
Abstract
Elucidating the genetic regulation of protein expression in specific disease states is important for understanding how genetic variation impact disease pathology. To this end, we conduct a large-scale genome-proteome-wide pQTL analysis on 2901 plasma proteins among 7626 healthy individuals and 28,064 patients across 42 disease statuses. We find 25,987 independent pQTL associations across 2224 regions, and investigate similarities and differences in their regulatory effects across various diseases and health states. We find that pQTL identified in specific diseases are more likely to be disease risk variants. We then integrate the association findings with Mendelian randomisation to identify 110 high-confidence causal proteins associated with 21 diseases, including Apolipoprotein(a) for cardiovascular diseases and angiotensin-converting enzyme for type 2 diabetes. Finally, we develop risk prediction models by integrating pQTL-derived polygenic risk scores and causal-protein-derived protein risk scores, which demonstrate good performance in discriminating populations at high risk for 21 disease types. These results indicate that disease state partly determines the impact of genetic variation on protein expression, implicating disease-related and disease-discordant pQTL associations as regulators of disease progression.
Similar content being viewed by others


Introduction
Translating the plethora of risk-associated genetic variants identified by genome-wide association studies (GWAS) into pathogenic mechanisms and treatments remains challenging. This difficulty arises from the ambiguity surrounding causal genes and the mapping of genes with poorly understood biology or unclear mechanisms of modulation. Addressing these challenges necessitates additional downstream analyzes and the integration of various types of omic data. Quantitative trait locus (QTL) mapping, which links genetic variants to intermediate molecular traits such as expression QTLs (eQTLs), can serve as invaluable tools for bridging the gap between genetic and phenotypic variation1. However, QTLs affecting mRNA levels are, on average, attenuated or buffered at the protein level2. In contrast, protein abundances, being more direct determinants of cellular functions, could play a more immediate role in shaping cellular activity and ultimately influencing phenotypic outcomes.
Proteomic studies provide valuable insights as proteins not only act as the effector molecules for most biological processes, serving as representations or indicators of physical condition, but also encompass the broadest spectrum of therapeutic targets. In recent years, emerging studies have integrated human genetics with high-throughput proteomics to systematically elucidate the association between genetic variants and plasma proteins3,4. With the expansion of population-scale and protein-coverage, an increasing number of protein quantitative trait loci (pQTL) have been identified5,6,7,8. However, previous large-scale population-based studies, including the UK Biobank (UKB) Proteomics Project8, were conducted in generally healthy participants or mixed populations with varying health statuses.
Considering that pathophysiological mechanisms differ by disease types, changes in the protein levels might be observable only within specific pathological states9. In support of this, a recent study demonstrated that the mobility of key proteins can be repressed when subjected to pathogenic signaling, leading to the dysregulation of diverse cellular processes10. These findings highlight the disease-specific feature of protein regulation, indicating that disease-related pQTLs present only under certain disease conditions. Consistently, analogous eQTL studies using human immune cells stimulated in vitro have identified eQTLs absent in resting cells but apparent upon cellular activation11,12. Therefore, the strategy used in previous studies to conduct pQTL studies in the general population limited their ability to finely uncover genetic associations that are specifically evident under pathological conditions. Systematic genome-proteome-wide pQTL analysis among individuals in specific health or disease states can offer a more comprehensive and precise understanding of the genetic architecture of proteins and underlying disease mechanisms. Moreover, investigating differences and similarities in pQTL regulatory effects across diseases and health states can reveal distinct genetic etiologies and shared therapeutic strategies, ultimately advancing precision medicine more effectively.
To address these gaps, we performed systematic proteogenomic association analyzes targeting 2901 proteins among 7626 healthy individuals and 28,064 patients across 42 disease statuses, and further provided insights into differences and similarities in pQTL regulatory effects between health and disease states. Through functional annotation and colocalization, we illustrate the underlying mechanism by which pQTLs influence protein abundance and contribute to disease. Integration of cohort study and MR reveals hundreds of high-confidence causal proteins. On this basis, we successfully constructed disease risk prediction models that integrate the pQTL-derived polygenic risk scores (pQTL-PRS) and causal-protein-derived protein risk scores (causal-ProRS), significantly enhancing the accuracy of risk assessments in multiple diseases. Finally, our comprehensive proteogenomic landscape and proteome-phenome associations are presented as a resource to support broader scientific community. Taken together, our findings shed light on the genetic etiology and are expected to accelerate the development of biomarkers, predictive models and therapeutics.
Results
A comprehensive atlas of pQTLs across healthy individuals and 42 diseases
In our quest to identify pQTLs, including those may exclusively manifest during specific health states (Supplementary Data 1), an average of 6.11 million genetic variants and 2901 proteins from an aggregate of 35,690 participants (7626 healthy subjects, 55 to 12,169 subjects across 42 diseases) were remained for subsequent analysis after quality control (Fig. 1A, B, Supplementary Fig. 1 and Supplementary Table 1, Supplementary Data 2). Then, we applied a linear model to test the association between plasma protein levels and genotypes in the above individuals, with adjustment for potential confounding factors (protein measurement variables, population structure and clinical status). With the Bonferroni-corrected threshold of P value < 1.72 × 10−11 (5 × 10−8/2901), we identified a total of 1,021,750 pQTLs-protein association pairs involving 533,975 pQTLs and 1856 proteins (Supplementary Data 3). Furthermore, to discover the independent signals associated with proteins, we performed conditional analyses with GCTA-COJO13, yielding a total of 25,987 independent pQTL associations across 2224 independent genetic regions, spanning from 4 regions in leukemia to 2130 regions in hypertension (Fig. 1B, Supplementary Data 3-4 and Supplementary Fig. 2A). The number of these independent associations varied from 4 in leukemia cancer to 6994 in hypertension (Fig. 1B–D and Supplementary Data 3). The diseases with more pQTL associations tended to have larger sample sizes (Fig. 1C). Additionally, ~59.24% of pQTLs were local acting (“cis” pQTLs) and 40.76% were distant acting (“trans”) (Fig. 1E).

A Identification framework of pQTLs and main results, created with BioRender.com. Identification of genetic variants associated with plasma protein levels across 43 disease types, using a Bonferroni-corrected significant threshold of P < 1.72 × 10−11. B The sample size of each disease type (bottom) and the number of independent pQTL associations (cis, darker; trans, lighter) identified for each disease (top). The numbers of cis- and trans-pQTLs are highlighted in gray. C The number of pQTL associations against sample size across 43 disease types. The R-squared and P-values of the linear regression model are also displayed. D A three-dimensional Manhattan plot displaying chromosomal position (x-axis) of pQTL associations (P < 1.72 × 10−11, z-axis) across 43 disease types (y-axis). Colors indicate disease types. P values were calculated by linear regression models, adjusted for covariates. E Genomic locations of pQTL associations across 43 disease types (cis-, purple; trans-pQTL, green). The x axis indicates positions of pQTL, and the y axis indicates the gene encoding the associated protein. F The figure depicts independent primary pQTL associations, color-coded to distinguish between those in linkage disequilibrium (LD; r² ≥ 0.1) with a previously identified pQTL (green, top) and those not previously reported (pink, bottom). A pie chart summarizes the relative proportions.
Specifically, nearly half (47.20%, 876 out of 1856) of the proteins are merely regulated by a single region (interquartile range, 1–3) (Supplementary Fig. 2B). In addition, 81.16% (1805 out of 2224) of these pQTL regions specifically associate with only one protein, whereas 22 genetic regions are found to regulate more than 10 proteins (Supplementary Fig. 2C). Of the 16,562 primary associations identified in our study, 12,109 (73.11%) were replicated in prior studies (r² ≥ 0.1) (Fig. 1F and Supplementary Data 5). Additionally, among these 2224 pQTL regions, 1977 regions (88.89%) have been corroborated by previously published research6,7,8, which highlights the reliability of our findings and the significance as a complement to existing resources.
Systematic characterization and functional annotation of pQTLs
In order to dissect the functional potential of pQTLs, we generated a set of non-pQTL variants by matching the number of variants in LD, minor allele frequency (MAF) and variant type to pQTLs for comparison (Supplementary Table 2). Consequently, we observed significant enrichment of pQTLs within missense and synonymous regions, compared with non-pQTLs (Fig. 2A, B). Notably, upon mapping these pQTLs to active transcription regions, we observed a marked increase within the binding sites of transcription factors (TFs) and histone modification marks, compared to non-pQTLs (Fig. 2C). Overall, these findings suggest that pQTLs might regulate protein levels not only by influencing translation and structure but also transcriptional regulation, which provides a potential clue linking allelic variation to protein levels.

A Genomic distribution of pQTLs (left) and matched non-pQTLs (right). B, C Enrichment analysis of cis-pQTLs compared with cis non-pQTLs among variants within functional category. Results were calculated using the two-tailed Fisher’s exact test. C or 7 epigenomic marks identified by ENCODE in each disease type. log2(OR) > 0 (OR > 1) indicates a positive association, and log2(OR)<0 (OR < 1) indicates a negative association. Categories significantly enriched for cis-pQTLs are indicated with an asterisk (two-tailed Fisher’s-exact test). D Explained variance in plasma abundances of proteins by identified pQTLs (cis, top; trans, middle; total, bottom). The data distribution is shown as a violin plot with an overlaid boxplot. The center line of the box presents as the median, the box limits indicate upper and lower quartiles and whiskers indicate the maximum and minimum. E MAF versus effect size for pQTL associations. (cis, purple; trans, green). F Bar chart showing pQTL classification based on GO term mapping of associated proteins. Yellow shades indicate cis-pQTLs and green shades indicate trans-pQTLs.
Next, we aimed to evaluate the contribution of pQTLs to target protein abundance by quantifying the proportion of variance in plasma protein concentrations explained by pQTLs. The results showed that pQTLs accounted for an average of 15.24% of the variance, ranging from 7.37% in hypertension to 70.24% in lung cancer (Fig. 2D and Supplementary Data 6). Interestingly, cis-pQTLs contributed significantly more to the variance (13.50%) compared to trans-pQTLs (1.81%). In addition, we noted that the phenotypic effect sizes of pQTLs were decreased within the increasing of MAF, and cis-pQTLs exhibited a stronger effect in comparison to trans-pQTLs (Fig. 2E). Therefore, our findings indicate that there may be a more remarkable contribution of cis-pQTLs to altered plasma protein levels and disease risk.
Furthermore, to elucidate the properties and classification of pQTL associations, we employed a tiered system that classified pQTL associations into protein-specific, pathway-specific and unspecific categories. For each disease type, an average of 83.14% (91.80% in cis, 48.87% in trans) of pQTL associations were assigned as protein-specific, while 4.58% (4.75% in cis, 5.51% in trans) as pathway-specific and a further 12.27% (4.45% in cis, 40.97% in trans) as unspecific (Fig. 2F and Supplementary Data 7), indicating that pQTLs exert their effects on the circulating proteome through diverse mechanisms. For example, the pathway-specific variant rs1761450, associated with LILRB1, LILRB2, LILRB5, LILRA3, and LILRA5 levels in hypertension, coordinately regulates immune receptor activity and cell surface receptor signaling pathways.
Insights into the genetic architecture of protein levels and common complexed diseases
To elucidate the underlying mechanisms through which pQTLs affect protein abundance under disease contexts and contribute to disease, we undertook a comprehensive evaluation of the shared genetic architecture of protein targets with gene expression, splicing and disease risk.
Given the disease- and tissue-specific nature of gene regulation, we restricted eQTL and sQTL data to disease-relevant tissues, enhancing the biological relevance and interpretability of our findings. Initially, we integrated plasma pQTL results with both expression quantitative trait loci (eQTLs) or splicing quantitative trait loci (sQTLs) from The Cancer Genome Atlas (TCGA) or Genotype-Tissue Expression Project (GTEx) using statistical colocalization (posterior probability > 0.7) for 1514 proteins with at least one cis-pQTL. Compelling evidence revealed that 34.68% (525/1,514) of proteins share a signal with their corresponding gene expression in at least one disease, with the majority of co-localized pQTLs and eQTLs showing effects in concordant direction (Fig. 3A and Supplementary Data 8). Significantly, around 20% of cis-pQTLs of prostate, breast, ovarian, and head and neck cancers were found to colocalized with cis-eQTLs in the corresponding tumor tissues, exhibiting effects in the same direction, which suggested the influence of gene expression on altered protein abundance (Supplementary Fig. 3A). As for another source of protein variation, cis-sQTLs co-localized with cis-pQTLs for 12.29% (186/1,514) of proteins, with the proportion over 30% in leukaemia (Fig. 3B, Supplementary Fig. 3B and Supplementary Data 9), highlighting the impact of altered splicing on plasma protein abundance. Altogether, our findings affirm that pQTLs can regulate protein abundance by modulating gene expression or splicing at the transcriptional level and reveal the synchronized alterations of genetic effects on protein levels in both plasma and corresponding disease tissues.

A, B Integration of gene expression and splicing quantitative trait loci (eQTLs and sQTLs). A Proteins ordered by the number of disease types for which at least one of the cis-pQTLs was also a cis-eQTL as determined by statistical colocalization (posterior probability > 0.7). Scatter plot illustrates effect size of genetic variants with colocalized signals in pQTLs and eQTLs. Colors represent disease types. B Same as A but considering cis-sQTLs. C Enrichment of pQTLs within GWAS loci across 42 diseases. Vertical bars represent the log2-transformed odds ratios (OR), with error bars indicating the 95% confidence intervals. D The contributions of pQTLs across diverse disease types to the heritabilities of corresponding diseases. The error bars represent the s.e.m. E, F Overview of colocalization between protein and disease GWAS traits across the genome. E The number of colocalized proteins (posterior probability > 0.7). Colors indicate categories of associations. Pink indicates higher protein abundance and increased risk, while green signifies the opposite. F Protein-disease network. The number of proteins (squares) and diseases (circles) with at least one connection are included. Effect directions are indicated by the line type (solid = higher protein abundance, increased risk, dashed = higher protein abundance, reduced risk). G Enrichment results of Reactome pathways for pGenes in each disease type, color-coded by log10(P). H pGene-Drug interaction pairs for 42 diseases based on the DGIdb database (https://www.dgidb.org/). I An example of the pGenes-Drug in hypertension. The width of the line between each gene and drug class indicates the number of interactions.
We next sought to investigate whether pQTLs could offer additional insights into disease susceptibility. As a result, we observed a robust enrichment of pQTLs among most (27 out of 42) disease GWAS loci (P < 5 × 10−8), indicating the promising roles of pQTLs in elucidating disease risk (Fig. 3C and Supplementary Data 10). To further quantify the contribution of pQTLs to disease-related variations, we employed the LD score regression model for a partitioned heritability analysis. Among the diseases examined, pQTLs explained an average of ~6.8% of the heritability (Fig. 3D). Notably, type 2 diabetes (T2D), breast cancer, prostate cancer, and glaucoma exhibited the highest proportions, with over 20% of the heritability explained by pQTLs (Fig. 3D), highlighting the connection between genetic risk factors, proteins and diseases.
The enrichment of pQTLs in disease-associated loci provides disease-specific insights into the overall impact of pQTLs. In order to reveal the extensive protein regulation network underlying complex diseases, we conducted colocalization analysis. This identified an average of 5, proteins per disease type sharing genetic determinants (posterior probability > 0.7), ranging from 1 (e.g., uterus cancer, ovary cancer, abdominal aortic aneurysm (AAA), liver, pernicious anaemia (PA), rheumatoid arthritis (RA)) to 20 (asthma). These findings demonstrate substantial co-localization between cis-pQTLs and disease loci (Fig. 3E and Supplementary Data 11). Of particular interest, we observed shared proteins among certain diseases, suggesting potential common mechanisms contributing to their pathogenesis (Fig. 3F). For example, apolipoprotein(a) (LPA), which promotes atherosclerotic lesions and thrombogenesis, was genetically associated with coronary heart disease (CHD), major adverse cardiac event (MACE) and peripheral artery disease (PAD) in the colocalization analysis, verifying the previously established mechanism connections of cardiometabolic disease (Fig. 3F). Moreover, the colocalization of apolipoprotein E (APOE) underscores the intricate interplay among renal disease, liver disease, CHD, chronic obstructive pulmonary disease, gout, and dementia, indicating a shared dysregulated lipid metabolism underlying these disease types across diverse systems (Supplementary Data 11).
To further elucidate the functional roles of pQTL-regulated protein-coding genes (pGenes) in human diseases, we performed pathway analysis on these genes. Immune system-related pathways were the most frequently enriched across diseases (Fig. 3G). Among the significantly enriched pathways, “Immunoregulatory interactions between a Lymphoid and a non-Lymphoid cell” recurred across nearly all disease types. This recurrence indicates that core immune pathways are common targets of genetic variation. Genetic perturbations in these networks appear to shape shared susceptibility patterns across diseases.
Considering the possible prognostic effect of pQTLs, we further performed survival analysis among patients across 36 different diseases (observed at least one specific death event) derived from the UKB cohort. Collectively, 969 independent survival-related pQTLs were identified (Supplementary Table 3). Furthermore, we amalgamated the pGenes with pharmacological information to assess the potential implications of our findings for drug development and repurposing. Through investigating the drug-gene interactions in the Drug Gene Interaction database (DGIdb), we identified a median of 580 drugs interacting with pGenes for each disease, with a maximum of 2707 interactions found in hypertension (Fig. 3H). Impressively, typical pGenes in hypertension, such as SMAD3, VEGFA, and APOE, exhibited interactions with a diverse range of drugs, including lipid-modifying agents, vasoprotectives, ophthalmologicals, and even antineoplastic agents (Fig. 3I). Overall, these results suggest that these disease-specific pGene-drug interactions could potentially broaden the horizons of clinical therapy.
Differences and similarities in pQTL regulatory effects between health and disease states
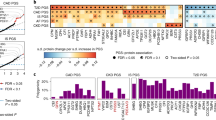
By considering the specific disease context, it is possible to more extensively resolve functional genetic variants that contribute to disease pathobiology. Similarly, a notable enrichment of pQTLs identified in various disease statuses within GWAS loci was observed across the majority (26 out of 27) of disease types, in contrast to pQTLs identified in health cohort (Supplementary Fig. 4A), which highlight the advantages of our pQTL detection method in the translation of GWAS findings into biological and possibly clinical insights. Next, to further explore how disease status influences the genetic regulation of plasma protein levels, we refitted each independent pQTL association using linear mixed effects (LME) models (“Methods”). We observed that the number of disease-related pQTLs increased with sample size, indicating that expanding the cohort would likely identify more associations. Across 27 diseases with at least 325 cases each, we identified 1806 disease-related pQTL associations (q-value < 0.05), ranging from 10 in Multiple Sclerosis (MS) to 360 in T2D (Fig. 4A and Supplementary Data 12). We also detected 7924 disease-biased pQTLs (q-value ≥ 0.05; not replicated in healthy cohort) and 13,844 non-disease pQTLs (q-value ≥ 0.05; shared with healthy cohort). All results are available on our public database. We noted the number of disease-related pQTLs correlated with sample size, suggesting expanded proteomic datasets for specific diseases will yield further discoveries (Supplementary Fig. 4B). Notably, disease-related pQTLs exhibited larger effect sizes than disease-biased and non-disease pQTLs (Fig. 4B). Compared to disease-biased and non-disease pQTLs, disease-related pQTLs exhibited a marked enrichment within missense regions (Fig. 4C). Several identified disease-related pQTL pairs are noteworthy. These include the rs77779567-CDCP1 pair (q-value = 3.92 × 10−4) linked to MACE (Supplementary Fig. 4C), which is associated with cardiac fibrosis development14; the rs4357365-EPHA4 pair (q-value = 8.30 × 10−3) associated with breast cancer (Supplementary Fig. 4D), which is upregulated in the breast cancer stem-like cells15; and the rs4592657-CDH15 (q-value = 1.20 × 10−5) pair related to CHD (Supplementary Fig. 4E), which potentially contributes to coronary repeat revascularization16. These findings highlight the potential of disease-type-hierarchical analyses in revealing regulatory effects that might be overlooked by less granular approaches.

A Bar and Scatter charts show the number and percentage of disease-related pQTL associations. B Comparison of absolute effect sizes of the disease-related pQTL associations (left), disease biased and non-disease associations (right). The center line of the box presents as the median, the box limits indicate upper and lower quartiles and whiskers indicate the maximum and minimum. ***P < 0.001 was calculated by a two-sided Mann-Whitney U test. C Genomic distribution of disease-specific (left), disease biased and non-disease (middle) pQTLs, alongside enrichment analysis comparing the two. Results were calculated using the two-tailed Fisher’s exact test. D Sankey plot displays the clustering results of 27 diseases based on pQTL analysis. From left to right, the figure shows the disease names, their conventional clinical classifications, and the corresponding clustering outcomes. E The results of Gene Ontology (GO) biological process (BP) enrichment proteins associated with the trans hotspot region chr8_77. F The results of Gene Ontology (GO) biological process (BP) enrichment of proteins related to cluster 7.
Furthermore, comparative analysis of pQTL effect sizes and directions across diseases may reveal distinct or shared underlying biological mechanisms and potential precision therapeutic targets. In total, we identified 2832 pQTL associations exhibiting significant cross-disease discordance (twofold or more difference in effect magnitude or opposite direction) across 27 diseases with at least 325 cases each (Supplementary Data 13). For instance, we found 23 pQTL associations with discordance between asthma and rhinitis. These include rs62217915-KIT (Supplementary Fig. 4F), which regulates mast cell proliferation and survival and correlates with asthma severity17, and rs8105105-SIGLEC6 (Supplementary Fig. 4G), a dynamically regulated mast cell marker18. These associations showed differential effects between individuals with asthma and rhinitis, highlighting genetic disparities and treatment implications between these conditions.
Additionally, we applied hierarchical clustering based on QTL profiles to uncover shared disease mechanisms, and grouped the 27 diseases into 8 clusters (Fig. 4D and Supplementary Table 4), which reveals both well-known connections between diseases and less anticipated similarities. For example, cluster 4 were made up of ophthalmic diseases including cataracts and glaucoma. (Fig. 4D). Common cardiometabolic diseases, including disorders of lipoprotein metabolism and other lipidaemias (DOLM), T2D, hypertension and its renal complications, also cluster together in terms of their overall pQTL association patterns, as do MACE and CHD. Notably, we identified trans-hotspot regions that were associated with multiple proteins yet confined to a single disease cluster, underscoring their shared genetic basis. For example, region chr8_77 is associated with nine proteins in trans, and crucially, these proteins are associated with either hypertension or DOLM, both mapping to cluster 6 (Supplementary Fig. 4H). By acting through multiple proteins in a specific disease context, this hotspot amplifies the co-morbidity pathological network, particularly in processes such as protein homeostasis, amino acid transport, and neuroendocrine and inflammatory signaling. This reveals an interconnected network of cardiovascular, metabolic, and immune dysregulation (Fig. 4E), offering crucial insights for understanding the genetic basis of these conditions and for identifying therapeutic targets. Intriguingly, 75% of clusters contained diseases from more than one disease category. Taking cluster 7 as an example, it included diseases from the respiratory system, digestive system and neurologic system. The results of pathway enrichment indicate that immune inflammation is the common thread connecting these four seemingly different diseases, and also reflect their respective pathological and physiological characteristics, such as tissue remodeling, barrier damage and repair, and neuroinflammation (Fig. 4F). These provide preliminary biological insights for re-understanding disease classification from biological profiles.
Establishing robust causal associations between proteins and diseases
The identification of causal proteins holds paramount significance in disease risk prediction and therapy development. To establish high-confidence causal relationships, we performed association analysis in an extensive cohort study and employed MR analysis to investigate the associations between plasma proteins and the incidence of diseases.
Leveraging the high-quality cohort consisting of 45,188 individuals from UKB, we systematically assessed the associations between 2905 plasma proteins and 32 disease endpoints (excluding 10 diseases with fewer than 250 incident cases in the years following blood sampling) with cox proportional hazard models (Supplementary Table 5). As a result, 2663 proteins exhibited significant associations with at least one incident, representing 25,940 associations in total with a false discovery rate (FDR) of less than 0.05 (Fig. 5A, B). Diseases such as DOLM, T2D, renal diseases, and CHD were associated with over 1500 proteins, whereas fewer associations were observed with melanoma, breast cancer, prostate cancer, and colorectal cancer, and no significant associations were found in rhinitis (Fig. 5A). Moreover, we observed a total of 2416 (90.72%) proteins that demonstrated associations with more than one disease (Fig. 5B), with the majority (66.13%) showing consistent associations across diverse diseases. For instance, GDF15, ELN, EDA2R, EDN1, and ACTA2 were identified to be positively associated with over 25 different diseases (Supplementary Figs. 5 and 6). However, some proteins displayed more distinct disease specificity. For example, vasorin (VASN) was primarily associated with the risk of renal diseases (HR = 1.17, 95% CI = 1.14–1.21), and a similar association was observed between TBC1 domain family member 17 (TBC1D17) and T2D (HR = 1.09, 95% CI = 1.04–1.14). Importantly, a substantial proportion (93.80%) of protein-disease associations remain robust in a sensitivity analysis excluding the first year of follow-up, indicating independence from clinically incipient cases at baseline. Similarly, 72.31% of protein-disease associations remained significant when restricting the controls and further adjusting for comorbidity status for each incident disease endpoint (“Methods”, all results are available on our public database).

A, B Protein-disease associations based on cohort study, using protein levels as exposure and disease onset as outcome. FDR < 0.05 was considered significant, taking into account the testing of 32 disease types tested for each protein. A Scatter plot shows the -log10(FDR) calculated from Cox models. Colors indicate disease types and numbers on top indicate number of significantly associated proteins (FDR < 0.05). Grey dots indicate associations not reaching significance. Positive associations are displayed in the upper panel and inverse associations in the lower. B Brick plot displays the ranking of proteins based on the number of associated incident endpoints, showing both positive associations (top) and inverse associations (bottom). Colors represent disease types, and proteins with multiple associations are annotated. C Causal proteins associated with 33 diseases identified by MR, including multiple proteins that are already targeted with drugs or marked as high-confident (annotated). Each colored block corresponds to a disease type, and each radius represents a protein. The height indicates OR, with red indicating hazardous effects and green indicating protective effects. D Bar charts illustrate the number of proteins associated with each disease. Left: Protein-disease associations evaluated by cox models; Right: Causal relationship identified by MR. The intersection reveals high-confidence proteins, both achieving FDR < 0.05.
Subsequently, in order to avoid potential biases introduced by disease-related regulatory changes, such as reverse causation or collider bias, we used cis-pQTLs identified from 7626 healthy individuals as instrumental variables for Mendelian randomization (MR) analysis. To mitigate the potential impact of horizontal pleiotropy on causal estimation, we further implemented the MR-Egger (P > 0.05) and MR-PRESSO (P > 0.05) methods. Using the Wald ratio or inverse-variance weighted (IVW) method, 639 significant putative causal associations (FDR < 0.05) were identified, with a mean of ~20 associations per disease (Fig. 5C and Supplementary Data 14). Genetically predicted elevated levels of 149 proteins were associated with increased disease risk, whereas 166 were protective. Additionally, 64 proteins exhibited opposing directions of association across different diseases. Furthermore, 68.37% (201/294) of MR associations instrumented by disease-derived pQTLs were also significant when instrumented by healthy-derived pQTLs, thereby reinforcing and expanding the MR results (Supplementary Data 14 and 15, and Supplementary Fig. 7). Notably, among the protein-disease associations not replicated in the healthy cohort, some proteins exhibited genetic effects exclusively under pathological conditions, representing promising therapeutic targets. For example, bridging integrator 2 (BIN2) is associated with an increased risk of hypertension (OR = 1.29, 95% CI = 1.14–1.46), a finding that aligns with its function in regulating platelet calcium signaling via the STIM1-IP3R axis and modulating a thrombo-inflammatory response which contributes to vascular dysfunction19. Collectively, the above results indicate that the MR of disease samples can serve as a supplement to the healthy cohort in terms of identifying plausible drug targets for risk mitigation and disease treatment.
To establish robust causal associations between proteins and diseases, we integrated evidence from association analysis and MR. Consequently, 110 disease-protein associations were supported by both analyzes and considered highly confident, ranging from 1 for cerebral stroke (CS), colorectal cancer, Depression, heart failure (HF), liver and rheumatoid arthritis (RA) to 23 for T2D (Fig. 5D, and Supplementary Data 16). Notably, these findings underscore several well-established associations between proteins and diseases, which are corroborated by additional lines of evidence. For example, our discovery associating elevated plasma lipoprotein(a) [Lp(a)], a key target in current cardiovascular disease (CVD) drug research, with an increased risk of seven CVDs (including MACE, CHD, AAA, CS, atrial fibrillation (AF), and PAD) suggests that elevated Lp(a) is a significant risk factor for CVD (Fig. 5D and Supplementary Data 16). Moreover, we found that elevated plasma protein convertase subtilisin/kexin type 9 (PCSK9) levels are associated with an increased risk of DOLM, MACE, and PAD, which aligns with the known therapeutic benefits of PCSK9 inhibitors (e.g., alirocumab, evolocumab) in hypercholesterolemia and CVD. This highlights the usefulness of this set of robust protein-disease associations in terms of disease etiology and drug discovery. Additionally, within this set of robust protein-disease associations, apart from those proteins currently under development as therapeutic targets, there exist other proteins supported by compelling experimental evidence that could emerge as promising drug targets. Our integrated analysis implicates secretoglobin 1A1 (SCGB1A1) (Fig. 5C, and Supplementary Data 16), a serum biomarker for obstructive lung diseases, in asthma susceptibility (OR = 0.95, 95% CI = [0.92–0.98]; HR = 0.88, 95% CI = [0.84–0.94]), and this directionality of effect between SCGB1A1 and asthma was recently supported by the experimental demonstration that lack of SCGB1A1 in the lung results in dramatically altered pulmonary function and structural alterations consistent with enhanced remodeling20. Separately, among proteins with MR evidence of association with T2D (Fig. 5C, and Supplementary Data 16), elevated plasma angiotensin-converting enzyme (ACE) levels were associated with an increased risk of T2D (OR = 1.07, 95% CI = 1.04–1.10; HR = 1.10, 95% CI = 1.06–1.15). This finding supports the potential repurposing of anti-hypertensive ACE inhibitors for T2D treatment21. Collectively, these findings highlight the broader utility of this dataset in uncovering disease etiology and accelerating the development of potential therapeutic targets.
Integration of plasma proteins with polygenic scores derived from pQTLs facilitates disease risk stratification
Given that pQTLs and robust causal proteins are closely related to the occurrence of diseases, we aimed to translate these findings into clinical applications by developing the PRS and ProRS. The PRS was constructed using pQTLs identified across various diseases, while the ProRS was derived from proteins robustly associated with disease causation.
Initially, we used logistic regression to assess the relationship between PRS, ProRS, their combination, and disease risk. The pQTLs-based PRS effectively stratifies the high-risk population for 12 diseases, including skin cancer, DOLM, venous thrombosis (VT) and asthma. The odds ratios (ORs) observed among individuals in the top quintile ranging from 1.13 (MACE) to 1.56 (colorectal cancer) compared to the bottom quintile (Fig. 6A, Supplementary Fig. 8, and Supplementary Data 17). ProRS, derived from robust causal proteins, demonstrates significant efficacy across 21 diseases with ORs ranging from 1.22 (HF) to 25.33 (T2D) (Fig. 6A, Supplementary Fig. 8, and Supplementary Data 17). By integrating PRS and ProRS into the PRS+ProRS model, we observed a similar yet enhanced stratification effect for 21 diseases compared to ProRS, with ORs ranging from 1.27 (HF) to 25.85 (T2D) (Fig. 6A, Supplementary Fig. 8, and Supplementary Data 17). In conclusion, both the pQTL-based PRS and the ProRS derived from robust causal proteins prove to be valuable in stratifying diseases risk.

A ORs for each quintile of the three risk scores (PRS, ProRS and PRS + ProRS), calculated via logistic regression analysis. Error bars indicate 95% CI. B Forest plots illustrate the Harrell’s C-index for predictive models including Age+Sex, Age+Sex+PRS,or Age+Sex+ProRS and Age+Sex+PRS+ProRS. Models were constructed using Cox proportional hazards regression, with normalization of each risk score. Error bars indicate 95% CI. C Decision curves for different groups: the four aforementioned predictive models (Age+Sex, Age+Sex+PRS, Age+Sex+ProRS, Age+Sex+PRS+ProRS), and the two reference lines (“Treat All” in black, “Treat None” in gray). There are exemplified with four diseases (skin cancer, DOLM, T2D, asthma), and other diseases are provided in the supplementary materials.
Next, we assessed the predictive efficacy of PRS and ProRS in diverse UKB cohorts representing corresponding diseases. We incorporated PRS, ProRS, and their combination with basic clinical factors (age and sex) into Cox proportional hazard models to develop prediction models for 21 disease endpoints. Harrell’s C-indexes, employed for evaluating model accuracy, demonstrate enhanced predictive capability when incorporating PRS and ProRS into the basic model (Age+Sex) (Fig. 6B, and Supplementary Data 18). The ProRS model demonstrated superior performance to the PRS. Furthermore, the combined PRS+ProRS model achieved comparable or slightly better performance than the ProRS alone, with an improvement of 0.17 observed in T2D (Fig. 6B, and Supplementary Data 18). These findings underscore the substantial potential of genetic and proteomic data in disease prediction and highlight the value of integrated models for precise risk assessment and management strategies.
Although the amalgamation of PRS with ProRS exhibited commendable efficacy in distinguishing at-risk populations, these results were not enough to advocate for their clinical application. To enhance the clinical interpretability of the statistics, we conducted a thorough analysis using decision curve analysis (DCA). Impressively, we observed that the incorporation of both PRS and ProRS into the basic model significantly enhances its clinical utility, with the joint model showing the highest clinical utility across most diseases (Fig. 6C, and Supplementary Fig. 9). Altogether, these results highlight the clinical value of combining plasma proteins with polygenic scores from pQTLs in identifying high-risk populations.
An open-access database of pQTL-Atlas
We have developed “pQTL-Atlas”, a user-friendly data portal (http://pqtl.whu.edu.cn), designed to facilitate the visualization, retrieval, and exploration of our findings for the biomedical research community (Fig. 7). The pQTL-Atlas platform comprises six primary modules: pQTLs, Survival-pQTLs, GWAS-pQTLs, Risk protein by cohort study, Causal protein by MR, and Drug target (Fig. 7A). Users can perform queries by selecting the disease type, protein, pQTL type, or by entering the single nucleotide polymorphism (SNP) ID of interest (Fig. 7B). In the Risk protein by cohort study and Causal protein by MR modules, users can identify risk or causal proteins associated with diseases by selecting a disease type or specific protein. The Drug Target module allows users to identify drug-gene pairs for diseases by selecting a disease type. The search results for each module are presented in a table that lists related information (Fig. 7C, D). Users can generate a box plot in the pQTLs module (Fig. 7E) or a Kaplan–Meier plot in the Survival-pQTLs module (Fig. 7F) by clicking the “Plot” button for each result item. Our database serves as a valuable resource for advancing the understanding of disease mechanisms, discovering protein biomarkers and prioritizing innovative drug targets, which is poised to significantly benefit the research community.

A Six functional modules are provided in pQTL-Atlas. B Query web interface in the ‘pQTLs’ module. C An example of query return table of the ‘pQTLs’ module. D Examples of query return tables from the ‘Risk Protein by cohort study’ module (upper panel), ‘Causal Protein by MR’ module (middle panel), and ‘Drug Target’ module (lower panel). E An example of box plot visualizing one pQTLs. The center line of the box presents as the median, the box limits indicate upper and lower quartiles and whiskers indicate the maximum and minimum. F An example of Kaplan–Meier plot (Cox’s proportional hazards regression) visualizing one survival associated pQTLs.
Discussion
Proteins play central roles in the vast majority of pathophysiological processes, bridging the gap between the genome and phenome. In this study, we present a characterization of regulatory genetic variants across plasma proteins in the human diseases. In total, we characterized 25,987 independent pQTL association pairs in 2901 plasma proteins across healthy and 42 diseases, in which 73.11% of primary associations were replicated, underscoring the reliability and significance of our findings as a valuable addition to existing resources. Building upon large-scale pQTL studies like the UK Biobank Pharma Proteomics Project (UKB-PPP) that characterized protein regulatory architecture in the general population, we conducted disease-stratified pQTL analysis to uncover disease-related regulatory mechanisms potentially masked in mixed population data. Using LME models, we dived deeper into the impact of disease status on the genetic regulation of plasma protein levels. This analysis identified 1806 disease-related pQTL associations and 7924 associations biased to particular diseases. Beyond identifying associations distinct to disease versus health states, we further explored differences and similarities across diseases, revealing 2832 pQTL associations with cross-disease discordance. These findings highlight distinct genetic etiologies and suggest potential treatment strategies. Additionally, by clustering diseases based on their pQTL profiles, we re-examined disease boundaries, leveraging disease convergence to reflect shared biological properties. These results provide a comprehensive view of how genetic variants are linked to plasma protein levels in specific disease contexts and suggest the broader utility of this dataset in elucidating disease mechanisms. Through functional annotation and colocalization, we illustrate the underlying mechanism by which pQTLs influence protein abundance and contribute to disease. In addition, through amalgamating the pGenes with pharmacological information, a median of 580 drugs interacting with pGenes for each disease were identified, thus providing promising therapeutic targets and extended applications for existing drugs. Critically, the capacity for disease and prediction models based on integrating pQTL-PRS and causal ProRS showcased markedly superior or comparable performance compared with established demographic variables across 21 disease endpoints. Our findings are publicly available, which we expect will help guide the development of future clinical diagnosis, prediction, and intervention strategies (Supplementary Fig. 10).
Protein expression regulation is influenced not only by genetic factors but also significantly by disease states. We identified 1806 disease-related pQTL associations, implying that the regulatory roles of the same protein can differ markedly depending on disease context. For instance, a MACE-associated pQTL, rs77779567, exhibits a significantly enhanced regulatory effect on plasma CDCP1 levels in hypertensive patients compared to healthy individuals. Previous studies suggest CDCP1 may modulate cardiac fibrosis and influence sST2 expression, a well-established prognostic biomarker for heart failure, thereby contributing to adverse cardiac remodeling and worse cardiovascular outcomes14. Similarly, rs4592657 regulates plasma CDH15 expression, with significantly stronger effects in MACE patients (more than double that in healthy individuals). These findings illustrate how human diseases substantially modify the regulatory impact of genetic variations on protein expression and underscore the importance of disease-associated pQTLs in providing unique insights into disease etiology and mechanisms.
Further investigation of cross-disease differences and similarities can reveal distinct genetic etiologies and shared treatment strategies, advancing precision medicine more effectively. We identified 2832 pQTL associations exhibiting significant cross-disease discordance, highlighting genetic disparities and treatment implications across conditions. Asthma and rhinitis, common respiratory diseases sharing etiological and pathogenic similarities, exemplify this. Notably, rs62217915-KIT and rs8105105-SIGLEC6 exert stronger effects in asthma than in rhinitis, underscoring the critical role of mast cell regulation specifically in asthma22. This finding aligns with the observed therapeutic efficacy of the KIT inhibitor like imatinib in asthma but not rhinitis17. Such discordant pQTLs provide a precise roadmap for developing disease-specific targeted therapies and genetic risk stratification. Furthermore, traditional disease classification, often based on clinical symptoms and phenotypic traits, may overlook shared molecular etiology. By clustering diseases based on pQTL profiles, we can redefine disorder boundaries by anchoring the convergence of diseases in their shared biological properties. Linking biologically related diseases could explain the co-occurrence of seemingly unrelated symptoms and enhance mechanistic understanding and therapeutic development.
Additionally, joint analyzes of pQTLs and disease risk loci facilitate to overcome the bottleneck in the translation of GWAS findings into biological and possibly clinical insights23. The significant enrichment of pQTLs within GWAS risk loci, as demonstrated by our study and othersʼ6,8,24,25,26, underscores the valuable utility of pQTLs in prioritizing candidate causal genes for diseases and traits. Notably, the pQTL mapping approach successfully identified plenty of biologically plausible candidate genes that were not implicated by other QTL mapping methods. For example, AHSG for osteoporosis, DUSP13 for atrial fibrillation, SERPING1 for gastro-oesophageal reflux disease and so on. As the pQTLs provide hints of mechanisms and a considerable proportion are deemed functional, they may contribute to elucidating some ‘missing heritability’ of complex diseases.
One of the principal challenges in observational studies investigating disease-associated proteins is identifying the causal proteins that can motivate therapeutic target discovery. To achieve this, we enable the identification of robust and high-confidence causal proteins associated with diseases through the integrated analysis of both effective methods for causal inference, cohort study and MR27,28, which offers a data-driven approach to drug discovery using population-level data. To estimate the effect of plasma protein levels unconfounded by disease status, we performed MR analysis using only samples from the healthy cohort. This robust set of protein-disease associations partially overlapped with established target-indication pairs in drug databases (e.g., LPA for CVDs and PCSK9 for DOLM, MACE, PAD), corroborating the principle that genetically supported targets enhance drug development success29. Furthermore, the identification of risk factors across disease categories presents opportunities for drug repurposing. For instance, MR evidence linking T2D to ACE supports the potential repurposing of anti-hypertensive ACE inhibitors for T2D treatment30. Beyond validating repurposing potential for known targets, our findings also revealed therapeutic candidates (e.g., SCGB1A1 for asthma), offering direction for future drug development.
Crucially, the study of both health and disease samples is complementary in terms of identifying plausible drug targets for risk mitigation and disease treatment. As previous studies suggest, genetic variations may exert effects only in pathological contexts, where risk alleles act as an “Achilles” heel’ and accelerate disease progression31. MR results from the disease population were broadly consistent with those from the healthy cohort. Additionally, the genetic effects on some proteins are only detectable under pathological conditions, representing promising therapeutic targets. For example, we observed an association between hypertension and BIN2, a protein known to regulate platelet calcium signaling via the STIM1-IP3R axis and modulate the thrombo-inflammatory response19. As this response contributes to vascular dysfunction in hypertension, BIN2 represents a promising target for future drug development. Collectively, these robust associations offer valuable insights into disease mechanisms and therapeutic target identification.
Additionally, our multi-disease profiling also suggests that integrating plasma proteomics with genomics holds promising predictive capabilities across a spectrum of diseases. There is compelling evidence that PRS complement conventional risk prediction, particularly in White populations, given the availability of extensive genotyped individual datasets32,33. Proteins not only mediate the phenotypic effects of genomic sequence diversity and various environmental factors but also reflect ongoing biological processes. Consequently, ProRS derived from large-scale proteomics data could, to a certain extent, supplement information regarding health status and disease risk, playing a significant role in disease prediction34,35,36. While genomics (PRS) and proteomics (ProRS) individually provide predictive efficacy34,35,36, our study establishes that the integrated model performs better than or comparably to those based on ProRS, PRS, or age and sex alone. This enhanced performance underscores its greater potential for clinical application. Furthermore, we highlight the potential for real-world application by harnessing the power of both proteomics and genomics. The integration of proteomic and genomic data, obtainable from a single blood draw, demonstrates significant potential for real-world clinical utility. This approach can enhance diagnostic support and refine multi-disease risk assessment. Even modest gains in predictive accuracy enable earlier intervention and improved patient stratification, thereby improving overall health outcomes and healthcare efficiency.
However, we also acknowledge several limitations that merit discussion. Firstly, in current plasma proteomic profiling, definitively determining whether detected proteins originate solely from their soluble forms or include contributions from cellular membranes remains challenging. This complexity further complicates the interpretation of effect directions in MR analyzes. In our study, we further corrected the effect direction using results from a cohort study. However, future research should consider employing powerful techniques such as flow cytometry to investigate genetic determinants of cell surface protein expression. These additional insights will enhance the interpretation of plasma pQTL studies. Secondly, for diseases with smaller cohorts, reduced statistical power may limit pQTL detection and preclude downstream analyzes such as causal inference and prediction model validation. Therefore, increasing cohort sizes in future studies will be essential for more comprehensive and statistically robust analyzes across a broader spectrum of diseases. Additionally, while validation is crucial in proteomics and disease prediction research, replication in other large prospective studies is currently not feasible due to the lack of comparable large-scale proteomic resources covering as many outcomes as our study. Lastly, we did not adjust for medication use in pQTL analyzes. This factor should be considered in future studies when precise medication timing and protein measurement data become available in UKB.
In summary, by integrating genetic with plasma proteomic data from healthy and disease-affected individuals, we conducted systematic proteogenomic analyzes that investigated cross-disease and health-state differences. This work provides a comprehensive understanding of the genetic architecture of plasma proteins and nominates putatively causal proteins for disease risk. These findings further enable precise risk prediction and offer insights that bridge target discovery and therapeutic development. Moving forward, this open-access proteogenomics atlas will serve as an invaluable resource, facilitating a deeper understanding of disease pathogenesis, enabling the interpretation of future GWAS findings, and promoting the effective development of biomarkers, predictive models, and therapeutic targets.
Methods
Ethics
The study design and conduct complied with all relevant regulations regarding the use of human study participants and was conducted in accordance with the criteria set by the Declaration of Helsinki. Ethics approval was granted by the North West Multi-Centre Research Ethics Committee. All participants provided informed consent via electronic signatures. Our research was carried out under UKB application number 94939.
Study design
The study mainly consisted of five steps (Supplementary Fig. 10): (1) a comprehensive genome-proteome-wide pQTL analysis of 2901 circulating proteins in 35,690 participants (7626 healthy individuals and 28,064 patients across 42 diseases); (2) functional annotation of pQTLs to elucidate their mechanisms of action on protein abundance; (3) a comparison of pQTL regulatory patterns between healthy and diseased states; (4) identification of high-confidence causal proteins for specific diseases by integrating cohort studies with MR; and (5) development of disease risk prediction models by integrating pQTL-derived PRS and causal-ProRS.
Data source
The UKB is a large-scale prospective cohort study that enlisted over 500,000 participants aged 37–73 years, who visited one of 22 assessment centers between 2006 and 2010. Participants completed an online questionnaire including questions about demographic characteristics (for example, age, sex, ethnicity), anthropometry (height, weight), lifestyle (for example, alcohol and smoke) and diet. Participants’ follow-up began at their initial visits to the UKB assessment centers (baseline), coinciding with the collection of blood samples and other clinical data. Follow-up was censored either upon disease diagnosis or at the last available date from hospital or general practitioner records, whichever came first (censored time).
Definition of cases in pQTL study
This study included 43 disease types at baseline, encompassing 42 specific diseases across 9 disease categories, as well as a healthy group. These disease cases were identified and categorized based on the ICD-10 codes (Supplementary Data 1), and were extracted from the first occurrence data (UKB categories 1712) and cancer diagnoses (UKB categories 40005-40006)34,37. The first occurrence data (October 2023 update) were derived from primary care records (category 3000), hospital inpatient records (category 2000), self-reported medical conditions (UKB field 20002), and death register records (fields 40001 and 40002).
The pQTL study only included prevalent cases, i.e., cases whose event date preceded the enrollment date. Participants without confirmed records in the database at baseline and within 6 months of follow-up for the 42 diseases and medications were included in the healthy population for pQTL analysis. The exclusion criteria for each disease were determined based on diagnoses extracted from self-reported clinical records.
Blood proteomics
The UKB-PPP consortium undertook proteomic profiling of blood plasma samples. These samples, collected from 54,219 UKB participants, were analyzed using the Olink™ Explore 3072 Proximity Extension Assay. This assay measures 2941 protein analytes, capturing 2923 unique proteins8. Proteins were measured across eight panels, which included cardiometabolic, cardiometabolic II, inflammation, inflammation II, neurology, neurology II, oncology, and oncology II proteins. Further details on sample selection, processing, and quality control information for the Olink assay can be found in previous publications8,38. Supplementary Data 2 provides a list of all proteins used in this study.
Genotyping and imputation
The UKB genotyping protocol, imputation and quality control have been described previously in detail39. In summary, UKB participants underwent genotyping using two similar arrays, the UKB Axiom array and the UK BiLEVE Axiom Array. Phasing was accomplished utilizing SHAPEIT3 in conjunction with the 1000 Genomes Project, while imputation was carried out with IMPUTE4 using the Haplotype Reference Consortium as the primary reference panel. We applied additional quality control procedures for each pQTL calculation: (1) genotype missing rate in the sample of less than 0.1; (2) MAF > 0.05; (3) SNP missing rate less than 0.05; (4) Hardy–Weinberg Equilibrium balance test P > 5 × 10−6. Quality control procedures were executed using PLINK (version 1.90) separately for each disease, resulting in an average of 6,107,244 SNPs per disease type remaining for analysis (Supplementary Fig. 1 and Supplementary Data 3).
Identification of pQTLs
We excluded participants with over 20% missing protein data or with official withdrawal status from the UKB. Proteins with a missing rate exceeding 10% were also excluded. The protein data used in our analysis were subjected to a rank-based inverse normal transformation method to alleviate the influence of outlier protein values. The final analytical cohort comprised 35,690 participants following exclusions for low-quality proteomic data, low-quality genetic data, missing covariate data, or official withdrawal from UKB. Supplementary Data 3 provides sample size for each disease. Protein quality control was conducted separately for each disease, resulting in the inclusion of 2895 to 2902 proteins, with an average of 2901 proteins analyzed (Supplementary Data 3). We adopted a generalized linear regression model MatrixEQTL in the genome-wide association study of the 2902 proteins across these 42 diseases and the healthy population. To control potential confounding factors, we adjusted for clinical status (age, sex), population structures (principal components, PC), and protein measurement variables (batch and time fasted at blood collection). PC of genetic ancestry were calculated using PLINK (version 1.90) on the imputed genetic dataset per disease type and we adjusted for the top ten PC in analysis. For the pQTLs found in each disease type, SNPs reaching the Bonferroni-corrected threshold of P < 1.72 × 10−11 (5 × 10−8 adjusted for 2901 unique proteins) were defined as pQTLs8. Figure 1A presents the pQTL study design and workflow. Cis-pQTLs were defined as a signal within 500 kb upstream or downstream of the protein-encoding gene, and all other signals were defined as trans-pQTLs, consistent with most previous studies7,25,40. A protein was classified as a pQTL-regulated protein (pProtein) for a specific disease if at least one significant pQTL was identified for that protein within the disease context.
Definition of pQTL regions
We defined a protein quantitative trait locus (pQTL) as a genetic locus significantly associated with protein abundance (P < 1.72 × 10−11). For each protein within every disease type, we: (1) collected all significant genetic variants and expanded region of ±500 kb around each variant, (2) iteratively merged overlapping regions until no overlap remained, and (3) designated the most significant variant in each resulting region as the sentinel variant. This window-merge procedure was implemented using BEDtools v.2.27.0. In addition, we treated the extended major histocompatibility complex (MHC) region (chr6: 25.5–34.0 Mb) as a single region.
To determine whether such a region were associated with multiple plasma proteins, we adopted the “LD-based clumping approach” by Sun et al.6. Regional sentinel variants in high LD (r2 > 0.8) with each other were combined together into a single region. Finally, we obtained 2224 independent regions across 43 disease types (Supplementary Data 4).
Comparison with reported pQTLs
To evaluate whether the independent pQTL associations were previously undescribed, we interrogated previous studies. The literature screening applied three criteria: (1) inclusion of studies profiling plasma/serum samples; (2) inclusion of associations reported in European-ancestry populations; and (3) exclusion of studies not assessing proteins. Through manual review of records published since 2018, we included 23 studies and documented their key characteristics, including detection method, sample size, number of proteins, and number of pQTLs, in Supplementary Data 5. We then manually extracted all reported association summaries (rsID, UniProt ID, P value, and cis/trans designation) from the main texts and supplementary materials. For each region, we contrasted our independent results with primary pQTL associations from previous studies, which were considered replicated when LD r2 ≥ 0.17.
Identification of survival-associated pQTLs
Survival-associated pQTLs were identified using the “survival” R package within the UKB population. Patients for each disease were selected based on identical ICD-10 codes for survival analysis. Criteria included removing participants with missing data (age, sex, genetics) and excluding individuals who opted out of the program. The MHC region (chr6:25.5–34.0 Mb) was excluded. The outcome was the observed occurrence of death from the disease. After individual-level quality control, 218,570 patients from 36 diseases remained for analysis (the seven diseases: PA, cataracts, glaucoma, depression, rhinitis, and gout were not observed in the mortality records) (Supplementary Table 3). Disease survival follow-up time was calculated from diagnosis to death or the last follow-up. Survival pQTLs were defined as those with a survival analysis result of P < 0.05 and a pQTL result of P < 1.72 × 1011 (Detailed results can be accessed online).
Enrichment analyses of pQTLs in regulatory elements
For comparative analysis, we generated matched non-pQTLs for each disease type by randomly selecting genomic variants using vSampler (http://www.mulinlab.org/vsampler/) based on the 1000 Genomes Project Phase 3 (1000G P3). These non-pQTLs were matched to the pQTLs based on the number of LD-associated variants, MAF, and variant type (Supplementary Table 2). All independent pQTLs and control variants were annotated using the SnpEff software41. Enrichment analyzes of pQTLs among each position region using a two-tailed Fisher’s exact test, with the following 2 × 2 table: columns; pQTLs and non-pQTLs, and rows; pQTLs within and not within the annotated region.
The functional file of histone modifications, including H3K4me1, H3K4me2, H3K4me3, H3K9ac, H3K27ac, H3K27me3, as well as transcription factor (TF) binding sites was downloaded from the ENCODE portal (https://www.encodeproject.org). BEDtools v.2.27.0 was used to find instances of SNPs overlapped with the peaks of regulatory elements. Enrichment analyzes of pQTLs within regulatory elements using a two-tailed Fisher’s exact test, with the following 2 × 2 table: columns; pQTLs and non-pQTLs, rows; pQTLs within and not within the regulatory element.
Variance explained
We estimated the variance explained by pQTLs for plasma levels of each protein with at least one associated pQTL. The models were fitted using three distinct genetic instrument sets: (1) the cis-pQTL, (2) trans-pQTLs, and (3) all pQTLs, with the respective model R² providing the estimate of explained variance.
Classification of pQTLs
Independent pQTL associations were classified following Pietzner et al.7. For each protein, Gene Ontology (GO) biological process terms were obtained from UniProt using all corresponding UniProt accessions. pQTLs were classified into three categories: (1) Specific-protein: associated with only one protein; (2) Specific-pathway: all associated proteins share a common GO term; (3) Non-specific: either no GO term encompasses all associated proteins, or at least one protein lacks GO annotation. This categorization was conducted independently for each disease context.
The eQTL-pQTL colocalization analysis
We performed pairwise statistical colocalization analyzes of cis-pQTLs with cognate cis-eQTL data from PancanQTL database42, GTEx v.843 (https://www.gtexportal.org/) and eQTLGen Consortium44 (https://www.eqtlgen.org/cis-eqtls.html). For each plasma protein sentinel cis-pQTL, we extracted results from its surrounding ±500 kb region and obtained the same genomic window from cis-eQTL data associated with the relevant tissue for the corresponding disease.
To perform the colocalization analysis, we utilized the coloc R package45. The package provides posterior probabilities (PP) that indicate the likelihood of various scenarios: PP0 (genetic locus not associated with either plasma protein or mRNA), PP1 (associated with protein abundance but not mRNA expression), PP2 (associated with mRNA only), PP3 (associated with both protein and mRNA, but with different causal variants), and PP4 (both protein and mRNA share a common causal variant). We considered PP4 > 0.7 as strong evidence of colocalization between cis-pQTLs and cis-eQTLs.
The sQTL-pQTL colocalization analysis
We performed pairwise statistical colocalization analyzes of cis-pQTLs identified in our study, comparing them with cognate cis-sQTL data in corresponding tumor tissues in TCGA from CancerSplicingQTL database46 (http://www.cancersplicingqtl-hust.com/) and disease-related tissues from GTEx v.843 (https://www.gtexportal.org/). The next steps aligned with the eQTL-pQTL colocalization analysis.
The pQTL-disease GWAS colocalization analysis
We explored evidence of shared genetic associations between variants that directly impact protein expression levels and diseases using the Coloc software package implemented in R45. Subsequently, we performed pairwise colocalization analyzes between cis-pQTLs and disease GWAS signals, applying default priors. We considered PP4 greater than 0.7 as indicative of shared genetic associations. The download of 42 GWAS summary statistics followed the procedure outlined in the MR section.
GWAS enrichment analyses
We obtained the GWAS summary statistics for each disease from OpenGWAS (https://gwas.mrcieu.ac.uk/datasets) and the GWAS catalog (https://www.ebi.ac.uk/gwas/downloads) (Supplementary Data 10), as detailed in the MR section. And selected SNPs that achieved genome-wide significance (P < 5 × 10⁻⁸) for subsequent enrichment analyzes. Enrichment analyzes of pQTLs among disease-related GWAS variants were performed by two-tailed Fisher’s exact test (with the 2 × 2 table (columns; pQTLs and non-pQTLs, rows; variants within and not within the GWAS variants). Then, to estimate the impact of pQTLs on disease heritability, we adopted Linkage Disequilibrium Score Regression (LDSC) using above summary.
Pathway enrichment analysis
For each disease, we performed enrichment analyzes for Gene Ontology (GO) biological process terms and Reactome pathways using the pQTL-associated proteins, with the set of genes encoding the Olink panel proteins serving as the background. The ClusterProfiler R package (v4.10.0)47 was employed to uncover over-represented biological processes based on the GO database. Then, Reactome pathway enrichment analysis was performed using the ReactomePA R package (v1.47.0)48.
Drug-gene interaction analysis
To identify potential drug targets that could be repurposed for disease treatment, we searched for target genes in the DGIdb database (https://www.dgidb.org/), which provides information on drug-gene interactions and druggable genes from various sources, including publications and databases. Medications were categorized using the Anatomical Therapeutic Chemical classification system, retrieved from the Kyoto Encyclopedia of Genes and Genomics (KEGG) drug section (https://www.genome.jp/kegg/drug/).
Assessment of disease status on independent pQTLs
As the method mentioned in previous studies49,50, we re-evaluated each pGene associated with the identified independent pQTLs using LME models within a combined cohort of both disease cases and healthy controls. We first fitted a null model (M0) in which protein levels were regressed age, sex, genetic principal components, batch, and sampling time, against a genotype-including model (M1). Associations where M1 outperformed M0 were considered supported. We further assessed an interaction model (M2) by including an interaction term between disease diagnosis (D) and genotype (G). A likelihood ratio test (LRT) was used to compare the M2 vs M1 models. A significant LRT result (q-value < 0.05) indicated that the more complex model provided a better fit for the SNP-gene association.
Associations of protein levels with disease onset
To investigate the association between plasma protein levels and disease onset in UKB participants, we standardized protein levels using a rank-based inverse normal transformation method. Proteins with a deletion rate over 40% were excluded, resulting in 2905 unique proteins for analysis. Exclusion criteria were applied to each endpoint, excluding samples with more than 50% missing data or with official withdrawal status from the UKB. Prevalent cases that occurred before or during the baseline assessment were excluded, as well as those recorded within the first 6 months after baseline. We utilized Cox proportional hazard models, adjusting for sex, age, smoking, drinking status and body mass index, to predict disease onset across endpoints with 250 or more events per protein (Model 1). Finally, analyzes were conducted for 32 diseases, involving a total of 45,188 participants (Supplementary Table 5). Statistically significant associations were reported when FDR was less than 0.05. Additionally, we conducted sex-specific analyzes for gender-specific diseases (breast cancer for females and prostate cancer for males), excluding sex as a covariate in the respective subsets. Sensitivity analyzes were performed by excluding individuals with disease onset within the first year (Model 2) and those with medication records to assess protein-disease risk associations (Model 3). To further assess the influence of comorbidity, we performed a sensitivity analysis with stringent quality controls (Model 4). Specifically, for each incident disease, we excluded individuals diagnosed with any condition from the same disease category prior to baseline. We also removed participants from the control group who developed an incident disease within the same category. Additionally, we adjusted for baseline multimorbidity from all other disease categories. Detailed results are available on our online platform.
Mendelian randomization analyses
To identify the proteins with causal roles in disease processes and potential pharmacological targets, we conducted MR analyzes. We used proteins with cis-pQTLs identified outside the MHC region (chr6:25.5-34.0 Mb) and that reached genome-wide significance (P < 5 × 10−8) as exposures. We obtained GWAS summary statistics from either OpenGWAS (https://gwas.mrcieu.ac.uk/datasets) or the GWAS catalog (https://www.ebi.ac.uk/gwas/downloads). When multiple datasets were available for a disease, we selected the one with the most cases, ensuring it met specific criteria: (1) adequate genotype data coverage at relevant loci, (2) European ancestry alignment with our pQTL study participants, and (3) available or computable effect estimates and standard error values (Supplementary Data 10). To mitigate weak instrument bias, we excluded SNPs with an F-statistic below 10.
Two-sample MR analyzes were conducted using the two-sample MR package (version 0.5.6) in R (version 4.1.0). PLINK (version 1.90) was employed to select independent variants as instrument variables (IVs) that reached genome-wide significant (P < 5 × 10−8) for each protein. LD clumping ensured SNP independence (10 MB, r2 < 0.001). For proteins with only one instrument, we used the Wald ratio method to estimate the log odds change in disease risk for per standard deviation (SD) increment of plasma protein levels (proxied by instrumental variables). Proteins with multiple instruments were analyzed using the IVW method. The IVW method assumes an intercept of 0 in the Mendelian randomization linear regression model, indicating no horizontal pleiotropy (i.e., SNPs do not simultaneously affect exposure and outcome). To assess potential pleiotropy impact, we scrutinized it further using the MR-Egger and MR-PRESSO methods. Additionally, we implemented other prevalent MR methods (weighted median, weighted mode, and simple mode) to enhance the robustness of our findings (Supplementary Data 14 and 15).
Definition of proteins with robust causal associations with diseases
To evaluate the robustness of these associations, we conducted additional checks by integrating the results from protein-disease associations with those obtained from MR. Proteins consistently showing associations in both analyzes were retained as “high-confidence proteins” (Supplementary Data 16).
Polygenic risk score development
PRS were generated using the C+T (clumping + thresholding) method, based on pQTLs identified for each disease type. These PRS were used to predict the risk of corresponding diseases in the UKB cohort. Specifically, we predicted the risk of 21 diseases in the UKB cohort, including participants who were free of the specific disease and related conditions at baseline. The PRS calculation followed a weighted model, as described below:
in which n means the number of variants, \({{SNP}}_{i}\) (0, 1, or 2) means the number of the risk alleles for the ith variant, and \({\beta }_{i}\) means the effect size of the risk alleles. Individuals are then additively scored in a weighted patten based on the number of risk alleles they carry for each variant in the PRS.
Protein risk score and composite risk score development
To compute proteomic and other composite risk scores, we adapted the PRS methodology with few modifications36,51. Specifically, we used a high-confidence set of proteins associated with each disease to generate protein risk scores. Coefficients for each protein were derived from Cox proportional hazards models, adjusted for all other proteins in the set. These coefficients, representing the multivariable-adjusted hazard ratio per log-unit increase in protein level, were used as weights in a weighted sum of the standardized protein levels to yield an individual protein risk score. Prior to modeling, all continuous variables were log-transformed and centered and scaled. Protein risk scores were then computed for all individuals following established procedures51. Our composite risk score model integrated all high-confidence proteins in a Cox model, along with the addition of the PRS. This resulted in coefficients for both proteins and PRS, ultimately generating a composite score using a weighted model.
Disease-risk prediction with different risk scores
Initially, we employed logistic regression analysis to evaluate the relationship between PRS, ProRS, and composite scores with disease risk. Each score (PRS, ProRS, or their combination) was used individually to rank individuals and divide them into equally sized risk subgroups (quantiles). Subsequently, to assess the predictive efficacy of the risk scores on disease outcomes, we constructed Cox models incorporating clinical features like gender and age. Four multivariable prediction models were developed with different combinations of PRS, ProRS, and clinical features as follows: Model 1 (clinical features), Model 2 (clinical features + PRS), Model 3 (clinical features + ProRS), and Model 4 (clinical features + PRS + ProRS). Survival outcomes were defined using follow-up time to event and the binary incident event indicator. For all disease outcomes, prevalent cases were excluded from the dataset before models were run. Additionally, the predictive performance was validated using Harrell’s C-index and DCA. These analyzes were conducted only for the 21 disease types with high-confidence proteins.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All results of pQTL associations, survival-pQTL associations, protein-disease associations, causal proteins for diseases, and drug target have been deposited through an interactive portal (https://pqtl.whu.edu.cn/pqtls) and are publicly available. Individual-level genetic and Olink proteomics data from the UKB are available at https://biobank.ndph.ox.ac.uk/ by application. This research has been conducted using the UKB Resource under application number 94939. The ICD-10 codes identified and categorized for the disease cases are listed in Supplementary Data 1. The accession codes for all publicly available cis-eQTL, cis-sQTL datasets, and GWAS summary datasets are provided in Supplementary Data 8, 9, and 10, respectively. ChIP-seq peaks and TF-binding sites data were downloaded from the ENCODE portal (https://www.encodeproject.org). The drug-gene interactions can be found from the DGIdb database (https://www.dgidb.org/). Source data are provided with this paper.
References
Barbeira, A. N. et al. Exploiting the GTEx resources to decipher the mechanisms at GWAS loci. Genome Biol. 22, 49 (2021).
Khan, Z. et al. Primate transcript and protein expression levels evolve under compensatory selection pressures. Science 342, 1100–1104 (2013).
Suhre, K., McCarthy, M. I. & Schwenk, J. M. Genetics meets proteomics: perspectives for large population-based studies. Nat. Rev. Genet. 22, 19–37 (2021).
Ferkingstad, E. et al. Large-scale integration of the plasma proteome with genetics and disease. Nat. Genet. 53, 1712–1721 (2021).
Emilsson, V. et al. Coding and regulatory variants are associated with serum protein levels and disease. Nat. Commun. 13, 481 (2022).
Sun, B. B. et al. Genomic atlas of the human plasma proteome. Nature 558, 73–79 (2018).
Pietzner, M. et al. Mapping the proteo-genomic convergence of human diseases. Science 374, eabj1541 (2021).
Sun, B. B. et al. Plasma proteomic associations with genetics and health in the UK Biobank. Nature 622, 329–338 (2023).
Niu, L. et al. Noninvasive proteomic biomarkers for alcohol-related liver disease. Nat. Med. 28, 1277–1287 (2022).
Dall’Agnese, A. et al. Proteolethargy is a pathogenic mechanism in chronic disease. Cell 8674, 01274–1 (2024).
Fairfax, B. P. et al. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science 343, 1246949 (2014).
Lee, M. N. et al. Common genetic variants modulate pathogen-sensing responses in human dendritic cells. Science 343, 1246980 (2014).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Liu, D. et al. Myocardial recovery in recent onset dilated cardiomyopathy: role of CDCP1 and cardiac fibrosis. Circ. Res. 133, 810–825 (2023).
Lu, H. et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat. Cell Biol. 16, 1105–1117 (2014).
Xiang, Q. et al. A potential role for the CDH13/CDH15 gene in repeat revascularization after first percutaneous coronary intervention. Pharmacogenomics 21, 91–99 (2020).
Cahill, K. N. et al. KIT Inhibition by Imatinib in Patients with severe refractory asthma. N. Engl. J. Med. 376, 1911–1920 (2017).
Smiljkovic, D. et al. Expression and regulation of Siglec-6 (CD327) on human mast cells and basophils. J. Allergy Clin. Immunol. 151, 202–211 (2023).
Volz, J. et al. BIN2 orchestrates platelet calcium signaling in thrombosis and thrombo-inflammation. J. Clin. Invest. 130, 6064–6079 (2020).
Zhai, J. et al. Club cell secretory protein deficiency leads to altered lung function. Am. J. Respir. Crit. Care Med. 199, 302–312 (2019).
Canter, D. & Frank, G. ACE inhibitors in the treatment of hypertension in the older patient. Eur. Heart J. 11, 33–43 (1990).
Joulia, R. et al. Mast cell activation disrupts interactions between endothelial cells and pericytes during early life allergic asthma. J. Clin. Invest. 134, e173676 (2024).
Schaid, D. J., Chen, W. & Larson, N. B. From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat. Rev. Genet. 19, 491–504 (2018).
Suhre, K. et al. Connecting genetic risk to disease end points through the human blood plasma proteome. Nat. Commun. 8, 14357 (2017).
Pietzner, M. et al. Genetic architecture of host proteins involved in SARS-CoV-2 infection. Nat. Commun. 11, 6397 (2020).
Gudjonsson, A. et al. A genome-wide association study of serum proteins reveals shared loci with common diseases. Nat. Commun. 13, 480 (2022).
Wang, X. & Kattan, M. W. Cohort studies: design, analysis, and reporting. Chest 158, S72–S78 (2020).
Sekula et al. Mendelian randomization as an approach to assess causality using observational data. J. Am. Soc. Nephrol. 27, 3253–3265 (2016).
Nelson, M. R. et al. The support of human genetic evidence for approved drug indications. Nat. Genet. 47, 856–860 (2015).
Shuey, M. M. et al. A genetically supported drug repurposing pipeline for diabetes treatment using electronic health records. eBioMedicine 94, 104674 (2023).
Ma, X. R. et al. TDP-43 represses cryptic exon inclusion in the FTD-ALS gene UNC13A. Nature 603, 124–130 (2022).
Elliott, J. et al. Predictive accuracy of a polygenic risk score-enhanced prediction model vs a clinical risk score for coronary artery disease. JAMA 323, 636–645 (2020).
Ma, Y. & Zhou, X. Genetic prediction of complex traits with polygenic scores: a statistical review. Trends Genet. 37, 995–1011 (2021).
You, J. et al. Plasma proteomic profiles predict individual future health risk. Nat. Commun. 14, 7817 (2023).
Eiriksdottir, T. et al. Predicting the probability of death using proteomics. Commun. Biol. 4, 758 (2021).
Lauber, C. et al. Lipidomic risk scores are independent of polygenic risk scores and can predict incidence of diabetes and cardiovascular disease in a large population cohort. PLoS Biol. 20, e3001561 (2022).
Buergel, T. et al. Metabolomic profiles predict individual multidisease outcomes. Nat. Med. 28, 2309–2320 (2022).
Elliott, P., Peakman, T. C. & Biobank, U. K. The UK Biobank sample handling and storage protocol for the collection, processing and archiving of human blood and urine. Int. J. Epidemiol. 37, 234–244 (2008).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Koprulu, M. et al. Proteogenomic links to human metabolic diseases. Nat. Metab. 5, 516–528 (2023).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92 (2012).
Gong, J. et al. PancanQTL: systematic identification of cis-eQTLs and trans-eQTLs in 33 cancer types. Nucleic Acids Res. 46, D971–D976 (2018).
GTEx Consortium The GTEx consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Võsa, U. et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 53, 1300–1310 (2021).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383 (2014).
Tian, J. et al. CancerSplicingQTL: a database for genome-wide identification of splicing QTLs in human cancer. Nucleic Acids Res. 47, D909–D916 (2019).
Yu, G., Wang, L.-G., Han, Y. & He, Q.-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Yu, G. & He, Q.-Y. ReactomePA: an R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst. 12, 477–479 (2016).
Nathan, A. et al. Single-cell eQTL models reveal dynamic T cell state dependence of disease loci. Nature 606, 120–128 (2022).
Haglund, A. et al. Cell state-dependent allelic effects and contextual Mendelian randomization analysis for human brain phenotypes. Nat. Genet. 57, 358–368 (2025).
Helgason, H. et al. Evaluation of large-scale proteomics for prediction of cardiovascular events. JAMA 330, 725 (2023).
Acknowledgements
We are grateful to all members who participated in the study, as well as all individuals who helped us complete the research. This work was supported by the Noncommunicable Chronic Diseases-National Science and Technology Major Project (2023ZD0501400), National Science Fund for Excellent Young Scholars (NSFC-82322058), Program of National Natural Science Foundation of China (NSFC-82103929), Young Elite Scientists Sponsorship Program by CAST (2022QNRC001), National Science Fund for Distinguished Young Scholars of Hubei Province of China (2023AFA046), and Fundamental Research Funds for the Central Universities (2042025kf0027) for J.T; Key Program of National Natural Science Foundation of China (NSFC-82130098), National Key R&D Program of China (2022YFA0806601, 2024YFC3405804) and Fundamental Research Funds for the Central Universities (2042022rc0026, 2042023kf1005) for X.M; Program of National Natural Science Foundation of China (NSFC-82373663), Program of Health Commission of Hubei Province (WJ2023M045) for Y.Z.
Author information
Authors and Affiliations
Contributions
J.T. conceived the study and revised the paper. Z.M., Y.Z., X.M., and J.T. supervised the whole project. Q.M. and C.H. organized the data, carried out the statistical analysis. Q.M., Y.Z., and C.H. wrote the first draft of the manuscript. Y.C. and Y.L. assisted in data analyses. C.H. and Y.L. implemented the web design. Q.M., H.C., and Y.L. designed and drew the figures. Q.M. and Y.C. critically revised the manuscript. C.N., M.Z., Z.L., C.C., Y.Z., F.W., M.J., Y.W., X.L., X.Y., C.H., and B.L. assisted in dataset curation. These authors jointly supervised this work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Zhenqiu Liu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ma, Q., Cai, Y., He, C. et al. Genetic profiling of the circulating proteome in common diseases suggests causal proteins and improves risk prediction. Nat Commun 17, 535 (2026). https://doi.org/10.1038/s41467-025-67238-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67238-x
