
Abstract
The omentum, a visceral adipose tissue with critical metabolic, immunological, and stem cell functions is the preferred site for ovarian cancer metastasis. However, its role in maintaining homeostasis and its responses to metastatic colonization remain incompletely understood. Using single-cell transcriptomics, we profile different anatomical regions of the omentum in patients with benign conditions and metastasis. We catalog the benign omentum and observe a stable cell type composition and a preserved stem and progenitor niche. Upon metastatic colonization, we report on increased immune heterogeneity and a concomitant reduction of mesothelial and progenitor cells. The lesser omentum, which is not routinely removed during surgical debulking, is identified as a premetastatic niche characterized by neutrophil infiltration, extracellular trap formation, and the presence of micrometastases. At established metastatic sites, resident cells exhibit cancer-associated phenotypes with regulatory, anti-adipogenic, and immunosuppressive properties. Cellular reprogramming across the omentum is associated with signaling profiles of tumor cells, suggesting potential influences on both proximal and distal tissue regions. This cell atlas illuminates the cellular and molecular determinants of organ homeostasis and reveals a high degree of plasticity and cellular reprogramming promoted by cancer colonization.
Similar content being viewed by others


Introduction
The omentum is an apron-like organ of folded peritoneum that extends from the stomach and covers the intestines. Dubbed ‘the policeman of the abdomen’ over 100 years ago for its ability to adhere to foreign bodies and sites of inflammation, the omentum plays a crucial role in maintaining abdominal homeostasis through tissue regeneration and countering infections1. As a visceral adipose tissue (VAT), it stores energy in the form of triglycerides and contributes to endocrine and metabolic health2. It is richly vascularized with an outer layer of mesothelial cells enclosing immune cells, adipocytes, stromal cells, and their associated stem cell niche3 within connective tissue. Immune cells are aggregated in fat-associated lymphoid clusters called ‘milky spots’ and mount antimicrobial, wound healing, and occluding responses1.
The omentum’s unique composition and physiological functions make it a preferential site for ovarian cancer metastasis and a key determinant of the metastatic tumor microenvironment (TME) within the peritoneal cavity4,5. Ovarian cancer is a heterogeneous disease comprising multiple subtypes6. Approximately 90% of ovarian cancers are of epithelial subtype, with high-grade serous adenocarcinoma of the ovary or fallopian tube (HGSOC) being the most commonly diagnosed one7. The 5-year overall survival rate for HGSOC remains around 46% and drops to 24% at late diagnosis (FIGO III/IV). Considering that the majority of HGSOC cases are diagnosed at advanced stages, often with extensive metastatic burden8 and that over 80% of patients experience disease recurrence7, this cancer type constitutes the most lethal gynecologic malignancy.
At diagnosis, practically all patients with FIGO III/IV HGSOC present with omental metastases. This dissemination can occur through transcoelomic spread, facilitated by invasion of the primary tumor into the peritoneal cavity, by ascitic fluid carrying tumor cells shed from the primary site, or via hematogenous9 or lymphatic10 routes. Despite its immunological functions5, the omentum appears unable to mount effective adaptive immune responses against infiltrating tumors, potentially facilitating metastatic colonization. For instance, cancer cells can be captured by milky-spot-embedded neutrophils11 and macrophages12,13, promoting metastasis. Once colonized, cancer cells benefit from immunosuppressive and wound healing environments and use adipocytes as an energy supply14.
Previous atlas-style VAT studies lacked specific aspects of cancer metastasis15 or single-cell resolution16. Moreover, knowledge of mesenchymal and adipose stem cell biology remains incomplete and is mostly based on murine or presorted studies17. Even less is known about mesothelial cell biology, including mechanisms of cell renewal, stem and progenitor markers, and differentiation hierarchies18,19. Additionally, research into the relationship between HGSOC and the TME did not exclusively focus on the omentum tissue, generally lacked healthy or benign reference samples, relied on sorted cell compartments20,21,22 or profiled only ascitic fluid23.
To better understand the cellular and molecular processes governing organ homeostasis and transformation upon ovarian cancer metastasis, we established a human omental cell atlas spanning both benign and metastatic settings. In this work, single-cell RNA sequencing (scRNA-seq) profiles a clinical cohort of 36 freshly dissociated omental tissue samples from patients with newly diagnosed benign disease, non-invasive serous tumors (borderline tumor), and HGSOC. Laparotomic or laparoscopic sampling across different anatomical sites enables regional comparisons within the benign/non-metastasized omentum and along the metastatic gradient, including distant (macroscopically tumor-free omentum), peritumoral (tissue adjacent to the tumor nodule, scraped from its surface), and tumor core (tumor nodule) omentum biopsies. Immunofluorescence validates expressed markers and elucidates spatial organization. The analysis of >110,000 cells identifies 12 cell types and more than 50 transcriptionally defined cell states, with specific enrichment observed in benign or metastatic samples. RNA velocity analysis provides insights into differentiation trajectories, while cell-cell communication analysis identifies key signaling pathways in both normal and metastasized omentum.
Results
The cellular landscape of the human omentum
We collected a cohort of fresh tissue biopsies containing a total of 36 samples derived from 15 patients (cohort 1) to characterize the changes that the omentum undergoes upon metastatic colonization (Fig. 1A, Supplementary Fig. 1A, and Supplementary Data 1). The entire treatment-naïve cohort consisted of age-matched samples derived from benign omentum and omentum with or without metastases. We first aimed to describe the ‘non-metastasized omentum’ and therefore examined 17 omentum samples from eight patients undergoing surgery for either benign disease or non-malignant tumor (patients 1–8). Regarding sampling location, we focused on the greater omentum (n = 8 samples), since this is the most frequent location of metastatic colonization in this disease. To investigate potential intra-organ variations, we collected samples from other locations within the omentum, namely distal to the former collected site (still greater omentum; n = 4 samples) as well as the lesser omentum (n = 4 samples). Subsequently, we collected an equally sized ‘omental metastasis’ subcohort including 19 samples from seven patients with metastatic disease (patients 9–15). This subcohort comprised three different omental regions: (i) macroscopically tumor-free sites distant to the omentum metastasis, and (ii) peritumoral region adjacent to the tumor, as well as (iii) metastatic tumors within the greater omentum. Biopsies were collected during surgery and immediately processed for scRNA-seq within two hours (Fig. 1A and Supplementary Fig. 1B). Due to their large size and fragility, adipocytes within the omentum are not compatible with scRNA-seq15. Therefore, we focused on the adipocyte-free fraction and depleted mature adipocytes during tissue dissociation via centrifugation. This procedure resulted in high sample quality with an average of 88.3% cell viability prior to single-cell capture (Supplementary Fig. 1C). Single cell RNA-seq libraries were collected and stored during the period of cohort acquisition and sequenced together to mitigate batch effects. After preprocessing and quality filtering, the final dataset was sequenced with 124,345 reads on average per cell and sample. It included 112,995 cells in total and on average 3139 cells per sample (Supplementary Fig. 1D, Supplementary Data 2).

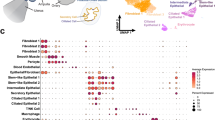
A Overview of anatomical sampling sites across two sub cohorts, ‘non-metastasized omentum’ and ‘omental metastasis’ from high-grade serous adenocarcinomas/ ovarian borderline tumor (36 samples derived from 15 patients). Figure elements partly created with BioRender (Jacob, F. (2025) https://BioRender.com/hh1ags). B UMAP of the entire cohort (36 samples derived from 15 patients), colored by cluster numbers. Cluster 7 showed a mixed cell type annotation with low marker expression and was regarded as noise. C Cohort UMAP with annotated major cell types and cell type composition bar plot of the omentum cohort. D Heatmap for selected marker gene expression by cluster-defining cell types detected in the entire cohort. E InferCNV heatmap of malignant cells from the “omental metastasis” cohort (n = 19 samples) showing CNV patterns across tumors. See also Supplementary Fig. 1, Supplementary Data 1 and 2. Source data are provided as a Source Data file.
In total, unsupervised clustering of all cells based on their transcriptomes identified 25 clusters (Fig. 1B). Automated cell type annotation24 combined with cell lineage marker expression revealed the following cell types (Fig. 1C, D, and Supplementary Fig. 1E): HGSOC/tumor cells (EPCAM, PAX8), T and NK cells (CD3E, KLRC1), mesothelial cells (ITLN1, UPK3B), mesenchymal/adipose stem and progenitor cells (CFD), monocytes and macrophages (LYZ, CD68), neutrophils (CSF3R, CXCR2), endothelial cells (VWF, PECAM1), lymphatic endothelial cells (LYVE1, PROX1), B cells (CD79A, MS4A1), plasma cells (SDC1, JCHAIN), plasmacytoid dendritic cells (pDCs) (IL3RA, LILRA4), and mast cells (KIT, CPA3). Cluster (cl.) 24 was classified as a mix of mesothelial and mesenchymal/adipose stem and progenitor cells (MSPCs) (Supplementary Fig. 1E). Based on its position between these cell types in the Uniform Manifold Approximation and Projection (UMAP) representation, enriched hallmark epithelial-to-mesenchymal transition (EMT) pathway together with expression of EMT (CDH2, TWIST1) and fibroblast (FN1, THY1) markers, we predicted cl. 24 cells to undergo EMT and hence labeled them ‘transdifferentiating’ cells (Fig. 1C and Supplementary Fig. 1F). Neutrophils had a low gene and UMI content, while tumor and mesothelial cells displayed the highest gene diversity and UMI content (Supplementary Fig. 1G, H). Finally, generally low cluster-specific mitochondrial gene content further confirmed high cell viability of the entire cohort (Supplementary Fig. 1I). Patient-specific clustering was not observed, except for tumor cells in the ‘omental metastasis’ subcohort (Supplementary Fig. 1J, K), which is further supported by patient-specific inferred copy number variation profiles uniquely observed in the cancer cell population (Fig. 1E). To validate the robustness of our major cell type annotations, we applied them to an independent scRNA-seq dataset (cohort 2) comprising solid tumor tissue samples collected from various anatomical sites despite differing sampling and processing protocols21. In general, CD45⁻ (non-immune) cells showed greater variability across sites compared to CD45⁺ (immune) cells, suggesting a conserved tumor immune landscape largely independent of tumor location. The omentum and upper quadrant (UQ) shared a similar cellular composition enriched in mesothelial, mesenchymal progenitor, and endothelial cells, while tumor cell fractions were lower in the UQ, consistent with its peripheral nature and regional enrichment of stromal and vascular elements (Supplementary Fig. 1L). Overall, the omentum cohort displayed a diverse cell type composition including T cells, mesothelial cells, MSPCs and rare cell types such as mast and pDCs. Importantly, fresh sampling allowed for capturing neutrophils, which are commonly lost during cryopreservation25.
The non-metastasized omentum is homogenous in cell type composition and transcriptional states across anatomical sites
Unsupervised clustering of samples by cell type composition revealed groups that primarily reflected the benign and metastatic settings (Fig. 2A). The ‘benign omentum’ and ‘omentum without metastasis’ groups showed similar cell type composition (Fig. 2B) and cell states (Fig. 2C), whereas the ‘omental metastasis’ cohort exhibited a distinctly altered cellular composition. Next, we analyzed the ‘non-metastasized omentum’ across different anatomical sites and found only minor differences in cell type composition (Supplementary Fig. 2A, B). Except for mesothelial cells, cell states were largely preserved in ‘non-metastasized omentum’ but shifted in metastasis (Supplementary Fig. 2C). Lastly, we performed a differential gene expression (DE) analysis across cell types, samples, and anatomical sites, and observed that samples were primarily clustered by cell type. Importantly, within each cell type, there was no clear clustering by anatomical location or patient, which was central to our analysis of the ‘non-metastasized omentum’ samples (Supplementary Fig. 2D). These data indicate that the ‘non-metastasized omentum’ displays homogeneous cell type composition and cell states across different anatomical sites and diagnosis, consisting mainly of mesothelial cells, MSPCs, and T cells. Thus, we concluded that ‘non-metastasized omentum’ samples may be grouped and serve as a reference dataset of benign omentum.

A Cell type composition heatmap per sample with clinical row annotations. B Sample composition barplots for benign omentum (n = 10 samples), omentum without metastasis (n = 7 samples), and omentum with metastasis (n = 19 samples) (HGSOC patient samples). c Kernel density heatmap showing cell distribution by metastatic status in UMAP space. Cells are grouped as ‘non-metastasized omentum’ (benign and non-metastatic) or ‘omental metastasis’. Color intensity indicates relative cell density within each hexagonal UMAP bin (75 bins). See also Supplementary Fig. 2. Source data are provided as a Source Data file.
Fundamental omentum tissue reprogramming during metastatic colonization
The comparison of cell type composition and transcriptional states between the main subcohorts revealed an extensive remodeling of the cellular environment in metastatic patient samples. This was marked by the loss of omentum-resident mesothelial cells and MSPCs, alongside an increased and diversified immune landscape (Fig. 2A and Supplementary Fig. 2A). We further investigated the altered cellular environment in ‘omental metastasis’ by comparing different sampling sites (Fig. 1A). Along the metastatic gradient towards the tumor core, cell type composition changed with increasing tumor and immune cell content while stromal cell fractions declined (Fig. 3A and Supplementary Fig. 3A). Notably, plasma cells and pDCs were detectable in ‘omental metastasis’ only (Supplementary Fig. 3A). B cells and endothelial cells were enriched in peritumoral samples while lymphatic endothelial cells disappeared at the tumor core. The transdifferentiating cell population was enriched in ‘omental metastasis’ samples and found mainly in peritumoral and tumor specimens. Although distant omentum samples were not in direct contact with the primary tumor or any metastases, they exhibited a unique cell type composition, characterized by a high neutrophil content and the presence of tumor cells (Fig. 3A and Supplementary Fig. 3A). A comparative analysis of ‘omental metastasis’ across samples, with a focus on distant sites, revealed that neither neutrophils nor tumor cells were specific to a single sample (Supplementary Fig. 3B). Notably, two distant samples contained more than 3% of tumor cells. In sample-specific UMAPs, these cells from patients P12 and P14 clustered distinctly and expressed tumor marker genes, thereby confirming correct cell identification (Supplementary Fig. 3C, D). To confirm and compare tumor cell presence in distant sites, we performed flow cytometry of matched and additional slow-frozen distant (n = 4), peritumoral (n = 6), and tumor (n = 4) tissue samples, which confirmed the presence of tumor cells in all distant samples in the range of 2.7–18.5% cell content (Supplementary Fig. 3E, F). In ‘omental metastasis’, cancer-associated phenotypes (Fig. 2C) were observed, most prominently at peritumoral and tumor core sites (Fig. 3A). In summary, our data revealed that, along the metastatic gradient, the omentum exhibits a fundamentally altered cellular composition. This is characterized by a gradual loss of stromal and mesothelial cells, an increase and diversification of immune cells, and transcriptional changes towards cancer-associated phenotypes. Distant samples of the ‘omental metastasis’ subcohort, which were expected to be cancer cell-free and showed no macroscopic sign of tumor manifestation, displayed characteristics of a premetastatic niche including neutrophil infiltration and the unexpected presence of micrometastases confirmed in multiple samples.

A Bubble plot for merged non-metastasized samples and ‘omental metastasis’ tissue samples comprising distant, peritumoral, and tumor core. Estimated effects of ‘Tissue Region’ on cell type composition based on GLM. The color gradient indicates the log2-transformed odds ratio (red, enrichment; blue, depletion), and bubble sizes indicate the Bonferroni-corrected –log10(P value). B UMAP visualization of the ‘omental metastasis’ cohort by tissue region. C UMAP visualization for lymphoid cells. D Matching cluster composition barplots by tissue region for lymphoid cells. E Stacked plot showing expression of gene signatures related to intratumoral CD8+ T cells states, including central memory (CM), effector memory (EM), terminally differentiated (TEMRA), progenitor exhausted (TPEX), exhausted (TEX) and naïve in non-metastasized samples and omental metastasis tissue samples using scRNA-seq. F Experimental design for flow cytometry analyses of phenotypic properties of CD8+ T cells in omental deposits of SO1 experimental syngeneic mouse models. Created in BioRender. Jacob, F. (2025) https://BioRender.com/hh1ags0. G Box plot showing percentage of TCF1+PD1+CD8+, TIM3+PD1+CD8+ and TIM3-PD1+CD8+ T cells in omental tumor samples of SO1 experimental models (nine mice per group). Box plot: lower quartile, median, upper quartile; whiskers, minimum and maximum. Statistical significance was calculated by a two-sided Mann-Whitney test. P-values are indicated. H UMAP visualization for myeloid cells. I Matching cluster composition barplots by tissue region for myeloid cells. J Neutrophil extracellular trap detection in omentum tissue distant to the tumor core. Myeloid UMAP plot showing activation marker ARG1 and NET marker PADI4 expression. K Confocal immunofluorescence stainings of matched distant tissue derived from FFPE blocks (‘omental metastasis’ subcohort) confirm neutrophil and NET presence in the omentum distant to the tumor core. Stainings were performed on two consecutive sections and yielded consistent results. Representative of n = 3 technical images obtained from n = 3 independent patient samples. MPO myeloperoxidase, CIT citrullinated histone H3, CK7 cytokeratin 7. See also Supplementary Figs. 3–5. Source data are provided as a Source Data file.
The immune microenvironment adopts pro-tumorigenic characteristics during metastatic colonization
The adaptation of the immune system within the ovarian cancer metastasis setting has been previously studied26. However, changes in the immune microenvironment along the metastatic gradient within the omentum remain uncertain. Our analysis of cell type composition in merged ‘non-metastatic’ and ‘omental metastasis’ tissue samples revealed a clear shift in cellular composition across regions. Tumor core and peritumoral areas showed enrichment of tumor cells and macrophages, while non-metastatic samples showed enrichment of mesenchymal progenitors and mesothelial cells. Regarding lymphoid populations, we observed an enrichment of T/NK cells in the tumor core. These findings reflect tissue-specific stromal and immune remodeling associated with metastasis formation and tumor proximity (Fig. 3A, B). Next, we focused our analysis on the lymphoid and myeloid compartments. Derived clusters were annotated using DE analysis-derived markers, along with the expression of classical and literature-derived lymphoid27,28,29,30,31,32,33,34,35,36 and myeloid37,38,39,40,41 markers, in combination with clinico-pathological information (Supplementary Figs. 4 and 5). The T cell compartment consisted of naïve-like (CCR7, SELL, cl. 6/7), transitional (cl. 5), activated effector (CD69, cl. 9), stressed (cl. 3/4), CD4/CD8 (cl.15), CD8 (cl. 1), activated inflammatory CD8 (IFNG, GZMB, cl. 8), early (TGFB1, GZMK, cl. 16), and late exhausted CD8 (TIGIT, LAG3, cl. 10), regulatory (IL2RA, CTLA4, cl. 13), and interferon-responsive T cells (ISG15, cl. 17) (Fig. 3C and Supplementary Fig. 4). The separated NK clusters consisted of conventional NK (PRF1, NKG7, cl. 2) and NK T-like cells (CD3E, KLRB1, cl. 12). No subclusters were found for B cells (cl. 11) and plasma cells (cl. 14). Along the metastatic gradient, lymphoid cells shifted from naïve-like and a more balanced CD4/CD8 T cell composition in benign samples to exhausted, regulatory, and interferon-responsive populations (Fig. 3D). Supporting this notion, we observed a significant increase in terminally exhausted (Tex) CD8+ T cell signature and a decrease in central memory (CM) CD8+ T cell signature in T cells in metastatic omental lesions compared to benign omentum (see methods), further confirming the impact of HGSOC cells on immune exclusion in the lymphoid compartment (Fig. 3E). To experimentally dissect the link between pro-tumorigenic omental microenvironment and T cell exhaustion, we harnessed a mouse experimental model of HGSOC Brca1-/-Trp53-/-/Myc/Hras SO1 cell line, to generate omental metastatic tumor in immunocompetent female C56BL/6 mice for subsequent flow cytometry analysis (Fig. 3f). Similar to our findings in human tumor samples, SO1 mouse omental ovarian tumors were associated with an increased density of effector TIM3-PD1+CD8+ T cells and terminally exhausted TIM3+PD1+CD8+ T cells over progenitor-like TCF1+PD1+CD8+ T cells in the ovarian cancer model (Fig. 3G and Supplementary Fig. 4G).
The myeloid compartment included several distinct cell types: CD1C-dendritic cells (CD1C, HLA-DRA, CLEC10A - cl. 1); tissue-resident, vascular-associated, phagocytic macrophages (LYVE1, FOLR2, STAB1 - cl. 2/5); inflammatory macrophages (IL1B, CXCL2 - cl. 6); fat-associated SPP1 macrophages (SPP1/osteopontin, APOE - cl. 7); a borderline tumor-specific macrophage population (cl. 12); nonclassical monocytes (FCGR3A, FCN1 cl. 3); monocytes (FCN1, VCAN, cl. 11); mature (S100P, cl. 13/8) and activated neutrophils (PADI4, ARG1, cl. 14/9); mast cells (cl. 10); and pDCs (cl. 4) (Fig. 3H and Supplementary Fig. 5A). The myeloid compartment composition showed stronger differences than the lymphoid compartment upon metastatic colonization (Fig. 3I). Samples from the ‘non-metastasized omentum’ subcohort contained mainly tissue-resident phagocytic macrophages and nonclassical CD16+ monocytes. Distant samples were specifically enriched in neutrophils, which showed gene signatures of activation (ARG1) and neutrophil extracellular trap (NET) formation (PADI4, Fig. 3J, Supplementary Fig. 5B, C). NET extrusion can result in cell rupture and is considered a form of cell death (NETosis)42. We confirmed NET presence by immunofluorescence (IF) staining in two patient-matched formalin-fixed, paraffin-embedded (FFPE) distant omentum tissue blocks (P10 and P14). Samples were positive for the NET markers citrullinated histone H3 (CIT) and myeloperoxidase (MPO), thus confirming NETs in these tissues (Fig. 3K). At the tumor core, tumor-infiltrating monocytes (TIMs, cl. 11) accumulated together with cDCs, pDCs and inflammatory macrophages (Fig. 3I). Consistent with previous findings40, pDCs had shifted from producing classical type I interferons to a more pro-tumorigenic role, with elevated TGFB1 and VEGFB expression (Supplementary Fig. 5F). At the tumor core, SPP1+ tumor-associated macrophages (TAMs) associated with HGSOC patients and a borderline tumor-specific TAM population were most abundant (Supplementary Fig. 5). While SPP1+ TAMs have also been described in other malignancies43,44,45, they were previously reported as a characteristic feature of TAMs in ovarian cancer37 and are known to have inflammatory, anti-adipogenic, and profibrotic functions46. Additionally, these TAMs expressed extracellular matrix (ECM)-degrading matrix metallopeptidases (MMPs), the hypoxia marker HIF1A, immunosuppressive LGALS1/3 and CD276, cytokines CXCL10/11, and angiogenic VEGFA (Supplementary Fig. 5C–F), indicating their role in tissue and immune regulation.
In summary, our data suggest that the lymphoid and myeloid immune compartments transitioned from their normal, tissue-resident states to acquiring a range of pro-tumorigenic characteristics, including regulatory, exhausted, angiogenic, anti-adipogenic, and immunosuppressive functions. This highlights a fundamentally altered omentum-immune microenvironment in ‘omental metastasis’. The accumulation of immune cells at metastatic sites, particularly plasma cells, TAMs, exhausted, and regulatory T cells, suggests a suppressed or failed attempt by the omentum to mount an effective immunological response to the tumor.
Mesothelial and progenitor cells give rise to a spectrum of transdifferentiating cells upon metastatic colonization
Prompted by the reduction of mesothelial and stromal cells in the ‘omental metastasis’ subcohort, and the presence of transdifferentiating cells bridging mesothelial cells and MSPCs (Figs. 1C and 2B) – a phenomenon also reported in long-read ovarian cancer metastasis to the omentum47 and single-nuclei adipose tissue15 studies - we next characterized this omentum cell compartment in more detail. Subclustering of mesothelial cells, MSPCs, and transdifferentiating cells identified a total of seven mesothelial, one transdifferentiating, and four MSPC subclusters (Fig. 4A and Supplementary Fig. 6). Cl. 11 and 12 are enriched with cells derived from metastatic patient samples (Supplementary Fig. 6A). Both mesothelial cells and MSPCs exhibited high plasticity, with cl. 1-4 and 9-11 showing diagnosis-associated subclustering (Supplementary Fig. 6B, C). Clusters were annotated using literature-derived3,17,48,49 and DE-derived marker gene expression (Supplementary Fig. 6 and 7A).

A UMAP with mesothelial and stromal cell subtyping annotated by cluster numbers in different colors. B Box plots showing flow cytometry data from dissociated single-cell suspensions derived from omental samples (ten benign omentum, ten peritumoral samples, and five metastasized/tumor core samples). Statistical significance was assessed using a two-sided unpaired Student’s t-test, and P-values are indicated. C Representative H&E images of benign, and metastasized omentum, and confocal immunofluorescence stainings of omental mesenchymal progenitor subpopulations of CD34+ preadipocyte (SDC2/CD362, CD34), CD34- preadipocyte (ENPEP/CD249, PDGFRa) location from mesothelial cell sheet (CD200) in benign omentum samples and diffuse localization on the metastasized omentum. Scale bar for H&E and IF images 50 µM. Images are representative of three independent samples; m mesothelial cells; t tumor cells D Velocity streams projected onto UMAP with tissue region annotation. E UMAP with sub clustering of transdifferentiating cells including subcluster annotation. f Transdifferentiating cell UMAP by tissue region and corresponding RUNX1 expression UMAP. G Representative confocal immunofluorescence images for tumor cells (EpCAM), macrophages (CD68), and RUNX1, with corresponding quantification (box plots). Primary tumor samples (n = 3) and metastases (n = 2) were derived from independent patients matching the scRNA-seq cohort, and each sample was quantified in three regions of interest, resulting in a total of 9622 cells analyzed. Box plots show the median (center line), interquartile range (box), whiskers (1.5× IQR), and individual jittered points. Scale bar: 50 µm. P-values were calculated using a two-sided Wilcoxon test. H Bubble plot showing Vázquez-García et al. scRNA-seq data (syn33521743) re-annotated with scROSHI using our ‘transdifferentiating cell’ signature. Dots indicate sampling site-specific enrichment of transdifferentiating subclusters. Sample sizes: Omentum n = 35; Adnexa n = 33; Ascites n = 25; Bowel n = 15; Peritoneum n = 15; Upper Quadrant (UQ) n = 8; Other n = 5. Color gradient indicates the log2-transformed odds ratio (red, enrichment; blue, depletion), and bubble sizes indicate the Bonferroni-corrected –log10(P value). I Box plot with RUNX1 expression on gene and protein level derived from bulk RNA-seq and matched mass spectrometry data, n refers to the number of samples (n = 109). P-values were calculated using the two-sided Wilcoxon test. J RUNX1 expression (left box plot) in an independent bulk RNA-seq cohort comparing primary tumors and matched omental metastases, along with the corresponding deconvoluted macrophage distribution (right box plot) in these samples (n = 43). P-values were calculated using the two-sided Wilcoxon test. In all box plots, the height of each box represents the interquartile range, with the thick line indicating the median and the box edges representing the first and third quartiles. Whiskers extend to the smallest and largest values within the 1.5 × interquartile range from the box, and data points beyond this range are shown individually as outliers. Jittered points are overlaid to display the distribution of individual observations. See also Supplementary Fig. 6–8. Source data are provided as a Source Data file.
Mesothelial cells are heterogeneous, highly plastic, and assume cancer-associated phenotypes upon metastatic colonization
Mesothelial cells were positive for MSLN, MUC16, UPK3B, CD200, microvilli-associated EZR, and the cytokine IL18. Additionally, WT1 was also shared with transdifferentiating cells (Supplementary Fig. 6D–G). Cl. 6 was defined as a benign omentum-associated mesothelial cell population (ITLN1, GATA4, and ADIRF). Cl. 1 and 4 were high in KRT8 and metallothionein genes reported to provide protection from oxidative stress and inflammation50. Cl. 11 was identified as cancer-associated mesothelial cells (CAMCs) that contained a unique gene expression profile (Supplementary Fig. 7A). CAMCs expressed inflammatory genes such as IL6, JUND, and NFKB1, including neutrophil-attracting chemokines CXCL1, CXCL2, and CXCL6. They also expressed aquaporin AQP1, lactotransferrin LTF, SLPI, and SDC4. In addition, these cells were enriched in cancer-associated long non-coding RNAs MTRNR2L8 and MTRNR2L12, and the hypoxia marker HIF1A. Complement components (C3 and C1R), EMT markers CD44, VIM, and TWIST2 were also expressed by CAMCs. These cells showed increased class I antigen presentation, and expression of various keratin-encoding genes (Supplementary Fig. 6D, E). Since the CAMCs (cl. 11) were enriched in distant and peritumoral sites (Supplementary Fig. 6A), we next investigated whether exposing mesothelial cells from ‘non-metastatic omentum’ to malignant ascites would induce CAMC-like features. To address this question, the most suitable cell surface markers, CD45- (for excluding immune cells) and CD200+ (Supplementary Fig. 7A), were used for quantification and isolation of human omentum-resident mesothelial cells. Flow cytometry analysis of 25 human samples, including ‘non-metastatic omentum’, peritumoral, and tumor core regions, revealed a trend-wise decrease in CD200+ and a significant increase in CD45+ cells toward the tumor core (Fig. 4B and Supplementary Fig. 7B), supporting the cell type composition changes observed in Fig. 2B. Isolated CD45-/CD200+ cells from ‘non-metastatic omentum’ samples maintained mesothelial-like cell morphology during the first passages (Supplementary Fig. 7C) and expressed CD200 and tight junction protein ZO-1 (Supplementary Fig. 7D). When cultured in a medium supplemented with malignant cell-free ascites, these isolated CD45-/CD200+ cells upregulated CAMC markers CXCL1 and CXCL2 compared to cultivation in normal medium (Supplementary Fig. 7E). IF analysis further supported the reduction of CD200+/ITLN1+ mesothelial cells in metastatic tissues (Fig. 4C). To investigate dynamic cell state transitions, we applied velocity analysis on mesothelial cells, MSPCs, and transdifferentiating cells (Fig. 4D and Supplementary Fig. 7F, G). Latent time projection revealed faster transcriptional changes in mesothelial cells, while MSPCs appeared later in latent time (Supplementary Fig. 7F). Removing either mesothelial cells or MSPCs from the projection did not alter the velocity vector streams allowing combined analysis of the three major cell types (Supplementary Fig. 7F). Mesothelial cell vectors revealed a single underlying differentiation trajectory across all subclusters roughly following from benign-associated mesothelial cells (cl. 6) to CAMCs (cl. 11). Marker gene analysis identified high ITLN1/omentin expression at the starting points of the vectors and increased expression of the antioxidant enzyme-encoding gene SOD2 toward the vector ends (Supplementary Fig. 6F), suggesting ITLN1 and SOD2 as markers of progenitor and differentiated mesothelial cells, respectively.
Mesenchymal stem cells follow a bifurcating differentiation trajectory into two preadipocyte lineages that disappear at metastatic sites
Cl. 5 and 7 of the four MSPC clusters (Fig. 4A and Supplementary Fig. 7A) shared progenitor features and adipogenesis markers (CFD, DCN, and PDGFRA). Cl. 7 included progenitor genes and markers related to ECM interaction (CD34, SDC2, MFAP5, and NT5E), ECM components (FBN1 and TNXB), remodeling (CD248, TIMP2, DDR2, and HTRA3), cell migration (TRIO), and TGFβ signal integration (TGFBR3). This cluster was labeled as CD34+ progenitors (Fig. 4A, Supplementary Fig. 7A, and 6D–F). Due to expression of CEBPA, PPARG, APOD, CXCL14, ALDH1A1, ENPEP, and PDGFRA, and absence of CD34, cl. 5 was considered as a CD34- progenitor population. Spatial localization by confocal immunofluorescence in ‘non-metastasized omentum’ revealed that SDC2/CD362+ cells (identifying CD34+ progenitors) are located underneath the mesothelial cell monolayer (CD200+ cells) (Fig. 4C). Their close proximity to mesothelial cells, together with an ECM gene signature, reinforces the notion that these cells play structural roles, potentially contributing to the basement membrane that separates the epithelium from the underlying stroma51. Conversely, CD34- progenitors (cl. 5) identified as ENPEP/CD249+ cells, were found close to the mesothelial cell monolayer as well as interstitially within the adipose tissue (Fig. 4C). In the ‘omental metastasis’, the specific localization of both rare SDC2/CD362+ and ENPEP/CD249+ cell types was diminished, likely due to the presence of tumor cells (EpCAM+) (Fig. 4C). However, these cells remained in close proximity to CD200+ mesothelial cells. Cl. 2 was transcriptionally similar to cl. 5 (Supplementary Fig. 6D, E) but shared gene expression with transdifferentiating cells at peritumoral sites such as for COL1A1 (Supplementary Fig. 6F). Thus, cl. 2 mainly contained further committed CD34- progenitor cells as well as cells adopting a cl. 12 phenotype. Cl. 3 contained cells with shared signatures from cl. 5 and 7 but uniquely expressed PLIN2 (Supplementary Fig. 6D–F). Velocity analysis revealed a common mesenchymal stem cell (MSC) population that was shared between cl. 5 and 7 (Fig. 4A, D), thus not revealed by cluster analysis alone. Here, MSC marker CD9, DCN, ENG, and THY1 expression were strongest at the velocity vector starting points, identifying these cells as multipotent tissue stem cells (Supplementary Fig. 6F). Furthermore, these cells expressed high levels of F3 and the osteogenesis inhibitor MGP52. The MSCs followed a bifurcating differentiation path into two adipocyte progenitor populations. Among the top velocity driver genes were the adipocyte differentiation transcription factor EBF1 and NEGR1, which were shared between both progenitor populations (Supplementary Fig. 7G). The CD34+-population (cl. 7) also expressed the neural cell adhesion molecule NTM and actin-binding ABLIM1, while the CD34--population (cl. 5 and 2) was positive for the lymphangiogenic marker SVEP1. The basement membrane component LAMA2 was expressed along the intersection of both progenitor populations (Supplementary Fig. 7G). Of note, patient-specific cl. 2, to which both progenitor lineages contributed, appeared last in the latent time projection (Supplementary Fig. 7F). This, together with its unique expression of the lipid droplet marker-encoding gene PLIN2, pointed towards further differentiation of CD34+ and CD34- progenitor cells towards a premature adipocyte state. In line with previous work defining adipose progenitors based on their CD34 protein expression53, the CD34+ and CD34- population would represent white and thermogenically potent beige-like progenitors, respectively. The nearly equal proportion of CD34+ to CD34- progenitors in benign VAT53 was confirmed (Supplementary Fig. 6G). However, the balance shifted towards increased CD34- proportion along the metastatic gradient, indicating that white progenitors were disproportionately lost in metastasis (Supplementary Fig. 6C, G). In summary, the omental stromal compartment contains multipotent tissue stem cells from which two adipocyte lineages emerge. Both white and beige-like progenitor cells share adipogenic differentiation capacities but are predominantly locked in their progenitor state. In metastatic colonization, the stem and progenitor niche seems to be disrupted, resulting in significant reduction of MSPCs, particularly CD34+-cells.
Transdifferentiating cells emerge from mesothelial and progenitor cells and their heterogeneity is shaped by the local tissue environment
Cancer-associated fibroblasts (CAFs) exert important pro-tumorigenic and immunomodulatory functions22. Cl. 12 (Fig. 4A) exhibited characteristics of CAFs, with expression profiles including collagen-encoding genes, POSTN, FN1, and FAP (Supplementary Fig. 6D–F). However, varied expression of these markers and overlap with mesothelial cells and MSPCs in metastasis, indicated heterogeneity within the transdifferentiating cell pool (Supplementary Fig. 6 and 7). Consistent with velocity-derived results, this suggests that cl. 12 potentially originated from both mesothelial cells and MSPCs (Fig. 4C and Supplementary Fig. 7A). Accompanied by DE- and literature-derived gene expression data54 (Supplementary Fig. 8A, B), further subclustering of cl. 12 revealed nine subtypes (Fig. 4E). Cl. 1 was enriched in benign samples and contained vascular smooth muscle cells (MCAM) and pericytes (RGS5) (Fig. 4E, Supplementary Fig. 8A, B). The remaining clusters formed one major fibroblast-like population. Cl. 9, specific to the ‘non-metastasized omentum’, shared transcriptional similarities with a mesothelial-like, omentum-specific, stromal cell population with anti-adipogenic function55. The remaining clusters were categorized as peritumoral (cl. 5-8) and tumor core (cl. 2-4) CAFs (Supplementary Fig. 8C, D). Transcriptional signatures suggested that peritumoral cells fell into the inflammatory CAF group (iCAF), characterized by CXCL12, LIF, and IL1R1 expression, while tumor core cells fell into the myofibroblast (myCAF) category, marked by POSTN, MMP11, TWIST1, FAP, and collagen-encoding genes (Supplementary Fig. 8D). Furthermore, CAMCs and CAFs shared a common gene expression cassette, which included keratins (KRT18, KRT19), the transcription factor RUNX1, and markers of immunosuppression (CD274, LGALS1), ECM remodeling (MMP2), and angiogenesis (VEGFA, MDK) (Supplementary Fig. 6D, E). Consistent with elevated RUNX1 expression in CAMCs and transdifferentiating cells (Fig. 4F and Supplementary Fig. 6F), a significant increase in RUNX1⁺ stromal-like cells was also observed in ‘omental metastasis’ samples by confocal IF analysis. Notably, this increase was not found in matched primary ovarian tumor tissue, which is not expected to harbor mesothelial cells or adipocytes (Fig. 4G). To further investigate the described clusters of the transdifferentiating cell population across different solid tumor tissues in patients with HGSOC, we applied these cell type signatures to scRNA-seq data derived from cohort 2. Approximately half of the nine clusters were significantly altered among the different anatomical sites (Fig. 4H and Supplementary Fig. 8E). Here, tansdifferentiating cell cl. 5 (inflammatory CAFs) and cl. 8 (peritumoral CAFs) appeared to be specific to the omentum and UQ, sites known to contain mesothelial cells. Moreover, RUNX1 expression across transdifferentiating cells was significantly elevated in the omentum compared to the remaining anatomical sites in this dataset (Supplementary Fig. 8F). We further validated RUNX1 expression in bulk RNA-seq and matched proteomics (n = 109 samples) data derived from cohort 3. RUNX1 expression was significantly increased in ‘omental metastasis’ compared to primary tumor or distant omentum tumor-free sites, both on RNA and protein level (Fig. 4I). The elevated RUNX1 expression observed in omental metastases, while macrophage levels differed only marginally, was further validated in cohort 4 (Fig. 4J). Apart from the expected absence of mesothelial cells in the primary tumor, lower RUNX1 levels in distant samples may indicate that CAF reprogramming and the mesothelial-to-mesenchymal transition (MMT) process require proximity to, or are accelerated by colonizing tumor cells (Fig. 4F). In summary, we describe a cell population that is increasingly found in metastatic colonization in the omentum, appearing to derive from both mesothelial and mesenchymal progenitor cells. These transdifferentiating cells display a high level of heterogeneity, with the different subpopulations being shaped by the local TME. Additionally, our data suggest that the transition of these resident cells into transdifferentiating cells may be a feature of the omentum, driven by the upregulation of the transcription factor RUNX1.
Omental tissue homeostasis is maintained by a balanced molecular crosstalk between resident stroma and immune cells
After characterizing the composition and changes in the omental microenvironment, we next aimed to identify mechanisms initiating the observed differences in distant samples and at metastatic sites. We applied cell-cell communication (CCC) analysis between major cell types using CellChat56, with a 0.5% cell type inclusion threshold (Methods, Supplementary Data 3). We first established a CCC interaction map in the ‘non-metastasized omentum’ subcohort to better understand the signaling involved in maintaining tissue homeostasis of the benign omentum. CCC aggregate networks between major cell types revealed that the stromal compartment plays a central role, both in the number and strength of interactions (Fig. 5A, B). Three outgoing CCC patterns were shared by cell types: 1) stromal cells primarily communicated via ECM and angiogenic pathways; 2) immune cells shared pathways of homing and adhesion, cytokine, and antigen presentation; and 3) endothelial cells sent signals related to vasoconstriction, adhesion, and cytokines (Fig. 5C). Similarly, three incoming CCC patterns were shared between cell types: 1) stromal cells, especially MSPCs, received growth signals including FGF, PDGF, and EGF; 2) immune cells received signals related to complement and cytokines, antigen presentation, and adhesion; and 3) endothelial cells received signals related to angiogenesis, adhesion, non-canonical Wnt, and insulin-like growth factor (IGF) (Fig. 5D). Mesothelial cells were unique in that they exhibited all three signaling patterns, indicating their broad biological functions and keystone communicator role. Specifically, they signaled through cytokine and adhesion pathways primarily with B cells, monocytes & macrophages, T/NK cells, and endothelial cells (Fig. 5E). Meanwhile, MSPCs interacted via collagen and other ECM pathways with various immune cells and the vasculature (Fig. 5F). MSPCs and transdifferentiating cells were critical in maintaining tissue homeostasis. Within the significant pathways of the ‘non-metastasized omentum’ subcohort, MSPCs were involved in ECM and basement membrane interactions (laminin, collagen, tenascin), hormone production (IGF) and stem cell maintenance (CD34, non-canonical Wnt, THY1), whereas mesothelial cells also contained angiogenic functions (ANGPTL) (Supplementary Fig. 9). CCC provided a global view of cellular interactions in the ‘non-metastasized omentum’, revealing that tissue homeostasis is regulated by an intricate set of pathways related to immune, ECM, hormonal, angiogenic, and developmental signaling.

Cell-cell-communication interactions between major cell types inferred by CellChat (‘non-metastasized omentum’ subcohort, n = 17). Aggregate communication networks of non-metastasized samples depicting the number (A) and the strength (B) of interactions between major cell types. A threshold of 0.5% cells per cell type out of the total was applied. Major outgoing (C) and incoming (D) patterns shared between cell types and their pathway contributions. Circos plots summarizing outgoing mesothelial cell (E) and MSPCs (F) communication pathways. See also Supplementary Fig. 9. Source data are provided as a Source Data file.
The TME mimics tumor signaling pathways
The establishment of cancer-associated phenotypes at distant sites indicates how the presence of cancer in the peritoneum exerts a far-reaching influence beyond the metastatic region. Here, we hypothesized that tumor cells influence the omentum via secretion of factors into the ascites or microenvironment that elicit long-range effects. To address this by an unsupervised approach, we assessed CCC patterns in the different tissue regions and quantified changes by differential pathway analysis (Methods). The comparison of tissue regions across all identified cell types did not differ in their interaction category composition (Supplementary Fig. 10A). However, we observed profound changes in the number and strength of interactions in ‘omental metastasis’ when comparing the three different sampling sites in this subcohort (Supplementary Fig. 10B). Averaged across all cell types, 5.34, 10.92, and 3.92 times more outgoing pathways (P-value < 0.05) were observed in the three ‘omental metastasis’ sites (distant, peritumor, and tumor) compared to benign ‘non-metastasized omentum’ tissue indicating peritumoral tissue as the site with particularly strong cellular crosstalk (Supplementary Fig. 10C). Next, we displayed the scaled contribution of all significant differential pathways between pairwise comparisons of the main sampling sites. This analysis revealed an increased contribution of the macrophage migration inhibitory factor (MIF), midkine (MDK), and collagen pathways in metastatic patient samples (Fig. 6A). Several pathways were unique to specific sampling sites (Fig. 6B). Tumor cells in the tumor core communicated primarily via MIF, MDK, amyloid precursor protein (APP), and to a small extent NECTIN2 with various cell types (Fig. 6C). They primarily received signals from ECM components such as collagens from transdifferentiating and mesenchymal progenitor cells (Fig. 6C), suggesting that they surrounded themselves with a pro-tumorigenic stroma. Along the metastatic gradient, tumor cells decreased their MIF but increased their midkine pathway signaling (Supplementary Fig. 10D). Individual pathway analysis (Fig. 6D) revealed that MIF was used by multiple cell types and signaled to pDCs, B cells, and to a lesser degree to macrophages and monocytes. In contrast, MK pathway signaling primarily originated from tumor cells and affected all cells in the TME mainly via the MDK-NCL interaction. Last, NECTIN signaling by endothelial, mesothelial, and to a lower extent by tumor cells was uniquely directed to NK/T cells (Supplementary Fig. 10E). Next, we investigated whether the TME contributes to tumor-specific signaling by performing a differential pathway analysis between distal and benign mesothelial, endothelial, progenitor, and transdifferentiating cell populations (Fig. 6E). MIF signaling was upregulated in all distal TME cells, APP signaling was upregulated in endothelial and progenitor but downregulated in transdifferentiating cells and MDK signaling was upregulated in endothelial and transdifferentiating cells (Fig. 6E). The combined MIF, MK, and APP pathway contribution averaged over endothelial, mesenchymal progenitors, transdifferentiating and mesothelial cells increased 29.1-fold (distant), 50.5-fold (peritumor), and 12.8-fold (tumor core) compared to benign tissue. Applying long-read scRNA-seq (cohort 5) to three matching patients’ tumor core and distal tissue samples (P1-P3 corresponding to P13-P15 in our data), we recently reported limitations of short-read sequencing47. Since genomically unstable tumor cells were particularly prone to inaccurately report on functional gene expression, we assessed and verified functional full-length transcript expression of relevant genes (MIF, MDK, APP, and NECTIN2) in the matching long-read scRNA-seq data (Supplementary Fig. 10E). Taken together, tumor cells, in collaboration with TME cells, modulated both proximal and distal omentum via a repertoire of signaling pathways particularly relying on MIF, midkine, APP, and NECTIN signaling.

A Cell type-agnostic pathway level barplot showing differential pathways for all pairwise comparisons between tissue locations (n = 36 samples). Both incoming and outgoing pathways are included. Dots under each pathway show the pairwise comparisons in which the pathway has a significant differential contribution (one-tailed permutation-based enrichment test with Benjamini–Hochberg false discovery rate correction, followed by an additional Bonferroni adjustment across comparisons; P-Value adjusted <0.05). B Probability and cell-type usage of pathways uniquely expressed in each tissue region cohort. The communication probability is a composite metric that aggregates gene expression and pathway structure, making it more suitable for connectivity comparison. C Chord plots for HGSOC outgoing and incoming signaling pathways at the metastatic core site. D Pathway centrality heatmap for MIF, midkine (MK), and NECTIN signaling, comparing the role of each cell type in four communication roles (sender, receiver, mediator or influencer) mediated by the pathway under analysis. Communication roles are scored by calculating the pathway network centralities for each cell type and reporting them to a relative scale defined as “importance”. E Differential expression of outgoing signaling pathways in distant versus benign comparison for mesothelial, endothelial, mesenchymal progenitor, and transdifferenting cells. See also Supplementary Fig. 10. Source data are provided as a Source Data file.
Discussion
Herein, we describe a human omental cell atlas in both benign and metastatic settings. Our analysis identified fifteen major cell types in total with over 50 transcriptionally distinct cell states, reconciling adipose tissue and ovarian cancer biology in the omentum. This unique data set, consisting of a ‘non-metastasized omentum’ and an ‘omental metastasis’ subcohort, each containing different anatomical sampling sites, allowed us to present a more unified model of organ homeostasis and reprogramming upon metastatic colonization.
The adipocyte-depleted ‘non-metastasized omentum’ subcohort exhibits homogenous and patient-independent cell type composition and transcriptional states across different anatomical sites primarily consisting of mesothelial cells, MSPCs, and T cells. This subcohort contained a population of mesothelial cell-derived mesenchymal-like cells that were transcriptionally similar to a population with anti-adipogenic function55, further confirming mesothelial cell involvement in adipose tissue regulation. Additionally, lymphatic endothelial cells were present, alongside a predominantly T cell-dominated immune compartment with small fractions of B cells and macrophages. Specific to this benign tissue was the presence of NK cells, while plasma cells and pDCs were absent. The majority of immune cells displayed tissue-resident, naïve, resting, or inflammatory phenotypes. Mesothelial cells, transdifferentiating cells, and MSPCs were identified as signaling hubs that orchestrate tissue homeostasis via ECM, immune, hormonal, angiogenic, and developmental signaling pathways.
The mesothelial cells covering the omentum appeared transcriptionally highly plastic, displaying varying levels of epithelial, activated/microvilli markers, as well as differences in mitochondrial gene and overall gene content. The cell plasticity likely reflects the wide spectrum of cytomorphological presentations typical for this cell type57. Mesothelial cells differentiated along a single pathway, transitioning from ITLN1hi progenitors to SOD2+ subtypes, thereby challenging the earlier hypothesis that they might originate from mesenchymal stem cells18. Adipocyte differentiation, which has been reported in mice58, was not observed, although it might occur under different conditions59. While ITLN1 is a known mesothelial marker, a key finding is that high ITLN1 expression seems to mark mesothelial progenitor-like cells, which are uniquely enriched in the benign omentum. ITLN1, a lectin and adipokine with anti-inflammatory and anti-microbial properties, is found in VAT and downregulated in certain cancers60,61. In ovarian cancer, ITLN1 suppresses tumor progression62 by (i) blocking lactotransferrin (LTF) binding to its receptor LRP1, which controls MMP1-mediated tumor invasion, and (ii) lowering nutrient availability through increased glucose uptake into adipocytes62. Moreover, ITLN1 has been shown to inhibit glycolysis and activate PPAR-γ63 suggesting that mesothelial cell-derived ITLN1 is a regulator of adipose tissue metabolism.
Our findings of the human mesenchymal progenitor cell population in the ‘non-metastasized omentum’ subcohort also provide a more fine-grained picture of the adipose stem and progenitor cell hierarchy, which is primarily established in mice17,64. Here, Dpp4 was identified as a murine adipose stem cell marker65, while DPP4 and MFAP5 were suggested as human equivalent17. In our data, however, MSCs with multi-lineage potential were defined by THY1, MGP, F3, FBLN1, and high DCN expression. Strong ECM component expression aligns with the concept of stem cells acting as architects of their own niche66. The expression of MGP in MSCs indicated an early inhibition of calcification and the osteogenic differentiation pathway52, which is regulated by ECM stiffness67. MGP and F3 overlap also pointed towards shared commonalities with adipocyte-regulatory cells68 indicating an uncommitted state. The stem cells followed a bifurcating differentiation trajectory. One population expressed CD34, MFAP5, DPP4, NTM, NT5E, and SDC2, indicating a strong contribution to and interaction with the ECM. These connective tissue progenitors resembled similar human adipose structural cells - termed by the literature as SWAT cells, which were under Wnt control and retained multipotency69. The second population was characterized by CD34-, PDGFRAhigh, CXCL12, CXCL14, APOD, PTN, LAMA2, SVEP1, ANGTP1, and ENPEP hinting at a different ECM composition, cytokine function, and vascular association. NTM and NEGR1 expression also indicated a role in sympathetic innervation and a preadipocyte-neuronal crosstalk70. Our data is also in line with functional studies of adipocyte progenitors distinguished by CD34 expression, which provides insight into their further differentiation53. Translated to our data, CD34+ progenitor cells would give rise to white adipocytes with different levels of lipid turnover, and CD34--progenitors would develop into thermogenically potent beige-like adipocytes. A third patient-specific population in our data contained mixed CD34 expression and increasing levels of the lipid droplet marker PLIN271. These cells appeared last in the latent time projection suggesting that this population represents further committed premature adipocytes starting to form lipid droplets71. This would support the possibility that cells from both trajectories indeed may give rise to adipocytes72. Latent time analysis also suggested that preadipocytes are predominantly arrested in their differentiation state, aligning with low de novo adipocyte differentiation55. Thus, preadipocytes may not represent a transient cell state en route to adipocyte maturation. Instead, as a resident stromal population within the connective tissue, they may play an important role in contributing to tissue architecture and function and only emigrate from their niche to become mature adipocytes upon receiving the appropriate signals. It is important to mention that our scRNA-seq protocol excluded mature adipocytes from downstream analysis, limiting direct conclusions about their role in omental metastasis. Prior studies have demonstrated that adipocytes provide fatty acids to support rapid tumor growth, underscoring the potential involvement of lipid metabolism in omentum metastasis14. Moreover, recent single-nucleus RNA-seq has emerged as the preferred approach for profiling mature adipocytes in adipose-rich tissues, as shown in mouse models of obesity-induced adipose tissue remodeling73.
Upon metastatic outgrowth (‘omental metastasis’ subcohort), the omentum undergoes a wide-ranging set of transformations beyond the metastatic site. Sampling at different sites revealed multiple pieces of supporting evidence for omental premetastatic niche reprogramming. Distant sites contained mainly naïve-like T cell populations, as well as low fractions of plasma cells, and angiogenic pDCs. A hallmark of the premetastatic niche at distant sites was high neutrophil infiltration, which underwent NETosis. NETs are known to enable cancer cell capture from the ascites and seem to be present in the omentum of early-stage ovarian cancer patients11. Another hallmark was the transition of normal mesothelial cells into CAMCs that displayed an altered transcriptional profile including the expression of neutrophil-attracting chemokines. Thus, beyond ascites components that induce neutrophil invasion in the omentum11, our work suggests a potential role of mesothelial cells in neutrophil attraction, as previously demonstrated in a murine peritonitis model74. The fact that NETs exert an important function in clearing the peritoneal cavity by capturing bacteria and contaminants suggests that tumor cells may take advantage of a naturally occurring tissue reaction for metastatic dissemination74. Indeed, micrometastases were found in half of the distant omentum HGSOC samples constituting 3–8% of the overall cell content. Whether neutrophil infiltration and activation are necessary for cancer cell colonization remains an open question. CAMCs also showed elevated expression of aquaporin 1, which is involved in water transport, indicating increased fluid dynamics into the abdomen and a role in ascites formation75,76. Another CAMC-specific gene was lactotransferrin (LTF). Simultaneous downregulation of ITLN1 highlights the counteracting roles of ITLN1 and LTF62.
In the ‘omental metastasis’ subcohort, we observed cellular differences that varied with the distance from the center of the metastasis. Along this gradient of increased tumor burden, the immune cell content increased and diversified, as evidenced by scRNA-seq and flow cytometry data. Precisely, this was the case for T cells, TAMs and TIMs37,77, plasma cells, and pDCs, which had shifted from their usual pro-inflammatory function of IFN production to the expression of angiogenic and pro-tumorigenic factors such as VEGFA and TGFB140. This switch aligns well with their known roles in the ovarian TME78. Notably, we also observed that the overall increase in the immune compartment came at the expense of a reduced mesothelial and mesenchymal compartment, which is suggestive of a cancer-induced disintegration of the protective barrier and disruption of the stem cell niche. Peritumoral samples derived from the ‘omental metastasis’ subcohort, exhibited transcriptional signatures indicating ongoing immune reaction and tissue modulation. These samples contained the highest level of inflammatory cell types with enrichment for iCAFs and TAMs, TIMs, mast cells, neutrophils, and early exhausted CD8 T cells. In contrast, the center of the metastasis contained the highest fraction of tumor cells and a larger macrophage pool. We also found SPP1 (osteopontin/OPN) expression being unique to HGSOC TAMs and specific to these tumor sampling sites indicative of inflammatory signaling, myofibroblast activation, angiogenesis, and inhibition of adipocyte differentiation79,80,81,82,83,84. This suggests that HGSOC-TAMs may play a role in adipocyte-fibroblast reprogramming and vascular remodeling. The remaining immune cells in the tumor core included pDCs, cDCs, late exhausted CD8 T cells, myCAFs, and almost no lymphatic system indicating a highly immunosuppressed environment. Interestingly, our study revealed that various mesenchymal cell types, including CAFs, pericytes, and VSMCs may originate from mesothelial and mesenchymal progenitor cells. Specifically, we found that transdifferentiating cells (i) shared a transcriptional program with differentiated mesothelial cells/CAMCs and overlapped in markers with cells from the CD34- progenitor lineage and (ii) retained mesothelial cell marker expression. Our data suggest that metastatic colonization in the omentum initiates mesothelial cells to undergo MMT, a process previously reported to lead to fibrosis in several organs, including the omentum85,86,87. This process may therefore function as the primary source of CAFs49,88,89. In addition, transcription patterns, velocity vectors, and latent time analysis revealed that CAFs do not emerge from progenitor mesothelial cells but from differentiated SOD2+ mesothelial cells, opposing the hypothesis of a multipotent mesothelial progenitor cell90. Thus, we report on a mesothelial cell hierarchy with ITLN1-progenitor cells differentiating first along the SOD2 gradient into CAMCs, which further de- and transdifferentiate via MMT into various fibroblast populations. Surprisingly, there was no evidence of structural CD34+-progenitor (SWAT cell) contribution to the CAF pool, which also disproportionately disappeared in metastasis. MSPC cells with shared transdifferentiating cell marker expression and thus considered en route to CAF transition were only found within the CD34- lineage in metastatic patients. Thus, a profibrotic SWAT cell fate does not seem to be favored in ovarian cancer91. We also found that the transcription factor RUNX1 is involved in the CAF transition. Elevated RUNX1 gene and protein expression was primarily detected in the omentum and not in the matched primary tumor site of the patients investigated, which notably do not contain mesothelial cells, MSPCs, or adipocytes. The specific expression of RUNX1 in the cancer-associated stroma supports the hypothesis that this transcription factor may promote the transdifferentiation into CAFs. RUNX1’s role in differentiation has been reported for myofibroblast differentiation92 and in inhibition experiments blocking differentiation of lung fibroblasts into myofibroblasts93. Moreover, RUNX1 was also shown to be upregulated in CAFs in breast cancer, where its gene signature correlated with poor outcomes and disease progression94. Its elevated expression in ER⁺ tumors – a feature also reported in ovarian cancer95 – highlights a potential subtype-specific role in tumor–stroma interactions. These findings further support the idea that RUNX1⁺ stromal cells in omental metastases may contribute to tumor progression and therapy resistance in ovarian cancer, potentially through CAF formation in the presence of cancer cells, warranting further functional investigation.
Following on the omentum remodeling capabilities by HGSOC upon colonization, we investigated underlying signaling pathways. CellChatDB provides a broad view of cellular crosstalks and revealed multiple signaling pathways used by tumor cells including the pleiotropic molecules MIF and MDK as well as APP and NECTIN2. We also observed that cells from the TME, in particular mesothelial, endothelial cells, MSPCs, and CAFs mimicked tumor-deployed signaling pathways, which may suggest cancer-derived exosome involvement in tissue reprogramming. Surprisingly, mimicry was not associated with proximity to the metastatic site and tumor cell content as it was lowest in cells residing in the tumor core of the ‘omental metastasis’ subcohort. MIF is known to be increased in various tumor types and supports tumor growth, reduces apoptosis, and exerts immunomodulatory roles96. In contrast to MIF, midkine was predominantly expressed by HGSOC cells making it a highly tumor-specific signaling molecule. It is a small heparin-binding growth factor implicated in supporting tumor growth, angiogenesis, migration, and inflammation (including neutrophil attraction and NETosis) via interaction with multiple receptors97,98. ‘Omental metastasis’ distant-specific agrin/AGRN and heparan sulfate proteoglycan/HSPG pathways also suggested MDK involvement in neuromuscular junction development99 and potential TRAIL-mediated protection from apoptosis100. Thus, MDK-mediated effects may be larger than predicted as these receptor interactions were not included in CellChatDB. In melanoma, midkine was associated with an inflamed but immune-evasive TME that defined poor patient prognosis and resistance to immune checkpoint blockade101. Commonly associated with Alzheimer’s disease, APP is also known to be upregulated in a variety of cancer types where it supports proliferation and motility102. Last, NECTIN2 interaction of tumor, endothelial, and mesothelial cells with TIGIT-expressing T cells identified an alternative checkpoint inhibitor opportunity with potential to remodel the immunosuppressive TME as demonstrated by preclinical and clinical findings103,104. This interaction was also observed in a larger clinical cohort of ovarian cancer covering different disease states with high NECTIN2 expression being associated with poor patient prognosis105. Moreover, for many of these pathways, therapeutic strategies are currently under investigation97,106,107, providing rationale for exploring more targeted and personalized treatments for this deadly disease.
These observations also have important clinical implications with the potential to alter best practices in patient treatment. The extent of residual tumor remaining after surgery is a critical determinant of clinical outcome108. From our findings of a fundamentally altered microenvironment and presence of cancer cells in macroscopically tumor-free distant omentum tissue follows that a reevaluation of the role of the omentum in ovarian cancer is needed. The entire omentum of advanced-stage HGSOC patients should be considered as a continual premetastatic niche that contains reservoirs of micrometastases invisible at the time of debulking surgery ultimately promoting early relapse. Therefore, our findings support evaluation to always remove the total omentum, not only its parts with visible metastases.
Methods
Resources
All consumables and equipment used in the study are listed in the resources (Supplementary Data 4).
Experimental model and subject details
Study cohorts
The study is based on newly generated data and the integration of publicly available datasets. All patients from the University Hospital Basel were female and provided preoperative informed consent to participate in this study (Ethics Committee of Northwest and Central Switzerland, EKNZ; BASEC IDs: 2017-01900 and 2023-00988, to V.H.S.), and also consented to the publication and sharing of de-identified individual-level data. Further details of all patient cohorts are described in more detail below.
Cohort 1: scRNA-seq patients with omentum samples (University Hospital Basel)
This study included 36 samples from 15 adult patients that underwent surgery at the University Hospital Basel, Switzerland.
Clinical information for scRNA-seq profiled patients is summarized in Supplementary Data 1. Patients at the time of surgery ranged from 39 to 89 of age, average 64.9 years. The ‘non-metastatic omentum’ subcohort included three endometrioid uterine cancer patients, two patients with uterus myomatosus, one patient with a cystadenofibroma of the ovary and one benign omentum. Samples in this subcohort were collected from various sites (Fig. 1a, b, Supplementary Fig. 1a, and Supplementary Data 1). Additionally, two serous ovarian borderline tumors (low-malignant potential) were included. The ‘omental metastasis’ subcohort or malignant omentum cohort included HGSOC from the ovaries (HGSOC, 3 patients) or fallopian tube (HGSTC, 3 patients). Samples in this cohort were collected from multiple sites (Fig. 1a, b, Supplementary Fig. 1a and Supplementary Data 1).
Cohort 2: scRNA-seq chemo-naive patients
Publicly available single-cell RNA-seq data21 were downloaded from Synapse (accession: syn33521743). Cells were re-annotated using scROSHI24, applying our in-house gene signature matrix derived from our own cohort to classify relevant cell types. To assess the impact of tissue region on cell type composition and gene expression, we employed a generalized linear model (GLM), following the approach described in the original study.
Cohort 3: Matched primary and omentum metastasis cohort (University Hospital Basel)
The study also includes a newly generated cohort (Personalized Health and Related Technologies; PHRT) consisting of HGSOC patient-derived samples and associated data included in this study fully adhered to ethical standards. A total of 109 samples derived from 47 high-grade serous ovarian cancer patients were processed. These samples represented diverse tissue types. Patients’ clinical characteristics are provided in Supplementary Data 5.
Cohort 4: Chemo-naive HGSOC profiled by bulk RNA-seq
A retrospective cohort of FFPE paired primary and metastatic tumor samples from chemo-naive HGSOC FIGO stage III–IV who underwent primary debulking surgery (2008–2014, University Hospital Hradec Kralove, Czech Republic) were analyzed109. The study was approved by the local ethics committee (201607S14P). Cohort-derived data served as an independent validation set to confirm key findings related to cancer-associated fibroblasts.
Cohort 5: Chemo-naive HGSOC profiled by long read scRNA-seq (University Hospital Basel)
This cohort comprises previously published long-read scRNA-seq data from human omental metastases47. Omental tissues were collected intraoperatively and promptly transferred on ice for dissociation. Raw sequencing files, along with associated analysis files (isoform GFF file, reads mapped to isoforms, and GENCODE annotation of novel isoforms), are available through the European Genome-phenome Archive (EGA) under accession number EGAS00001006807.
Methods
Sample collection, tissue dissociation, and processing
Tissue samples were immediately collected at the time of surgery and transferred on ice to the Department of Biomedicine, University Hospital Basel for tissue processing. Tissues were cut into small pieces and dissociated in digestion solution (1 mg/mL collagenase/Dispase [Sigma-Aldrich, cat. no. 10269638001], 1 unit/mL DNase I [NEB, cat. no. M0303], 10% fetal bovine serum (FBS) in DMEM [Sigma-Aldrich, cat. no. D8437-500mL]) for 30 min at 37 °C. The omentum is a VAT primarily composed of adipocytes. To focus on the non-adipose cell fraction, adipocytes were separated by centrifugation and the cell pellet was collected (Supplementary Fig. 1A). Red blood cell lysis (RBC) was performed using MACS red blood cell lysis solution (cat. no. 130-094-183). Subsequently, the cell pellet was resuspended into MACS dead cell removal microbeads (cat. no. 130-090-101) and was loaded into the autoMACS Pro Separator (Miltenyi Biotec, cat. no. 130-092-545) to remove dead cells. Cells were counted and resuspended in PBS supplemented with 1% bovine serum albumin (BSA, Sigma-Aldrich, cat. no. F7524) and transferred to the Genomics Facility Basel for immediate single-cell sequencing processing. Cells were collected (350 g, 5 min), resuspended in 1 mL washing buffer consisting of PBS (Ca2+/Mg2+-free, Gibco, cat. no. 10010-015) and 1% BSA (Sigma-Aldrich, cat. no. A7906) and sequentially filtered through 100 µm (Falcon, cat. no. 352360) and 40 µm (Falcon, cat. no. 352340) cell strainers to remove cell clumps. If the cell pellet appeared still red, RBC lysis was performed following the manufacturer’s instructions (Roche, cat. no. 11814389001). Apoptotic and dead cells were removed by immunomagnetic cell separation using the Annexin Dead Cell Removal Kit (StemCell Technologies, cat. no. 17899) and EasySepTM Magnet (StemCell Technologies, cat. no. 18000). Whenever required, dead cell removal was repeated to increase cell viability. Next, cells were washed with a resuspension buffer (PBS with 0.05% BSA), spun down and resuspended in a resuspension buffer. Cell numbers and viability were assessed at each step on a Cellometer K2 Image Cytometer (Nexcelom Bioscience, cat. no. Cellometer K2) using ViaStain AOPI Staining Solution (Nexcelom Bioscience, cat. no. CS2-0106-5mL) and PD100 cell counting slides (Nexcelom Bioscience, cat. no. CHT4-PD100-003). Fresh sample processing resulted in overall high sample quality with an average 88.3% cell viability (final count before droplet encapsulation and scRNA-seq processing - Supplementary Fig. 1B, Supplementary Data 1 and 2).
Single-cell RNA sequencing
Optimal cell concentrations were set according to 10x Genomics protocols (700–1200 cells/µL). Cells were loaded and processed using the 10x Genomics Chromium platform with the Next GEM Single Cell 3’ Reagent Kit v3.1 on 10x Genomics Chromium Single Cell Controller (10x Genomics, cat. no. PN-120263). 4500 cells were targeted per sample. Gene expression (GEX) libraries were amplified and the quality of cDNA traces and sequencing libraries was evaluated on a 5200 Fragment Analyzer System (Agilent Technologies). Libraries were pooled and sequenced on the Illumina NovaSeq 6000 platform with addition of 1% PhiX at recommended sequencing depth (30,000–50,000 reads/cell, Supplementary Data 1 and 2).
SO1 mouse tumor models
Female C57BL/6 mice (8–12 weeks old) were obtained from the Institute of Microbiology, Czech Academy of Sciences (Prague, Czech Republic) and housed in individually ventilated cages. Mice had ad libitum access to irradiated Teklad Global 18% Protein Rodent Diet and sterile prefilled water bottles. Environmental conditions were maintained at 21 ± 2 °C, 50 ± 10% humidity, with a 12 h light/dark cycle. Animals were randomly assigned to experimental groups; blinding was not applied.
Murine ovarian cancer Trp53-/-, BRCA1-/-, Myc, Hras SO1 cells (kindly provided by Dr. Sandra Orsulic, UCLA) were cultured in DMEM (Sigma) supplemented with 10% fetal bovine serum (FBS; Gibco), 100 U/mL penicillin, and 100 µg/mL streptomycin (Gibco) at 37 °C with 5% CO₂. Mycoplasma contamination was routinely ruled out using the LookOut® PCR Detection Kit (Millipore Sigma). For tumor induction, 1 × 10⁶ SO1 cells were mixed 1:1 with cold Cultrex BME (Biotechne) and injected intraperitoneally into the fat pad near the ovary. Mice were regularly monitored for disease progression and toxicity (e.g., >20% weight loss, lethargy, furruffling). Euthanasia was performed upon reaching humane endpoints, including >50% weight gain due to ascites or signs of distress. All procedures were approved by the Institutional Animal Care and Use Committee of the Czech Academy of Sciences and conducted in accordance with local and EU guidelines.
Flow cytometry analysis of murine samples
Fresh tumor specimens were processed to obtain viable mononuclear cells. On day 20, CD8⁺ T-cell phenotypes were characterized using a combination of fluorescently labeled extracellular and intracellular antibodies, together with matching isotype controls. Staining was performed for 20 min at 4 °C in the dark, followed by washing and data acquisition on a Fortessa flow cytometer (BD Biosciences). The resulting data were analyzed using FlowJo software (BD Biosciences).
Immunofluorescence
After deparaffinization, heat-induced (98 °C) antigen retrieval was performed with a citrate buffer (pH 6.0) (Sigma-Alrich, cat. no. C9999), and slides were incubated with Triton X-100 (Roth, cat. no. 3051.3) 0.25% in PBS for 5 min, washed in PBS for another 5 min followed by incubation with blocking solution (5% FBS with 1% BSA in PBS 1% Triton-X 100) for 1 h. After blocking, samples were incubated with one of the listed primary antibodies (Supplementary Data 4) diluted in 1% BSA 0.1% Triton-X 100 in PBS and incubated overnight at 4 °C. Primary antibodies were detected using goat anti-mouse or anti-rabbit IgG (H + L) Alexa fluor 488 or 647 (Cell signaling Technology, Bioconcept) secondary antibodies diluted 1:500 in 1% BSA 0.1% Triton-X 100 in PBS for 3 h in the dark at room temperature. Slides were mounted using ProLong® Gold antifade reagent (Cell Signaling Technology, Bioconcept) and a coverslip. Images were taken using the confocal microscope Nikon CSU-W1, analyzed with Image J (2.3.0/1.53q) and Qupath (0.3.0) software, and developed scripts for cell detection and annotations. Further analyses were performed by R/Bioconductor.
Flow cytometry, isolation, and culture of CD200+/CD45- mesothelial cells
Primary mesothelial cells were isolated from tumor-free omentum of ‘non-metastasized omentum’ patients by fluorescence-activated cell sorting. Briefly, omentum tissues were cut into small pieces and digested with collagenase/dispase as previously described to create a single-cell suspension. After the removal of adipocytes and red blood cells, cells were stained with anti-human CD45 antibody conjugated with FITC (Biolegend, cat. no. 304006) and anti-human CD200 antibody conjugated with PE/Cy7 (Biolegend, cat. no. 329212) for 1 h on ice in the dark and subsequently stained with DAPI to measure dead cells. The cells were analyzed using the CytoFLEX Flow Cytometer (Beckman Colter) or sorted for DAPI-/CD45-/CD200+ (mesothelial) and DAPI-/CD45-/CD200- (stromal-like) cells using the BD FACSAria Cell Sorter (BD Bioscience). FlowJo v10 (Becton Dickinson) software was used for further analysis of the flow cytometry data.
Exposure of isolated mesothelial cells to ascites fluid followed by RT-qPCR analysis
DAPI-/CD45-/CD200+ mesothelial (CD45-/CD200+) and stromal (CD45-/CD200-) cells were cultured in DMEM/F-12 (Sigma-Alrich, cat. no. D8062) supplemented with 20% FBS (Sigma-Alrich, cat. no. F7524), 1% of antibiotic antimycotic solution (Sigma-Alrich, cat. no. A5955) at 37 °C in a 95% humidified atmosphere containing 5% CO2. After in vitro expansion, the mesothelial cell population was verified by cell morphology and immunoblotting with anti-CD200 (Cell Signaling Technology, cat. no. 23451) and anti-ZO-1 (Cell signaling Technology, cat. no. 8193) antibodies. For ascites exposure experiment, three primary mesothelial populations isolated from three benign patients’ omentum were used and had undergone less than three passages after sorting. 2:1 mixture of cell-free ascites collected from patients with ovarian cancer and culture medium described above was added to the mesothelial population. After 4 days of culture, RNA was extracted using the ReliaPrep RNA Cell Miniprep System (Promega, cat. no. A2891) and was reverse transcribed using the iScript Reverse Transcription Supermix for reverse transcription-quantitative polymerase chain reaction (RT-qPCR - Bio-Rad Laboratories, cat. no. 1708841 A). RT-qPCR was performed on CXCL1 (Forward 5’-AGT GGC ACT GCT GCT CCT-3’, Reverse 5’-TGG ATG TTC TTG GGG TGA AT-3’), CXCL2 (Forward 5’-CGC CCA AAC CGA AGT CAT AG-3’, Reverse 5’-AGA CAA GCT TTC TGC CCA TTC T-3’), and reference genes SDHA (Forward 5’-TGG GAA CAA GAG GGC ATC TG-3’, Reverse 5’-CCA CCA CTG CAT CAA ATT CAT G-3’) on a QuantStudio™ 5 Real-Time PCR System (ThermoFisher Scientific).
Bulk RNA sequencing (PHRT)
Biobanked RNAlater (Thermo Fisher Scientific, cat. No. AM7021) samples were homogenized, and an average weight aliquot of 54 mg was used for total RNA isolation. RNA extraction was performed using the RNeasy Plus Universal Mini Kit (50) (Qiagen, cat. no. 73404), incorporating PhasemakerTM Tubes (Thermo Fisher Scientific, cat. No. A33248). RNA sample quantification was performed using an Invitrogen Qubit 4 fluorometer with the Invitrogen Qubit RNA BR Assay Kit, while RNA sample integrity was assessed using an Agilent Fragment Analyzer with the RNA Kit. The mean RIN of the entire cohort was 8.6 ranging from 6.2 to 10. RNA-seq libraries were prepared using 200 ng of total RNA and the Illumina TruSeq Stranded mRNA Library Prep kit, following the manufacturer’s protocol. Library quality control and quantification were performed using an Invitrogen Qubit 4 fluorometer with the Invitrogen Qubit dsDNA 1X HS Assay kit for quantification, and an Agilent Fragment Analyzer with the Agilent NGS Fragment Kit for quality control. Sequencing was performed on an Illumina NovaSeq 6000 platform, generating 100 bp paired-end reads (Health 2030 Genome Center, Geneva, Switzerland). RNA-seq data were processed and analyzed using the nf-core/rnaseq pipeline (version 3.8.1)110. The pipeline encompassed several key steps, beginning with initial quality assessment and adapter trimming of raw reads. Processed reads were then mapped to the GRCh38 primary assembly (GRCh38.primary_assembly.genome.fa, obtained from the Gencode website) using STAR aligner111. The alignment was guided by the Gencode v41 primary annotation (gencode.v41.primary_assembly.annotation.gtf). Following alignment, Salmon112 was employed to quantify transcript abundances, with the --rf-stranded (reverse/forward) parameter specified to account for the stranded nature of the TruSeq library preparation, ensuring correct strand-specific quantification of transcripts. Throughout the pipeline, several quality control measures were implemented. These included FastQC for assessment of sequence quality metrics, featureCounts for quantification of reads mapped to different genomic features and biotypes (e.g., protein-coding genes, lncRNAs, miRNAs), and DESeq2 for Principal Component Analysis (PCA) and sample similarity analysis to identify potential batch effects or outliers. Additionally, Picard MarkDuplicates was used to identify PCR and optical duplicates (duplicates were marked but not removed), Samtools generated alignment statistics and quality metrics, and RSeQC provided comprehensive quality control of alignment results, including read distribution, gene body coverage, and splice junction analysis. The nf-core/rnaseq pipeline ensured standardized and reproducible processing of all samples.
Proteomics using mass spectrometry (PHRT)
Samples for proteotyping were prepared using the PreOmics iST kit (PreOmics GmbH, Martinsried, Germany, cat. no. P.O.00027). Cells underwent lysis, digestion, and cleanup in accordance with the manufacturer’s guidelines. An additional sonication step, consisting of three 30 s pulses using a VialTweeter (Dr. Hielscher, Tottweil, Germany), supported the lysis. The samples were then resuspended to a concentration of 1 μg/μL for mass spectrometry (MS) measurements. Analysis was performed on a Q Exactive HF-X mass spectrometer (Thermo Fisher Scientific, Reinach) equipped with an Easy-nLC 1200 (Thermo Fisher Scientific, Reinach, Switzerland).
Peptide separation occurred on a C18 50 cm EASY-Spray™ HPLC column (Thermo Fisher Scientific, 2 µm, 100 Å, 75 µm i.d., cat. no. ES903). The mobile phase A was HPLC-grade water with 0.1% formic acid, while mobile phase B comprised 80% HPLC-grade acetonitrile with HPLC-grade water and 0.1% (v/v) formic acid. Peptides were eluted at a flow rate of 200 nL/min using a non-linear gradient from 4 to 52% mobile phase B over 117 min.
For data-independent acquisition (DIA) on the HF-X, the isolation windows were set to 15 m/z, covering a mass range of m/z 400–1210. A total of 54 DIA scan windows were recorded at a resolution of 30,000 with an automatic gain control (AGC) target value of 1e6113,114. Fragmentation by high-energy collisional dissociation was set to 28% normalized collision. Full MS spectra were recorded at a resolution of 120,000 with a normalized AGC target of 3e6 and maximum injection time of 50 ms.
Data were analyzed using Spectronaut v15 (Biognosys, Schlieren, Switzerland). MS1 values were used for quantification with a peptide and a protein Qvalue cut-off of 0.01. Quantitation was set to mean, Qvalue identified without missing value imputation. Data were searched against a Uniprot human library (release January 2022) together with standards and common contaminants in standard DIA with a sample-specific spectral library generated in house using ph-fractionated samples from OVCAR3, SKOV3 and CD3, CD14 and CD138 pooled and sorted primary cells supplemented with a library generated via Pulsar v15 (Biognosys) from the PHRT HGSOC sample DIA files.
Quantification and statistical analysis
In the following, we briefly describe the overall steps of the sequencing data analysis. The main processing steps are part of the scRNA-seq analysis workflow scAmpi115, enriched with in-house information on selected pathways and marker genes used for cell type classification.
scRNA-seq read mapping and quality control
Raw reads were mapped to the GRCh38 reference genome using 10x Genomics Cell Ranger 6.0.1 to infer read counts per gene per cell. Since samples were sequenced on the NovaSeq platform, we performed index-hopping removal using a method developed by Griffiths et al. 116. After the mapping, the resulting UMI counts were quality controlled and cells and genes filtered to remove known contaminants and potential doublets. Doublet detection was performed using the tool scDblFinder117. Cells were removed if more than 50% of their reads mapped to mitochondrial genes or if they expressed fewer than 400 different genes. Genes were removed if they were not protein-coding or if they were expressed in less than 20 cells. Subsequently, counts were normalized and corrected for cell-cycle effects and library size using sctransform118.
Unsupervised clustering and cell type annotation
Similar cells were grouped based on unsupervised clustering using Phenograph119. An automated cell type classification was performed independently for each cell using scROSHI24. Cell type annotation was performed using lists of cell type-defining, highly expressed marker genes. Final cluster-based cell type annotation refinement was performed manually using literature-defined markers. Marker genes were selected from previous publications120 and were based on in-house knowledge on expected tumor markers. Literature-based marker lists were curated to only select genes that are actually expressed in the cohort. All cell type marker gene lists are included on GitHub (refer to Section Data availability).
Pathway analysis and visualization
Based on the gene expression profile of each cell, a gene set analysis was performed using GSVA121 to identify pathways with increased or decreased expression. Pathway analysis as well as gene expression results were visualized using Uniform Manifold Approximation and Projection (UMAP)122.
Cohort analysis
For a combined analysis and visualization, all samples were pooled across experiments, followed by sctransform normalization of the raw counts to account for library size and additionally including “patient” as a co-variable to the model, to account for the expected patient-specific batch effect. Subsequently, pathway analysis and gene expression analysis and visualization were performed as described above.
scVelocity analysis
We performed a velocity analysis using a combination of Velocyto123 and scVelo124. Briefly, we used Velocyto to retrieve the spliced and unspliced read counts, then we used scVelo to calculate the velocity vectors, velocity streams, and latent time. Results include the velocity stream correction obtained by the differential kinetic analysis used to detect overlapping velocity patterns in the genes. To allow a detailed investigation of the kinetics of individual cell types, we repeated the velocity analysis on subsetted data, by removing either the mesothelial cells or MSPCs, respectively.
Cell-cell-communication analysis using CellChat
We investigated the cell-cell communication network between cell types in each tissue region using CellChat56. To concentrate on sufficiently represented cell types, we included in the analysis cell types accounting for at least 0.5% of the total amount of cells. We performed the analysis including the complete human interaction database provided by CellChat. The pairwise comparison between communication networks of different tissue regions is based on the communication probability. Aggregation of modulated pathways throughout pairwise comparisons was performed using custom scripts. P-value correction during aggregation was performed using Bonferroni correction.
Differential expression
We performed gene-level differential expression analyses using Seurat FindMarkers125. We visualized the top 5 genes for each possible pairwise comparison between the available conditions.
InferCNV analysis
To infer large-scale chromosomal copy number variations (CNVs) in malignant cells, we used the inferCNV R package (v1.20). The input consisted of raw counts derived from the omentum single-cell RNA-seq dataset. Cell annotations, distinguishing malignant from non-malignant cell types, were generated using scROSHI24. The gene order was provided based on genomic coordinates from the hg38 reference genome. An infercnv object was created using CreateInfercnvObject(), without specifying reference groups (ref_group_names = NULL) to allow full CNV inference across the tumor cell population. The analysis was run using the run() function with parameters optimized for 10x Genomics data (cutoff = 0.1), and additional options for denoising (denoise = TRUE), clustering by groups, and HMM-based CNV prediction (HMM = TRUE). Diagnostic plots were also generated during the run (diagnostics = TRUE). The resulting CNV profiles were visualized using a customized heatmap function, which includes enhanced row annotations. Gains and losses in chromosomal regions are displayed in red and blue, respectively, reflecting relative expression shifts inferred to result from underlying copy number changes.
Mapping CD8 + T cell functional states across ovarian tumor regions
To characterize intratumoral CD8 + T cell states, single-cell RNA-seq data were preprocessed and integrated using Seurat and Harmony for batch correction. CD8 + T cells were identified based on annotation with SingleR using the BlueprintEncodeData reference and subsequently isolated for downstream analysis. Functional phenotypes of CD8 + T cells were inferred using ProjecTILs with the CD8T_human_ref_v1 reference, allowing classification into defined states including central memory (CM), effector memory (EM), TEMRA, progenitor exhausted (Tpex), exhausted (Tex), and naïve-like cells. State-specific distributions across anatomical sites (benign, distant, peritumoral, and tumor-associated omentum) were visualized using UMAP and projected into the reference atlas space. Cell state composition per region was summarized using stacked barplots and alluvial plots generated with ggplot2. To complement single-cell analyses, RUNX1 expression was compared across primary tumors and matched omental metastases and the expression difference was evaluated using the Wilcoxon test and visualized alongside deconvoluted macrophage proportions.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Single-cell RNA-seq data generated from human omentum tissue samples at the time of diagnosis (Cohort 1) have been deposited in GEO under accession code GSE286057. Publicly available single-cell RNA-seq data (Cohort 2)21 were obtained from Synapse under accession code syn33521743. Cohort 3 data are available from GEO under accession code GSE289067 (bulk RNA-seq). Raw bulk RNA-seq data are available under controlled access through EGA (accession: https://ega-archive.org/studies/EGAS50000001424), governed by the Ovarian Cancer Data Access Committee (DAC: EGAC00001003014). Matched proteomics data are available from MassIVE under accession code MSV000096327 (proteomics, [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE289067] and from MassIVE under accession code MSV000096327 [https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=4e2f1efaea2e42ad91b21850f5a8b490]). Bulk RNA-seq data (Cohort 4) for RUNX1 expression and deconvolution were derived from Lanickova et al. 109. Cohort 5 raw sequencing files and associated analysis files (isoform GFF file, reads associated with isoforms, and the Gencode annotation of novel isoforms) from long-read scRNA-seq47 are available through the EGA under accession code EGAS00001006807. All other data supporting the findings of this study are available within the article, its Supplementary Information, or the Source Data file. Source data are provided with this paper.
Code availability
The scAmpi workflow for GEX analysis is available on GitHub: https://github.com/ETH-NEXUS/scAmpi_single_cell_RNA. Customized scripts for the velocity, CCC, subtyping, and DE analysis, as well as the cell type marker lists are available on GitHub: https://github.com/ETH-NEXUS/single-cell_omentum_analysis/.
References
Platell, C., Cooper, D., Papadimitriou, J. M. & Hall, J. C. The omentum. World J. Gastroenterol. 6, 169–176 (2000).
Ronti, T., Lupattelli, G. & Mannarino, E. The endocrine function of adipose tissue: an update. Clin. Endocrinol. 64, 355–365 (2006).
Vijay, J. et al. Single-cell analysis of human adipose tissue identifies depot and disease specific cell types. Nat. Metab. 2, 97–109 (2020).
Yeung, T.-L. et al. Cellular and molecular processes in ovarian cancer metastasis. a review in the theme: cell and molecular processes in cancer metastasis. Am. J. Physiol., Cell Physiol. 309, C444–C456 (2015).
Meza-Perez, S. & Randall, T. D. Immunological functions of the omentum. Trends Immunol. 38, 526–536 (2017).
Veneziani, A. C. et al. Heterogeneity and treatment landscape of ovarian carcinoma. Nat. Rev. Clin. Oncol. 20, 820–842 (2023).
Matulonis, U. A. et al. Ovarian cancer. Nat. Rev. Dis. Prim. 2, 16061 (2016).
Siegel, R. L., Miller, K. D., Fuchs, H. E. & Jemal, A. Cancer statistics, 2021. CA Cancer J. Clin. 71, 7–33 (2021).
Pradeep, S. et al. Hematogenous metastasis of ovarian cancer: rethinking mode of spread. Cancer Cell 26, 77–91 (2014).
Harter, P. et al. Pattern and clinical predictors of lymph node metastases in epithelial ovarian cancer. Int. J. Gynecol. Cancer 17, 1238–1244 (2007).
Lee, W. et al. Neutrophils facilitate ovarian cancer premetastatic niche formation in the omentum. J. Exp. Med. 216, 176–194 (2019).
Krishnan, V. et al. Omental macrophages secrete chemokine ligands that promote ovarian cancer colonization of the omentum via CCR1. Commun. Biol. 3, 524 (2020).
Etzerodt, A. et al. Tissue-resident macrophages in omentum promote metastatic spread of ovarian cancer. J. Exp. Med. 217, e20191869 (2020).
Nieman, K. M. et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 17, 1498–1503 (2011).
Emont, M. P. et al. A single-cell atlas of human and mouse white adipose tissue. Nature 603, 926–933 (2022).
Norreen-Thorsen, M. et al. A human adipose tissue cell-type transcriptome atlas. Cell Rep. 40, 111046 (2022).
Ferrero, R., Rainer, P. & Deplancke, B. Toward a consensus view of mammalian adipocyte stem and progenitor cell heterogeneity. Trends Cell Biol. 30, 937–950 (2020).
Herrick, S. E. & Mutsaers, S. E. Mesothelial progenitor cells and their potential in tissue engineering. Int. J. Biochem. Cell Biol. 36, 621–642 (2004).
Liu, Y. et al. Transition of mesothelial cell to fibroblast in peritoneal dialysis: EMT, stem cell or bystander? Perit. Dial. Int. 35, 14–25 (2015).
Deng, Y. et al. Single-Cell RNA-sequencing atlas reveals the tumor microenvironment of metastatic high-grade serous ovarian carcinoma. Front. Immunol. 13, 923194 (2022).
Vázquez-García, I. et al. Ovarian cancer mutational processes drive site-specific immune evasion. Nature 612, 778–786 (2022).
Hornburg, M. et al. Single-cell dissection of cellular components and interactions shaping the tumor immune phenotypes in ovarian cancer. Cancer Cell 39, 928–944 (2021).
Izar, B. et al. A single-cell landscape of high-grade serous ovarian cancer. Nat. Med. 26, 1271–1279 (2020).
Prummer, M. et al. scROSHI: robust supervised hierarchical identification of single cells. NAR Genom. Bioinform. 5, lqad058 (2023).
Brooks, C. R., van Dalen, C. J., Hermans, I. F. & Douwes, J. Identifying leukocyte populations in fresh and cryopreserved sputum using flow cytometry. Cytom. B Clin. Cytom. 84, 104–113 (2013).
Jiménez-Sánchez, A. et al. Heterogeneous tumor-immune microenvironments among differentially growing metastases in an ovarian cancer patient. Cell 170, 927–938 (2017).
van der Leun, A. M., Thommen, D. S. & Schumacher, T. N. CD8 + T cell states in human cancer: insights from single-cell analysis. Nat. Rev. Cancer 20, 218–232 (2020).
Castellanos-Rueda, R., Di Roberto, R. B., Schlatter, F. S. & Reddy, S. T. Leveraging single-cell sequencing for chimeric antigen receptor T cell therapies. Trends Biotechnol. 39, 1308–1320 (2021).
Szabo, P. A. et al. Single-cell transcriptomics of human T cells reveals tissue and activation signatures in health and disease. Nat. Commun. 10, 4706 (2019).
Chtanova, T. et al. T follicular helper cells express a distinctive transcriptional profile, reflecting their role as non-Th1/Th2 effector cells that provide help for B cells. J. Immunol. 173, 68–78 (2004).
Saravia, J., Chapman, N. M. & Chi, H. Helper T cell differentiation. Cell. Mol. Immunol. 16, 634–643 (2019).
Sharonov, G. V., Serebrovskaya, E. O., Yuzhakova, D. V., Britanova, O. V. & Chudakov, D. M. B cells, plasma cells and antibody repertoires in the tumour microenvironment. Nat. Rev. Immunol. 20, 294–307 (2020).
Pende, D. et al. Killer Ig-like receptors (KIRs): their role in NK cell modulation and developments leading to their clinical exploitation. Front. Immunol. 10, 1179 (2019).
Khan, M., Arooj, S. & Wang, H. NK cell-based immune checkpoint inhibition. Front. Immunol. 11, 167 (2020).
Victora, G. D. et al. Identification of human germinal center light and dark zone cells and their relationship to human B-cell lymphomas. Blood 120, 2240–2248 (2012).
Nutt, S. L., Hodgkin, P. D., Tarlinton, D. M. & Corcoran, L. M. The generation of antibody-secreting plasma cells. Nat. Rev. Immunol. 15, 160–171 (2015).
Cheng, S. et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell 184, 792–809 (2021).
Veglia, F., Sanseviero, E. & Gabrilovich, D. I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 21, 485–498 (2021).
Ma, R.-Y., Black, A. & Qian, B.-Z. Macrophage diversity in cancer revisited in the era of single-cell omics. Trends Immunol. 43, 546–563 (2022).
Zhou, B., Lawrence, T. & Liang, Y. The role of plasmacytoid dendritic cells in cancers. Front. Immunol. 12, 749190 (2021).
Trim, W. V. & Lynch, L. Immune and non-immune functions of adipose tissue leukocytes. Nat. Rev. Immunol. 22, 371–386 (2022).
Thiam, H. R., Wong, S. L., Wagner, D. D. & Waterman, C. M. Cellular mechanisms of netosis. Annu. Rev. Cell Dev. Biol. 36, 191–218 (2020).
Mei, S. et al. Single-cell and spatial transcriptomics reveal a tumor-associated macrophage subpopulation that mediates prostate cancer progression and metastasis. Mol. Cancer Res. https://doi.org/10.1158/1541-7786.MCR-24-0791. (2025)
Dong, L. et al. SPP1 + TAM regulates the metastatic colonization of CXCR4+ metastasis-associated tumor cells by remodeling the lymph node microenvironment. Adv. Sci. 11, e2400524 (2024).
Kale, S. et al. Osteopontin signaling upregulates cyclooxygenase-2 expression in tumor-associated macrophages leading to enhanced angiogenesis and melanoma growth via α9β1 integrin. Oncogene 34, 5408–5410 (2015).
Kahles, F., Findeisen, H. M. & Bruemmer, D. Osteopontin: a novel regulator at the cross roads of inflammation, obesity and diabetes. Mol. Metab. 3, 384–393 (2014).
Dondi, A. et al. Detection of isoforms and genomic alterations by high-throughput full-length single-cell RNA sequencing in ovarian cancer. Nat. Commun. 14, 7780 (2023).
Lavie, D., Ben-Shmuel, A., Erez, N. & Scherz-Shouval, R. Cancer-associated fibroblasts in the single-cell era. Nat. Cancer 3, 793–807 (2022).
Huang, H. et al. Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer. Cancer Cell 40, 656–673 (2022).
Dai, H., Wang, L., Li, L., Huang, Z. & Ye, L. Metallothionein 1: a new spotlight on inflammatory diseases. Front. Immunol. 12, 739918 (2021).
Pozzi, A., Yurchenco, P. D. & Iozzo, R. V. The nature and biology of basement membranes. Matrix Biol. 57–58, 1–11 (2017).
Yuan, W. et al. Reduced APPL1 impairs osteogenic differentiation of mesenchymal stem cells by facilitating MGP expression to disrupt the BMP2 pathway in osteoporosis. J. Biol. Chem. 299, 104823 (2023).
Raajendiran, A. et al. Identification of metabolically distinct adipocyte progenitor cells in human adipose tissues. Cell Rep. 27, 1528–1540 (2019).
Davidson, S. et al. Fibroblasts as immune regulators in infection, inflammation and cancer. Nat. Rev. Immunol. 21, 704–717 (2021).
Ferrero, R. et al. A human omentum-specific mesothelial-like stromal population inhibits adipogenesis through IGFBP2 secretion. Cell Metab. 36, 1566–1585 (2024).
Jin, S. et al. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 12, 1088 (2021).
Shidham, V. B. The panorama of different faces of mesothelial cells. Cytojournal 18, 31 (2021).
Westcott, G. P. et al. Mesothelial cells are not a source of adipocytes in mice. Cell Rep. 36, 109388 (2021).
Lansley, S. M. et al. Mesothelial cell differentiation into osteoblast- and adipocyte-like cells. J. Cell. Mol. Med. 15, 2095–2105 (2011).
Zhao, A. et al. Omentin-1: a newly discovered warrior against metabolic related diseases. Expert Opin. Ther. Targets 26, 275–289 (2022).
Paval, D. R., Di Virgilio, T. G., Skipworth, R. J. E. & Gallagher, I. J. The emerging role of intelectin-1 in cancer. Front. Oncol. 12, 767859 (2022).
Au-Yeung, C.-L. et al. ITLN1 modulates invasive potential and metabolic reprogramming of ovarian cancer cells in omental microenvironment. Nat. Commun. 11, 3546 (2020).
Zhang, Y. et al. Omentin-1 induces mechanically activated fibroblasts lipogenic differentiation through pkm2/yap/pparγ pathway to promote lung fibrosis resolution. Cell. Mol. Life Sci. 80, 308 (2023).
Maniyadath, B., Zhang, Q., Gupta, R. K. & Mandrup, S. Adipose tissue at single-cell resolution. Cell Metab. 35, 386–413 (2023).
Merrick, D. et al. Identification of a mesenchymal progenitor cell hierarchy in adipose tissue. Science 364, eaav251 (2019).
Fuchs, E. & Blau, H. M. Tissue stem cells: architects of their niches. Cell Stem Cell 27, 532–556 (2020).
Panciera, T., Azzolin, L., Cordenonsi, M. & Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 18, 758–770 (2017).
Schwalie, P. C. et al. A stromal cell population that inhibits adipogenesis in mammalian fat depots. Nature 559, 103–108 (2018).
Palani, N. P. et al. Adipogenic and SWAT cells separate from a common progenitor in human brown and white adipose depots. Nat. Metab. 5, 996–1013 (2023).
Duregotti, E. et al. Reduced secretion of neuronal growth regulator 1 contributes to impaired adipose-neuronal crosstalk in obesity. Nat. Commun. 13, 7269 (2022).
Itabe, H., Yamaguchi, T., Nimura, S. & Sasabe, N. Perilipins: a diversity of intracellular lipid droplet proteins. Lipids Health Dis. 16, 83 (2017).
Gao, Z., Daquinag, A. C., Su, F., Snyder, B. & Kolonin, M. G. PDGFRα/PDGFRβ signaling balance modulates progenitor cell differentiation into white and beige adipocytes. Development 145, dev155861 (2018).
So, J. et al. Robust single nucleus RNA sequencing reveals depot-specific cell population dynamics in adipose tissue remodeling during obesity. BioRxiv https://doi.org/10.1101/2024.04.08.588525. (2024)
Jackson-Jones, L. H. et al. Stromal cells covering omental fat-associated lymphoid clusters trigger formation of neutrophil aggregates to capture peritoneal contaminants. Immunity 52, 700–715 (2020).
Yang, J. H., Shi, Y. F., Chen, X. D. & Qi, W. J. The influence of aquaporin-1 and microvessel density on ovarian carcinogenesis and ascites formation. Int. J. Gynecol. Cancer 16, 400–405 (2006).
Ji, H.-L. & Nie, H.-G. Electrolyte and fluid transport in mesothelial cells. J. Epithel. Biol. Pharmacol. 1, 1–7 (2008).
Coulis, G. et al. Single-cell and spatial transcriptomics identify a macrophage population associated with skeletal muscle fibrosis. BioRxiv https://doi.org/10.1101/2023.04.18.537253. (2023)
Yang, Y., Yang, Y., Yang, J., Zhao, X. & Wei, X. Tumor microenvironment in ovarian cancer: function and therapeutic strategy. Front. Cell Dev. Biol. 8, 758 (2020).
Chen, Q. et al. An osteopontin-integrin interaction plays a critical role in directing adipogenesis and osteogenesis by mesenchymal stem cells. Stem Cells 32, 327–337 (2014).
Moreno-Viedma, V. et al. Osteopontin-deficient progenitor cells display enhanced differentiation to adipocytes. Obes. Res. Clin. Pract. 12, 277–285 (2018).
Zeyda, M. et al. Osteopontin is an activator of human adipose tissue macrophages and directly affects adipocyte function. Endocrinology 152, 2219–2227 (2011).
Hoeft, K. et al. Platelet-instructed SPP1+ macrophages drive myofibroblast activation in fibrosis in a CXCL4-dependent manner. Cell Rep. 42, 112131 (2023).
Huang, R. et al. SPP1 derived from silica-exposed macrophage exosomes triggers fibroblast transdifferentiation. Toxicol. Appl. Pharmacol. 422, 115559 (2021).
Dai, J. et al. Osteopontin induces angiogenesis through activation of PI3K/AKT and ERK1/2 in endothelial cells. Oncogene 28, 3412–3422 (2009).
Pascual-Antón, L. et al. Mesothelial-to-mesenchymal transition and exosomes in peritoneal metastasis of ovarian cancer. Int. J. Mol. Sci. 22, 11496 (2021).
Wang, J. et al. Fibrosis of mesothelial cell-induced peritoneal implantation of ovarian cancer cells. Cancer Manag. Res. 10, 6641–6647 (2018).
Mutsaers, S. E. et al. Mesothelial cells in tissue repair and fibrosis. Front. Pharmacol. 6, 113 (2015).
Rynne-Vidal, A., Jiménez-Heffernan, J. A., Fernández-Chacón, C., López-Cabrera, M. & Sandoval, P. The mesothelial origin of carcinoma associated-fibroblasts in peritoneal metastasis. Cancers 7, 1994–2011 (2015).
Sandoval, P. et al. Carcinoma-associated fibroblasts derive from mesothelial cells via mesothelial-to-mesenchymal transition in peritoneal metastasis. J. Pathol. 231, 517–531 (2013).
Lucas, P. A. Stem cells for mesothelial repair: an understudied modality. Int. J. Artif. Organs 30, 550–556 (2007).
Greenhill, C. Defining subpopulations of human adipose tissue progenitor cells. Nat. Rev. Endocrinol. 19, 498 (2023).
Kim, W. et al. RUNX1 is essential for mesenchymal stem cell proliferation and myofibroblast differentiation. Proc. Natl. Acad. Sci. USA 111, 16389–16394 (2014).
Dubey, S., Dubey, P. K., Umeshappa, C. S., Ghebre, Y. T. & Krishnamurthy, P. Inhibition of RUNX1 blocks the differentiation of lung fibroblasts to myofibroblasts. J. Cell. Physiol. 237, 2169–2182 (2022).
Halperin, C. et al. Global DNA methylation analysis of cancer-associated fibroblasts reveals extensive epigenetic rewiring linked with RUNX1 upregulation in breast cancer stroma. Cancer Res. 82, 4139–4152 (2022).
Heinzelmann-Schwarz, V. et al. Letrozole may be a valuable maintenance treatment in high-grade serous ovarian cancer patients. Gynecol. Oncol. 148, 79–85 (2018).
Nobre, C. C. G. et al. Macrophage migration inhibitory factor (MIF): biological activities and relation with cancer. Pathol. Oncol. Res. 23, 235–244 (2017).
Filippou, P. S., Karagiannis, G. S. & Constantinidou, A. Midkine (MDK) growth factor: a key player in cancer progression and a promising therapeutic target. Oncogene 39, 2040–2054 (2020).
Neumaier, E. E., Rothhammer, V. & Linnerbauer, M. The role of midkine in health and disease. Front. Immunol. 14, 1310094 (2023).
Kadomatsu, K. & Muramatsu, T. Midkine and pleiotrophin in neural development and cancer. Cancer Lett. 204, 127–143 (2004).
Ohuchida, T. et al. Midkine protects hepatocellular carcinoma cells against TRAIL-mediated apoptosis through down-regulation of caspase-3 activity. Cancer 100, 2430–2436 (2004).
Cerezo-Wallis, D. et al. Midkine rewires the melanoma microenvironment toward a tolerogenic and immune-resistant state. Nat. Med. 26, 1865–1877 (2020).
Pandey, P. et al. Amyloid precursor protein and amyloid precursor-like protein 2 in cancer. Oncotarget 7, 19430–19444 (2016).
Guan, X. et al. Anti-TIGIT antibody improves PD-L1 blockade through myeloid and Treg cells. Nature 627, 646–655 (2024).
Nutsch, K. et al. TIGIT and PD-L1 co-blockade promotes clonal expansion of multipotent, non-exhausted antitumor T cells by facilitating co-stimulation. Nat. Cancer 5, 1834–1851 (2024).
Chai, C. et al. Single cell analysis of epithelial, immune, and stromal signatures and interactions in human ovarian cancer. Res. Sq. https://doi.org/10.21203/rs.3.rs-3024248/v1. (2023)
O’Reilly, C., Doroudian, M., Mawhinney, L. & Donnelly, S. C. Targeting MIF in cancer: therapeutic strategies, current developments, and future opportunities. Med. Res. Rev. 36, 440–460 (2016).
Lozano, E. et al. Nectin-2 Expression on malignant plasma cells is associated with better response to TIGIT blockade in multiple myeloma. Clin. Cancer Res. 26, 4688–4698 (2020).
du Bois, A. et al. Role of surgical outcome as prognostic factor in advanced epithelial ovarian cancer: a combined exploratory analysis of 3 prospectively randomized phase 3 multicenter trials: by the Arbeitsgemeinschaft Gynaekologische Onkologie Studiengruppe Ovarialkarzinom (AGO-OVAR) and the Groupe d’Investigateurs Nationaux Pour les Etudes des. Cancers de. l’Ovaire. Cancer 115, 1234–1244 (2009).
Lanickova, T. et al. Chemotherapy drives tertiary lymphoid structures that correlate with ICI-responsive TCF1 + CD8 + T cells in metastatic ovarian cancer. Clin. Cancer Res. 31, 164–180 (2025).
Ewels, P. A. et al. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 38, 276–278 (2020).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Xuan, Y. et al. Standardization and harmonization of distributed multi-center proteotype analysis supporting precision medicine studies. Nat. Commun. 11, 5248 (2020).
Forny, P. et al. Integrated multi-omics reveals anaplerotic rewiring in methylmalonyl-CoA mutase deficiency. Nat. Metab. 5, 80–95 (2023).
Bertolini, A. et al. scAmpi-A versatile pipeline for single-cell RNA-seq analysis from basics to clinics. PLoS Comput. Biol. 18, e1010097 (2022).
Griffiths, J. A., Richard, A. C., Bach, K., Lun, A. T. L. & Marioni, J. C. Detection and removal of barcode swapping in single-cell RNA-seq data. Nat. Commun. 9, 2667 (2018).
Germain, P.-L., Lun, A., Garcia Meixide, C., Macnair, W. & Robinson, M. D. Doublet identification in single-cell sequencing data using scDblFinder. F1000Res. 10, 979 (2021).
Hafemeister, C. & Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296 (2019).
Levine, J. H. et al. Data-driven phenotypic dissection of AML reveals progenitor-like. Cells Correl. Prognosis. Cell 162, 184–197 (2015).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinforma. 14, 7 (2013).
McInnes, L., Healy, J. & Melville, J. UMAP: uniform manifold approximation and projection for dimension reduction. arXiv https://doi.org/10.48550/arxiv.1802.03426. (2018)
La Manno, G. et al. RNA velocity of single cells. Nature 560, 494–498 (2018).
Bergen, V., Lange, M., Peidli, S., Wolf, F. A. & Theis, F. J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 38, 1408–1414 (2020).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021).
Acknowledgements
We are grateful to the Department of Biomedicine Microscopy Facility (Mike Abanto, Loïc Sauteur, and Ewelina Bartoszek), Histology Facility (Diego Calabrese), and Flow Cytometry Core Facility (Cecile Cumin). We further acknowledge Monica Núñez and Natalie Rimmer from the Ovarian Cancer Research group for their support throughout this project. We wish to thank Shane Vondelin Van Breda for scientific discussions and sharing resources. We also thank Ina Nissen and Elodie Burcklen from the Genomics Facility Basel of the University of Basel and the Department of Biosystems Science and Engineering (ETH Zurich) for technical support and sequencing. This project received funding from the grant #2017-510 of the Strategic Focal Area “Personalized Health and Related Technologies” of the ETH Domain, grant #171037, #207975, and #220706 of the Swiss National Science Foundation, Swiss Cancer League (KFS-5389-08-2021), and from the RIVKIN Pilot Study Award.
Author information
Authors and Affiliations
Contributions
Conceptualization: U.L., F.J., C.B., and V.H.S. Methodology: U.L., C.Y.L., R.C., F.J., and C.B. Project Administration: U.L., C.B., F.J., and V.H.S. Investigation: U.L., R.C., C.Y.L., M.C., A.H., F.S., F.L., S.G., J.F., A.D., N.B., and F.J. Resources: C.K., C.M.S., C.B., J.F., and V.H.S. Visualization: U.L., C.Y.L., M.C., R.C., A.H., F.L., A.D., M.H., F.S., J.F., and F.J. Supervision: F.S., C.B., F.J., and V.H.S. Funding Acquisition: U.L., C.B., F.J., and V.H.S. Writing - Original Draft: U.L. and F.J. Writing - Review & Editing: all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lischetti, U., Liang, CY., Carrara, M. et al. Ovarian cancer metastasis to the human omentum disrupts organ homeostasis and induces fundamental tissue reprogramming. Nat Commun 17, 849 (2026). https://doi.org/10.1038/s41467-025-67557-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67557-z
