
Abstract
The cerebral cortex comprises diverse excitatory and inhibitory neuron subtypes, each with distinct laminar positions and connectivity patterns. Yet, the molecular logic underlying their precise wiring remains poorly understood. To identify ligand–receptor (LR) interactions involved in cortical circuit assembly, we tracked gene expression dynamics in mice across major neuronal populations at 17 developmental stages using single-cell transcriptomics. This generated a comprehensive atlas of LR-mediated communication between excitatory and inhibitory neuron subtypes, capturing known and novel interactions. Notably, we identified NEOGENIN-1 as the principal receptor for CBLN4 during the perinatal period, mediating synapse formation between somatostatin-expressing interneurons and glutamatergic neurons. We also identified members of the cadherin superfamily as candidate regulators of perisomatic inhibition from parvalbumin-expressing basket cells onto deep and superficial excitatory neurons, exerting opposing effects on synapse formation. These findings suggest a context-dependent role for cadherins in synaptic specificity and underscore the power of single-cell transcriptomics for decoding the molecular mechanisms of cortical wiring.
Similar content being viewed by others


Introduction
The mammalian cerebral cortex is a six-layered structure at the brain’s surface that supports the most complex cognitive functions. These functions rely on highly orchestrated and evolutionarily conserved developmental processes that establish precise patterns of communication between two major classes of neurons: excitatory glutamatergic neurons (GlutNs) and inhibitory GABAergic neurons (GABANs). Both GlutNs and GABANs are further subdivided into numerous transcriptionally defined subtypes, a diversity largely revealed by single-cell RNA sequencing (scRNA-seq). Dozens of GlutN and GABAN subtypes have been identified1,2,3,4,5, each characterized by distinct morphological, electrophysiological, and molecular properties, preferential laminar localization, specific synaptic partners and selective targeting of subcellular compartments. This precise organization establishes the intricate cortical wiring that underpins cortical computation. Disruption of connectivity between even a single pair of neuron subtypes at a given developmental stage can impair circuit function and contribute to neurodevelopmental disorders6. Elucidating how subtype-specific connections are established and regulated is therefore essential for understanding both cortical development and disease.
In the past decade, major advances have been made in mapping the spatial organization and synaptic connectivity rules of cortical neuron subtypes. In particular, high-resolution studies have delineated the cellular and subcellular wiring patterns between GlutN and GABAN subtypes in the adult mouse cortex, revealing remarkable specificity, stereotypy, and evolutionary conservation of these connections across mammalian species7,8,9,10. Despite these insights, the molecular mechanisms that guide corticogenesis and ultimately produce this intricate architecture remain poorly understood. Only a few studies have begun to identify the specific molecules that govern the interactions and developmental processes shaping cortical circuit assembly11,12,13.
It is likely that neuron-neuron communications via ligand-receptor (LR) interactions play a key role, as these interactions are critical for the development of many tissues11,12,13. This hypothesis is supported by several studies suggesting that GlutNs may non-cell autonomously influence the recruitment of specific GABAN subtypes. Indeed, GlutN subtypes settle into their final positions earlier than the synchronically generated GABANs14, and altering specific GlutN subtype identities during development modifies the allocation and synaptic connectivity of corresponding GABAN subtypes15,16,17.
In this study, we integrated newly generated and publicly available scRNA-seq datasets from the developing mouse cortex to investigate the molecular logic of cortical wiring through LR inference. We first mapped gene expression dynamics across glutamatergic and GABAergic neuron subtypes over development, with particular focus on the perinatal period when circuit assembly intensifies. Using these data, we constructed a comprehensive atlas of putative LR-mediated signaling between neuronal subtypes. We then validated this atlas through functional experiments, showing that it recapitulates known interactions and identifies further interactions. For example, we confirmed the role of Cbln4 in inhibitory synapse formation onto excitatory neurons and identified Neogenin-1 as its likely cortical receptor. We also uncovered cadherin superfamily members exerting opposing effects on perisomatic inhibition in deep versus superficial excitatory neurons. All data are available for interactive exploration at https://sclrsomatodev.online/.
Results
Comprehensive single neuron transcriptomics atlas covering mouse somatosensory cortex development
To infer LR-mediated cell-cell interactions during cortical development, we cross-referenced scRNA-seq information from all cortical neuron subtypes across multiple stages with publicly available LR information.
First, we generated a comprehensive scRNA-seq transcriptomic dataset of all cortical neurons throughout somatosensory corticogenesis. We conducted scRNA-seq and snRNA-seq at six key stages within the underexplored P0-P30 circuit wiring period: P0-P2 (radial migration and laminar allocation of GABANs18,19), P5-P8 (programmed cell death of GlutNs and GABANs20,21,22), and P16-P30 (circuit refinement and synaptogenesis completion). We integrated previously published scRNA-seq data covering earlier embryonic (E11.5-E18.5) and adult stages (P53-P102)3,23,24,25,26,27,28, including ganglionic eminences (GEs), to capture early transcriptional signature of future cortical GABANs25,26,27,28 (see “Methods”). Our analysis spans 17 time points from E11.5 to adulthood (Fig. 1A).

A Sankey diagram depicting the experimental paradigm for data collections and integration of published datasets. Yao*: Yao et al., 2021; J. Di Bella*: J. Di Bella et al., 2021, Telley*: Telley et al., 2019; R.Lee*: R.Lee et al., 2022; Bandler: Bandler et al., 2021; Mayer*: Mayer et al., 2018; Mi*: Mi et al., 2018; SSp primary somatosensory cortex, SSs supplemental somatosensory cortex, MGE medial ganglionic eminence, dMGE dorsal MGE, vMGE ventral MGE, CGE caudal ganglionic eminence, LGE lateral ganglionic eminence, 10X 10X Genomics, SSv4 Smart-seq version 4, SSv2 Smart-seq version 2, C1 Fluidigm C1. B UMAP visualization of post-mitotic neurons from the 17 time points after integration. Cells are colored by cell-type assignment. C UMAP visualization is colored by age, study, region, RNA-seq method, sequencing platform, and gene markers. D Left: Sankey plot representing the different hierarchical levels of cell type assignment; middle: heatmap representing the fraction of each cell type per age; right: heatmap representing some cell type-specific genes. “Normalized expression” represents the 25% trimmed mean log2(CPM + 1) expression normalized by row. See also Figs. S1–S4. Associated source data are provided.
After stringent quality control and filtering (Supplementary Fig. 1A, B and “Methods”), we identified postmitotic neuronal cell types and tracked their transcriptional dynamics. For final cell type nomenclature, we used the most comprehensive resource of transcriptomic cell-types in the somatosensory cortex that existed before December 2023, i.e., Yao et al.3 (hereafter referred to as AllenRef21), corresponding to the adult stage (Supplementary Fig. 1A). Assigning cellular identities in early developmental stages proved difficult given that transcriptional signatures of specific cell-types evolve drastically during development23,26. To overcome this difficulty, we developed a hierarchical pipeline assigning identities at increasing resolution levels, similar to the method used by the Allen Institute3 (Supplementary Fig. 1A).
First, we assigned cell identities at the class level (Glutamatergic, GABAergic, Non-Neuronal, Immature/Migrating, dorsal pallium progenitor and subpallium progenitor)3. For datasets covering early development (up to P5), we used the perinatal somatosensory cortex dataset from Di Bella et al.24, the largest published at this stage, and the early ganglionic eminence (GE) GABANs dataset from Bandler et al.27, representing neurons fated for the cortex. For P8 to P30, as cells were transcriptionally similar to the adult reference, we used the adult dataset3 directly as the reference (Supplementary Fig. 1A). In order to investigate LR interactions mediating circuit wiring between neuronal subtypes, we focused our downstream analyses on post-mitotic GlutNs and GABANs (Fig. 1A, B). In total, 182,084 high-quality post-mitotic neurons were analyzed. To account for technical variation arising from different studies, RNA-seq methods (scRNA-seq and snRNA-seq) and sequencing platforms (Fig. 1C), we integrated all data using the Seurat SCT workflow (Supplementary Fig. 1C). The integrated dataset was split into classes, i.e., GlutNs and GABANs. For each class, cell identities were further delineated at increasing resolutions; first at the subclass level, and then at the supertype level in each subclass, as defined in AllenRef213. To identify biologically meaningful cell-types, we leveraged the correspondence provided by Yao et al. between supertypes from scRNA-seq studies and morpho-electro-connectomic types (mec-types) from patch-seq studies1,2,3,29 (Supplementary Fig. 2, S3). Pooling supertypes belonging to the same mec-type allowed us to reach a biologically meaningful cell-type annotation (Supplementary Fig. 4). Ultimately, we identified 159 290 cells, representing 27 cell-types (Fig. 1D) including 11 GlutN cell-types grouped in 3 families – intratelencephalic (IT), extratelencephalic (ET), Other GlutNs - and 16 GABAN cell-types grouped in 5 families - Lamp5, Sst, Pvalb, Vip and Other GABANs - (Fig. 1B–D, Supplementary Fig. 4). All intermediate steps of the label transfer procedure, including bootstrapped prediction scores and cell-wise assignment files, are available in the Zenodo folder, ensuring full transparency and reproducibility of the classification process.
In the UMAP embedding of all neurons, each GlutN and GABAN cell-type exhibited a largely continuous temporal gradient of transcriptional variation across corticogenesis, with gene expression profiles of individual cells correlating with mouse developmental age (Fig. 1B, C).
Temporal transcriptional dynamics
To track the temporal maturation of each neuronal cell-type in a continuous rather than discrete manner, single cells were ordered along a continuous trajectory on the basis of their transcriptional profile30 (see “Methods”) (Supplementary Fig. 5A, Supplementary Fig. 6A, B). This “pseudo-maturation” axis correlated well with the actual age of the cells (Supplementary Fig. 5A, Supplementary Fig. 6A, B), indicating gradual transcriptional variation over neuronal differentiation.
For each cell type, genes showing significant variation along the “pseudo-maturation” axis were identified and used to illustrate temporal gene dynamics in six waves (Supplementary Fig. 5A, Supplementary Fig. 6A, B). During early development (E11.5-E18.5), most cell-types displayed transcriptional signatures related to cell-intrinsic properties (Supplementary Fig. 5B, Supplementary Fig. 6C), shifting later to programs controlling cell–cell and cell–environment interactions (Supplementary Fig. 5B, Supplementary Fig. 6C)31,32. These findings reveal the molecular transitions through which each neocortical cell type shifts from intrinsic programs to extrinsic, interaction-driven programs essential for integration into the cortical network.
Spatial transcriptional gradients
In adult mice, glutamatergic IT neurons exhibit a spatial gradient of transcriptional variation along the cortical sheet3,33. We investigated whether similar laminar transcriptional gradients exist within each neuronal family between embryonic day 18.5 (E18.5) and postnatal day 30 (P30). To do so, we cross-referenced our datasets with Patch-seq1,2 and MERFISH33 data to determine the exact normalized laminar position of each cell type within its family, providing information about their relative positions (Supplementary Figs. 2, S3).
In parallel, for each neuronal family and at each developmental stage, we fitted a curve along the UMAP transcriptional continuum and defined this axis as a “pseudo-layer” score. This approach allowed us to assess whether transcriptional variation along the pseudo-layer axis corresponded to the actual laminar organization of cell types, which would reflect the presence of spatial transcriptional gradients across cortical layers.
Strikingly, the spatial gradient previously described for adult IT neurons33 was already present at all earlier developmental stages examined, from E18.5 to P30 (Supplementary Fig. 7A). Similar gradients were also observed for all other neuronal families analyzed; namely ET, Sst, Pvalb, Vip, and Lamp5 (Supplementary Fig. 7–S9). In summary, our results show that all major neuronal families, both glutamatergic and GABAergic, exhibit transcriptional gradients aligned with cortical lamination, consistently across development from E18.5 to P30.
We identified genes showing significant variation along the pseudo-layer axis and used them to define six distinct spatial transcriptional gradients. In GlutNs, the upper waves, corresponding to upper-layer (UL) displayed gene expression patterns enriched for features commonly observed in earlier developmental states, particularly involving cell-intrinsic properties (Supplementary Fig. 7B and D). In contrast, the lower waves, corresponding to deep-layer (DL) GlutNs, showed comparatively reduced expression of these features. This is consistent with UL GlutNs being born later than DL GlutNs, due to cortical inside-out patterning23. In contrast, for GABANs, environment-sensing programs were already active by E18.5 (Supplementary Fig. 8B and D), supporting the hypothesis that cortical GABANs mature later than their earlier-settled GlutN counterparts and depend on cues from these excitatory neurons to reach their final positions and establish proper connectivity15,16,17,34.
Spatiotemporal transcriptional landscapes
Based on the pseudo-maturation and pseudo-layer scores, cells of each family were embedded within 2D graphs to generate spatiotemporal transcriptional landscapes of gene expression (Fig. 2A–D and Fig. S10). For any gene queried within a given family, the 2D transcriptional map shows temporal dynamics along the x axis and spatial dynamics on the y axis (Fig. 2B–D). This confirmed the spatiotemporal expression of genes known to be involved in cortical layer patterning (Cux1, Fezf2, Reln, Fig. 2B, C) or in neuropeptidergic system maturation in specific neuronal families (Fig. 2D).

A UMAP visualization of all neurons color coded with cell family labels. B Top: 2D map in which cells are embedded according to their pseudo-maturation (x-axis) and pseudo-layer (y-axis) scores for the IT family. Bottom: A generalized additive model (GAM) was applied to generate 2D maps, i.e., “transcriptional landscapes”, for the spatiotemporal expression of genes throughout cortical wiring; Cux1 expression is represented as an example. C Transcriptional landscapes for Fezf2 in ET (top) and for Reln in Other GlutNs (bottom). D Transcriptional landscapes for representative genes in each GABAN family. E Example of transcriptional landscapes for genes implicated in GABAN migration (Cxcl12, Ackr3 and Cxcr4), and in synaptogenesis (Nlgn1 and Nrxn1). F Gene map obtained by performing a PCA on all the significantly expressed genes colored by cell family (left) and by spatiotemporal cluster (right). Average transcriptional landscapes of each cluster are displayed around the gene map, annotated with the corresponding top KEGG ontology label (Supplementary Data S8). G Varying spatiotemporal clusters for the Cadherin gene family among 3 neuronal families. CP cortical plate, IZ intermediate zone, MZ marginal zone, SVZ subventricular zone, VZ ventricular zone, WM white matter. See also Figs. S5–S16. Associated source data are provided.
To validate our approach, we examined whether LR transcriptional landscapes matched known roles in migration or synaptogenesis. We focused on CXCL12 and its receptors CXCR4 and ACKR3, which are essential for GABAN tangential migration35,36. As previously reported, CXCL12 is expressed perinatally by meninges37, immature GlutNs38, and possibly Cajal-Retzius (CR) cells39, while CXCR4/ACKR3 are co-expressed on migrating GABANs40,41, with CXCR4 also present in CR cells40. CXCL12 downregulation postnatally is thought to trigger GABAN radial migration and cortical plate invasion35,36,42. Our data aligned with these patterns: Cxcl12 was enriched in CR cells, and Cxcl12, Cxcr4, and Ackr3 expression was restricted to E18.5–P0, consistent with a role in migration but not in synaptogenesis (Fig. 2E).
It is well established that the formation of synapses requires specific adhesion molecules, including the NRXN1-NLGN1 LR-pair (Fig. 2E)43,44. Our landscapes showed NRXN1 and NLGN1 expression across all cell types during developmental windows consistent with synaptogenesis (Fig. 2E), supporting their role in synapse formation.
Applying PCA and subsequent k-means clustering enabled us to define 15 archetypal spatiotemporal transcriptional landscapes (Fig. 2F). Some genes mapped to early or late developmental trajectories, whereas others showed layer-specific or combined spatiotemporal patterns. To functionally interpret these clusters, we performed KEGG enrichment analysis and annotated each spatiotemporal cluster with its dominant ontology term (Fig. 2F; Supplementary Data S8), revealing programs ranging from ribosome biogenesis and spliceosome activity to calcium signaling and neuroactive ligand–receptor pathways. Intriguingly, members of the Cadherin gene family were distributed across all 15 clusters (Fig. 2G), underscoring their broad spatiotemporal diversity throughout cortical development.
We developed a Shiny application, scLRSomatodev, (https://sclrsomatodev.online/) that enables interactive exploration of spatiotemporal gene expression patterns across cortical neuronal subtypes, using various visual representations (Supplementary fig. 11).
An atlas for inferring LR interactions between neuronal subtypes over corticogenesis
We hypothesized that LR interactions between neuronal subtypes, especially between GlutNs and GABANs, are key to shaping stereotypical cortical wiring. If this hypothesis proved correct, we would expect highly correlated transcriptional landscapes between GlutN-expressed ligands and their cognate GABAN receptors, and vice versa. To test this, we assembled LR_DB_2025, the most comprehensive curated LR database to date. It integrates existing LR datasets with manually added pairs from published studies (Fig. 3A & Fig. S12, Supplementary Data S1, see “Methods”), yielding 8,789 curated LR pairs involving 1421 ligands and 1233 receptors. Each molecule was further classified by molecular function and known involvement in neurodevelopmental processes (see “Methods” & Supplementary Data S2).

A Curated ligand–receptor database containing 8789 LR pairs compiled from 109 sources (LR_DB_2025). To increase biological relevance for our study, we generated a brain-focused subset (LR_DB_2025_Brain) by retaining only ligands and receptors expressed in our single-cell dataset, resulting in 5341 LR pairs. See Supplementary Data S1 and S2 for full listings. B Correlations between transcriptional landscapes of neurodevelopmental process-associated ligands in ITs and receptors in GABANs, suggesting a ligand–receptor code for neuronal adhesion and synaptogenesis that involves the cadherin family of adhesion molecules. (Left) bars show the mean correlation between IT ligand and GABAN receptor landscapes for random ligand–receptor (LR) pairs, ADAM metalloproteases, cadherins and Ig-like cell-adhesion molecules. (Right) bars show the mean correlation for LR pairs assigned to neurodevelopmental categories (brain-associated, migration, cell death, differentiation, cell adhesion and synaptogenesis). Data are presented as mean values, statistical significance was assessed by comparing each category to its respective reference group (Left: random non-LR gene pairs; Right: “Brain associated” category) using a two-sided z-test based on Fisher’s r-to-z transformation, with no adjustment for multiple comparisons. Exact P values were: cadherins vs random LR pairs, P = 0.00125; cell adhesion vs “ Brain associated ”, P = 2.215e-05; synaptogenesis vs “ Brain associated ”, P = 7.0357e-04. Asterisks indicate significance levels: P < 0.05 (*), P < 0.01 (**), P < 0.001 (***). C Simplified method for the inference of LR-mediated interactions. TF, transcription factor; TG, target gene; S_inter, score for intercellular communication; S_intra, score for intracellular signaling. See Figs. S13–14. D Heatmaps illustrating the number of inferred LR interactions that persist across at least two consecutive ages for all 729 cell-type pairs. From left to right and top to bottom: all possible interactions; interactions unique to a single cell-type pair, widely shared interactions (by >400 pairs) and very rare interactions (shared by <10 pairs). Associated source data are provided.
For LR pairs in the cadherin superfamily, known to play key roles in neurodevelopment, we observed strong correlations between ligand expression in GlutN neurons and receptor expression in GABANs (Fig. 3B). This underscores the importance of cadherin-mediated interactions in GlutN-GABAN communication during development and supports previous hypotheses implicating cadherins in areal specialization34,45 and synaptic specificity46. LR pairs tied to migration, differentiation, cell death, cell adhesion, and synaptogenesis (Supplementary Data S2, Methods) exhibited varying degrees of spatiotemporal correlation (Fig. 3B). Synaptogenesis displayed the strongest LR pair correlation, suggesting that this process relies heavily on intercellular communication. In contrast, LR pairs associated with processes such as cell death or differentiation were less consistently correlated, possibly reflecting a greater reliance on cell-intrinsic mechanisms or limitations in the completeness of GO annotations for these pathways (Fig. 3B).
To identify ligand-receptor pairs driving intercellular interactions shaping cortical wiring, we built a comprehensive LR atlas inferring high-confidence interactions between neuronal cell types from E18.5 to adulthood, covering the full period of cortical circuit formation in mice. This approach integrates temporal cell-by-gene expression matrices with our curated LR database (Fig. 3C, Supplementary Fig. 13; see “Methods”).
First, we implemented and adapted the scSeqComm method47 to our dataset. This method computes an intercellular ligand–receptor (LR) score for each source–target cell-type pair, based on the relative expression levels of ligands in source cells and receptors in target cells. The core scSeqComm tool was not modified; intercellular scores were calculated exactly as described in the original publication and returned for all pairs across all cell-type combinations.
To improve the biological plausibility of predicted interactions and increase stringency, we introduced an additional cell pair score, applied independently of any particular LR pair. This score enabled prioritization of LR interactions according to known developmental timing and the final connectivity patterns between the cell-types. Specifically, the cell-pair score represents the (normalized) sum of (i) a developmental score, reflecting the relative timing at which each cell type populates the cortex and could transiently interact, (Fig. 3C and Fig. S13, Supplementary Data S3), and (ii) a connectivity score, estimating the likelihood of synaptic connections inferred from adult cortical architecture, derived from morphological reconstructions of ~1000 neurons within a cortical column from adult Patch-seq studies48 (Fig. 3C, Figs. S13–14, Supplementary Data S4). For LRs with documented downstream genetic signaling from the receptor, our pipeline quantified intracellular signaling activity in target cells by integrating information from public regulatory gene databases47 (see “Methods”). This yielded an intracellular signaling score (Fig. 3C and Fig. S13), which helped assess the robustness of inferred LR interactions and in determining the preferred receptor(s) for ligands with multiple potential targets.
Predicted LR interactions can be explored at https://sclrsomatodev.online/ (Supplementary Fig. 16), where users can visualize both the number and identities of interactions between any of the 729 neuronal cell-type pairs across seven developmental stages covering cortical wiring (E18.5 to adulthood). Interaction counts are shown as heatmaps (Fig. 3D, Fig. S16A), while specific LR pairs and associated GO pathways are displayed as dot plots (Supplementary Fig. 16B). LR pair predictions were generally consistent between scRNA-seq and snRNA-seq (Supplementary Fig. 15), demonstrating modality consistency. An exception is observed for certain short transcripts, which are underdetected in snRNA-seq, as previously reported. For instance, Neurod2, Slc12a5, and Dlx2 (~1 kb) show reduced detection in snRNA-seq at P8, whereas larger genes such as Cux1 (~340 kb) are comparable across both modalities.
To determine which molecular codes underlie the specificity of LR-mediated cell positioning and connectivity, we systematically analyzed LR predictions across 729 neuronal cell-type combinations from E18.5 to adulthood. We quantified LR interactions maintained across at least two consecutive stages and categorized them as “unique” (1 cell pairs), “widely shared” (≥400 cell pairs), or “rarely shared” (≤10 cell pairs) (Fig. 3D). While some cell pairs showed up to ~600 predicted LR interactions, uniquely used LR pairs were extremely rare. Widely shared LRs represented less than 10% of all inferred interactions. In contrast, rarely shared LRs showed more variability, with up to 60 detected in CR–CR interactions. These findings suggest that cortical neurons employ a combinatorial code of common and rare ligand–receptor interactions, rather than unique pairs, to establish subtype-specific communication and connectivity. To directly evaluate whether LR expression patterns can distinguish neuronal subtypes, we developed a machine-learning framework that generates single-cell replicates for each SOURCE → TARGET cell-type pair across developmental stages. Each example encodes ligand × receptor product values, and the model is tasked with predicting the correct cell-type pair identity (Supplementary Fig. 17). While the prediction task is extremely challenging (>600 classes), many individual cell-type pairs achieved high ROC–AUC values, demonstrating strong discriminability and reinforcing the idea of a combinatorial LR code (Supplementary Fig. 17A). We benchmarked models trained on five LR feature sets: all significant pairs, widely shared pairs, rarely shared pairs, unique-only pairs, and random non-LR pairs (Supplementary Fig. 17B, C). Across developmental stages, classifiers trained on unique-only, shared-only, or random pairs performed at or near chance, whereas those trained on rare-only pairs consistently provided predictive performance above chance. The highest accuracy was obtained when combining feature sets (all significant = rare + shared + occasional unique) with moderately strong performance for the rarely shared pairs, supporting our conclusion that specificity is encoded by combinatorial patterns rather than by shared or unique pairs alone. This trend was maintained across all developmental stages and remained consistent even when one cell type was held constant while varying the other (Supplementary Figs. S18–S20).
These findings suggest that a combinatorial LR code underlies neuronal specificity. A key question is whether such codes are determined by laminar position, class identity, or whether fate-intrinsic factors, such as the progenitor domain of origin of interneurons, also impose additional constraints. To address this, we further quantified the distribution of interactions between GlutNs and GABANs, as well as within each neuronal class. This analysis revealed progressive differences between superficial and deep layers across development (Figs. S21, S22). To investigate whether the origin of interneurons influences their predicted LR interactions with glutamatergic partners, we grouped subtypes by MGE, CGE, or POA origin, which revealed a temporal shift from GE-specific programs at embryonic/early postnatal stages to predominantly shared modules at P8–P30, followed by renewed specificity in adulthood (Supplementary Fig. 23A–C). GE-specific modules were enriched for partner recognition and integration processes at early stages, while shared modules were dominated by synaptic organization ontologies during peak synaptogenesis. These results show that LR communication is shaped not only by class and laminar identity (Figs. S21, S22), but also by interneuron progenitor domain (Supplementary Fig. 23), adding a fate-dependent dimension to cortical wiring.
Key roles for LR interactions during synaptogenesis and in neurodevelopmental diseases
We analyzed the temporal progression of LR predictions between GlutNs and GABANs as source and target cells, respectively. The total number of predicted LRs increased over time (Fig. 4A), consistent with the gradual establishment of synaptic connectivity. Among GABAN subtypes, Sst and Pvalb neurons were the first to receive a high number of LR interactions from GlutNs (Fig. 4A), while Lamp5, Vip, and other GABAN populations showed a later increase. Notably, Pvalb cells initially exhibited fewer LRs than Sst neurons but progressively reached comparable levels to Sst neurons, consistent with studies showing that Sst neurons precede Pvalb neurons in cortical network formation and in agreement with recent findings49.

A Number of LR pairs predicted to be utilized per GABAN family as target cells, from E18 to adulthood. B Number of predicted LR interactions between IT and GABAN cell-types as source and target cells, respectively, from E18 to adulthood. C Percentage of LR pairs of five main neurodevelopmental ontologies and of the cadherin LR family that are predicted as utilized between IT and GABAN cell-types as source and target cells, respectively, from E18 to adulthood. D Percentage of LR pairs with both ligand and receptor associated with neurodevelopmental diseases that are predicted as utilized between IT and GABAN cell-types as source and target cells, respectively, from E18 to adulthood. E Intersections between LR pairs of the 4 diseases studied. F Interaction network showing the LR pairs associated with each disease. G Gene ontologies associated with inferred, disease-associated LR pairs. Associated source data are provided.
Next, we focused our analysis at the cell-type level, using IT neurons as source cells and GABAN subtypes as targets. The number of predicted LR interactions increased over time (Fig. 4B), consistent with the global trend (Fig. 4A). However, we noted only subtle differences in the temporal profiles of upper layer (UL) and deep layer (DL) GABANs. LR interactions with UL GABANs were low at early stages but showed a gradual increase over time. DL GABANs showed a marginally higher level of LR engagement early on, though this difference was very subtle (Fig. 4B). While such a pattern could be compatible with earlier maturation of DL neurons, the effect size is minimal, and we interpret this as a possible depth-related trend, although additional analyses will be needed to confirm it.
We calculated the proportion of LRs associated with the five key neurodevelopmental processes. Migration, differentiation, and cell death were sparsely predicted, suggesting that our atlas captures fewer LR interactions associated with these processes. This could indicate a stronger reliance on cell-intrinsic programs, but may also reflect underrepresentation of relevant signaling interactions in current GO terms or from the limitations of transcriptomic inference. In contrast, synaptogenesis-related LRs, and to a lesser extent those involved in adhesion, were consistently and robustly utilized from E18.5 through adulthood (Fig. 4C). Notably, Cadherins showed sharp increases in inferred activity from P8 onward, underscoring their possible role in shaping cortical wiring.
We investigated the engagement of LR pairs associated with neurodevelopmental disorders during cortical development. LRs from our curated database were categorized based on their disease associations: autism, epilepsy, schizophrenia (SCZ), and intellectual disability (ID) (Supplementary Data S5). While the proportion of active SCZ- and ID-linked LRs remained consistently low across all developmental stages (Fig. 4D), those associated with autism and epilepsy showed markedly higher engagement, peaking at postnatal day 8 (P8) and remaining elevated into adulthood. In some instances, up to 50% of autism- or epilepsy-associated LRs were active at specific time points (Fig. 4D). These findings suggest that the etiology of autism and epilepsy may be more closely related to LR-mediated intercellular communications in cortical circuits than that of SCZ and ID. Several of the highly utilized LRs were implicated in multiple disorders (Fig. 4E). Notably, CDH13 and CDH22, two cadherins, were the only significantly utilized LRs shared between autism and SCZ (Fig. 4F). Overall, gene ontology analysis revealed that predicted disease-associated LRs were enriched in neurodevelopmental biological processes (Fig. 4G).
Validation of the LR prediction atlas: Nrg3-Erbb4
We queried our LR atlas to validate known interactions between GlutNs and GABANs, focusing on the NRG3-ERBB4 pair, which is recognized for mediating the development of excitatory synapses onto GABANs and is linked to neurodevelopmental disorders50 (Fig. 5A). Consistent with the literature, transcriptional landscapes confirmed NRG3 expression in GlutNs and the restriction of ERBB4 expression to GABANs (Fig. 5B). NRG3 and ERBB4 expression levels peaked in the second half of the developmental timeline, coinciding with synaptogenesis (Fig. 5B). Our atlas predicts significant NRG3-ERBB4 interactions from E18.5-P0 through P30, with GlutNs werving as the NRG3 source and most GABANs as ERBB4-expressing targets (Fig. 5C). Across all developmental stages, the intracellular signaling score for NRG3-ERBB4 was highest when target cells were Pvalb BCs and Vip|L2/3 − L5 BP/BTCs, two GABAN subtypes known to critically depend on ERBB4 signaling for the formation of excitatory synapses (Fig. 5D)12,13,51. Overall, our atlas supports a role for the NRG3-ERBB4 interaction in driving excitatory synapse formation onto specific GABAN subtypes during corticogenesis.

A Diagram illustrating Nrg3-Erbb4 interaction between GlutNs and GABANs. B Nrg3 and Erbb4 transcriptional landscapes in GlutN and GABAN families. C Heatmap depicting predictions of Nrg3-Erbb4 mediated interactions between GlutN and GABAN cell-types. D Left: Predicted Nrg3-Erbb4 mediated cell-cell interactions at P8 and P30. GlutNs (blue) are sources of Nrg3 and GABANs are targets expressing Erbb4. Right: Schematic representation illustrating the predicted interaction strength among three example cell pairs, highlighting preferred connectivity patterns based on computational predictions, which validates the 3 cited references. E Diagram illustrating CBLN4-mediated inhibitory synapse formation from Sst cells to GlutNs in the cortex. F Transcriptional landscapes of Cbln4 expression across different neuron families. G Heatmap with LR pairs predicted to involve CBLN4 as a ligand. H Predicted CBLN4-mediated cell-cell interactions at P4-P5 and P8. Pathways on the left, CBLN4-receptor pairs on the right. I Proximity ligation assay (PLA) for interactions between CBLN4 and GLUD1, and between CBLN4 and NEO1, in layers L1 and upper L2. A magnified view of a GFP-positive neuronal process with PLA puncta (red) is shown on the lower right. Scale bar: 10 µm. The PLA experiment was performed independently in 4 biological replicates, each yielding similar results. J Summary of the experimental findings related to investigations on CBLN4. S_inter, intercellular score; S_intra, intracellular score.
NEO1 is the primary CBLN4 receptor at Martinotti-glutamatergic developing synapses
To explore whether our atlas could illuminate previously incompletely understood intercellular communications, we focused on CBLN4, a ligand that facilitates synapse formation between Sst neurons axon terminals and the apical dendrites of GlutNs in the mouse cortex11,52 (Fig. 5E). Which precise Sst subtype expresses CBLN4, and which receptor(s) mediate its effects on GlutNs, remain open questions52,53 (Fig. 5F). Our transcriptional landscape analyses revealed that among the 4 identified Sst cell-types (Sst|L2/3-L5 fan-MC, Sst|L4 IVC, Sst|L5 T − MC, and Sst|L5/L6 NMCs), only the Sst|L2/3-L5 fan-MCs exhibited clear Cbln4 expression (Fig. 5G). Our LR interactome atlas predicted that CBLN4 is involved in relatively few intercellular interactions during synaptogenesis (P5 to P30), specifically between Sst|L2/3-L5 fan-MCs as source cells and select GlutN subtypes (L2/3 IT, L4 IT | SSC, L4/5 IT | PC, L5 IT and L5 PT) as target cells (Fig. 5G). Unexpectedly, most GABAN cell-types were also predicted as target cells. Intercellular signaling scores further indicated that among the three known CBLN4 receptors (DCC, GLUD1 and NEO1), only GLUD1 and NEO1 were predicted to be involved (Fig. 5H). Intercellular scores rose from P4 to P8, consistent with CBLN4’s known role in synapse formation during this window11.
We used intracellular scores to assess whether GLUD1 or NEO1 predominates as the CBLN4 receptor in cortex. Although GLUD1 has been identified as a CBLN4 receptor in GlutNs at P21–P3011,52, our data suggest that NEO1 signaling is more active during peak synaptogenesis (P4–P8) in CBLN4-receiving neurons (Fig. 5H). NEO1-associated genes were enriched for axon guidance and TGF-β pathways. To test for direct interactions at P8, we performed in situ PLA and observed strong CBLN4–NEO1 signals in cortical layers L1–2, while CBLN4–GLUD1 interactions were minimal or absent (Fig. 5I). These results underscore the atlas’s ability to identify key LR pairs and their cell-type specificity.
CDH13 and PCDH8 mediate perisomatic inhibition in deep and superficial layers
To determine whether our LR atlas can help identify novel LR pairs essential for cortical wiring, we focused on the cadherin family of adhesion molecules. Cadherins were prioritized because they are thought to play such a role45,46,54 and because our atlas revealed extensive spatiotemporal diversity in S1 (Fig. 2G).
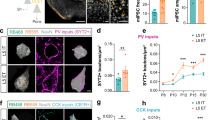
The atypical cadherin Cdh13, genetically associated with neurodevelopmental disorders55,56, exhibited a distinct and widespread expression pattern across cortical neuronal subtypes. In transcriptional landscapes, Cdh13 was detected in all GABAergic neuron types except Lamp5 neurons, with the highest expression levels observed in MGE-derived Sst and Pvalb populations (Fig. 6A). It was also broadly expressed across GlutN subtypes, with enrichment in DL subtypes. During corticogenesis, Cdh13 expression in DL GlutNs was sustained, whereas in MGE-derived GABAergic neurons, it peaked during the wiring period (P4–P30), suggesting a temporally coordinated role in circuit formation (Fig. 6A). These spatiotemporal dynamics point to a potential role for CDH13 in mediating homophilic interactions between MGE-derived GABAergic neurons and DL GlutNs. Consistent with this hypothesis, our LR inference analysis predicted that the vast majority of CDH13–CDH13 interactions occur between Sst/Pvalb interneurons and DL (L5 and L6) GlutNs (Fig. 6B, C, only ITs visualized as sources). These predictions are consistent with recent evidence implicating CDH13 in perisomatic inhibition of L5 subtypes by Pvalb basket cells (BCs)57. To experimentally validate this interaction, we performed in utero electroporation to knock down Cdh13 in L5 GlutNs and assessed perisomatic innervation by Pvalb BCs. Cdh13 knockdown significantly reduced the area of GlutN somata contacted by SYT2⁺ Pvalb BC boutons (Fig. 6C), indicating that postsynaptic CDH13 is required for proper perisomatic inhibition. Notably, chandelier cell (CHC) synapses onto the axon initial segment were unaffected (Fig. 6D), highlighting the specificity of CDH13 function and supporting our atlas-based predictions.

A Transcriptional landscapes of Cdh13 expression. B Heatmap of predicted CDH13–CDH13 interactions between GlutN ITs and GABAN types, shared by at least two consecutive ages. C Predicted utilization of CDH13–CDH13 interactions across ITs and GABANs at P4–P5 and P30; interaction strength is quantified by the inter_diff score. D In vivo knock-down of CDH13 in L5 GlutNs (left). Perisomatic (middle) and axon initial segment (right) inhibitory inputs from Pvalb BC and CHC boutons onto L5 GlutNs were quantified as SYT2 coverage. Statistical analysis was performed using Two-sided Mann-Whitney tests with biological replicates: n = 4 mice per electroporation condition (shCtl, shCdh13). Unit of analysis: individual electroporated L5 GlutN somata; technical replicates: 36 somata (shCtl) and 37 somata (shCdh13). Exact P value for perisomatic SYT2 coverage: P = 0.0227 *p < 0.05. No correction for multiple comparisons. Data are presented as median values in box plots, which show the median (centre line), 25th–75th percentiles (box), and whiskers representing minimum and maximum values within 1.5× the interquartile range (IQR). E Transcriptional landscapes of Pcdh8 expression. F Heatmap of predicted PCDH8–PCDH8 interactions between GlutN ITs and GABAN types shared by at least two consecutive ages. G Predicted utilization of PCDH8–PCDH8 interactions at P1–P2 and P4–P5. H In vivo knock-down of PCDH8 in L2/3 ITs (left). Perisomatic (middle) and axon initial segment (right) inhibitory inputs from Pvalb BC and CHC boutons onto L2/3 ITs were quantified as SYT2 coverage. Statistical analysis was performed using Two-sided Mann-Whitney tests with biological replicates: n = 4 mice per electroporation condition (shCtl, shPcdh8). Unit of analysis: individual electroporated L2/3 IT somata; technical replicates: 72 somata (shCtl) and 66 somata (shPcdh8). Exact P value for perisomatic SYT2 coverage: P = 0.028 *p < 0.05. No multiple-comparison correction. Data are presented as median values in box plots, with the median (centre line), 25th–75th percentiles (box), and whiskers indicating minimum and maximum values within 1.5× IQR. Associated source data are provided.
We next focused on PCDH8, whose spatial expression pattern appeared largely complementary to that of CDH13, with enrichment in GlutNs and Pvalb neurons of the UL (Fig. 6E). In line with this expression landscape, our LR atlas predicted that, aside from a few additional putative interactions, PCDH8–PCDH8 signaling would predominantly occur between L2/3 intratelencephalic (IT) neurons and Pvalb basket cells (BCs) (Fig. 6F, G). To test this prediction, we performed in utero electroporation of a Pcdh8 shRNA at E15.5 to knock down PCDH8 specifically in L2/3 IT neurons and assessed their perisomatic innervation by Pvalb BCs. Strikingly, Pcdh8 knockdown significantly increased SYT2⁺ bouton coverage of L2/3 IT somata, indicating that PCDH8 negatively regulates perisomatic inhibition in this circuit (Fig. 6H). This effect contrasts sharply with CDH13 function in L5 neurons, which acts as a positive regulator of Pvalb BC-mediated inhibition.
Thus, our LR atlas supports the conclusion that distinct cadherins differentially regulate perisomatic inhibition in a layer-dependent manner.
Discussion
Seminal studies in the last decades have shown that stereotyped cortical circuit wiring in mammals is regulated by LR interactions between GlutNs and GABANs15,16,17. More specifically, these data suggest that a molecular code, established by LR interactions between early-settling GlutNs and later-arriving GABANs, orchestrates circuit assembly. To identify LR pairs critical for GlutN–GABAN interactions and cortical circuit formation, we leveraged high-throughput single-cell transcriptomics. Our LR predictions, along with gene expression visualizations, are available at https://sclrsomatodev.online/.
We began by characterizing the transcriptomic profiles of the major neuronal classes across cortical development. This dataset, generated through extensive integration and meticulous annotation, is provided as a publicly accessible reference for exploring the dynamic transcriptional landscape of corticogenesis (https://sclrsomatodev.online/). For our analysis of LR interactions, we focused on the critical E18–P30 window, during which GABANs migrate to their target layers and form synaptic connections with GlutNs and other GABAergic partners. We developed computational tools to assess spatiotemporal expression of all genes in main cortical neuron types and to infer the number and identities of significant LR pairs that may govern cortical wiring. These tools can be applied to: (1) validate established interactions (e.g., NRG3 – ERBB4), (2) extend knowledge of known LR interactions (e.g., Cbln4 – Neo1 for Sst MC -> GlutN interaction) and (3) identify novel LR pairs involved in the formation of specific connections (e.g., the CDH13 and PCDH8 cases).
LR-mediated intercellular interactions are fundamental processes that shape the development and function of most biological tissues. Before the development of scRNA-seq, studying the molecular underpinnings of cell-cell interactions was low-throughput, restricted to a short selection of genes or proteins and a limited number of cell-type pairs. The emergence of scRNA-seq has enabled the development of numerous computational tools to infer cell-cell communication. While many existing methods predict LR interactions based solely on the expression levels of the LR pair across cell types58,59,60, a subset of more recent approaches may better approximate biological ground truth by also incorporating the downstream responses elicited in receptor-expressing cells47,61,62,63. For this study, we benchmarked most available methods and selected scSeqComm47, one of the most recent tools capable of inferring both intercellular and intracellular signaling. The intracellular score provided an additional layer of reliability for inferred interactions, and custom thresholds can be set for both intercellular and intracellular scores to modulate confidence. To further increase the reliability of the inferred interactions, we curated a comprehensive LR database by integrating existing published databases and incorporating additional LR pairs from the literature.
Our LR predictions indicate that neuronal connections are determined by specific combinations of LRs with distinct specificities. Notably, a core group of approximately 40 broadly expressed LRs mediate interactions in over 50% of cell-type pairs, complemented by a larger subset of LRs with varying degrees of specificity. Importantly, LRs unique to single neuronal connections are extremely rare, suggesting that cortical wiring is predominantly shaped by a combination of shared and context-dependent LRs rather than by unique or isolated interactions. Our predictive modeling analysis further strengthens the “combinatorial code” hypothesis. Classifiers trained on distinct LR feature subsets revealed that neither shared nor unique LR pairs alone could reliably discriminate cell types. Instead, rare context-dependent interactions provided the key discriminatory signal, with their predictive value maximized when combined with shared pairs. These results suggest that synaptic specificity emerges from the combined deployment of common and rare LR modules, rather than from exclusive dependence on unique interactions. This combinatorial logic echoes classical molecular code theories in neural development and provides a quantitative framework for how LR diversity encodes cell-type-specific connectivity.
Cadherin superfamily members are differentially expressed across subtypes of cortical excitatory and inhibitory neurons34,45, suggesting that a combinatorial cadherin code could guide the structured assembly of cortical circuits. Our data support this idea by demonstrating, with unprecedented resolution, that cadherins exhibit highly cell-type–specific and developmentally dynamic expression patterns (Fig. 4C). Building on predictions from our LR atlas, we experimentally identified two cadherins, CDH13 and PCDH8, as regulators of perisomatic inhibition by Pvalb basket cells (BCs) in deep and superficial layers, respectively (Fig. 6). Specifically, we found that CDH13–CDH13 interactions are crucial for Pvalb BC-mediated perisomatic inhibition of deep-layer GlutNs. Our atlas also shows that this interaction is not required for BCs expressing Sncg/CCK, which is consistent with recent findings57. In contrast, in superficial layers, PCDH8 negatively regulates Pvalb BC innervation, demonstrating that cadherins can either promote or restrict synapse formation depending on the context. This duality highlights the predictive power of our LR atlas, particularly in uncovering inhibitory or repulsive interactions, an underappreciated dimension of forebrain circuit development. Our atlas also predicts a higher number of LR interactions between excitatory neurons and Sst or Pvalb interneurons compared to Vip or Lamp5 populations, which is consistent with these findings. This may indicate that Sst and Pvalb rely more strongly on non-cell-autonomous signals for their integration, although alternative explanations should be considered, including differences in cell abundance or sampled developmental stages, which may not fully capture the wiring periods of other interneuron subtypes.
Beyond laminar and cell-class distinctions, our data indicate that the progenitor domain of interneurons (MGE, CGE, or POA) imposes an additional layer of specificity on predicted LR interactions. We found that GE-derived interneurons exhibit dynamic shifts from early GE-specific LR modules, enriched in partner recognition and integration processes, to shared programs during peak synaptogenesis (P8–P30), and back to fate-specific modules in adulthood (Supplementary Fig. 23). These findings suggest that cortical wiring is not only structured by laminar position and cell-class identity but also by developmental lineage, underscoring the combinatorial logic of fate- and stage-dependent LR programs.
Interestingly, the suppressive role of PCDH8 in superficial layers mirrors that of another protocadherin, PCDH18, which limits Sst neuron connectivity with GlutNs11. Given that both Cdh13 and Pcdh8 are genetically linked to neurodevelopmental disorders55,56,64, our findings raise the possibility that layer-specific disruption of Pvalb BC-mediated inhibition may contribute to the etiology of these conditions.
It remains unclear how generalizable our somatosensory cortex-focused analysis is to other cortical areas. Most GABAergic clusters defined in the Allen Institute taxonomy are shared across isocortical regions, and the relative proportions of cells within these clusters are largely consistent across areas3. For glutamatergic cell types, there is a modest, gradual transcriptomic variation across regions, and some degree of regional specificity is observed, particularly at the cluster level and in isocortical areas located at the rostral and caudal extremes3. In contrast, adjacent cortical areas largely share the same clusters. Importantly, our analysis is anchored at the supertype level; a higher-order classification that is more stable than individual clusters and broadly conserved across cortical areas. As described in Yao et al.3, supertypes are consistently observed across the isocortex, with only a few known exceptions, such as the L5 PT and Car3 supertypes. Because the cell types in our study were defined at the supertype level, we expect that our findings are likely to generalize to neighboring cortical regions.
Nonetheless, because our analyses were restricted to the somatosensory cortex, extrapolation to other cortical areas should be approached cautiously. In addition to neurons, non-neuronal cells also play a role in the early stages of cortical circuit assembly. For example, blood vessels and ventral oligodendrocyte precursor cells regulate the tangential migration of GABANs from the subpallium to the cortical plate through specific CXCL12-SEMA6A/B-PLXNA3 unipolar contact repulsion65. Future studies should leverage single-cell transcriptomics to systematically investigate neuron-non-neuronal cell communication to further explore these interactions.
Limitations of the approach—considerations for Future Improvements in Ligand-Receptor Atlases
Despite the robustness and comprehensiveness of our approach, certain limitations should be acknowledged. Reliance on mRNA levels as proxies for protein abundance can be misleading due to post-transcriptional regulation, and even when ligand and receptor proteins are expressed, their functional interaction depends on correct trafficking and subcellular localization, which single-cell RNA-seq cannot capture. Advances in single-cell proteomics may soon allow integration of such spatial information into developmental atlases. Our focus on glutamatergic and GABAergic neurons also excludes non-neuronal cell types such as astrocytes, oligodendrocytes, and microglia, which likely contribute to cortical circuit assembly and should be incorporated in future analyses. Interpretations based on GO terms remain provisional, as they may underrepresent intercellular contributions to broader processes such as cell death or differentiation. Finally, the use of an adult-derived connectivity matrix constrains predictions to mature architectures, providing a conservative but potentially incomplete view that may overlook transient developmental connections.
Currently, our understanding of the spatial relationship between cortical cell types is largely limited to adult stages66,67, which impedes precise inference of timely LR mediated cell-cell interactions at specific developmental stages, whether transient or stable. The advent of spatial transcriptomic technologies with high sensitivity, throughput, and resolution promises to bridge this gap in the near future.
Methods
Animals
Mice (mus musculus) were group housed (2–5 mice/cage) with same-sex littermates on a 12-hour light-dark cycle with access to food and water ad libitum. They were bred and maintained on a mixed SVeV-129/C57BL/6 N background. Animal experiments were carried out in accordance with European Communities Council Directives and approved by French ethical committees (Comité d’Ethique pour l’expérimentation animale no. 14; permission number: 62-12112012, Apafis #21683- 2019073011285386v4). Mice were housed on a 12-hour light–dark cycle at 21–23 °C and 40–60% humidity. Sample sizes were not predetermined by statistical methods; rather, they were guided by standards commonly used in single-cell transcriptomics and developmental neurobiology, ensuring sufficient cell numbers and biological replicates to achieve robust clustering, stable ligand–receptor inference, and reliable validation of observed phenotypes. For tissue collection, mice were deeply anesthetized and euthanized in accordance with institutional and governmental guidelines. Adult and postnatal mice were euthanized by intraperitoneal injection of pentobarbital (150 mg/kg). Loss of reflexes was confirmed prior to tissue collection. For histological analyses, animals were transcardially perfused with 0.9% saline followed by 4% paraformaldehyde. For experiments requiring fresh tissue for single-cell isolation, brains were rapidly dissected following euthanasia.
Single-cell isolation
Male and Female mice brains were dissected submerged in an ice-cold bubbled artificial cerebrospinal fluid (ACSF) with carbogen (95% O2 and 5% CO2). Our ACSF consisted of NaCl (7.32 g/L), KCl (0.26 g/L), NaH2PO4,H2O (0.165 g/L), Cacl2,2H2O (0.438 g/L), MgCl2,6H2O (0.264 g/L), D(+)-Glucose (1.98 g/L), NaHCO3 (2.1 g/L), and acid kynurenic (0.567 g/L). Brains were then sliced into a 300 µm (P0 and P2 mice) or 500 µm (P5, P8 and P30 mice) coronal sections with a vibratome (Leica). Somatosensory cortex area was dissected under a binocular loop. Enzymatic digestion was then processed by using pronase (Septomyces Argeus at 1 mg/mL) during 25 min at room temperature (RT) for P0 to P8 datasets. Cells were dissociated and triturated into single cell suspension in a solution consisting of ACSF, 1% FCS and DNAse (1 µl/10 mL). Trituration was carried out by using 3 glass pasteur pipets prepared at 3 different diameters. For P30 datasets, we used the Worthington Papain Dissociation System to carry out the enzymatic digestion and the cell dissociation following the manufacturer instructions.
Single-nuclei isolation
Dissection of the somatosensory cortex was achieved by following the same procedure as described for single-cell isolation. The dissected somatosensory cortices were transferred immediately into 500 µl of Hibernate™-E Medium (#A12476-01), then frozen for 3 min in isopentane pre-cooled to −80 °C. The samples were subsequently stored at −80 °C for long-term preservation. To process the tissue after conservation, the medium was first removed from the Eppendorf tube. Chilled 0.1X NP40 Lysis Buffer was then added in a volume of 500 µl, and the tissue was immediately homogenized using a Pellet Pestle with 15 strokes. The homogenized samples were incubated on ice for 5 min. The suspension was then pipette-mixed 10 times using a wide-bore pipette tip and incubate for 10 min on ice to ensure proper lysis. Following lysis, 500 µl of chilled wash buffer was added to the suspension, and the mixture was pipette-mixed 5 times using a regular-bore pipette tip. The suspension was passed through a 30 µm cell strainer into a 50 ml tube to remove debris. The filtered suspension was subsequently transferred to a 1.5 ml tube for centrifugation. Samples were centrifuged at 950 × g for 10 min at 4 °C. After centrifugation, the supernatant was carefully removed to avoid disrupting the nuclei pellet, which was retained for further analysis.
cDNA Amplification and library construction
10xv3 libraries were sequenced on Illumina HiSeq 4000. For single-nuclueus RNAseq, nuclei suspensions were adjusted following 10X recommendation. For GEM generation and barcoding, we utilized the Chromium Next GEM Single Cell 3’ Reagent Kits, following the manufacturer’s protocol. In summary, the prepared single-cell suspensions were loaded onto a Chromium Next GEM Chip G, along with the appropriate reagents, and processed using the chromium controller to encapsulate individual cells into GEMs. After GEM-RT incubation, cDNA was recovered and amplified through a series of cleanup and amplification steps, including SPRIselect bead-based purification. The amplified cDNA was then subjected to fragmentation, end repair, A-tailing, adaptor ligation, and sample index PCR to construct the final 3’ gene expression libraries. The constructed libraries were sequenced 10xv3 libraries were sequenced on Illumina HiSeq 4000.
Sequencing data processing
Sequencing reads were aligned to the mouse pre-mRNA reference transcriptome (mm10) using the 10x Genomics CellRanger pipeline (version 3.1.0 or 6.1.1) with default parameters.
External datasets
All external datasets3,23,24,25,26,27,28 were incorporated exactly as provided by the original publications. In the Di Bella et al.24 dataset, cells that were annotated as low-quality cells, doublets and Red blood cells were discarded. Because only the somatosensory cortex (SS CTX) was dissected in our experiments, we selected from the Allen Institute dataset only cells coming from the primary somatosensory cortex (SSp) for 10x data, and from both SSp and SSs for Smart-seq v4 data. To ensure that clusters were representative of the SS CTX, we discarded clusters containing fewer than 5 cells within a given subclass (as defined by Yao et al. 2021b) and retained only subclasses with at least two clusters. Furthermore, for supertypes containing fewer than 100 cells, we supplemented them—where possible—to reach 100 cells passing quality control (see Methods) by randomly selecting cells from cortical areas most correlated with the SS CTX (primary motor, MOp; secondary motor, MOs; frontal pole, FRP; and auditory areas). Exceptions were made for the CR Trp73, Meis2, Astro Gfap Apoe, and PVM Mrc1 supertypes, for which all cortical areas were used due to the low total cell numbers. The resulting dataset from Yao et al.3,68 constitutes our reference for cell-type assignment (hereafter referred to as AllenRef21).
Quality control
In order to retain only high-quality cells in all datasets, cells that passed the following criteria were kept (see Supplemental information QC metrics):
-
Cells with a mitochondrial gene percentage ≤10%.
-
Cells with a log10 (number of detected genes) within three double median absolute deviations (doubleMAD) of the population median, with a minimum low threshold of 500 genes.
-
Cells with a log10 (number of detected UMIs) above 3 doubleMADs of the population median.
-
We plotted log10 (nFeature RNA) versus log10 (nCount RNA) and fitted a linear regression model between these variables. Cells falling below an offset of −0.09 were retained.
-
Doublets in scRNA-seq datasets were identified using Scrublet69, with an expected doublet rate of 10%. The number of simulated doublets was set to twice the number of observed transcriptomes, and eight neighbors were used to construct the KNN classifier for observed and simulated doublets.
Assigning cell identity
Cells that passed QC criteria were used for the following analysis. Key steps to determining cell identity consisted of (Fig. S1):
Assigning Broad class identity
An in-house artificial neural network (ANN) was used to determine the identity of the cells at the class level for E11.5 to P5 datasets. We selected the datasets from Di Bella et al.24 and Bandler et al.27 as the training set for the ANN as they encompassed all broad cell classes relevant to our studies. Cells from Di Bella et al.24 were organized into five classes: GABAergic (Interneuron), Glutamatergic (CR, UL CPN, Layer 4, DL CPN, SCPN, NP, CThPN, Layer 6b), Immature/Migrating (Immature neurons, migrating neurons), Non-Neuronal (Astrocytes, Oligodendrocytes, Microglia, Cycling glial cells, Ependymocytes, Endothelial cells, VLMC, Pericytes) and Dorsal Pallium Progenitor (Apical progenitors, Intermediate progenitors). Cells from Bandler et al. (2021) were organized into two classes: GABAergic and SubPallium Progenitor. The weights derived from this model (hereafter referred to as the class PAB21 model) were independently applied to datasets from E11.5 to P5, excluding those used as reference.
For P8 to P30 datasets we used the map_sampling function of the R package scrattch.hicat to train a centroid classifier, randomly selecting 80% of marker genes. Test data were mapped to the AllenRef21 reference set at the class level (GlutNs, GABANs and NN classes). Classification was bootstrapped 1000 times to estimate robustness. Cells with prediction probabilities below 0.5 + 1/(number of class)² were considered undetermined.
Seurat Louvain graph-based clustering was initially performed on each dataset independently at the top level (k.param=round(sqrt(Number of cells)), annoy.metric = “cosine” in the FindNeighbors function and resolution=1 in the FindClusters function of the Seurat R package). One or more additional rounds of clustering were performed to resolve subclusters within candidate major clusters, with cluster heterogeneity evaluated using the Silhouette Score computed via a modified ReclusterCells function from the R package SCISSORs70 (Leary et al., 2021)). Cluster identities were assigned based on the most frequent prediction obtained either from the class PAB21 model or using scrattch.hicat. A cluster was assigned a given identity only if the difference between the most and second-most frequent predicted identities exceeded 40%. For downstream analyses, only cells predicted as Glutamatergic or GABAergic were retained. In GE datasets, only GABAergic cells were retained, as other postmitotic classes were considered potential contaminants.
Determining future cortical GABANs in the ganglionic eminences datasets
Since not all ganglionic eminence (GE)-derived cells migrate towards the cortex, we aimed to identify the migratory population. Using the differentially expressed (DE) branch analysis by Mayer et al.26, we identified the common DE genes for each branch within each GE and predicted cell identities by applying the assign_cell function from the R package MetaMarkers71. Cells were then clustered using the method described above, and the most frequently predicted branch identities were assigned to each cluster. As branch 1 was identified as giving rise to most future cortical cells26, only cells assigned to branch 1 were retained for downstream analysis.
Integration of all studies
To identify homologous cell types across 10X, Drop-seq, SSv4, SSv2, C1 scRNA-seq and snRNA-seq datasets, both from this study and from external studies, datasets were integrated using Seurat’s SCTransform integration workflow. For the integration analysis, 3’000 variable genes were selected. k.anchor= 5 and k.filter= 150 were the parameters set for the FindIntegrationAnchors function used to identify anchors between the datasets. For Yao et al. (2021b), only cells from the SS CTX were integrated (supplementary Fig. 1c).
Assigning subclass and supertype identities
Datasets were split into glutamatergic and GABAergic classes.
Glutamatergic neurons
For the E11.5 to P5 datasets, the in-house ANN was initially trained with the GlutNs part of the Di Bella et al. dataset24 using their defined cell-type labels (referred to as the ctPA21 model). The resulting model weights were then independently applied to each dataset from E11.5 to P5. The clustering module described above was performed on the entire E11.5 to P5 dataset. Cluster identities were assigned by comparing predictions from the ctPA21 model, and the most frequent predicted identity was designated as the cluster identity.
Correspondence between cell types annotations of Di Bella et al.24 and subclass annotation of Yao et al. (2021b) was achieved as followed: UL CPN, Layer 4, DL CPN: IT, SCPN: L5 PT, NP: L5 NP, CThPN: L6 CT, Layer 6b: L6b, Cajal-Retzius cells: CR.
Next, we aimed to distinguish IT cells among L2/3 IT, L4/5 IT, L5 IT, L6 IT, or Car3 subclass identities. We used the map_sampling function of the scrattch.hicat package as previously described, utilizing the IT cells from the AllenRef21 dataset as the reference set.
For P8 to P30 datasets, subclass identities were directly assigned using the map_sampling function of the scrattch.hicat package, as these time points are less far transcriptionally from the adult dataset. After applying our clustering module to the entire dataset, we assigned final subclass identities to clusters based on the most frequently predicted identity. To ensure accuracy, the difference between the top predicted subclass and the second most predicted subclass within each cluster had to exceed 20%. If this difference was less than 20%, we assigned a mixed subclass identity. For the adult dataset, we retained the original subclass labels.
Finally, for each unique subclass, we assigned the supertype level of Yao et al.3 by using the same procedure used to assign subclass labels with the map_sampling function.
GABAergic neurons
For datasets sampled from SS CTX, we assigned the subclass labels by using the same procedure as described above with scrattch.hicat. Once we obtained the subclass labels in the SS CTX datasets, we used the MapQuery function from the R package Seurat v4 (with weigh.reduction = “cca” parameter) to transfer these labels to datasets sampled from the GEs using the SS CTX subclass labels. Subclass labels were assigned by comparing each cell to the cluster defined by the clustering module. For GE cells whose subclass remained undetermined, typically less mature cells, we performed an additional round of the MapQuery function, using the subclass labels of more mature GE cells as reference. The final subclass labels were assigned by comparison to the clusters obtained with the clustering module. The same procedure was applied to determine supertype labels for each subclass. Because the Pvalb Lpl supertype (which corresponds to the Pvalb FS BC) was distributed across the full cortical thickness (Supplementary Fig. 2e), we deepened our analysis in order to discriminate between upper and lower Pvalb FS BC. This was achieved by using the map_sampling function, with clusters of the Pvalb Lpl supertype in our AllenRef21 dataset serving as reference.
Assigning cell-type names
Cell-type names were assigned by incorporating morphology, electrophysiological properties, and/or connectivity (mec-type) associated with each final identity. Mec-types were determined using patch-seq and connectivity studies1,2,3,29, which showed that many transcriptomic neuron types are correlated with known cortical neuron types by integrating transcriptomic clustering with patch-seq datasets (simultaneous electrophysiological recording and morphology reconstruction) and large-scale connectivity studies. These multimodal approaches demonstrated reproducible correspondences between molecularly defined subtypes and canonical cortical neuron classes. This integrative framework allowed us to anchor transcriptomic identities to established cortical cell types with known functional and anatomical properties. Final cell-type names were assigned by pooling original cell-type labels that shared a common mec-type.
Classification of cell-types into families
We classified our 27 identified cell-types into 8 families: IT family encompassing L2/3 IT, L4 IT | SSC, L4/5 IT | PC, L5 IT, L6 IT cell-types; ET family encompassing L5 PT, L6 CT, L6b cell-types; Other GlutNs encompassing CR, L5/6 NP and Car3|Claustrum-like cell-types; Lamp5 encompassing Lamp5 | L1 A7C/CNC and Lamp5 | L1-L5 NGC cell-types; Vip encompassing Vip|L2/3-L4 BP/BTC, Vip|Uncharacterized and Vip|L2/3-L5 BP/BTC cell-types; Sst encompassing Sst|L2/3-L5 fan-MC, Sst|L4 IVC, Sst|L5 T-MC and Sst|L5/L6 NMC cell-types; Pvalb encompassing Pvalb|L2/3 CHC, Pvalb|L2/3-L4 FS BC, Pvalb|L4/5 FS BC and Pvalb|L5/6 FS BC cell-types; Other GABANs encompassing Sncg|CCK BC, Sst|FS-like and Sst Chodl|LPC cell-types.
Pseudo-maturation score analysis
Pseudo-maturation analysis was performed from the earliest time point at which a given cell-type was observed up to P30. For each cell-type, we first performed an integration designed to preserve the developmental trajectory of the cells by using the R package FastMNN72,73 with prop.k set to 0.1 for most cell types and 0.4 for those with a large number of cells (L2/3 IT, L6 CT). Pseudo-maturation scores were calculated by first performing k-means clustering (k = 2) and then by using the R package slingshot (Street et al., 2018) which fits principal curves to identify lineages within each cell-type directly on the mutual nearest neighbor graph generated from the integration. Maturation directionality along each lineage was established by specifying a starting cluster. The resulting pseudo-maturation scores were normalized between 0 and 1. At each age, cells with normalized pseudo-maturation scores exceeding ±3 double median absolute deviations (doubleMADs) from the population median were considered outliers and discarded. After outlier removal, pseudo-maturation scores were scaled into three bins spanning ages E11.5-E17.5, E18.5-P5, and P8-P30 corresponding to intervals [0;1/3], [1/3;2/3] and [2/3;1] respectively. Cells in the E18.5–P5 and P8–P30 intervals were further subdivided into 6 and 3 equally sized bins, respectively.
Temporal gene wave computation
Genes differentially expressed along this inferred maturation axis were identified using the differentialGeneTest function of the R package Monocle 2 (v2.18.0)74 using parameters”fullModelFormulaStr = ˜sm.ns(pseudomaturation, df = 3)+study.RNAseq.method.platform“, and”reducedModelFormulaStr=˜study.RNAseq.method.platform”. We maintained genes with q-values less than 0.05 for downstream analysis. Genes with similar expression dynamics were grouped in 6 clusters using partition around medoids on the smoothed expression profiles of the significantly differentially expressed genes.
Pseudo-layer score analysis
Pseudo-layer analysis was performed from E18.5 to P30 by pooling the 2 nearest time points 2 by 2 from E18.5 to P5. Furthermore, this analysis was done independently for each family except for the Other GlutNs and Other GABANs families as they included cell-types that were not related to each other. UMAP was computed for IT and ET families at each defined time point, with Seurat SCT integration applied beforehand when necessary. For the Sst, Pvalb, Vip and Lamp5 families, all GABANs present at each defined time point were jointly embedded using UMAP, with dataset integration performed when necessary. After generating unified embedding, families were subset while retaining their original UMAP coordinates. Pseudo-layer scores were calculated following the same procedure used for the pseudo-maturation described above. Briefly, k-means clustering was applied (k = 3 or 2, depending on the dataset), except for the ET family, for which predefined cell-type annotations were used in place of clustering, designating L5 PT as the starting state and L6b as the terminal state. Lineage trajectories were inferred with slingshot30. The pseudo-layer score was normalized between 0 and 1. Cells with normalized values above or below three doubleMADs of the population median were considered as outliers. Once outliers were discarded, the pseudo-layer score was renormalized between 0 and 1. To enable comparisons both across time points and across families, the pseudo-layer scores were further scaled using the 5th percentile and the 95th percentile of each cell-type distribution (as determined in Supplementary Fig. 2e and Supplementary Fig. 3e). In addition, the median score of each cell-type was aligned to the corresponding median position determined in Supplementary Fig. 2e and Supplementary Fig. 3e, with the overall scaling set such that the 95th percentile of the L6b distribution equaled 1.
Spatial gene gradient computation
Genes differentially expressed along this inferred layer axis were identified using Monocle 2 using parameters “fullModelFormulaStr =˜sm.ns(pseudolayer, df = 3)+orig.ident”, and “reducedModelFormulaStr=˜orig.ident”. Genes with similar expression dynamics were grouped in 6 clusters.
Determining the distribution of cell types within a cortical column
We took advantage of two patch-seq studies from Gouwens et al.1 and Scala et al.2 and a multiplexed error-robust fluorescence in situ hybridization (MERFISH) study33. For each subclass, we applied the map_sampling function of scrattch.hicat to independently map cells of the 2 patch-seq studies to our reference AllenRef21 by taking the supertype level of the matching subclass (Supplementary Fig. 2a, Supplementary Fig. 2b). We assigned each of the clusters defined in these studies to the most frequent predicted supertype identity. For the MERFISH dataset, which comprised only 254 genes, the limited gene coverage prevented us from reliably identifying the corresponding supertype of the AllenRef21. We therefore took advantage of the published confusion matrix between this MERFISH dataset and 7 scRNAseq and snRNAseq datasets29,68 to guide cell-type correspondence (Supplementary fig. 2d). For each subclass, we applied the map_sampling function to the 7 datasets and assigned supertype of AllenRef21 to the cluster defined in Callaway et al.29 (Fig. 2c). The confusion matrix between MERFISH dataset and the 7 datasets allowed us to find the corresponding supertype in the MERFISH dataset. Clusters whose predicted subclass did not match their original subclass assignment were excluded from further analysis. Finally, supertype labels were merged into cell types defined in the section above to obtain the distribution of our cell-types along the cortical thickness (Supplementary Fig. 2e). For certain cell-types, no corresponding populations were identified in the patch-seq and MERFISH studies. We overcame this issue by assigning laminar distribution of these cell-types as follows:
CR: we took 100 random values encompassed in the L1 ( < 0.07) to reconstruct the distribution.
Vip|L2/3 − L4 BP/BTC is a mixed cell-type corresponding to the Vip Mypc1 and Vip Lmo1 supertypes in AllenRef21. We therefore pooled cells corresponding to these two cell-types to reconstruct the distribution of this cell-type.
We further applied the map_sampling function from the scrattch.hicat R package to each “Pvalb FS BC” clusters identified in the two patch-seq and MERFISH studies, using AllenRef21 cluster labels to determine layer specific Pvalb FS BC (Supplementary Fig. 3a, Supplementary fig. 3b, Supplementary Fig. 3c). The resulting AllenRef21 cluster assignments were then consolidated into three cell types, defined on the basis of their corresponding distribution extrapolated from the normalized soma depth of patch-seq and MERFISH studies: Pvalb | L2/3-L4 FS BC (114 Pvalb/113 Pvalb and 114 Pvalb clusters), Pvalb | L4/5 FS BC (115 Pvalb, 116 Pvalb/115 Pvalb, and 117 Pvalb/116 Pvalb clusters), and Pvalb | L5/6 FS BC (111 Pvalb, 112 Pvalb, 116 Pvalb, 116 Pvalb/112 Pvalb, 117 Pvalb, and 119 Pvalb).
Transcriptional landscape analysis
For each family, cells were independently embedded in a 2D graph based on their pseudo-maturation and pseudo-layer scores from E18.5 to P30. Gene expression values were represented as Log2(CPM + 1). For all genes expressed in at least five cells, we fitted a generalized additive model using the gam function from the mgcv R package, modelling expression as a function of both the pseudo-maturation and the pseudo-layer axis. For each family, to prevent oversmoothing of expression profiles, one third of the total number of cells were artificially added with log2(CPM + 1) expression values set to zero for all genes. As pseudo-layer score was not computed for other GlutNs and other GABANs families, an artificial pseudo-layer score was assigned to each cell included in these families according to their relative position determined in Supplementary Fig. 2e. As described above, the gam function was applied to these two families. In a given family, genes were considered significantly expressed if and only if for at least one cell-type and at least one age the number of cells was ≥5, the gene was expressed in more than 20% of the cells and its mean expression was higher or equal to the median of the median of the log2(CPM + 1) values across all cell-types at that age. Significant genes were used to perform a PCA on their smoothed expression landscape. The resulting PCA space was discretized using a k-mean clustering, with k = 15 determined by the gap statistic method. Average smoothed expression profiles were subsequently computed for each cluster. Enriched genes were split by cluster within each sheet and converted from mouse gene symbols to Entrez IDs using clusterProfiler and org.Mm.eg.db. KEGG pathway enrichment was performed for each (family, cluster) gene set using enrichKEGG, with the background universe defined as all Entrez IDs present across all clusters. Enriched pathways were retained at FDR ≤ 0.05. KEGG functional hierarchy (level 1 and level 2) was retrieved directly from KEGG via KEGGREST and assigned to each enriched pathway. For each (family, cluster), we defined a wave-level functional label as the KEGG level 2 category most significantly and frequently enriched within that cluster. Representative pathways (top 3 by FDR) were recorded for interpretability. Global spatiotemporal wave labels were derived analogously by aggregating enrichment across all neuronal families and selecting, for each cluster, the dominant KEGG level 2 category and its most recurrent enriched pathways (Supplementary Data S8 and Fig. 2F).
Gene ontology analysis
We used the clusterProfiler75,76 R package (v4.0) to find enriched biological processes in gene sets by using the enrichGO function. Gene ontology analyses were applied for each wave identified along the pseudo-maturation axis, and along the pseudo-layer axis.
Construction of the ligand-receptor database
LR_DB_2025 is the result of integrating and curating 109 existing public databases, to which we manually added 203 LR pairs based on literature (Supplementary Data S1). To the best of our knowledge, it is the largest curated LR database. Public databases were found by using some R and python packages, in particular OmnipathR77, singleCellSignalR59, CellPhoneDB58, NATMI60, CellCall17, scMLnet62 and CytoTalk63. We retained only ligand–receptor pairs corresponding to strictly intercellular interactions. LRs referenced in LR_DB_2025 were curated with supporting PMIDs documenting the evidence for each the interaction (Supplementary Data S1). We assigned each LR to a category and a family. Categories for LRs included in Omnipath were taken directly from the database, whereas categories for additional LRs were manually curated. In total, there are 20 distinct ligand categories and 10 distinct receptor categories. The LR family was attributed by using the HUGO Gene Nomenclature Committee (HGNC) resource and to a lesser extent the Uniprot database. Overall, 344 distinct ligand families and 313 distinct receptor families were identified (Supplementary Data S1).
Construction of the transcription regulatory database
The transcription factor (TF) database is the result of combining the merged mouse TRRUST v278 and “high” confidence RegNetwork79 TFs provided by the scSeqComm R package47 with the transcription regulatory database from Omnipath, retaining only entries supported by at least one literature reference.
Receptor-Transcription factor a priori association
Directed graphs of Reactome and KEGG signaling pathways, available from the GitLab repository associated with the scSeqComm package47, were used to compute the score of a-priori association between a receptor and a TF using the compute tfactor PPR function of scSeqComm.
LR landscape correlations in specific LR categories or LR neurodevelopmental ontologies (Fig. 3B)
We analyzed LR pairs where ligands showed significant transcriptional landscapes in IT neurons and where receptors exhibited significant expression in GABANs. LRs belonging to neurodevelopmental ontologies were identified using the MSigDB resource via the msigdbr R package80,81, focusing on gene sets from the H, C2, C5, and C8 ontologies, while excluding the “cellular component (CC)” ontology. Using specific keywords, we identified LRs belonging to six neurodevelopmental ontologies: Migration (2008 LRs), Cell Death (1,135 LRs), Differentiation (3158 LRs), Cell Adhesion (961 LRs), Synaptogenesis (554 LRs), and Brain-associated processes (1243 LRs), the latter encompassing nervous system functions not directly tied to development. LRs identified in these ontologies formed a new database: LR_DB_2025_Brain-Dev-Ontologies. We calculated the average correlation between ligand and receptor expression landscapes for LRs of specific categories (Fig. 3B, left plot) or neurodevelopmental ontologies (Fig. 3B, right plot), with ITs as source cells and GABANs as target cells. Statistical significance was assessed using Fisher Z-transformation.
Inferring ligand-receptor interactions
To infer ligand-receptor interactions between all cell-type pairs, we used the R package scSeqComm47 using log2(CPM + 1) of expression values. The interaction analysis was conducted from E18.5 to P30 by pooling the two nearest time points two-by-two from E18.5 to P5 and at the cell-type level. Only cell-types with at least five cells were kept for analysis. scSeqComm processed each LR pair referenced in LR_DB_2025 and computed intercellular and intracellular scores (S_inter and S_intra respectively) for all pairs of cell-types. For each ligand and receptor, scSeqComm assigned a score between 0 and 1 that quantified how strongly their average expression in a given cell type exceeded the expression levels expected by chance for randomly selected genes in the same cell type. The S_inter score was equal to the minimum value obtained between the ligand and the receptor implicated in the LR pair. For each known biological signaling pathway and a given receptor, S_intra was computed to quantify the evidence that the receptor in a given cell-type activated intracellular signaling within that pathway. We also computed the S_inter_diff score allowed by scSeqComm which represents an alternative version of the default scSeqComm intercellular signaling score. For each gene G in the input matrix, gene expression levels of G were normalized by the average expression level of gene G across all cell-types before computing the intercellular scores. This score aimed to prioritize ligands (receptors) that behaved differently across cell-types, thereby highlighting LR pairs enriched in particular cell-type interactions. To reduce false positives and increase the robustness of our approach, we generated a cell-type score that combined two metrics, incorporating prior knowledge about the likelihood of interaction between two cell-types:
-
A connectivity score: We constructed a cortical column in 3D of the SS cortex including our identified cell-types. To determine the distribution of our cell types within a cortical column, we used the cross-reference approach described above with Gouwens et al.1, Scala et al.2 Zhang et al.33. To gain access to the morphology of our cell-types, morphological reconstructions in SWC format (https://download.brainimagelibrary.org/3a/88/3a88a7687ab66069/) from Scala et al.2 were used. Only three of our identified cell-type did not have matched cell- type morphology: Vip Uncharacterized, Car3 Claustrum-like and CR. For Vip Uncharacterized, corresponding morphological reconstruction from Vip | L2/3-L4 BP/BTC and Vip | L2/3-L5 BP/BTC cell-types were used. Some studies suggest that Car3 Claustrum-like cells were IT cell-types predominantly located in L629,82, consequently L6 IT cells were used as proxies for Car3 Claustrum-like morphologies. For CR cells, a mouse neocortex morphological reconstruction was obtained from the neuromorpho.org website (<https://neuromorpho.org/neuroninfo.jspneuronname=AnstoetzNCCajalRetzius 9>). This CR morphological reconstruction was rescaled in order to match the morphological reconstruction scale of Scala et al. (2020). The constructed 3D cortical column encompassed 1 000 cells. The proportion of cell types was set, as closely as possible, to reflect their reported abundances in the somatosensory cortex based on the literature83,84,85,86,87,88,89,90,91 (80%GlutNs: 28.25% L2/3 IT, 23.25% L4 IT (85% L4 IT | SSC (60% strictly SSC morphology, 25% star pyramidal cell morphology (SPC)), 15% L4/5 IT | PC), 16.25% L5 (80% L5 PT, 20% L5 IT), 29.25% L6 (85% L6 CT, 10% L6 IT, 5% L6b), 1% CR, 1% L5/6 NP, 1% Car3|Claustrum-like; 20% GABANs: 45% Pvalb (42% Pvalb|FS BC (45% Pvalb|L2/3-L4 FS BC, 29% Pvalb|L4/5 FS BC, 26% Pvalb|L5/6 FS BC), 3% Pvalb|L2/3 CHC), 28% Sst (15% Sst MC (50% Sst|L5 T-MC, 50% Sst|L2/3-L5 fan-MC), 6% Sst|L4 IVC, 5% Sst|L5/L6 NMC, 2% Sst|FS-like), 2% Sst Chodl|LPC, 10% Vip (60% Vip|L2/3-L4 BP/BTC, 20% Vip|L2/3-L5 BP/BTC, 20% Vip|Uncharacterized), 6% Sncg|CCK BC, 9% Lamp5 (44% Lamp5 L1 A7C/CNC, 56% Lamp5 L1-L5 NGC). The centroid corresponding to the position of the soma for each reconstructed cell was calculated by using the nGauge python package92. All the reconstructed cells were embedded in a 3D cortical cortical column by using the natverse R package93 by positioning the soma according to the normalized cortical depth provided in2. If more cells than provided in2 were needed, a random normalized cell soma depth was selected between the 5th and the 95th percentile of the cell-type distribution along the cortical column. Values of soma position were adjusted in such a way that the cortical column had a cortical depth of 1 500 µm (Y axis) and the X and Z axis had a length of 150 µm. X and Z coordinates were randomly taken between 0 and 150 for each cell. Once the 1 000 cells were embedded in the reconstructed cortical column, the potential synapses between each cell and the 999 other cells were computed by using the potential_synapses function of the natverse package that implements the method of Stepanyants and Chklovskii48. This method created a technical artifact by facilitating potential synapses between cells belonging to the same cell-type as they were similarly distributed within the cortical column. All the potential synapses involving 2 cells belonging to the same cell-type were assigned as NA values. A connectivity score between 2 cell-types was obtained as follows:
where:
\(-a:{cell}\,a\)
\(-b:{cell}\,b\)
\(-{potS}:{Potential\; synapses}\)
\({where}:\)
\(-{{ct}}_{j}:{cell}-{type}\,j\)
\(-{{ct}}_{k}:{cell}-{type}\,k\)
\({-n}_{c{t}_{j}}:{Number}\,{of}\,{cells}\,{belonging}\, {to}\,{cell}-{type}\,j\)
\({-n}_{c{t}_{k}}:{Number}\,{ofcells}\,{belonging}\,{to}\,{cell}-{type}\,k\)
Due to the technical artifact affecting cell pairs belonging to the same cell-type, a medium connectivity score was assigned:
-
A development score: Some cell types colonize the cortex before others. For example, GlutNs colonize the cortex before GABANs and deep-layer GlutNs before superficial layers GlutNs. We attributed a development score between cell-types to reflect the degree of maturity of the different cell-types across cortical development. These scores span values from 0.1 to 1 (Supplementary Data S3).
These 2 scores allowed us to define a cell-type score:
\({where}:\)
-
-ctj : cell – type j
-
\(- {{ct}}_{k}\) : \({cell}-{type}\,k\)
-
-m : Total number of cell – types in the processed dataset
A ligand-receptor pair (LR)i between 2 cell-types pairs was considered significant if and only if:
where:
-
–\({\left({LR}\right)}_{i}\) : LR pair i
-
–n : Total number of LR pairs in LR_DB_2025 detected in the processed dataset
-
–l : subunit l composing the ligand L
-
–r : subunit r composing the receptor R
-
–M : Total number of subunits composing the ligand L or the receptor R
-
–\({w}_{c{t}_{{score}}}:{weight}\,{applied}\,{to}\,{the}\,{cell}-{type}\,{score}\). \({If\; the\; ligand\; L\; of\; the}\;{\left({LR}\right)}_{i}\;{was}\;{{{\bf{not}}}}\; {{{\bf{a}}}}\; {{{\bf{secreted}}}}\)
\({{{\boldsymbol{molecule}}}}\,{then}\,{{{{\boldsymbol{w}}}}}_{{{{\boldsymbol{c}}}}{{{{\boldsymbol{t}}}}}_{{{{\boldsymbol{score}}}}}}=0.6\);
\({else}\,{if}\,{the}\,{ligand}\,L\,{of}\,{the}{({LR})}_{i}{was}\,a\,{{{\boldsymbol{secreted}}}}\,{{{\boldsymbol{molecule}}}}\,{then}\)
\({{{{\boldsymbol{w}}}}}_{{{{\boldsymbol{c}}}}{{{{\boldsymbol{t}}}}}_{{{{\boldsymbol{score}}}}}}={{{\bf{0}}}}{{{\boldsymbol{.}}}}{{{\bf{4}}}}\)
-
–\({w}_{{inte}{r}_{{score}}}:{weight}\,{applied}\,{to}\,{the}\,{ligand}-{receptor}\,{interaction}\,{\hskip 49pt}{score}.\) \({If}\,{the}\,{ligand}\,L\,{of}\,{the}\,{({LR})}_{i}\,{was}\)
\({{{\boldsymbol{not}}}}\,{{{\boldsymbol{a}}}}\,{{{\boldsymbol{secreted}}}}\,{{{\boldsymbol{molecule}}}}\,{then}\,{{{{\boldsymbol{w}}}}}_{{{{\boldsymbol{inte}}}}{{{{\boldsymbol{r}}}}}_{{{{\boldsymbol{score}}}}}}={{{\bf{0.4}}}}\);
\({else}\,{if}\,{the}\,{ligand}\,L\,{of}\,{the}\,{({LR})}_{i}\,{was}\,{{{\boldsymbol{a}}}}\,{{{\boldsymbol{secreted}}}}\)
\({{{\boldsymbol{molecule}}}}\,{then}\,{{{{\boldsymbol{w}}}}}_{{{{\boldsymbol{inte}}}}{{{{\boldsymbol{r}}}}}_{{{{\boldsymbol{score}}}}}}={{{\bf{0.6}}}}\)
-
–\({Pc}{t}_{L}^{c{t}_{j}}:{Ligand\; L\; percentage\; expression\; in\; cell}-{type\; j}\)
-
–\({Pc}{t}_{R}^{c{t}_{k}}:{Receptor\; R\; percentage\; expression\; in\; cell}-{type\; k}\)
In order to prioritize cell-type specific interactions, we retained only significant LR pairs that had a S_inter_diff score above the population median minus 1.5 x DoubleMAD for each cell-type pair and at each age.
In the case of homophilic interaction, i.e., LR pairs in which the same molecule acted as both ligand and receptor, the mean S_inter values could differ according to the direction of the interaction between two cell-types. Therefore, the threshold described was evaluated in both directions to consider the LR pair between two cell-types as significant.
For LRs with secreted ligands, we gave a lower weight to the cell pair score and a higher weight to the LR intercellular score to account for their ability to act over greater distances. To further reduce false positives, only LR pairs that were significantly present across contiguous developmental stages were included (Supplementary Fig. 13).
Definition and use of intercellular score thresholds
As detailed above, each ligand–receptor interaction was associated with an intercellular score \({S}_{{{\rm{inter}}}}\), a cell-type score combining the developmental score and the anatomical connectivity score of the two cell types, and ligand-type–specific weights (0.6/0.4 for membrane ligands and 0.4/0.6 for secreted ligands) that determine how the cell-type score and the \({S}_{{{\rm{inter}}}}\) score contribute to the thresholding step. To clarify how these components were used to establish significance, we applied a two-step thresholding procedure. First, for each LR–cell-type pair, we compared the weighted observed value (cell-type score × weight + \({S}_{{{\rm{inter}}}}\) × weight) with the corresponding weighted expected value (threshold derived from the same LR pair and its mean \({S}_{{{\rm{inter}}}}\) across all cell-type pairs). An interaction was retained only when its observed value exceeded this threshold, ensuring that LR pairs were accepted only when their intercellular score was stronger than expected given the developmental and anatomical plausibility of the interacting cell types. Second, LR pairs were required to pass an independent expression filter in which all ligand and receptor subunits had to be detected in more than 15% of cells in their respective cell types. Among the remaining interactions, only those with an \({S}_{{{\rm{inter}}}{\_}{{\rm{diff}}}}\) score above the median minus 1.5×DoubleMAD were kept ensuring cell-type specificity. Together, these steps convert continuous scSeqComm scores into a strict binary classification (significant vs. non-significant) while leaving the underlying scores unchanged.
Machine learning framework to test the combinatorial code hypothesis
To assess whether LR expression patterns can distinguish cell-type pairs, we implemented a machine learning classification framework in R/PyTorch. For each developmental stage, source→target cell-type pairs were expanded into single-cell replicates by subsetting a fixed number of cells from each source and target cell-type. For every replicate, we generated a ligand–receptor feature matrix in which each entry corresponded to the product of the normalized RNA counts of a ligand in the source cell and its cognate receptor in the target cell. This encoding captured the potential interaction strength at the single-cell level and yielded a standardized feature set across all pairs. These matrices were then used to train classifiers tasked with predicting the correct source→target identity. Each example was encoded as ligand × receptor expression products (log1p, z-scaled). Five LR feature sets were evaluated: (i) all significant pairs; (ii) shared-only pairs ( ≥ 300 pairs); (iii) rare-only pairs ( ≤ 10 pairs); (iv) unique-only pairs (1 pair); and (v) size-matched random non-LR pairs. Train/test splits were made by cells (no source or target cell seen in training was included in testing). We trained a multi-layer perceptron (MLP) classifier with and without weight pruning, optimizing with Adam and early stopping. Model performance was assessed by test accuracy, ROC–AUC, and macro-F1 across >600 possible cell-type pairs. While overall accuracy was necessarily modest due to the difficulty of the task, rare-only feature sets consistently exceeded chance, and combining feature sets (all significant) achieved the best performance, consistent with a combinatorial LR code.
Analysis on ganglionic eminences (Supplementary Fig. 23)
To characterize ligand–receptor (LR) interactions within the ganglionic eminences (GE), we combined curated LR pairs from LR_DB_2025 with ontology annotations derived from MSigDB. After filtering out complexes, unique ligand–receptor couples were intersected with neurodevelopmental gene sets to identify processes relevant to GE maturation. We classified interactions according to their progenitor origin (VZ → MGE, VZ → CGE, VZ → POA) and monitored their persistence across contiguous developmental stages. Using UpSet plots and clustered heatmaps, we quantified the proportion of LR pairs that were specific versus shared between domains and traced their evolution across ages. Pairwise overlap was further assessed with Jaccard indices to measure the stability of interactions between domains. Enrichment analysis of LR-associated ontologies was then performed separately for specific and shared interactions, enabling the identification of domain- and age-dependent signaling programs. Enrichment analysis of LR-associated ontologies was performed using the clusterProfiler R package. First, ligand–receptor pairs were collapsed into ontology terms by mapping both ligands and receptors to MSigDB biological process gene sets (C5 collection), creating a custom TERM2GENE table. This annotation was joined to LR pairs detected in each GE domain and age group. LR pairs were then split into “specific” (present in only one domain) and “shared” (present in two or more domains) subsets, and enrichment was tested independently within each subset using the enricher function, with all detected LR pairs as the background universe. Significant terms were defined by adjusted p-value < 0.05. For visualization, the most enriched terms were displayed as dot plots, separated by domain (VZ–MGE, VZ–CGE, VZ–POA), developmental stage (E18.5–P0, P1–P2, P4–P5), and interaction type (specific vs shared), enabling identification of signaling programs enriched at distinct ages or across domains.
General analysis of inferred LR interactions (Fig. 4)
We analyzed the percentage of LR pairs associated with neurodevelopmental processes and disorders predicted by our atlas, focusing on their utilization at specific time points and between defined cell- pairs. In Fig. 4C, LRs were categorized into six neurodevelopmental processes using an approach similar to the landscape analysis (Fig. 3B). However, in this case, we included all significant LRs from the LR_DB_2025 database, as determined by intercellular and cell pair scores, regardless of their landscape significance or prior classification within brain ontologies (Fig. 3B). This analysis identified 2576 LRs associated with neuronal migration, 1494 with neuronal cell death, 3921 with differentiation/morphogenesis, 1391 with cell recognition/adhesion, 613 with synaptogenesis, and 238 specific to the Cadherin family. These groups were further stratified by developmental time points (E18.5-P0 to adulthood) and cell families (Pvalb, Sst, Vip, Lamp5, and Other GABANs). We used curated neurodevelopmental disorder gene lists to identify LRs implicated in specific conditions (Fig. 4D-F). We found 29 LRs associated with epilepsy94, 104 with intellectual disorders (from the ITHACA database: https://id-genes.orphanet.app/ithaca/), 14 with schizophrenia95, and 100 with autism96 (from the SFARI Gene database, accessed December 2024) (Supplementary Data S5). For Fig. 4F, we visualized the disease-associated LR pairs as an interaction network using the ggraph and igraph R packages. In Fig. 4C and D to enable fair comparison across ontologies with unequal numbers of possible ligand–receptor pairs or Diseases, the number of significant interactions was normalized by the total number of LR combinations within each ontology or diseases category.
ShRNAs
RNAi experiments were conducted using shRNAs targeting the coding sequence of Mus musculus Cdh13 and Pcdh8 (GenBank accession number NM_019707 and NM_021543) based on the following criteria (http://www.promega.com/siRNADesigner/program) :
-
-the sequence must start with either a Cysteine (C) or Guanine (G)
-
-It must have more than 50% G or C bases
-
-No more than 3 consecutive base repetitions in the sequence
The sequences chosen to design oligonucleotides (Supplementary Data S7) for shRNA genesis recognized nucleotides 1521-1541 of Cdh13 coding sequence and nucleotides 2302-2322 of Pcdh8 coding sequence. BLAST searches against Mus musculus databases confirmed the specificity of each target. As negative controls, we used corresponding non-targeting shRNAs with the same nucleotide sequence except in four positions. These shRNAs were subcloned into the mU6pro vector (gift from Dr J. LoTurco) and validated in vitro using classical western blot assays (Supplementary Data S7).
In utero electroporations
Timed pregnant C57BL6/J females were anesthetized with isoflurane (75% for induction and 2 to 2.5% for surgery) at E13.5 to trace DL neurons, at E15.5 to trace superficial layer neurons. The uterine horns were exposed. A volume of 1–2 µL of small hairpin RNA-expressing DNA plasmid (shRNA against Cdh13 or against Pcdh8 vs their respective control shRNAs, 1.5 µg/µl) was mixed with pCAG-GFP plasmids (1 µg/µl) and Fast Green (2 mg/ml, Sigma) for further injection into the lateral ventricle of each embryo with a pulled glass capillary and a microinjector (Picospritzer II, General Valve Corporation, Fairfield, NJ, USA). Electroporation was then conducted by discharging a 4000 µF capacitor charged to 27 V using a BTX ECM 830 electroporator (BTX Harvard Apparatus, Holliston, MA, USA). Five electric pulses (5 ms duration) were delivered at 950 ms intervals using electrodes. Embryos were allowed to be born and develop before being sacrificed at P28, and 50 µm coronal brain slices were cut using a sliding microtome (Microm).
Histology and immunostainings
Mice were perfused transcardially with ice-cold 4% paraformaldehyde (in PBS). Brains were removed and post-fixed overnight at 4 °C with the same fixative. Coronal sections were cut at 50 µm thickness using a sliding microtome (Microm). Briefly, for immunofluorescence experiments, free-floating sections were blocked and permeabilized for 2 h in a blocking buffer composed of 10% Normal Bovine Serum, 0.2% Triton X-100 (Sigma) in PBS. Primary antibodies, diluted in blocking solution and added overnight at 4 °C, were as follows: rabbit anti-Parvalbumin (1:1000, Swant), chicken anti-GFP (1:500, Aves), rabbit anti-Synaptotagmin 2 (SYT2) (1:100, DSHB), mouse IgG2a anti-SYT2 (DSHB, 1:100), and rabbit anti-SPTBN4 (Thermofisher, 1:1000). Corresponding fluorescently labeled secondary antibodies (AlexaFluor, Invitrogen) were added for 2 h in blocking solution at room temperature. Hoechst was added in PBS for 10 min, and sections were mounted on microscope slides that were coversliped using Mowiol solution (Sigma).
Proximity ligation assay
60 μm sagittal sections were treated for rabbit anti-CBLN4 (Invitrogen, PA5-36472) and anti-Mouse GLUD1 (Proteintech, 67026-1-Ig) or anti-CBLN4 and goat anti-NEO1 (Biotechne, AF1079) co-immunolabeling before application of Rabbit Plus and Mouse Minus or Rabbit Plus and Goat Minus probes, respectively (Merck, Duolink). Experiments were then done according to the manufacturer’s instructions.
Image acquisition
Images were obtained from 50 µm thick sections using a Zeiss LSM-800 confocal microscope. Electroporated zones were imaged with a 10X objective (Plan-Apochromat, Numerical aperture 0.3) to provide an overall view of all electroporated cells. For the proximity ligation assay, we imaged layers 1 and 2 with an oil-immersed 40X objective using mosaic tiles, or with an oil-immersed 63X objective (Olympus, Numerical aperture 1.4) for GFP colocalization with puncta. For synaptic analysis, GFP+ GlutNs located in deep or superficial cortical layers were imaged with the 63X objective. A 3X digital zoom was applied to achieve a lateral and z-axis resolution of 85 nm. The Z-stack was adjusted for each slice to ensure complete imaging of the entire neuron for subsequent 3D reconstruction. Laser power and detection filter settings were optimized based on the staining quality of each slice.
Image analysis
All cell analyses were performed on GFP-electroporated neurons found in the DL of the S1 cortex. All images were blinded using “blind analysis tool” plugins in ImageJ. 3D-Image reconstructions and analyses were performed with IMARIS 9.9.0 software. First, a zoomed crop was done on the GFP+ soma compartment. Then, to assess Pvalb FS BC cell input onto GFP-expressing electroporated cells, the Syt2+ presynaptic boutons physically contacting the GFP+ soma of pyramidal electroporated cells were analyzed. First, spots (diameter 0.6 µm) corresponding to individual Syt2+ presynaptic boutons were created by using the create spots function. The GFP+ soma was then reconstructed using the create surface tool. The density of Syt2+ synaptic spots contacting the GFP+ soma’s surface was then measured using the object-object statistic tool with the filter “shortest distance from soma.” To analyze the mean volume of presynaptic Syt2+ puncta, these puncta were modelized using the create surface tool followed by the filter “shortest distance from soma” adjusted to 0 to isolate only the Syt2+ surfaces contacting the GFP+ soma surface.
Statistics
All statistical tests are described in the figure legends. Statistical methods to predetermine sample size were not used. Unless otherwise stated, all values represent the averages of independent experiments ± SEM. Shapiro-Wilk or Anderson-Darling test was used to test the normality of the data. Statistical significance for comparisons of one variable was determined by student’s t-test using two- tailed distribution for two normally distributed groups, and by Mann-Whitney non-parametric test when distributions were not normal. For proportion comparisons, χ² test was applied. Differences were considered significant when p-value < 0.05. All statistical analyses were performed with R and Rstudio or with Prism 8.0.2 software (GraphPad).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw sc/snRNA-seq data generated in this study are available in the ArrayExpress database under accession E-MTAB-16260, and bulk RNA-seq data are available under accession E-MTAB-16355 (https://www.ebi.ac.uk/biostudies/arrayexpress/studies). Processed data, QC outputs, and derived RDS files are deposited on Zenodo (https://zenodo.org/records/11634657). Interactive exploration of inferred signaling networks is available through the scLRSomatoDev Shiny application at https://sclrsomatodev.online/. Source data are provided with this paper.
Code availability
All scripts used for data preprocessing, quality control, ligand–receptor inference, ontology annotation, and enrichment analysis were written in R and Python. The complete codebase, including custom functions and documentation, is publicly available on Zenodo (https://zenodo.org/records/11634657). Additional documentation and tutorials are provided online: Documentation: https://cortical-interactome.github.io/scLRSomatoDev-Docs/ Video tutorials: https://www.youtube.com/playlist?list=PLyfGSyn6Q6UY82ccuHRZQmRchVx6DskfJ.
References
Gouwens, N. W. et al. Integrated morphoelectric and transcriptomic classification of cortical GABAergic cells. Cell 183, 935–953.e19 (2020).
Scala, F. et al. Phenotypic variation of transcriptomic cell types in mouse motor cortex. Nature 598, 144–150 (2021).
Yao, Z. et al. A taxonomy of transcriptomic cell types across the isocortex and hippocampal formation. Cell 184, 3222–3241.e26 (2021).
Yao, Z. et al. A high-resolution transcriptomic and spatial atlas of cell types in the whole mouse brain. Nature 624, 317–332 (2023).
Zhang, M. et al. Molecularly defined and spatially resolved cell atlas of the whole mouse brain. Nature 624, 343–354 (2023).
del Pino, I. et al. Erbb4 deletion from fast-spiking interneurons causes schizophrenia-like phenotypes. Neuron 79, 1152–1168 (2013).
Jiang, X. et al. Principles of connectivity among morphologically defined cell types in adult neocortex. Science 350, aac9462 (2015).
Motta, A. et al. Dense connectomic reconstruction in layer 4 of the somatosensory cortex. Science 366, eaay3134 (2019).
Campagnola, L. et al. Local connectivity and synaptic dynamics in mouse and human neocortex. Science 375, eabj5861 (2022).
Schneider-Mizell, C. M. et al. Cell-type-specific inhibitory circuitry from a connectomic census of mouse visual cortex. bioRxiv 6, 2023.01.23.525290 (2023).
Favuzzi, E. et al. Distinct molecular programs regulate synapse specificity in cortical inhibitory circuits. Science 363, 413–417 (2019).
Fazzari, P. et al. Control of cortical GABA circuitry development by Nrg1 and ErbB4 signalling. Nature 464, 1376–1380 (2010).
Bernard, C. et al. Cortical wiring by synapse type–specific control of local protein synthesis. Science 378, eabm7466 (2022).
Pla, R., Borrell, V., Flames, N. & Marín, O. Layer acquisition by cortical GABAergic interneurons is independent of reelin signaling. J. Neurosci. 26, 6924–6934 (2006).
Lodato, S. et al. Excitatory projection neuron subtypes control the distribution of local inhibitory interneurons in the cerebral cortex. Neuron 69, 763–779 (2011).
Ye, Z. et al. Instructing perisomatic inhibition by direct lineage reprogramming of neocortical projection neurons. Neuron 88, 475–483 (2015).
Wester, J. C. et al. Neocortical projection neurons instruct inhibitory interneuron circuit development in a lineage-dependent manner. Neuron 102, 960–975.e6 (2019).
Miyoshi, G. & Fishell, G. GABAergic interneuron lineages selectively sort into specific cortical layers during early postnatal development. Cerebral Cortex 21, 845–852 (2011).
Baudoin, J.-P. et al. Tangentially migrating neurons assemble a primary cilium that promotes their reorientation to the cortical plate. Neuron 76, 1108–1122 (2012).
Southwell, D. G. et al. Intrinsically determined cell death of developing cortical interneurons. Nature 491, 109–113 (2012).
Wong, F. K. et al. Pyramidal cell regulation of interneuron survival sculpts cortical networks. Nature 557, 668–673 (2018).
Priya, R. et al. Activity regulates cell death within cortical interneurons through a calcineurin-dependent mechanism. Cell Reports 22, 1695–1709 (2018).
Telley, L. et al. Temporal patterning of apical progenitors and their daughter neurons in the developing neocortex. Science 364, eaav2522 (2019).
Di Bella, D. J. et al. Molecular logic of cellular diversification in the mouse cerebral cortex. Nature 595, 554–559 (2021).
Mi, D. et al. Early emergence of cortical interneuron diversity in the mouse embryo. Science 360, 81–85 (2018).
Mayer, C. et al. Developmental diversification of cortical inhibitory interneurons. Nature 555, 457–462 (2018).
Bandler, R. C. et al. Single-cell delineation of lineage and genetic identity in the mouse brain. Nature 601, 404–409 (2022).
Lee, D. R. et al. Transcriptional heterogeneity of ventricular zone cells in the ganglionic eminences of the mouse forebrain. eLife 11, e71864 (2022).
BRAIN Initiative Cell Census Network (BICCN) et al. A multimodal cell census and atlas of the mammalian primary motor cortex. Nature 598, 86–102 (2021).
Street, K. et al. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19, 477 (2018).
Mandai, K., Rikitake, Y., Mori, M. & Takai, Y. Nectins and nectin-like molecules in development and disease. Curr. Top. Dev. Biol. 112, 197–231 (2015).
Friedman, L. G., Benson, D. L. & Huntley, G. W. Cadherin-based transsynaptic networks in establishing and modifying neural connectivity. Curr Top. Dev. Biol. 112, 415–465 (Elsevier, 2015).
Zhang, M. et al. Spatially resolved cell atlas of the mouse primary motor cortex by MERFISH. Nature 598, 137–143 (2021).
Libé-Philippot, B. et al. Auditory cortex interneuron development requires cadherins operating hair-cell mechanoelectrical transduction. Proc. Natl. Acad. Sci. USA 114, 7765–7774 (2017).
Li, G. et al. Regional distribution of cortical interneurons and development of inhibitory tone are regulated by Cxcl12/Cxcr4 signaling. J. Neurosci. 28, 1085–1098 (2008).
López-Bendito, G. et al. Chemokine signaling controls intracortical migration and final distribution of GABAergic interneurons. J. Neurosci. 28, 1613–1624 (2008).
Borrell, V. & Marín, O. Meninges control tangential migration of hem-derived Cajal-Retzius cells via CXCL12/CXCR4 signaling. Nat. Neurosci. 9, 1284–1293 (2006).
Tiveron, M.-C. et al. Molecular interaction between projection neuron precursors and invading interneurons via stromal-derived factor 1 (CXCL12)/CXCR4 signaling in the cortical subventricular zone/intermediate zone. J. Neurosci. 26, 13273–13278 (2006).
Causeret, F., Moreau, M. X., Pierani, A. & Blanquie, O. The multiple facets of Cajal-Retzius neurons. Development 148, dev199409 (2021).
Stumm, R. K. et al. CXCR4 regulates interneuron migration in the developing neocortex. J. Neurosci. 23, 5123–5130 (2003).
Wang, Y. et al. CXCR4 and CXCR7 Have distinct functions in regulating interneuron migration. Neuron 69, 61–76 (2011).
Tanaka, D. H. et al. CXCR4 is required for proper regional and laminar distribution of cortical somatostatin-, calretinin-, and neuropeptide y-expressing gabaergic interneurons. Cerebral Cortex 20, 2810–2817 (2010).
Graf, E. R., Zhang, X., Jin, S.-X., Linhoff, M. W. & Craig, A. M. Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell 119, 1013–1026 (2004).
Krueger, D. D., Tuffy, L. P., Papadopoulos, T. & Brose, N. The role of neurexins and neuroligins in the formation, maturation, and function of vertebrate synapses. Curr. Opin. Neurobiol, 22, 412–422 (2012).
Sun, Y.-C. et al. Integrating barcoded neuroanatomy with spatial transcriptional profiling enables identification of gene correlates of projections. Nat. Neurosci. 24, 873–885 (2021).
Basu, R., Taylor, M. R. & Williams, M. E. The classic cadherins in synaptic specificity. Cell Adhesion Migration 9, 193–201 (2015).
Baruzzo, G., Cesaro, G. & Di Camillo, B. Identify, quantify and characterize cellular communication from single-cell RNA sequencing data with scSeqComm. Bioinformatics 38, 1920–1929 (2022).
Stepanyants, A. & Chklovskii, D. Neurogeometry and potential synaptic connectivity. Trends Neurosci. 28, 387–394 (2005).
Mòdol, L., Moissidis, M., Selten, M., Oozeer, F. & Marín, O. Somatostatin interneurons control the timing of developmental desynchronization in cortical networks. Neuron 112, 2015–2030.e5 (2024).
Exposito-Alonso, D. et al. Subcellular sorting of neuregulins controls the assembly of excitatory-inhibitory cortical circuits. eLife 9, e57000 (2020).
Batista-Brito, R. et al. Developmental dysfunction of vip interneurons impairs cortical circuits. Neuron 95, 884–895.e9 (2017).
Fossati, M. et al. Trans-synaptic signaling through the glutamate receptor delta-1 mediates inhibitory synapse formation in cortical pyramidal neurons. Neuron 104, 1081–1094.e7 (2019).
Liakath-Ali, K., Polepalli, J. S., Lee, S.-J., Cloutier, J.-F. & Südhof, T. C. Transsynaptic cerebellin 4–neogenin 1 signaling mediates LTP in the mouse dentate gyrus. Proc. Natl. Acad. Sci. USA. 119, e2123421119 (2022).
Lv, X. et al. Patterned cPCDH expression regulates the fine organization of the neocortex. Nature 612, 503–511 (2022).
Lesch, K.-P. et al. Molecular genetics of adult ADHD: converging evidence from genome-wide association and extended pedigree linkage studies. J. Neural Transm. 115, 1573–1585 (2008).
Sanders, S. J. et al. Multiple recurrent de novo CNVs, Including duplications of the 7q11.23 williams syndrome region, are strongly associated with autism. Neuron 70, 863–885 (2011).
Jézéquel, J. et al. Cadherins orchestrate specific patterns of perisomatic inhibition onto distinct pyramidal cell populations. Nat. Commun. 216, 4481 (2023).
Efremova, M., Vento-Tormo, M., Teichmann, S. A. & Vento-Tormo, R. CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat. Protoc. 15, 1484–1506 (2020).
Cabello-Aguilar, S., Fau, C., Lacroix, M. & Colinge, J. SingleCellSignalR: inference of intercellular networks from single-cell transcriptomics. Nucleic Acids Res. 48, e55 (2020).
Hou, R., Denisenko, E., Ong, H. T., Ramilowski, J. A. & Forrest, A. R. R. Predicting cell-to-cell communication networks using NATMI. Nat. Commun. 11, 5011 (2020).
Browaeys, R., Saelens, W. & Saeys, Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods 17, 159–162 (2020).
Cheng, J., Zhang, J., Wu, Z. & Sun, X. Inferring microenvironmental regulation of gene expression from single-cell RNA sequencing data using scMLnet with an application to COVID-19. Brief. Bioinform. 22, 988–1005 (2021).
Hu, Y., Peng, T., Gao, L. & Tan, K. CytoTalk: De novo construction of signal transduction networks using single-cell transcriptomic data. Sci. Adv. 7, eabf1356 (2021).
Butler, M., Rafi, S., Hossain, W., Stephan, D. & Manzardo, A. Whole exome sequencing in females with autism implicates novel and candidate genes. IJMS 16, 1312–1335 (2015).
Lepiemme, F. et al. Oligodendrocyte precursors guide interneuron migration by unidirectional contact repulsion. Science 376, eabn6204 (2022).
Zhang, M. et al. A molecularly defined and spatially resolved cell atlas of the whole mouse brain. Nature 624, 343−354 (2023)
Yao, Z. et al. A high-resolution transcriptomic and spatial atlas of cell types in the whole mouse brain. Nature 624, 317–33 2023)
Yao, Z. et al. A transcriptomic and epigenomic cell atlas of the mouse primary motor cortex. Nature 598, 103–110 (2021).
Wolock, S. L., Lopez, R. & Klein, A. M. Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst. 8, 281–291.e9 (2019).
Leary, J. et al. Sub-cluster identification through semi-supervised optimization of rare-cell silhouettes (SCISSORS) in single-cell RNA-sequencing. Bioinformatics 39, btad449 (2021).
Fischer, S. & Gillis, J. How many markers are needed to robustly determine a cell’s type? iScience. 25, 103378 (2022).
Haghverdi, L., Lun, A. T. L., Morgan, M. D. & Marioni, J. C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 36, 421–427 (2018).
Luecken, M. D. et al. Benchmarking atlas-level data integration in single-cell genomics. Nat. Methods 19, 41–50 (2022).
Qiu, X. et al. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982 (2017).
Yu, G., Wang, L.-G., Han, Y. & He, Q.-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS: A J. Integrative Biol. 16, 284–287 (2012).
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2, 100141 (2021).
Türei, D. et al. Integrated intra- and intercellular signaling knowledge for multicellular omics analysis. Mol. Syst. Biol. 17, e9923 (2021).
Han, H. et al. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 46, D380–D386 (2018).
Liu, Z.-P., Wu, C., Miao, H. & Wu, H. RegNetwork: an integrated database of transcriptional and post-transcriptional regulatory networks in human and mouse. Database 2015, bav095 (2015).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550 (2005).
Castanza, A. S. et al. Extending support for mouse data in the molecular signatures database (MSigDB). Nat. Methods 20, 1619–1620 (2023).
Peng, H. et al. Morphological diversity of single neurons in molecularly defined cell types. Nature 598, 174–181 (2021).
Tamamaki, N. et al. Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J. Comparative Neurol. 467, 60–79 (2003).
Staiger, J. F. et al. Functional diversity of layer iv spiny neurons in rat somatosensory cortex: quantitative morphology of electrophysiologically characterized and biocytin labeled cells. Cerebral Cortex 14, 690–701 (2004).
Ma, Y., Hu, H., Berrebi, A. S., Mathers, P. H. & Agmon, A. Distinct subtypes of somatostatin-containing neocortical interneurons revealed in transgenic mice. J. Neurosci. 26, 5069–5082 (2006).
Rudy, B., Fishell, G., Lee, S. & Hjerling-Leffler, J. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev. Neurobiol. 71, 45–61 (2011).
Tremblay, R., Lee, S. & Rudy, B. GABAergic interneurons in the neocortex: from cellular properties to circuits. Neuron 91, 260–292 (2016).
Nigro, M. J., Hashikawa-Yamasaki, Y. & Rudy, B. Diversity and connectivity of layer 5 somatostatin-expressing interneurons in the mouse barrel cortex. J. Neurosci. 38, 1622–1633 (2018).
Lim, L., Mi, D., Llorca, A. & Marín, O. Development and functional diversification of cortical interneurons. Neuron 100, 294–313 (2018).
Scala, F. et al. Layer 4 of mouse neocortex differs in cell types and circuit organization between sensory areas. Nat. Commun. 10, 4174 (2019).
Billeh, Y. N. et al. Systematic integration of structural and functional data into multi-scale models of mouse primary visual cortex. Neuron 106, 388–403.e18 (2020).
Walker, L. A. et al. nGauge: Integrated and extensible neuron morphology analysis in python. Neuroinform 20, 755–764 (2022).
Bates, A. S. et al. The natverse, a versatile toolbox for combining and analysing neuroanatomical data. eLife 9, e53350 (2020).
Genes4Epilepsy. An epilepsy gene resource - Oliver - Epilepsia - Wiley Online Library. https://onlinelibrary-wiley-com.insb.bib.cnrs.fr/doi/10.1111/epi.17547 (2023).
Merikangas, A. K. et al. What genes are differentially expressed in individuals with schizophrenia? a systematic review. Mol. Psychiatry 27, 1373–1383 (2022).
Abrahams, B. S. et al. SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol. Autism 4, 36 (2013).
Acknowledgements
We thank Julien Prados (UNIGE) for providing his Torch model for artificial neural network (ANN)-based cell type identification, members of the Cardoso laboratory for their input, and the Molecular and Cellular Biology Facility (PBMC), the Animal Core Facility and the Imaging Facility (inMagic) INMED platforms. This work was supported by the Institut National de la Santé et de la Recherche Médicale (INSERM), the Agence Nationale de la Recherche with ANR-13-JSV4-0006 SynD2 and ANR-23-CE16-0021 CALIN (A.d.C.), NeuroMarseille ICR+ Grant 2021 (A.d.C.), Fondation pour la Recherche sur le Cerveau ‘Développement et vieillissement’ (A.d.C.), Fondation Lejeune (A.d.C.), European Community 7th Framework programs (Development and Epilepsy; Strategies for Innovative Research to improve diagnosis, prevention and treatment in children with difficult to treat Epilepsy [DESIRE], Health-F2-602531-2013 (A. R., C.C.), and by an Excellence Initiative of Aix–Marseille University/A*MIDEX grant (CALIN-R24002AA) of the French ‘Investissements d’Avenir’ programme (C.C., A.d.C.). Research in the Telley laboratory was supported by ERC starting grant CERDEV_759112 and a SNSF grant 31003A_182676/1.
Author information
Authors and Affiliations
Contributions
A.d.C. and R.M. initiated the study. A.d.C. and L.T. conceptualized and supervised the study. A.d.C., L.T. and R.M. designed and conceptualized the experiments. A.d.C. L.T and R.M. performed most experiments. R.M. analyzed most experiments, supervised by L.T. and A.d.C. T.D.N. performed analyses for Figs. 3 and 4 and revision analysis. R.M. and L.S generated the Shiny App scLRSomatodev (https://sclrsomatodev.online/). C.C., A.G., L.C. and V.B. performed and analyzed shRNA design/production and in utero electroporations. E.P. validated shRNAs and performed proximity ligation assays. A.R participated in project management and help in manuscript writing and corrections. A.d.C. L.T. and R.M. wrote the manuscript with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mathieu, R., Draia-Nicolau, T., Corbières, L. et al. Uncovering the molecular logic of cortical wiring between neuronal subtypes across development through ligand–receptor inference. Nat Commun 17, 1298 (2026). https://doi.org/10.1038/s41467-025-68059-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-68059-8
