.Z. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jin Hoe Huh and the other anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
He, L., Yao, Y., You, Y. et al. Versatile molecular tools enabling customizable DNA methylation editing in Arabidopsis. Nat Commun 17, 251 (2026). https://doi.org/10.1038/s41467-025-66933-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-66933-z
