
Abstract
The cell-free biogenesis of the protein translation machinery is essential for the creation of a self-regenerating synthetic cell. Here, we demonstrate the autonomous and simultaneous biogenesis of all thirty proteins of the translation machinery of E. coli in a reconstituted transcription-translation system. We first establish self-regeneration of every translation protein by determining the threshold concentration required for its own synthesis from a synthetic gene coding for the protein, thereby demonstrating the functionality of all nascent proteins, separately. Simultaneous biogenesis of multiple translation proteins at their threshold results in delayed synthesis below detection levels. To achieve self-regeneration of multiple translation proteins, we induce boundary-free compartmentalization of the reaction by immobilizing the genes on a surface at high density. The co-localization of genes, molecular machinery, messenger RNA and nascent proteins at the surface create sufficient conditions to catalyze the simultaneous self-regeneration of sub-groups up to all thirty translation proteins, as measured by total internal reflection fluorescence on the surface. Our approach provides mechanistic insight and presents a general methodology for the biogenesis of cellular machines toward autonomous synthetic systems.
Similar content being viewed by others

Introduction
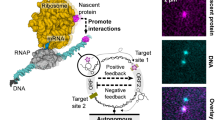
Recreating the conditions for autonomous biogenesis of the protein translation machinery is fundamental to our understanding of living systems and is essential for the establishment of minimal cell models1. A major step forward was the cell-free reconstitution of genetically encoded protein synthesis, defining the minimal set of purified protein factors and enzymes that support multiple translation cycles2 (Fig. 1a). This cell-free bacterial platform, termed the PURE system, has been shown throughout the years to support the synthesis of a myriad of proteins3,4,5,6,7,8. Finding conditions that enable the minimal translation system to exhibit biogenesis and self-regeneration is the next essential milestone toward an autonomous protein-based artificial cell9,10. We focus on the development of a general approach for the autonomous co-synthesis of thirty protein factors and enzymes, including twenty aminoacyl transfer RNA (tRNA) synthetases (aaRS) and ten translation factors (Fig. 1a), that allows for the nascent proteins to actively integrate back into the system that created them (Fig. 1b).

a Schematic overview of the 30 protein factors and enzymes involved in protein translation, including initiation factors (IF), aminoacyl-tRNA synthetases (aaRS), elongation factors (EF), and release/recycling factors (RF). b Scheme illustrating the concept of gene expression with positive feedback from self-synthesis of translation proteins. c A PURE reactions with component at threshold concentrations, GFP expression is undetectable prior to biogenesis (i), but becomes detectable after the self-synthesis of translation machinery (ii), indicating functional activity of newly synthesized proteins. A PURE with saturating concentrations of all components shows indistinguishable GFP reporter expression before (iii) and after (iv) biogenesis. d Fluorescence intensity (FI) time traces of an eGFP reporter gene in a full PURE reaction with and without the genes of the translation proteins. Mean ± STD (n = 3). e Numerical simulation of AsnRS expression in PURE at different initial AsnRS concentrations. Max. synthesis rate vs. initial AsnRS0 for reactions with (biogenesis) and without (no biogenesis) functional nascent AsnRS. The gray band denotes the threshold regime, where rates are highly sensitive to small changes. f FI(t) and its second derivative, FI″(t), for PURE reactions at different initial ValRS concentrations with and without the ValRS gene. FI and FI″ are normalized to the maxima of a full PURE control. Dashed lines mark t = argmax FI″(t). g Peak synthesis rate (maximum of dFI/dt) as a function of initial ValRS concentration. Rates are normalized to the control maximum. Horizontal markers indicate means that are connected with a dotted line (n = 3 different and independent titration series, see Supplementary Fig. 7). h tmax is the time of max. FI″(t) as a function of ValRS from the same data as in (g). i Average normalized peak rate across all 30 translation proteins ± their genes at threshold (n = 3 independent experiments). A protein Pi is marked with an asterisk when purified Pi was added to kick-start biogenesis (Supplementary Table 1). Data is provided in the Source Data file.
Intuitively, the demand for the co-synthesis of thirty different proteins at sufficiently high concentrations calls for a reconstituted system that operates at the highest possible capacity. While improved versions of the PURE system with increased protein yields have been established11,12,13, they contain high concentrations of the initial machinery. Therefore, the contribution of newly synthesized translation factors and enzymes would have little observable effect when the system operates at saturating synthesis rates (Fig. 1c, right panel). This challenge could be resolved by diluting the reaction after sufficient proteins had been synthesized, to allow identification of the nascent components. Using this approach, the self-regeneration of T7 RNA polymerase, two kinases and RF1 and 12 aaRS was demonstrated using a serial dilution experiment of two cycles14. In a separate study, all 20 aaRS were simultaneously self-regenerated from their corresponding genes in a serial dilution series of up to 20 cycles15. Furthermore, the self-regeneration of up to seven aaRS was achieved within a PURE system diluted using a microfluidic device16.
Here, we demonstrate an alternative autonomous approach, starting with a system that operates at its threshold level under extremely diluted conditions of initial PURE translation proteins. In this scenario, functional nascent translation proteins are synthesized from their genes and accumulate very slowly, feeding back into the system until a threshold concentration is crossed, producing a significant rise in activity due to self-amplification (Fig. 1c, left panel). For such a scenario to succeed, a mechanism to catalyze and enhance the reaction at low machinery concentrations during the initial stages is required. Recently, we presented a cell-free scenario for the localization of gene expression reactions at surface-immobilized high-density gene brushes as boundary-free compartments17,18, leading to the accumulation of nascent RNA and protein products and increasing their local concentration to facilitate macromolecular interactions. We demonstrated co-translation assembly of T4 bacteriophage structural modules and reconstituted the biogenesis of functional E. coli RNA polymerase19. In addition, by encoding 26 different ribosomal proteins and assembly factors and one scaffold ribosomal RNA in such localized reaction centers, we were able to demonstrate the biogenesis, hierarchical order, and assembly timeline of the E. coli small ribosomal subunit20. Here, we utilize this approach, immobilizing all the genes coding for the thirty protein factors and enzymes of the translation machinery in a DNA brush to demonstrate their autonomous co-synthesis and functional integration back into the machinery that created them. Other scenarios to confine and co-localize the reaction components, for example, by packing multiple genes as continuous synthetic genomes in enclosed compartments, could also be envisioned using the same general principles21,22,23,24.
As part of our general self-regeneration strategy, we first determine in bulk solution reactions for each translation protein (Pi) separately the threshold concentration below which the synthesis rate of a reporter green fluorescence protein (GFP) is strongly reduced. Upon addition of the genes coding for the corresponding translation protein, we detect an increase in the reporter signal due to crossing the threshold concentration by self-synthesis. When self-regeneration for individual or multiple proteins can not be demonstrated in a bulk solution, we adopt the approach of localized reaction centers. We immobilize the corresponding genes as dense DNA brushes, add the dilute PURE reaction mixture with each protein at its threshold concentration, and use total internal reflection fluorescence (TIRF) microscopy for sensitive detection of a surface-trapped GFP reporter. At the threshold level, the minimal system is unable to support GFP synthesis unless all genes coding for the translation proteins are included in the DNA brush, allowing us to define the transition point to cell-free biogenesis of all thirty translation proteins simultaneously from the bottom up.
Results
A self-regeneration assay for the protein translation machinery
The thirty protein factors and enzymes that are essential for in vitro protein translation belong to four biochemically distinct subgroups: twenty aaRSs, each of which covalently charges a tRNA with its cognate amino acid; and four initiation, three elongation and three termination factors which together comprise a total of ten proteins involved in the three stages of the translation cycle (Fig. 1a). We introduced all thirty translation genes and the GFP reporter gene to a PURE bulk reaction (Fig. 1b) and monitored the fluorescence intensity time trace, FI(t), against control reaction with dummy genes coding for unrelated proteins of a similar synthesis load as the translation proteins (Fig. 1d). Since there was no significant difference between the two fluorescence time traces, we were unable to deduce the synthesis and functionality of the nascent translation machinery. We verified that each of the translation machinery proteins was synthesized from its gene in the minimal protein translation system (Supplementary Fig. 1). Therefore, the functionality of the nascent translation proteins may have been masked because the initial PURE reaction was already saturated at the reaction conditions (Fig. 1c).
We reasoned that a very dilute PURE mix in which all the translation machinery proteins were at a very low initial concentration would be a more suitable scenario for self-regeneration, as it allows for a bottom-up buildup of all components and provides a definitive manifestation of the activity of nascent machinery (Fig. 1c). To support this scenario, we performed numerical simulations of protein synthesis in the PURE system25 with different initial concentrations of all thirty translation factors (Supplementary Fig. 2) and found a dilution regime where the translation rate is more sensitive to protein concentrations than in a full PURE. We then simulated the biogenesis of one of the translation proteins (Pi), asparaginyl-tRNA synthetase (AsnRS), for two different scenarios: (i) with biogenesis, that is, the nascent AsnRS proteins are active and feed back into the pool of initial AsnRS, or (ii) without biogenesis, that is, the nascent proteins are synthesized but are inactive and just accumulate (Fig. 1e). Consistent with autocatalytic biogenesis, once nascent AsnRS exceeded a threshold, its maximum synthesis rate increased sharply compared with non-autocatalytic production. For higher initial concentration of AsnRS, the translation rates at both scenarios converged and plateaued, coinciding with the saturating regime of the full PURE reaction (Fig. 1d and Supplementary Fig. 2).
Empirical determination of threshold conditions for self-regeneration
Based on the simulation, we sought to determine empirically the threshold concentration of each Pi in an initial PURE composition at which GFP fluorescence could be barely detected. The addition of genes coding for this Pi should result in its synthesis and accumulation to a concentration sufficiently above the threshold to produce GFP, indicating self-regeneration. We first set 30 parallel PURE reactions, each with one of the proteins removed completely (ΔPi), expecting to obtain no GFP synthesis since each Pi component should be essential for PURE activity2. For some ΔPi reactions, we obtained negligible GFP signals as expected, but for others (i.e., for all the initiation and termination factors and some aaRSs), there were substantial GFP signals that most likely originated from Pi contamination. This background protein expression level was highly dependent on the source of the added ribosomes suggesting they were the main source of contamination (Supplementary Fig. 3 and 4). Mass spectrometry analysis demonstrated directly that the ribosome preparations indeed included translation machinery proteins (Supplementary Fig. 5). The commercial preparation of ribosomes (see “Methods”) had the overall lowest residual activity for most of the ΔPi, excluding the initiation and release factors, which had substantial residual activity throughout all ribosome preparations (Supplementary Fig. 4). To minimize masking of the self-regeneration activity, we therefore chose to use the commercial preparation of ribosomes in the rest of the study. In one batch reaction, our full PURE mix with the commercial ribosomes produced around 1600 nM of GFP with a max. rate of 14 nM min−1 (Supplementary Fig. 6).
We then introduced the gene coding for Pi at different concentrations in ΔPi-PURE and monitored the GFP expression rate. If no increase was observed, we titrated purified Pi back into the ΔPi-PURE mix to determine the threshold and dynamic range for a GFP response and then reintroduced the gene at that threshold concentration. For example, at an initial Valyl-tRNA synthetase (ValRS) concentration of 1 nM, the addition of the ValRS gene resulted in a significant boost in the synthesis rate of the reporter protein compared to the reaction without the added ValRS gene, indicating that the nascent ValRS was functional and integrated back into the PURE machinery (Fig. 1f, g and Supplementary Fig. 7). This analysis allowed us to observe ValRS self-regeneration for initial concentrations between 0.25 to 2.5 nM (Fig. 1f, g). Notably, we detected an inflection point in the GFP synthesis rate at about 20 to 30 min, even without the aaRS genes, indicating low rates of protein synthesis in the initial PURE reaction (Fig. 1f). In the presence of aaRS genes, a second peak appeared after a significant delay (Fig. 1f and Supplementary Fig. 7), which we attribute to the activity of nascent ValRS contributing both to GFP and its own synthesis. The time of the second peak (tmax) decreased as the initial ValRS concentration increased (Fig. 1h and Supplementary Fig. 7).
We repeated this analysis for the other twenty-nine translation proteins and summarized the effect of supplementing the gene on the protein synthesis rate at the threshold concentration in Fig. 1i. For each of the twenty aaRS we found threshold conditions that establish their self-synthesis and functionality, similarly to the ValRS analysis. For ten of the aaRSs (Arg, Asn, Cys, Glu, His, Lys, Met, Thr, Trp, and Tyr), the residual activity in a ΔPi,-PURE was enough to kick-start the self-synthesis upon addition of the corresponding gene (Fig. 1h and Supplementary Fig. 8), resulting in increased maximal GFP synthesis rate (max. rate), and implying that the nascent aaRS was functional. Beyond a certain gene concentration, often around 1 nM, the max. rate decreased again, which could be attributed to a “competition” effect, where increasing the DNA of one gene reduces the expression of another gene (GFP), most likely due to the sharing of resources26 (Supplementary Fig. 9). Specifically, for ArgRS, we found that its synthesis required the addition of a protein chaperone mix (see Methods) which we thus added to all reactions (Supplementary Fig. 10). For the other ten aaRSs (Ala, Asp, Gln, Gly, Ile, Leu, Phe, Pro, Ser, and Val) (Fig. 1h and Supplementary Fig. 11), the threshold concentration was found to be, on average, between 1 and 2% of the concentration of the aaRS in a full reaction, often reaching sub-nanomolar concentration, with GlyRS being the exception, requiring 42% of its full reaction concentration (Supplementary Table 1).
For the three elongation factors, we recalled that their concentration in a full PURE reaction is at the micromolar range. Accordingly, the threshold concentration and dynamic range of two elongation factors, EF-G and EF-Ts, were found to be about ten times higher compared to the threshold concentration of the aaRSs (Supplementary Figs. 12 and 13). Still, we could demonstrate their self-regeneration upon addition of their respective genes, observing an increase in the yield and the max. rate and a decrease in the tmax (Fig. 1i and Supplementary Figs. 12 and 13). For EF-Tu, we could not differentiate between the functionality of initial or nascent EF-Tu, most likely due to the broad dynamic range of the GFP response function to EF-Tu concentration, from a few nanomolar to several micromolar (Fig. 1i and Supplementary Fig. 14). In addition to EF-Tu, we could not demonstrate at this stage self-regeneration of some of the translation initiation and termination factors (IF1, IF3, MTF, RF1, and RF3), as individual removal of each from the reaction did not significantly reduce the protein synthesis rate (Supplementary Fig. 4).We only observed some indication for self-regeneration for IF2 and RRF (Supplementary Fig. 15).
Self-regeneration of groups of initiation and release factors
The self-regeneration in bulk solution for the initiation and termination factors in corresponding ΔPURE versions was masked by substantial contaminations far beyond threshold conditions. (Supplementary Figs. 4 and 5). We thus hypothesized that we could create threshold conditions by removing multiple factors simultaneously from the initial PURE reaction (Fig. 2a). Indeed, the max. rate dropped for the simultaneous deletions of all four initiation factors (IF1 + 2 + 3 + MTF) or for the mutual deletion of the release factors (RF1 + RF3 + RRF) and was restored in the presence of their respective genes (Fig. 2b). By using permutations of the four- and three-member initiation (IF1, IF2, IF3, and MTF) and release groups (RF1, RF3, and RRF), respectively, we were able to dissect the contribution of each protein (Fig. 2c, d and Supplementary Fig. 16). The difference in max rates with and without their respective genes (Δkmax) showed an increase for all combinations except for Δ(IF1 + MTF) and Δ(RF1 + RF3), but their functionality could be deduced from the rest of the combinations. Interestingly, in combining the initiation factors from one to four, we observed an extension in tmax, from a few min to 40 min (Fig. 2c). We did not observe such extension in tmax as more release factors were deleted, but the Δkmax for Δ(RF1 + RF3) increased slightly compared to the single deletions (Fig. 2d), and the other group deletions were dominated by RRF contribution. Despite substantial contaminations of these protein factors from the ribosomes in the initial reaction, we could conclude from the bulk solution reactions that the seven nascent initiation and release factors were participating in GFP synthesis upon their self-synthesis.

a Top: IF assembly on the ribosomal subunit during early stages of translation. Bottom: Translation termination with release and recycling factors. b Top: FI(t) from ΔIF-PURE reactions (ΔIFs and ΔMTF), with or without the corresponding IF genes. For each IF: [IF DNA] = 0.1 nM; [GFP DNA] = 0.4 nM. Bottom: Biogenesis assay in ΔRF-PURE, (ΔRF1, ΔRF3, and ΔRRF) ± RF genes. For each RF: [RF DNA] = 0.1 nM; [GFP DNA] = 0.4 nM. All FI values are normalized to the maximum FI of a full PURE reaction. Mean ± STD (n = 3, independent experiment). c Bottom: Difference between the normalized max. rate ± their corresponding genes (Δkmax) of ΔPURE for all the permutations of the three IFs and MTF (minimum n = 3 independent experiments). Dotted horizontal lines: guide to the eye for comparing the recovery levels of each subgroups. Top: the time point of the max. FI″(t), tmax, indicating the moment the threshold concentration is crossed due to biogenesis. d Bottom: Δkmax for ΔPURE reaction with all the permutations of a grouping of RF1, RF3, and RRF± their corresponding gene with n = 3 independent experiments. Top: corresponding times max. FI”(t) Source data are provided as a Source Data file.
Self-regeneration of EF-Tu at surface-localized reaction centers
Demonstrating self-regeneration of EF-Tu was challenging, and we therefore sought evidence that nascent EF-Tu synthesized in the PURE was functional by separating the reaction into two consecutive steps (Fig. 3a) (see Methods). In the first step, we added the EF-Tu gene to a PURE reaction with lowered EF-Tu concentration, and after incubation, allowing for sufficient EF-Tu synthesis to occur (reaction 1), we transferred a small volume fraction to a fresh ΔEF-Tu-PURE reaction supplemented with GFP genes (reaction 2). We observed that the GFP signal in reaction 2 increased with the EF-Tu gene concentration in reaction 1, thus confirming that nascent EF-Tu synthesized in a PURE reaction was functional (Fig. 3b, c and Supplementary Fig. 17). Self-regeneration of EF-Tu was most likely hidden by the high threshold concentrations necessary for efficient protein synthesis in a one-step coupled system.

a Two-step functionality assay. R1: expression of EF-Tu in PURE at threshold. R2: serial transfer of 2.5 µl (R1) into 7.5 µl ΔEF-Tu PURE containing a GFP gene (R2). b FI(t) of R2 after spiking from R1 with varying EF-Tu gene concentrations. R2 normalized to the max FI of a full PURE control. Conditions: [EF-Tu]0 = 800 nM; [GFP DNA] = 0.4 nM. (replicates: Supplementary Fig. 17). c Max rate of FI(t) in R2 versus EF-Tu DNA in R1; normalized to full PURE; mean values (n = 3, independent experiments). d Schematic of a DNA brush with EF genes and surface-immobilized anti-HA antibodies that capture GFP-HA. e Three identical DNA brushes with a hexagonal pattern for traps. f TIRF microscopy set-up. g TIRF images of EF-Tu self-synthesis from DNA brushes. Top: brush with 2% GFP-HA genes (DNA-labeled) and 98% dummy genes (unlabeled). Bottom: brush with 20% EF-Tu (DNA-labled), 2% GFP-HA, and 78% dummy genes (leftmost image is a red/green overlay). Increased GFP-HA accumulation is seen at HA traps around brushes containing EF-Tu genes. PURE with [EF-Tu] = 1 µM. h Avg. GFP FI(t) from TIRF imaging of DNA brushes with different fractions of EF-Tu genes. i Mean fold change of GFP FI ± s.e.m from DNA brushes (normalized to GFP signal from “0%” brush) versus total DNA FI (EF-Tu + GFP DNA) across brushes (n = 4 clusters from two slides) at t = 60 or 120 min according to expression kinetics. Replicates: Supplementary Fig. 18. j TIRF images comparing brushes with 2% GFP-HA + 98% dummy genes (top) versus an EF-gene mix (bottom; 20% total EFs with gene ratio [Ts]:[G]:[Tu] = 1:2:3; 78% dummy). PURE: [EF-Ts]0 = 100 nM, [EF-G]0 = 25 nM, [EF-Tu]0 = 1 µM. k Average GFP FI(t) for brushes with indicated fractions of the EF-gene mix (replicates: Supplementary Fig. 18). l Mean fold change of GFP FI ± s.e.m from DNA brushes (normalized to the GFP signal from the “0%” brush) at t = 90 min versus DNA FI (EFs + GFP DNA) across brushes (n = 5 clusters from three slides). Scale bars, 200 µm. Source data are provided as a Source Data file.
To increase the efficiency of EF-Tu self-regeneration at reduced threshold concentrations, we created localized reaction centers of EF-Tu genes immobilized on a surface as high-density DNA brushes within a large reservoir of nutrients (Fig. 3d). This design served two purposes: first, to overcome the capacity barrier imposed by the limited nutrient supply and reduced EF-Tu concentration; and second, to spatially co-localize EF-Tu synthesis to the site of GFP synthesis, where EF-Tu activity is required. The genes were arranged in clusters of three identical DNA brushes (Fig. 3d, e). Within each cluster the brushes contained a different fraction of EF-Tu genes, from 0 to 80%, and in addition, a 2% gene fraction coding for GFP fused to a Hemagglutinin tag (GFP-HA). The surface surrounding the DNA brushes was patterned with HA-specific antibodies for trapping nascent GFP-HA on the surface for sensitive detection by TIRF imaging (Fig. 3d–f). The overall DNA density and, thus, gene expression rate were kept constant by supplementing brushes with DNA coding for an unrelated (dummy) gene17 (see Methods). Upon addition of a PURE reaction mixture initially containing 1 µM purified EF-Tu, we observed, over the course of 1–2 hours, accumulation of GFP-HA on the surface in the vicinity of the DNA brushes. This demonstrates, in a single reaction, that the nascent EF-Tu synthesized from the brushes is functional in the PURE system and essential for GFP-HA synthesis (Fig. 3g–i, Supplementary Fig. 18 and Supplementary Movie 1). The GFP-HA signal increased in response to larger fractions of EF-Tu genes up to 40%, with a slight decrease at 80% most likely due to resource competition or inhibiting byproducts.
Simultaneous self-regeneration of sub-groups of translation machinery proteins
Encouraged by the demonstration of self-regeneration of EF-Tu when encoded in DNA brushes, we attempted simultaneous biogenesis of all three elongation factors encoded in the same DNA brushes, with the ratio of the respective genes set by the results from the single self-regeneration assays (Supplementary Table 3). Upon addition of a translation reaction with purified EF-Ts, EF-G, and EF-Tu set at threshold concentrations, GFP-HA signals accumulated on the surface surrounding DNA brushes in response to the increase of EFs genes fraction, with a maximum GFP signal at 40% and a sharp drop at 80% (Fig. 3j–l, Supplementary Fig. 18 and Supplementary Movie 2). We concluded that the localization of the translation machinery at the DNA brushes co-localized the synthesis of all the nascent elongation factors in the vicinity of the brushes at sufficient concentrations to self-amplify their own synthesis as well as GFP synthesis18.
Towards self-regeneration of all twenty aaRSs simultaneously, we recalled that they comprise 20 distinct enzymes (Fig. 4a). This diversity motivated us to investigate whether specific parameters within this group might influence the self-regeneration process. To this end, we compared the max. rate in the self-regeneration assays (Fig. 1 and Supplementary Fig. 8) with respect to various physical and biochemical parameters—including kinetic rate constants, number of cognate amino acids, protein or substrate amino acid size and amino acid hydrophobicity27 (Supplementary Figs. 19 and 20). We looked for correlations between physical parameters of the aaRS and Δkmax, the difference between the GFP max. synthesis rate with and without the aaRS gene, and found a moderate negative correlation between Δkmax and the length of the aaRS genes as well as the hydrophobicity of the substrate amino acid, and moderate positive correlation between Δkmax and the gel band intensity for its own expression (Fig. 4b and Supplementary Fig. 20). We did not observe a correlation between Δkmax and the number of amino acids in the reporter protein. The only GFP sequence effect we may have observed was for the ΔTrpRS-PURE reactions, where residual GFP production was higher than for other aaRS deletions. Since GFP contains only a single Trp, the residual GFP signal might be attributed to misincorporation at this position. However, adding the TrpRS gene still boosted the rate substantially, supporting the conclusion that TrpRS could be self-regenerated. We were encouraged that there was no apparent obstacle to simultaneous self-regeneration of all twenty aaRSs, except for their load on the PURE reaction. Indeed, when attempting co-synthesis of all twenty aaRSs in bulk reaction, the GFP signal was reduced compared to a bulk reaction without the aaRSs genes (Fig. 4c and Supplementary Fig. 21). The combined synthesis yield of all the twenty aaRSs was most likely not sufficient to cross the threshold concentrations required to demonstrate self-biogenesis.

a Schematic representation of aminoacylation: aaRS enzymes charge tRNAs with their cognate amino acids. b Difference in maximum GFP synthesis rates (Δkmax) at threshold with and without each aaRS gene plotted versus aaRS length (amino acids). Kendall’s rank correlation (see Methods) indicates a moderate negative correlation. Colors distinguish initial concentrations C0 = 0 and C0 > 0. Data are mean ± s.e.m., n = 3.c GFP FI(t) while expressing 18 aaRS genes (all except GlyRS and PheRS) in a bulk PURE reaction with three different threshold concentrations. Left: Cmin < 5 nM: AlaRS = 3.0, AspRS = 2.0, GlnRS = 0.8, IleRs = 0.5, LeuRS = 2.0, MetRs = 0.5, ProRs = 1, SerRS = 1, ValRs = 1. Middle and right: Cmin = 5 nM or 10 nM for all aaRS. [aaRS-DNA] = 0.1 nM (except for MetRS-DNA = 0.4 nM), [DNAGFP] = 0.4 nM (for replicates see Supplementary Fig. 21). d Scheme: DNA brush immobilized on a surface localizing the purified (dark blue) translation proteins and nascent (yellow) aaRS. e Prism-TIRF images of a hexagonal array of GFP-HA traps (green) and three DNA brushes (white dashed circles). Top: brush containing 2% GFP gene and 98% non-interacting dummy gene (DNA unlabeled). Bottom: 20% aaRS gene mix containing all 20 aaRS genes (Supplementary Table 3), 2% GFP gene, and 78% dummy genes; aaRS DNA is labeled (leftmost image is a red/green overlay). GFP-HA accumulates around brushes encoding aaRS genes. Scale bar, 200 µm. f Top: average GFP FI(t) from brushes with varying aaRS fractions (replicates in Supplementary Fig. 22). Bottom: mean fold change of GFP FI ± s.e.m from DNA brushes (normalized to the signal from “0%” brush) at t = 120 min ±s.e.m. as a function of total DNA FI (aaRS + GFP DNA) across brushes (n = 6 brush clusters from two slides). Source data are provided as a Source Data file.
Inspired by the biogenesis of the elongation factors in the localized-reaction centers, we created DNA brushes that contain different fractions of all the twenty genes of the aaRS, and a 2% constant GFP-HA fraction (Fig. 4d). We added a PURE reaction with ten aaRSs completely removed and ten at their threshold concentrations. We observed GFP accumulation in the proximity of the DNA brushes dependent on the amount of aaRS genes in the brush (Fig. 4e, f; Supplementary Fig. 22 and Supplementary Movie 3). We concluded that co-localizing the synthesis of all the nascent aaRSs in the vicinity of the DNA brushes yielded sufficient apparent concentrations to support self and GFP synthesis, thus demonstrating simultaneous biogenesis of twenty aaRSs in one minimal translation reaction, which was not possible in a bulk solution.
Direct observation of nascent protein localization to gene brushes
To gain mechanistic insight into the biogenesis of the translation machinery catalyzed by DNA brushes, we created C-terminal GFP fusions of several translation proteins (see Supplementary Data File 1). The transient accumulation of nascent protein fusions at the vicinity of their DNA brushes could be detected by the localized TIRF illumination of the surface, implying their participation in gene expression18 (Fig. 5c). We first tested the functionality of C-terminal GFP fusions in bulk solution reactions using mScarlet as a reporter protein (Fig. 5a). It required a more concentrated PURE mix to achieve comparable expression levels to their non-fusion versions (see Supplementary Table 4). We observed activity for the GFP fusions of the three EFs, as well as those of the GluRS, CysRS, and IF3. For RRF-GFP, we detected lower activity than for the other candidates, and for IF1 and RF1 fusions, we were not able to discriminate between nascent and backgound activity (Supplementary Figs. 23 and 24). We proceeded to immobilize the genes with the GFP fusions as DNA brushes, each GFP fusion gene in its own brush. We monitored the TIRF signal at the DNA brush compared to the overall fluorescence signal (epi) and observed a strong localization and accumulation of IF1-GFP, IF3-GFP and RF1-GFP to the DNA brushes, as evident by a strong signal at the three brush clusters which was persistent with time (Fig. 5b, c and Supplementary Fig. 25). The DNA brush localization for RRF-GFP, GluRS-GFP, CysRS-GFP and EF-GFPs was mainly observed at the beginning of the reaction (first 20 minutes), after which the TIRF signal at the brushes reached the signal of the surroundings. No specific localization to DNA brushes could be detected for three control proteins, GFP, MBP-GFP and T4-gp25-GFP, even at initial stages of the reaction. We thus concluded that the IF3-, RRF-, GluRS-, CysRS-, and the EFs-GFP fusions are localized to the brush due to their function in translation. Although we did not detect a boost in activity for IF1-and RF1-GFP fusions in bulk solution, we still observed strong localization to their DNA brushes, which we attribute to their RNA-binding activity that was most likely not hampered by the GFP fusion (Supplementary Fig. 25). This hypothesis requires further validation. Active site mutants of IF1 and RF1 fused to GFP also localized to DNA brushes, suggesting that, indeed, localization of these two candidates is due to unspecific RNA binding28,29. Control experiments with purified GFP allowed us to roughly estimate the protein concentrations in the TIRF images, by converting the intensity of the signal at the brush according to the calibration curve (Supplementary Fig. 26). Within the DNA brush, we estimate the GFP fusions of IF1, IF3, and RRF to reach the micromolar range measured within the first hour, while GFP fusions of RF1, CysRS, GluRS, EF-Tu, EF-Ts and EF-G, levels are 10-to-100 fold lower. The localization of the translation protein factors and not those of the control proteins provided direct evidence that protein synthesis from a DNA brush enhances self-regeneration by creating high local concentrations.

a Top: Schematic of the functionality assay for translation proteins fused to GFP with the reporter protein mScarlet. Bottom: GFP and mScarlet FI(t), showing expression of a functional EF-G with a C-terminal GFP fusion versus a non-functional control (dummy protein fused to GFP) and the corresponding reporter expression (replicates see Supplementary Fig. 23). b Scheme: translation proteins were fused to GFP and expressed from surface-immobilized DNA brushes. TIRF microscopy, illuminates a ~100 nm region above the surface, was used to visualize protein localization at the DNA brushes (height ~100 nm). c Mean GFP fluorescence signal averaged over 20 min from TIRF microscopy of DNA brushes encoding various translation protein–GFP fusions. Below: average time-resolved fluorescence traces from three DNA brushes within each cluster. Mean ± s.e.m. (n = 3 DNA brush cluster). “Brush” denotes mean GFP FI within the region of interest (ROI) of the DNA brush; “background” represents mean FI from a neighboring ROI lacking DNA; “epi” corresponds to epifluorescence, which detects GFP throughout the bulk volume above the surface. A second data set in Supplementary Fig. 25. Scale bars = 200 µm. Source data are provided as a Source Data file.
Simultaneous self-regeneration of all thirty translation proteins
We attempted the biogenesis of all thirty translation proteins simultaneously in a single reaction by immobilizing mixed DNA brushes on a glass surface (Fig. 6a, b) with relative gene fractions based on the individual self-regeneration (Fig. 1h) and previous DNA brush experiments (Figs. 3 and 4). On the same surface different DNA brushes had an increasing ratio of translation genes, ranging from zero to 90%, keeping constant the GFP-HA fraction at 2% and the overall DNA density by supplementing brushes with DNA coding for an unrelated gene. Our initial attempt to use an initial PURE reaction composed of all the threshold concentrations failed to support GFP-HA synthesis and capture on the surrounding surface (Supplementary Fig. 27), suggesting that these conditions were too stringent. Only with the addition of 25 nM of each IFs and RFs to the initial PURE reaction, with all other Pi at their threshold concentration, we could observe accumulation of GFP-HA on the surface surrounding brushes coding for the translation proteins but not next to brushes with unrelated genes (Fig. 6c–e; Supplementary Fig. 28; Supplementary Tables 2 and 3 and Supplementary Movie 4). To improve the signal-to-background ratio for the measurement of the maximal GFP accumulation rate, we doubled the initial protein concentrations from a 100-fold dilution of the full PURE levels to a 50-fold dilution, with the exception of EF-Tu, which remained at a two-fold dilution from the full level in both versions. The increase in initial protein concentrations accelerated the initial protein synthesis rate (Supplementary Fig. 28). The GFP accumulation rate for the 50-fold diluted PURE peaked at about 3 h compared to a full PURE reaction, which peaked already at 50 min (Supplementary Figs. 29 and 30).

a Scheme: biogenesis for all translation factors from a minimal translation system (see Supplementary Table 2). b Gene ratios within the DNA brush (see Supplementary Table 3). c TIRF microscopy images of the synthesis of all translation proteins from surface-immobilized DNA brushes. Top row: DNA brush with a gene ratio of 2% coding for GFP-HA and 98% for a non-translation (“dummy”) protein (see b)(DNA not labeled). Bottom row: DNA brushes with a gene ratio of 90% coding for all translation genes (DNA-labeled), 2% coding for GFP-HA and 8% for dummy protein (see b). Upon exposure to a protein synthesis reaction at threshold, surface-bound GFP-HA accumulates around DNA brushes only with translation genes. Scale bar = 200 µm. d Average GFP FI(t) for DNA brushes with different fractions of translation genes. Repeats in Supplementary Fig. 28. e Mean fold change of GFP FI ± s.e.m from DNA brushes with different amount of translation genes (normalized to the signal from “0%” brush) at = 6 h and its s.e.m. as a function of the DNA FI (translation protein-DNA + GFP DNA) for different DNA brushes (n = 6 DNA brush cluster from two different slides). Source data are provided as a Source Data file.
Discussion
We devised a bottom-up self-regeneration scenario and demonstrated that all thirty proteins constituting the translation machinery of E. coli could be synthesized from their cognate genes in a functional form in the PURE system, both individually and simultaneously. To do that, we employed two general strategies: (i) We reduced the initial protein factor(s) to the threshold concentration at which the fold-change in protein synthesis rate per unit of nascent factor was maximized. (ii) As a consequence of the initial low rates due to the threshold concentrations, we employed a mechanism to catalyze and enhance the efficiency of protein synthesis. Specifically, we utilized gene brushes as reaction centers within a large reservoir of resources, co-localizing the gene expression machinery, enhancing both the efficiency of the reaction and the sensitivity of detection. Other artificial cell designs that cluster genes on large plasmids22,30 or by physical anchoring to specific sites, e.g., to membranes31 could be utilized as well.
The gene clustering at the DNA brushes creates confinement, keeping machinery at the site of action against entropy and mirrors natural strategies, such as bacterial transcription and translation foci32, or co-translational regulation at bacterial operons33. By expressing some of the translation proteins with a C-terminal GFP fusion, we could directly gain support for this scenario by observing their high local concentration at the DNA brush by TIRF microscopy. The thickness of the DNA brush at the salt conditions of the PURE is about 100–120 nm, which is in the same order of magnitude as the exponential decay length of the evanescent wave used in the TIRF microscopy34,35. This means that the TIRF signal captures only the molecules within or in very close proximity to the DNA brush and thus directly reports on the relative concentration of molecules around the brush. For every fusion protein that remained active, we observed an increased localization signal within the first 20 min of the reaction. This early peak signal at the brush diminished as the surrounding solution filled with diffusing nascent proteins; nevertheless, it showed that nascent factors remained transiently at their own gene cluster compared to unrelated control fusion proteins. The strong accumulation of IF1 and RF1 wild type and active-site mutants at the DNA brush, which persisted for several hours, can most likely be attributed to their intrinsic, nonspecific RNA-binding activity, especially for IF1, rather than to translating ribosomes29.
To provide a rough estimate of the local concentration of the translation machinery at the DNA brush, we used the GFP signals from TIRF imaging in the more concentrated PURE version (see Supplementary Table 4) and previous characterizations of the DNA brush. The density of genes in the DNA brush for an average gene length (2000 base pairs) was determined previously to be roughly 100–1000 genes per square micron34,35,36. Thus, the concentration of genes within the DNA brush is about 1–10 μM, which is three orders of magnitude higher than the gene concentration in a bulk solution reaction (mostly about 1 nM). Using previous characterization of transcription from DNA brushes, we can assume a transcription rate of about one molecule per gene per every few minutes, placing the local mRNA concentration also within a micromolar range. Given that the intensity of TIRF signal at the DNA brush of expressed GFP reaches about 1 µM within the first hour (Fig. 5c) and that each ribosome does not complete more than two translation cycle in the PURE37, we can assume that the concentration of translating ribosomes around the brush is also in the micromolar range. Then, estimating that on average at least one of each translation factor interacts with a ribosome, the local concentration of each translation factor is expected to be comparable to those of the ribosomes. Although the concentrated PURE mix supplies near-saturating translation components in the bulk, we observe transient accumulation of nascent GFP-fusion proteins at the DNA brush, indicating a positive feedback loop between local synthesis of translation proteins and localized translation. Notably, while the local concentration of nascent proteins (as reflected by the TIRF signal at brush) varied among constructs, it correlated with the global concentration of nascent proteins (as measured by the epifluorescence signal).
In defining the threshold concentration for each component, we observed a non-additive effect as the threshold for several proteins simultaneously was higher than the sum of the individual ones, and tmax was extended (Fig. 2c). As the self-regenerating system grows with the number of components, there is a necessity for a cost-benefit analysis for every additional factor, such as chaperones38, modifying enzymes39,40 or additional factors41,42, that are added to the 30 translation proteins and the ribosome, as the benefit of improved efficiency will have to surpass the costs of its own biogenesis. This is reflected in our choice to not include RF2 in the biogenesis reaction since in principle a minimal system requires only one release factor. Although some of the genes have a RF2 dependent stop codon (see Methods), we did not observe an effect on their biogenesis since the contamination levels of RF2 coming from the ribosomes were sufficient (Supplementary Fig. 5).
The prevailing notion is that a system can be called self-regenerating only when all original machinery is fully replaced by nascent components. Practically, for the PURE system, this prevailing notion cannot be simply executed. A full PURE reaction contains ~10,500 nM translation factors, neglecting the ribosomes and tRNA molecules, whereas a single batch produces only ~1600 nM total protein at a maximum of ~ 14 nM min−1. In the PURE at threshold, the total translation protein concentration was reduced to 1360 nM, which is about 13% of full PURE, and although it has lower yields and max. synthesis rates (~10%) than the full system, it offers a bottom-up strategy for constructing a fully self-regenerating in vitro protein synthesis platform. Other approaches, which can be integrated with ours, focus on increasing the enzymatic efficiency of the protein components through protein engineering43, by substitution with more active components that function better in a dilute system15, or by creating conditions that prolong the reaction with continuous feeding of nutrients and removal of toxic waste products44,45,46. DNA brushes may create an alternative scenario with a local concentration of nascent proteins higher than the machinery that created them.
While determining the threshold concentrations for the translation proteins, we found significant carry-over contamination from the ribosome, which can have direct implications for self-regenerating protein synthesis. For example, the twenty aaRSs reached their threshold at only pico-to nanomolar levels, which is low enough to allow for biogenesis over several generations despite low yields in the PURE system, as demonstrated by other studies15,16. Hence, the purity of the ribosomes can change the results of the self-regeneration capabilities of PURE components, and specifically the aaRS. In our study, GlyRS stood out as the aaRS with the highest threshold concentration of around 40% of the full levels. Recently, GlyRS was found to have the highest ratio between the activity of the in situ expressed and purified enzyme of all 20 aaRS15. Thus, the high threshold concentration may originate from a low activity of the purified GlyRS.
The high threshold concentration of EF-Tu, which exceeded that of all other 29 proteins by a factor of ten to a hundred, is another consideration at the other extreme of self-regeneration. EF-Tu is one of the most abundant proteins in prokaryotes, comprising about 6% of the total protein of an E. coli47. In the full PURE, it makes up about 21% of all translation proteins, and in the PURE at threshold, it is about 74%, highlighting its importance at minimal conditions and the limitations it imposes for self-regeneration, which were alleviated by the DNA brush localization.
In agreement with recent findings of lower yield of proteins expressed in the PURE system above a molecular weight of ~82 kDa12 and observed accumulation of truncated protein products22, we observed a moderate negative correlation between the length of the aaRS and the efficiency of the biogenesis process rather than a dependence on the amino acid composition, at least as long as the amino acid occurrence within a protein sequence is greater than one. Surprisingly, the biogenesis of ArgRS required a chaperone mix, whereas other studies have demonstrated ArgRS biogenesis without any additional factors15,16. We cannot explain this discrepancy but acknowledge a difference in the operational temperature, which can influence protein folding; 30 °C and 34 °C, instead of 37 °C, which was used in this study. While we can exclude the necessity of the chaperone mix for the biogenesis of 12 other aaRSs, we did not verify its requirements for the biogenesis of the remaining 7 aaRS and the IFs, EFs, and RFs.
The threshold concentrations for the 30 translation proteins, the ratio of the different genes in the DNA brush and the localization effect supporting their biogenesis, present a promising foundation and starting point to build on for the creation of a self-regenerating in vitro protein-based system. Ongoing efforts towards the biogenesis of the ribosome in a minimal reconstituted system20,44,48, the creation of synthetic tRNA molecules49,50 and the replication of genetic material51,52,53,54 in addition to the current results, open avenues towards a minimal protein synthesis system that can self-regenerate all of its components.
Methods
PURE system preparation
Protein purification
The PURE system was prepared according to Shimizu et al.55: E. coli BL21(DE3) pLyS strain was used for protein expression. All plasmids encoding PURE proteins were obtained from Y. Shimizu. Plasmids were transformed and amplified in E. coli DH5α. except for plasmids coding for AlaRS, AsnRS, ThrRS, IF1-3, EF-G, RRF, CK, T7RNAP, and PheRS, which were transformed to E. coli JM109 due to their expression vector (pQE30). All plasmids were subsequently transformed in E. coli BL21 (DE3) pLyS for protein expression. All cultures were grown at 37 °C, 250 rpm. Overnight cultures were grown in 5 mL of LB medium (LBX0102, Formedium, UK) with 100 μg/mL of ampicillin or 50 μg/mL of kanamycin. Each strain was inoculated in a flask with 1 L of LB and grown to OD600 = 0.6 before induction with 0.1 mM of isopropyl β-D-1-thiogalactopyranoside (IPTG from Applichem, Germany) for 3 h, and then harvested after centrifugation and stored at −80 °C.
Protein purification buffer compositions are as follows. Buffer A: 50 mM HEPES-KOH (8042391, Bio-Lab, IL) pH 7.6, 1 M ammonium chloride (3384-12, Avantor, USA), 10 mM MgCl2 and 7 mM 2-mercaptoethanol (b-ME, AlfaAesar, ThermoFisher Scientific, USA, only added immediately before use). Buffer B: 50 mM HEPES-KOH pH 7.6, 100 mM KCl (7447-40-7, Merck, DE), 10 mM MgCl2, 500 mM imidazole (GB9580, Glentham Life Sciences, UK) and 7 mM b-ME. HT Buffer: 50 mM HEPES-KOH pH 7.6, 100 mM KCl, 10 mM MgCl2, and 7 mM b-ME. Protein storage buffer: 50 mM HEPES-KOH pH 7.6, 100 mM KCl, 10 mM MgCl2, 30% glycerol (007120223300, Bio-Lab, IL) and 7 mM b-ME.
The cell pellets were re-suspended in 30 mL of buffer A and lysed by sonication for 3 min on ice (Sonics Vibra-Cell VC 750; probe tip diameter: 13 mm; 20 s:20 s pulse; 70% amplitude). Cell debris was removed by centrifugation (25,000 cf, 30 min, 4 °C). The protein was then purified from the supernatant either in a gravity-flow column or with an FPLC (Äkta avant, Cytiva, USA). For the gravity-flow method, the supernatant was mixed with 2–3 mL of equilibrated resin (PureCube, Ni-NTA agarose, Cube Biotech) and incubated for up to 2 h at 4 °C. After the incubation, lysate was loaded on a column (Econo-Pac, Bio-Rad, USA). The column was washed with 30 mL of a wash buffer (95% buffer A, 5% buffer B) and eluted with 15 mL of an elution buffer (10% buffer A, 90% buffer B). For the FPLC method, the supernatant was loaded onto a 5 mL HisTrap FF Crude column (Cytiva, USA). The column was then washed with 20 column volume of wash buffer (95% buffer A, 5% buffer B). Proteins were eluted by a linear 20 column volume gradient (5 to 100% buffer B) with a flow rate of 1 ml/min. Following either method, the elution fraction was dialyzed twice against HT buffer followed by once against protein storage buffer. Protein purity was assessed with SDS-PAGE gel (4–20% GeBaGel, Gene Bio-Application, IL) and Coomassie staining (InstantBlue, Abcam, UK). The protein solution was concentrated using a Vivaspin 20 centrifugal concentrator (molecular cut-off depending on the protein, Cytiva, USA). Protein concentrations were estimated by absorbance at 280 nm using calculated protein extinction coefficients. Proteins were then stored at −80 °C in aliquots.
Ribosome purification
Ribosomes were purified from the E. coli strain A19 (no antibiotics). The cells (1 l culture) were grown from an overnight culture to the OD of 1.2 at 37 °C, centrifuged (8000 x g, 30 min at 4 °C) and resuspended in 45 ml in ribosome buffer A (70 mM KCl, 10 mM MgCl2, 10 mM Tis-HCl, pH 7.4 + 7 mM b-mercaptoethanol) Afterwards, cells were lysed using a French press (Constant Systems Cell Disruptor, England, 12,000 PSI). Protease Inhibitor Mix (1 mM PMSF, 0.4 mM Benzamidine, 0.06 mM Benzamide) is added to the lysate and we centrifugate again (30,000 × g, 1 h, 4 °C). We then continued with following three different published protocols.
(1) Anion Exchange Chromatography, as described in Trauner et al.56 using a quaternary amine monolithic column (CIMmultus QA 80 ml, 2 µm, BIA separations) connected to an FPLC (Äkta avant, Cytiva, USA). Briefly, the lysate was filtered and loaded to the column, washed (15 ml/min, 2 x CV) with ribosome buffer A and then with additional 25% ribosome buffer B (ribosome buffer A + 1 M NH4Cl). Ribosome were eluted by a gradient of buffer B (25–65%, 10 ml/min, 80 min) and fractions were collected and concentrated on a Vivaspin 20 3kDA MWCO with a subsequent buffer exchange to the ribosome storage buffer(20 mM Hepes pH 7.6, 30 mM KCl, 6 mM MgAce, 7 mM b-mercaptoethanol).
(2) Hydrophobic interaction chromatography followed by ultracentrifugation was performed according to Shimizu et al.55. After growth, celll pellets were dissolved in suspension buffer (10 mM Hepes-KOH, pH 7.6, 50 mM KCl, 10 mM MgAce, 7 mM 2-mercaptoethanol) and lysed, 1.5 M ammonium sulfate is added the lysate, and after centrifugation (20,000 x g, 20 min) the supernatant is filtered (0.45 mm). Then the sample is applied to a 5 mL HiTrap Butyl FF column (Cytiva, USA) connected to an FPLC (Äkta avant, Cytiva, USA), washed, eluted (20 mM Hepes-KOH, pH 7.6, 0.75 M ammonium sulfate, 10 mM MgAce, 7 mM 2-mercaptoethanol) and their buffer was exchanged the to ribosome storage buffer. The ribosomes were layered on a sucrose cushion buffer (10 Tris, pH = 8, Mg(OAc)2 14 mM, KOAc 60 mM, Sucrose 1.1 M, b-ME 6 mM) and pelleted using a Ti-70 rotor at 311,000 x g (55,000 rpm), 4 °C for 17 h.
(3) For the third purification method, we followed the protocol of Halfon et al.57 we first used an ultracentrifuge with a sucrose cushion buffer like previously mentioned, followed by sucrose gradient ultracentrifugation on a 10–40% sucrose gradient with the same buffer 10 Tris, pH = 8, Mg(OAc)2 14 mM, KOAc 60 mM, b-ME 6 mM for 16 h at 62,000 x g in a swing SW-28 rotor (19,000 rpm). Ribosome fractions were collected according to A260 values and pelleted at 200,000 x g (45,000 rpm) using a Ti-70 rotor for 19 h. After purification, the ribosomes from each method were dissolved in or dialyzed against 1× Ribosome buffer (20 mM Hepes, pH = 7.6, 30 mM KCl, 6 mM Mg(OAc)2, 7 mM 2-mercaptoethanol) and stored in aliquots at −80 °C.
Mass spectrometry
We measured the protein compositions of three different batches of commercial ribosome solution from (Gene frontier) and three different samples of self-purified ribosomes (see Ribosome purification) (n = 3, different batches for commercial ribosome, and n = 3 from the same batch for self-purified ribosomes). In between runs, we added blanks to further minimize potential carry-over between samples. The samples were subjected to tryptic digestion using an S-trap The resulting peptides were analyzed using Bruker Aurora column on nanoflow liquid chromatography (nanoElute2) coupled to high resolution, high mass accuracy mass spectrometry (timsTOF Pro), in DIA mode. Raw data was processed with Spectronaut software version 18.1. The data was searched against the E. coli proteome database (“Ecoli_UP_Jan25.fasta”), appended with common lab protein contaminants. The quantitative comparisons were calculated using Perseus v1.6.2.3. The intensity values were log-transformed. Missing intensity values were replaced by a zero value. For convenience reasons, all values (intensities and ratios) were transformed back from their log values (since missing values were replaced by zero values. When transferring from log, you will see “1” values).
In addition to fold change values (using geometric means), we performed a student’s t test for these two groups. The resulting p values can help assess the statistical significance of the observed differences in protein abundance. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE58 partner repository with the dataset identifier PXD071320.
Buffer preparation
PURE buffer was prepared by stepwise addition of the following solutions, in the order they are listed here, starting with the volume of water necessary to reach the desired concentrations. The final concentrations are listed in the PURE assembly below. The concentrations listed here are for a 2× buffer stock: 100 mM HEPES-KOH pH 7.6 200 mM K-glutamate, 20 mM Mg(OAc)2, 4 mM ATP, 4 mM GTP, 2 mM CTP, 2 mM UTP, 108 units of A280/mL tRNA, 40 mM creatine phosphate, 2 mM DTT, 20 µg/mL folinic acid, 4 mM spermidine, 0.6 mM each amino acid (prepared according to ref. 59). The solution was aliquoted and stored at −80 °C. The optimal Mg(OAc)2 for GFP expression was determined by doing an initial Mg(OAc)2 scan between 10 and 30 mM and varied slightly between 16 and 22 mM between buffer preparation batches.
PURE assembly
The PURE system was mixed on ice. The protein solution contained (following Shimizu et al.55) except for RF2: 704 nM AlaRS, 30 nM ArgRS, 408 nM AsnRS, 119 nM AspRS, 23 nM CysRS, 59 nM GlnRS, 230 nM GluRS, 85 nM GlyRS, 17 nM HisRS, 381 nM IleRS, 41 nM LeuRS, 109 nM LysRS, 27 nM MetRS, 134 nM PheRS, 154 nM ProRS, 38 nM SerRS, 83 nM ThrRS, 29 nM TrpRS, 12 nM TyrRS, 16 nM ValRS, 1.036 µM IF1, 405 nM IF2, 456 nM IF3, 636 nM EF-G, 2.255 µM EF-Tu, 1.588 µM EF-Ts, 247 nM RF1, 162 nM RF3, 458 nM RRF, 568 nM MTF, 89 nM CK, 64 nM NDK, 50 nM T7 RNA P., 132 nM MK, 30 nM PPiase; The buffer and energy solution (see “Buffer Preparation”). The ribosomes: 2 µM of commercial (Solution III of PUREfrex 2.1, Gene Frontier, JP) or purified ribosomes. We added to each reaction 1.25%(v/v) GroE mix Gene Frontier, JP and 2.5%(v/v) DnaK mix Gene Frontier, JP, which results in the following concentrations of chaperones: DnaK = 2.5 µM, DnaJ = 0.5 µM, GrpE = 0.5 µM, GroEL = 250 nM and GroES = 500 nM PURE assembly was mostly done by hand pipetting. Initial concentrations of single PURE proteins were diluted by hand in protein storage buffer and then dispensed with a liquid handling robot (iDOT, Dispendix Germany) into a premixed ΔPi-PURE. For the ribosome purity test (Supplementary Fig. 3), we pre-mixed solutions containing 5 proteins by hand, and then we assembled the PURE with an Echo 525 (Labcyte, USA).
DNA preparation
For expression in PURE, all translation genes were placed under a T7 promoter and a strong ribosome binding site (RBS). All genes were cloned into pET-plasmids using Gibson Assembly (NEBuilder HiFi Assembly Master Mix, E2621, NEB, USA). The translation genes have either UGA or UAA stop codons. The UGA stop codon requires RF2, and although RF2 was not added to the PURE mix, its carry-over contamination from the ribosomes was sufficient (Supplementary Fig. 5). Thus there was no observed difference in biogenesis (Δkmax) for genes harboring UGA vs. UAA stop codons. For primers and sequences see Supplementary Data File 1. Plasmids were amplified in E. coli DH5α and purified using Wizard SV-Gel Miniprep (Promega, USA), and DNA concentrations were determined using a NanoDrop (NanoPhotometer, Implen, USA). Linear double-stranded DNA fragments were amplified from plasmids with polymerase chain reaction (PCR) with KAPA HotStart ready mix (07958935001, Roche, Switzerland), using a reverse primer conjugated to biotin35 and a forward primer that was either unmodified or conjugated to an ATTO 647 fluorophore on its 5′ end (Integrated DNA Technologies, USA), and purified with the Wizard SV-Gel and PCR Clean-Up System (Promega, USA).
For surface immobilization, DNA was mixed with streptavidin (S4762, Sigma-Aldrich) at a 1.4:1 streptavidin: DNA ratio in 1× phosphate-buffered saline (PBS, 02-023-5A, Sartorius, Germany) and 7% glycerol (Bio-Lab, Israel), forming a DNA-streptavidin conjugate. The correct amplification of DNA fragments and DNA-streptavidin conjugates was verified with 1% agarose gel electrophoresis. The solution deposited on the DASIY-treated surface to create DNA brushes always has a total gene concentration of 150 nM. According to the experiment, this solution was composed of different gene mixes (Supplementary Table 3).
Bulk solution experiments
Bulk expression experiments in the PURE system were conducted at 37 °C in a ClarioStar plate reader (BMG Labtech, Germany). DNA was added to the PURE reaction prepared as described above. Volumes of 10 µL were pipetted in a black optical-bottom 384-well plates (Nalge Nunc International, USA), sealed with a sticky transparent foil (SealPlate, Excel Scientific, USA), and spun down at 1000 x g for 30 s. GFP fluorescence was measured with an excitation filter of 470/15 nm, a dichroic filter of 491 nm, and an emission filter of 515–20 nm. Every experiment contained a well with full PURE with the same GFP-DNA concentration for normalization purposes between experiments. For repeats of the same experiment, the critical components of the PURE reaction, such as the protein threshold concentration or the gene DNA, were always added separately to each reaction.
Gel electrophoresis analysis of fluorescently labeled nascent proteins
A more concentrated PURE protein and energy/buffer solution was mixed following the composition provided by PURE version 2 by Kazuta et al.11. Commercial ribosomes (Solution III of PUREfrex 2.1, Gene Frontier, JP) were used according to their published manual. For each PURE gene, 1 nM of DNA and 4%(v/v) Green-Lys tRNA (FluoroTect GreenLys, Promega, USA) were added to the assembled PURE and incubated for 4 h at 37 °C in a PCR thermocycler (Bio-Rad, USA). The reactions were incubated for 2 min at 60 °C and assessed with SDS-PAGE (160 V 1 h) gel (4–20% GeBaGel, Gene Bio-Application, IL) and imaged with a Typhoon FLA 9000 (GE, USA).
Glass surface experiments
Slide preparation
Fused silica slides (24 x 24 x 1 mm, UQG Optics, UK) were coated with a biocompatible photosensitive monolayer34 of a polymer formed by a polyethylene glycol backbone with a protected amine and a triethoxysilyl group at either end. The slides were first cleaned in boiling ethanol (830109326, 96%, Gadot Group, IL) for 10 min, followed by base piranha cleaning (1:1:4 H2O2 (000855032300, Bio-Lab, IL):NH3 (105432, Merck, DE):H2O) at 70 °C for 10 min. The slides were incubated for 20 min with a 1 mg ml−1 concentration of polymer dissolved in dried toluene (244511, Sigma-Aldrich, USA), then rinsed with toluene (Bio-Lab, Israel) and dried. Surface amines were then deprotected with UV exposure (UV-KUB, FR, 365 nm, 2.5 J/cm2) through a custom photomask with an array of 30 µm hexagons (CAD/Art Services. USA). The slides were immediately incubated with 0.5 mg/ml biotin 3-sulfo-N-hydroxysuccinimide ester (EZ-link NHS biotin, 20217, Thermo Fisher Scientific, USA) in 0.2 M borate-buffered solution pH 8.6 (Thermo Fisher Scientific) for 30 min, then rinsed with water and dried. The slides were then fixed on custom fused silica prisms (Zell Quarzglas und Technische Keramik, Germany) with Frame-Seal Slide Chambers adhesive (Bio-Rad, USA). Frame-Seal Slide Chambers of size 15 × 15 mm, with a volume of 65 µL, were applied to the polymer-treated side of the fuseSource data and code: autonomous biogenesis of all thirty translatid-silica slides to act as reaction chambers for the DNA brushes.
DNA deposition
The biotinylated surfaces of the wells were patterned with nano-liter DNA-SA droplets using the GIX Microplotter II (Sonoplot Inc., USA) and incubated overnight at room temperature and 50–65% humidity to allow DNA brush build-up through biotin-streptavidin binding. We defined 12 positions for DNA brushes on each chip, ensuring at least a 3 mm difference between positions. We spotted three different identical DNA brushes at each position to increase reproducibility (also referred to as a cluster) and increase GFP signal strength. In each experiment, we titrated the concentration of the PURE genes in the DNA brush cluster in question (e.g., 0, 20, 50, and 90%). The dilutions of the PURE genes were done with a control gene that codes for a non-interacting protein so that the total DNA concentration remained at 150 nM. On each glass slide, we repeated the titration series of DNA brush clusters at least twice.
Capture antibodies immobilization
Biotinylated anti-HA antibodies (50 µg/ml, High Affinity, 12158167001, Roche, Sigma-Aldrich) were mixed with streptavidin (S4762, Sigma-Aldrich) at a concentration ratio of 1.5:1 in 1× PBS and incubated for 30 min at 4 °C, after which the mix was diluted to 5 µg/ml of antibodies in 1× PBS. 200 µl of this solution was applied to the surface of the chip after the overnight DNA incubation and incubated for 1 h at 4 °C. Afterwards, the surface was washed several times with 1× PBS, then with 50 mM HEPES pH7.4.
Protein expression from DNA brushes
The HEPES buffer was washed and exchanged with a PURE mix in four consecutive washes of 50 µL each. The well was then sealed with a glass coverslip. The slide and attached prism were immediately positioned on the microscope stage and kept at 17 °C with a temperature-controlled holder. The temperature was increased to 37 °C to start gene expression. Different experiments on a chip were repeated at least two times.
TIRF imaging
Prior to imaging, the space between the prism and the slide was filled with index-matching liquid (Cargille, USA). The wells were imaged with a custom-made Total Internal Reflection Fluorescence (TIRF) system. An upright microscope (Olympus BX51WI, JP) was equipped with a motorized stage (Scientifica, UK) and a laser system for TIRF excitation. Two lasers (OBIS 488-150 LS and OBIS 647 LX, Coherent, USA) were coupled into a single-mode optical fiber (Oz optics, CAN), after which the beam was collimated and directed to the prism with a goniometer (Thorlabs, USA) at an angle of total internal reflection on the surface of the chamber, creating an evanescent wave inside the chamber. Images were acquired using an Andor iXon Ultra camera (Andor Technology, UK) and a 10× Olympus objective. The set-up was controlled through a custom-made LabView code (National Instruments, USA). The DNA signal was measured with the red laser at the beginning of the acquisition at 17 °C. The GFP signal time series was measured in the green laser afterwards at 37 °C.
Data processing and analysis
Plate reader data
Automated data processing and analysis were implemented with Python v3.7, and data was generally handled with the pandas (v2.2.3) package. For bulk solution experiments, fluorescence intensity time traces were normalized:
where FIi(t) is the fluorescence intensity time trace for sample i, minFIi is the minimum fluorescence intensity value of sample i (background level), and maxFIfull is the maximal value (only after the signal has saturated with time ~4–8 h) of a full PURE reaction from the same experiment with the same concentration of reporter gene. The FI(t) was smoothed by applying the Savitzky–Golay filter (imported from scipy (v.1.14.1) package with a window length = 7 and a polynomial order = 0) before the first rate is calculated. The normalized max. rate is calculated as the max. rate of the sample divided by the max. rate of a full PURE sample from the same experiment. The second rate is calculated by applying the Savitzky–Golay filter to the first-rate and then calculating its rate again. The normalized second rate is calculated by dividing the max. of the 2nd rate of sample i, by the max. of the 2nd rate of a full sample.
Δkmax is defined as the difference between the normalized max. rate in the presence of the gene and the normalized max. rate in the absence of the gene.
Kendall’s rank correlation coefficient was calculated with the kendalltau function with variant b in the scipy (v.1.14.1) package. For categorical variables (e.g., AaRS: “I” or “II”), the rank-biserial correlation r was calculated with:
where U is calculated by the Mann–Whitney U test from the scipy (v.1.14.1) package, and n1 and n2 are the sample sizes of the groups that belong to class I or II.
Schematics and illustrations were made with Inkscape.
TIRF microscopy data
For TIRF microscopy images, we averaged the GFP signal of the whole image for each time frame. The background signal was determined to be the green channel signal from one of the first 5 frames and then subtracted from the GFP signal of all other time frames. For experiments with a very low GFP signal compared to the background, we defined two regions of interest (ROI) with the same area. The first ROI-1 is nine circles positioned in the center of the hexagons that contain the antibody traps for GFP-HA (specific GFP signal), and the second ROI-2 is nine circles positioned in areas between hexagons, where the density of the antibodies is significantly reduced (background). The circle positions are chosen to be close to each other within the image to avoid differences in illumination due to the alignment of the laser. The background-subtracted GFP signal for each time frame is then calculated by the mean intensity of ROI-1 minus ROI-2.
For time series of GFP expression from DNA brushes, t = 0 is set to the time point when the temperature on the prism holder reached 37 °C, determined using a temperature sensor. Image acquisition started after the focus plane had been adjusted at 37 °C. This time difference was measured and considered in the data analysis. The TIRF signal from the labeled DNA in the DNA brushes was measured by defining two ROIs of the same area, the first ROI-1, including the DNA brushes (DNA-specific signal), and the second ROI-2, excluding the DNA brushes (background).
The normalization of the GFP signal across glass slides for the TIRF microscopy data was conducted in the following way. For each gene titration series using DNA brushes (e.g., 0%, 10%, 20% …), we used the GFP FI signal from DNA brush without translation genes (“0%”) to normalize the GFP FI signal from the other DNA brushes within this gene titration series reporting on the fold-change in GFP SI signal compared to the “0%” DNA brush. Then, the average and s.e.m. for the fold changes is calculated from the each genetic compositions of DNA brush.
Statistics and reproducibility
No statistical method was used to predetermine sample size, but it was on par with standard biophysical measurements. In plate reader experiments, each reaction was repeated at least three times, and it was made sure that the reactions for the repeats were not prepared or measured on the same day as the first measurement. For TIRF microscopy data, each silica slide contained at least three DNA brushes per cluster, and each cluster was repeated at least once per silica slide. In addition, each measurement was repeated on at least two different silica slides. Data that was determined to be heavily affected by technical problems was discarded after discussion between authors. As standard procedure in our field of study, the experiments were not randomized, and the investigators were not blinded to allocation during experiments and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The source data generated in this study have been deposited in the Zenodo60 database under accession code 17546983: https://doi.org/10.5281/zenodo.17546983. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD071320 [CODE] (https://www.ebi.ac.uk/pride/archive/projects/PXD071320). The source data of the Supplementary Figs. is provided in the Supplementary Information/Source Data file. Source data are provided with this paper.
Materials availability
Requests for materials should be addressed to Roy H. Bar-Ziv and Shirley S. Daube.
Code availability
The code used in this study is deposited in the Zenodo60 database at https://doi.org/10.5281/zenodo.17546983.
References
Noireaux, V., Maeda, Y. T. & Libchaber, A. Development of an artificial cell, from self-organization to computation and self-reproduction. Proc. Natl. Acad. Sci. USA 108, 3473–3480 (2011).
Shimizu, Y. et al. Cell-free translation reconstituted with purified components. Nat. Biotechnol. 19, 751–755 (2001).
Niwa, T. et al. Large-scale analysis of macromolecular crowding effects on protein aggregation using a reconstituted cell-free translation system. Front. Microbiol. 6, 1113 (2015).
Awai, T., Ichihashi, N. & Yomo, T. Activities of 20 aminoacyl-tRNA synthetases expressed in a reconstituted translation system in Escherichia coli. Biochem. Biophys. Rep. 3, 140–143 (2015).
Berhanu, S., Ueda, T. & Kuruma, Y. Artificial photosynthetic cell producing energy for protein synthesis. Nat. Commun. 10, 1325 (2019).
Giaveri, S. et al. Integrated translation and metabolism in a partially self-synthesizing biochemical network. Science 385, 174–178 (2024).
Wei, E. & Endy, D. Experimental tests of functional molecular regeneration via a standard framework for coordinating synthetic cell building. Preprint at https://doi.org/10.1101/2021.03.03.433818 (2021).
Kohyama, S., Merino-Salomón, A. & Schwille, P. In vitro assembly, positioning and contraction of a division ring in minimal cells. Nat. Commun. 13, 6098 (2022).
Luisi, P. L., Ferri, F. & Stano, P. Approaches to semi-synthetic minimal cells: a review. Naturwissenschaften 93, 1–13 (2006).
Forster, A. C. & Church, G. M. Towards synthesis of a minimal cell. Mol. Syst. Biol. 2, 45 (2006).
Kazuta, Y., Matsuura, T., Ichihashi, N. & Yomo, T. Synthesis of milligram quantities of proteins using a reconstituted in vitro protein synthesis system. J. Biosci. Bioeng. 118, 554–557 (2014).
Li, J. et al. Dissecting limiting factors of the protein synthesis using recombinant elements (PURE) system. Translation 5, e1327006 (2017).
Ganesh, R. B. & Maerkl, S. J. Towards self-regeneration: exploring the limits of protein synthesis in the protein synthesis using recombinant elements (PURE) cell-free transcription–translation system. ACS Synth. Biol. 13, 2555–2566 (2024).
Libicher, K. & Mutschler, H. Probing self-regeneration of essential protein factors required for in vitro translation activity by serial transfer. Chem. Commun. 56, 15426–15429 (2020).
Hagino, K., Masuda, K., Shimizu, Y. & Ichihashi, N. Sustainable regeneration of 20 aminoacyl-tRNA synthetases in a reconstituted system toward self-synthesizing artificial systems. Sci. Adv. 11, eadt6269 (2025).
Lavickova, B., Laohakunakorn, N. & Maerkl, S. J. A partially self-regenerating synthetic cell. Nat. Commun. 11, 6340 (2020).
Daube, S. S., Bracha, D., Buxboim, A. & Bar-Ziv, R. H. Compartmentalization by directional gene expression. Proc. Natl. Acad. Sci. USA 107, 2836–2841 (2010).
Levy, M. et al. Boundary-free ribosome compartmentalization by gene expression on a surface. ACS Synth. Biol. 10, 609–619 (2021).
Vonshak, O. et al. Programming multi-protein assembly by gene-brush patterns and two-dimensional compartment geometry. Nat. Nanotechnol. 15, 783–791 (2020).
Levy, M., Falkovich, R., Daube, S. S. & Bar-Ziv, R. H. Autonomous synthesis and assembly of a ribosomal subunit on a chip. Sci. Adv. 6, eaaz6020 (2020).
Libicher, K., Hornberger, R., Heymann, M. & Mutschler, H. In vitro self-replication and multicistronic expression of large synthetic genomes. Nat. Commun. 11, 904 (2020).
Doerr, A., Foschepoth, D., Forster, A. C. & Danelon, C. In vitro synthesis of 32 translation-factor proteins from a single template reveals impaired ribosomal processivity. Sci. Rep. 11, 1898 (2021).
Dora Tang, T. Y., van Swaay, D., deMello, A., Ross Anderson, J. L. & Mann, S. In vitro gene expression within membrane-free coacervate protocells. Chem. Commun. 51, 11429–11432 (2015).
Sokolova, E. et al. Enhanced transcription rates in membrane-free protocells formed by coacervation of cell lysate. Proc. Natl. Acad. Sci. USA 110, 11692–11697 (2013).
Shimizu, Y., Tanimura, N. & Matsuura, T. ePURE_JSBML: a tool for constructing a deterministic model of a reconstituted Escherichia coli protein translation system with a user-specified nucleic acid sequence. Adv. Biol. 7, 2200177 (2023).
Borkowski, O. et al. Cell-free prediction of protein expression costs for growing cells. Nat. Commun. 9, 1457 (2018).
Ganesh, R. B. & Maerkl, S. J. Biochemistry of aminoacyl tRNA synthetase and tRNAs and their engineering for cell-free and synthetic cell applications. Front. Bioeng. Biotechnol. 10, 918659 (2022).
Diago-Navarro, E., Mora, L., Buckingham, R. H., Díaz-Orejas, R. & Lemonnier, M. Novel Escherichia coli RF1 mutants with decreased translation termination activity and increased sensitivity to the cytotoxic effect of the bacterial toxins Kid and RelE. Mol. Microbiol. 71, 66–78 (2009).
Croitoru, V. et al. RNA chaperone activity of translation initiation factor IF1. Biochimie 88, 1875–1882 (2006).
Shepherd, T. R. et al. De novo design and synthesis of a 30-cistron translation-factor module. Nucleic Acids Res. 45, 10895–10905 (2017).
Nikolov, V., Lipowsky, R. & Dimova, R. Behavior of giant vesicles with anchored DNA molecules. Biophys. J. 92, 4356–4368 (2007).
Bakshi, S., Choi, H. & Weisshaar, J. C. The spatial biology of transcription and translation in rapidly growing Escherichia coli. Front. Microbiol. 6, 636 (2015).
Tian, T. & Salis, H. M. A predictive biophysical model of translational coupling to coordinate and control protein expression in bacterial operons. Nucleic Acids Res. 43, 7137–7151 (2015).
Buxboim, A. et al. A single-step photolithographic interface for cell-free gene expression and active biochips. Small 3, 500–510 (2007).
Buxboim, A., Daube, S. S. & Bar-Ziv, R. Ultradense synthetic gene brushes on a chip. Nano Lett. 9, 909–913 (2009).
Bracha, D., Karzbrun, E., Daube, S. S. & Bar-Ziv, R. H. Emergent properties of dense DNA phases toward artificial biosystems on a surface. Acc. Chem. Res. 47, 1912–1921 (2014).
Kempf, N. et al. A novel method to evaluate ribosomal performance in cell-free protein synthesis systems. Sci. Rep. 7, 46753 (2017).
Niwa, T., Kanamori, T., Ueda, T. & Taguchi, H. Global analysis of chaperone effects using a reconstituted cell-free translation system. Proc. Natl. Acad. Sci. USA 109, 8937–8942 (2012).
Heurgué-Hamard, V., Champ, S., Engström, A., Ehrenberg, M. & Buckingham, R. H. The hemK gene in Escherichia coli encodes the N(5)-glutamine methyltransferase that modifies peptide release factors. EMBO J. 21, 769–778 (2002).
Pierson, W. E. et al. Uniformity of peptide release is maintained by methylation of release factors. Cell Rep. 17, 11–18 (2016).
Ude, S. et al. Translation elongation factor EF-P alleviates ribosome stalling at polyproline stretches. Science 339, 82–85 (2013).
Li, J., Gu, L., Aach, J. & Church, G. M. Improved cell-free RNA and protein synthesis system. PLoS ONE 9, e106232 (2014).
Doi, Y., Ohtsuki, T., Shimizu, Y., Ueda, T. & Sisido, M. Elongation factor Tu mutants expand amino acid tolerance of protein biosynthesis system. J. Am. Chem. Soc. 129, 14458–14462 (2007).
Li, J. et al. Cogenerating synthetic parts toward a self-replicating system. ACS Synth. Biol. 6, 1327–1336 (2017).
Lavickova, B., Grasemann, L. & Maerkl, S. J. Improved cell-free transcription–translation reactions in microfluidic chemostats augmented with hydrogel membranes for continuous small molecule dialysis. ACS Synth. Biol. https://doi.org/10.1021/acssynbio.2c00453 (2022).
Karzbrun, E., Tayar, A. M., Noireaux, V. & Bar-Ziv, R. H. Programmable on-chip DNA compartments as artificial cells. Science 345, 829–832 (2014).
Furano, A. V. Content of elongation factor Tu in Escherichia coli. Proc. Natl. Acad. Sci. USA. 72, 4780–4784 (1975).
Aoyama, R. et al. In vitro reconstitution of the Escherichia coli 70S ribosome with a full set of recombinant ribosomal proteins. J. Biochem. 171, 227–237 (2021).
Hibi, K. et al. Reconstituted cell-free protein synthesis using in vitro transcribed tRNAs. Commun. Biol. 3, 350 (2020).
Miyachi, R., Shimizu, Y. & Ichihashi, N. Transfer RNA synthesis-coupled translation and DNA replication in a reconstituted transcription/translation system. ACS Synth. Biol. https://doi.org/10.1021/acssynbio.2c00163 (2022).
Sakatani, Y., Yomo, T. & Ichihashi, N. Self-replication of circular DNA by a self-encoded DNA polymerase through rolling-circle replication and recombination. Sci. Rep. 8, 13089 (2018).
van Nies, P. et al. Self-replication of DNA by its encoded proteins in liposome-based synthetic cells. Nat. Commun. 9, 1583 (2018).
Abil, Z. et al. Darwinian evolution of self-replicating DNA in a synthetic protocell. Nat. Commun. 15, 9091 (2024).
Nishizawa, C., Aburaya, S., Kosaka, Y., Sugase, K. & Aoki, W. Optimizing in vitro expression balance of central dogma-related genes using parallel reaction monitoring. J. Biosci. Bioeng. 138, 97–104 (2024).
Shimizu, Y., Kanamori, T. & Ueda, T. Protein synthesis by pure translation systems. Methods 36, 299–304 (2005).
Trauner, A., Bennett, M. H. & Williams, H. D. Isolation of bacterial ribosomes with monolith chromatography. PLoS ONE 6, e16273 (2011).
Halfon, Y. et al. Exit tunnel modulation as resistance mechanism of S. aureus erythromycin resistant mutant. Sci. Rep. 9, 11460 (2019).
Perez-Riverol, Y. et al. The PRIDE database at 20 years: 2025 update. Nucleic Acids Res. 53, D543–D553 (2024).
Grasemann, L., Lavickova, B., Elizondo-Cantú, M. C. & Maerkl, S. J. OnePot PURE cell-free system. JoVE, 62625. https://doi.org/10.3791/62625 (2021).
Schwarz-Schilling, M. Autonomous biogenesis of all thirty proteins of the Escherichia coli translation machinery, [Data set]. Zenodo, https://doi.org/10.5281/zenodo.17546983 (2025).
Acknowledgements
The authors thank André Rivalta and Ada Yonath for help with the ribosome purification, Galit Cohen and Haim Barr from the G-INCPM for help with the ECHO operation, Amir Pri-Or from the De Botton Protein Profiling institute of the Nancy and Stephen Grand Israel National Center for Personalized Medicine for the mass spectrometry measurements and Nimrod Barak for preliminary experiments with the GFP-fusions and Ohad Vonshak and Viktoriia Belousova for useful discussions. This research was supported by the United States Office of Naval Research (R.B.Z. and S.S.D. Award N62909-22-1-2042) and the Isak Ferdinand and Dwosia Artmann Research Fund for Biological Physics (R.B.Z.). The work of M.S.S. was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, Projektnummer 456967983). A.D. acknowledges funding from the EMBO postdoctoral fellowship, award number: ALTF 131-2020. Y.B. is the incumbent holder of the Beatrice Barton Research Fellowship.
Author information
Authors and Affiliations
Contributions
R.H.B.-Z., S.S.D., and Y.S. conceived the project. R.H.B.-Z., S.S.D., and M.S.S. designed the experiments. M.S.S., A.D., and Y.B. prepared the PURE system. Y.S. provided the PURE plasmids. A.D. provided the reporter genes. M.S.S. performed the experiments, ran the simulations and analyzed the data. M.S.S. and N.A. designed and performed the TIRF microscopy experiments. I.C. performed the functionality test for the GFP fusions. M.S.S. and I.C. planned and performed the TIRF experiments with the GFP fusions. M.S.S., S.S.D., and R.H.B.-Z. wrote the manuscript. All authors reviewed and discussed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Shunshi Kohyama and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Schwarz-Schilling, M., Cohen, I., Dupin, A. et al. Autonomous biogenesis of all thirty proteins of the Escherichia coli translation machinery. Nat Commun 17, 1028 (2026). https://doi.org/10.1038/s41467-025-67772-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-67772-8
