
Abstract
Lysosomal and endoplasmic reticulum (ER) Ca2+ release mutually influence each other’s functions. Recent work revealed that ER-located ryanodine receptor(s) (RyR(s)) Ca2+ release channels suppress autophagosome turnover by the lysosomes. In familial Alzheimer’s disease, inhibiting RyR hyperactivity restored autophagic flux by normalizing lysosomal vacuolar H+-ATPase (vATPase) levels. However, the mechanisms by which RyRs control lysosomal function and how this involves the vATPase remain unknown. Here, we show that RyRs interact with the ATP6v0a1 subunit of the vATPase, contributing to ER-lysosomal contact site formation. This interaction suppresses RyR-mediated Ca²⁺ release, leading to reduced lysosomal exocytosis. Pharmacological inhibition of RyR activity mimics these effects on lysosomal exocytosis. Retaining lysosomes inside cells via RyR inhibition increases ER-lysosomal contact site formation, rendering lysosomes more available for autophagic flux. In summary, these findings establish RyR/ATP6v0a1 complexes as ER-lysosomal tethers that dynamically and Ca2+ dependently regulate the intracellular availability of lysosomes to participate in autophagic flux.
Similar content being viewed by others

Introduction
Macroautophagy (hereafter referred to as autophagy) is an important catabolic pathway that sustains energy production ensuring the turnover of damaged organelles, misfolded proteins, and protein aggregates1. During autophagy, damaged cellular components are encapsulated within autophagosomes, double-membrane organelles, which subsequently fuse with lysosomes, enabling the degradation and recycling of their content2,3. Given that lysosomes control the final stages of this degradative process, any disruption in lysosomal function, commonly observed in a broad spectrum of human diseases, inevitably impairs autophagic flux4.
Lysosomes also serve as important intracellular Ca2+ stores that contribute to physiological Ca2+ signaling. In turn, proper lysosomal function is dependent on local Ca2+ signaling events. Lysosomes harbor several Ca2+-transport systems. Lysosomal Ca2+ efflux occurs via two pore channels (TPC1/TPC2) and transient receptor potential mucolipin 1 (TRPML1)5. The activity of these channels controls key lysosomal functions such as trafficking, fusion events, lysosomal exocytosis and lysosomal pH6. Ca2+/H+ exchanger (in nonplacental mammals) and TMEM165 have been identified as important, pH-dependent Ca2+-import systems in lysosomes7. Moreover, lysosomes are physically and functionally connected to the endoplasmic reticulum (ER), the major intracellular Ca2+-storage organelle. These contact sites enable bidirectional cross-talk between the two organelles, thereby coordinating their biological functions8,9. However, compared to ER-mitochondrial contact sites, the mechanisms and proteins contributing to ER-lysosomal tethering remain poorly understood. Yet, it is clear that ER-lysosomal contact sites are important and serve as hotspots for local and bidirectional Ca2+ signaling events between the ER and the lysosomes10,11,12. At the ER, two major families of intracellular Ca2+-release channels reside: inositol 1,4,5-trisphosphate receptors (IP3R)13 and ryanodine receptor(s) (RyR(s)), which are both activated via Ca2+-induced Ca2+ release14,15. The role of these channels, particularly for IP3Rs, at ER-lysosomal contact sites has only recently emerged. Indeed, it is well-established that IP3Rs are located at ER-lysosomal contact sites and contribute to Ca2+ transfers to lysosomes10,16. However, for RyR, their contribution to ER-lysosomal contact site formation, role in local ER-lysosomal Ca2+ signaling and impact on lysosomal function remains poorly understood especially at the molecular level. Nevertheless, the interplay between RyR function and autophagy, advocates for an important and direct role at ER-lysosomal contact sites17,18,19.
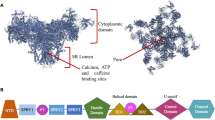
RyRs are large sarcoplasmic/endoplasmic reticulum (SR/ER)-resident Ca2+ release channels, particularly present in excitable cells. Three RyR isoforms exist with unique abundancies among specialized tissues. In neurons and brain, RyR2 is the most abundantly expressed isoform20. Here, RyR2-mediated Ca2+ release is important for memory formation21. In contrast, deregulated RyR-mediated Ca2+ release has been implicated as an early event driving the pathogenesis of several neurodegenerative diseases including (familial) Alzheimer’s disease ((F)AD)22,23,24,25,26,27,28. Elevated neuronal RyR-protein levels, together with associated excessive RyR activity, in various FAD mouse models and cell lines have been found to increase the production of amyloid beta (Aβ) from amyloid precursor protein (APP). These effects on Aβ can be reversed by pharmacological inhibitors of RyR channels22,23,24,25,26,27,28. In addition, post-translational modifications of RyR2 that evoke channel leakiness were reported to occur in the brain of AD patients and in mouse models, which in turn increased Aβ production and resulted in cognitive defects27,28.
Our own work in rat dissociated-hippocampal neurons revealed that spontaneous RyR-mediated Ca2+ release impairs the turnover of autophagosomes by lysosomes thus inhibiting autophagic flux which could potentially impair clearance of autophagic and lysosomal cargo including for instance Aβ17. This impaired clearance of Aβ was recently confirmed by another group through the introduction of a loss of function RyR2 gating mutation in a FAD mouse background19. Reduced RyR2 activity resulted in upregulation of autophagic flux, thereby increasing the clearance of Aβ. Yet, how exactly RyR activity controls autophagosome turnover in health and disease remains poorly understood. A recent study demonstrated that caffeine, which triggers RyR-mediated Ca2+ responses, results in the alkalization of lysosomal pH and interferes with the fusion of autophagosomes and lysosomes18. The same study showed, in both a FAD mouse model and human induced pluripotent stem cell (hiPSC)-derived neurons from AD patients, that lysosomal pH is elevated compared to control conditions. This elevated lysosomal pH is caused by a reduced abundance of the lysosomal vacuolar H+-ATPase (vATPase) complex, including the ATP6v0a1 subunit. Both lysosomal pH and vATPase-protein levels could be restored by inhibiting RyR activity. Interestingly, reduced ATP6v0a1-protein levels have been previously linked to FAD via presenilin 1 (PS1) at the ER29,30. FAD-associated mutations in PS1 impair delivery of ATP6v0a1 to the lysosomes impacting the formation of functional vATPase complexes and subsequently lysosomal pH and function.
In this study, we set out to unravel how RyRs control lysosomal function at the molecular level. We demonstrate that, RyRs act as ER-lysosomal tethering proteins by physically and functionally interacting with ATP6v0a1, thereby contributing to ER-lysosomal contact site formation. In doing so, RyRs directly and dynamically control lysosomal availability for either the autophagic pathway, when RyRs are inhibited, or lysosomal exocytosis, when RyRs are active, by regulating ER-lysosomal contact site formation in a highly flexible fashion.
Results
RyRs physically and dynamically interact with ATP6v0a1
Inhibiting FAD-associated excessive RyR-mediated Ca2+ release normalized ATP6v0a1-protein levels18, whereby increased ATP6v0a1 restored lysosomal vATPase function and thus rescued FAD-associated defects in lysosomal function and autophagic flux18. These results suggest a close correlation between RyR activity and vATPase expression/function. To unravel how these two protein-complexes affect each other’s function, we first asked whether endogenous RyRs could interact with ATP6v0a1 subunit in physiological systems. Therefore, co-immunoprecipitation assays were set up using cortical samples from 6 month-old-mice. In lysates from wild type (WT) cortical samples, the ATP6v0a1 co-immunoprecipitated with the RyR, indicating that endogenous RyRs and ATP6v0a1 formed a protein complex (Fig. 1a). When performing the same experiment in age and sex matched cortical samples from a triple transgenic (3xTg) FAD mouse model (also used in ref. 18), the amount of ATP6v0a1 co-immunoprecipitating with the RyR was significantly reduced (Fig. 1a, b). To corroborate these findings, similar experiments were performed in human embryonic kidney (HEK)293 T-REx™ cells ectopically expressing RyR2 (further named T-Rex RyR2 cells). These cells overexpress RyR2, the primary isoform present in brain, under the control of a tetracycline-inducible promotor. In these cells, transiently overexpressed Flag-tagged ATP6v0a1 (ATP6v0a1-Flag) specifically co-immunoprecipitated with RyR2, pulled down using a specific RyR2 antibody but not with a non-specific immunoglobulin G (IgG) (Fig. 1c). To validate that the ATP6v0a1-Flag pulldown by the RyR2 antibody was dependent on the presence of the RyR2 protein, similar co-immunoprecipitation experiments were performed using lysates from HEK293 cells transiently overexpressing ATP6v0a1 but lacking RyR expression (Suppl. Fig. 1a). Using these HEK293 cell lysates, neither RyR nor ATP6v0a1-Flag could be detected in the co-immunoprecipitated fraction, validating the antibody does not recognize ATP6v0a1-Flag. Combined, these co-immunoprecipitation results indicate that RyR2 and the ATP6v0a1 subunit form a protein complex. Next, protein-protein docking of the RyR2 and vATPase complex, including the ATP6v0a1 subunit, was performed (Fig. 1d, e). This docking analysis supported our biological results, indicating that the RyR via its cytosolic region could interact with multiple subunits of the vATPase complex (Table 1), including the N-terminal cytosolic domain of the ATP6v0a1 subunit. Of note, based on this docking analysis, the ER and lysosomal membranes would be 166 angstrom (or about 16.6 nm) apart, which is within the distance range to form lysosomal-organellar membrane contact sites. To assess whether the entire vATPase complex could interact with RyR2, co-immunoprecipitation of RyR2 was performed using lysates prepared from cerebral cortex samples obtained from WT 6 month-old-mice (Suppl. Fig. 1b). Using these cortical lysates, immunoprecipitating the RyR2 resulted in co-purification of ATP6v0a1 and ATP6v1b2 subunits, with the latter not directly involved in RyR binding according to our predicted protein docking model. Considering the interaction surface of RyR2 is at the cytosolic side of the ER membrane and ATP6v0a1 at the cytosolic side of the lysosome membrane, the logical interaction surface is situated in the cytosol. Based on the molecular docking models, we next designed smaller fragments of both the ATP6v0a1 and RyR2 covering the presumed interaction sites. A schematic overview of the predicted binding sites on full-length RyR2 and ATP6v0a1 and the smaller fragments designed is depicted in Fig. 1f. We asked whether the first 433 amino acids (aa) of ATP6v0a1, comprising the N-terminal cytosolic domain and the first transmembrane domain, was sufficient to interact with RyR2 channels. We therefore generated 3xFLAG-ATP6v0a11-433 and found that this N-terminal fragment could be co-immunoprecipitated with RyR2 (Fig. 1g). Next, a 3xHA-tagged 400 aa long fragment (aa 1450-1850 of RyR2, 3xHA-RyR2400aa) spanning the predicted binding site of both ATP6v0a1 and ATP6v1g1 was designed (Fig. 1f). Flag pulldowns were performed in cell lysates from HEK293 cells transiently overexpressing both 3xHA-RyR2400aa and ATP6v0a1-Flag (Fig. 1h). These pulldowns revealed a clear interaction between the RyR2 domain and full length ATP6v0a1. Finally, a smaller 3xFlag-tagged 300 amino acid long fragment of the ATP6v0a1 was also designed (aa 1-300, 3xFlag-ATP6v0a11-300) (Fig. 1f). This fragment contains the predicted RyR2 binding site but lacks the first transmembrane domain, excluding potential transmembrane region interactions. Flag pulldowns performed in cell lysates from HEK293 cells transiently overexpressing both 3xHA-RyR2400aa and 3xFlag-ATP6v0a11-300 revealed that these fragments interact with each other, indicating that these regions are sufficient for binding (Fig. 1i). Taken together, these experiments validate the protein-protein docking and suggest RyR2 and ATP6v0a1 directly interact.

a Representative immunoblot of co-immunoprecipitation experiments using cortical lysates from 6 month-old wild-type (WT) and 3xTg FAD mice, probed for RyR and ATP6v0a1. b Densitometric quantification of ATP6v0a1 co-immunoprecipitated with RyR, normalized to immunoprecipitated RyR levels, relative to WT. Each data point represents an independent experiment (n = 5). Mean ± s.d. shown. Two-tailed one-sample t-test: P = 0.041; t = 2.971; d.f. = 4; (* P < 0.05). c Representative immunoblot of co-immunoprecipitation in T-Rex RyR2 cells treated with tetracycline ( + TET) to induce RyR2 expression, transiently expressing ATP6v0a1-Flag. Blots were probed for RyR and Flag. Experiments were performed independently five times (n = 5). d, e Surface representations of protein–protein docking analysis showing RyR2 (blue), lysosomal vATPase (green), and ATP6v0a1 (violet). Side (d, left) and top (d, right) views of predicted interacting protein complexes and membrane spacing. Side views (e) highlight predicted interacting residues (red) on RyR2 and vATPase. f Linearized schematic of RyR2 and ATP6v0a1 showing predicted interaction regions (red). Additional predicted interaction sites for ATP6v1a1 and ATP6v1g1 on RyR2 are indicated. Constructs used for interaction studies include RyR2400aa (aa 1450–1850) and ATP6v0a1 fragments (first 433 and 300 aa). g Representative immunoblot of co-immunoprecipitation in T-Rex RyR2 cells ( + TET) transiently expressing 3xFlag-ATP6v0a11–433, probed for RyR and Flag. Experiments were performed three times independently (n = 3). h, i Representative immunoblots of Flag pulldown assays in HEK293 cells expressing empty vector, ATP6v0a1-Flag or 3xFlag-ATP6v0a11–300, and 3xHA-RyR2400aa. Blots were probed for HA and Flag. Experiments were performed three times independently (n = 3). Molecular weights (kDa) are indicated for all immunoblots. Source data are provided as a Source Data file.
Next, we examined whether ATP6v0a1 could also impact the activity of RyR2 channels and vice versa. For this, single-cell Ca2+ measurements were setup in T-Rex RyR2 cells. An empty vector control or the ATP6v0a1-Flag were co-transfected together with lysosomal associated membrane protein 1 (LAMP1)-GCaMP6S, a genetically encoded Ca2+ indicator directed to the lysosomal membrane to measure peri-lysosomal Ca2+ signals (Fig. 2a). Following induction of RyR2 expression with tetracycline, these cells displayed large spontaneous cytosolic Ca2+ oscillations reaching the lysosomal membranes. These oscillations are silenced upon removing extracellular Ca2+ (Fig. 2a) or when the specific RyR blocker ryanodine (30 µM) was added for 24 hr (Suppl. Fig. 2a). In addition to measuring these spontaneous Ca2+ oscillations, caffeine was used to trigger RyR2-mediated Ca2+ release. Overexpression of the ATP6v0a1-Flag resulted in reduced spontaneous and caffeine-induced Ca2+ release compared to the empty vector control (Fig. 2a, b). The ER-store content was also assessed by measuring the amount of Ca2+ released by thapsigargin, an irreversible sarco/endoplasmic reticulum ATPase inhibitor, in Fura2-AM-loaded T-Rex RyR2 cells in the absence of extracellular Ca2+ (Fig. 2c). No difference in the thapsigargin releasable Ca2+ pool was observed comparing empty vector control to ATP6v0a1-Flag overexpressing cells (Fig. 2d). The latter indicates that the ER Ca2+-store content is not affected by ATP6v0a1 presence, thereby excluding an indirect impact of ATP6v0a1 on RyR2-mediated Ca2+ responses. Hence, we propose that ATP6v0a1 inhibition of RyR2-mediated Ca2+ release is mediated through a direct effect of ATP6v0a1 on RyR2. Next, lysosomal pH measurements were setup in order to identify whether RyR2 affected lysosomal pH and if this was modulated by ATP6v0a1 overexpression. To this end, fluorescein isothiocyanate (FITC)-dextran and rhodamine-dextran were loaded via endocytosis into the lysosomal lumen. FITC-dextran, which is quenched at low pH, serves as the pH sensor, whereas the pH-insensitive rhodamine-dextran serves as a measure for lysosomal dye loading31,32. The ratio of FITC/rhodamine signal provides a good indication of lysosomal pH. In these experiments, the empty vector control or the ATP6v0a1-Flag constructs were co-transfected with Blue Fluorescent Protein (BFP) to identify transfected cells. The experiments were performed in the presence and absence of RyR2 overexpression ( ± TET) (Fig. 2e). No significant differences in lysosomal pH (FITC-dextran intensities) were observed among the different conditions (Fig. 2f). However, quantifying the amount of rhodamine dextran loaded per transfected cell showed that upon RyR2 overexpression (+TET conditions), lysosomal dye loading was significantly reduced. This decrease could be rescued by overexpressing ATP6v0a1-Flag, which did not have an effect in the absence of RyR2 (-TET conditions) (Fig. 2g).

a Representative single-cell Ca²⁺ traces from T-Rex RyR2 cells treated with tetracycline ( + TET) inducing RyR2 overexpression. Cells were transiently transfected with LAMP1-GCaMP6S and either empty vector (pCMV24, left) or ATP6v0a1-Flag (right). Traces are shown as F/Fmin, where Fmin is the average of five recordings prior to caffeine addition. After extracellular Ca2+ removal, 500 µM caffeine was applied to trigger RyR2-mediated Ca2+ release. b Quantification of spontaneous Ca2+ release (area under the curve (AUC) of first 120 sec; blue) and caffeine-induced Ca2+ release (AUC, 60 s post-caffeine; red), normalized to empty vector control. Each symbol represents an independent experiment (spontaneous: pCMV24, n = 12; ATP6v0a1, n = 10; caffeine: n = 17 each). Mean ± s.d. shown. Two-tailed unpaired Student’s t-tests: spontaneous, P = 0.0011; t = 3.791; d.f. = 20; caffeine, P = 0.0019; t = 3.380; d.f. = 32 (** P < 0.01). c Average cytosolic Ca2+ traces from Fura-2 AM–loaded T-Rex RyR2 cells (+TET), transiently expressing mCherry with either empty vector or ATP6v0a1-Flag. Traces are shown as fluorescence ratio (520 nm emission after 340/380 nm excitation). After extracellular Ca2+ chelation with 3 mM BAPTA, 1 µM thapsigargin was added to deplete ER Ca2+ stores. d Quantification of thapsigargin-releasable ER Ca2+ content (AUC post-thapsigargin), normalized to empty vector control. Each symbol represents an independent experiment (n = 7). Mean ± s.d. shown. Two-tailed unpaired Student’s t-test: P = 0.224; t = 1.287; d.f. = 12. e Representative fluorescence microscopy images of T-Rex RyR2 cells ±TET, loaded with FITC-dextran and Rhodamine-dextran via endocytosis. Cells were co-transfected with BFP and either empty vector or ATP6v0a1-Flag. Scale bar, 20 µm. f, g Quantification of lysosomal pH (FITC/Rhodamine ratio, (f) and dye loading (Rhodamine/BFP area, (g) normalized to -TET empty vector control. Each symbol represents an independent experiment (n = 16), averaging ≥4 images. Mean ± s.d. shown. One-way ANOVA with Tukey’s post hoc test: f P = 0.1052; F = 2.135; d.f. = 63; P < 0.0001; F = 14.77; d.f. = 63; (** P < 0.01 and **** P < 0.0001). Source data are provided as a Source Data file.
To elucidate whether ATP6v0a1 binding to RyR2 was responsible for RyR inhibition and subsequent augmented lysosomal dye loading, we designed competition experiments using 3xHA-RyR2400aa as a decoy for ATP6v0a1. Since 3xHA-RyR2400aa interacts with ATP6v0a1, we hypothesized that this RyR2 fragment could be used to occupy the RyR2-binding site on ATP6v0a1, thereby interfering with the binding of full-length RyR2 to ATP6v0a1. To test this, a co-immunoprecipitation assay was setup in T-Rex RyR2 cells treated with tetracycline and transiently overexpressing ATP6v0a1-Flag in absence (empty vector control) or presence of 3xHA-RyR2400aa (Fig. 3a). The expression of 3xHA-RyR2400aa reduced the amount of ATP6v0a1-Flag in the RyR2-immunoprecipitated sample by about 40% in comparison to empty vector condition, indicating this fragment, at least in part, disrupts RyR2-ATP6v0a1-complex formation (Fig. 3a, right). Next, we asked whether preventing the ATP6v0a1-Flag binding to RyR2 also had functional effects. Single-cell Ca2+ measurements were performed in T-Rex RyR2 cells treated with tetracycline, co-overexpressing LAMP1-GCaMP6s together with the empty vectors, ATP6v0a1-Flag + empty vector or ATP6v0a1-Flag + 3xHA-RyR2400aa. Caffeine-evoked Ca2+ release was quantified in the absence of extracellular Ca2+ as a readout for RyR2 activity (Fig. 3b). Quantification of these Ca2+ responses indicated that ATP6v0a1-Flag reduced RyR2 activity compared to empty vector conditions (Fig. 3c). The inhibition of RyR2 activity by ATP6v0a1 was partially alleviated by co-expressing the 3xHA-RyR2400aa fragment, indicating that RyR2400aa could functionally prevent RyR2 modulation by ATP6v0a1 (Fig. 3c). Next, we examined the impact of the RyR2400aa fragment on regulating lysosomal dye loading (Fig. 3d, e). Therefore, a similar experiment as performed in Fig. 2e was setup in T-Rex RyR2 cells treated with or without tetracycline. Rhodamine dextran was loaded in these cells via endocytosis following co-transfections of p2a-BFP together with the empty vectors, ATP6v0a1-Flag + empty vector or ATP6v0a1-Flag + RyR2400aa-p2a-BFP (Fig. 3d). Quantifying the rhodamine signal per transfected cell (BFP positive cells) indicated that the RyR2400aa construct reversed the effects of ATP6v0a1-Flag on lysosomal dye loading in RyR2-expressing cells (Fig. 3e).

a Left: Representative immunoblot of co-immunoprecipitation experiments using T-Rex RyR2 cells treated with tetracycline (+TET) inducing RyR2 overexpression. Cells were transfected with empty vector, and ATP6v0a1-Flag, or 3xHA-RyR2400aa as indicated. RyR2 was immunoprecipitated and blots were probed for RyR, Flag, and HA. Right: Densitometric quantification of ATP6v0a1-Flag co-immunoprecipitated with RyR2, normalized to immunoprecipitated RyR2 relative to empty vector control. Each data point represents an independent experiment (n = 3). Mean ± s.d. shown. Two-tailed one-sample t-test: P = 0.0267; t = 5.997; d.f. = 2 (* P < 0.05). Molecular weights (kDa) are indicated. b Average traces ± s.e.m. of intracellular Ca²⁺ measurements using LAMP1-GCaMP6S in T-Rex RyR2 cells (+TET), transfected with empty vectors, ATP6v0a1-Flag + empty vector, or ATP6v0a1-Flag + 3xHA-RyR2400aa. Traces are shown as F/Fmin, where Fmin is the average of five recordings prior to caffeine addition. After extracellular Ca2+ removal, 500 µM caffeine was applied to trigger RyR2-mediated Ca2+ release. c Quantification of caffeine-induced Ca2+ release (AUC, 60 s post-caffeine). Each symbol represents an independent experiment (n = 5), averaging ≥ 2 technical repeats. Mean ± s.d. shown. Repeated measures one-way ANOVA with Tukey’s post hoc test: P = 0.0005; F = 84.59; d.f. = 16 (** P < 0.01). d Representative fluorescence microscopy images of T-Rex RyR2 cells ±TET, loaded with Rhodamine-dextran. Cells were co-transfected with p2a-BFP and either empty vector, ATP6v0a1-Flag, or ATP6v0a1-Flag + RyR2400aa-p2a-BFP. Scale bar, 20 µm. e Quantification of lysosomal dye loading (Rhodamine/BFP area). Each symbol represents an independent experiment (control n = 8; ATP6v0a1-Flag and ATP6v0a1+RyR2400aa n = 16), averaging ≥ 4 images. Mean ± s.d. shown. One-way ANOVA with Tukey’s test: P = 0.003; F = 10.31; d.f. = 39 (** P < 0.01 and *** P < 0.001). f Representative fluorescence microscopy images of T-Rex RyR2 cells ±TET, loaded with FITC and Rhodamine dextran, treated with DMSO or 30 µM ryanodine (Rya). Scale bar, 20 µm. g, h Quantification of lysosomal dye loading (g) and pH (h). Each symbol represents an independent experiment (DMSO -TET n = 19; +TET n = 19; +TET+Rya n = 12), averaging ≥4 images, normalized to DMSO. Mean ± s.d. shown. One-way ANOVA with Tukey’s test: g P < 0.0001; F = 52.84; d.f. = 49; **** indicates P < 0.0001; h P = 0.0218; F = 4.154; d.f. = 49; * indicates P < 0.05.
Since ATP6v0a1 overexpression resulted in the inhibition of RyR2-mediated Ca2+ release, we wondered whether pharmacological inhibition of RyR2 activity was sufficient for this phenomenon. An inhibitory concentration of ryanodine (Rya, 30 µM applied for 24 hr in T-Rex RyR2 cells, validated in Suppl. Fig. 2a) prevented the reduced lysosomal rhodamine dextran loading caused by RyR2 overexpression and normalized the lysosomal rhodamine dextran levels to control levels observed in non-RyR2 expressing cells (Fig. 3f, g). In these experiments, a modest but significant increase in pH was observed following RyR2 overexpression which was not reversed following ryanodine treatment (Fig. 3h).
Taken together, these experiments indicate that ATP6v0a1 functionally interacts with RyR2, thereby inhibiting RyR2 activity and increasing lysosomal dye uptake. Furthermore, pharmacological inhibition of RyR2-mediated Ca2+ release was sufficient to counteract and normalize the decreased lysosomal dye loading.
Inhibition of RyRs stimulates autophagic flux while inhibiting lysosomal exocytosis in hiPSC-derived cortical neurons
In the previous experiments, RyR2 expression and activity were linked to decreased lysosomal dye loading without major impacts on the lysosomal pH. Changes in dye loading could be influenced by several factors, including lysosomal enzyme activity or changes in handling of lysosomal cargo. Since the T-Rex RyR2 cells display high, potentially non-physiological, RyR2-protein levels, we wished to explore these mechanisms in a physiological cell system expressing endogenous RyR2 proteins. Hence, cortical neurons were differentiated from a control hiPSC line using established protocols33. For all experiments, cortical neurons were differentiated and used between day 52 and 56 in vitro. The cortical lineage of the neurons was verified by immunofluorescence staining for microtubule-associated protein 2 (MAP2) (neuronal marker34) and special AT-rich sequence-binding protein 2 (SATB2) (cortical lineage35) (Fig. 4a). Since pharmacological inhibition of RyR2 was sufficient to impact lysosomal dye loading thereby mimicking the effect of ATP6v0a1 overexpression, we used ryanodine to further explore these mechanisms in a physiologically relevant model. In these cortical neurons, we first validated our previous observations in primary dissociated hippocampal neurons from rat showing that inhibiting spontaneous RyR-mediated Ca2+ release stimulates autophagic flux at the level of the lysosomes17. A reliable way to measure autophagy is to quantify LC3-II levels. LC3-II is incorporated in autophagosome membranes and as such is a good marker for the final steps of the autophagy pathway. However, upon fusion with the lysosomes, LC3-II associated with autophagosomes is also degraded. Thus, to measure autophagic flux, LC3-II-proteins levels need to be measured in the absence and presence of lysosomal inhibitors such as bafilomycin A136. hiPSC-derived cortical neurons were treated for 24 hr with DMSO or ryanodine in the presence and absence of bafilomycin A1, added during the last 3 hr of treatment. Immunoblot stainings were performed for LC3-II and used for quantification (Fig. 4b). Inhibition of endogenous/spontaneous RyR activity using ryanodine significantly reduced LC3-II-protein levels (Fig. 4c). However, in the presence of bafilomycin A1, LC3-II-protein levels were elevated in ryanodine treated conditions compared to DMSO control (Fig. 4d), combined these results suggest an increased autophagic flux when blocking RyR (Fig. 4c, d). Of note, RyR levels were not affected by the treatments (Fig. 4e). To confirm increased lysosomal turnover of autophagosomes occurs upon ryanodine treatment, a green fluorescent – red fluorescent protein (GFP-RFP)-LC3 assay was set up. GFP-RFP-LC3 was transiently introduced in the cortical neurons treated with DMSO or ryanodine for 24 hr (Fig. 4f). In this assay, autophagosomes are both GFP- and RFP-positive whereas upon fusion of autophagosomes with lysosomes the acidic lysosomal pH quenches the GFP signal, thereby rendering autolysosomes only RFP positive. Quantification was performed by determining in each transfected cell whether the RFP-positive puncta were also GFP positive, analyzed as the ratio of GFP-positive puncta over the RFP-positive puncta (Fig. 4g). This ratio decreased upon treating the neurons with ryanodine, signifying a drop in autophagosomes caused by increased fusion with lysosomes, suggesting an increased lysosomal turn-over of the autophagosomes thus confirming the LC3-II results. Next, we set out to determine whether the initiation and/or early phases of autophagy were affected when RyR activity was blocked. AMPK and p70S6 kinase activity were measured as readout for nutrient sensing pathways, which can induce autophagy in response to nutrient deprivation or declining energy levels. Importantly both pathways are also sensitive to alterations in intracellular Ca2+ levels making them highly relevant to evaluate37. Our results show that both AMPK (Fig. 5a) and p70S6 kinase activity (Fig. 5b) were unaltered following ryanodine treatment. Next, changes in early autophagic markers were monitored. In contrast to LC3-II, early autophagic markers such as Beclin 1-protein levels and ATG13 activity were not altered upon treatment with ryanodine (Fig. 5c, d). To further evaluate the initial phases of the autophagic pathway FIP200 and WIPI2 puncta were imaged (Fig. 5e). FIP200 is part of the mammalian ULK1 complex, whose activation is one of the initial steps in autophagy induction. WIPI2 accumulates at the limiting membranes involved in the initial formation of omegasomes and autophagosomes. FIP200 and WIPI2 were immunofluorescently labeled in the cortical neurons following 24 hr treatments with DMSO or ryanodine (Fig. 5e). Analysis of the number of FIP200 and WIPI2 puncta per cell revealed no difference between control and ryanodine treatment (Fig. 5f). These results further confirm that ryanodine treatment does not affect the early phases of the autophagic pathway. In the context of FAD, excessive RyR activity has been shown to contribute, at least in part, to the production of Aβ from APP, and reducing RyR activity also leads to reduced Aβ production18,19,22,23,24,25,26,27,28. We next set out to address whether in our model ryanodine treatment also reduces APP levels by affecting lysosomal turnover of the protein. To test this, hiPSC-derived cortical neurons were exposed to ryanodine with or without proteasomal (bortezomib) or lysosomal (bafilomycin A1) inhibitors. APP-protein levels were quantified via immunoblot (Fig. 5g). Densitometric analysis of the APP signal showed that ryanodine treatment reduced APP levels and that this could be reversed by inhibiting lysosomal but not proteasomal function, confirming that ryanodine promoted APP turnover via lysosomal pathways (Fig. 5h). Of note, all treatments were limited to three hr, as prolonged bafilomycin A1 or bortezomib treatments impacted cell viability. Taken together these results indicate that inhibition of spontaneous RyR-mediated Ca2+ release stimulated autophagic flux by increasing lysosomal turnover of autophagosomes without altering early stage of the autophagic process, thereby increasing turnover of autophagosomal and lysosomal cargo. These results are in agreement with our previous finding in dissociated hippocampal neurons validating these hiPSC-derived cortical neurons as a suitable model for our studies. Finally, single-cell cytosolic Ca2+ measurements were performed in the hiPSC-derived cortical neurons to validate the presence of ryanodine sensitive (and thus RyR-mediated) Ca2+ responses (Suppl. Fig. 2b). A recombinant adeno-associated viral vector (rAAV) was used to transduce GCaMP6S, a genetically encoded Ca2+ indicator, in the hiPSC-derived cortical neurons. These neurons displayed both spontaneous and caffeine-induced Ca2+ release, with the latter being evidence for the presence of endogenous and active RyR channels (Suppl. Fig. 2b). Moreover, treatment for 24 hr with ryanodine almost completely suppressed spontaneous Ca2+-release events and severely reduced caffeine-evoked Ca2+ transients (Suppl. Fig. 2c). Overall, these data validated the presence of endogenous RyR channels in hiPSC-derived cortical neurons that are spontaneously active.

a Representative confocal microscopy images of fixed hiPSC-derived cortical neurons differentiated for 52 days (DIV52), immunostained for MAP2 (red) and SATB2 (green). Experiments were performed three times independently (n = 3). Scale bar: 20 µm. b Representative immunoblot of hiPSC-derived cortical neurons treated for 24 hr with DMSO or 30 µM ryanodine (Rya), stained for RyR, vinculin, and LC3. Where indicated, 100 nM bafilomycin A1 ( + BM) was added during the final 3 hr. c–e Densitometric quantification of LC3-II (n = 6, c), LC3-II with bafilomycin ( + BM) (n = 6, d), and RyR (n = 5, e). Each symbol represents an independently performed experiment. Values were normalized to their respective loading controls and plotted relative to DMSO controls. Data are shown as mean ± s.d. Two-tailed one-sample t-tests were used to assess significance versus DMSO controls for (c) P = 0.0142, t = 5.264, df = 5, for (d) P = 0.0405, t = 2.747, df = 5 and for (e) P = 0.9463, t = 0.07164, df = 4, (* P < 0.05). f Representative fluorescence microscopy images of fixed DIV52 hiPSC-derived cortical neurons transfected with GFP-RFP-LC3 and treated for 24 hr with DMSO or 30 µM Rya. Nuclei were counterstained with Hoechst. Scale bar: 10 µm. g Quantification of GFP/RFP-LC3 puncta ratio per transfected cell as a measure of autophagic flux. Each data point represents the average GFP/RFP-LC3 ratio from one independent experiment (n = 7), averaging ≥5 images. Mean ± s.d. are shown. A two-tailed unpaired Student’s t-test was used to assess significance: P = 0.0393, t = 2.312, df = 12. * indicates (*P < 0.05). Source data are provided as a Source Data file.

a–d top Representative immunoblots of hiPSC-derived cortical neurons treated for 24 hr with DMSO or 30 µM ryanodine (Rya), stained for (P-)AMPK (a), (P-)p70S6 kinase (b), Beclin 1 and vinculin (c), or (P(Ser355))-ATG13 (d). a–d bottom: Densitometric quantification of the above immunoblots plotted relative to DMSO. Each data point represents an independent experiment (n = 5). Data are shown as mean ± s.d. Two-tailed one sample t-tests were performed to assess significant differences relative to DMSO (for (a) P = 0.6108, t = 0.5513 df = 4, for (b) P = 0.3314, t = 1.104 df = 4 for (c) P = 0.2979 t = 1.196 df = 4 for (d) P = 0.3542, t = 1.047 df = 4). e Representative fluorescence microscopy images of hiPSC-derived cortical neurons immunolabeled for FIP200 and WIPI2, co-stained with Hoechst. Scale bar: 10 µm. f Quantification of FIP200 and WIPI2 puncta per cell. Each data point depicts the average number of puncta per cell of one independent repetition (n = 6). Data shown as mean ± s.d. Two-tailed unpaired t-tests were performed FIP200, P = 0.3217, t = 1.043, df = 10; WIPI2, P = 0.1824, t = 1.433, df = 10. g Representative immunoblot of neurons treated for 3 hr with DMSO or 30 µM Rya ± bortezomib (Bort, 10 µM) and/or bafilomycin A1 (BM, 100 nM), labeled for vinculin and APP. h Densitometric quantification of APP normalized to loading controls and plotted relative to DMSO. Each symbol represents an independent repetition (n = 7). A two-tailed one-sample t-test (DMSO vs Rya) was performed: P = 0.0077, t = 3.932, df = 6. A one-way ANOVA with Tukey multiple comparison testing (with significance level adjusted to p < 0.025 to account for multiple testing) was performed to test for significant differences between the Rya condition and Rya + Bort or Rya + BM condition (P = 0.0006, F = 11.39, df = 20, *,** and ***p < 0.025, **p < 0.01 and p < 0.001 respectively). Molecular weights (kDa) are indicated on all blots. Source data are provided as a Source Data file.
Next, lysosomal pH and dye loading were quantified in the hiPSC-derived cortical neurons (Fig. 6a). Treatment with ryanodine (24 hr) did not change lysosomal pH (Fig. 6b, left), while treatment with bafilomycin A1 (3 h) served as a positive control, demonstrating the expected alkalizing effect on lysosomal pH. Similarly to our observations in the T-Rex RyR2 cells, lysosomal dye loading was significantly upregulated following ryanodine treatment compared to the DMSO control (Fig. 6b, right).

a Representative fluorescence microscopy images of hiPSC-derived cortical neurons loaded with FITC and Rhodamine dextran and stained with Hoechst. Cells were treated for 24 hr with DMSO or 30 µM ryanodine (Rya); bafilomycin A1 (100 nM) was applied for 3 hr. Scale bar, 20 µm. b Quantification of lysosomal pH (FITC/Rhodamine, left) and dye loading (Rhodamine/cell, right), normalized to DMSO. Each data point represents an independent experiment (pH: DMSO n = 23, Rya n = 22; dye loading: DMSO n = 22, Rya n = 21), averaging ≥ 4 images. Mean ± s.d. shown. One-way ANOVA with Tukey’s test: pH, P < 0.0001; F = 157.3; d.f. = 58; dye loading, P < 0.0001; F = 11.84; d.f. = 56; (**** P < 0.0001). c Representative fluorescence microscopy images of fixed hiPSC-derived cortical neurons treated for 24 hr with DMSO or Rya, loaded with DQ-red BSA prior to fixation and stained with Hoechst. Scale bar, 20 µm. d Quantification of DQ-red BSA signal per cell, normalized to DMSO. Each data point represents an independent experiment (n = 5), averaging ≥ 5 images. Mean ± s.d. shown. Two-tailed unpaired Student’s t-test: P = 0.4032; t = 0.877; d.f. = 9. e Representative fluorescence microscopy images of fixed, non-permeabilized hiPSC-derived neurons immunolabeled for plasma membrane LAMP1 (LAMP1 PM) and stained with Hoechst, treated for 24 hr with DMSO or Rya. Scale bar, 20 µm. f Quantification of LAMP1 PM signal per cell, normalized to DMSO. Each data point represents an independent experiment (DMSO n = 10; Rya n = 9), averaging ≥5 images. Mean ± s.d. shown. Two-tailed unpaired Student’s t-test: P = 0.0002; t = 4.618; d.f. = 17; *** P < 0.001. g, h Quantification of secreted glucosaminidase activity in hiPSC-derived neurons (g) and T-Rex RyR2 cells (h), normalized to DMSO -TET controls. Each data point represents an independent experiment (g: n = 13; h: n = 5), averaging ≥3 internal replicates. Mean ± s.d. shown. g: two-tailed unpaired t-test, P = 0.0197; t = 2.499; d.f. = 24; h: repeated measures ANOVA, P = 0.0124; F = 7.991; d.f. = 14; P < 0.05. i Representative 3D-SIM Z stack images of fixed T-Rex RyR2 cells co-expressing GFP-Sec61β (green) and LAMP1-RFP670 (red), treated for 24 hr with DMSO or Rya. One 200 nm plane and volume renders are shown. Scale bar, 5 µm. j Quantification of lysosomal localization within the ER network. Distance of lysosomal volumes to ER edge was calculated and normalized to total lysosomal volume. Each data point represents an independent experiment (DMSO n = 5; Rya n = 6), averaging ≥5 stacks per condition each time. Cumulative Gaussian curves ± s.d. were fitted through means of relative lysosomal volume at each distance. Two-tailed sum-of-squares F test: P < 0.0001; F = 104.4; d.f. = 1118. k Representative 3D-SIM images of fixed, permeabilized hiPSC-derived neurons stained for LAMP1 (green), RyR (red), and Hoechst (blue). Scale bar, 10 µm. l Nearest-neighbor distance analysis of RyR and LAMP1 signals from (k). Each data point represents the average ± s.e.m. from six independent experiments (n = 6), each averaging ≥ 7 images. Gaussian curve fit applied. Two-tailed sum-of-squares F test: mean, P = 0.0013; F = 10.47; amplitude, P < 0.0001; F = 288; d.f. = 786. Source data are provided as a Source Data file.
We then set out to identify the underlying cause of this increased lysosomal dye loading. To determine whether RyR inhibition affected lysosomal enzyme activity, a DQTM-red bovine serum albumin (BSA) assay was setup (Fig. 6c). This assay is based on a BSA variant that is heavily labeled with BODIPY-TR-X, resulting in its self-quenching. The DQTM-red BSA dye was loaded into the neurons via endocytosis, ultimately sequestering in the lysosomal compartment. Once in the lysosomal lumen, the BSA fraction will be degraded by proteolytic lysosomal enzymes and the BODIPY-TR-X is released, alleviating the self-quenching effects on the probe. Hence, BODIPY-TR-X fluorescence serves as a measure for lysosomal enzyme activity. Inhibition of RyR with ryanodine did not alter BODIPY-TR-X fluorescence compared to control, indicating that the proteolytic activity of lysosomal enzymes was unaffected (Fig. 6d). Hence, lysosomal enzyme activity is not impacted by blocking RyR activity in hiPSC-derived neurons and thus cannot account for the observed changes in lysosomal dye loading.
A second mechanism by which lysosomal dye loading can be reduced is through increased lysosomal exocytosis, thereby dispersing the lysosomal content in the extracellular medium. To measure lysosomal exocytosis, plasma membrane located LAMP1 was immunofluorescently labeled using an antibody targeting a luminal epitope of LAMP1 in non-permeabilized hiPSC-derived cortical neurons (Fig. 6e)38,39. Quantification of plasmalemmal LAMP1 revealed a significant decline in the fluorescent signal corresponding to plasma membrane located LAMP1 upon treatment with ryanodine, indicating decreased lysosomal exocytosis upon RyR inhibition (Fig. 6f). To further strengthen this observation, a second approach was taken in which N-acetyl-β-d-glucosaminidase activity, an enzyme located in the lysosomes, was measured in the medium of hiPSC-derived cortical neurons as a measure of lysosomal exocytosis. For this, a 4-methylumbelliferyl N-acetyl-β-D-glucosaminide probe was applied40. 4-methylumbelliferyl by itself is highly fluorescent at low pH but is quenched by the covalent coupling to N-acetyl-β-D-glucosaminide. Hence, cleavage of the glucosaminide moiety by the extracellularly present lysosomal-derived N-acetyl-β-D-glucosaminidase enzyme results in freeing of the 4-methylumbelliferyl fraction, alleviating its quenching and enabling its fluorescent properties. Thus, measuring the fluorescence of 4-methylumbelliferyl in the extracellular medium serves as a readout for extracellular N-acetyl-β-d-glucosaminidase activity and lysosomal exocytosis. Using this approach, the extracellular N-acetyl-β-D-glucosaminidase activity was measured in the medium of hiPSC-derived cortical neurons treated with ryanodine for 24 hr. Compared to the DMSO control, extracellular N-acetyl-β-D-glucosaminidase enzyme activity was dampened upon inhibition of RyR activity (Fig. 6g). We corroborated these findings in T-Rex RyR2 cells, demonstrating that extracellular N-acetyl-β-D-glucosaminidase activity increased upon RyR2 expression (comparing -TET to +TET conditions) and that this elevated extracellular lysosomal enzyme activity in RyR2-expressing cells could be reversed by the addition of the RyR inhibitor ryanodine (Fig. 6h). To further support these findings, lysosomal localization relative to the outer ER edge was studied. T-Rex RyR2 cells, treated with tetracycline, were transiently co-transfected with GFP-SEC61β and LAMP-1-RFP670 labeling the ER (green) and the lysosomes (red) (Fig. 6i). Following a 24 hr treatment with either DMSO or ryanodine the cells were fixed and imaged via 3D structured illumination microscopy (SIM). Z-stacks were imaged and used to create volume renders to quantify lysosomal localization with respect to the outer edge of the ER network, which was determined using the volume render of GFP-SEC61β. For this analysis, only lysosomes localized within the ER network were included. The distance of each lysosomal volume, given by the LAMP1-RFP670 volume render, was determined to the nearest outer ER edge. Next, the summed lysosomal volume within incremental 100 nm intervals, moving further away from the outer ER edge, was determined and put relative to the total lysosomal volume providing a normalized volume distribution for each 100 nm interval. These values were plotted as a cumulative frequency distribution through which a cumulative Gaussian curve was fitted (Fig. 6j). This analysis revealed that upon ryanodine treatment the mean lysosomal distance from the outer ER edge increased, moving the lysosomes further away from the outer ER edge, and thus the plasma membrane, compared to the DMSO control. Finally, the subcellular localization of lysosomes in relation to RyR was assessed in hiPSC-derived cortical neurons using super-resolution microscopy. In hiPSC-derived cortical neurons, control and ryanodine-treated conditions, endogenous RyR and LAMP1 were stained via immunofluorescence. 3D-SIM was used to visualize the localization of RyR and LAMP1 puncta at sub 100 nm range (Fig. 6k). Nearest neighbor distances were quantified in order to assess the proximity of LAMP1, and thus lysosomes, to RyR. This approach indicated that RyR and LAMP1 puncta resided in close proximity already in control treated cells. However, treatment with ryanodine decreased the mean distance between RyR and LAMP1 and increased the number of puncta at this distance compared to the vehicle control (Fig. 6l). Taken together, our results indicate that the increased dye loading observed in lysosomes evoked by RyR inhibition is not due to decreased lysosomal enzyme activity but rather a decrease in lysosomal exocytosis. This corresponds with a positioning of lysosomes further away from the plasma membrane and closer to the RyR channels and by extension deeper within the ER network upon RyR inhibition.
Inhibiting RyR activity simulates ER-lysosomal contact site formation by increasing the interaction between RyR and the vATPase
The results above indicate an opposing role for spontaneous RyR activity in lysosomal functions in autophagy versus lysosomal exocytosis. Indeed, spontaneous RyR activity restricted lysosomal turnover of autophagosomes, thus inhibiting autophagic flux (Figs. 4, 5), but it promoted lysosomal exocytosis (Fig. 6). In addition, we found that RyRs interacted physically and functionally with the ATP6v0a1 subunit of the vATPase (Figs. 1–3), thus potentially acting as a tethering mechanism for ER-lysosomal contact site formation. By regulating the localization of lysosomes at the ER, RyRs could direct whether lysosomes become available for either autophagy or exocytosis. To test this hypothesis, we established a number of independent approaches. First, transmission electron microscopy (TEM) experiments were performed in hiPSC-derived cortical neurons treated for 24 hr with DMSO or ryanodine (Fig. 7a). Quantifying the number of lysosomes in close contact (< 30 nm) with the ER relative to the total number of lysosomes imaged indicated that, RyR inhibition increased the number of lysosomes in close contact with the ER (Fig. 7b). Next, ER-lysosomal contact site formation was assessed by loading the neurons with LysoTrackerTM Green DND-26 and BrightER, labeling the lysosomes in green and the ER in red. Airyscan microscopy was used to image a single confocal plane at 1 Hz for 10 s to observe lysosomal contacts with the ER over time in vehicle-treated and ryanodine-treated cells (Fig. 7c). Manders overlap coefficients were calculated to quantify the overlap of both signals. This approach revealed an increase in the overlap of the lysosomal compartment with the ER upon inhibition of RyR activity (Fig. 7d), suggesting an increase in ER-lysosomal contact sites to occur when RyR are inhibited. To directly measure ER-lysosomal contact sites, we developed a SplitFAST sensor. SplitFAST probes have been previously reported to be able to reversibly assemble and have recently been used to monitor the dynamics of membrane contact sites between ER and mitochondria41,42. Here, a split greenFAST tag was used. A schematic representation of the approach is given in Fig. 7e. This approach relies on the reversible recomplementation of both the N- and C-terminus of the greenFAST probe allowing a chemical dye, in this case TFLime, to bind to the complete tag. Only when bound to the recomplemented FAST tag the dye shows strong fluorescent capacities. In Fig. 6i the GFP-SEC61β and LAMP1-RFP670 signals clearly displayed a degree of overlap validating the use of SEC61β and LAMP1 as bona fide partners to detect ER-lysosomal contact sites. Using the split greenFAST tag instead of the GFP and RFP670 tags further enhances the resolution and specificity by which these contact sites can be imaged. The C-terminal part of the greenFAST was fused to the N-terminal cytosolic end of SEC61β, previously shown to be suitable for labeling the ER43. The N-terminal greenFAST fragment was attached to the C-terminal cytosolic end of LAMP1 to target it to the lysosomes. A linker sequence between LAMP1 and the greenFAST tag was included to allow flexible movement of the tag in the 20 nm range. The length of the linker was based on the molecular docking prediction that ER and lysosomal membranes would be about 17 nm apart following binding of the RyR to the lysosomal vATPase (Fig. 1d). Transfecting the split greenFAST probe in hiPSC-derived cortical neurons showed green puncta positioned between the ER and lysosomes labeled with ER-Tracker™ Blue-White DPX and LysoTrackerTM Red DND-99 respectively (Suppl. Fig. 3a–c), indicating the probe can be used in these neurons to detect ER-lysosomal contact site formation. Following a 24 hr ryanodine treatment, neurons transfected with the split greenFAST probe were fixed and imaged via 3D-SIM (Fig. 7f). Z-stacks were taken which were used to create volume renders to quantify ER-lysosomal contact sites in 3D. Quantifying both the number of contact sites (Fig. 7g) and the contact site volume (Fig. 7h) per cell revealed that inhibiting RyR activity significantly increased ER-lysosomal contact site formation. To address whether inhibiting RyR activity could impact the proximity of RyR and ATP6v0a1 subunit. endogenous RyR and the ATP6v0a1 subunit were labeled via immunofluorescence and compared between vehicle- and ryanodine-treated conditions using 3D-SIM imaging (Fig. 7i, j). The obtained Z-stacks were transformed to volume renders, allowing to quantify the overlap/co-localization of the RyR and ATP6v0a1 signal in 3D. As expected, inhibiting the RyR increased the volumetric overlap between both stains, indicating an increased co-localization of the two proteins (Fig. 7k). These results indicate that RyR and ATP6v0a1 reside in close proximity in neuronal cells and that inhibiting spontaneous RyR activity enhanced ER-lysosomal contact site formation, likely by increasing the interaction between the RyR and the v-ATPase.

a Transmission electron microscopy (TEM) images of hiPSC-derived cortical neurons following 24 h treatment with DMSO or 30 µM ryanodine (Rya). Scale bar, 200 nm. b Quantification of ER-lysosome contacts in TEM images, defined as membrane proximity <30 nm. Each experiment the number of lysosomes in contact with ER was normalized to the total lysosomes imaged per condition. Each data point represents and independent experiment (n = 4), with > 150 lysosomes analyzed in total per condition. Mean ± s.d. shown. Two-tailed unpaired Student’s t-test: P = 0.0019; t = 5.282; d.f. = 6; **P < 0.01. c Representative Airyscan live-cell images (imaging 10 s sequences at 1 Hz) of neurons stained with LysoTracker™ Green DND-26 (green) and BrightER (red). Scale bar, 5 µm. d Manders coefficients quantifying signal overlap between LysoTracker and BrightER. Each data point (n = 4) represents an independent experiment averaging ≥ 5 sequences. Mean ± s.d. shown. Two-tailed paired Student’s t-test of lysosome-ER overlap P = 0.0368; t = 3.598; d.f. = 3, and ER-lysosome overlap P = 0.7815, t = 0.3032, df = 3, *P < 0.05. e Schematic of the split greenFAST approach used to detect ER-lysosome contact sites. f 3D-SIM Z stack images of fixed hiPSC-derived neurons showing split greenFAST probe signal and Hoechst nuclear stain. One 200 nm plane and volume renders are shown. Scale bar, 10 µm. g, h Quantification of ER-lysosome contact site number (g) and total contact volume per cell (h), normalized to DMSO. Each data point (n = 5) represents an independent experiment averaging ≥5 stacks. Mean ± s.d. shown. Two-tailed unpaired Student’s t-tests: g P = 0.0218; t = 2.841 df = 8; h: P = 0.0184; t = 2.952; d.f. = 8; *P < 0.05. i, j 3D-SIM Z stack images of fixed, permeabilized hiPSC-derived neurons immunolabeled for RyR (green) and ATP6v0a1 (red) following 24 hr treatment with Rya or DMSO. One 200 nm plane and volume renders are shown. Scale bar, 10 µm. k Quantification of overlap volume (yellow) between RyR and ATP6v0a1 for each treatment, normalized to total stain volume relative to DMSO. Data points (n = 5 DMSO, n = 4 Rya) represent an independent experiment averaging ≥5 stacks. Mean ± s.d. shown. Two-tailed unpaired Student’s t-test: P = 0.0130; t = 3.305; d.f. = 7; *P < 0.05. Source data are provided as a Source Data file.
To further investigate the importance of the RyR2 ATP6v0a1 interaction in regulating lysosomal secretion, additional experiments were setup. First, lysosomal localization relative to the outer ER edge was studied in the presence of overexpressed ATP6v0a1-Flag. T-Rex RyR2 cells were treated with tetracycline and were transiently co-transfected with empty vector or ATP6v0a1-Flag together with GFP-SEC61β and LAMP1-RFP670 labeling the ER (green) and the lysosomes (red) (Fig. 8a). After 48 hr following transfection the cells were fixed and imaged via 3D-SIM. Z-stacks were imaged and were used to create volume renders to quantify lysosomal localization with respect to the outer edge of the ER. The analysis was performed as described in Fig. 6j. The fitted cumulative Gaussian curve showed that ATP6v0a1-Flag overexpression positioned the lysosomes further away from the outer ER edge (Fig. 8b), phenocopying our results obtained with ryanodine treatment (Fig. 6j). Next, exploiting the insights obtained from the RyR2400aa-based competition experiments in Fig. 3, we setup similar experiments in hiPSC-derived cortical neurons. For these experiments the p2a-BFP and RyR2400aa-p2a-BFP constructs were used avoiding the need for co-transfections. Single-cell Ca2+ measurements using Cal520 were set up in hiPSC-derived cortical neurons, transfected with either p2a-BFP or RyR2400aa-p2a BFP (Fig. 8c). Caffeine was used to trigger RyR-mediated Ca2+ release. Quantifying the RyR-mediated Ca2+ release in BFP-positive cells showed that the RyR2400aa construct increased RyR-mediated Ca2+ release (Fig. 8d), indicating that RyR2400aa can alleviate the inhibitory impact of endogenous ATP6v0a1 on RyR2 activity. Finally, lysosomal exocytosis was measured by monitoring plasma membrane located LAMP1 as performed in (Fig. 6e) in hiPSC-derived cortical neurons overexpressing either p2a-BFP or RyR2400aa-p2a-BFP (Fig. 8e). Quantification of plasmalemmal LAMP1 in BFP-positive cells revealed a significant increase in the fluorescent signal corresponding to plasmalemmal LAMP1 when the RyR2 fragment was present (Fig. 8f). Collectively, these results suggest that ATP6v0a1 by binding to the RyR2 suppresses endogenous RyR activity thereby at the same time positioning lysosomes to partake in autophagic flux while limiting their availability for lysosomal exocytosis.

a Representative 3D-SIM Z stack images of fixed, tetracycline-treated T-Rex RyR2 cells transiently co-expressing either empty vector or ATP6v0a1-Flag, together with GFP-Sec61β (green, ER marker) and LAMP1-RFP670 (red, lysosomal marker). A single plane (200 nm) and volume renders are shown. Scale bar, 5 µm. b Quantification of lysosomal positioning within the ER network. For each lysosomal volume, the distance to the ER edge was calculated relative to total lysosomal volume. The experiment was performed six times independently (n = 6) averaging ≥ 5 stacks per condition each time. Cumulative Gaussian curves ± s.d. were fitted through means of relative lysosomal volume at each distance. Statistical significance was assessed using a two tailed sum-of-squares F test (P < 0.0001; F = 102.8; d.f. = 1220). c Representative traces (mean ± s.e.m.) of cytosolic Ca²⁺ dynamics in Cal520-loaded hiPSC-derived cortical neurons transiently expressing either p2a-BFP or RyR2400aa-p2a-BFP. Traces were normalized to F/Fmin, where Fmin corresponds to baseline fluorescence prior to stimulation with 10 mM caffeine evoking RyR-mediated Ca2+ release. d Quantification of caffeine-induced Ca2+ release amplitude in BFP-positive neurons. Each data point represents an independent repetition (n = 7, averaging ≥ 2 technical repeats), normalized to the DMSO control mean for each experimental day. Data are presented as mean ± s.d. Two-tailed paired Student’s t-test (P = 0.0247; t = 2.979; d.f. = 6; *P < 0.05). e Fluorescence microscopy images of fixed, non-permeabilized hiPSC-derived cortical neurons immunolabeled for plasma membrane-localized LAMP1 (LAMP1 PM), following transient expression of p2a-BFP or RyR2400aa-p2a-BFP. Scale bar, 20 µm. f Quantification of LAMP1 PM signal relative to p2a-BFP control. Only BFP-positive regions were analyzed. Each data point reflects an independent experiment (n = 10), averaging ≥ 10 images per replicate. Data are shown as mean ± s.d. Statistical significance was determined using a two-tailed unpaired Student’s t-test (P = 0.0044; t = 3.256; d.f. = 18; **P < 0.01). g Schematic summary of experimental findings. Created with BioRender.com. Source data are provided as a Source Data file.
Discussion
The main conclusion of this work is that RyR channels reside at ER-lysosomal contact sites, thereby steering lysosomal trafficking and function through localized Ca2+-signaling nanodomains. As such, RyR channels control the proximity of lysosomes to the ER in a highly dynamic fashion through interorganellar protein complexes involving the ATP6v0a1 subunit of the vATPase.
We presented multiple lines of evidence for this conclusion obtained in different cell models including T-Rex RyR2 and cortical neurons, either primary from mice or differentiated from hiPSCs. RyR2 channels could directly bind to the N-terminal cytosolic region of the ATP6v0a1 subunit. RyR2 and ATP6v0a1 mutually influence Ca2+ signaling at lysosomal and ER compartments, respectively. RyR2 evokes Ca2+ signaling near lysosomal membranes, while ATP6v0a1 suppresses RyR2-mediated Ca2+ fluxes from the ER Ca2+ stores. Furthermore, RyRs reside in close proximity of lysosomes and are localized in proximity of ATP6v0a1 proteins. As such, RyR channels contribute to ER-lysosomal contact sites in a RyR-activity-dependent manner, as pharmacological RyR inhibition tightens ER-lysosomal contacts. As biological consequence of this finding, we postulate that endogenous and spontaneously active RyR channels direct lysosomal trafficking (exocytosis) and function (autophagy), as application of RyR inhibitors limits lysosomal exocytosis, while promoting the late lysosomal-dependent steps in autophagy. We therefore propose that endogenous RyR channels function as dynamic, Ca2+-signaling-dependent, ER-lysosomal tethering proteins that control the availability of lysosomes in cells, thereby steering lysosomal outcomes towards lysosomal exocytosis (when RyR are active) versus autophagy (when RyR are closed).
This study shows that RyR channels directly interact with the lysosomal vATPase. Protein docking analysis predicts multiple subunits of the vATPase to contribute to the interaction with RyR (Fig. 1d, e and Table 1), whereby ATP6v0a1 is one of the major interacting subunits. Immunoprecipitation assays indicated that ATP6v0a1 interacts with the RyR and is sufficient to inhibit RyR-mediated Ca2+ fluxes (Figs. 1 and 2). Based on the protein docking, we could narrow down the binding site on ATP6v0a1 to the first 300 aa and 400 aa in the central domain of RyR2 (aa 1450-1850) (Fig. 1h, i). Disrupting the binding between RyR2 and ATP6v0a1 using this 400-aa long RyR2 fragment increased RyR2-mediated Ca2+ release (Fig. 3). These results complement and align well with a recent study showing that excessive FAD-associated RyR-mediated Ca2+ release impairs lysosomal function18. In that work, the observed lysosomal dysfunction was attributed to a lower expression of the lysosomal vATPase, ultimately limiting autophagic flux, which both could be reversed by inhibiting RyR activity18. Combining these results with our own findings indicates that the interaction between RyRs and vATPases may locally regulate RyR activity, thereby determining lysosomal function. Intracellular Ca2+ signals, depending on their spatio-temporal properties, have been reported to have both inhibitory and stimulatory effects on autophagic flux37. Our own previous work shows that spontaneous RyR-mediated Ca2+ release inhibited autophagic flux by reducing autophagosomal turnover via lysosomes17. The latter was also confirmed in the context of FAD, hallmarked by increased basal RyR hyperactivity18,19. The data presented in Fig. 4 confirm these findings, indicating that inhibiting RyR-mediated Ca2+ release in hiPSC-derived cortical neurons stimulated autophagic flux at the level of autophagosome-autolysosome fusion. Moreover, this enhanced autophagic flux is accompanied by an inhibition of lysosomal exocytosis (Fig. 6). Although ryanodine treatment did not increase early autophagy markers (Fig. 5), prolonged treatment with ryanodine in the presence of bafilomycin A1 resulted in elevated LC3-II levels (Fig. 4d). This observation suggests that ryanodine may promote LC3-II lipidation through non-canonical autophagy pathways44. The mechanisms underlying this effect remain unclear and warrant further investigation. The ER-Golgi intermediate compartment (ERGIC) is known to serve as primary membrane source for LC3 lipidation. While ERGIC is important for the PI3K-dependent generation of pre-autophagosomal membranes by recruiting ATG14, an early autophagosome marker45, LC3 lipidation at Golgi complex membranes can also occur independently of ULK1 activation44. Moreover, this Golgi-associated non-canonical LC3 lipidation appears to depend on vATPase activity, although its contribution to our findings requires additional study46.
There are a number of possible manners in which RyR-mediated Ca2+ release can regulate lysosomal exocytosis. It is known that lysosomal exocytosis is regulated by increased peri-lysosomal Ca2+47. As such RyR-mediated Ca2+ release, which reaches the peri-lysosomal area (Fig. 2a), may directly stimulate lysosomal exocytosis. Besides this mode of action, an indirect effect of RyR-mediated Ca2+ release is also possible. Lysosomal Ca2+ release via for instance TRPML1 has been shown to trigger lysosomal exocytosis48. TRPML1-mediated Ca2+ release activates lysosomal large-conductance Ca2+-activated K+ channels (BKCa) which provide K+ as counterion to sustain further TRPML1-mediated Ca2+ release and subsequent lysosomal exocytosis49,50. Hence, it is possible that RyR-mediated Ca2+ signals reaching the peri-lysosomal environment activate BKCa channels, thereby sustaining TRPML1 activity and thus driving lysosomal exocytosis. However, further research is required to elucidate the involvement of TRPML1 channels, and other lysosomal Ca2+ channels such as TPC2, in RyR-controlled lysosomal exocytosis. From our results, we however can postulate that the spatio-temporal properties of RyR-mediated Ca2+ release impact lysosomal exocytosis. Inhibiting endogenous RyR activity, not only in ectopic expression models but also in physiological systems such as neurons, limits lysosomal exocytosis. Of note, the effect of RyR inhibition was much more profound in T-Rex RyR2 cells compared to the hiPSC-derived neurons (Fig. 6g, h). A critical difference between these two cells types is the RyR-protein abundances and thus the manifestation of spontaneous RyR-mediated Ca2+ release. In the neurons, these spontaneous Ca2+ events appear less frequent, less regular and with lower amplitudes (Suppl. Fig. 2b). However, in the T-Rex RyR2 cells display large rhythmic Ca2+ transients (Fig. 2a)51, further underscoring the importance of the spatio-temporal properties of RyR activity on lysosomal exocytosis. Although our results show that RyR2-mediated Ca2+signals reach the peri-lysosomal region, it remains to be established whether the Ca2+ is transferred into the lysosomal lumen and thus is taken up by the lysosomes, e.g. via TMEM165 lysosomal Ca2+-import systems. However, at the same time, it is also possible that Ca2+-dependent sensor on the lysosomal membranes could be responsible for regulating lysosomal exocytosis. Elucidating whether Ca2+ released by RyR2 actually needs to be taken up by lysosomes in order to affect their function will be an interesting avenue for future research.
Cellular lysosomal positioning is critical for their proper functioning and affects for instance autophagy and lysosomal exocytosis52. One way of positioning organelles is to physically tether them to each other, possibly resulting in membrane contact site formation. At these contact sites, Ca2+ signaling plays important roles. Ca2+-release channels have been shown to reside at such nanodomains and act as tethers to promote membrane contact site formation. Both IP3Rs and RyRs haven been shown to interact with voltage dependent anion channels at the mitochondria resulting in the formation of ER-mitochondrial membrane contact sites53,54,55,56. At the level of ER-lysosomal contact sites, the contribution of ER-located Ca2+-release channels in tethering lysosomes has not yet been fully resolved. Previous studies have focused on IP3Rs, showing that they can mediate ER-originating Ca2+ signals towards the lysosomes as well as amplify lysosome-originating Ca2+ signals10,11,12,16. However, IP3Rs have not been implicated in acting as tethers for ER-lysosomal contact site formation16. Here, we demonstrate that RyR activity controls ER-lysosomal membrane contact site formation (Fig. 7) and that RyR channels may act as tethers for these contact sites by binding to the lysosomal vATPase (Fig. 1). By regulating ER-lysosomal contact site formation RyRs may direct the availability of lysosomes for autophagic flux or lysosomal exocytosis. Inhibiting spontaneous RyR-mediated Ca2+ release tightens ER-lysosomal contacts and moreover favors the presence of ATP6v0a1 in close proximity of the RyR channels. Interestingly, ATP6v0a1 can directly interact with RyR channels and suppress its Ca2+-flux properties. Together, these results indicate that under physiological conditions spontaneous RyR-mediated Ca2+ release prime lysosomes for exocytosis, whereas non-active channels recruit lysosomes to ER membranes, likely through interaction with ATP6v0a1, thereby promoting lysosomal availability for autophagy. Our competition experiments in neurons support this hypothesis. Overexpression of the RyR2400aa fragment, which interferes with the RyR2-ATP6v0a1-protein complex, increased RyR2-mediated Ca2+ release and lysosomal exocytosis (Fig. 8c–f), indicating that endogenous RyR2-ATP6v0a1 complexes control lysosomal availability and thus function. Since ATP6v0a1 inhibits RyR-mediated Ca2+ release, we postulate that the RyR-ATP6v0a1 complex operates as a positive feedback loop, whereby ATP6v0a1 keeps RyR channels closed, thereby favoring the availability of lysosomes for autophagy over lysosomal exocytosis.
The pathophysiological implications of the RyR-ATP6v0a1 nexus are still unclear. Excessive RyR-mediated Ca2+ release22,23,24,25,26,27,28, reduction in ATP6v0a1-protein levels18,29,30, lysosomal dysfunction, reduced autophagic flux4,57,58,59,60 and alterations in membrane contact site formation/function61,62,63,64,65 have been previously reported as hallmarks in the onset of FAD. Elucidating whether these alterations are due to alterations in RyR-ATP6v0a1 complexes will further our understanding of the early phases of AD pathogenesis, potentially opening up avenues for treatment. Previous studies already indicated that inhibition of FAD-associated RyR-mediated Ca2+ release is beneficial in rescuing lysosomal dysfunction, associated reduced autophagic flux18,19 and reduced Aβ burden23,25,26. The results here also support this as blocking RyR activity increased lysosomal degradation of APP (Fig. 5g, h). Lysosomal vATPase-protein levels are also known to be downregulated in FAD conditions18,29. Here, in our work, we demonstrate that RyR-ATP6v0a1-complex formation was reduced in cortical samples of a 3xTg FAD mouse model compared to the WT control (Fig. 1a, b). An important question to answer will be whether this reduced interaction causes, at least in part, the observed excessive RyR-mediated Ca2+ release. Furthermore, it will be important to assess whether FAD-associated excessive RyR-mediated Ca2+ release deregulates ER-lysosomal contact site formation, thereby evoking excessive lysosomal exocytosis, a phenomenon that could directly contribute to the deposit and accumulation of Aβ in the extracellular environment.
In summary (Fig. 8g), our results suggest that spontaneously active RyR channels stimulate lysosomal exocytosis. Non-active RyR channels are available to interact with the vATPase, increasing ER-lysosomal contact site formation positioning lysosomes for autophagy. The interaction stabilizes the inhibition of the RyR channel favoring lysosomal availability for autophagy over exocytosis. Inhibition of RyR-mediated Ca2+ release drives this pathway towards stimulation of autophagic flux, stimulating the formation of ER-lysosomal membrane contact sites by increasing the interaction between RyRs and the vATPase.
Methods
Reagents and antibodies
Unless otherwise specified, all chemicals were purchased from Merck. Primary antibodies were Anti- Flag M2 HRP conjugated (A8592, Merck, 1:2000), anti-Vinculin (#V-9131, Merck, 1:5000), anti-phospho-AMPK (2535S, Cell signaling technology, 1:1000), anti-AMPK (2603S, Cell signaling technology, 1:1000), anti-phospho-p70 S6 kinase (9234S, Cell signaling technology, 1:1000), anti-p70 S6 kinase (9202, Cell signaling technology, 1:1000) and anti-LC3 (2775S, Cell signaling technology, 1:1000), anti-phospho-ATG13 (46329S, Cell signaling technology, 1:1000), anti-ATG13 (13273S, Cell signalling technology, 1:1000) anti-Lamp1 (H4A3, Developmental Studies Hybridoma Bank, University of Iowa, 1:100), anti-RyR (34 C) (Developmental Studies Hybridoma Bank, University of Iowa, 1:100 immunofluorescence, 1:1000 immunoblot), anti-Beclin-1 (sc-48341, Santa Cruz biotechnology 1:500), anti-SATB2 (ab51502, Abcam, 1:500), anti-ATP6v0a1 (ab237611, Abcam, 1:200 immunofluorescence, 1:1000 immunoblot) and anti-MAP2 (188002, Synaptic Systems, 1:500) anti-HA tag HRP conjugated (26183-HRP, Thermo Fisher Scientific, 1:1000), anti-FIP200 (10069-1-AP, proteintech,1:200) anti-WIPI2 (MCA5780GA, Biorad, 1:200). A rabbit polyclonal antibody targeting RyR2 was produced by LifeTein directed against the following epitope CKPEFNNHKDYAQEK of the mouse RyR2 sequence with a cysteine added to the N-terminal end (1:200)27.
Mouse model
Isolated cortex from 6 month old age and sex matched WT mice [C57BL6/J9] and 3xTg FAD (APPswe, tauP301L, and PS1M146VKI66) were used in this study. Mice were housed at the RFUMS Biological Resource Facility in accordance with institutional animal care and use committee (IACUC) regulations and kept on a 12:12 h light/dark cycle with food and water available ad libitum. Both sexes were used in this study.
Cell lines and neuronal differentiation
T-Rex RyR2 cells were kindly gifted by Dr. Wayne Chen (University of Calgary, Canada) originally described in ref. 51. HEK293 cells were obtained from the Leibniz Institute DSMZ (ACC 305). The T-Rex RyR2 cells were cultured at 37 °C in the presence of 5% CO2 whereas the HEK293 cells were incubated at 10% CO2. Both cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (D5796, Thermo Fisher Scientific) supplemented with 10% fetal calf serum, 1% non-essential amino acids (11140-035, Thermo Fisher Scientific), 4 mM glutamax, (35050038, Thermo Fisher Scientific) 100 units/mL penicillin and 100 µg/mL streptomycin (P4333, Merck). Cells were routinely checked for the absence of mycoplasma infection. When indicated tetracycline (1 µg/µl) was added for 48 hr with daily medium changes. Cortical neural precursor cells (NPC) were generated from Gibco™ Episomal hiPSC Line (A18945; Thermo Fisher Scientific), which was tested to be mycoplasma negative, using protocol described by Shi et al.33. The hiPSCs were plated on human Matrigel coated 6-well plates (354277, Corning) in mTESR (85850, StemCell) with Revitacell (A2644501, Life Technologies). hiPSCs cultures were maintained in E8 flex medium (E8 basal medium (Gibco) complemented with E8 supplement Flex A2858501 and 5 U/ml Penicillin-Streptomycin) and split twice a week with 0.5 mM EDTA. When the hiPSC colonies reached 90% confluency, NPC induction was done by dissociating the hiPSC colonies by accutase (A6964, Merck) and seeding 2.5 million single cell suspension per well of a 6-well plate in neural induction media (NIM) comprising of neural maintenance medium (NMM) complemented with the dual-SMAD inhibitors SB431542 (10 µM) (1614, Tocris) and LDN193189 (1 µM) (130-106-540, Miltenyi). The NMM comprises of 1:1 mixture of N-2 medium consisting of DMEM/F-12 (31331-028), 10 mM GlutaMAX (35050038), 1× N-2 supplement (17502-048), 100 μM nonessential amino acids (11140-035), 100 μM 2-mercaptoethanol (31350010), and B-27 medium consisting of Neurobasal (21103-049), 1× B-27 supplement (17504-044), all from Thermo Fisher Scientific, 50 U/ml penicillin and 50 mg/ml streptomycin (P4333) 5 μg/ml insulin (I9278), 0.5 mM sodium pyruvate (S8636) from Merck). Media changes with NIM were performed every day for 11 days. At day 12, the neuroepithelial cells were collected with Dispase II (04942078001, Merck) and cultured for additional 4 days with NMM spiked with 20 ng/mL bFGF (BT-FGFB-AFL, R&D Systems). Further, rosette-forming neuroepithelial cells were passaged twice with Dispase II (one each after 5-7 days) to purify the NPC population. The cultured NPC were dissociated as single cells using accutase on DIV (day in vitro) 33 and cryopreserved for further experimental use. Differentiation to cortical neurons was performed on human Matrigel coated plates or coverslips by culturing the neuronal precursor cells in NMM. Briefly, NPC were thawed and seeded on human Matrigel coated 6 well plates at 1 million per well in NMM with revitacell. Medium was replaced the next day, to medium without revitacell after which medium changes were performed twice every week. After 1 week of differentiation, single cell suspensions were obtained by accutase treatment and cells were reseeded for the final differentiation on human matrigel coated plates or coverslips at 20000 cells/cm² in neuronal maintenance medium supplemented with revitacell. Medium was replaced the next day, to medium without revitacell after which the medium changes were performed twice a week. The hiPSC-derived cortical neurons were differentiated for two more weeks after which experiments were performed in the following week. All experiments were performed on neurons differentiated for between 52 and 56 days.
Plasmids and constructs
The p4-Neo-mRFP-GFP-LC3 plasmid was a kind gift from Dr. Patrizia Agostinis (KU Leuven). The pFLAG-CMV-5a vector expressing the ATP6v0a1-Flag (accession number ATP6v0a1: AF218249) construct was a kind gift of Dr. Wim Annaert (KU Leuven, Belgium)67. This plasmid was used as template to subclone the first 433 amino acids in a pCMV24 vector to obtain the 3xFlag ATP6v0a11-433 construct. Cloning was performed using NotI and HindIII restriction enzymes and the following primers forward: GTGACGAAGCTTGGGGAGCTTTTCCGGAGTG, reverse: GTAGTAGCGGCCGCTCACTGGGAGAGGATCCGGC. The 3xFlag ATP6v0a1-300 construct was obtained by introducing a stop codon after amino acid 300 of the 3xFlag ATP6v0a11-433 truncating the latter construct. Site directed mutagenesis was performed using the following primers forward: CAAGGTGCGGAAGATGTAGGCTATCTACCATAC, reverse: GTATGGTAGATAGCCTACATCTTCCGCACCTTG. The 3 x HA tagged RyR2400aa construct, corresponding to amino acid 1450-1850 of RyR2 (accession number RyR2: X98330) was cloned, in a pcDNA3.1 vector using NotI and EcorV restriction enzymes via codon optimized gBlockTM of the following sequence: GCCCTCTAGACTCGAGCGGCCGCATGTACCCGTATGATGTTCCGGATTACGCTGGCTACCCATACGACGTCCCAGACTACGCTGGCTACCCATACGACGTCCCAGACTACGCTCATCAGTATGACACAGGCTTTGACTTGGACAGAGTTCGCACAGTAACAGTTACTCTAGGAGATGAAAAAGGAAAAGTGCATGAAAGCATCAAACGCAGCAACTGCTATATGGTATGTGCGGGTGAGAGCATGAGCCCCGGGCAAGGACGCAACAATAATGGACTGGAGATTGGCTGTGTGGTGGATGCTGCCAGCGGGCTGCTCACATTCATTGCCAATGGCAAGGAACTGAGCACATACTATCAGGTGGAACCGAGTACAAAATTATTTCCTGCGGTTTTTGCACAAGCTACAAGTCCCAATGTTTTCCAGTTTGAGTTGGGAAGAATAAAGAATGTGATGCCTCTCTCGGCGGGATTATTCAAGAGTGAGCACAAGAACCCCGTGCCGCAGTGCCCCCCGCGCCTCCACGTGCAGTTCCTGTCACACGTCCTGTGGAGCAGAATGCCCAACCAGTTTTTGAAGGTAGATGTGTCTCGAATAAGTGAACGCCAAGGCTGGTTGGTGCAGTGTTTGGATCCTCTGCAGTTCATGTCTCTTCATATCCCTGAGGAAAACAGATCTGTTGACATCTTAGAGTTGACAGAGCAGGAGGAATTGCTGAAATTTCACTATCACACTCTCCGGCTCTACTCAGCCGTCTGTGCTCTTGGGAACCACCGGGTGGCCCATGCCCTGTGCAGCCATGTGGATGAACCTCAGCTCCTCTATGCCATTGAGAACAAGTACATGCCTGGTTTGCTGCGTGCTGGCTACTATGACCTGCTGATTGACATCCACCTGAGCTCCTATGCCACTGCCAGGCTCATGATGAACAACGAGTACATTGTCCCCATGACGGAGGAGACGAAGAGCATCACCCTGTTCCCTGATGAGAACAAAAAACACGGCCTTCCAGGGATCGGCCTCAGCACCTCCCTCAGGCCACGGATGCAGTTTTCCTCCCCCAGTTTTGTAAGCATTAGTAATGAATGTTACCAGTACAGTCCAGAGTTCCCACTGGACATCCTCAAGTCCAAAACCATACAGATGCTGACAGAAGCTGTTAAAGAGGGCAGTCTTCATGCCCGGGACCCAGTTGGAGGGACTACTGAATTCCTCTTTGTACCTCTCATCAAGCTTTTCTATACCCTGCTGATCATGGGCATCTTTCACAACGAGGACTTGAAGCACATCTTGCAGTTGATTGAGCCCAGTGTGGAACAAAAACTCATCTCAGAAGAGGATCTGTGAGATATCTGCAGAATTCCACCACACTG. The split greenFAST probe for detecting ER-lysosomal contact sites was obtained by sequential cloning of gBlocks™ (from integrated DNA technologies) containing the sequence of C-term splitFAST-SEC61 P2A, using NotI and EcoRV, and LAMP1-N-term greenFast, using EcoRV and HindIII in a pcDNA3.1 vector. gBlock™ sequences were codon optimized and were as follows TACGATGCGGCCGCATGGGTGACAGCTATTGGGTCTTTGTGAAACGGGTGTCCGGAGGAGGCGGCAGCGGCGGAGGGGGATCCCCTGGTCCGACCCCCAGTGGCACTAACGTGGGATCCTCAGGGCGCTCTCCCAGCAAAGCAGTGGCCGCCCGGGCGGCGGGATCCACTGTCCGGCAGAGGAAAAATGCCAGCTGTGGGACAAGGAGTGCAGGCCGCACAACCTCGGCAGGCACCGGGGGGATGTGGCGATTCTACACAGAAGATTCACCTGGGCTCAAAGTTGGCCCTGTTCCAGTATTGGTTATGAGTCTTCTGTTCATCGCTTCTGTATTTATGTTGCACATTTGGGGCAAGTACACTCGTTCGGCTACTAACTTCAGCCTGCTGAAGCAGGCTGGAGACGTGGAGGAGAACCCTGGACCTGATATCGTGGGCCGG AND CCGGCCCTGATATCATGGCGGCGCCTGGATCTGCGCGACGCCCGCTGTTGCTTTTGCTCCTCCTGCTTCTCTTGGGCCTTATGCATTGCGCTAGTGCGGCCATGTTTATGGTCAAGAATGGTAACGGGACAGCGTGCATAATGGCGAATTTCAGTGCGGCTTTTTCAGTGAACTATGATACCAAGAGCGGACCCAAAAACATGACGTTTGACCTGCCAAGTGACGCCACAGTTGTTTTGAATCGGTCTTCCTGTGGAAAAGAGAACACCAGTGACCCAAGTCTCGTTATCGCTTTCGGCAGAGGACACACCCTGACCCTCAATTTCACGAGAAATGCAACACGGTATTCAGTACAATTGATGTCATTCGTATATAACTTGTCAGATACGCATCTGTTCCCTAACGCGAGCTCCAAGGAGATCAAGACAGTTGAGTCCATTACCGACATACGAGCTGATATAGATAAAAAATATAGGTGTGTTTCAGGAACCCAGGTACACATGAATAATGTTACAGTAACATTGCATGACGCGACTATTCAGGCTTACCTTAGTAACTCTTCTTTTAGTCGAGGAGAGACCCGCTGCGAACAGGACAGACCCTCCCCCACGACCGCACCACCTGCCCCCCCTAGCCCGTCACCCAGCCCAGTTCCTAAATCTCCGTCTGTGGATAAGTACAATGTCTCCGGTACCAACGGGACTTGCCTCCTCGCATCTATGGGCCTCCAATTGAATCTCACATATGAACGCAAAGATAATACTACCGTAACCAGACTGCTCAATATAAATCCTAACAAAACAAGCGCAAGTGGAAGCTGTGGAGCTCACCTTGTCACCTTGGAATTGCATTCTGAAGGCACGACGGTCTTGCTGTTCCAGTTCGGGATGAATGCCTCCAGCAGCAGGTTCTTTCTCCAAGGTATCCAGCTTAATACTATACTCCCAGACGCCAGGGACCCCGCGTTCAAAGCGGCAAATGGGAGTTTGAGAGCGTTGCAGGCGACTGTCGGAAATTCCTACAAGTGCAATGCAGAAGAACACGTGCGGGTCACCAAGGCGTTCTCCGTTAATATCTTCAAGGTCTGGGTTCAGGCGTTCAAAGTGGAAGGCGGTCAGTTCGGAAGTGTCGAAGAGTGCCTGTTGGACGAGAACAGTATGCTTATACCAATAGCGGTGGGGGGTGCACTGGCGGGCTTGGTTCTGATCGTCCTCATTGCGTACCTCGTCGGTCGCAAGAGAAGCCATGCGGGTTACCAGACAATCGAATTCGACGTTGGTGGTGGCGGATCAGAAGGAGGCGGTAGCGGGGGCCCTGGTTCGGGAGGGGAAGGTTCTGCTGGGGGAGGGAGCGCTGGCGGGGGGTCTCGGGGCGACGGCGGCAGCGGCGGCGGCAGCGGCCGGGAGCATGTTGCCTTTGGCAGTGAGGACATCGAGAACACTCTGGCCAAAATGGACGACGAACAACTGGATGGGTTGGCCTTTGGCGCAATTCAGCTCGATGGTGACGGGAATATCCTGCAGTACAATGCTGCTGAAGGAGACATCACAGGCAGAGATCCCAAACAGGTGATTGGGAAGAACTTCTTCAAGGATGTTGCAACTGGAACGGATTCTCCCGAGTTTTACCGCAAATTCAAGGAAGGCGTAGCGTCAGGGAATCTGAACACCATGTTCGAATGGATGATACCGACAAGCAGGGGACCAACCAAGGTCAAGGTGCACATGAAGAAAGCCCTTTCCTGAAAGCTTACTAGC. pExp_CAG:EBFP2 (BFP expressing plasmid) was a gift from Ron Weiss (Addgene plasmid # 51790)68. GFP- SEC61β was a gift from Christine Mayr (Addgene plasmid #121159)43. To produce the LAMP1-RFP670 construct, LAMP1 was first cloned in the pcDNA vector using NotI and EcorV using a codon optimized gBlockTM. The sequence corresponding to LAMP1 in the above gBlockTM was utilized for this cloning. Following the introduction of LAMP1 in this vector, RFP670 was cloned in frame with LAMP1 using EcorI and HindIII as restriction enzyme in combination with the following primers forward: GCGGTAGAATTCGCGTAGCAGGTCATGCCTCTGG, reverse: GCTAGTAAGCTTTTAGCTCTCAAGCGCGGTGATCC. The pmiRFP670-N1, a gift from Vladislav Verkhusha (Addgene plasmid # 79987)69, was used as template. The pcDNA LAMP1-GCaMP6S construct was a kind gift from Dr. Lee Haynes (University of Liverpool, United Kingdom)70. GCaMP6S was cloned in pAAV-TF-CaMKII 0.4-intron vector generating the pAAV-TF-CaMKII 0.4-intron GCaMP6S construct, using BamHI and Eco47III as restriction enzymes and the following primers forward: GCGGTAGGATCCGCCACCATGACTGGTGGACAGCAAATG and reverse: GCTAGTAGCGCTTCACTTCGCTGTCATCATTTG using the LAMP1-GCaMP6s construct as a template. The RyR2400aa-p2a-BFP construct was developed by first introducing p2a-BFP in a pcDNA3.1 vector, resulting in the p2a-BFP vector, using EcorV and HindIII as restriction enzymes in combination with codon optimized gBlockTM: GCGGGCGATATCATGGCTACTAACTTCAGCCTGCTGAAGCAGGCTGGAGACGTGGAGGAGAACCCTGGACCTGTGAGCAAGGGCGAGGAGCTGTTCACCGGGGTGGTGCCCATCCTGGTCGAGCTGGACGGCGACGTAAACGGCCACAAGTTCAGCGTGAGGGGCGAGGGCGAGGGCGATGCCACCAACGGCAAGCTGACCCTGAAGTTCATCTGCACCACCGGCAAGCTGCCCGTGCCCTGGCCCACCCTCGTGACCACCCTGAGCCACGGCGTGCAGTGCTTCGCCCGCTACCCCGACCACATGAAGCAGCACGACTTCTTCAAGTCCGCCATGCCCGAAGGCTACGTCCAGGAGCGCACCATCTTCTTCAAGGACGACGGCACCTACAAGACCCGCGCCGAGGTGAAGTTCGAGGGCGACACCCTAGTGAACCGCATCGAGCTGAAGGGCGTCGACTTCAAGGAGGACGGCAACATCCTGGGGCACAAGCTGGAGTACAACTTCAACAGCCACAACATCTATATCATGGCCGTCAAGCAGAAGAACGGCATCAAGGTGAACTTCAAGATCCGCCACAACGTGGAGGACGGCAGCGTGCAGCTCGCCGACCACTACCAGCAGAACACCCCCATCGGCGACGGCCCCGTGCTGCTGCCCGACAGCCACTACCTGAGCACCCAGTCCGTGCTGAGCAAAGACCCCAACGAGAAGCGCGATCACATGGTCCTGCTGGAGTTCCGCACCGCCGCCGGGATCACTCTCGGCATGGACGAGCTGTACAAGTAAAAGCTTGTGACG. Following this the same gBlockTM encoding 3xHA-RyR2400aa was cloned in frame before p2a-BFP using NotI and EcorV.
Production of recombinant adeno associated viral vectors
pAAV-TF-CaMKII 0.4-intron GCaMP6S was used to produce rAAV2/7 expressing GCaMP6S under the control of a neuronal promotor. rAAV2/7 vectors were produced by the Leuven viral vector core as reported earlier71,72 with minor modifications. Subconfluent, low (<50) passage adherent HEK 293 T cells (ATCC, Manassas, VA, USA) were transfected using a 25 kDa linear polyethylenimine solution using the AAV-TF, AAV rep/cap and pAdDeltaF6 plasmids in the ratio of 1:1:1. Productions were performed using an 8-layer Celldisc (2000 cm2 growth area, Greiner Bio-One). 293 T cells were seeded at 350 × 106 cells per production in DMEM (Gibco, Fisher Scientific) with 2% fetal calf serum. The next day, 260 mg of pAAV-TF plasmid, 260 mg of rep/cap construct and 260 mg of pAdDeltaF6 plasmid were mixed in 20 ml of PBS. An equal volume containing 8.7 ml of 13 µM polyethylenimine solution and 11.3 ml of PBS was added slowly to the DNA mixture. Following incubation at room temperature, the DNA-polyethylenimine complex was added to the 293 T cells. After 24 hr of incubation at 37 °C in a 5 % CO2 humidified atmosphere, the medium was replaced with Optimem (Gibco, Fisher Scientific) without serum. The medium was harvested and replaced 5 consecutive days after transient transfection and concentrated using tangential flow filtration (TFF).
Recombinant rAAV purification
The TFF concentrated supernatant was purified using an iodixanol step gradient. Iodixanol (60 %) was diluted with phosphate-buffered saline (PBS) containing a final concentration of 1 M NaCl in final 20, 30 and 40 % (w/v) solutions. A discontinuous gradient was prepared by carefully underlayering the concentrated supernatant with 5 ml of 20 % iodixanol, 3 ml of 30 % iodixanol, 3 ml of 40 % iodixanol and 3 ml of 60 % iodixanol. The gradient was centrifuged in a Beckman Ti-70 fixed angle rotor (Analis, Gent, Belgium) at 60 000 x g for 2 hr. Gradient fractions were collected in 250 µl aliquots and real-time PCR was performed to identify fractions containing AAV vector using a primer probe set for the BGHpolyA sequence (primer sequences, 5’-TCTAGTTGCCAGCCATCTGTTGT-3’ and 5’-TGGGAGTGGCACCTTC CA-3’; probe sequence, 5’-TCCCCCGTGCCTTCCTTGACC-3’). The pooled positive fractions were centrifuged in a Vivaspin 6 (PES, 100,000 MWCO, Sartorius AG, Goettingen, Germany) using a swinging bucket rotor at 3000 x g. The iodixanol of the pooled fractions was replaced by a five times exchange with PBS. The final sample was aliquoted and stored at -80 °C. Final titers of the rAAV stocks were determined by real-time PCR analysis for genomic copy determination (vg/ml) and Ruby-stained sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis for vector purity.
Transfections
T-Rex RyR2 cells were transfected 48 hr before use, using the TransIT-X2 Dynamic Delivery System (MIR 6004, Mirius) according to the manufacturer’s protocol. A ratio of 2 µl to 1 µg DNA of TransIT-X2 reagent was maintained for all experiments. For co-transfections, the GCaMP6S, GFP, RFP or BFP (-tagged) proteins were included at a ratio of 1:3 compared to the non-tagged constructs. In doing so the fluorescent markers could be utilized to identify transfected cells. Similarly, hiPSC-derived cortical neurons were transfected with the split greenFAST probe for detecting ER-lysosomal contact sites, GFP-RFP-LC3, p2a-BFP or RyR2400aa-p2a-BFP. Also here, a ratio of 2 µl of TransIT-X2 reagent to 1 µg DNA was maintained. In addition, 6 hr post-transfection neuronal maintenance medium was replaced to neuronal maintenance medium supplemented with 0.5 mM kynurenic acid73.
Cell lysis and immunoblot
T-Rex RyR2 cells and hiPSC-derived cortical neurons were treated as indicated in the figures. Following treatments, the cells were mechanically detached and pelleted in PBS-/- by spinning down (300 x g) for 5 min at 4 °C. After removing PBS cells were lysed in a CHAPS based lysis buffer (50 mM Tris-HCl pH 7.5, 100 mM NaCl, 2 mM EDTA, 50 mM NaF, 1 mM Na3VO4, 1 % CHAPS and protease inhibitor tablets (04693132001, Merck)) (CHAPS lysis buffer) for 30 min with head over head mixing. Following lysis, cell lysates are spun down (4000 x g) for 5 min at 4 °C and the supernatant was collected. Protein concentration was determined via Bradford analysis and protein samples were prepared at 1 µg/µl in 1X NuPAGE LDS Sample Buffer (NP0007, Thermo Fisher Scientific) followed by heating for 5 min at 95 °C. Following heating, samples were loaded on NuPAGETM 4-12 % Bis-Tris gels (NP0322BOX, Thermo Fisher Scientific). Immunoblotting using the indicated antibodies was performed as in ref. 74.
Co-immunoprecipitation assays
Cortex from 6-month-old age and sex matched WT mice [C57BL6/J9] and 3xTg FAD (APPswe, tauP301L, and PS1M146VKI66) mice were homogenized, using an eppendorf douncer, and lysed for 30 min with head over head mixing in CHAPS lysis buffer. Following lysis, cell lysates are spun down (4000 x g) for 5 min at 4 °C. The supernatant was collected and protein concentration was determined. Five µg of C34 anti-RyR antibody or a mouse control IgG (SC-2025, Santa Cruz) was immobilized on 20 µl Protein G DynabeadsTM (10004D, Thermo Fisher Scientific) according to the manufacturers protocol. Next, the antibody coated beads were incubated with 800 µg of cortical lysates overnight with head over head mixing. The next day, the immune complexes were washed twice with the CHAPS lysis buffer. Elution of the immune complexes was obtained by 30 min incubation with continuous shaking at room temperature in 50 µl elution buffer (50 mM Tris pH8, 0.2 % sodium dodecyl sulfate and 0.1 % Tween). After collecting the eluted fraction NuPAGE LDS Sample Buffer (1X final concentration) was added and samples were heated for 5 min at 70 °C before immunoblot analysis. Similar co-immunoprecipitation experiments were setup in T-Rex RyR2 cells and HEK293 cells. In these experiments 5 µg of the rabbit polyclonal RyR2 antibody or a normal Rabbit IgG (CR1, Sino Biological) was immobilized to the protein G Dynabeads and incubated with 100 µg of protein lysate of T-Rex RyR2 cells or HEK293 cells overexpressing the indicated constructs. Following the overnight incubation the immune complexes were washed five times. Elution and immunoblotting was performed as described above. The Flag-pulldowns in HEK293 cell lysates were performed by incubating anti-DYKDDDDK labelled beads (651503, biolegend) with 100 µg of total cell lysate overexpressing the indicated constructs overnight at 4 °C. The next days the complexes were washed 5 times and eluted by boiling in LDS Sample buffer prior to immunoblot analysis.
Molecular docking analysis
The cryo-electron microscopy structures of RyR2 (PDB entry: 5GOA75) and vATPase (PDB entry: 7U4T76) were first downloaded from the Protein Data Bank. Next, ribonuclease kappa, renin receptor, SidK, mTOR-associated protein MEAK7, and N-acetyl-β-D-glucosamine molecules were removed from the vATPase complex. Protein preparation was carried out using the Protein Preparation Wizard (Schrödinger Release 2024-1, Schrödinger, LLC, New York, NY, USA, 2024) with the default settings: bond orders were assigned using CCD database, missing hydrogens were added, termini were capped, the missing side chains were modelled with Prime, and het protonation states (pH 7.0 ± 2.0) were modelled with Epik77. Protein-protein docking of RyR2 and vATPase was performed using the server version of LightDock78,79, which was optimized for membrane-associated protein assemblies80. Membrane-embedded structures were obtained from the Orientations of Proteins in Membranes (OPM) database81. For RyR2, only chain A was used due to system size limitations, while for vATPase, the entire assembly was included. The Membranator module in LightDock was used to define the cytosolic and transmembrane regions of vATPase, since only the cytosolic region was used for docking and thus reducing the system size. Docking between RyR2 and vATPase was performed using LightDock’s default settings, residue restraints were employed in which the cytosolic residues of ATP6v0a1 were chosen to guide the docking to RyR2. The algorithm generated 200 docking complexes, ranked according to an implementation of the DFIRE scoring82 function used by default in Lightdock. Docking complexes were visualized. The highly scored docking complex, with non-overlapping parallel membranes, was then used to reconstitute the full RyR2-vATPase docking complex for visualization. Visualization of docking complexes and detection of interacting hotspot residues between both docking partners were preformed using PyMOL. A distance cut-off of 3.6 Å between any atoms on the interface between the docking partners was used for detecting hotspot residues in both partners.
Single cell Ca2+ measurements
T-Rex RyR2 cells were co-transfected with LAMP1-GCaMP6S and either empty vector (pCMV24), ATP6v0a1-Flag, ATP6v0a1-Flag + empty vector or ATP6v0a1-Flag + RyR2400aa vector at a 1:3 ratio (LAMP1-GCaMP6S to other constructs). Just before measuring, the medium was changed to a modified Krebs-Ringer solution (135 mM NaCl, 6.2 mM KCl, 1.2 mM MgCl2, 12 mM HEPES, pH 7.3, 11.5 mM glucose and 2 mM CaCl2). Additions were performed as indicated in the figure. Only LAMP1-GCaMP6S positive cells were measured using a Nikon eclipse Ti2 inverted fluorescence microscope (Nikon) equipped with a 20 ×0.5 NA Plan Fluor DIC N2 air objective, excitation filter FF01-378/474/554/635 and dichroic mirror FF409/493/573/652-Di02 and emission filter 515/30 (Semrock). Excitation was performed at 470 nm using a CoolLed pR-4000 (CoolLed). Acquisition of the emitted fluorescent signal was performed at 0.5 Hz using a pco.edge 4.2bi sCMOS camera (pCO). For ER-store content measurements, T-Rex RyR2 cells co-transfected with mCherry and either the empty vector of ATP6v0a1-Flag vector at a 1:3 ratio were loaded with 1 µM Fura-2AM (21023, AAT Bioquest) for 30 min in modified Krebs-Ringer solution. Next, de-esterification of the dye was allowed to occur by incubating the cells in Fura-2AM free modified Krebs-Ringer solution for 30 min at room temperature. Additions were performed as indicated in the figure. Only mCherry positive cells were measured using the above Nikon microscope equipped with an additional CoolLed pE-340Fura system, a 409 nm blocking edge BrightLine® long-pass filter (Semrock) and a 595/31 emission filter (Semrock). This setup allows measuring mCherry signal for selecting cells at excitation 550 emission 580 nm together with Fura measurements exciting at 340/380 measuring emission at 520 nm. For the single cell Ca2+ measurements in the hiPSC-derived cortical neurons, GCaMP6S was transduced via rAAV2/7 1 week prior to the measurements. At least 5 days before measuring the neurons, medium was changed to BrainPhysTM medium without phenol red (05791, Stemcell technologies) supplemented with the NeuroCult™ SM1 Neuronal Supplement (05711, Stemcell technologies). Following medium changes and all treatments including the additions on the microscope were performed in this medium. GCaMP6S positive neurons were measured on the above Nikon microscope using the same setup as for the LAMP1-GCaMP6S measurements. In addition, Cal520 was used as Ca2+ indicator in the single cell Ca2+ measurements performed on neurons overexpressing either p2a-BFP or RyR2400aa-p2a-BFP. For this 1 µM of Cal520-AM was loaded for 30 min at 37 °C in the neurons cultured in BrainPhysTM medium without phenol red. Following a wash step, de-esterification of the dye was allowed to occur by incubating the cells in Cal520-AM free BrainphysTM medium for 30 min at 37 °C. Subsequent additions on the microscope were also performed in this medium. The same Nikon microscope and setup as described above was used for these measurements. FIJI software83 was utilized for image analysis. After background subtraction, thresholding in combination with particle analysis was used to determine regions of interest (ROI) which were either GCaMP6S, mCherry or BFP positive. If Thresholding was not feasible due to cells being in to close proximity, ROI were drawn manually. Fluorescence changes were obtained as mean intensities for each ROI which were normalized as F/Fmin for the GCaMP6S and Cal520 measurements or the ratio of emission at 520 nm following excitation at 340 and 380 nm (F340/F380) for the Fura-2AM measurements. AUC were obtained using GraphPad Prism 10 software. Amplitudes from caffeine responses were obtained by subtracting the baseline fluorescence (average of 3 measurements before caffeine addition) from the peak response.
GFP-RFP-LC3 measurements
hiPSC-derived cortical neurons, seeded and differentiated on human Matrigel coated cover slips (625995, Chem Lab) were transiently transfected with p4-Neo-mRFP-GFP-LC3 plasmid. After 48 hrs the cells were treated for 24 hr with DMSO or Ryanodine (30 µM). Following this, the coverslips were washed once with PBS containing MgCl2 and CaCl2 (PBS+/+) and fixed for 15 min at room temperature using 4 % paraformaldehyde (J19943.K2, Thermo scientific). Following fixation, the coverslips were washed twice with PBS+/+ of which the last one was supplemented with 2 µg/ml Hoechst 33342 dye, coverslips were mounted using ProLongTM Glass Antifade mountant (P36980, Thermo Fisher Scientific). After 24 hr of curing at room temperature the mounted coverslips were kept at 4 °C until imaged. The coverslips were imaged using the above Nikon microscope. The FIJI software was used to quantify the ratio of GFP over RFP positive puncta in each transfected cell.
Lysosomal pH measurements
All lysosomal pH measurements were performed in phenol red free medium. When T-Rex RyR2 cells were measured 48 hr before measuring, the medium was changed to phenol red free FluoroBrite™ DMEM medium (A1896701, Thermo Fisher Scientific), supplemented with 10 % fetal calf serum, 1% non-essential amino acids, 4 mM glutamax, 100 units/mL penicillin and 100 µg/mL streptomycin. In hiPSC-derived cortical neurons, medium was changed at least 5 days prior to measuring to NeuroCult™ SM1 Neuronal Supplement supplemented BrainPhysTM medium without phenol red. In all experiments, 24 hr before measuring, phenol red free medium supplemented with FITC-dextran 10000 MW (50 mg/ml) and rhodamine-dextran 10000 MW (25 mg/ml) was added to the cells allowing to load the dextran conjugated dyes via endocytosis. Three hr before measuring the cells were chased in dye free medium allowing the dyes to sequester in the lysosomes. If indicated DMSO or ryanodine (30 µM) (ALX-630-062-M005, Enzo Life Sciences) was included during both the incubation and the chase phase of the dextran dye loading to obtain a 24 h treatment. Bafilomycin A1 (100 nM) (sc-201550A, Santa Cruz) treatment and loading of a cell permeable Hoechst 33342 dye (2 µg/ml) was performed during the 3 hr chase. The cells were imaged a Nikon eclipse Ti2 inverted fluorescence microscope equipped with a 40 ×1.3 NA Plan Fluor oil objective with excitation filter FF01-378/474/554/635 and dichroic mirror FF409/493/573/652-Di02 and emission filters 515/30 and 590/31 (Semrock). A CoolLed pR-4000 was used to excite at 380 nm, 470 nm and 580 nm measuring emission at 440 nm, 520 nm and 580 nm for BFP (or Hoechst), FITC and rhodamine signals respectively via a pco.edge 4.2bi sCMOS camera. At least four images were taken per measured chamber. In the T-Rex RyR2 cells FIJI software was utilized to quantify the intensities of FITC and rhodamine in BFP positive cells following background subtraction. In the hiPSC-derived cortical neurons the nuclear Hoechst 33342 stain was used to determine the total amount of neurons measured per image. Lysosomal pH was obtained by making the ratio of FITC over rhodamine fluorescence (FFITC/Frhodamine)31,32. Lysosomal dye loading was obtained by dividing the rhodamine fluorescence by either the BFP area or the total cell count (Hoechst stain).
Lysosomal enzyme activity
hiPSC-derived cortical neurons, seeded and differentiated on Matrigel coated cover slips were treated for 24 hr with DMSO or Ryanodine (30 µM). Three hr before the end of the treatment DQ™ Red BSA (0.01 mg/ml) together with Hoechst 33342 dye (2 µg/ml) was added to all conditions. Next, coverslips were washed once with PBS containing MgCl2 and CaCl2 (PBS+/+) and fixed for 15 min at room temperature in 4 % paraformaldehyde. Following fixation, the coverslips were washed twice with PBS+/+ and mounted using ProLongTM Glass Antifade mountant. After 24 hr of curing at room temperature the mounted coverslips were kept at 4 °C until imaged. Imaging was performed using a Axio Observer Z1 fluorescent microscope (Zeiss) equipped with a Plan-Neofluar 40 ×1.3 NA oil objective, with 62 HE BFP GFP HcRed filter set (Zeiss). Excitation was performed using a Lambda DG4 Xenon-lamp based system equipped with excitation filters 387/15 and 585/35 for exciting Hoechst 33342 at 380 nM and DQ™ Red BSA at 590 nm. Fluorescent signals were captured at 450 nm and 620 nm for Hoechst 33342 and DQ™ Red BSA DQ respectively. At least 10 images were obtained for each measured coverslip. Data analysis was performed using FIJI. After background subtraction, the intensity of the DQ-Red BSA signal was determined per cell (obtained from the Hoechst dye).
Lysosomal exocytosis assays
Plasma membrane LAMP1 labeling
hiPSC-derived cortical neurons, seeded and differentiated on human Matrigel coated cover slips (625995, Chem Lab) were treated for 24 hr with DMSO or Ryanodine (30 µM) or were transiently transfected with p2a-BPP or RyR2400aa-p2a-BFP. After the treatments, the coverslips were washed once with ice cold PBS+/+ and incubated with anti-Lamp1 (H4A3), targeted against a luminal epitope on LAMP1, in PBS+/+ containing 10 % BSA for 1 h at 4 °C. After primary antibody treatment, the coverslips were washed twice with PBS+/+ and fixed with 4 % paraformaldehyde for 15 min at room temperature followed by two wash steps with PBS+/+. Next, the coverslips were incubated with secondary goat anti mouse-Alexa FluorTM 488 (A11017, Thermo Fisher Scientific) in PBS+/+ containing 10 % BSA for 1 h at room temperature. After washing twice with PBS+/+, of which the last one was supplemented with 2 µg/ml Hoechst 33342 dye, coverslips were mounted using ProLongTM Glass Antifade mountant. After 24 hr of curing at room temperature the mounted coverslips were kept at 4 °C until imaged. Imaging was performed using the above mentioned Nikon eclipse Ti2 inverted fluorescence microscope equipped with a 40 ×1.3 NA Plan Fluor oil objective with excitation filter FF01-378/474/554/635 and dichroic mirror FF409/493/573/652-Di02 and emission filters 515/30. A CoolLed pR-4000 was used to excite at 380 nm (Hoechst) and 470 nm (Alexa FluorTM 488). A pco.edge 4.2bi sCMOS camera was used to register emitted fluorescence at 450 nm and 520 nm. Data analysis was performed using FIJI. Following background subtraction Alexa FluorTM 488 intensity was determined per cell given by the nuclear Hoechst 33342 stain.
Secreted glucosaminidase activity assay
All glucosaminidase activity assays were performed in phenol red free medium using. When T-Rex RyR2 cells were measured 48 hr before measuring medium was changed to phenol red free FluoroBrite™ DMEM medium (A1896701, Thermo Fisher Scientific), supplemented with 10 % fetal calf serum, 1 % non-essential amino acids, 4 mM glutamax, 100 units/mL penicillin and 100 µg/mL streptomycin. In the hiPSC-derived cortical neurons medium was changed at least 5 days prior to measuring to NeuroCult™ SM1 Neuronal Supplement supplemented BrainPhysTM medium without phenol red. Cells were seeded in human Matrigel coated 12 well plates and were treated as indicated in the figures. After the treatments 50 µl of the medium or fresh medium as background control was transferred to a black 96 well plate (655090, Greiner) in triplicate for each condition. 200 µl of 4-methylumbelliferyl N-acetyl-β-D-glucosaminide (69585, Merck) solubilized to 150 µg/ml in 0.1 M acetate buffer pH 4,5 was added to the medium in the 96 well plate right before measuring. The 96-well plate was read out every 10 min for 3 hr using a Flexstation 3 (Molecular devices) at 37 °C, at excitation 360 emission 450. Secreted glucosaminidase was determined by determining the amplitude of the 4-methylumbelliferyl intensity and subtracting the same value obtained in the fresh medium as background control. All obtained values are normalized to the total amount of cells in the 12 well counted after enzymatic detachment (trypsin or accutase).
Immunofluorescence imaging
hiPSC-derived cortical neurons were seeded on human Matrigel coated coverslips for immunofluorescence stainings and treated as indicated in the figures. Following treatment, the cells were washed once with PBS-/- before fixation for 15 min with 4 % paraformaldehyde at room temperature. Following fixation, the cells were washed once more with PBS+/+ before being permeabilized for 15 min using filter-sterilized PBS+/+ supplemented with 1 % BSA and 0.2 % triton X. After washing twice, the coverslips are incubated in blocking solution (PBS+/+ + 1% BSA + 10% goat serum) for 1 hr at room temperature prior to overnight incubation with primary antibodies (as indicated in the figures) solubilized in PBS+/+ supplemented with 1 % BSA and 0.1 % triton X at 4 °C. The next day, coverslips are washed 3 times with PBS+/+ + 1 % BSA followed by a 1-h incubation at room temperature with suitable fluorescently labelled secondary antibodies (goat anti mouse-Alexa FluorTM 488 and goat anti rabbit-Alexa FluorTM 555 (A21430, Thermo Fisher Scientific)). After this, the coverslips were washed three times with PBS+/+ + 1 % BSA with an addition of Hoechst 33342 dye (2 µg/ml) if required during the final wash step. Finally, the coverslips were mounted using the ProLongTM Glass Antifade mountant. After 24 hr of curing at room temperature the mounted coverslips were kept at 4 °C until imaged. For MAP2 and SATB2 imaging, confocal images were captured using a 63 × 1.4 NA oil immersion objective on a Zeiss LSM510 confocal microscope with laser excitation at 543 nm and emission detection with a LP560 filter and with laser excitation at 488 nm and emission detection at 530 nm using a BP505-530 filter. FIP200, WIPI2 and Hoechst combination stains were imaged using the above defined Nikon eclipse Ti2 inverted fluorescence microscope equipped with a 40 × 1.3 NA Plan Fluor oil objective. Quantification of the FIP200 and WIPI2 puncta per cell was performed using FIJI. RyR, LAMP1 and RyR, ATP6v0a1 combination stains were imaged using 3D-SIM and 3D-SIM Z-stacks respectively (see below).
Structured illumination microscopy (SIM)
For SIM imaging a Nikon N-SIM S inverted microscope in combination with a 100 X TIRF oil (NA 1.49) objective and a SIM405/488/561/640 excitation filter was used. Hoechst 33342 or Alexa FluorTM 555 were measured following excitation with a 405 and 561 laser in combination with a DAPI, EM580 emission filter (Nikon) respectively. Split green fast or mouse-Alexa FluorTM 488 were measured following excitation with a 488 nm laser line in combination with an EM520 emission filter (Nikon). RFP670 was measured following excitation with a 640 nm laser line and in combination with a Cy5 (Nikon) emission filter When 3D-SIM Z-stacks were imaged, nine images with 200 nm step size in Z-axis were obtained for each stack. Reconstructing the images was performed using the Nikon NIS software. Further image analysis was performed using FIJI. Nearest neighbor distances were obtained using the MOSAIC interaction analysis plugin for FIJI84,85, following adjustment of brightness settings to the same level for all conditions measured in one experimental day. This plugin provides for each 3D-SIM image nearest neighbor distance in pixels between two imaged channels as a normal distribution plot. For each image the mean and standard deviations of these normal distribution plot was used to plot the data points for the bell-shaped curves in the relevant distance range taken that 1 pixel = 32 nm. The average of these data points, at least 7 images on one cover slip, was taken as one independent repeat. Next, the average of all coverslips ± SEM was determined and used to fit a Gaussian distribution curve. Analysis of the 3D-SIM Z-stacks was performed by first adjusting the brightness settings to the same level for all conditions measured in one experimental day. Next, the VolumeJ86 FIJI plugin was utilized to generate volume renders of each Z-stack. When analyzing the split greenFAST probe thresholding and particle analysis, both maintained at the same values for all experiments, was used to determine the total volume and number of separate fluorescent puncta indicative for ER-lysosomal contact site formation. The overlap between the ATP6v0a1 and RyR channel was quantified using particle analysis and volumetric quantification on volume renders of Z-stack acquisitions.
Lysosomal localization imaging
T-Rex-RyR2 cells were grown on coverslips and transiently co-transfected with GFP-SEC61β and LAMP1-RFP670 together with empty vector or ATP6v0a1-Flag at a (1:3 ratio) or were treated for 24 hr with DMSO or 30 µM ryanodine. 48 hr following the transfections the coverslips were washed with PBS+/+ and fixed with 4 % paraformaldehyde for 15 min at room temperature followed by two wash steps with PBS+/+ and mounting using ProLongTM Glass Antifade mountant. 3D-SIM Z-stacks were imaged and volume renders of the GFP and RFP670 channels were generated as described above. The volume render of GFP-SEC61β was used to determine the outer ER edge as an ROI in Fiji. Using the Exact Euclidean Distance Transform (3D) command a distance map relative to the outer ER edge ROI is generated. Next, using the LAMP1-RFP670 volume render, lysosomal volumes were determined as separate ROIs. The distance of each of these ROI was measured on the generated distance map providing for each separate ROI the distance to the ER edge. Finally, lysosomal volume within incremental 100 nm intervals, moving further away from the ER edge were determined and put relative to the total lysosomal. This provides a normalized lysosomal volume distribution for each 100 nm interval. These values were plotted as a cumulative volume corresponding to each interval through which a cumulative Gaussian curve was fitted.
Airyscan microscopy
hiPSC-derived cortical neurons, seeded and differentiated on human Matrigel coated 8 well chambers (80807-90, Ibidi) were treated for 24 hr with DMSO or Ryanodine (30 µM). LysotrackerTM green DND26 (L7526, ThemoFisher Scientific) and BrigthtER (Idylle) were added to the medium of the cells and returned to the incubator for 30 min. Image acquisition was performed using an inverted Zeiss LSM880 with fast airyscan microscope in combination with a 60 X objective (NA 1.4, oil). The setup was controlled by ZEN black (Carl Zeiss Microscopy GmbH). LysotrackerTM green DND26 (L7526, ThemoFisher Scientific) and BrigthtER (Idylle) were excited with an argon or 561 laser line respectively and collected with BP495-550 + LP570 emission filter. Live imaging was performed at 1 Hz for 10 s. For reconstructing the images, ZEN black (Carl Zeiss Microscopy GmbH) was used. Image analysis was performed with FIJI. First, background was subtracted from each channel after which brightness was adjusted to the same level for all conditions measured in one experimental day. Manders coefficients were determined using the JACOP FIJI plugin. In this plugin, the threshold values were also kept identical between conditions measured on the same day.
Transmission electron microscopy (TEM)
Neurons were grown on human matrigel coated Aclar 33 C embedding film (7.8 mil thickness, 50425-25, electron microscopy sciences (EMS)). Following treatments neurons were fixed by adding single strength fixative solution (2.5 % glutaraldehyde (GA) (16200, EMS) and 4% paraformaldehyde (PFA); (15714, EMS) in 0,2 M sodium cacodylate buffer, pH 7,2 (12300, EMS) and stored overnight at 4 °C. For the next step, samples were washed three times with 0,1 M sodium cacodylate buffer and stained with ferrocyanide-reduced osmium tetroxide (19151, EMS, USA) for 1 h at 4 °C. After three washes with dH2O, the samples were incubated overnight in 0,5% uranyl acetate (02624-AB, SPI supplies) at 4 °C. The next day, samples were washed with dH2O, stained en bloc with lead aspartate and dehydrated in an ascending series of ethanol solutions. Cells were dehydrated completely with ethanol and infiltrated with resin (Agar 100, AGR1043, Agar Scientific)/etha,ol mixtures. The following day, the cells were embedded in 100% epoxy resin by inverted beem® capsules covering the cells. Ultrathin sections of 70 nm were cut from the embedded resin blocks using a Leica Ultracut S ultramicrotome (Leica Microsystems, Germany) and collected on 200 mesh grids (01800 N, Van Loenen Instruments). Micrographs were taken with a JEOL TEM 1400-LaB6 (Jeol, Japan) operated at 80 kV and equipped with a 11 Mpxl EMSIS Quemesa camera.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 10 software. The software was used to identify potential outlier values which were excluded. All datasets were normally distributed, assessed by Kolmogorov-Smirnov or Shapiro-Wilk test. The normally distributed data were analysed with two-tailed one sample t-tests, student’s t-tests and ANOVA tests depending on the number of variables analysed. Additional information on the performed statistical tests can be found in the figure legends.
Ethical approval
All experiments, the use of the different cell lines, stem cell lines and mouse brain samples were approved by the Research Ethical Committee of UZ/KU Leuven (protocol numbers S64379 and S63808). For this study no stem cells were harvested or derived from donors directly as the used stem cell line was commercially available. Isolation of the cortical samples was performed in the lab of Dr. Stutzmann and was approved by the NIH and the Institutional Animal Care and Use Committee at RFUMS (protocol # TB23-09).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The authors declare that all data supporting the findings of the study are available without restriction in this article and its supplementary files. The data from this study was deposited to the research data repository of KU Leuven under accession code https://doi.org/10.48804/IMNFSS or can be obtained from the corresponding authors. Source data are provided with this paper.
References
Park, H., Kang, J. H. & Lee, S. Autophagy in neurodegenerative diseases: a hunter for aggregates. Int. J. Mol. Sci. 21, 3369 (2020).
Decuypere, J. P., Bultynck, G. & Parys, J. B. A dual role for Ca2+ in autophagy regulation. Cell Calcium 50, 242–250 (2011).
Wu, N. et al. Autophagy in aging-related diseases and cancer: principles, regulatory mechanisms and therapeutic potential. Ageing Res Rev. 100, 102428 (2024).
Bustamante, H. A. et al. Interplay between the autophagy-lysosomal pathway and the ubiquitin-proteasome system: a target for therapeutic development in Alzheimer’s disease. Front Cell Neurosci. 12, 126 (2018).
Hu, M. et al. The ion channels of endomembranes. Physiol. Rev. 104, 1335–1385 (2024).
Riederer, E., Cang, C. & Ren, D. Lysosomal ion channels: what are they good for and are they druggable targets? Annu Rev. Pharm. Toxicol. 63, 19–41 (2023).
Zajac, M. et al. A mechanism of lysosomal calcium entry. Sci. Adv. 10, eadk2317 (2024).
Morgan, A. J. Ca2+ dialogue between acidic vesicles and ER. Biochem. Soc. Trans. 44, 546–553 (2016).
Penny, C. J., Kilpatrick, B. S., Eden, E. R. & Patel, S. Coupling acidic organelles with the ER through Ca2+ microdomains at membrane contact sites. Cell Calcium 58, 387–396 (2015).
Yuan, Y. et al. Two-pore channel-2 and inositol trisphosphate receptors coordinate Ca2+ signals between lysosomes and the endoplasmic reticulum. Cell Rep. 43, 113628 (2024).
Lopez Sanjurjo, C. I., Tovey, S. C. & Taylor, C. W. Rapid recycling of Ca2+ between IP3-sensitive stores and lysosomes. PLoS ONE 9, e111275 (2014).
Lopez-Sanjurjo, C. I., Tovey, S. C., Prole, D. L. & Taylor, C. W. Lysosomes shape Ins(1,4,5)P3-evoked Ca2+ signals by selectively sequestering Ca2+ released from the endoplasmic reticulum. J. Cell Sci. 126, 289–300 (2013).
Prole, D. L. & Taylor, C. W. Structure and function of IP(3) receptors. Cold Spring Harb. Perspect. Biol. 11, a035063 (2019).
Kilpatrick, B. S., Eden, E. R., Schapira, A. H., Futter, C. E. & Patel, S. Direct mobilisation of lysosomal Ca2+ triggers complex Ca2+ signals. J. Cell Sci. 126, 60–66 (2013).
Calcraft, P. J. et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 459, 596–600 (2009).
Atakpa, P., Thillaiappan, N. B., Mataragka, S., Prole, D. L. & Taylor, C. W. IP(3) receptors preferentially associate with er-lysosome contact sites and selectively deliver Ca2+ to lysosomes. Cell Rep. 25, 3180–3193 e3187 (2018).
Vervliet, T. et al. Basal ryanodine receptor activity suppresses autophagic flux. Biochem. Pharm. 132, 133–142 (2017).
Mustaly-Kalimi, S. et al. Protein mishandling and impaired lysosomal proteolysis generated through calcium dysregulation in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 119, e2211999119 (2022).
Zhang, H., Knight, C., Chen, S. R. W. & Bezprozvanny, I. A gating mutation in ryanodine receptor Type 2 rescues phenotypes of Alzheimer’s disease mouse models by upregulating neuronal autophagy. J. Neurosci. 43, 1441–1454 (2023).
Santulli, G., Lewis, D., des Georges, A., Marks, A. R. & Frank, J. Ryanodine Receptor Structure and Function in Health and Disease. Subcell. Biochem. 87, 329–352 (2018).
Hidalgo, C. & Paula-Lima, A. RyR-mediated calcium release in hippocampal health and disease. Trends Mol. Med. 30, 25–36 (2024).
Bruno, A. M. et al. Altered ryanodine receptor expression in mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging 33, 1001 e1001–1001 e1006 (2012).
Chakroborty, S. et al. Stabilizing ER Ca2+ channel function as an early preventative strategy for Alzheimer’s disease. PLoS ONE 7, e52056 (2012).
Chan, S. L., Mayne, M., Holden, C. P., Geiger, J. D. & Mattson, M. P. Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons. J. Biol. Chem. 275, 18195–18200 (2000).
Liu, J. et al. The role of ryanodine receptor type 3 in a mouse model of Alzheimer's disease. Channels (Austin) 8, 230–242 (2014).
Oules, B. et al. Ryanodine receptor blockade reduces amyloid-beta load and memory impairments in Tg2576 mouse model of Alzheimer disease. J. Neurosci. 32, 11820–11834 (2012).
Lacampagne, A. et al. Post-translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer’s disease-like pathologies and cognitive deficits. Acta Neuropathol. 134, 749–767 (2017).
Bussiere, R. et al. Amyloid beta production is regulated by beta2-adrenergic signaling-mediated post-translational modifications of the ryanodine receptor. J. Biol. Chem. 292, 10153–10168 (2017).
Bagh, M. B. et al. Misrouting of v-ATPase subunit V0a1 dysregulates lysosomal acidification in a neurodegenerative lysosomal storage disease model. Nat. Commun. 8, 14612 (2017).
Lee, J. H. et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141, 1146–1158 (2010).
Bright, N. A., Davis, L. J. & Luzio, J. P. Endolysosomes are the principal intracellular sites of acid hydrolase activity. Curr. Biol. 26, 2233–2245 (2016).
Johnson, D. E., Ostrowski, P., Jaumouille, V. & Grinstein, S. The position of lysosomes within the cell determines their luminal pH. J. Cell Biol. 212, 677–692 (2016).
Shi, Y., Kirwan, P. & Livesey, F. J. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat. Protoc. 7, 1836–1846 (2012).
Bernhardt, R. & Matus, A. Light and electron microscopic studies of the distribution of microtubule-associated protein 2 in rat brain: a difference between dendritic and axonal cytoskeletons. J. Comp. Neurol. 226, 203–221 (1984).
MuhChyi, C., Juliandi, B., Matsuda, T. & Nakashima, K. Epigenetic regulation of neural stem cell fate during corticogenesis. Int. J. Dev. Neurosci. 31, 424–433 (2013).
Klionsky, D. J. et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 17, 1–382 (2021).
Bootman, M. D., Chehab, T., Bultynck, G., Parys, J. B. & Rietdorf, K. The regulation of autophagy by calcium signals: do we have a consensus? Cell Calcium 70, 32–46 (2018).
Reddy, A., Caler, E. V. & Andrews, N. W. Plasma membrane repair is mediated by Ca2+-regulated exocytosis of lysosomes. Cell 106, 157–169 (2001).
Andrews, N. W. Detection of lysosomal exocytosis by surface exposure of Lamp1 luminal epitopes. Methods Mol. Biol. 1594, 205–211 (2017).
Rodriguez, A., Webster, P., Ortego, J. & Andrews, N. W. Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J. Cell Biol. 137, 93–104 (1997).
Tebo, A. G. & Gautier, A. A split fluorescent reporter with rapid and reversible complementation. Nat. Commun. 10, 2822 (2019).
Garcia Casas, P. et al. Simultaneous detection of membrane contact dynamics and associated Ca2+ signals by reversible chemogenetic reporters. Nat. Commun. 15, 9775 (2024).
Ma, W. & Mayr, C. A membraneless organelle associated with the endoplasmic reticulum enables 3’UTR-mediated protein-protein interactions. Cell 175, 1492–1506 e1419 (2018).
Codogno, P., Mehrpour, M. & Proikas-Cezanne, T. Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nat. Rev. Mol. Cell Biol. 13, 7–12 (2011).
Ge, L., Melville, D., Zhang, M. & Schekman, R. The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. Elife 2, e00947 (2013).
Gao, Y. et al. Golgi-associated LC3 lipidation requires V-ATPase in noncanonical autophagy. Cell Death Dis. 7, e2330 (2016).
Tsunemi, T. et al. Increased lysosomal exocytosis induced by lysosomal ca2+ channel agonists protects human dopaminergic neurons from alpha-synuclein toxicity. J. Neurosci. 39, 5760–5772 (2019).
Kim, M. S., Muallem, S., Kim, S. H., Kwon, K. B. & Kim, M. S. Exosomal release through TRPML1-mediated lysosomal exocytosis is required for adipogenesis. Biochem. Biophys. Res. Commun. 510, 409–415 (2019).
Cao, Q. et al. BK Channels alleviate lysosomal storage diseases by providing positive feedback regulation of lysosomal Ca2+ r.elease. Dev. Cell 33, 427–441 (2015).
Wu, Y. et al. Lysosomal potassium channels. Cell Calcium 102, 102536 (2022).
Liu, Y. et al. The CPVT-associated RyR2 mutation G230C enhances store overload-induced Ca2+ release and destabilizes the N-terminal domains. Biochem. J. 454, 123–131 (2013).
Pu, J., Guardia, C. M., Keren-Kaplan, T. & Bonifacino, J. S. Mechanisms and functions of lysosome positioning. J. Cell Sci. 129, 4329–4339 (2016).
Hajnoczky, G., Csordas, G. & Yi, M. Old players in a new role: mitochondria-associated membranes, VDAC, and ryanodine receptors as contributors to calcium signal propagation from endoplasmic reticulum to the mitochondria. Cell Calcium 32, 363–377 (2002).
Garcia-Perez, C., Hajnoczky, G. & Csordas, G. Physical coupling supports the local Ca2+ transfer between sarcoplasmic reticulum subdomains and the mitochondria in heart muscle. J. Biol. Chem. 283, 32771–32780 (2008).
Min, C. K. et al. Coupling of ryanodine receptor 2 and voltage-dependent anion channel 2 is essential for Ca(2)+ transfer from the sarcoplasmic reticulum to the mitochondria in the heart. Biochem. J. 447, 371–379 (2012).
Szabadkai, G. et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 175, 901–911 (2006).
Salminen, A. et al. Impaired autophagy and APP processing in Alzheimer’s disease: the potential role of Beclin 1 interactome. Prog. Neurobiol. 106-107, 33–54 (2013).
Arnst, N. et al. Mitochondrial Ca(2+) signaling and bioenergetics in Alzheimer’s disease. Biomedicines 10, 3025 (2022).
Fedeli, C., Filadi, R., Rossi, A., Mammucari, C. & Pizzo, P. PSEN2 (presenilin 2) mutants linked to familial Alzheimer disease impair autophagy by altering Ca(2+) homeostasis. Autophagy 15, 2044–2062 (2019).
Rossi, A. et al. Defective mitochondrial pyruvate flux affects cell bioenergetics in alzheimer’s disease-related models. Cell Rep. 30, 2332–2348 e2310 (2020).
Area-Gomez, E. et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 31, 4106–4123 (2012).
Filadi, R. et al. Presenilin 2 modulates endoplasmic reticulum-mitochondria coupling by tuning the antagonistic effect of mitofusin 2. Cell Rep. 15, 2226–2238 (2016).
Vrijsen, S. et al. Inter-organellar communication in parkinson’s and alzheimer’s disease: looking beyond endoplasmic reticulum-mitochondria contact sites. Front. Neurosci. 16, 900338 (2022).
Zampese, E. et al. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. USA 108, 2777–2782 (2011).
Bretou, M. et al. Accumulation of APP C-terminal fragments causes endolysosomal dysfunction through the dysregulation of late endosome to lysosome-ER contact sites. Dev Cell 59, 1571−1592.e9 (2024).
Oddo, S. et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39, 409–421 (2003).
Coen, K. et al. Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J. Cell Biol. 198, 23–35 (2012).
Davidsohn, N. et al. Accurate predictions of genetic circuit behavior from part characterization and modular composition. ACS Synth. Biol. 4, 673–681 (2015).
Shcherbakova, D. M. et al. Bright monomeric near-infrared fluorescent proteins as tags and biosensors for multiscale imaging. Nat. Commun. 7, 12405 (2016).
Helassa, N., Nugues, C., Rajamanoharan, D., Burgoyne, R. D. & Haynes, L. P. A centrosome-localized calcium signal is essential for mammalian cell mitosis. FASEB J. 33, 14602–14610 (2019).
Challis, R. C. et al. Systemic AAV vectors for widespread and targeted gene delivery in rodents. Nat. Protoc. 14, 379–414 (2019).
Van der Perren, A. et al. Efficient and stable transduction of dopaminergic neurons in rat substantia nigra by rAAV 2/1, 2/2, 2/5, 2/6.2, 2/7, 2/8 and 2/9. Gene Ther. 18, 517–527 (2011).
Sun, M., Bernard, L. P., Dibona, V. L., Wu, Q. & Zhang, H. Calcium phosphate transfection of primary hippocampal neurons. J. Vis. Exp. 12, 50808 (2013).
Vervliet, T. et al. Cardiomyocyte differentiation from human induced pluripotent stem cells is delayed following knockout of Bcl-2. J. Cell Sci. 136, jcs260216 (2023).
Peng, W. et al. Structural basis for the gating mechanism of the type 2 ryanodine receptor RyR2. Science 354, aah5324 (2016).
Wang, L., Wu, D., Robinson, C. V. & Fu, T. M. Identification of mEAK-7 as a human V-ATPase regulator via cryo-EM data mining. Proc. Natl. Acad. Sci. USA 119, e2203742119 (2022).
Shelley, J. C. et al. Epik: a software program for pK(a) prediction and protonation state generation for drug-like molecules. J. Comput Aided Mol. Des. 21, 681–691 (2007).
Jimenez-Garcia, B., Roel-Touris, J. & Barradas-Bautista, D. The LightDock server: artificial Intelligence-powered modeling of macromolecular interactions. Nucleic Acids Res. 51, W298–W304 (2023).
Jimenez-Garcia, B. et al. LightDock: a new multi-scale approach to protein-protein docking. Bioinformatics 34, 49–55 (2018).
Roel-Touris, J., Jimenez-Garcia, B. & Bonvin, A. Integrative modeling of membrane-associated protein assemblies. Nat. Commun. 11, 6210 (2020).
Lomize, A. L., Todd, S. C. & Pogozheva, I. D. Spatial arrangement of proteins in planar and curved membranes by PPM 3.0. Protein Sci. 31, 209–220 (2022).
Zhou, H. & Zhou, Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci. 11, 2714–2726 (2002).
Rueden, C. T. et al. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinforma. 18, 529 (2017).
Shivanandan, A., Radenovic, A. & Sbalzarini, I. F. MosaicIA: an ImageJ/Fiji plugin for spatial pattern and interaction analysis. BMC Bioinforma. 14, 349 (2013).
Helmuth, J. A., Paul, G. & Sbalzarini, I. F. Beyond co-localization: inferring spatial interactions between sub-cellular structures from microscopy images. BMC Bioinforma. 11, 372 (2010).
Abramoff, M. D. & Viergever, M. A. Computation and visualization of three-dimensional soft tissue motion in the orbit. IEEE Trans. Med. Imaging 21, 296–304 (2002).
Acknowledgements
This project was funded via the Research Foundation—Flanders (FWO; project numbers G0E7520N and G081821N (to G.B.), project number FWO-SBO-S001221N-OrganID (to C.V.) and fellowship number: 12ZG121N (to T.V.) and 209833/1S03424N (to K.A.)); Stichting Alzheimer Onderzoek (SAO IP3 receptor) (to G.B.), Central European Leuven Strategic Alliance (CELSA/23/031) (to G.B. and T.T.), Global Seed Fund (GSF/26/056) (to G.B.), AKUL/19/34 to (G.B.), the National Institutes of Health (NIH grant: R01 AG065628) (to G.S.) and the Slovenian Research Agency (Grant No. P1-0208) (M.S. and T.T.). G.B. is member of the FWO Scientific Research Network CaSign (W0.014.22N). We would like to thank Anja Florizoone, Rita La Rovere and Katlijn Vints for the excellent technical support. We are grateful to Wayne S.R. Chen (University of Calgary, Canada) and Wim Annaert (KU Leuven, Belgium) for kindly providing the HEK293 T-Rex RyR2 cells and ATP6v0a1-Flag construct, respectively. We want to thank the collaborators of the VIB BioImaging Core Leuven for their valuable support and assistance to the research presented in this paper. AAV vectors were produced by the Leuven Viral Vector Core (KU Leuven, Belgium). The authors gratefully acknowledge the Electron Microscopy team of the VIB-KULeuven Bio Imaging Core for their support and assistance in this work.
Author information
Authors and Affiliations
Contributions
T.V. conceptualized the study. T.V., C.V.D.H., G.S., T.T., C.V, G.B. designed and provided the necessary methodology and tools used in this study. T.V., J.L and K.A. performed the neuronal differentiations. C.V.D.H. developed and purified the used viral vectors. T.T. and M.S. performed protein docking analysis. T.L. performed electron microscopy experiments. G.S. isolated and provided mouse cortical samples. T.V. performed all other experiments. T.V. performed data analysis with help of J.L. T.V. wrote the manuscript with help and input from all authors. All authors read and approved the manuscript. T.V. and G.B. supervised and acquired funding.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Thierry Galli and the other anonymous reviewers for their contribution to the peer review of this work. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Vervliet, T., Loncke, J., Sever, M. et al. Inactive ryanodine receptors sustain lysosomal availability for autophagy by promoting ER-lysosomal contact site formation. Nat Commun 17, 1293 (2026). https://doi.org/10.1038/s41467-025-68054-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-68054-z
