
Abstract
The formation of the female gamete is a complex developmental process that begins with the differentiation of the Megaspore Mother Cell (MMC) within the ovule. SPOROCYTELESS/NOZZLE (SPL/NZZ) is the principal regulator of the MMC formation, as mutations in the SPL/NZZ gene lead to the failure of the MMC differentiation. Nonetheless, the SPL/NZZ-dependent regulatory pathway governing the MMC development remains largely unknown. Using a multi-omics approach, we identify direct SPL/NZZ targets and their downstream network. We discover that SPL/NZZ interacts with ovule-identity MADS-domain transcription factor complexes to regulate the expression of common target genes. By integrating the omics data with the analysis of either complementation or mutant lines, we describe a comprehensive regulatory mechanism in which SPL/NZZ controls the differentiation of the MMC by acting on an auxin-dependent downstream network.
Similar content being viewed by others

Introduction
In flowering plants, sporogenesis starts with the differentiation of female and male meiocytes: Megaspore Mother Cells (MMCs) within ovules and Pollen Mother Cells (PMCs) within anthers1,2. In Arabidopsis thaliana, the master regulator of meiocytes differentiation is SPOROCYTELESS/NOZZLE(SPL/NZZ), since mutations in the SPL/NZZ gene result in complete female and male sterility due to the absence of both the MMC and the PMC1,2. SPL/NZZ belongs to the SPEAR family, which consists of 5 genes3,4 in A. thaliana. The SPEAR proteins have a nuclear localisation signal at the N-terminus, the SPL motif in the middle of the protein, and an EAR motif at the C-terminus. It has been reported that, through the EAR motif, SPL/NZZ recruits co-repressors, like TOPLESS (TPL), that mediate the transcriptional repression of target genes by interacting with histone deacetylases (HDAs)3,4,5,6,7.
SPL/NZZ also has an additional Myc-type helix-loop-helix domain that could mediate SPL/NZZ homodimerization2,4. Interestingly, SPL/NZZ was initially described as a MADS-like protein, since it contains a sequence of 17 amino acids similar to the MADS-domain2.
According to earlier reports, SPL/NZZ can interact with two families of transcription factors (TFs): the YABBY (YAB) and the CINCINNATA-like TEOSINTE BRANCHED/CYCLOIDEA/PCF (TCP). SPL/NZZ interaction with these TFs mediates the transcriptional repression of their targets4,5,8.
Despite its importance for plant reproduction, the mechanisms of SPL/NZZ transcriptional regulation, as well as its downstream network, are still poorly understood. Among the factors involved in SPL/NZZ regulation, auxin signalling was shown to be a positive regulator of SPL/NZZ expression in anthers, as demonstrated by the reduced SPL/NZZ transcript accumulation in the taa1 tar2-2 mutant9. AGAMOUS (AG) is one of the few factors described to regulate SPL/NZZ expression in both ovules and stamens. Indeed, in flowers at stages 11–1310, AG binds a CArG-box in the SPL/NZZ 3′ UTR to promote its expression in integuments and chalaza of the ovule. Here, the SPL/NZZ expression pattern is also regulated by the RdDM pathway, since SPL/NZZ is ectopically expressed in the drm1drm2 mutant11.
In addition to its function in MMC and PMC differentiation, SPL/NZZ is required for ovule patterning by negatively regulating the expression of factors such as AINTEGUMENTA (ANT), BELL1 (BEL1), INNER-NO-OUTER (INO), and PHABULOSA (PHB)12,13. Indeed, besides the absence of the MMC, spl/nzz mutant ovules are characterised by an impairment in integuments growth and a longer funiculus12.
Concerning the SPL/NZZ downstream network, SPL/NZZ plays a crucial role in the auxin signalling pathway, as the auxin-response reporter line DR5v2 and the auxin efflux carrier PIN-FORMED1 (PIN1) exhibit decreased expression in the spl/nzz mutant ovule14,15.
This study employs a multi-techniques approach, integrating Co-immunoprecipitation coupled with mass spectrometry (Co-IP/MS), Chromatin Immunoprecipitation and sequencing (ChIP-seq), and RNA sequencing (RNA-seq), to comprehensively identify direct SPL/NZZ targets and their downstream network. Our findings reveal the interaction between SPL/NZZ and ovule identity MADS-domain transcription factor complexes, thereby regulating the expression of common target genes, such as ANT. This activity influences the auxin signalling, which was previously suggested as a primary pathway necessary for the MMC differentiation15. We propose a multistep model in which MMC development relies on an SPL/NZZ-dependent auxin downstream network.
Results
SPL/NZZ and MADS-domain transcription factors interact to form multimeric complexes
To gain insight into the network regulated by SPL/NZZ, essential for the differentiation of the MMC, we identified potential SPL/NZZ partners during early stages of ovule development, before and at the MMC stage, by performing co-immunoprecipitation followed by mass spectrometry (Co-IP/MS). To do this, we took advantage of the pSPL/NZZ::SPL/NZZ:GFP construct, previously used to complement the spl-1 mutant allele11.
As previously described11, at ovule stage 2-I16, SPL/NZZ-GFP accumulated in the most apical cells of the L1 layer (Supplementary Fig. 1A). From stages 2-II to stage 2-V, associated with meiosis end, the SPL/NZZ-GFP accumulation domain comprised the L1 layer of the whole nucellus (Supplementary Fig. 1B–D). At later stages, the SPL/NZZ-GFP signal decreased without changing its pattern (Supplementary Fig. 1E, F).
We introduced the pSPL/NZZ::SPL/NZZ:GFP construct into the pAP1::AP1:GR ap1cal system for synchronised induction of flower development17,18. We performed DEX treatment on the inflorescences and evaluated the SPL/NZZ-GFP expression at different days after induction (DAI), to select samples prior to and at the MMC stage for collection. At 3 DAI, SPL/NZZ-GFP was mainly visible in another primordia, a result in line with SPL/NZZ activity in anther morphogenesis2,19 (Supplementary Fig. 1G–I). At 9 DAI, SPL/NZZ-GFP expression in anthers was almost absent (Supplementary Fig. 1J). By contrast, despite the partial loss of synchronisation at this later time point after AP1-GR induction, most flowers showed ovules at stage 2-I, where we could clearly observe a strong and specific nucellar SPL/NZZ-GFP accumulation (Supplementary Fig. 1K, L).
Therefore, we used three independent biological replicates of inflorescences at 9 DAI for Co-IP/MS, employing an anti-GFP antibody20. We found 660 proteins that were statistically enriched (log2 intensity difference > 0; FDR ≤ 0.05) in the IP samples with respect to the input controls (Supplementary Data 1). As expected, SPL/NZZ itself was found as one of the most enriched proteins (Fig. 1A, and Supplementary Data 1).

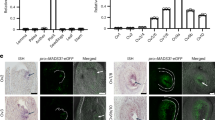
A Vulcano plot showing, with purple dots, the TFs statistically enriched in the SPL/NZZ Co-IP/MS dataset. MADS-domain TFs, SPL/NZZ and TCP22 have been highlighted with labels. The experiment has been performed using three independent biological replicates of SPL/NZZ:GFP AP1:GR ap1cal inflorescences at 9 DAI. Proteins with a positive enrichment (log2 Intensity difference > 0) and FDR ≤ 0.05 were considered significant. The full list of SPL/NZZ interactors identified by Co-IP/MS can be found in Supplementary Data 1. B SPL/NZZ-MADS-domain TFs (AG, STK and SEP3) direct interaction by yeast-two-hybrid assay. Interactions are tested on media depleted of histidine (-W -L -H), or histidine and adenine (-W -L -H -A). For each interaction on the different media, yeast has been spotted at four different concentrations according to a serial dilution (OD 0.5; 1:10; 1:100; 1:1000). C SPL/NZZ form complexes with STK, SEP3 and AG by yeast-three-hybrid assays. Interactions are tested on a medium depleted of histidine (-W -L -H -A). For each interaction, yeast has been spotted at four different concentrations according to a serial dilution (OD 0.5; 1:10; 1:100; 1:1000). D–I SPL/NZZ interaction with SEP3 and STK tested by FRET-FLIM. D GFP fluorescence imaged from a nucleus expressing SEP3Δ-GFP. E GFP and mCHERRY fluorescence imaged from a nucleus expressing SEP3Δ-GFP+SPLΔ-mCHERRY. F GFP and mCHERRY fluorescence imaged from a nucleus expressing SEP3Δ-GFP-mCHERRY. G GFP fluorescence imaged from a nucleus expressing STK-GFP. H GFP and mCHERRY fluorescence imaged from a nucleus expressing STK-GFP+SPLΔ-mCHERRY. I Box plot representing the GFP lifetime of nuclei expressing SEP3Δ-GFP, SEP3Δ-GFP+SPL/NZZΔ-mCHERRY, SEP3Δ-GFP-mCHERRY, STK-GFP and STK-GFP+SPL/NZZΔ-mCHERRY, as calculated by FILM combined with a phasor plot to distinguish different lifetime populations. Measurements have been performed on 5, 5, 5, 11 and 5 different nuclei expressing SEP3Δ-GFP, SEP3Δ-GFP+SPL/NZZΔ-mCHERRY, SEP3Δ-GFP-mCHERRY, STK-GFP and STK-GFP+SPL/NZZΔ-mCHERRY, respectively. Source data are provided as a Source Data file. Asterisks over boxes represent the statistical significance as determined by student’s t-test, two-sided distribution, homoscedastic, confronting SEP3Δ-GFP with SEP3Δ-GFP+SPL/NZZΔ-mCHERRY and SEP3Δ-GFP-mCHERRY, or STK-GFP with STK-GFP+SPL/NZZΔ-mCHERRY. **p ≤ 0.01; ***p ≤ 0.001. Exact P values for each comparison can be found in the Source Data file. Box-plots elements correspond to: centre line = median; X = average; box limits = interquartile range; whiskers = lowest and highest values in the 1.5 interquartile range. Single measures are represented with dots in the boxes. Scale Bars: 10 µm.
To obtain an overview regarding the function of the proteins enriched in the SPL/NZZ Co-IP/MS, we performed Molecular Function and Biological Process GO terms enrichment analysis. The most enriched categories were associated with RNA polymerase activity, DNA binding and transcription (Supplementary Data 1). Moreover, we found enrichment in Biological Process categories such as floral and gynoecium development, regulation of transcription and RNA processing (Supplementary Fig. 2A, and Supplementary Data 1).
Since SPL/NZZ binds to the DNA by its association with transcriptional factor (TF) complexes4,5, we focused our attention on the TFs identified among the SPL/NZZ partners. We found 56 TFs as putative SPL/NZZ interactors. Quite strikingly, among the TCPs, only TCP22 was statistically enriched in our list, while none of the YABBY TFs were present (Supplementary Data 1). The MADS-domain TF family was the most represented in our dataset, corresponding to almost 20% of all the TFs found (Fig. 1A, and Supplementary Data 1). This list of potential MADS-domain TFs interacting with SPL/NZZ included SEEDSTICK (STK), SHATTERPROOF2 (SHP2), SEPALLATA1 (SEP1), SEPALLATA2 (SEP2), SEPALLATA3 (SEP3) and AGAMOUS (AG), all proteins previously associated with ovule specification and development21,22,23.
Based on these results, we verified whether SPL/NZZ could directly interact with MADS-domain TFs in yeast. Since SPL/NZZ contains an EAR-motif, which could negatively impact GAL4 transcriptional activation functionality, we used an SPL/NZZ variant lacking the EAR-motif sequence (SPL/NZZΔ). We initially tested the interaction between SPL/NZZΔ, fused with a GAL4 activation domain (AD) or DNA binding domain (BD) with AG, SEP3Δ23 or STK. We found positive results for the SPL/NZZΔ(AD) - AG(BD), SPL/NZZΔ(AD) - STK(BD) and SEP3Δ(AD) - SPL/NZZΔ(BD) interactions (Fig. 1B, and Supplementary Fig. 2B). It was previously shown that MADS-domain proteins generate multimeric complexes to regulate target genes22,24. Based on this, we performed yeast-three-hybrid assays. We initially evaluated the formation of complexes using SPL/NZZΔ fused to the AD, SEP3Δ to the TFT and either STK or AG to the BD, finding positive results for the SPL/NZZΔ(AD) - SEP3Δ(TFT) - STK(BD) and SPL/NZZΔ(AD) - SEP3Δ(TFT) - AG(BD) interactions (Fig. 1C). It is worth noting that, even though SPL/NZZΔ(AD) did not directly interact with SEP3Δ(BD) (Supplementary Fig. 2B) and only modestly with STK (Fig. 1B), the presence of all three components enhanced yeast growth, suggesting the formation of a more stable complex (Fig. 1C). We then tested the same interactions by fusing SPL/NZZΔ with the GAL4 BD, reconfirming the establishment of complexes with either STK and SEP3Δ or AG and SEP3Δ (Supplementary Fig. 2C). Concerning the screening with SPL/NZZΔ fused with the TFT, we found that the presence of SPL/NZZΔ(TFT) did not enhance the already described SEP3-AG interaction22 (Supplementary Fig. 2D).
As we confirmed that SPL/NZZ can interact with the ovule identity MADS-domain TFs, we analysed STK, AG and SEP3 expression in the nucellus at stages 2-I, 2-II, concurrently with the MMC differentiation. STK and SEP3 were expressed in the nucellus, as shown by the study of GFP-fusion-protein reporter lines, with a pattern comparable to that of SPL/NZZ25,26,27 (Supplementary Fig. 2E–H). By contrast, AG was not expressed in the nucellus (Supplementary Fig. 2I, J)27. To further corroborate the SPL/NZZ direct interaction with SEP3 and STK, we performed Förster Resonance Energy Transfer-fluorescence lifetime imaging (FRET-FLIM)28 assay in planta. We transiently expressed, in tobacco leaves, SEP3Δ-GFP or STK-GFP fusion proteins either alone or in association with SPL/NZZΔ-mCHERRY (Fig. 1D–I, and Supplementary Fig. 3A–E). By imaging nuclei expressing solely SEP3Δ-GFP (Fig. 1D) or STK-GFP (Fig. 1G), we could measure, through FLIM, a GFP lifetime of around 2.3 and 2.7 ns, respectively (Fig. 1I, and Supplementary Fig. 3A, D). When SEP3Δ-GFP or STK-GFP were co-expressed with SPL/NZZΔ-mCHERRY (Fig. 1E, H), we measured a statistically significant reduction in the GFP lifetime because of FRET occurring between GFP and mCHERRY (Fig. 1I, and Supplementary Fig. 3B, E). Since FRET could occur only when donor and acceptor proteins are at a distance lower than 10 nm, this experiment confirms SEP3 and STK ability to physically interact with SPL/NZZ in plant cells.
It was already reported that STK-SEP3 can form a tetramer29 while SPL/NZZ can form a homodimer4. Using AlphaFold330, we predicted in silico the possible formation of SPL/NZZ-SEPs-STK complexes containing, respectively, two copies of SEP (1, 2, or 3), STK and SPL/NZZ. AlphaFold3 preliminary predictions could fold SPL/NZZ only from amino acid 61 to amino acids 91, forming an alpha helix, a protein portion that contains the 8 amino acids corresponding to the SPL-motif. Indeed, the SPL-motif was originally predicted to be part of an alpha helix spanning from amino acids 64 to amino acid 80, which has some homology to the first helix of MADS-domain TFs2. For these reasons, we considered only the SPL/NZZ region spanning from amino acid 61 to 91 for further predictions. All the different simulations predicted the formation of complexes with good confidence levels, assessing the reliability of the results obtained (Supplementary Fig. 4A). Regarding the docking features of SPL/NZZ to the MADS-domain TFs tetramer, the different simulations indicated that SPL/NZZ contacts the K-box of STK and SEPs (Supplementary Fig. 4B). Finally, we performed binding affinity analyses31,32,33, retrieving Gibbs free energy (ΔG) values and dissociation constants (Kd) that confirm the thermodynamic stability of the SPL/NZZ-MADS complexes (Supplementary Fig. 4C).
SPL/NZZ and the MADS-domain TFs share common targets
To identify the direct targets of SPL/NZZ, we used the pSPL/NZZ::SPL/NZZ:GFP AP1:GR ap1cal line to perform a ChIP-seq experiment. Three biological replicates of inflorescence at 9 DAI were used for chromatin immunoprecipitation using an anti-GFP antibody34,35. After input and IP samples sequencing, peaks were considered significant with a fold enrichment (FE) > 0, FDR ≤ 0.05 and if present in at least 2 out of 3 biological replicates. This approach retrieved a list of 203 peaks, corresponding to 197 different genes (Supplementary Data 2). Around 80% of the peaks mapped in genomic regulatory regions, such as promoters, supporting the reliability of the results obtained (Fig. 2A). Among SPL/NZZ-bound genes, we found genes previously linked to SPL/NZZ function as ANT 12, YUCCA6 (YUC6)36 and PHB37 (Supplementary Data 2). Interestingly, also SPL/NZZ itself was present in the list, indicating the possible existence of a feedback loop involved in SPL/NZZ regulation.

A Bar plot showing the SPL/NZZ ChIP-seq peaks localisation along the genome. Around 80% of the identified peaks fall within transcriptional regulatory sequences, such as promoters, 5′ UTRs, first exons, first introns, and 3′ UTRs. The experiment has been performed on three biological replicates of SPL/NZZ:GFP AP1:GR ap1cal inflorescences at 9 DAI. Peaks were considered significant with FE > 0, FDR ≤ 0.05 and if present in at least 2 biological replicates out of 3. The full list of SPL/NZZ targets identified by ChIP-seq can be found in Supplementary Data 2. B The CArG-box motif is the only enriched TF DNA sequence binding-site identified among the SPL/NZZ ChIP-seq peaks. The analysis has been performed with the XTREME tool from the MEME suite. C Venn diagram showing the intersection between the SPL/NZZ ChIP-seq peaks and the SEP3 ChIP-seq peaks. 185 SPL/NZZ peaks, representing more than 90% of the total peaks, are overlapping with SEP3 peaks. D SPL/NZZ ChIP-seq and SEP3 ChIP-seq overlapping peaks visualisation along the A. thaliana genome. All the significant SPL/NZZ ChIP-seq peaks are visualized in green. SEP3 ChIP-seq peaks overlapping with SPL/NZZ ones are shown in magenta. The full list of SPL/NZZ-SEP3 common targets can be found in Supplementary Data 2. E wild type, spl-1 and pSPL/NZZ::MADSas ovules at stage 2-IV. Even if with a milder effect with respect to the spl-1 case, pSPL/NZZ::MADSas fails in the differentiation of the MMC, that could be clearly identified in the wild type nucellus. F Bar plot showing the mean ± SEM of the percentage of ovules developing the MMC in wild type, spl-1, pSPL/NZZ::MADSas L1 (shp1,2,STK/STK), pSPL/NZZ::MADSas L2 (shp1,2,STK/STK) and pSPL/NZZ::MADSas L3 (shp1,2,stk). The analysis has been performed on 5, 5, 5, 9 and 4 different pistils, respectively, for the wild type (122 ovules observed in total), spl-1 (110 ovules observed in total), pSPL/NZZ::MADSas L1 (113 ovules observed in total), pSPL/NZZ::MADSas L2 (208 ovules observed in total), pSPL/NZZ::MADSas L3 (67 ovules observed in total). Source data are provided as a Source Data file. Letters above the bars indicate homogenous categories with p ≤ 0.05 as determined by one-way ANOVA with post-hoc Tukey HSD test. Exact P values for each comparison can be found in the Source Data file. Single measures are represented with dots in the bars. nu nucellus, fu funiculus, ii inner integument, oi outer integument, in integument, MMC megaspore mother cell. Scale bar = 20 µm.
SPL/NZZ does not have a DNA-binding domain. This implies that SPL/NZZ binds to its direct targets via TF complexes in which it is recruited. To characterise which TFs mostly relate to SPL/NZZ activity, we performed a TF DNA-motif enrichment analysis. We used the MEME suite38 to analyse the 203 peak sequences retrieved from the SPL/NZZ ChIP-seq experiment. The only statistically enriched TF DNA binding site identified was the CArG-box consensus sequence, to which the MADS-domain TFs bind (Fig. 2B)39. In addition, enrichment analysis against the Transcriptional Factor Target lists from both the AGRIS server40,41,42 and the PlantGSAD database43 revealed that SPL/NZZ targets were enriched in genes targeted by MADS-domain TFs such as AG and SEP3 or floral homeotic factors such as APETALA2 (AP2) (Supplementary Data 2).
If SPL/NZZ binding to the DNA largely depends on its association with MADS-domain TF complexes, not only SPL/NZZ and MADS-domain TFs should target the same genes, but they should bind these genes in the same genomic sequences. Based on this assumption, we determined the overlap of peaks obtained by the SPL/NZZ ChIP-seq with the ones obtained from a SEP3 ChIP-seq dataset44. The already available SEP3 ChIP-seq was performed using the same induction system and tissue used for the SPL/NZZ ChIP-seq and on a similar stage of development (8 DAI). Over 90% of SPL/NZZ peaks (185 out of 203, corresponding to 182 different genes) overlapped with peaks in the SEP3 dataset, strongly supporting the cooperation between SPL/NZZ and SEP3 for binding to the DNA, at least at the developmental stages used for the ChIP-seq experiments. (Fig. 2C, D, and Supplementary Data 2).
Our data indicate that SPL/NZZ could cooperate with the MADS-domain TFs SEP1, 2, and 3, as well as STK and SHPs for MMC development. This outcome was also predictable, as it has been shown that MADS-domain TFs of classes C, D and E form multimeric complexes to establish ovule domains and define cell identity23. Regrettably, these transcription factors exhibit significant redundancy and multiple mutants, as the shp1 shp2 stk ag/+ one, displays ovules homeotically changed into leaves21,22,23, whereas the sep1 sep2 sep3 mutant has all floral organs homeotically changed into sepals45. Therefore, to investigate the potential relevance of these MADS-domain TFs in MMC differentiation, we created a pSPL/NZZ::MADSas construct to operate the post-transcriptional gene silencing of SEP genes in the nucellus (Supplementary Fig. 5A). We subsequently introduced this construct into shp1/shp1 shp2/shp2 stk/STK plants, intending to inhibit the activity of many ovule-identity MADS-domain TFs within the nucellus. We obtained three independent transformant lines expressing the pSPL/NZZ::MADSas (Supplementary Fig. 5B). Specifically, lines 1 and 2 resulted shp1 shp2 STK/STK, whereas line 3 was classified as a triple shp1 shp2 stk mutant. Although the phenotype was much less penetrant with respect to that reported in spl-1, the transgenic plants shp1 shp2 stk pSPL/NZZ::MADSas and shp1 shp2 STK/STK pSPL/NZZ::MADSas showed a ratio of 25% to 45% of ovules that failed to produce the MMC (Fig. 2E, F, and Supplementary Fig. 5C).
Altogether, these data support that SPL/NZZ and MADS-domain TFs cooperate, forming a multimeric complex for the differentiation of the MMC.
SPL/NZZ regulates the auxin signalling network
Having identified potential direct SPL/NZZ-SEP3 target genes, we focused on their biological function to determine the mechanisms involved in MMC differentiation. To do so, we performed a Biological Process GO terms enrichment analysis using the SPL/NZZ-SEP3 common targets (Supplementary Fig. 6A, and Supplementary Data 2). Besides categories associated with reproductive organs differentiation, the most enriched terms were related to auxin metabolism and biosynthesis, suggesting a direct link between SPL/NZZ and auxin (Supplementary Fig. 6A, and Supplementary Data 2).
To identify the genes whose expression was modified in the absence of SPL/NZZ, and the pathways associated with SPL/NZZ activity, we performed RNA-seq on wild-type (Col-0) and spl-1 mutant pistils at stage 946,47. Differential expression analysis retrieved a list of 6877 differentially expressed genes (DEGs) (FDR ≤ 0.05) (Supplementary Data 3).
We performed a Biological Process GO term enrichment analysis on the 3502 downregulated genes, revealing enrichment in terms associated with chromatin remodelling and floral development (Supplementary Fig. 6B, and Supplementary Data 3). By contrast, Biological Process GO term enrichment analysis done on the 3375 upregulated genes confirmed an impairment in auxin biosynthesis, metabolism and signalling (Supplementary Fig. 6C, and Supplementary Data 3). We also observed enrichment in categories involved in cell cycle regulation, cell wall modification and callose deposition, processes known to be required for the MMC differentiation48. To further dissect the activity of SPL/NZZ over its direct targets, we analysed the SPL/NZZ-SEP3 common target list in light of the DEGs obtained from the spl-1 RNA-seq. We found that 77 SPL/NZZ-SEP3 targets were also differentially expressed in the spl-1 pistil with respect to the wild type (Supplementary Data 3). Nicely, these include genes involved in ovule development, such as ANT 49, LUG50, AP251, CUC352, IPT114, SEP322, AGL15 53 and FUL54. Interestingly, all the auxin biosynthetic genes present in the spl-1 RNA-seq, including also the genes targeted directly by SPL/NZZ, were upregulated (Supplementary Data 3). Key players of auxin biosynthesis, such as YUCCA 2, 4, 6 and TAA1, which are normally expressed in the ovule chalaza55, showed enhanced expression. This result is in line with SPL/NZZ proposed activity as a transcriptional repressor, which is also supported by the reduced expression of YUC2 and 4 in the spl-D dominant mutant36. After biosynthesis in the chalaza, auxin is polarly transported towards the primordium apex mainly by PIN1, generating a region of high signalling output at the nucellus tip55,56,57. Auxin is then directed toward the L2 layer by PIN3 and PIN1 action1,57. Since it was already reported that PIN1 was drastically downregulated in spl-1 ovules14, as well as the auxin response15, we decided to investigate in more detail auxin transport and response in wild-type and spl-1 ovules to evaluate their impact on the spl-1 phenotype.
Focusing on wild-type ovules, we observed that the PIN1-GFP signal increased in the nucellus from stage 2-I, the stage in which the differentiation of the MMC initiates, to stage 2-III, associated with the complete differentiation of the MMC (Fig. 3A, B, M). Likewise, we measured a significant increase in the nucellar DR5v258 signal passing from stage 2-I to stage 2-III (Fig. 3E, F, N). This dynamic in PIN1 and DR5v2 expression during MMC differentiation could suggest the requirement of auxin transport and signalling to support the MMC identity acquisition. Indeed, even though we observed that PIN1-GFP accumulated similarly in wild-type and spl-1 ovules at stage 2–I (Fig. 3A, C, M), at stage 2-III, PIN1-GFP signal was drastically reduced in the spl-1 mutant (Fig. 3B, D, M), as previously shown14,15. Likewise, the DR5v2 signal observed between wild-type ovules at stages 2-I and 2-III was severely reduced in the spl-1 background (Fig. 3E–H, N). Besides PIN1, PIN3 is also involved in auxin transport in the nucellus. Despite this, the pPIN3::PIN3:GFP57,59 signal was localised at the very tip of ovule primordia in spl-1, and no differences were observed compared to the wild type (Supplementary Fig. 7A–D).

pPIN1::PIN1:GFP expression in wild type (A, B) and spl-1 (C, D) ovules at stages 2-I (A, C) and 2-III (E, H).DR5v2 expression in wild type (E, F) and spl-1 (G, H) ovules at stages 2-I (E, G) and 2-III (F, H). R2D2 reporter line in wild type (I, J) and spl-1 (K, L) ovules at stage 2-III, showing mDII-tdTOMATO (I, K) and DII-VENUS (J, L) accumulation. I–L, M Line graph showing the mean ± SEM of PIN1-GFP signal intensity in the L1 layer of wild type and spl-1 ovules at stages 2-I and 2-III. Measurements have been performed on 5 wild type and 5 spl-1 different ovules at stage 2-I and on 12 wild type and 10 spl-1 different ovules at stage 2-III. Source data are provided as a Source Data file. Asterisks represent the statistical significance as determined by student’s t-test, two-sided distribution, homoscedastic, confronting the mutant with the wild-type condition at each time point. ***p ≤ 0.001. Exact P values for each comparison can be found in the Source Data file. Single measures are represented with dots in correspondence to the line ends. N Line graph showing the mean ± SEM of the tdTOMATO signal intensity, from the DR5v2 reporter, in the L1 layer of wild-type and spl-1 ovules at stages 2-I and 2-III. Measurements have been performed on 6 wild-type and 4 spl-1 different ovules at stage 2-I and on 5 wild-type and 5 spl-1 different ovules at stage 2-III. Source data are provided as a Source Data file. Asterisks represent the statistical significance as determined by student’s t-test, two-sided distribution, homoscedastic, confronting the mutant with the wild-type condition at each time point. ***p ≤ 0.001. Exact P values for each comparison can be found in the Source Data file. Single measures are represented with dots in correspondence to the line ends. nu nucellus, ch chalaza, fu funiculus, ii inner integument, oi outer integument, in integument, MMC megaspore mother cell. Scale Bars = 20 µm.
Downregulation of PIN1 expression and DR5v2 response suggests that auxin is accumulated at lower levels in the spl-1 nucellus. Indeed, by using the R2D2 reporter line58, we detected a higher accumulation of DII-VENUS in the spl-1 nucellus with respect to the wild type (Fig. 3I–L, and Supplementary Fig. 7E–I).
Considering the behaviour of PIN1 and DR5v2, together with the fact that auxin transport was previously suggested to be required for MMC development15, we believed that PIN1 downregulation could be the main cause of MMC absence in the spl-1 mutant ovule.
Restoring the auxin transport is sufficient to rescue the MMC differentiation in spl-1 ovules
Even if molecularly SPL/NZZ is a repressor of transcription, during MMC differentiation, SPL/NZZ works as a positive modulator of PIN1 expression. This implies that SPL/NZZ activity over PIN1 is indirect and involves, potentially, the direct targeting of negative regulators of PIN1. Indeed, PIN1 was not present in the list of SPL/NZZ direct targets. Taking into consideration this information, among SPL/NZZ-SEP3 direct targets, we searched for factors which could be genetically associated with SPL/NZZ activity, and which could work as intermediates of SPL/NZZ action over PIN1. During ovule patterning, SPL/NZZ was shown to be required for the repression of chalazal factors such as ANT, BEL1, INO and PHB in the nucellus12,37,49. As mentioned above, ANT is a direct target of the SPL/NZZ-MADS-domain TF complex. Particularly, in both SPL/NZZ and SEP3 ChIP-seq datasets, we found a peak containing 4 CArG-box sequences at around -3.6 Kb from ANT TSS (Supplementary Fig. 8A). Absence of SPL/NZZ function determines ANT ectopic expression in the nucellus12 (Supplementary Fig. 8B–I). Our attention was caught by ANT as a putative intermediary of SPL/NZZ action over PIN1, especially due to the previous description of the ability of ant mutation to rescue MC differentiation in spl/nzz mutant background12. Considering the centrality of auxin transport and response for the MMC differentiation, we speculated that ANT could act as a repressor of PIN1, contributing to PIN1 downregulation in the spl-1 nucellus. Even though we did not find PIN1 among ANT direct targets60, we observed that PIN1 was ectopically expressed in the chalaza and funiculus of ant.4 mutant ovules with respect to the wild type (Supplementary Fig. 8J–Q). This suggests that, in ovules, ANT acts as an indirect local repressor of PIN1.
Based on our present and previously reported observations15, we hypothesised that restoring polar auxin transport in the spl-1 ovule might be sufficient to recover the MMC differentiation. Therefore, we developed the pSPL/NZZ::PIN1 construct, and we transformed it into spl-1 mutant plants. Four distinct spl-1 lines expressing the pSPL/NZZ::PIN1 construct were characterised (Supplementary Fig. 9A).
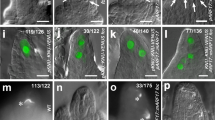
In contrast with the spl-1 situation (Fig. 4F–J, P), in spl-1 pSPL/NZZ::PIN1, ovule developmental defects were almost completely restored (Fig. 4A–E, K–P, and Supplementary Fig. 9B). The MMC correctly developed (Fig. 4C, D, M, N) and completed the megasporogenesis (Fig. 4E, O). Indeed, fertile ovules were formed and fertilised by pollen produced by the same plants, showing that the pSPL/NZZ:PIN1 had also rescued the PMC defects (Supplementary Fig. 9C). Interestingly, spl-1 pSPL/NZZ:PIN1 ovules also showed a rescue of spl-1 integuments defect (Fig. 4G–J, L–O). Indeed, ovules could correctly differentiate inner and outer integuments (Fig. 4G–J, L–O). Complementation of inner integuments development suggests that SPL/NZZ is important for the proper PIN1 localisation and auxin transport during inner integuments formation. Indeed, regulation of auxin transport was shown to be involved in integuments development61. Nevertheless, similarly to the spl-1 mutant, the funiculus length in spl-1 pSPL/NZZ::PIN1 ovules remained statistically longer with respect to the wild type (Supplementary Fig. 10A, B). We hypothesised that this phenotype could be related to the fact that genes involved in funiculus differentiation remained deregulated in spl-1 pSPL/NZZ::PIN1 ovules. For instance, it was shown that ANT ectopic expression is directly associated with the differentiation of a longer funiculus62. By ISH, we observed that ANT remained ectopically expressed in both nucellus and funiculus of spl-1 pSPL/NZZ::PIN1 ovules (Supplementary Fig. 10C). The ectopic expression of ANT in spl-1 pSPL/NZZ::PIN1 not only provides a hierarchical view in which PIN1 is placed downstream ANT but also represents a plausible explanation for the elongated funiculus of spl-1 pSPL/NZZ::PIN1 ovules.

Wild type ovules at stage 1-II (A), 2-I (B), 2-III (C), 2-IV (D) and 2-V (E). At stage 2-III, the MMC is differentiated (C) while callose depositions can be observed at stage 2-IV (D). At stage 2-V (E), the MMC meiosis ends as denoted by the formation of the two meiotic plates. spl-1 ovules at stage 1-II (F), 2-I (G), 2-III (H), 2-IV (I) and 2-V (J). In the spl-1 mutant the MMC do not differentiate (H, I). spl-1 pSPL/NZZ::PIN1 ovules at stage 1-II (K), 2-I (L), 2-III (M), 2-IV (N) and 2-V (O). Restoring PIN1 expression in the spl-1 nucellus is sufficient to drive the MMC differentiation (M, N). The MMC can be observed at stage 2-III (M). As in the wild-type situation (D), the MMC is surrounded by callose depositions at stage 2-IV (N) and correctly goes through the meiotic process (O). P Bar plot showing the mean ± SEM of the percentage of ovules developing the MMC, in wild type, spl-1 and four spl-1 pSPL/NZZ::PIN1 independent lines. The analysis has been performed on 8, 7, 8, 6, 8 and 9 different pistils, respectively, for the wild type (202 ovules observed in total), spl-1 (238 ovules observed in total), spl-1 pSPL/NZZ::PIN1 L1 (294 ovules observed in total), spl-1 pSPL/NZZ::PIN1 L2 (196 ovules observed in total), spl-1 pSPL/NZZ::PIN1 L3 (274 ovules observed in total) and spl-1 pSPL/NZZ::PIN1 L4 (364 ovules observed in total). Source data are provided as a Source Data file. Letters above the bars indicate homogenous categories with p ≤ 0.05 as determined by one-way ANOVA with post-hoc Tukey HSD test. Exact P values for each comparison can be found in the Source Data file. Single measures are represented with dots in the bars. nu nucellus, ch chalaza, fu funiculus, ii inner integument, oi outer integument, in integument, MMC megaspore mother cell. Blue arrowheads indicate the callose depositions. Green arrowhead indicates the meiotic plates. Scale bar = 20 µm.
The inhibition of ARFs activity phenocopies the spl-1 mutation
The results described above demonstrate the need for SPL/NZZ to regulate the auxin accumulation in the nucellus, which is essential for MMC differentiation. According to the auxin response model, auxin accumulation enhances Aux/IAAs degradation, releasing the ARFs from Aux/IAAs repressing activity63. This leads to the regulation of the ARFs target genes expression. Based on this model, we hypothesised that the impairment of the MMC differentiation in the spl-1 mutant could be ascribed to the post-translational suppression of ARFs function. Indeed, lower auxin accumulation in spl-1 may inhibit Aux/IAAs degradation, affecting ARFs activity and, thus, MMC differentiation. Accordingly, ovules containing an Aux/IAA isoform resistant to the auxin-dependent proteasomal degradation might mimic the spl-1 phenotype. Single-cell RNA-seq, performed using ovules between stages 2-I and 2-III, revealed that Aux/IAA 1, 2, 3, 4, 8, 9, 13, 16 and 26 were expressed in nucellar clusters64. To select the appropriate Aux/IAA to repress the ARFs activity in the nucellus, we chose the one that was previously reported to interact with several nucellar-expressed ARFs65,66,67, and for which a dominant auxin-resistant allele was available. We chose the Aux/IAA3/SHY268 dominant mutant allele shy2.669. shy2.6 carries a mutation in the degron motif, impairing its ability to interact with the SCF/TIR1 complex at threshold auxin levels69, remaining constitutively bound to ARFs PB1 domain69. Consequently, we generated the pSPL/NZZ::shy2.6 construct to express shy2.6 in the nucellus of wild-type ovules.
We introduced the pSPL/NZZ::shy2.6 construct into wild-type plants, obtaining three independent lines expressing the transgene (Supplementary Fig. 11A). Even if with a lower penetrance with respect to the spl-1 mutant, in all the lines we observed an impairment in MMC differentiation (Fig. 5A–M). To confirm that the observed phenotype was caused by the inhibition of ARFs activity rather than other factors, such as a disruption of the polar auxin transport, we examined the expression of pPIN1::PIN1:GFP in pSPL/NZZ::shy2.6 plants. The expression of PIN1-GFP in pSPL/NZZ:shy2.6 resembled that of the wild type, suggesting that the observed phenotype was probably caused by an impairment in ARFs activity (Supplementary Fig. 11B–E).

Wild type ovules at stage 1-II (A), 2-I (B), 2-II (C) and 2-III (D). spl-1 ovules at stage 1-II (E), 2-I (F), 2-II (G) and 2-III (H). pSPL/NZZ:shy2.6 ovules at stage 1-II (I), 2-I (J), 2-II (K) and 2-III (L). shy2.6 expression in the wild-type nucellus impair the MMC differentiation, mimicking the spl-1 phenotype (G, H, K, L). M Bar plot showing the mean ± SEM of the percentage of ovules showing an MMC in wild type, spl-1 and pSPL/NZZ::shy2.6 lines. The analysis has been performed on 3, 4, 8, 9 and 5 different pistils, respectively, for the wild type (113 ovules observed in total), spl-1 (157 ovules observed in total), pSPL/NZZ::shy2.6 L1 (356 ovules observed in total), pSPL/NZZ::shy2.6 L2 (378 ovules observed in total) and spl-1 pSPL/NZZ::shy2.6 L3 (202 ovules observed in total). Source data are provided as a Source Data file. Letters above the bars indicate homogenous categories with p ≤ 0.05, as determined by one-way ANOVA with post-hoc Tukey HSD test. Exact P values for each comparison can be found in the Source Data file. Single measures are represented with dots in the bars. nu nucellus, ch chalaza, fu funiculus, ii inner integument, oi outer integument, in integument, MMC megaspore mother cell. Scale bars = 20 µm.
ARFs activity is required for the MMC differentiation
Collectively, our data support a scenario in which the MMC differentiation is mediated by the post-translational regulation of the ARFs action. SHY2 can interact with all the class A ARFs, including ARF5/MP, ARF6, ARF7, ARF8 and ARF19. Concerning the repressor ARFs, SHY2 was shown to interact with ARF4 and ARF965,66,67. According to an ovule single-cell RNA-seq dataset64, MP, ARF6, ARF8 and ARF9 are the only SHY2-interacting ARFs expressed in the nucellus. Consequently, we analysed MP, ARF6, ARF8 and ARF9 expression in both wild-type and the spl-1 ovules. We used previously published reporter lines to study MP, ARF6 and ARF8 domains of transcription and protein accumulation70. Concerning ARF9, as no functional protein reporter lines were available, we studied its expression pattern by in-situ hybridisation.
In ovule primordia, the levels of both ARF6-VENUS and ARF8-VENUS were presumably controlled at the post-translational stage, supporting previously reported observations53 (Supplementary Fig. 12E–L). Indeed, although both genes were transcribed in ovules64 (Supplementary Fig. 12E, I, G, K), ARF6 was faintly visible solely in a limited number of cells (Supplementary Fig. 12F, H), while ARF8 was not visible (Supplementary Fig. 12J, L). By contrast, the MP-VENUS protein accumulated in the nucellus, as previously shown70,71 (Supplementary Fig. 12A–D). The three ClassA ARFs exhibited comparable expression and protein accumulation in both wild-type and spl-1 ovules (Supplementary Fig. 12A–L). Concerning ARF9, we detected its transcript accumulation in both wild type and spl-1 nucellus at stage 1-II (Supplementary Fig. 13A, C). At stage 2-III, the ARF9 transcript was accumulated in the MMC and the integuments of wild-type ovules (Supplementary Fig. 13B). By contrast, in the spl-1 mutant background, ARF9 transcript appeared miss-localised in both the nucellus and the chalaza (Supplementary Fig. 13D). This could be mainly due to the fact that, in a wild-type ovule, ARF9 transcript accumulates in the MMC and the integuments, both structures that are impaired in the spl-1 background. Despite this, by analysing the spl-1 RNA-seq, we observed an overall decrease in ARF9 expression level in the spl-1 pistil (Supplementary Data 3).
Given their expression patterns, MP and ARF9 were selected as ARFs potentially involved in the downstream auxin response, required for MMC differentiation. According to our hypothesis, the absence of the MMC in the spl-1 mutant was associated with a decrease in the nucellar auxin response, caused by the stabilisation of Aux/IAAs and the consequential ARFs inhibition. Indeed, as previously described63,70,72,73, ARFs activity is post-translationally regulated by the balance between ARFs and Aux/IAAs relative abundance. For this reason, we hypothesised that increasing MP or ARF9 levels in the spl-1 background could overcome the repression operated by the Aux/IAAs, restoring the MMC presence. Therefore, we generated the pSPL/NZZ::MP and the pSPL/NZZ::ARF9 constructs that we introduced in the spl-1 mutant. We obtained two independent spl-1 pSPL/NZZ::MP lines, while we found three spl-1 pSPL/NZZ::ARF9 independent lines (Supplementary Fig. 14A, B). In spl-1 pSPL/NZZ::MP plants, we observed that ~10% of ovules developed an MMC (Fig. 6A–F, I). However, these cells did not undergo meiosis, suggesting only a partial acquisition of the MMC identity (Supplementary Fig. 14C). By contrast, the majority of spl-1 pSPL/NZZ::ARF9 ovules differentiated an MMC, which could also proceed into sporogenesis (Fig. 6G, H, J, and Supplementary Fig. 14D), indicating the direct involvement of ARF9 in the MMC differentiation.

Wild type ovules at stage 2-I (A) and 2-III (B). spl-1 ovules at stage 2-I (C) and 2-III (D). spl-1 pSPL/NZZ::MP ovules at stage 2-I (E) and 2-III (F). spl-1 pSPL/NZZ::ARF9 ovules at stage 2-I (G) and 2-III (H). While increasing MP expression only partially restores the MMC differentiation, pSPL/NZZ::ARF9 rescues the MMC differentiation in spl-1. I Bar plot showing the mean ± SEM of the percentage of ovules showing an MMC in wild type, spl-1 and spl-1 pSPL/NZZ::MP lines. The analysis has been performed on 5, 8, 12 and 9 different pistils for the wild type (150 ovules observed in total), spl-1 (304 ovules observed in total), spl-1 pSPL/NZZ::MP L1 (362 ovules observed in total) and spl-1 pSPL/NZZ::MP L2 (270 ovules observed in total). Source data are provided as a Source Data file. Letters above the bars indicate homogenous categories with p < 0.05, as determined by one-way ANOVA with post-hoc Tukey HSD test. Exact P values for each comparison can be found in the Source Data file. Single measures are represented with dots in the bars. J Bar plot showing the mean ± SEM of the percentage of ovules showing an MMC in wild type, spl-1 and spl-1 pSPL/NZZ::ARF9 lines. The analysis has been performed on 4, 4, 5, 4, 4 different pistils for the wild type (157 ovules observed in total), spl-1 (131 ovules observed in total), spl-1 pSPL/NZZ::ARF9 L1 (193 ovules observed in total), spl-1 pSPL/NZZ::ARF9 L2 (115 ovules observed in total) and spl-1 pSPL/NZZ::ARF9 L3 (177 ovules observed in total). Source data are provided as a Source Data file. Letters above the bars indicate homogenous categories with p ≤ 0.05, as determined by one-way ANOVA with post-hoc Tukey HSD test. Exact P values for each comparison can be found in the Source Data file. Single measures are represented with dots in the bars. nu nucellus, ch chalaza, fu funiculus, ii inner integument, oi outer integument, in integument, MMC megaspore mother cell. Scale bars = 20 µm.
Altogether, our results highlight the regulation of the ARFs action as a key step of the SPL/NZZ downstream response during MMC differentiation.
Discussion
Although the mechanisms necessary to restrict the acquisition of the MMC identity to a single cell have been addressed74, the regulatory network controlling MMC differentiation is still largely unknown. Since SPL/NZZ is a major factor controlling MMC development, we initially focused on the identification of SPL/NZZ-interacting partners in planta. Even though previous works highlighted the TCP TFs as the main SPL/NZZ partners4,5, in our Co-IP/MS dataset, the MADS-domain TFs resulted in the most abundant TF family. The differences in SPL/NZZ partners identified by the Co-IP/MS and by the previous screenings of SPL/NZZ interactors could be partially explained by our use of the AP1:GR/ap1cal inducible system. This system allowed us to find SPL/NZZ partners within a constrained developmental time window. Additionally, while our experiments were performed in planta, the previously reported interactors were identified by yeast-two-hybrid screenings4,5. We identified only one TCP family member among SPL/NZZ partners. Taking into consideration that a limited number of SPL/NZZ target genes were also identified as TCP targets (Supplementary Data 2), we suggest that complexes including SPL/NZZ and TCPs may function predominantly in other developmental stages or tissues, with respect to the ones analysed in our experiments. On the contrary, several MADS-domain transcription factors were enriched in the SPL/NZZ Co-IP/MS data. Indeed, the genomic sequences bound by SPL/NZZ-containing complexes comprised the CArG-box consensus DNA sequence, typically used by MADS-domain TFs for DNA binding.
We highlighted SEP3 as a TF putatively involved in SPL/NZZ recruitment to the DNA, at least in ovules from stage 2-I to 2-III, since these were the stages from which we had isolated SPL/NZZ-GFP to perform both ChIP-seq and Co-IP/MS. Indeed, SPL/NZZ shared more than 90% of its targets with SEP3, and SPL/NZZ and SEP3 bound these genes at the same genomic positions. To gather additional evidence supporting the cooperation between MADS-domain TFs and SPL/NZZ during MMC differentiation, we developed transgenic lines exhibiting reduced expression of multiple ovule-identity MADS-box genes in the SPL/NZZ expression domain. This strategy was crucial to overcome the redundancy of the ovule identity MADS-domain TFs, and because multiple mutations in these genes result in organ homeotic modifications, hindering the differentiation of ovules. Indeed, higher-order mutants such as sep1 sep2 sep3 have flower organs converted into sepals45 and stk shp1 shp2 ag/+ develop leaves instead of ovules21,23. The transgenic plants obtained by post-transcriptionally silencing SEP genes in the shp1 shp2 stk mutant, using the SPL/NZZ promoter, showed ovules with defects in MMC differentiation. In these transgenic plants, we also observed additional defects resulting from the broader role of these MADS-domain TFs in ovule differentiation and development with respect to SPL/NZZ activity (Supplementary Fig. 5D).
The MADS-domain TFs STK, AG and SEP3 were shown to be fundamental for ovule development21,22,23, forming complexes that regulate ovule identity. For instance, the interaction among STK, AG, SEP3 and BEL1 was described as of pivotal importance to determine chalaza identity and to correctly differentiate the integuments23. Taking into consideration the present and the previously reported findings, it became evident that MADS-domain TF complexes interact either with SPL/NZZ or BEL1 to control the nucellar and chalazal development, respectively. Indeed, the role of SPL/NZZ in repressing chalaza identity genes in the nucellus, such as ANT, BEL1 and PHB, was already reported12,37. At the same time, SPL/NZZ ectopic expression was associated with the differentiation of multiple MMC-like cells11.
We show that the key mechanism for the MMC differentiation likely relies on the post-translational regulation of ARFs activity, associated with the nucellar auxin accumulation.
Re-establishing the auxin transport in the spl-1 nucellus, by expressing PIN1 under the SPL/NZZ promoter, was sufficient to restore the MMC differentiation and sporogenesis (Fig. 7D). At the same time, the expression of the auxin-insensitive shy2.6 mutant protein mimicked the spl-1 phenotype (Fig. 7E). We propose that SPL/NZZ activity ensures the correct nucellar auxin level and response by indirectly regulating the expression of PIN1 (Fig. 7A).

A In the wild type, SPL/NZZ interacts with the ovule-identity MADS-domain complex to inhibit ANT and other chalazal genes expression, facilitating auxin accumulation in the nucellus and subsequently enabling the ARF-mediated auxin response required to promote the MMC differentiation. B In the spl/nzz mutant, ANT and other chalazal genes are expressed in the nucellus, leading to the downregulation of PIN1 and to an auxin subthreshold concentration, which disables the post-translational activation of the ARFs, necessary for MMC formation. C In the ant spl/nzz mutant, the inhibition of PIN1 does not take place, allowing the formation of the MMC even in the absence of SPL/NZZ. D Expressing PIN1 under the control of the SPL/NZZ promoter in the spl/nzz mutant, is sufficient to restore the proper auxin threshold in the nucellus and the ARF-mediated auxin response required for the MMC differentiation. E The expression of the non-degradable shy2.6 isoform in the wild-type nucellus leads to the post-translational deactivation of the ARFs, resulting in the absence of the MMC formation. F Increasing ARF levels in the spl/nzz mutant can overcome the repressive state caused by Aux/IAAs over-accumulation, determining the MMC differentiation.
Likely, SPL/NZZ, acting together with SEP3 and other MADS-domain TFs, promotes PIN1 expression by repressing ANT and other chalaza genes in the nucellus. According to this, ANT ectopic expression in the spl/nzz nucellus, together with other chalaza-specific factors, could cause a downregulation of PIN1 and, consequently, the impaired nucellar auxin accumulation (Fig. 7B). Indeed, it was reported that the ant mutation partially rescues the lack of the MMC in the spl/nzz mutant12, suggesting that the nucellar auxin level could also be restored in the spl/nzz ant double mutant (Fig. 7C). To define whether ANT expression in the nucellus was sufficient to control PIN1, we transformed wild-type plants with a pSPL/NZZ::ANT construct. Even though we obtained 3 independent lines expressing the construct (Supplementary Fig. 15A), we did not observe defects in MMC development (Supplementary Fig. 15B, C). This result is not unexpected since, in spl/nzz mutants, several genes involved in the acquisition of the chalazal identity are ectopically expressed8,12,49. Therefore, there are other factors not expressed in the wild-type nucellus, which remain unknown to date, that could work together or in parallel with ANT to impair MMC differentiation when expressed in the nucellus (as in the spl mutant). According to our data, the SPL/NZZ-dependent repression of such factors should contribute to the establishment of the nucellar auxin response, which is central for MMC differentiation. The repression of chalazal genes could act on the spatiotemporal expression of PIN1. Indeed, besides ANT, BEL1 was also reported to be involved in PIN1 repression14. While ANT regulation over PIN1 is indirect, no information regarding BEL1 action is available to date.
It is interesting to notice that until stage 2-I, when the MMC starts its differentiation, PIN1 expression can be observed even in the spl-1 ovule. In the wild type, starting from stage 2-I until 2-III, the MMC undergoes important modifications. These include a sensible enlargement in cellular and nuclear dimensions, the deposition of callose, and the expression of cellular-specific markers48. These cellular changes are associated with an increase in PIN1-GFP signal in the nucellus L1 and with an increase in auxin signalling output. By contrast, the spl-1 nucellus, after stage 2-I, showed a significant reduction in both PIN1 expression and a severely reduced auxin response.
Interestingly, before stage 2-I one or multiple potential MMC initials have been reported to be present75. The nucellar phenotypic plasticity is reduced until only one MMC precursor is specified at stage 2-I75. The plasticity in the identification of potential MMC candidates is also supported by the eviction of the histone marker HTR13-GFP that, until stage 2-I, occurs in multiple nucellar cells. Removal of HTR13-GFP in one single nucellar cell becomes prominent from stage 2-II, supporting the clear differentiation of the MMC from the other cells at this stage76.
According to these reported findings, the process of MMC development could be divided into two main steps: the first, during which the nucellar phenotypic plasticity is reduced and leads toward the specification of a single MMC at around stage 2-I. The second step, during which the MMC differentiates, starts after stage 2-I and ends at stage 2-III. According to our results, the SPL/NZZ-dependent regulation of PIN1 is of pivotal importance for MMC differentiation.
Wild-type and spl-1 ovules, at precocious developmental stages, present similar phenotypes, allowing the morphological identification of one or multiple potential MMC candidate cells even in the mutant background (Supplementary Fig. 16A–D). This suggests that, although the MMC does not differentiate, putative MMC precursors could be formed in the spl-1 mutant. This observation is also corroborated by previous published studies1,2,5, supporting a scenario in which the archespore specification could take place independently from SPL/NZZ action.
The PIN1 expression during ovule development is involved in several regulatory pathways, specific to each ovule developmental phase, such as ovule primordia formation52 and integuments formation and outgrowth, and it is likely that SPL/NZZ activity over PIN1 is temporally restricted to specific processes such as the MMC differentiation.
Previous works already highlighted the connection between the auxin signalling and the MMC initiation. For instance, ARF3 and ARF17 silencing were shown to be required to repress the formation of multiple MMC cells15,77.
According to our findings, auxin accumulation in the nucellus may trigger the degradation of the Aux/IAAs, leading to ARFs activation to support the MMC development (Fig. 7A). Indeed, we observed that inhibiting the ARFs activity in a wild-type situation, by expressing the auxin-insensitive shy2.6 mutant protein, mimicked the spl-1 phenotype (Fig. 7E). Among the ARFs expressed in the nucellus, we highlighted MP and ARF9 as putatively involved in the MMC differentiation. We observed that increasing ARF9 levels could partially rescue the MMC development in spl-1 (Fig. 7F). According to this, the MMC development might rely on the cooperation between more ARF types. Even though we focused on MP and ARF9, since they both interact with SHY2, we could not exclude the possibility that multiple ARFs could be involved in this process. Indeed, based on ovule single-cell RNA-seq data64, other repressor ARFs, such as ARF1, ARF3 and ARF18, are expressed in the nucellus during MMC differentiation, although they do not interact with SHY2. Based on our experiments, we propose that the auxin signalling, and therefore ARFs action, represents the endpoint of SPL/NZZ activity. Despite this, the complete description of how ARFs promote the MMC differentiation is still hindered by the limited knowledge regarding some aspects, such as ARF9 interacting partners and target genes. This would require additional work that we believe could represent a new and exciting field of study.
In conclusion, our data provide a regulatory mechanism (Fig. 7), in which SPL/NZZ is required for the establishment of the nucellar auxin pattern, needed for the post-translational regulation of ARF TFs responsible for MMC differentiation and development. It will be of great interest to investigate the SPL/NZZ downstream network controlling PMC differentiation. Although we have not investigated this process in detail, we speculate that a similar mechanism could also control PMC formation. Although the SPL/NZZ-interacting MADS-domain partners might be different in anthers with respect to ovules, the pSPL/NZZ::PIN1 and pSPL/NZZ::ARF9 constructs also rescued pollen formation in the spl-1 mutant, confirming, as previously suggested9, the importance of auxin response also for the male germline.
Methods
Plant material and growing conditions
A. thaliana Columbia-0 plants were grown in controlled conditions under a long-day photoperiod (16 h light 150 µm.m−2.s−1 / 8 h dark) at 22 °C. spl-1 mutant in the Col-0 background was previously described14. The pSPL/NZZ::SPL/NZZ:GFP line was previously described11. The AP1:GR/ap1cal inducible system was described in ref. 17. shp1, shp2, stk/STK plants were described in ref. 21. ant.4 mutant in the Ler background was previously described78,79. pSTK:STK:GFP, pSEP3::SEP3:GFP, pAG::AG:GFP were previously described25,26,27. The spl-1 pPIN1::PIN1:GFP line was described in ref. 14. pPIN3::PIN3:GFP 59, DR5v2 58 and R2D2 58 lines were manually crossed with spl-1 heterozygous plants.
Generation of transgenic lines
To obtain the pSPL/NZZ::SPL/NZZ:GFP pAP1::AP1:GR ap1cal inducible line, pAP1::AP1:GR ap1cal plants17 were transformed with the pSPL/NZZ::SPL/NZZ:GFP construct via the floral dip technique using the Agrobacterium tumefaciens strain GV3101.
SPL/NZZ:GFP AP1:GR ap1cal plants were induced with dexamethasone treatment directly on the inflorescence-like meristem for 9 days. After the third and the ninth dexamethasone treatment, the expression of SPL/NZZ-GFP was controlled with a confocal microscope.
The pSPL/NZZ::MADSas, pSPL/NZZ::PIN1, pSPL/NZZ::shy2.6, pSPL/NZZ::MP, pSPL/NZZ::ARF9 and pSPL/NZZ::ANT constructs were obtained with the multisite gateway cloning technique. MADSas, PIN1, shy2.6, MP, ARF9 and ANT CDS sequences were amplified using the Q5 High-Fidelity DNA polymerase (NEB) and cloned into the pDONR221 plasmid, according to the BP II protocol (Invitrogen, cat. number: 11789020). Multi-site LR reactions were performed using the pDONR221 P1-P4 containing the SPL/NZZ promoter sequence11; the pDONR221 containing either MADSas, PIN1, shy2.6, MP, ARF9 or ANT; the pDONR221 P3-P2 carrying the SPL/NZZ 3’UTR sequence11 and the destination vector pH7m34GW 80, according to the LR II Plus protocol (Invitrogen, cat. number: 12538120). Thanks to the floral dip technique using the Agrobacterium tumefaciens strain GV3101, pSPL/NZZ::PIN1, pSPL/NZZ::MP and pSPL/NZZ::ARF9 were transformed into the SPL/NZZ spl-1 heterozygous background. Four, two and three T2 independent transformant lines were analysed for each transformation, respectively. pSPL/NZZ::MADSas was transformed into the shp1, shp2, stk/STK background. Three T1 independent transformant lines were analysed. pSPL/NZZ::shy2.6 and pSPL/NZZ::ANT were transformed into wild-type Col-0 plants. Three T1 independent transformant lines were analysed for each transformation. Primers used are listed in Supplementary Data 4.
RT-qPCR expression analysis
For each sample, 5 inflorescences pooled together (comprising flowers spanning from stage 1 to stage 12) were used for total RNA extraction. RNA extraction was performed using the Macherey-Nagel NucleoSpin RNA plus kit (Item number: 740984.50) according to the manual instructions. 500 ng of DNA-clean total RNA for each sample was retrotranscribed using the Bio-Rad iScript™ Reverse Transcription Supermix for RT-qPCR (Bio-Rad, cat number: 1708840). cDNA was used for RT-qPCR using the Bio-Rad iTaq Universal SYBR Green Supermix (Bio-Rad, cat number: 1725120). ACTIN8 was used as a housekeeping normaliser for relative normalised expression calculation. Expression levels were expressed as mean ± SEM among three technical replicates. Primers used are listed in Supplementary Data 4.
Analysis of ovule morphology by confocal and optical microscopy
Ovule morphology was evaluated by both confocal and optical widefield microscopy. For confocal microscopy, pistils were manually dissected from the flowers and mounted in a solution of 10 µg/ml Renaissance2200 (Renaissance Chemicals, SR2200). Samples were immediately imaged using a Nikon AX confocal microscope. Renaissance2200 was excited at 405 nm, and the emission signal was detected between 440 and 470 nm. For widefield microscopy, inflorescences were collected in a solution of 9:1 ethanol/acetic acid. After 24 h at 4 °C, inflorescences were transferred to a solution of 70% ethanol. Pistils were manually dissected and mounted in chloral hydrate clearing solution before microscopic imaging. Analysis of the percentage of ovules developing the MMC in wild type, spl-1, pSPL/NZZ::MADSas, spl-1 pSPL/NZZ::PIN1, pSPL/NZZ::shy2.6, spl-1 pSPL/NZZ::MP, spl-1 pSPL/NZZ::ARF9 and pSPL/NZZ::ANT was performed on ovules at stages 2-II, 2-III.
For fluorescent reporter lines signal imaging, pistils were mounted in the Renaissance2200 solution and imaged using a Nikon AX confocal microscope. mTQ was excited with a 445 nm laser and detected at 480–490 nm. GFP was excited with a 488 nm laser and detected at 520-540 nm. VENUS was excited with a 514 nm laser and detected at 530–560 nm. tdTOMATO was excited with a 514 nm laser and detected at 570-630 nm. Imaging was performed with sequential settings to eliminate signal crosstalk. Confocal images were analysed with the Fiji software81. For PIN1-GFP and DR5v2 signal quantification, the Renaissance2200 channel, showing the cellular organisation, has been used to define a region of interest (ROI) comprising the whole L1 layer of the nucellus. The mean GFP and tdTOMATO signals were subsequently measured in the ROI. The R2D2 signal was analysed as previously described70. The tdTOMATO channel has been used to generate a binary mask indicating the nucellar nucelli showing R2D2 expression. The VENUS and the tdTOMATO mean signals in the single nuclei were subsequently measured.
Co-IP/MS material generation and data analysis
IP-MS experiments were performed as described before20. Essentially, nuclei were isolated from the unfixed plant tissues. Proteins, including native protein complexes, were extracted from nuclear extracts and immunoprecipitated with the GFP antibody using μMACS GFP Isolation Kit (Miltenyi Biotec, cat. number: 130-091-288). Immunoprecipitated proteins were released from the beads with 8 M urea. As control, protein extracts from before the immunoprecipitation were used and processed with the FAST method82. Eluted proteins were reduced with DTT, alkylated with IAA and digested with Trypsin/Lys-C Mix, Mass Spec Grade (Promega, cat. number: V5071). Resulting peptides were desalted, dried and resuspended in a loading solution. Peptide concentration was estimated using the NanoDrop One A205 method. Samples were analysed using the EASY-nLC 1200 (Thermo Fisher ScientificTM) coupled to the Q Exactive Plus mass spectrometer (Thermo Fisher ScientificTM). Peptides were loaded onto the Acclaim PepMap 100 C18 analytical column and separated with a linear solvent B gradient of 5% to 40% over 70 min and 40% to 50% over 5 min. Tandem MS spectra (MS/MS) were acquired in the data-dependent acquisition (DDA) mode using a TOP15 approach.
The RAW peptide sequencing data obtained from the Q Exactive Plus were processed by the MaxQuant (version 1.6.14.0) software. Settings of the MaxQuant were mostly kept default with “label-free quantification (LFQ)”, “iBAQ” and “Match between runs” options on. The protein database was A. thaliana UniProt UP000006548 without protein isoforms. After the MaxQuant analysis, the output proteinGroups.txt file was taken for further filtering and statistical analysis in the Perseus (version 1.6.15.0) software. The protein LFQ intensity data were log2-transformed. Proteins identified by only modified peptides (“Only identified by site”), by reverse sequence database (“Reverse”) and protein contaminants (“Potential Contaminant”) were removed from the analysis. Proteins with LFQ intensity values in at least two biological replicates and proteins with at least two unique peptides were kept. Missing protein LFQ intensity values were replaced only in the input samples by the imputation from the left arm of the LFQ intensity normal distribution (0.3 width, 1.8 down shift, separately for each column) to emulate background protein level. Relative protein amount differences were evaluated by two-sample t-tests with permutation-based FDR correction and S0 parameter set to 1.0. The GO term enrichment analyses were performed with clusterProfiler83, with default parameters. The statistical significance of enriched categories was determined by the Benjamini–Hochberg (False Discovery Rate-FDR) test84, considering significant only categories with FDR ≤ 0.05. GO term enrichment analyses were performed on R studio, version 4.4.1.
ChIP-seq material generation and data analysis
ChIP-seq experiments were performed as described before34,35. Specifically, plant tissues were fixed in 1% formaldehyde solution, frozen in liquid nitrogen and ground to a fine powder. Nuclei were isolated through a sequential wash with buffer containing 0.5% Triton X-100 and lysed followed by chromatin sheering by sonication. After chromatin clearing, immunoprecipitation was performed with the GFP antibody (Abcam, ab290) followed by incubation with protein A magnetic beads (Thermo Fisher ScientificTM, cat. number: 88845). The immunoprecipitated chromatin was released from the beads and de-crosslinked. The input sample, corresponding to the protein extract from before the immunoprecipitation, was also de-crosslinked and used as a ChIP control. The DNA was purified and concentrated using DNA Clean & Concentrator-5 kit (Zymo Research, cat. number: D4004) following manufacturer’s instructions and eluted with 20 μl of the DNA Elution Buffer. The DNA sequencing libraries for both ChIP and Input samples were prepared using ThruPLEX DNA-Seq Kit (Takara Bio, cat. number: R400674) and SMARTer DNA Unique Dual Index Kit (Takara Bio, cat. number: R400697) following manufacturer’s instructions. DNA double size selection 100–700 bp was performed using SPRIselect beads (Beckman Coulter, cat. number: B23317). DNA libraries were sequenced on the Illumina sequencer.
Raw data were trimmed using Trimmomatic 0.3985 with the following parameters: ILLUMINACLIP :1:30:6, LEADING: 3, TRAILING: 3, HEADCROP: 10, AVGQUAL: 20, MINLEN: 35. Mapping was performed using Bowtie 2 (version 2.5)86 on TAIR10 genome. Duplicated reads were removed using SAMtools (version 1.7)87. The peak calling was performed with MACS2 (version 2.2.9.1)88 with -g 118459858. Peaks enrichment statistical significance was evaluated according to the Benjamini–Hochberg (FDR) test. Peaks were merged using BEDTools (version 2.26.0)89. Only peaks present in at least 2 out of 3 biological replicates were considered. Peaks annotation was performed with ChIPseeker90 with the following parameters: tssRegion comprised between −3000 and 0 and the genomicAnnotationPriority to 5UTR, 3UTR, Exon, Intron, Promoter, Downstream and Intergenic. The GO term enrichment analysis was performed with clusterProfiler83, with default parameters. The statistical significance of enriched categories was determined by the Benjamini–Hochberg (FDR) test, considering significant only categories with FDR ≤ 0.05. Peak annotation and the GO term enrichment analysis were performed on R studio, version 4.4.1. The consensus sequence enrichment analysis was performed using the MEME suite38 with default parameters. The analysis of common peaks between the SPL/NZZ ChIP-seq and the SEP3 ChIP-seq has been performed by using the command subsetByOverlaps on R studio, version 4.4.1.
In-silico prediction of protein complex structures
Protein complex structure predictions were performed with AlphaFold3 algorithm with default parameters. Protein sequences and domains annotation of SPL/NZZ (AT4G27330), STK (AT4G09960), SEP1 (AT5G15800), SEP2 (AT3G02310) and SEP3 (AT1G24260) were retrieved from the Uniprot database. AlphaFold3 models were analysed with the PyMOL software. The complexes confidence-level representations are based on the confidence files provided by AlphaFold3 algorithm. The binding-affinity analysis and the visualisation of the SPL/NZZ binding interface with the MADS tetramer was performed with the Prodigy webserver31,32,33 using default parameters, at a temperature of 22 °C.
RNA-seq material generation and data analysis
Three biological replicates of wild-type and spl-1 pistils at stage 947 were collected in the NucleoProtect RNA (Macherey-Nagel, cat. number: 740750.500) and stored at 4 °C for 1 day. RNA extraction was performed using the NucleoSpin RNA kit (Macherey-Nagel).
RNA was sequenced with the Novaseq X, at a depth of 30 M reads with a paired-end sequencing.
Raw data were trimmed using Trimmomatic 0.3985 with the following parameters: ILLUMINACLIP :1:30:6, LEADING: 3, TRAILING: 3, HEADCROP: 10, AVGQUAL: 20, MINLEN: 35. Mapping was performed with STAR91, with --alignIntronMax 10000 on TAIR10 genome. Counts were obtained with featureCounts92 (version 2.0.1), using Araport11 annotation.
Counts were filtered with HTSFilter93 and the DEGs were obtained using DESeq294, considering statistically differentially expressed DEGs with an FDR ≤ 0.05, as determined by the Benjamini–Hochberg (FDR) test. The GO terms enrichment analyses were performed with clusterProfiler83. Only categories with an FDR ≤ 0.05, according to the Benjamini–Hochberg (FDR) test were considered statistically significant. DEGs evaluation and GO terms enrichment analyses were performed on R studio, version 4.4.1.
In-situ hybridisation
In-situ hybridisation was performed as described in refs. 54,95, with some modification. Briefly, developing inflorescences were fixed in Formaldehyde/Ethanol/Acetic Acid (3.7%/50%/5%) solution, dehydrated and embedded in paraffin. Embedded inflorescences were sectioned to 8 µm thickness with a microtome. Dewaxing in histoclear was followed by rehydration and acidification in 0.2 M HCl for 20 min. Neutralisation occurs in 2X SSC for 10 min, followed by an incubation for 30 min with 1 μg/ml Proteinase K at 37 °C. Glycine (2 mg/ml) was used to incubate sections for 5 min to block Proteinase K, followed by 10 min of post-fixation in 4% formaldehyde. Washing in PBS and dehydrating via an ethanol series occurs before the application of the hybridisation solution (100 μg/ml tRNA; 6X SSC; 3% SDS; 50% formamide, containing ~100 ng/μl of antisense DIG-labelled RNA probe). Samples were hybridised overnight at 48 °C and the next day washed in SSC 2X/formamide 50% solution for 2 times for 1 h each. Antibody incubation (Anti-Digoxigenin-AP, Fab fragments, Roche, cat. number: 11093274910) and colour detection (BCIP/NBT Colour Development Substrate, Promega, cat. number: S3771) were performed according to the manufacturer’s instructions. ANT, PIN1 and ARF9 anti-sense probes were obtained from retrotranscribed RNA extracted from wild-type inflorescences. Primers used are listed in Supplementary Data 4.
Yeast-two- and three-hybrid assays
Yeast-two- and three-hybrid assays were performed following the protocol from ref. 96 with some modifications.
Both yeast-two- and three-hybrid assays were performed at 28 °C using a single AH109 yeast strain via co-transformation, or AH109 and Y187 yeast strains through co-transformation and mating.
The interactions were tested in selective media lacking leucine, tryptophan, histidine and adenine. AG and STK full-length CDS and SPL/NZZ and SEP3Δ shorter versions (SPL/NZZΔ and SEP3Δ, respectively) were cloned into Gateway vector GAL4 system (pGBKT7 and pGADT7, Clontech; pTFT197). Primers used are described in Supplementary Data 4.
FRET-FLIM
To test SPL/NZZ, SEP3 and STK interaction by FRET-FLIM, SPL/NZZΔ, SEP3Δ and STK CDS sequences, without the stop codons, were cloned into the pDONR207 plasmid according to the BP II protocol (Invitrogen, cat. number: 11789020). Thanks to LR II Plus reaction (Invitrogen, cat. number: 12538120), SEP3Δ and STK CDS were introduced into the pAB117 (provided by Rüdiger Simon, HHU Düsseldorf), which allow the C-terminus fusion of the GFP. SPL/NZZΔ was introduced in the pAB118 (provided by Rüdiger Simon, HHU Düsseldorf), which allows the C-terminus fusion of the mCHERRY. SEP3Δ was also cloned into the pAB119 plasmid (provided by Rüdiger Simon, HHU Düsseldorf) which allow the C-terminus fusion with both GFP and mCHERRY. Plasmids were transformed in the Agrobacterium tumefaciens strain GV3101. Tobacco leaf infiltration was performed as previously described98. The effect of FRET between GFP and mCHERRY was measured by FLIM of the GFP, using a Nikon ECLIPSE Ni-E A1 confocal equipped with a pulsed laser at 485 nm and taking advantage of the SymPhoTime 64 software (PicoQuant, https://www.picoquant.com/). Each acquisition has been performed reaching a photon count per pixel spanning from 30 to 50. Acquisition of coumarin6 (0.1 mM EtOH 100%), was used as a reference. Coumarin6 has excitation and emission spectra similar to the GFP ones and has a known photon lifetime of around 2.5 ns. The evaluation of its decay was used, through a phasor transformation function, to calculate the Instrument Response. The FLIM results obtained by the SymPhoTime 64 software were corrected with the Instrument Response thanks to the FLIM-Phasor analysis software99 (FlimLabs, https://www.flimlabs.com), generating a phasor plot in which the different pixels of an image are clustered in relation to their lifetime. This step allows the identification of the nuclei and the evaluation of the GFP photon decay.
Statistical information
The SPL/NZZ Co-IP/MS experiment has been performed on three independent biological replicates of pSPL/NZZ::SPL/NZZ:GFP pAP1::AP1:GR ap1cal inflorescences at 9 DAI. Proteins with LFQ intensity values in 2 out of 3 biological replicates and with at least two unique peptides were kept. Relative protein amount differences were evaluated by two-sample t-tests with permutation-based FDR correction and S0 parameter set to 1.0.
The SPL/NZZ ChIP-seq experiment has been performed on three independent biological replicates of pSPL/NZZ::SPL/NZZ:GFP pAP1::AP1:GR ap1cal inflorescences at 9 DAI. Peaks with an FDR ≤ 0.05 and present in at least 2 out of 3 biological replicates were considered. Statistical significance was evaluated according to the Benjamini–Hochberg (FDR) test.
Statistical significance of SPL/NZZ target genes enrichment against AGRIS and PlantGSAD transcriptional factor target lists has been evaluated with the ShinyGo100 webtool, and only categories with an FDR ≤ 0.05 were considered significant.
The spl-1 vs wild type RNA-seq experiment has been performed on three independent biological replicates of wild type and spl-1 pistils at stage 9 of development. Statistical significance of spl-1 vs wild-type DEGs was evaluated with the Benjamini–Hochberg (FDR) test and only genes with an FDR ≤ 0.05 were considered significant.
Statistical significance of Gene Ontology enrichment analyses on the results obtained from the SPL/NZZ Co-IP/MS, the SPL/NZZ ChIP-seq and the spl-1 vs wild type RNA-seq, was evaluated according to the Benjamini–Hochberg (FDR) test. Categories with an FDR ≤ 0.05 were considered significant.
In the FRET-FLIM experiment to test the SEP3-SPL/NZZ interaction, statistical significance of the reduction in the GFP lifetime has been evaluated according to the results of the student’s t-test, two-sided distribution, homoscedastic, comparing the GFP lifetime in nuclei expressing SEP3Δ-GFP with the one measured in nuclei expressing SEP3Δ-GFP + SPL/NZZΔ-mCHERRY. The comparison between SEP3Δ-GFP and SEP3Δ-GFP-mCHERRY has been used as a positive control for the experiment. Comparisons with a p ≤ 0.05 were considered significant. student’s t-test calculation has been performed with Microsoft Excell. In the FRET-FLIM experiment to test the STK-SPL/NZZ interaction, statistical significance of the reduction in the GFP lifetime has been evaluated according to the results of the student’s t-test, two-sided distribution, homoscedastic, comparing the GFP lifetime in nuclei expressing STK-GFP with the one measured in nuclei expressing STK-GFP + SPL/NZZΔ-mCHERRY. The comparison between SEP3Δ-GFP and SEP3-GFP-mCHERRY has been used as a positive control for the experiment. Comparisons with a p ≤ 0.05 were considered significant. student’s t-test calculation has been performed with Microsoft Excell. For PIN1-GFP, DR5v2, and R2D2 signal intensity measurements, statistical significance has been evaluated according to the results of the student’s t-test, two-sided distribution, homoscedastic, comparing the mutant with the wild-type. Comparisons with a p ≤ 0.05 were considered significant. student’s t-test calculation has been performed with Microsoft Excell. For the morphological analyses of ovules differentiating an MMC, as well as funiculus length measurements, statistical significance has been evaluated according to the results of one-way ANOVA with post-hoc Tukey HSD test. Comparisons with a p ≤ 0.05 were considered significant. The analysis has been performed with the webtool: https://astatsa.com/OneWay_Anova_with_TukeyHSD/. Exact p-values can be found in the Source Data.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data generated in this study are available from the corresponding author upon reasonable request. Source Data underlying Figs. 1–6 and Supplementary Fig. 5, 7, 9, 10, 11, 14, 15 are provided as a Source Data file. Lists of SPL/NZZ interactors and target genes, as well as differentially expressed genes in spl-1 with respect to the wild type, can be found in the Supplementary Data 1–3. Primers used are listed in Supplementary Data 4.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE101 partner repository with the dataset identifier PXD070409. ChIP-seq raw data have been deposited to the NCBI Sequence Read Archive (SRA) under the accession PRJNA1303074. RNA-seq raw data have been deposited to the NCBI Sequence Read Archive (SRA) under the accession PRJNA1365841. Source data are provided with this paper.
References
Schiefthaler, U. et al. Molecular analysis of NOZZLE, a gene involved in pattern formation and early sporogenesis during sex organ development in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 96, 11664–11669 (1999).
Yang, W. C., Ye, D., Xu, J. & Sundaresan, V. The SPOROCYTELESS gene of Arabidopsis is required for initiation of sporogenesis and encodes a novel nuclear protein. Genes Dev. 13, 2108–2117 (1999).
Tao, Q. et al. The TIE1 transcriptional repressor links TCP transcription factors with TOPLESS/TOPLESS-RELATED corepressors and modulates leaf development in Arabidopsis. Plant Cell 25, 421–437 (2013).
Chen, G.-H., Sun, J.-Y., Liu, M., Liu, J. & Yang, W.-C. SPOROCYTELESS is a novel embryophyte-specific transcription repressor that interacts with TPL and TCP proteins in Arabidopsis. J. Genet. Genomics 41, 617–625 (2014).
Wei, B. et al. The molecular mechanism of SPOROCYTELESS/NOZZLE in controlling Arabidopsis ovule development. Cell Res. 25, 121–134 (2015).
Szemenyei, H., Hannon, M. & Long, J. A. TOPLESS mediates auxin-dependent transcriptional repression during Arabidopsis embryogenesis. Science 319, 1384–1386 (2008).
Wang, L., Kim, J. & Somers, D. E. Transcriptional corepressor TOPLESS complexes with pseudoresponse regulator proteins and histone deacetylases to regulate circadian transcription. Proc. Natl. Acad. Sci. USA 110, 761–766 (2013).
Sieber, P., Petrascheck, M., Barberis, A. & Schneitz, K. Organ polarity in Arabidopsis. NOZZLE physically interacts with members of the YABBY family. Plant Physiol. 135, 2172–2185 (2004).
Zheng, Y. et al. Auxin guides germ-cell specification in Arabidopsis anthers. Proc. Natl. Acad. Sci. USA. 118, e2101492118 (2021).
Ito, T. et al. The homeotic protein AGAMOUS controls microsporogenesis by regulation of SPOROCYTELESS. Nature 430, 356–360 (2004).
Mendes, M. A. et al. The RNA-dependent DNA methylation pathway is required to restrict SPOROCYTELESS/NOZZLE expression to specify a single female germ cell precursor in Arabidopsis. Development 147, dev194274 (2020).
Balasubramanian, S. & Schneitz, K. NOZZLE regulates proximal-distal pattern formation, cell proliferation and early sporogenesis during ovule development in Arabidopsis thaliana. Development 127, 4227–4238 (2000).
Schneitz, K., Baker, S. C., Gasser, C. S. & Redweik, A. Pattern formation and growth during floral organogenesis: HUELLENLOS and AINTEGUMENTA are required for the formation of the proximal region of the ovule primordium in Arabidopsis thaliana. Development 125, 2555–2563 (1998).
Bencivenga, S., Simonini, S., Benková, E. & Colombo, L. The transcription factors BEL1 and SPL are required for cytokinin and auxin signaling during ovule development in Arabidopsis. Plant Cell 24, 2886–2897 (2012).
Huang, J. et al. Specification of female germline by microRNA orchestrated auxin signaling in Arabidopsis. Nat. Commun. 13, 1–14 (2022).
Vijayan, A. et al. A digital 3D reference atlas reveals cellular growth patterns shaping the arabidopsis ovule. eLife 10, 1–38 (2021).
O’Maoiléidigh, D. S. & Wellmer, F. A floral induction system for the study of early Arabidopsis flower development. Methods Mol. Biol. 1110, 307–314 (2014).
Ó’Maoiléidigh, D. S. et al. Control of reproductive floral organ identity specification in arabidopsis by the C function regulator AGAMOUS. Plant Cell 25, 2482–2503 (2013).
Zhao, F. et al. Phosphorylation of SPOROCYTELESS/NOZZLE by the MPK3/6 Kinase is required for anther development. Plant Physiol. 173, 2265–2277 (2017).
Smaczniak, C. Immunoprecipitation-mass spectrometry (IP-MS) of protein-protein interactions of nuclear-localized plant proteins. Methods Mol. Biol. 2698, 163–181 (2023).
Pinyopich, A. et al. Assessing the redundancy of MADS-box genes during carpel and ovule development. Nature 424, 85–88 (2003).
Favaro, R. et al. MADS-box protein complexes control carpel and ovule development in Arabidopsis. Plant Cell 15, 2603–2611 (2003).
Brambilla, V. et al. Genetic and molecular interactions between BELL1 and MADS box factors support ovule development in Arabidopsis. Plant Cell 19, 2544–2556 (2007).
Espinosa-Soto, C., Immink, R. G. H., Angenent, G. C., Alvarez-Buylla, E. R. & de Folter, S. Tetramer formation in Arabidopsis MADS domain proteins: analysis of a protein-protein interaction network. BMC Syst. Biol. 8, 9 (2014).
Mizzotti, C. et al. SEEDSTICK is a master regulator of development and metabolism in the Arabidopsis seed coat. PLoS Genet. 10, e1004856 (2014).
Smaczniak, C. et al. Characterization of MADS-domain transcription factor complexes in Arabidopsis flower development. Proc. Natl. Acad. Sci. USA 109, 1560–1565 (2012).
Urbanus, S. L. et al. In planta localisation patterns of MADS domain proteins during floral development in Arabidopsis thaliana. BMC Plant Biol. 9, 1–16 (2009).
Somssich, M. & Simon, R. Studying Protein–Protein Interactions In Planta Using Advanced Fluorescence Microscopy BT - Plant Genomics: Methods and Protocols. (ed. Busch, W.) 267–285 (Springer New York, New York, NY, 2017). https://doi.org/10.1007/978-1-4939-7003-2_17.
Mendes, M. A. et al. MADS domain transcription factors mediate short-range DNA looping that is essential for target gene expression in Arabidopsis. Plant Cell 25, 2560–2572 (2013).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Honorato, R. V. et al. Structural biology in the clouds: the WeNMR-EOSC ecosystem. Front. Mol. Biosci. 8, 729513 (2021).
Xue, L. C., Rodrigues, J. P., Kastritis, P. L., Bonvin, A. M. & Vangone, A. PRODIGY: a web server for predicting the binding affinity of protein–protein complexes. Bioinformatics 32, 3676–3678 (2016).
Vangone, A. & Bonvin, A. M. J. J. Contacts-based prediction of binding affinity in protein–protein complexes. eLife 4, e07454 (2015).
van Mourik, H., Muiño, J. M., Pajoro, A., Angenent, G. C. & Kaufmann, K. Characterization of in vivo DNA-binding events of plant transcription factors by ChIP-seq: experimental protocol and computational analysis. Methods Mol. Biol. 1284, 93–121 (2015).
Kaufmann, K. et al. Chromatin immunoprecipitation (ChIP) of plant transcription factors followed by sequencing (ChIP-SEQ) or hybridization to whole genome arrays (ChIP-CHIP). Nat. Protoc. 5, 457–472 (2010).
Li, L.-C. et al. SPOROCYTELESS modulates YUCCA expression to regulate the development of lateral organs in Arabidopsis. New Phytol. 179, 751–764 (2008).
Sieber, P. et al. Pattern formation during early ovule development in Arabidopsis thaliana. Dev. Biol. 273, 321–334 (2004).
Bailey, T. L. et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–8 (2009).
Folter, S. de & Angenent, G. C. <em>trans</em> meets <em>cis</em> in MADS science. Trends Plant Sci. 11, 224–231 (2006).
Yilmaz, A. et al. AGRIS: the Arabidopsis gene regulatory information server, an update. Nucleic Acids Res. 39, D1118–D1122 (2011).
Palaniswamy, S. K. et al. AGRIS and AtRegNet. a platform to link cis-regulatory elements and transcription factors into regulatory networks. Plant Physiol. 140, 818–829 (2006).
Davuluri, R. V. et al. AGRIS: Arabidopsis Gene Regulatory Information Server, an information resource of Arabidopsis cis-regulatory elements and transcription factors. BMC Bioinformatics 4, 25 (2003).
Ma, X. et al. PlantGSAD: a comprehensive gene set annotation database for plant species. Nucleic Acids Res. 50, D1456–D1467 (2022).
Pajoro, A. et al. Dynamics of chromatin accessibility and gene regulation by MADS-domain transcription factors in flower development. Genome Biol. 15, R41 (2014).
Pelaz, S., Ditta, G. S., Baumann, E., Wisman, E. & Yanofsky, M. F. B and C floral organ identity functions require SEPALLATA MADS-box genes. Nature 405, 200–203 (2000).
Manrique, S. et al. HISTONE DEACETYLASE19 controls ovule number determination and transmitting tract differentiation. Plant Physiol. 194, 2117–2135 (2024).
Smyth, D. R., Bowman, J. L. & Meyerowitz, E. M. Early flower development in Arabidopsis. Plant Cell 2, 755–767 (1990).
Pinto, S. C. et al. Germline β-1,3-glucan deposits are required for female gametogenesis in Arabidopsis thaliana. Nat. Commun. 15, 5875 (2024).
Balasubramanian, S. & Schneitz, K. NOZZLE links proximal-distal and adaxial-abaxial pattern formation during ovule development in Arabidopsis thaliana. Development 129, 4291–4300 (2002).
Liu, Z., Franks, R. G. & Klink, V. P. Regulation of gynoecium marginal tissue formation by LEUNIG and AINTEGUMENTA. Plant Cell 12, 1879 (2000).
Byzova, M. V. et al. Arabidopsis STERILE APETALA, a multifunctional gene regulating inflorescence, flower, and ovule development. Genes Dev. 13, 1002–1014 (1999).
Galbiati, F. et al. An integrative model of the control of ovule primordia formation. Plant J. 76, 446–455 (2013).
Fernandez, D. E. et al. The embryo MADS domain factor AGL15 acts postembryonically: inhibition of perianth senescence and abscission via constitutive expression. Plant Cell 12, 183–197 (2000).
Ferrándiz, C., Gu, Q., Martienssen, R. & Yanofsky, M. F. Redundant regulation of meristem identity and plant architecture by FRUITFULL, APETALA1 and CAULIFLOWER. Development. 734, 725–734 (2000).
Robert, H. S., Crhak Khaitova, L., Mroue, S. & Benková, E. The importance of localized auxin production for morphogenesis of reproductive organs and embryos in Arabidopsis. J. Exp. Bot. 66, 5029–5042 (2015).
Benková, E. et al. Local, efflux-dependent auxin gradients as a common module for plant organ formation. Cell 115, 591–602 (2003).
Ceccato, L. et al. Maternal control of PIN1 is required for female gametophyte development in Arabidopsis. PLoS ONE 8, 2–8 (2013).
Liao, C.-Y. et al. Reporters for sensitive and quantitative measurement of auxin response. Nat. Meth. 12, 207–210 (2015).
Zádníková, P. et al. Role of PIN-mediated auxin efflux in apical hook development of Arabidopsis thaliana. Development 137, 607–617 (2010).
Krizek, B. A., Blakley, I. C., Ho, Y.-Y., Freese, N. & Loraine, A. E. The Arabidopsis transcription factor AINTEGUMENTA orchestrates patterning genes and auxin signaling in the establishment of floral growth and form. Plant J. 103, 752–768 (2020).
Kelley, D. R., Arreola, A., Gallagher, T. L. & Gasser, C. S. ETTIN (ARF3) physically interacts with KANADI proteins to form a functional complex essential for integument development and polarity determination in Arabidopsis. Development 139, 1105–1109 (2012).
Krizek, B. A. Ectopic expression of AINTEGUMENTA in Arabidopsis plants results in increased growth of floral organs. Dev. Genet. 25, 224–236 (1999).
Weijers, D. & Wagner, D. Transcriptional responses to the Auxin hormone. Annu. Rev. Plant Biol. 67, 539–574 (2016).
Hou, Z. et al. High-throughput single-cell transcriptomics reveals the female germline differentiation trajectory in Arabidopsis thaliana. Commun. Biol. 4, 1149 (2021).
Piya, S., Shrestha, S. K., Binder, B., Neal Stewart, C. & Hewezi, T. Protein-protein interaction and gene co-expression maps of ARFs and Aux/IAAs in Arabidopsis. Front. Plant Sci. 5, 1–9 (2014).
Vernoux, T. et al. The auxin signalling network translates dynamic input into robust patterning at the shoot apex. Mol. Syst. Biol. 7, 508 (2011).
Wanamaker, S. A. et al. CrY2H-seq: a massively multiplexed assay for deep-coverage interactome mapping. Nat. Methods 14, 819–825 (2017).
Tian, Q., Uhlir, N. J. & Reed, J. W. Arabidopsis SHY2/IAA3 inhibits auxin-regulated gene expression. Plant Cell 14, 301–319 (2002).
Knox, K., Grierson, C. S. & Leyser, O. AXR3 and SHY2 interact to regulate root hair development. Development 130, 5769–5777 (2003).
Cavalleri, A. et al. Auxin-dependent post-translational regulation of MONOPTEROS in the Arabidopsis root. Cell Rep. 43, 115083 (2024).
Cucinotta, M. et al. Alternative splicing generates a MONOPTEROS isoform required for ovule development. Curr. Biol. 31, 892–899.e3 (2021).
Chandler, J. W. Auxin response factors. Plant Cell Environ. 39, 1014–1028 (2016).
Guilfoyle, T. J. The PB1 domain in auxin response factor and aux/IAA proteins: a versatile protein interaction module in the auxin response. Plant Cell 27, 33–43 (2015).
Pinto, S. C., Mendes, M. A., Coimbra, S. & Tucker, M. R. Revisiting the female germline and its expanding toolbox. Trends Plant Sci. 24, 455–467 (2019).
Hernandez-Lagana, E. et al. Organ geometry channels reproductive cell fate in the Arabidopsis ovule primordium. eLife 10, e66031 (2021).
Hernandez-Lagana, E. & Autran, D. H3.1 Eviction marks female germline precursors in Arabidopsis. Plants 9, 1322 (2020).
Su, Z. et al. The THO complex non-cell-autonomously represses female germline specification through the TAS3-ARF3 module. Curr. Biol. 27, 1597–1609.e2 (2017).
Losa, A., Colombo, M., Brambilla, V. & Colombo, L. Genetic interaction between AINTEGUMENTA (ANT) and the ovule identity genes SEEDSTICK (STK), SHATTERPROOF1 (SHP1) and SHATTERPROOF2 (SHP2). Sex. Plant Reprod. 23, 115–121 (2010).
Baker, S. C., Robinson-Beers, K., Villanueva, J. M., Gaiser, J. C. & Gasser, C. S. Interactions among genes regulating ovule development in Arabidopsis thaliana. Genetics 145, 1109–1124 (1997).
Karimi, M., De Meyer, B. & Hilson, P. Modular cloning in plant cells. Trends Plant Sci. 10, 103–105 (2005).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Wiśniewski, J. R., Zougman, A., Nagaraj, N. & Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 (2009).
Yu, G., Wang, L.-G., Han, Y. & He, Q.-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300 (1995).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Yu, G., Wang, L.-G. & He, Q.-Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383 (2015).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Rau, A., Gallopin, M., Celeux, G. & Jaffrézic, F. Data-based filtering for replicated high-throughput transcriptome sequencing experiments. Bioinformatics 29, 2146–2152 (2013).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21 (2014).
Ambrose, B. A. et al. Molecular and genetic analyses of the silky1 gene reveal conservation in floral organ specification between eudicots and monocots. Mol. Cell 5, 569–579 (2000).
Gietz, D., Jean, A. S., Woods, R. A. & Schiestl, R. H. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 20, 1425 (1992).
Gutierrez-Cortines, M. E. & Davies, B. Beyond the ABCS: Ternary complex formation in the control of floral organ identity. Trends Plant Sci. 5, 471–476 (2000).
Costa, A. et al. Ca2+-dependent phosphoregulation of the plasma membrane Ca2+-ATPase ACA8 modulates stimulus-induced calcium signatures. J. Exp. Bot. 68, 3215–3230 (2017).
Vallmitjana, A. et al. GSLab: open-source platform for advanced phasor analysis in fluorescence microscopy. Bioinformatics 41, btaf162 (2025).
Ge, S. X., Jung, D., Jung, D. & Yao, R. ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 36, 2628–2629 (2020).
Perez-Riverol, Y. et al. The PRIDE database at 20 years: 2025 update. Nucleic Acids Res. 53, D543–D553 (2025).
Acknowledgements
AC and LC were supported by MIUR PRIN2017 (20175R447S); CA was supported by MIUR UNIMI PhD fellowship. SM was supported by postdoctoral fellowships from Fundación Alfonso Martín-Escudero (Madrid, Spain) and UNIMI. AM was supported by a predoctoral grant from UNIMI. Research in the Kaufmann lab is funded by DFG grants 512328399, 512328399, 458750707. The Kaufmann lab wishes to thank the Dresden Concept Genome Centre for support with NGS sequencing. Mass Spec was performed using a Q Exactive Plus funded by DFG (428987924). The authors acknowledge support from the University of Milan through the APC initiative. We thank Francesca Lopez for contributing to preliminary plants characterisation. We thank Stefano Buratti for technical support during the FRET-FLIM experiment. We thank Rosanna Petrella and Simona Masiero for the helpful discussion. Part of this work was performed at the Unitech NoLimits, an advanced imaging facility established by the University of Milan. We thank Laura Luoni and Plant Platform, an advanced facility for plant developmental studies established by the University of Milan.
Author information
Authors and Affiliations
Contributions
A.C., C.A., S.M., and L.C. designed the research. A.C., C.A., G.B., C.S., C.M., S.M., A.R., M.S., A.M., V.G., X.X. performed the experiments. C.A., C.S., A.C., G.B., C.M., L.C., and K.K. analysed the data. A.C., C.A., L.C., G.B., C.S., and S.M. wrote the manuscript. L.C. and K.K. supported the research.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Dazhong Zhao and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cavalleri, A., Astori, C., Manrique, S. et al. SPOROCYTELESS/NOZZLE cooperates with MADS-domain transcription factors to regulate an auxin-dependent network controlling Megaspore-Mother-Cell differentiation. Nat Commun 17, 683 (2026). https://doi.org/10.1038/s41467-025-67343-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67343-x
