
Abstract
The vagus nerve (VN) is a major component of the parasympathetic nervous system that regulates glucose and energy homeostasis. However, the specific molecular signaling pathways within the VN that regulates this homeostasis remain unclear. Here, we show that vagal neuron-specific deletion of phosphatase and tensin homolog (Pten), the endogenous negative regulator of PI3K, led to increased vagal output. Intriguingly, dopaminergic signaling genes were upregulated, correlating with elevated sympathetic nerve density and increased norepinephrine levels in adipose tissue of vagal Pten-deficient mice. These mice were also protected against high-fat diet-induced obesity, insulin resistance, and systemic inflammation. To investigate insulin-specific PI3K signaling within the VN, we generated mice with vagal neuron-specific insulin receptor deletion that resulted in exacerbation of metabolic defects, which was rescued by concomitant Pten deletion. In summary, we show that insulin-PI3K-PTEN axis within vagal neurons is essential in optimizing the autonomic output that determines peripheral inflammatory and metabolic homeostasis.
Similar content being viewed by others


Introduction
The autonomic nervous system (ANS) plays a key role in regulating all bodily functions, including blood pressure, body temperature, digestion, respiration, cardiac output, and metabolism1. The major branches of the ANS are the parasympathetic nervous system (PNS) and the sympathetic nervous system (SNS), which together contribute to maintaining physiological homeostasis1. The PNS is generally known to promote the ‘rest and digest’ response, in contrast to the ‘fight or flight’ response that is mediated by the SNS. Although the PNS and SNS are traditionally held to oppose each other in a reciprocal manner, emerging evidence indicates that these branches may also work together to achieve optimal homeostasis in response to various conditions, such as inflammation or metabolic stress1,2,3. Interestingly, cells of these two branches of the ANS also share essential transcriptional signatures during development, including activation of the paired-liked homeobox 2B (Phox2B) gene4. Postnatally, however, Phox2B is primarily expressed in parasympathetic cholinergic neurons in the VN and in the regulatory center in the dorsal vagal complex (DVC), which consists of the nucleus tractus solitarius and dorsal motor nucleus of vagus5. The VN is the major branch of the PNS that relays important cues from the periphery via vagal afferent fibers to the DVC, which in turn relays signals to other neuronal centers such as the hypothalamus6,7. Responding signals are then sent via vagal efferent fibers that innervate peripheral organs to regulate metabolic actions, such as insulin secretion8 and hepatic gluconeogenesis9. Recent work has also shown that the VN mediates the ‘anti-inflammatory reflex’ to modulate peripheral inflammation10. Conversely, VN dysfunction leads to the breakdown of metabolic and inflammatory homeostasis.
Decline in vagal function is a hallmark of aging and a strong determinant of both insulin resistance and type 2 diabetes (T2D)11,12,13. Population studies have indicated that decreased vagal tone and dysregulated sympathovagal balance can precede T2D onset14,15. These findings reflect the critical importance of the ANS in the maintenance of glucose and energy homeostasis6. Other work has demonstrated that increased vagal function, through vagus nerve stimulation (VNS), can improve glucose homeostasis16,17. Therefore, strategies to promote and maintain vagal function may be a promising therapeutic avenue for T2D16,17. However, the molecular signaling pathways within the neurons of the VN that determine its function in the regulation of metabolism remain elusive.
In this report, we show that VN output was enhanced in mice in which PI3K signaling in Phox2B-expressing neurons within the VN was increased by deleting PI3K’s endogenous negative regulator, Pten. These mutant vagal neurons showed elevated expression of dopaminergic genes, along with increased sympathetic nerve density and norepinephrine (NE) concentrations in the white adipose tissue (WAT). These mutant mice were protected against high-fat diet (HFD)-induced obesity, insulin resistance, and inflammation. In contrast, deletion of insulin receptor (Ir) in Phox2B-expressing neurons disrupted vagal output and exacerbated HFD-induced metabolic and inflammatory abnormalities. Strikingly, these alterations were completely rescued by concomitant Pten deletion. Our work demonstrates that PI3K-mediated insulin signaling in Phox2B-expressing neurons is essential for regulating autonomic output in peripheral tissues, and thus for maintaining optimal metabolic and inflammatory homeostasis.
Results
Increased Pten and decreased vagal output in mice after high-fat diet
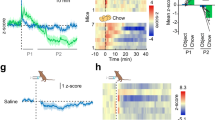
Decreased vagal activity has been shown to precede the onset of T2D14, but the specific molecular signaling pathways within the VN that drive diabetes pathogenesis remain unclear. Previous work demonstrated that Pten expression was increased in peripheral metabolic organs in various models of T2D18,19,20. In line with other tissues and cell types, Pten gene expression was also increased in the VN of wildtype C57Bl6 mice in a model of T2D induced by 3 months of HFD-feeding starting at 6 weeks of age, compared to continuing on control normal chow diet (NCD) for 3 months (Fig. 1A, B). We next measured vagal output in response to HFD by directly stimulating the cervical VN using cuff electrodes (Fig. 1C, left panel) and determining the current threshold required for triggering bradycardia21. We found diminished vagal output in response to HFD as reflected by their increased bradycardia stimulation threshold (Fig. 1C, right panel).

A Experimental schematic: Mice were weaned at 4 weeks of age, and at 6 weeks of age, were switched to either HFD or continued on NCD for 12 weeks ending at 4.5 months of age, at which time ITT, GTT and other metabolic tests were performed. B Relative Pten gene expression levels in the VN of C57Bl6 WT mice fed on continued NCD or HFD for 3 months (n = 5 for NCD-fed C57Bl6 WT mice and n = 8 for HFD-fed WT C57Bl6 mice). C Bradycardia stimulation threshold for WT mice fed on either NCD or HFD for 3 months (n = 4 for P-PTEN WT mice and P-PTEN KO mice). D Top: Diagram of tdTomato gene construct after Cre-mediated recombination in Phox2B-expressing cells. Bottom: Representative images of tdTomato expression in the indicated tissues of NCD-fed P-PTEN WT and P-PTEN KO mice. Results are representative of 6 mice for both groups after 4 weeks of age. E Left: Representative coronal sections of the brainstem showing NTS/DMV at the base of the 4th ventricle (4 V) from 4-week-old P-PTEN WT and P-PTEN KO mice and co-stained to detect immunofluorescence of pS6 (green) and tdTomato (red). Scale bars, 50 µm. Right: Quantitation of pS6 staining (n = 4 mice for both groups of mice). F Bradycardia stimulation threshold for P-PTEN WT and P-PTEN KO mice fed on either NCD or HFD for 3 months (NCD: n = 6 for P-PTEN WT mice and n = 4 for P-PTEN KO mice; HFD: n = 6 for P-PTEN WT mice and p = 4 for P-PTEN KO mice). G Umap representation of the cell clusters within the VN as determined by snRNAseq. H Volcano plot showing the most downregulated (left) and upregulated (right) genes in P-PTEN KO vs. P-PTEN WT mice fed on HFD for 2 weeks. Each group represents pooled VN (12) from 6 mice/genotype. I Left: Representative images of fluorescent co-staining to detect myelin (MBP), neuronal bundles (NeuroH) and myelinated type A fibers (NPY) of the VN in P-PTEN WT and P-PTEN KO mice fed on NCD for 3 months. Scale bars, 50 µm. Right: Quantitation of MBP, NeuroH and NPY staining (MBP: n = 4 for both groups; NeuroH: n = 4 for P-PTEN WT mice and n = 3 for P-PTEN KO mice NPY: n = 3 for both groups). Data are the mean ± SEM. p-values for (B, C, E, and I) were determined by unpaired, two-tailed Student’s t test. Unadjusted p-values are shown for H in the figure for clarity; multiple comparisons were applied with the Holm-Sidak method, and adjusted p-values are provided in data source file.
Enhanced vagal output in mice with Pten-deficiency in vagal neurons
To investigate the significance of Pten induction in the VN in response to HFD, we used the cre-loxP system to generate Phox2B cre+-Ptenflox/flox (denoted hereafter as P-PTEN KO) mice. In these conditional mutants, Pten can be deleted specifically in the Phox2B-expressing neurons that represent cholinergic parasympathetic neurons in adult mice5 (Fig. 1D, top). To validate the specificity of cre activity in these neurons, we used the Tdtomato reporter line and showed cre to be active exclusively in the DVC and VN of P-PTEN KO mice (Fig. 1D, bottom) and Pten specifically deleted in their VN and not in other tissues (Supplementary Fig. S1). Consistent with the enhanced PI3K signaling caused by the Pten deficiency specifically in these neurons, the DVC of P-PTEN KO mice showed increased expression of phospho-S6, a downstream target of the PI3K-AKT pathway compared to control Phox2B cre+-Ptenwt/wt (denoted hereafter as P-PTEN WT) mice (Fig. 1E).
We next measured vagal output in P-PTEN KO and P-PTEN WT mice under NCD and HFD-fed conditions. Under NCD-fed conditions, P-PTEN KO mice exhibited enhanced vagal output compared to P-PTEN WT littermate controls, as assessed by their decreased bradycardia stimulation threshold. Remarkably, after 3 months of HFD, P-PTEN KO mice maintained their enhanced vagal output in contrast to P-PTEN WT littermate controls that showed a decline in vagal output as evidenced by increased bradycardia stimulation threshold (Fig. 1F). Thus, PI3K signaling and its regulation by PTEN is critical for maintaining optimal VN output.
Molecular changes induced by vagal Pten deletion
To investigate the molecular changes in the VN that are regulated by Pten, we used an unbiased approach and performed single-nucleus RNA sequencing (snRNAseq) of cervical VNs-containing nodose and jugular ganglia (from left and right VN respectively) isolated from P-PTEN KO and P-PTEN WT littermate mice. They were fed an HFD for two weeks to capture early molecular changes of metabolic stress. ~ 7500 nuclei isolated from pooled VNs (12 VN from 6 mice per genotype) revealed at least nine distinct cellular clusters, including neurons, microglia, and astrocytes (Fig. 1G), consistent with a previous report22. Indeed, we confirmed Pten deletion in P-PTEN KO mice was indeed restricted to neuronal populations within the VN, and did not affect non-neuronal clusters (Fig. S2A, red circle). Differential gene expression analysis of these neuronal clusters revealed 281 downregulated and 336 upregulated genes in P-PTEN KO mice compared to P-PTEN WT littermate controls (Fig. 1H). Notably, the most prominently upregulated genes in the mutant VN included those associated with neuronal development and size (Tmsb10 and Prph), neurite and axonal growth (Tub1a1, Tpp3, Stmn2, Sncg, Prph), and anti-oxidation (Uchl1) (Fig. 1H), results that we validated by qRT-PCR (Fig. S2B, C).
In keeping with the increased expression of genes linked to neuronal growth and development, we also observed an increase in myelin in the VN of P-PTEN KO mice compared to P-PTEN WT controls, as indicated by increased staining for myelin basic protein (MBP), which are primarily expressed within myelin-producing oligodendrocytes (Fig. 1H, I). Concomitantly, large diameter Neurofilament H (NeuroH)-positive nerve bundles staining for all myelinated nerve bundles and myelinated type A nerves expressing Neuropeptide Y (NPY) were increased in cross-sections of P-PTEN KO VN (Fig. 1I). These richly myelinated nerves are capable of high conductivity, with elevated firing action potential at an increased rate23. Thus, P-PTEN KO mice show improved vagal output along with genetic and histological changes that are associated with enhanced nerve function. These results indicate that Pten in vagal neurons is an essential regulator of neuronal growth and myelination.
Increased sympathetic innervation in the adipose tissue of P-PTEN KO mice
Further examination of our snRNAseq analysis of neuronal clusters within the VN of P-PTEN KO and WT mice revealed remarkable transcriptional changes for factors involved in the catecholamine biosynthetic pathway (Fig. 1H). Genes affected included dopa decarboxylase (Ddc) and dopamine beta-hydroxylase (Dbh), which encode key enzymes responsible for the dopamine synthetic pathway. Specifically, Ddc converts L-DOPA to dopamine24, while Dbh converts dopamine to NE25, and dopamine and NE are classical neurotransmitters of the SNS. As noted above, although the SNS and PNS may be reciprocally opposed in their functions to maintain physiological homeostasis1, these two autonomic branches may also be co-regulated by common upstream C1 neurons, and thereby may cooperate to regulate some processes such as inflammation2,3.
To investigate the significance of altered sympathetic neurotransmitter transcription in Pten-deficient vagal neurons, we assessed for sympathetic innervation in the perigonadal white adipose tissue (pWAT) of P-PTEN KO and P-PTEN WT mice by immunostaining for tyrosine hydroxylase (TH), a widely used marker for sympathetic nerves. Given that TH can be produced endogenously by adipocytes in addition to sympathetic nerves26, we utilized Adipo-Clear27 to visualize the 3D morphology of TH-expressing sympathetic nerves. Remarkably, we found increased sympathetic nerve density in pWAT of P-PTEN KO mice as determined by immunostaining of TH (Fig. 2A, B). Consistent with their increased TH nerve density, NE was also increased in pWAT of P-PTEN KO mice compared to WT controls in both chow- and HFD-fed groups (Fig. 2C). Furthermore, TH expression by western blot was also increased in pWAT of HFD-fed P-PTEN KO mice compared to HFD-fed WT controls (Supplementary Fig. S3). Thus, P-PTEN KO mice that exhibit elevated vagal output show enhanced sympathetic innervation in their pWAT compared to control P-PTEN WT mice.

A Representative 3D images of pWAT prepared from 4.5-month-old P-PTEN WT and P-PTEN KO mice on NCD (n = 3 for P-PTEN WT mice and n = 4 for P-PTEN KO mice) and immunostained for tyrosine hydroxylase (TH; blue) post-clearing. Images in the bottom panels are higher magnification renderings of the tissue in the insets in the top panels. Top scale bars, 2000 µm; bottom scale bars, 300 µm; (B) and their quantification. C Norepinephrine levels in pWAT isolated from 4.5-month-old P-PTEN WT and P-PTEN KO mice on NCD (n = 7 for all groups). D Representative whole mount images of pWAT prepared from P-PTEN WT and P-PTEN KO mice fed a HFD for 3 months starting at 6 weeks of age and immunostained to detect CD206 (red). Scale bar, 50 µm. E, F Quantification of the (E) size and (F) number per slide of adipocytes in (D) (n = 6 for both P-PTEN WT and P-PTEN KO mice). G Representative MRI images of coronal sections of P-PTEN WT and P-PTEN KO mice fed an HFD for 3 months starting at 6 weeks of age. H Weights of the indicated adipose tissues as percentages of body weight for P-PTEN WT and P-PTEN KO mice fed a HFD for 3 months after 6 weeks of age (n = 19 for P-PTEN WT mice and n = 10 for P-PTEN KO mice). I, J Relative gene expression of the indicated (I) anti- and (J) pro-inflammatory gene markers in pWAT of mice in (H) (anti-inflammatory genes: n = 11 for P-PTEN WT and n = 6 for P-PTEN KO mice; pro-inflammatory genes: n = 10 for P-PTEN WT mice and n = 5 for P-PTEN KO mice). K Representative “cytometry by time of flight” (CyTOF) analysis showing dot plots for macrophage polarity and fibrogenic PDGFRα+-CD9hi fractions, and (L) spanning-tree progression analysis (SPADE) of SVF from pWAT after 3-months of HFD-fed pWAT. M Relative gene expression of the indicated markers of adipose-related processes in pWAT from mice after 3 months of HFD (Adipogenesis/Beiging Genes: n = 13 for P-PTEN WT mice and n = 9 for P-PTEN KO mice per group; Lipolysis genes: n = 13 for P-PTEN WT mice, n = 6 for P-PTEN KO mice; Lipogenesis genes: n = 8 for P-PTEN WT mice and n = 6 for P-PTEN KO mice). Data are the mean ± SEM. P-values were determined for (B, E and F) by unpaired, two-tailed Student’s t test. Unadjusted p-values are shown for (C, H, I, J, and M) in the figure for clarity; multiple-comparisons were applied with the Holm-Sidak method, and adjusted p-values are provided in data source file.
Anti-inflammatory state in the adipose tissue of P-PTEN KO mice
Activation of sympathetic nerves in adipose tissue enhances energy expenditure and lipolysis28. When we compared the pWATs of P-PTEN WT and P-PTEN KO mice after 3 months of HFD feeding, we found a greater number of smaller adipocytes (Fig. 2D, E) and an increase in adipocyte numbers in PTEN-deficient mice (Fig. 2F). These mice had reduced adiposity, which was clearly visible by MRI (Fig. 2G), and confirmed by a reduction in adipose depot weights even after normalization to total body weight (Fig. 2H). This lower adiposity in males was similarly present in female P-PTEN KO mice compared to P-PTEN WT littermate counterpart controls (Supplementary Fig. S4A).
We next measured markers of inflammation in various WAT depots of HFD-fed P-PTEN WT and P-PTEN KO mice. pWAT of PTEN mutant mice showed increased gene expression of several anti-inflammatory (Fig. 2I) and decreased pro-inflammatory genes (Fig. 2J). In support of these findings, analysis of the stromal vascular fraction (SVF) of pWAT by “cytometry by time of flight” (CyTOF) showed increased CD206+ anti-inflammatory and decreased CD11C+ pro-inflammatory macrophage populations in P-PTEN KO mice compared to P-PTEN WT controls (Fig. 2K, top). In addition, we detected fewer pro-fibrogenic CD9high PDGFRA+ adipose progenitors29 in pWAT of P-PTEN KO mice (Fig. 2K, bottom). “Spanning-tree progression analysis” (SPADE) further demonstrated the overall prominence of the anti-inflammatory phenotype of macrophages in the SVF of PTEN mutant compared to WT control mice (Fig. 2L).
In addition to genes involved in inflammation, we examined markers of adipogenesis (PPARγ and adiponectin), that are known to promote insulin sensitivity, increased in the adipose depots of HFD-fed P-PTEN KO mice compared to P-PTEN WT controls (Fig. 2M). In line with the increased sympathetic innervation in the pWAT of P-PTEN KO mice (Fig. 2B), there was also increased expression of genes associated with beiging (Cox8B and Cidea) with no difference in Ucp-1 (Fig. 2M). Increase in gene markers of lipolysis (Atgl and Hsl), but no differences in those of lipogenesis (Fasn and Srebp-1C) (Fig. 2M) were observed. Together, these data show that HFD-fed P-PTEN KO mice exhibit enhanced sympathetic innervation in their adipose tissue that is accompanied by gene markers of increased anti-inflammatory state and insulin sensitivity.
P-PTEN KO mice are protected against HFD-induced metabolic abnormalities
We next assessed whole-body metabolic parameters in P-PTEN WT and P-PTEN KO mice fed an NCD or HFD. Upon HFD feeding, both male (Fig. 3A) and female (Supplementary Fig. S4B) mutant mice were protected against weight gain compared to their respective P-PTEN WT littermate controls. Weights of individual organs in all mutants (apart from adipose depots) were unchanged when normalized to total body weight (Fig. 4B, Supplementary Fig. S4C). The mutant mice also showed no differences in body weight compared to P-PTEN WT controls when fed an NCD (Supplementary Fig. S5A). This protection against HFD-induced weight gain in P-PTEN KO mice was not associated with changes in food intake (Supplementary Fig. S5B, C). Interestingly, these mutant mice did exhibit an increase in average meal duration (Supplementary Fig. S5D), which is known to be regulated by vagal function30 and associated with weight loss31. Furthermore, these P-PTEN KO mice showed no differences in stool mass or its lipid content (Supplementary Fig. S5E, F), indicating that altered food absorption was also not likely to be a contributing factor to their reduced body weight.

A, B Body and organ weights normalized to total body weights of P-PTEN WT and P-PTEN KO male mice fed HFD for 3 months (Body weights: n = 14 for P-PTEN WT mice and n = 11 for P-PTEN KO mice; Organ weights: n = 22 for P-PTEN WT mice and n = 10 for P-PTEN KO mice). C–G Daily non-normalized metabolic caging data for the mice fed HFD for 3 months, including: (C) energy expenditure, (D) oxygen consumption (VO2), (E) carbon dioxide production (VCO2), (F) respiratory exchange ratio, and (G) locomotion. Dark and light refer to the 12 h on/off light cycle (Energy Expenditure/RER/VO2/VCO2: n = 7 for both groups of mice; Locomotion: n = 8 for both groups of mice). H, I (H) Glucose and (I) insulin tolerance tests (and (J) fasting serum insulin of mice fed a HFD for 3 months (A, B) (GTT: n = 13 for P-PTEN WT mice and n = 12 for P-PTEN KO mice; ITT: n = 14 for P-PTEN WT and n = 12 for P-PTEN KO mice, n = 8-10 per group; baseline insulin: n = 8 for P-PTEN WT mice and n = 10 for P-PTEN KO mice). Data are the mean ± SEM. p-values for (J) were determined by an unpaired, two-tailed Student’s t test. Unadjusted p-values are shown for (B–I) in the figure for clarity; multiple comparisons were applied with the Holm-Sidak method, and adjusted p-values are provided in the data source file.

A Representative images of liver sections P-PTEN WT and P-PTEN KO mice (n = 6 /group) fed a HFD for 3 months, stained with H&E (top) or Oil red O (bottom). Scale bars, 30 µm.; (B) and triglyceride and (C) NE levels of their livers (Triglyceride: n = 10 for P-PTEN WT and n = 8 for P-PTEN KO mice; Norepinephrine: n = 7 for NCD P-PTEN WT and P-PTEN KO mice and n = 8 for HFD P-PTEN WT and P-PTEN KO mice). D–F Relative gene expression of (D) anti-inflammatory, (E) pro-inflammatory, and (F) lipid metabolic markers in their livers and (G–I) analytes found in their serum (Metabolic genes: n = 17 for P-PTEN WT mice and n = 10 for P-PTEN KO mice; Anti-inflammatory genes: n = 15 for P-PTEN WT mice and n = 9 for P-PTEN KO mice; Pro-inflammatory genes: n = 14 for P-PTEN WT mice and n = 11 for P-PTEN KO mice; Multiplex: adiponectin: n = 4 for P-PTEN WT mice and n = 5 for P-PTEN KO mice; and n = 7 per group for all other analytes). Data are the mean ± SEM. p-values for (B, G, H and I) were determined by unpaired, two-tailed Student’s t test. Unadjusted p-values are shown for (C–F) in the figure for clarity; multiple comparisons were applied with the Holm-Sidak method and adjusted p-values are provided in data source file.
We next examined various metabolic and behavioral parameters that may contribute to their body weight differences. We found that HFD-fed P-PTEN KO mice showed increased energy expenditure as measured by elevated VO2 consumption and VCO2 production (Fig. 3C–E). These differences were accompanied by an increase in the respiratory exchange ratio (RER) (Fig. 3F), which indicates a shift in energy source that favors carbohydrates over lipids, and opposes the energy fuel shift efficiency and weight gain that normally occurs with HFD-feeding32. We also measured plasma lipid metabolites and found that HFD-P-PTEN KO mice showed a decrease in total fatty acids that was primarily due to reduced saturated fatty acids and decreased 16:0 palmitic acid (Fig. S6A–C). These results are in contrast to the increase in these lipid metabolites that typically occurs with HFD-feeding, which are known to promote obesity and insulin resistance33. Finally, the mutant mice on HFD had a higher basal body temperature and were more cold-tolerant than P-PTEN WT controls (Supplementary Fig. S6D), in contrast to worsening cold intolerance that is linked to obesity34. These findings are in line with the increased sympathetic innervation in the adipose tissue of P-PTEN KO mice (Fig. 2B).
Enhanced vagal function is also associated with elevated physical activity35,36. Interestingly, P-PTEN KO mice exhibited an increase in locomotion (Fig. 3G), which likely contributed (at least in part) to their enhanced energy expenditure (Fig. 3C). We next asked if the overall increase in energy expenditure in the mutant mice was due to their enhanced locomotion or rather to tissue-intrinsic factors driving an increase in energy expenditure. To address this question, we measured respiration ex vivo in the quadriceps muscles of NCD- or HFD-fed P-PTEN WT and P-PTEN KO mice by stimulating various mitochondrial complexes in the mitochondrial respiratory chain with different concentrations of ADP and succinate37,38. We detected no differences in respiration for any mitochondrial complex at any ADP or succinate concentration (Supplementary Fig. S7). These results indicate that the increased energy expenditure in HFD-fed P-PTEN KO mice most likely results from their increased locomotion, rather than from intrinsic factors determining tissue respiration.
Finally, we assessed glucose homeostasis in NCD- and HFD-fed P-PTEN WT and P-PTEN KO mice. P-PTEN KO mice showed improved glucose levels during glucose and insulin tolerance tests (GTT and ITT) as early as 6 weeks of age compared to P-PTEN WT controls (Supplementary Fig. S8A-B), and further improved after 3 months of either NCD (Supplementary Fig. S8C, D) or HFD (Fig. 3H, I). Fasting insulin levels were also lower in HFD-fed P-PTEN KO mice, indicating enhanced peripheral insulin sensitivity (Fig. 3J). These changes in males were also present in female P-PTEN-KO mice (Supplementary Fig. S9A, B). Thus, P-PTEN KO mice exhibit enhanced autonomic output and are protected against HFD-induced metabolic defects.
Anti-inflammatory state in P-PTEN KO mice
The VN drives the “anti-inflammatory reflex” to modulate systemic inflammation, and also innervates key metabolic organs, including the liver and skeletal muscle, to modulate metabolic regulation1,10. We found that P-PTEN KO mice were substantially protected against HFD-induced fatty liver, as shown by decreased lipid droplets and triglyceride content in their liver compared to those in livers of HFD-fed P-PTEN WT littermate controls (Fig. 4A, B). As found in pWAT of P-PTEN KO mice, NE levels were also increased in the livers of these mice (Fig. 4C), but there were no changes in TH protein expression (Supplementary Fig. S10). We also observed increased several anti-inflammatory and decreased pro-inflammatory gene markers (Fig. 4D, E), as well as reductions in markers of lipogenesis (Pparγ, Srebp-1C, Fasn) in livers of mutant mice (Fig. 4F). In keeping with these favorable metabolic and inflammatory changes, we also found improved glucose levels during pyruvate tolerance test at 6 weeks of age, indicating improved hepatic gluconeogenesis (Supplementary Fig. S11).
The anti-inflammatory state was also found in the subcutaneous WAT, BAT, skeletal muscle (Supplementary Fig. S12A–F), and systemically, with increased levels of the anti-inflammatory (IL-4 and IL-10), and decreased levels of the pro-inflammatory cytokines (CCL2 and IL-6), in the serum of P-PTEN KO mice (Fig. 4G, H). Lastly, increased adiponectin and decreased leptin were observed in the serum of these P-PTEN KO mice (Fig. 4I), in line with their enhanced insulin sensitivity and decreased adiposity. However, we found no major changes in both phospho- or total AKT protein expression in the skeletal muscle or the liver of P-PTEN KO mice compared to P-PTEN WT control mice (Supplementary Fig. S13A–C).
Essential role of vagal insulin signaling in metabolic homeostasis
We next investigated whether insulin signaling within Phox2B-expressing neurons was responsible for the overall favorable metabolic and anti-inflammatory state in P-PTEN KO mice. Insulin is a major metabolic hormone that binds to the Insulin receptor (Ir) and utilizes the PI3K pathway39. However, many other factors or hormones can also activate PI3K signaling, including leptin and glucagon-like peptide (GLP)−140,41. Therefore, to examine the role of insulin signaling per se within the VN, we generated mice with insulin receptor (Ir) deletion specifically in Phox2B-expressing neurons (Phox2Bcre+-Irflox/flox; denoted hereafter as P-IR KO mice), as well as control Phox2Bcre+-Irwt/wt (denoted as P-IR WT) mice. These P-IR KO mice showed decreased vagal output as evidenced by an increased bradycardia stimulation threshold upon direct stimulation of the VN (Fig. 5A). This result demonstrates the essential role of insulin signaling in determining vagal output under basal conditions. Furthermore, these mice showed a decrease in phospho-S6 within the DVC (Fig. 5B). P-IR KO mice on NCD showed no differences in body weight compared to P-IR WT controls (Supplementary Fig. S14A) but did exhibit spontaneous glucose intolerance and resistance to the glucose-lowering effects of insulin during GTT and ITT, respectively, as early as 6 weeks of age (Supplementary Fig. S14B, C).

A Bradycardia stimulation threshold values for 4.5-month-old P-IR WT and P-IR KO mice on NCD (n = 3 for P-IR WT mice and n = 5 for P-IR KO mice). B Left: Representative coronal sections of the brainstem showing NTS/DMV at the base of the 4th ventricle (4 V) and co-stained to detect immunofluorescence of pS6 (green) and tdTomato (red). Scale bars, 50 µm. Right: Quantitation of the pS6 staining (n = 4 mice for both groups). C–F (C) Body weights, (D) glucose tolerance test, (E) insulin tolerance test, (F) organ weights as a percentage of body weight, and (G) adipose tissue depot weights in P-IR WT and P-IR KO male mice fed a HFD for 3 months (Body weight : n = 7 for P-IR WT mice and n = 10 for P-IR KO mice; GTT, n = 6 for P-IR WT mice and n = 7 for P-IR KO mice; ITT, n = 5 for P-IR WT and n = 8 for P-IR KO mice; organ and adipose weights: n = 5-6 for P-IR WT mice and n = 7-8 for P-IR KO mice). H–J Relative gene expression of the (H) anti-inflammatory, (I) pro-inflammatory, and (J) metabolic markers in pWAT from mice after 3 months of HFD (C–G). (anti-inflammatory/pro-inflammatory genes: n = 7 for P-IR WT mice and n = 8 for P-IR KO mice; metabolic genes: n = 8 for P-IR WT mice and n = 10 for P-IR KO mice). Data are the mean ± SEM. p-values for (A, B) were determined by unpaired, two-tailed Student’s t test. Unadjusted p-values are shown for (C–J) in the figure for clarity; multiple-comparisons were applied with Holm-Sidak method and adjusted p-values are provided in data source file.
We next examined the metabolic parameters in P-IR KO mice in response to HFD. P-IR KO mice showed exacerbated weight gain compared to their P-IR WT counterparts (Fig. 5C). This greater body weight gain was accompanied by a modest reduction in energy expenditure and an increase in food intake during the dark period of the diurnal cycle, but no increase in locomotion compared to controls (Supplementary Fig. S14D–F). These P-IR KO mice were more glucose intolerant and had higher glucose levels during ITT reflecting more insulin resistance compared to their P-IR WT counterparts (Fig. 5D, E). The increased body weight gain in P-IR KO mice was accompanied by greater adiposity and organ weights compared to littermate controls when normalized to total body weight (Fig. 5F, G). Moreover, in contrast to P-PTEN KO mice, these metabolic abnormalities were associated with some decreased anti-inflammatory and similar pro-inflammatory gene expression in peripheral tissues in the pWAT (Fig. 5H, I), with a similar trend in the liver, BAT, and skeletal muscle (Supplementary Fig. S15A–F). No differences in expression of total TH protein levels (Supplementary Fig. S16) or gene markers of adipogenesis, lipolysis, or beiging were observed in the pWAT of P-IR KO mice (Fig. 5J). The findings in males were not present in female P-IR KO mice with respect to exacerbated body weight gain, glucose intolerance and resistance to glucose-lowering effects of insulin during GTT and ITT, respectively to glucose (Supplementary Fig. S17A–C). Together, these data support an important role for insulin signaling in the VN to regulate the vagal output critical for maintaining metabolic and inflammatory homeostasis.
Concomitant Pten deletion rescues the metabolic defects in P-IR KO mice
To assess whether enhancing PI3K signaling could overcome the metabolic defects present in P-IR KO mice, we generated Pten/Ir double KO mice (denoted hereafter as DKO mice) lacking both PTEN and IR specifically in Phox2B-expressing neurons. Remarkably, concomitant Pten deletion in P-IR KO mice restored normal vagal output under NCD-fed conditions (Fig. 6A), while also protecting the animals from the body weight gain during HFD-feeding (Fig. 6B). This decreased body weight gain of DKO mice was accompanied by an increase in energy expenditure compared to P-IR KO mice with no changes in locomotion and daily food intake (Supplementary Fig. S18A–C). The exacerbations in HFD-induced insulin resistance and glucose intolerance in P-IR KO mice were also abolished in DKO mice (Fig. 6C, D), as well as the increased adipose tissue weights observed in P-IR KO mice (Fig. 6E, F). These findings in males were recapitulated in female DKO mice, which showed reduced HFD-induced body weight gain and improved glucose and insulin tolerance compared to female P-IR KO mice (Supplementary Fig.. S19A–C).

A Bradycardia stimulation threshold values for 4.5-month-old P-IR WT, P-IR KO PTEN-KO and DKO mice on NCD (n = 3 for P-IR WT mice, n = 5 for P-IR KO mice, n = 3 for P-PTEN KO mice, n = 5 for DKO mice). B–F (B) Body weights, (C) glucose tolerance test, (D) insulin tolerance test, (E) organ weights as a percentage of body weight, and (F) adipose tissue depot weights as a percentage of body weight in P-IR WT, P-IR KO, PTEN-KO and DKO male mice fed a HFD for 3 months (Body weights: n = 7 for P-IR WT mice, n = 9 for P-IR KO mice, n = 8 for P-PTEN KO mice and n = 12 for DKO mice; GTT: n = 7 for P-IR WT mice, n = 7 for P-IR KO mice, n = 9 for P-PTEN KO mice, n = 11 for DKO mice; ITT: n = 8 for P-IR WT mice, n = 8 for P-IR KO mice, n = 10 for P-PTEN KO mice and n = 9 for DKO mice; Organ weights: n = 6 for P-IR WT mice, n = 6 for P-IR WT mice, n = 10 for P-IR KO mice, n = 9 for P-PTEN KO mice and n = 9 for DKO mice; Adipose weights: n = 5 for P-IR WT mice, n = 8 for P-IR KO mice, n = 9 for P-IR KO mice and n = 8 for DKO mice). G, H Relative gene expression of (G) anti- and pro-inflammatory, and (H) metabolic markers in pWAT of mice after 3 months of HFD (anti-inflammatory: n = 12 for P-IR WT mice, n = 11 for P-IR KO mice, n = 4 for P-PTEN Ko mice and n = 8 for DKO mice; pro-inflammatory: n = 12 for P-IR WT mice, n = 12 for P-IR KO mice, n = 9 for P-PTEN KO mice and n = 12 for DKO mice; metabolic: n = 13 for P-IR WT mice, n = 13 for P-IR KO mice and n = 12 for DKO mice); (I–K) and their levels of (I) anti-inflammatory, (J) inflammatory and (K) metabolic markers in serum (adiponectin: n = 4 for P-IR WT mice and n = 5 for all other groups; n = 6 for P-IR WT mice, n = 7 for P-IR KO mice, n = 8 for P-PTEN KO mice and n = 7 for DKO mice for all other analytes). Data are the mean ± SEM. p-values were determined for (B–I) by two-way ANOVA multiple comparison with Tukey’s correction. p-values were determined for (A, J, K and L) by one-way ANOVA multiple comparison with Tukey’s correction.
To further study the effects of restoring vagal output in DKO mice, we measured metabolic and inflammatory changes in DKO mice after HFD. We observed increased expression of some anti-inflammatory genes along with decreased expression of pro-inflammatory markers compared to P-IR KO control mice (Fig. 6G, H). Metabolic genes associated with adipogenesis and lipolysis were also at least partially restored in the pWAT of DKO mice compared to that of P-IR KO mice (Fig. 6I). In line with these findings, serum levels of anti-inflammatory cytokines were increased and pro-inflammatory cytokines were decreased in DKO mice compared to those of P-IR KO mice (Fig. 6J, K). Lastly, serum levels of adiponectin and leptin were also at least partially restored in DKO mice compared to those of P-IR KO mice (Fig. 6L).
Together, these results offer genetic evidence that preserving PI3K signaling by disrupting its endogenous brake, Pten, is both essential for enhancing vagal output and sufficient for overcoming the metabolic defects of vagal insulin resistance.
Discussion
T2D and obesity are growing global epidemics despite recent advances in therapy42. Novel treatment paradigms, including non-pharmacological strategies, are therefore needed. One such potential approach would be to harness the VN to either prevent or treat T2D10,43, based on the observation that the decline in vagal function that occurs with aging11,12,13 is both associated with worsening glucose homeostasis44 and predicts T2D onset14. However, despite a large body of collective evidence supporting the importance of the VN’s role in metabolic regulation, the intracellular molecular signaling pathways within the VN that control vagal function remain poorly defined.
In this study, we demonstrate that insulin-PI3K signaling within the VN is a key molecular pathway regulating optimal vagal output. We show in mice that, upon the metabolic challenge caused by HFD, a rise in Pten occurs in the VN that is associated with disrupted PI3K signaling in the DVC and the VN.
To determine whether increased Pten and the consequent disruption in PI3K signaling in the VN neuronal population might cause VN dysfunction and the metabolic abnormalities induced by HFD, we generated P-PTEN KO mice in which Pten was deleted exclusively in parasympathetic cholinergic Phox2B-expressing neurons located in the DVC and VN. snRNAseq analyses of the VN neuronal population revealed a neuroprotective signature involving genes governing neuron development and size, as well as neurite and axonal growth. The VN in these animals showed increased myelination and enhanced vagal output. Interestingly, these features were linked to elevated expression of Dbh and Ddc, genes related to the dopaminergic synthesis pathway that is traditionally associated with the SNS. In line with these transcriptional changes, the WAT of these mutant mice showed increased sympathetic nerve density as measured by TH immunostaining, along with increased NE concentrations indicative of enhanced sympathetic innervation. Interestingly, this elevated autonomic output in P-PTEN KO mice was accompanied by an anti-inflammatory phenotype within the adipose tissue that protected against the metabolic abnormalities induced by HFD-feeding. We also observed an anti-inflammatory state in peripheral metabolic tissues, including the skeletal muscle, liver, and also systemically in the serum. Glucose homeostasis and insulin sensitivity were also improved. As a result, P-PTEN KO mice were able to resist HFD-induced weight gain and exhibited increased energy expenditure due largely to greater locomotion.
Multiple hormones and growth factors utilize the PI3K signaling pathway. Given the beneficial effects of enhancing PI3K signaling by deleting Pten in Phox2B+ neurons, we wanted to focus on insulin-mediated PI3K signaling in the VN. Mice in which insulin signaling was disrupted by Ir deletion specifically in Phox2B+ neurons showed defective efferent vagal output as assessed by an increase in the stimulation threshold for bradycardia. These mice also showed enhanced peripheral insulin resistance, glucose intolerance, and a pro-inflammatory state in peripheral metabolic tissues and serum, and these abnormalities were further exacerbated upon HFD-feeding. These metabolic defects in P-IR KO mice illustrate the essential role of insulin signaling in maintaining the vagal output needed to ensure proper regulation of metabolic and inflammatory homeostasis.
Since many signaling branches, including those involving PI3K and MAPK, operate in response to insulin, we generated DKO mice lacking both Ir and Pten in Phox2B-expressing neurons. We analyzed these double mutants to assess whether enhancing the PI3K pathway by deleting Pten could overcome the defects present in P-IR KO mice. Indeed, Pten deletion was able to rescue the abnormalities of P-IR KO mice since DKO mice exhibited enhanced vagal output. Our work thus provides genetic evidence that PI3K signaling that mediates insulin action in these neurons is crucial for regulating vagal output and metabolic homeostasis.
Insulin and its downstream signaling pathways are highly evolutionarily conserved from mammals down to lower organisms such as Drosophila melanogaster and Caenorhabditis elegans45,46. In these lower organisms, insulin is predominantly produced in the nervous system45,46, and the ANS may execute the primitive neuronal functions that regulate the fundamental metabolic processes of the organism. Thus, insulin action within the ANS could represent an important concept of metabolic regulation that originated in the insulin-producing cells of primitive organisms. Indeed, our previous study has shown that, in mice with Pten-deficiency in rat insulin promoter-expressing neurons led to improved glucose homeostasis with increased insulin sensitivity and an anti-inflammatory state20. In our current study, we focused on the effects of Pten deletion in cholinergic parasympathetic neurons without the confounding presence of rat insulin promoter-expressing cells in the pancreas.
Our snRNAseq analysis of Pten-deficient vagal neurons revealed an upregulation of genes encoding enzymes that are involved in the synthesis of epinephrine and NE, and are generally associated with the SNS. This observation of increased NE in peripheral metabolic tissues of P-PTEN KO mice that exhibit enhanced vagal output may further challenge the conventional dogma of the reciprocal inhibitory roles of the SNS and PNS1. Our work supports a recent demonstration that C1 neurons located in the medulla have key regulatory effects on the PNS as well as on the SNS, which are both involved in the regulation of inflammation in peripheral tissues such as the kidney and spleen3,4. Technological advances in molecular characterization at the single-cell level and cell fate tracking have shown that the relationship between these two seemingly opposing autonomic functions may be more complex than originally defined47.
While Phox2B is specifically expressed in parasympathetic cholinergic neurons in adult mice5, during embryonic development, these neurons give rise to the entirety of the ANS, including the SNS4. Thus, the upregulation in Pten-deficient vagal neurons of genes encoding enzymes involved in NE synthesis suggests the intriguing possibility that autonomic neurons can be reprogrammed to a novel cell fate that can promote both sympathetic and parasympathetic output in a unique manner not previously described. Accordingly, we found that Pten-deficient Phox2B-expressing neurons showed a distinct transcriptional signature associated with not only enhanced vagal output but also upregulation of sympathetic output in the adipose tissue.
The VN is known to activate the anti-inflammatory reflex to regulate peripheral inflammation through the vagal efferent fibers10,20. Our P-PTEN KO mice exhibited increased vagal efferent output as assessed by classical induction of bradycardia48,49. It has been reported that insulin can promote neurite extension and myelination in vitro50, and that treatment with an insulin-sensitizing drug such as metformin can reduced demyelination in the spinal cord51. In line with a role for insulin in promoting myelination, we showed that the Pten-deficient neuronal population in the VN exhibited transcriptional changes in genes involved in neurite growth and myelination, and that the VN of P-PTEN KO mice showed histological changes that were conducive to improved vagal output. These neuronal changes were accompanied by macrophage polarization towards an anti-inflammatory state in WAT. However, while bradycardia induction is a widely used classical functional readout of vagal output, it is an indirect measurement of efferent vagal function. As such, future studies are warranted to investigate whether there is involvement from both the afferent and efferent arms of the VN, using optogenetics or DREAD receptors2 to stimulate the different arms of the VN. Further examination of the SNS would also increase our understanding the role of Phox2B-expressing neurons in both branches of the ANS.
In conclusion, our study has shown that vagal insulin-PI3K signaling is crucial for regulating peripheral metabolism, inflammation and adiposity, likely through the control of VN output. Enhancement of vagal insulin-PI3K signaling through Pten deletion led to increased vagal output and protection against HFD-induced diabetes, obesity, and inflammation. In contrast, insulin resistance within the VN, as evidenced in P-IR KO mice, resulted in reduced vagal output that exacerbated metabolic defects and inflammation. These defects were abolished upon concomitant Pten deletion, which restored vagal PI3K signaling and vagal output. Our results show that PI3K signaling is the main insulin-responsive pathway involved in determining the vagal output that governs metabolic homeostasis. Our work thus provides potential insights into molecular mechanisms that may lead to new strategies for enhancing vagal output. Applications of such strategies to manipulate vagal nerve stimulation may provide non-pharmacologic therapies to combat T2D and obesity.
Methods
Experimental animal models and diets
To generate Pten deficiency specifically in Phox2B-expressing neurons, Ptenflox/flox mice were bred with Phox2Bcre+ mice to generate Phox2Bcre+-Ptenwt/flox mice, which were intercrossed to generate Phox2Bcre+-Ptenwt/wt (denoted as P-PTEN WT) littermate control mice and Phox2Bcre+-Ptenflox/flox (denoted as P-PTEN KO) mutant mice. Another cohort of Phox2Bcre+-Ptenwt/flox mice was crossed with Rosa26 tdTomatoflox/flox reporter mice (The Jackson Laboratory) to generate Phox2Bcre+-Ptenwt/wt-tdTomatowt/flox controls and Phox2Bcre+-Ptenflox/flox-tdTomatowt/flox mice. To generate mice with insulin receptor (Ir) deficiency specifically in the Phox2B-expressing neurons, Irflox/flox mice were bred with Phox2Bcre+ mice to generate Phox2Bcre+-Irwt/flox heterozygous mice, and these were intercrossed to generate Phox2Bcre+-Irwt/wt (P-IR WT) littermate controls and Phox2Bcre+-IRflox/flox (P-IR KO) mutant mice. To generate mice with double loss of Pten and Ir in Phox2B-expressing neurons, Phox2Bcre+-Ptenflox/flox mice were crossed with Irflox/flox mice to generate Ir/Pten double heterozygotes, which were then intercrossed to generate Phox2Bcre+-Irflox/flox-Ptenwt/flox breeders. These breeders were intercrossed to generate Phox2Bcre+-Irflox/flox-Ptenflox/flox (DKO) mutant mice. All mice were bred on to the C57B6 background. C57B6/J mice were purchased from the Jackson Laboratory. Data from males and females were collected and analyzed separately as indicated.
All mice were housed in a pathogen-free facility with a 12 h light/dark cycle from 6 AM – 6 PM with humidity controlled between 40–60%. Animals had free access to water and standard irradiated rodent normal chow diet (NCD; 5% fat, Harland Teklad) or high-fat diet (HFD; 60% of energy from fat, Bioserv). Animals were fed an HFD for 3 months prior to sacrifice. All animal experiments were approved by the Animal Resource Center at the Toronto General Hospital Research Institute (TGHRI).
For mouse genotyping, ear notch samples were digested using an alkaline lysis buffer followed by the addition of neutralization buffer. Primer sequences for genotyping PCR are listed in Supplementary Table 1. Target DNA was amplified in a thermocycler (Mastercycler, Pro S, Eppendorf) using a mixture of primers, DNA sample and DNA Fast extract (2x His-red taqmix; Wisent). Cycling conditions are available upon request. All PCR products were electrophoresed on 2% agarose gels containing Sybr safe created from BioShop agarose powder.
RNA isolation and quantitative real-time (qRT)-PCR
Prior to tissue collection, each mice was fasted overnight and subsequently euthanized by cervical dislocation following 5 minutes of CO2 exposure. VN tissue collection was performed on mice euthanized with 10 minutes of CO2 exposure. Total RNA was isolated from flash-frozen sWAT, pWAT, BAT, VN, quadriceps, and liver tissues using TRIzol Reagent (Invitrogen). cDNA was synthesized by reverse transcription of mRNA using the M-MLV reverse transcriptase enzyme (Invitrogen) following DNAse digestion (Thermo Fisher). qRT-PCR was performed using specific primers (see Supplementary Table 2) and LUNA universal qPCR Master Mix (M3003, New England Biolabs) on a 7900HT Fast Real-Time PCR System (Applied Biosystems). Relative gene expression levels were calculated using the delta CT method and normalized to 18S for all adipose and liver tissues, and to β-actin for VN and quadriceps muscle. Samples with multiple melt curve peaks or standard deviations greater than 0.5 per triplicate were excluded from the results to remove primer and pipetting error. Primer sequences for qRT-PCR are listed in Supplementary Table 2.
Vagal nerve recording
In vivo VN recordings were performed on mice fed for 3 months on either NCD or HFD starting at 6 weeks of age. All VN surgeries and procedures were approved by the Department of Comparative Medicine Animal Facility at the University of Toronto in accordance with the Ontario Animal Research Act (Toronto, Ontario, Canada). Anesthesia was induced by nasal delivery of 5% isoflurane and subsequently maintained using a gas mask delivering 2–2.5% isoflurane (O2 flow rate: 1 L/min). Heart rate and O2 saturation levels were monitored throughout the experiment. Electrocardiography (ECG) was used to measure heart rate. For bradycardia threshold experiments, the right VN was accessed, and a 150 µm nerve cuff electrode with two contact sites was applied onto the VN and attached to a stimulator. To initiate bradycardia, a brief biphasic pulse (0.05 interpulse period 1 x multiplier, 0.3 pulse duration 10−3x multiplier, 1 train burst width 100 x multiplier) was applied to the VN at varying current strengths as described49. Bradycardia was defined as a 10% decrease in heart rate.
Brain and VN immunofluorescent imaging
For brain and VN immunofluorescent imaging, each mouse was euthanized after 10 min of exposure in CO2 chambers and was perfused twice with 4% PFA and immunostained with the protocol shown below52. Tissues were washed with PBS and embedded in OCT with 30% sucrose for cryoprotection and subsequently sectioned at 20 µm thickness with a cryostat. Antigen retrieval was then performed in 10 mM sodium citrate buffer, and brain slice were blocked for 1 hr at room temperature using 5% donkey serum (Sigma). Primary antibodies were then incubated overnight at 4 °C. Tissues were then washed 3 x with PBST, and a secondary antibody, donkey anti-rabbit Alexa Fluor 488, 555 or 647 (Thermo Fisher; 1:2000 dilution) was applied. Sections were then counterstained and mounted onto a slide with mounting medium (Abcam), and fluorescence was imaged using a Nikon A1R confocal microscope system. Antibodies against phospho-s6 (pS6; 1:500, CST), tdTomato (1:500, Rockland), and DAPI (1:2000, Invitrogen) were used on the DVC. Rostral sections of the DVC were acquired and regional sections within DVC was examined for total fluorescence consistent with well established literature5,53,54. Antibodies against MBP (1:50, CST), NPY (1:200, Ab-clonal), and NeuroH (1:50, Ab-clonal) were used. Images were quantified with ImageJ to assess the fluorescence of the antibody across the total vagal area55.
Single nucleus RNA sequencing and pathway analysis
Mice were fed on HFD for 2 weeks starting at 6 weeks of age. Mice were fasted for 16 hours overnight and euthanized after 10 minutes of exposure in CO2 chambers before the entire left and right cervical VNs containing the nodose and jugular ganglia were harvested (12 VN collected from 6 mice per sample). For euthanization, Tissue samples were processed at the Princess Margaret Genomics Center (Toronto, Ontario), where ~7500 nuclei were extracted per sample, and snRNAseq was performed using the 10x Genomics 3’ v3 Reagent Kit and the 10x Cell Ranger software version 3. Each sample was sequenced using 100 bp pair-ended protocol and multiplexed to obtain ~40 million reads/sample with reads mapped to GRCh38. The Seurat56 and Harmony57 programs were used in RStudio version 4.2.2 to annotate and cluster different cell populations and to generate the list of genes enriched within the neuronal population. The raw data from snRNAseq are available upon request.
Whole-mount immunostaining and tissue clearing of pWAT
Tissues were collected from 16 hour overnight fasted mice that were euthanized with cervical dislocation following 5 minutes of CO2 exposure. Adipose tissue clearing and immunostaining were performed using the Adipo-Clear protocol58. Briefly, pWAT was harvested and fixed in 4% PFA overnight at 4 °C. Samples underwent dehydration through an ascending methanol gradient (20, 40, 60, 80, and 100%) in H2O containing 0.3 M glycine and 0.1% Triton X-100 (B1N), followed by delipidation with dichloromethane (DCM) and bleaching in 5% hydrogen peroxide (H2O2) in PBS. Rehydration was achieved using a reverse methanol gradient (80%, 60%, 40%, 20%) in B1N buffer. Samples were blocked in PBS containing 0.1% Triton X-100, 0.05% Tween 20, and 2 µg/mL Heparin (PTxwH) overnight before incubation with anti-tyrosine hydroxylase (1:1000, Millipore, AB1542) for 5 days at RT. Samples were then incubated in secondary antibody Alexa-647 (1:1000, Invitrogen, A-21448) for 5 days at RT. Immunostained samples were embedded in 1% agarose, dehydrated in an increasing methanol gradient in water (25%, 50%, 75%, 100%), and cleared via incubation in ethyl cinnamate (ECI; Tokyo Chemical Industry).
3D imaging and quantification of TH immunostaining of pWAT
pWAT samples were imaged on a light-sheet microscope (UltraMicroscope Blaze, Miltenyi Biotec) under the 1 x objective lens, using ethyl cinnamate (ECI; Tokyo Chemical Industry) imaging solution. Individual tiles and z-stacks were reconstructed, and additional analyses were performed using Imaris x 64 software (version 10.1.0). The surface function with machine learning segmentation was used to reconstruct a 3D model of the sympathetic nerves from images of TH immunostaining. TH density was then calculated by selecting at least 5 cubed regions of interest (of fixed size) within each sample and quantifying the volume occupied by the sympathetic nerves as a ratio of the volume occupied by the cubed regions of interest.
Biochemical analyses of the serum, liver and pWAT
For tissue NE measurements, 100 mg of liver or pWAT was homogenized in 1 mL of PBS containing protease and phosphatase inhibitor (CST#5872 S). NE was measured using a competitive calorimetric NE ELISA kit (ab287789, Abcam).
For serum insulin measurements, blood was collected from mice that had been fasted for 16 hours overnight, and insulin was measured using a mouse insulin ELISA kit (Cat. #90080, Crystal Chem). Liver triglycerides were measured in frozen liver homogenates using a triglyceride calorimetric assay kit (Cat. #10010303, Cayman Chemical). All assays were performed using commercially available kits with duplicate samples according per manufacturer’s instructions.
Western blotting
Tissue lysates were collected from 3-month HFD-fed mice after a 16 h overnight fast, which were euthanized with cervical dislocation following 5 min of CO2 exposure. Tissue lysates were mechanically homogenized for 3 cycles at 15 s intervals in ice-cold RIPA buffer (Thermo Fisher) containing protease and phosphatase inhibitors (CST#5872S), and centrifuged for 10 minutes at 14,000 × g at 4 °C to remove the lipid layer. Protein lysates (30 µg) were fractionated by 10% sodium dodecyl sulfate-polyacrylamide (SDS-PAGE) gel electrophoresis followed by transfer onto ethanol-hydrated polyvinylidene fluoride membranes using the Trans-Blot Turbo Transfer System (Bio-Rad) for 10 min. Membranes were blocked for 1 h with 5% non-fat dry milk, and then treated with antibodies against either tyrosine hydroxylase (1:1000, AB#152; Sigma Aldrich), or p-AKT (1:1000, Phospho-Akt (Ser473) (D9E) XP® Rabbit mAb #4060; CST), or total AKT (1:1000, Akt Antibody #9272; CST) at 4 °C with gentle shaking overnight. Membranes were washed with TBST for 15 min total (3 × 5 min) and subsequently treated with anti-rabbit (CST#7074; 1:5000; CST) IgG horseradish-peroxidase (HRP)-conjugated secondary antibody for 1 h with gentle shaking. Membranes were incubated in chemiluminescent ECL-plus (PerkinElmer Inc., Waltham, MA, USA) at RT for between 30 s and 3 min, and imaged using MicroChemi 4.2 (TH; DNR Bio-Imaging Systems, Mahale HaHamisha, Jerusalem, Israel). GAPDH served as the protein loading control. Quantification of protein expression was performed by densitometry using the ImageJ (version 1.54 J) component of Fiji software59.
Histology and whole mount imaging
Mouse organs were harvested from 3-month HFD-fed mice after a 16 h fast, each mice was euthanized with cervical dislocation following 5 min of CO2 exposure, and sections were fixed in either 4% paraformaldehyde (PFA) in 0.1 M PBS (pH 7.4) or 10% formalin. Sections of liver, subcutaneous white adipose tissue (sWAT), perigonadal (pWAT), and brown adipose tissue (BAT) were stained with hematoxylin and eosin (H&E). Images of whole mounts of pWAT were obtained by treating in 1% PFA overnight and staining with anti-CD206 and LipidTox far-red (1:2000, Thermo Fischer H34477) as previously described60. Liver samples were either stored in 4% PFA overnight for sectioning and subsequent H&E staining or embedded in optimal cutting temperature (OCT) medium prior to flash-freezing and conveying to the STTARR facility. All H&E- and Oil red O-stained sections were imaged using an Olympus light microscope. Adipose images were quantified with ImageJ by averaging data collected from manual counting across different regions of adipose sections61.
Magnetic resonance imaging (MRI) analysis
MRI to assess body composition was provided by the Spatio-Temporal Targeting and Amplification of Radiation Response (STTARR) facility at TGHRI (Toronto, Ontario, Canada). A 7 Tesla BioSpec MRI system (Bruker Canada, Milton, Ontario, Canada) was used with a B-GA12 gradient system and a quadrature volume resonator RF transceiver coil. Mice were sedated with 2% isoflurane and placed in a prone position, and coronal sections were acquired to compare lean and fat tissue areas.
Cytometry by Time of Flight (CyTOF) of stromal vascular fraction of pWAT
pWAT depots were collected from overnight fasted mice that were euthanized with cervical dislocation following 5 minutes of CO2 exposure. pWAT from 3-month HFD-fed mice were pooled (2-3 mice per sample) and dissociated into single-cell suspensions using collagenase to obtain the adipose stromal vascular fraction (SVF). Cells in the adipose SVF were washed with 2 mL of cell staining media (CSM; Biolegend) and pelleted by centrifugation at 300 x g for 5 min. Fc receptors were blocked by resuspending the cells in 25 mL purified anti-mouse CD16/CD32 antibodies (1:100, BD Biosciences) for 10 min at room temperature (RT). The same volume of heavy metal-tagged antibody cocktail was prepared in CSM, added to the cell suspension, and incubation continued for 30 min at RT. After washing in 2 mL of PBS, antibody-labeled cells were resuspended in 100 μL PBS and 100 μL of cisplatin viability stain (1 μL of 10 mM (Cisplatin; Standard Biotools) into 5 mL of PBS), incubated for 5 min at RT, and quenched with 2 mL CSM. After washing in 3 mL of CSM and collected by centrifugation for 5 minutes at 600 x g, cells were stained with 1 mL 100 nM Cell-ID Iridium intercalator (#201192 A, Standard Biotools) overnight at 4 °C. On the day of data acquisition, cells were washed with CSM and then PBS. Cell pellets were resuspended in Maxpar Cell Acquisition Solution containing EQ normalization beads (#201237, Fluidigm). Data were collected using a Helios instrument (Standard Biotools) containing the Helio system to collect CyTOF data. Live singlets were pre-gated using 191Ir-DNA1, 193Ir-DNA2, 140Ce-EQ, Event-Length, Center, Offset, and 195Pt-Cisplatin. SPADE (Spanning-tree Progression Analysis of Density-normalized Events) algorithms from Cytobank (www.cytobank.org) were used to visualize the various cellular compositions by clustering cells with similar protein expression into specific nodes62.
Metabolic caging studies
For parameters examined using a metabolic cage system (CLAMs, Columbus Instruments, Columbus OH), each experimental animal was placed into an individual metabolic cage and allowed to acclimatize for 24 hours prior to data collection. Metabolic caging data were collected over the next 24 h post-acclimatization following the dark (7PM-7AM) and light (7AM-7PM) cycle of the animal facility. Energy expenditure, locomotion, meal duration and food intake were measured in a manner consistent with well established 24 h protocols63,64,65. O2 consumption and CO2 emission values, alongside food intake measurements, were unnormalized using the web-based CalR analysis tool66.
Body temperature measurements and acute cold stress test
Body temperatures were measured between 9 and 10 AM using a mouse rectal temperature probe (Kent Scientifics). For cold exposure, the CLAMS cage temperature was set to 4 °C, after which the mouse body temperatures were recorded hourly for 6 h.
Fecal and serum lipid content
Prior to sample collection, each experimental animal was placed into an individual standard cage with free access to food and water. Stool samples were collected over 3 consecutive nights at 9:30 AM each morning from cages containing fresh Tekklad Diamond Dry Cellulose Bedding and flash-frozen in dry ice. Total lipids were extracted from stools using adapted methods from Folch et al.67. Briefly, stools were softened by soaking in 1.75 ml 0.88% potassium chloride for 24 h prior to the addition of 2 ml methanol and 4 ml chloroform (final volume 0.8:1:2). Heptadecanoic acid (17:0), an internal standard, was added just prior to lipid extraction for quantification of fatty acid concentrations. Stool homogenates were centrifuged at 1000 x g for 10 min at room temperature to obtain the total lipid extract. Stool lipids were methylated with boron-trifluoride in methanol at 100 °C for 1 h. Fatty acid methyl esters (FAMEs) were quantified using a gas chromatograph equipped with a flame ionization detector (Varian 430 GC, Bruker). Using a DB-FFAP column (30 m x 0.25 mm i.d. x 0.25 µm film thickness), FAMEs were separated by the following method: oven set at 50 °C for 1 min, then ramped up to 130 °C at a rate of 30 °C/min, then to 175 °C at 10 °C/min, and then to 230 °C at 5 °C/min, where this temperature was held for 9.5 min. The temperature was finally ramped up to 240 °C at a rate of 50 °C/min and held for 13 minutes. Total lipid content was quantified by adding the contents of the known and unknown fatty acid peaks. For serum lipid measurements, serum was collected from mice fasted for 16 h and lipid metabolite species were quantified with gas chromatography68.
Preparation of permeabilized muscle fibers
This technique was adapted from previous methods for muscle. Briefly, mice were euthanized with CO2, and the quadriceps muscles were excised and placed immediately into ice-cold BIOPS buffer containing 50 mM MES hydrate, 7.23 mM K2EGTA, 2.77 mM CaK2EGTA, 20 mM imidazole, 0.5 mM dithiothreitol, 20 mM taurine, 5.77 mM ATP, 15 mM PCr, and 6.56 mM MgCl2·6 H2O (pH 7.2)69. Muscle fiber bundles were permeabilized with 40 µg saponin (Sigma Aldrich) in BIOPS on a platform rotor for 30 min at 4 °C. Bundles were then washed for 15 min at 4 °C in Buffer Z containing 105 mM K-MES, 30 mM KCl, 10 mM KH2PO4, 5 mM MgCl2 • 6H2O, 1 mM EGTA, and 5 mg/ml BSA38,70.
Mitochondrial respiration
Mitochondrial respiration of permeabilized fiber bundles was assessed by measuring high-resolution oxygen consumption in 2 mL Buffer Z supplemented with 20 mM creatine to saturate mitochondrial creatine kinase activity37. O2 consumption was measured using an Oroboros Oxygraph-2K (Oroboros Instruments, Corp., Innsbruck, Austria) while stirring at 37 °C. 5 µM blebbistatin was added to prevent muscle fiber contraction by rigor in response to ADP. Each chamber was oxygenated with 100% pure O2 to an initial concentration of 350 μM, and experiments were completed before chamber [O2] reached 150 μM. Complex I-supported respiration was stimulated using 5 mM pyruvate and 2 mM malate (NADH; State II respiration), followed by titration of ADP concentrations (State III) from physiological ranges (25–100 µM) to supraphysiological (500 µM), and saturating to stimulate maximal coupled respiration (1000 µM, 5000 µM). Subsequently, 10 mM succinate (FADH2) was added and titrated to stimulate complex II-supported electron flow. Polarographic O2 was measured in 2 s intervals and expressed as picomoles per second per milligram of tissue.
In vivo metabolic studies
Glucose tolerance test (GTT) and pyruvate tolerance tests (PTT) were performed on mice fasted for 16 h overnight between 6 PM and 10 AM. Glucose (1 g/kg body weight) or sodium pyruvate (1.5 g/kg body weight) was injected intraperitoneally. Insulin tolerance test (ITT) was performed on mice fasted for 4 h between 9 AM and 1 PM. Insulin lispro (Lilly; 0.5 U/kg body weight) was injected intraperitoneally. Blood glucose measurements were taken at 0, 15, 30, 45, 60 and 120 min after glucose or insulin injection using a GE200 glucometer. GTTs and ITTs were performed at 6 weeks of age and after 3 months of feeding on NCD or HFD.
Multiplex analysis
Mice were euthanized with CO2 and blood collected by cardiac puncture was transferred to blood collection tubes (SKU #365978, Becton Dickinson). Tubes were centrifuged, and serum (10–25 µL) was subjected to either mouse endocrine multiplex assay (adiponectin and leptin; MADCYMAG-72K, Millipore Sigma) or mouse cytokine/chemokine assays (IL4, IL10, IL-6, CCL2; MCYTOMAG-70K-PMX, Millipore Sigma), as per the manufacturer’s instructions. Absorbance data were generated and analyzed via best-fit line analysis using Excel.
Statistics
All data presented in this study are shown as the mean ± standard error of the mean (SEM). Data comparing two groups were analyzed using Student’s t test for normally distributed data (determined by a Shapiro-Wilk normality test) or by the Mann‒Whitney test (for two group datasets that are not normally distributed). Multiple-comparisons test with the Holm-Sidak with an alpha of 0.05 are applied One-way ANOVA with Tukey’s post-hoc test was used to analyze data that was obtained from 3 or more groups and was normally distributed. The Kruskal-Wallis test with Dunn’s correction was used for comparing multiple groups containing non-normally distributed data. Two-way ANOVA with Tukey’s post-hoc test was used for experiments comparing diet and genotyping conditions. All statistical tests were conducted using GraphPad Prism 8. Data presented in figures are represented with an unadjusted p-value for clarity, with the adjusted p-value shown in the raw data source file.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw nuclear-RNA seq data is uploaded onto NCBI GEO gene expression omnibus under accession series GSE309905. Source data are provided in this paper.
References
Jänig, W. Integrative Action of the Autonomic Nervous System: Neurobiology of Homeostasis. (Cambridge University Press, 2006).
Tanaka, S. et al. Vagus nerve stimulation activates two distinct neuroimmune circuits converging in the spleen to protect mice from kidney injury. Proc. Natl. Acad. Sci. USA 118, e2021758118 (2021).
Abe, C. et al. C1 neurons mediate a stress-induced anti-inflammatory reflex in mice. Nat. Neurosci. 20, 700–707 (2017).
Pattyn, A., Morin, X., Cremer, H., Goridis, C. & Brunet, J.-F. The homeobox gene Phox2b is essential for the development of autonomic neural crest derivatives. Nature 399, 366–370 (1999).
Rossi, J. et al. Melanocortin-4 receptors expressed by cholinergic neurons regulate energy balance and glucose homeostasis. Cell Metab. 13, 195–204 (2011).
Aklan, I. et al. NTS Catecholamine neurons mediate hypoglycemic hunger via medial hypothalamic feeding pathways. Cell Metab. 31, 313–326.e5 (2020).
Sohn, J.-W., Elmquist, J. K. & Williams, K. W. Neuronal circuits that regulate feeding behavior and metabolism. Trends Neurosci. 36, 504–512 (2013).
Makhmutova, M. et al. Pancreatic β-Cells communicate with vagal sensory neurons. Gastroenterology 160, 875–888.e11 (2021).
Pocai, A. et al. Hypothalamic KATP channels control hepatic glucose production. Nature 434, 1026–1031 (2005).
Pavlov, V. A. & Tracey, K. J. The vagus nerve and the inflammatory reflex—linking immunity and metabolism. Nat. Rev. Endocrinol. 8, 743–754 (2012).
Laucius, O. et al. Effect of aging on vagus somatosensory evoked potentials and ultrasonographic parameters of the vagus nerve. J. Clin. Neurosci. 90, 359–362 (2021).
Low, P. A., Opfer-Gehrking, T. L., Proper, C. J. & Zimmerman, I. The effect of aging on cardiac autonomic and postganglionic sudomotor function. Muscle Nerve 13, 152–157 (1990).
Poon, A. K. et al. Insulin resistance and reduced cardiac autonomic function in older adults: the Atherosclerosis Risk in Communities study. BMC Cardiovasc. Disord. 20, 217 (2020).
Lee, D. Y. et al. Decreased vagal activity and deviation in sympathetic activity precedes development of diabetes. Diabetes Care 43, 1336–1343 (2020).
Pal, G. K. et al. Effects of gender on sympathovagal imbalance, prehypertension status, and cardiovascular risks in first-degree relatives of type 2 diabetics. Am. J. Hypertens. 27, 317–324 (2014).
Payne, S. C. et al. Differential effects of vagus nerve stimulation strategies on glycemia and pancreatic secretions. Physiol. Rep. 8, https://doi.org/10.14814/phy2.14479 (2020).
Payne, S. C. et al. Blood glucose modulation and safety of efferent vagus nerve stimulation in a type 2 diabetic rat model. Physiol. Rep. 10, https://doi.org/10.14814/phy2.15257 (2022).
Wang, L. et al. Deletion of Pten in pancreatic B-Cells protects against deficient B-Cell mass and function in mouse models of type 2 diabetes. Diabetes 59, 3117–3126 (2010).
Birnbaum, Y. et al. PTEN Upregulation may explain the development of insulin resistance and type 2 diabetes with high dose statins. Cardiovasc. Drugs Ther. 28, 447–457 (2014).
Wang, L. et al. Pten deletion in RIP-Cre neurons protects against type 2 diabetes by activating the anti-inflammatory reflex. Nat. Med. 20, 484–492 (2014).
Thompson, G. W. et al. Bradycardia induced by intravascular versus direct stimulation of the vagus nerve. Ann. Thorac. Surg. 65, 637–642 (1998).
Wang, J. et al. Distinct and common expression of receptors for inflammatory mediators in vagal nodose versus jugular capsaicin-sensitive/TRPV1-positive neurons detected by low input RNA sequencing. PLOS ONE 12, e0185985 (2017).
Perge, J. A., Niven, J. E., Mugnaini, E., Balasubramanian, V. & Sterling, P. Why do axons differ in caliber?. J. Neurosci. 32, 626–638 (2012).
Maneckjee, R. & Baylin, S. B. Use of radiolabeled monofluoromethyl-Dopa to define the subunit structure of human L-Dopa decarboxylase. Biochemistry 22, 6058–6063 (1983).
Park, D. H., Kashimoto, T., Ebstein, R. P. & Goldstein, M. Purification and immunochemical characterization of dopamine β-hydroxylase from human pheochromocytoma. Mol. Pharmacol. 12, 73 (1976).
Vargovic, P. et al. Adipocytes as a new source of catecholamine production. FEBS Lett. 585, 2279–2284 (2011).
Chi, J. et al. Three-dimensional adipose tissue imaging reveals regional variation in beige fat biogenesis and PRDM16-dependent sympathetic neurite density. Cell Metab. 27, 226–236.e3 (2018).
Russo, B., Menduni, M., Borboni, P., Picconi, F. & Frontoni, S. Autonomic nervous system in obesity and insulin-resistance-the complexinterplay between leptin and central nervous system. Int. J. Mol. Sci. 22, 5187 (2021).
Marcelin, G. et al. A PDGFRα-Mediated switch toward CD9high adipocyte progenitors controls obesity-induced adipose tissue fibrosis. Cell Metab. 25, 673–685 (2017).
Yang, Z.-J. et al. Influence of anterior subdiaphragmatic vagotomy and TPN on rat feeding behavior. Physiol. Behav. 51, 919–926 (1992).
Simon, S. L. et al. Body mass index and variability in meal duration and association with rate of eating. Front. Nutr. 9, 941001 (2022).
Marvyn, P. M., Bradley, R. M., Mardian, E. B., Marks, K. A. & Duncan, R. E. Data on oxygen consumption rate, respiratory exchange ratio, and movement in C57BL/6J female mice on the third day of consuming a high-fat diet. Data Brief. 7, 472–475 (2016).
Pakiet, A. et al. The effect of a high-fat diet on the fatty acid composition in the hearts of mice. Nutrients 12, 824 (2020).
Young, J. B. & Landsberg, L. Diminished sympathetic nervous system activity in genetically obese (ob/ob) mouse. Am. J. Physiol. Endocrinol. Metab. 245, E148–E154 (1983).
Korsak, A. et al. Immediate and sustained increases in the activity of vagal preganglionic neurons during exercise and after exercise training. Cardiovasc. Res. 119, 2329–2341 (2023).
Machhada, A. et al. Vagal determinants of exercise capacity. Nat. Commun. 8, 15097 (2017).
Walsh, B. et al. The role of phosphorylcreatine and creatine in the regulation of mitochondrial respiration in human skeletal muscle. J. Physiol. 537, 971–978 (2001).
Perry, C. G. R. et al. Inhibiting myosin-ATPase reveals a dynamic range of mitochondrial respiratory control in skeletal muscle. Biochem. J. 437, 215–222 (2011).
Schultze, S. M., Hemmings, B. A., Niessen, M. & Tschopp, O. PI3K/AKT, MAPK and AMPK signalling: protein kinases in glucose homeostasis. Expert Rev. Mol. Med. 14, e1 (2012).
Buteau, J. et al. Glucagon-like peptide-1 prevents beta cell glucolipotoxicity. Diabetologia 47, 806–815 (2004).
Plum, L. et al. Enhanced leptin-stimulated Pi3k activation in the CNS promotes white adipose tissue transdifferentiation. Cell Metab. 6, 431–445 (2007).
Ahmad, E., Lim, S., Lamptey, R., Webb, D. R. & Davies, M. J. Type 2 diabetes. Lancet 400, 1803–1820 (2022).
Waise, T. M. Z., Dranse, H. J. & Lam, T. K. T. The metabolic role of vagal afferent innervation. Nat. Rev. Gastroenterol. Hepatol. 15, 625–636 (2018).
Kalyani, R. R. & Egan, J. M. Diabetes and altered glucose metabolism with aging. Endocrinol. Metab. Clin. North Am. 42, 333–347 (2013).
Leinwand, S. G. & Chalasani, S. H. Neuropeptide signaling remodels chemosensory circuit composition in Caenorhabditis elegans. Nat. Neurosci. 16, 1461–1467 (2013).
Rulifson, E. J., Kim, S. K. & Nusse, R. Ablation of insulin-producing neurons in flies: growth and diabetic phenotypes. Science 296, 1118–1120 (2002).
Le Douarin, N. M. & Dupin, E. The “beginnings” of the neural crest. Dev. Biol. 444, S3–S13 (2018).
Yoo, P. B. et al. High-resolution measurement of electrically-evoked vagus nerve activity in the anesthetized dog. J. Neural Eng. 10, 026003 (2013).
Yoo, P. B. et al. Modulation of heart rate by temporally patterned vagus nerve stimulation in the anesthetized dog. Physiol. Rep. 4, e12689 (2016).
Pham, V. M. & Thakor, N. Insulin enhances neurite extension and myelination of diabetic neuropathy neurons. Korean J. Pain. 35, 160–172 (2022).
Gilbert, E. A. B. et al. Metformin treatment reduces inflammation, dysmyelination and disease severity in a mouse model of multiple sclerosis, experimental autoimmune encephalomyelitis. Brain Res. 1822, 148648 (2024).
Son, J. E. et al. Irx3 and Irx5 in Ins2-Cre+ cells regulate hypothalamic postnatal neurogenesis and leptin response. Nat. Metab. 3, 701–713 (2021).
Tao, J. et al. Highly selective brain-to-gut communication via genetically defined vagus neurons. Neuron 109, 2106–2115 (2021).
Bellusci, L. et al. Interactions between Brainstem Neurons That Regulate the Motility to the Stomach. J. Neurosci. 42, 5212–5228 (2022).
Duplancic, R. & Kero, D. Novel approach for quantification of multiple immunofluorescent signals using histograms and 2D plot profiling of whole-section panoramic images. Sci. Rep. 11, 8619 (2021).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021).
Korsunsky, I. et al. Fast, sensitive, and accurate integration of single cell data with harmony. Nat. Methods 16, 1289–1296 (2018)
Chi, J., Crane, A., Wu, Z. & Cohen, P. Adipo-Clear: a tissue clearing method for three-dimensional imaging of adipose tissue. J. Vis. Exp. 137, 58271 (2018).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Jiang, Y. et al. Visualization of 3D white adipose tissue structure using whole-mount staining. J. Vis. Exp. https://doi.org/10.3791/58683 (2018).
Luk, C. T. et al. FAK signalling controls insulin sensitivity through regulation of adipocyte survival. Nat. Commun. 8, 14360 (2017).
Lee, J. H. et al. Characterization of adipose depot-specific stromal cell populations by single cell mass cytometry. iScience 25, https://doi.org/10.1016/j.isci.2022.104166 (2021).
Pak, H. H. et al. Fasting drives the metabolic, molecular and geroprotective effects of a calorie-restricted diet in mice. Nat. Metab. 3, 1327–1341 (2021).
Hu, S. et al. Dietary fat, but not protein or carbohydrate, regulates energy intake and causes adiposity in mice. Cell Metab. 28, 415–431 (2018).
Liu, X. et al. Activation of GPR81 by lactate drives tumour-induced cachexia. Nat. Metab. 6, 708–723 (2024).
Mina, A. I. et al. CalR: A web-based analysis tool for indirect calorimetry experiments. Cell Metab. 28, 656–666 (2018).
Folch, J., Lees, M. & Sloane Stanley, G. H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 226, 497–509 (1957).
Lai, K. Z. H. et al. Association of serum very-long-chain saturated fatty acids with changes in insulin sensitivity and β-cell function: the prospective metabolism and islet cell evaluation (PROMISE) cohort. Diabetes 72, 1664–1670 (2023).
Veksler, V. I., Kuznetsov, A. V., Sharov, V. G., Kapelko, V. I. & Saks, V. A. Mitochondrial respiratory parameters in cardiac tissue: A novel method of assessment by using saponin-skinned fibers. Biochim. Biophys. Acta BBA Bioenerg. 892, 191–196 (1987).
Doerrier, C. et al. Harmonization of experimental procedures to assess mitochondrial respiration in human permeabilized skeletal muscle fibers. Free Radic. Biol. Med. 223, 384–397 (2024).
Acknowledgements
This work was supported by operating grants from Diabetes Canada (OG-3−22-5681-MW) and Canadian Institutes of Health Research (PJT-159505). Dr. Minna Woo holds the Soham and Shaila Ajmera Family Chair in Molecular Diabetes Research. We thank Dr. Mary Saunders for scientific editing of the manuscript.
Author information
Authors and Affiliations
Contributions
Y.Z.L., J.R., P.Y., and M.W. designed the experimental studies. Y.Z.L., J.R., P.Y., and E.P. conducted the experiments. C.B. conducted the mitochondrial respiration experiments. J.E.S. performed neuronal imaging. J.H.L. performed the whole mount staining and CyTOF experiments. N.V. performed adipose 3D TH immunostaining. C.T.C conducted the stool lipid analysis. T.W.M. and A.S. provided the Ptenfl/fl mice. Y.Z.L and M.G. analyzed the data. R.B., F.B., C.H., and H.S. reviewed the results. Y.Z.L. and M.W. wrote and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
All authors declare no competing interests relevant to this work. T.W.M. holds equity in Treadwell Therapeutics Inc.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, Y.Z., Son, J.E., Vo, N. et al. PTEN regulates vagal-insulin signaling to optimize autonomic output determining peripheral inflammatory and metabolic homeostasis. Nat Commun 17, 774 (2026). https://doi.org/10.1038/s41467-025-67192-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67192-8
