
Abstract
The interplay between the nervous and reproductive systems, encapsulated by the brain-gonad axis, is pivotal for reproduction. However, it remains largely unclear how hormones released from reproductive system actively regulate male sexual behaviors and mating strategies. Here we find that sexual priming induces an elevated sexual drive in both male mice and Drosophila, triggered by the surge of circulating steroid hormones 20-hydroxyecdysone (20E) in flies and testosterone in mice, respectively. In Drosophila, 20E is released from the male reproductive system upon sexual priming, which is controlled by a descending brain-gonad pathway composed of P1 neurons, Dsk neurons, and Crz/CrzR neurons. In return, 20E activates P1 neurons to elevate and sustain male flies’ mating drive. The enhanced mating drive not only fosters consequent mating success but also makes full use of the reproductive materials prepared during the pre-ejaculatory emission stage, e.g., seminal proteins which would otherwise be potentially harmful to males’ gonads. Collectively, our study offers insights into how hormones regulate males’ reproductive strategies.
Similar content being viewed by others


Introduction
Male animals’ mating motivation is actively regulated by their mate’s availability/quality and sexual experience. During courtship, males integrate female signals and environmental cues to sustain the appropriate level of mating drive1,2,3. In mice, key brain regions like the MPOA (medial preoptic area) and BNST (bed nucleus of the stria terminalis) control males’ mating drive. On the other hand, amygdala circuits responding to environmental and social factors negatively regulate mating drive4,5,6,7. In Drosophila, male-specific P1 neurons integrate female stimuli to identify the target mate and trigger vigorous courtship rituals8. Meanwhile, males adjust their mating drive by monitoring their internal states such as nutrition and prior sexual experience9,10,11.
Mating drive drops with ejaculation. Failure to transition from sexual arousal to sexual satiety may lead to an elevated sexual drive state, characterized by intensified mating drive11. This elevated sexual drive state is an analogy to food hunger to some extent. Notably, mice in starvation exhibit a tolerance for aversive tastes12. It’s thus an intriguing question whether and how males in elevated mating drive adjust their mating drive and pickiness in choosing their mates.
The interplay between the nervous and reproductive systems has been extensively investigated for development13. This brain-gonad axis regulates gonadal steroidogenesis and the development of gametes, such as oogenesis and spermatogenesis, through the release of gonadotropins from the brain14. Gonadal steroid hormones, in return, participate in the maturation of the brain and the emergence of sexually dimorphic behavior15. However, the bi-directional communication of the brain-gonad axis in governing sexual behaviors during adulthood remains elusive.
Hormones likely play a key role in regulating males’ mating behavior, as their levels fluctuate during sexual arousal. For example, testosterone levels increase in their plasm after men watch erotic movies or male mice interact with female mice16,17. Similarly, the hormone 20-hydroxyecdysone (20E) in Drosophila increases in the male hemolymph with intense courtship behavior18. In addition, testosterone receptor ARs (Androgen receptors) are widely expressed in sexual-related brain areas of mice19,20,21. 20E receptors, EcR and DopEcR, are both found in the fly neurons expressing fruitless (fru)+ and doublesex (dsx)+ 22, two transcription factors developmentally shaping the sexual neural circuits that control sexual behaviors23. Thus, the expression pattern of hormone receptors in the male brain suggests that hormones may regulate sexual behavior by modulating the activity of sex-related neural circuits.
Here, we found that both male mice and flies transform into an elevated mating drive state and increase their mating drive after experiencing sexual priming, e.g., sexual arousal without ejaculation. During the priming of male flies, 20E released from the reproductive system is triggered by the brain-gonad axis, including P1 neurons and their downstream Dsk and Crz neurons. In return, 20E activates P1 neurons to promote mating drive. Meanwhile, males adjust their allocation of reproductive materials after priming to avoid the waste of prepared reproductive materials. Our findings shed light on the dynamic regulation of mating drive and reproductive strategies by hormones.
Results
Male animals prefer females with high fecundity
Male animals deliberately choose their mates, and they tend to court females with high fecundity24. We first aimed to quantify males’ mate preference in both flies and mice. For flies, w1118 and CS males were allowed to court with either optimal (5~ 7 d old, with highest fecundity) or aged (35–45 d old, with lower fecundity) virgin females (Fig. 1A). During the first 5 min of the courtship assay, males showed higher mating drive to the optimal females compared with aged females (Fig. 1B). Similarly, wildtype CS males exhibited higher mating drive to the optimal females than aged females (Fig. 1C). These results imply that young females are optimal mates for males as they may have higher fecundity than sub-optimal aged females.

A Schematic illustration of the 5-min courtship assay for male flies. One optimal female or one aged female was introduced into the chamber in the presence of one male. B w1118 male flies performed more courtship actions towards optimal females than aged females. N = 16. Error bars represent ± SEM. Two-tailed unpaired t test, p = 0.0009. C CS male flies performed more courtship actions to optimal females than aged females. N = 17. Error bars represent ± SEM. Two-tailed unpaired t test, p = 0.0023. D Schematic of the 30 min courtship assay for male mice. One estrus optimal or aged female was introduced into the cage in the presence of one male. E Male mice showed the higher mating intensity to optimal females than aged females. Ethogram: left, optimal females; right, aged females. F Male mice exhibited reduced mating intensity towards aged females compared to optimal females. N = 6. Error bars represent ± SEM. Two-tailed unpaired t test, p = 0.0296. G Schematic of the priming assay for male flies. Each male courted a genital-sealed female for 10 min before its courtship was tested. H w1118 Male flies displayed comparable courtship intensity towards both optimal and aged females. N = 15. Error bars represent ± SEM. Two-tailed unpaired t test. I After experiencing 10 min priming, CS male flies showed the same courtship index between optimal and aged females. N = 15. Error bars represent ± SEM. Two-tailed unpaired t test. J Schematic of the priming assay for male mice. Males were allowed to interact with females for up to 5 times intromission and were separated from the female before ejaculation. And then, the courtship behavior of the mice was tested. K Male mice showed the same mating intensity between optimal females and aged females after priming. Ethogram: left, optimal females; right, aged females. L Male mice increased their mating intensity to aged female mice after they experienced priming behavior. N = 6. Error bars represent ± SEM. Two-tailed unpaired t test. ns, not significant. *p < 0.05. **p < 0.01, ***p < 0.001, ****p < 0.0001.
In parallel, we found male mice showed decreased mating intensity (Fig. 1D–F) and towards aged female mice (25–29 weeks) compared with optimal female mice (8–10 weeks). This “mate taste” cross taxa underscores males’ ability to assess and select mates based on female’s reproductive potentials.
Males’ mate preference is regulated by sexual priming
Male animals should be able to adjust their mating strategies to adapt to various environments and mate availability. We wondered whether “mate taste” is regulated by sex experience, such as sexual arousal. A male fly was sexually primed by grouping and vigorously courting with a genitalia-sealed virgin female for 10 min (Fig. 1G). Then, they were allowed to court different female mates. We found that w1118 male flies after priming showed the elevated mating drive to sub-optimal mates (aged females) (Fig. 1H). Similarly, primed CS males performed higher mating drive to the aged females (Fig. 1I). Notably, the courtship index in primed flies to the optimal females was not further increased, suggesting that these optimal mates already elicit plateaued mating intensity that cannot be revealed by the one-on-one courtship assay. We then performed a mating competition assay for two males to a single optimal female and found that primed males showed higher competitiveness compared to a control male (Supplementary Fig. S1A, B). The results imply that priming causes an increase in males’ mating drive, and primed males no longer show a significantly different courtship intensity towards optimal or aged females.
Similarly, male mice were primed by pairing them with estrus females until they exhibited sexual behaviors (sniffing, mounting, and five times intromissions) without ejaculation (Fig. 1J). Primed male mice showed higher mating intensity to aged female mice (Fig. 1K, L), demonstrating an elevated mating drive on sub-optimal mates. Interestingly, although the mating intensity index to optimal females did not further increase by priming, the reduced latency to mounting behavior argued that primed mice still exhibit a higher mating drive to optimal females (Supplementary Fig. S1E).
The results above suggest that the sex experience of sexual priming without ejaculation induces a stronger mating drive, thus dampening the males’ choosiness to sub-optimal mates. It’s worth noting that in both primed male flies and mice, neither the courtship latency index nor the mating intensity towards the optimal females appears to increase. This may due to the one-on-one mating assays failed to reveal the increase to mating drive to optimal females. In this case, males engage in the elevated mating drive state with the modificatory “sexual taste”.
Steroid hormones boost males’ mating drive
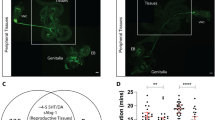
What’s the molecular and neural basis of the priming-induced mating drive elevation? In Drosophila, the 20-hydroxyecdysone (20E) is the major steroid hormone25. The levels of 20E in the hemolymph were reported to surge during intense courtship behavior18. We here demonstrated that the levels of 20E in the hemolymph were higher in primed males (Fig. 2A), suggesting that 20E may play a role in the priming induced sexual drive increase. Then, we pinpointed the source of 20E by looking at the expression pattern of the gene dare, which encodes the Drosophila homolog of vertebrate adrenodoxin reductase (AR) and is essential for the synthesis of steroid hormones26. The dare-GAL4 expression was found across many tissues, such as the intestine and male reproductive system, including the testes (TE), seminal vesicle (SV), anterior ejaculatory duct (AED), and the epithelium of the EB (Supplementary Fig. S2A). Its expression in the nervous system is marginal (Supplementary Fig. S2A). Furthermore, ELISA analysis confirmed the presence of 20E in the gut and the male reproductive system but not in the nervous system of naive males (Fig. 2A and Supplementary Fig. S2B). In contrast, only the male reproductive system showed a decrease in 20E levels after 10 min of priming (Fig. 2A and Supplementary Fig. S2B), indicating that 20E was released from the male reproductive organs during priming.

A The levels of 20E in the hemolymph were increased, and in the MRS were decreased after males experienced 10 min priming. In the hemolymph: Naive, N = 6; Primed, N = 6, each data point representing 20 flies. In the MRS: Naive, N = 4; Primed, N = 5, each data point representing 10 flies. MRS, male reproductive system. Error bars represent ± SEM. Two-tailed unpaired t test, in hemolymph: p < 0.0001; in MRS, p = 0.0106. B The levels of testosterone in the serum were increased in primed male mice compared with naïve male mice. N = 6. Error bars represent ± SEM. Two-tailed unpaired t test, p = 0.0033. C Scheme of 20E (3 mM, 5 hr) feeding assay. D w1118 Male flies fed with 20E displayed a similar courtship index towards both optimal females and aged females. N = 15. Error bars represent ± SEM. Two-tailed unpaired t test. E CS male flies fed with 20E showed the same courtship index between optimal females and aged females. N = 9. Error bars represent ± SEM. Two-tailed unpaired t test. F Schematic illustrating male mice being injected with 500 ng of testosterone subcutaneously in the back and then resting for 30 min. G Male mice injected with testosterone showed enhanced mating motivation towards aged female mice. Left, optimal females. Right, aged females. H Male mice injected with testosterone showed the same mating intensity to optimal females and aged females. N = 6. Error bars represent ± SEM. Two-tailed unpaired t test. ns, not significant. *p < 0.05. **p < 0.01, ***p < 0.001, ****p < 0.0001.
To directly test whether 20E enhanced males’ mating drive and made them less picky, we fed male flies with 20E (3 mM) for 5 h (Fig. 2C) and found that both CS and w1118 males fed with 20E showed a higher mating drive to the sub-optimal mates (Fig. 2D, E) and stronger courtship competitiveness to optimal females (Supplementary Fig. S1C, D). We thus concluded that sexual priming and 20E feeding had a similar effect in boosting male flies’ sexual drive.
The steroid hormone testosterone levels in the serum of mice and humans are also increased along with sexual arousal/priming16,17. We also observed that the serum testosterone levels of the mice were increased after they were primed with the estrus female (sniffing, mounting, and five times intromissions without ejaculation) (Fig. 2B). To test whether the increased testosterone modulates male mice’s mating intensity, we subcutaneously injected 500 ng testosterone in male mice (Fig. 2F). Thirty minute after injection, male mice performed more vigorous and consistent mating behavior to aged females comparable to optimal mates (Fig. 2G, H). Moreover, male mice injected with testosterone showed a reduced mounting latency to both optimal and aged female mice (Supplementary Fig. S1E), similar to the male mice which were sexually primed (Supplementary Fig. S1E). By contrast, vehicle control of corn oil injection alone does not enhance mating motivation (Supplementary Fig. S6). Taken together, we found that steroid hormones of both rodents and insects are released from their reproductive system and accumulated in the hemolymph or plasm during sexual arousal/priming, converting males into elevated mating drive states.
20E activates P1 neurons to regulate male flies’ mating drive
How do steroid hormones elevate the mating drive for male animals? In Drosophila, male-specific P1 neurons control males’ mating drive8. We thus asked whether 20E activates P1 neurons to enhance mating drive. Two 20E receptors were identified in flies, with EcR as a nuclear complex and DopEcR as a G-protein coupled receptor (GPCR)27. We then conducted RNAi knockdown for ECR or DopECR in P1 neurons (labeled by R71G01-GAL4) (Fig. 3A). Knocking down ECR but not DopECR in P1 neurons even resulted in decreased mating drive towards optimal mates (Fig. 3B), indicating that 20E and ECR may be required for the developmental and normal function of P1 neurons. In contrast, knocking down dopECR in P1 neurons blocked the priming-induced higher mating drive to aged females (Fig. 3C) and reduced the priming-induced stronger courtship competitiveness to optimal females (Supplementary Fig. S2C). Thus, we reasoned that 20E released during priming activates P1 neurons through DopECR to elicit a higher mating drive to sub-optimal mates.

A Subcellular localization of ECR and DopECR. B Knocking down ECR but not DopECR in P1 neurons by an R71G01-GAL4 driver caused a defect in the male courtship index. R71G01-Gal4/+ naive, N = 10; UAS-EcR-RNAi/+ naive, N = 10; R71G01-Gal4 > UAS-EcR-RNAi primed, N = 13; UAS-DopEcR-RNAi/+ naive, N = 10; R71G01-Gal4 > UAS-DopEcR-RNAi primed, N = 10. Error bars represent ± SEM. Two-tailed unpaired t test, column A vs column C, p = 0.0005; column B vs column C, p = 0.0312. C Knocking down DopECR in P1 neurons blocked priming-induced enhanced mating drive of males to age females. R71G01-Gal4/+ naive, N = 36; R71G01-Gal4/+ primed, N = 33; UAS-DopEcR-RNAi/+ naive, N = 34; UAS-DopEcR-RNAi/+ primed, N = 31; R71G01-Gal4 > UAS-DopEcR-RNAi naive, N = 31; R71G01-Gal4 > UAS-DopEcR-RNAi primed, N = 33. Error bars represent ± SEM. Two-tailed unpaired t test, column A vs column B, p = 0.0043; column C vs column D, p = 0.0247. D Intersectional expression between DopECR neurons driven by DopECR-GAL4 and P1 neurons driven by R71G01-LexA or fru+ neurons driven by fruLexA. The white arrows indicate P1 neurons. Scale bars represent 50 μm. Green, GFP antibody; magenta, nc82 antibody. E GCaMP7s signals in P1 neurons were increased with the concentration of 20E (above, left). The traces of fluorescence changes after 20E or AHL adding (below). The dashed line indicates the time when we added 20E or AHL to the samples. The GCaMP7s fluorescence change ratios in 20E added and control group (above, right). Scale bars represent 50 μm. AHL, N = 8; 10−1 nM 20E, N = 9; 10−2 nM 20E, N = 7; 10−3 nM 20E, N = 8. Error bars represent ± SEM. Two-tailed unpaired t test, column A vs column B, p < 0.0001; column A vs column C, p < 0.0001; column A vs column D, p = 0.0003. F 20E could not induce Ca2+ response in P1 neurons after knocking down DopECR in these neurons (above, left). The traces of fluorescence changes after 20E or AHL adding (below). The dashed line indicates the time when 20E or AHL was added. The GCaMP7s fluorescence change ratios in 20E added and control groups (above, right). Scale bars represent 50 μm. AHL, N = 10; 10−1 nM 20E, N = 7; 10−2 nM 20E, N = 8; 10−3 nM 20E, N = 9. Error bars represent ± SEM. Two-tailed unpaired t test. ns, not significant. *p < 0.05. **p < 0.01, ***p < 0.001, ****p < 0.0001.
We then used GCaMP7s to monitor P1 neurons’ activity and found that these neurons were activated by 10 min priming of males (Supplementary Fig. S2D). Importantly, knockdown of DopECR in P1 neurons abolished the priming-induced activation (Supplementary Fig. S2E). We thus proposed that P1 neurons’ activity may be upregulated by 20E. However, this ex vivo experimental setting may not be able to fully capture the activity of P1 neurons. To further test the hypothesis that 20E can activate P1 neurons via dopECR, we performed the following experiments. Firstly, to confirm that DopECR is indeed expressed in P1 neurons, we used an intersectional strategy to label the overlapping neurons between DopECR-GAL4 and FruLexA or R71G01-LexA and found that the intersectional neurons included the male-specific P1 neurons (Fig. 3D). Secondly, To gain direct evidence that 20E activates P1 neurons, we performed ex vivo calcium imaging on P1 neurons. The calcium level of P1 neurons exhibited a dosage-dependent increase with the concentration of 20E from 10−1 nM to 10−3 nM, a range comparable with the physiological condition (Fig. 3E and Supplementary Fig. S2F)18. In contrast, when we knocked down DopECR using RNAi in P1 neurons, 20E could no longer activate P1 neurons (Fig. 3F and Supplementary Fig. S2G). Thus, we demonstrated that 20E is released by the male reproductive system during priming, activating P1 neurons through its receptor dopECR. Thus, we have demonstrated that 20E released from the reproductive system can act on the brain to regulate mating drive.
A brain-gonad axis controls 20E released from the male reproductive system
We next asked how the 20E release from the male reproductive system was controlled during the sexual priming. As an integrative center in mating behavior, P1 neurons are actively involved in the priming stage and are likely to play an important role in regulating the activity of the male reproductive system. Here, we found that thermo-genetically activating P1 neurons with dTPRA1 for 10 min could induce 20E release from the male reproductive system without any sexual behavior (Fig. 4A), suggesting that P1 neurons control the secretion of 20E from the male reproductive system during priming.

A Activation of P1 neurons induced the emission of 20E from the male reproductive system. N = 4. Each data point represents 10 flies. MRS, male reproductive system. Error bars represent ± SEM. One-way ANOVA, p = 0.0002. B Blocking Crz neurons blocked the emission of 20E from the male reproductive system. Crz-Gal4/+ naive, N = 4; Crz-Gal4/+ primed, N = 5; UAS-Kir2.1/+ naive, N = 4; UAS-Kir2.1/+ primed, N = 5; Crz>Kir2.1 naive, N = 4; Crz>Kir2.1 primed, N = 4. Each data point represents 10 flies. MRS, male reproductive system. Error bars represent ± SEM. Two-tailed unpaired t test, column A vs column B, p = 0.0050; column C vs column D, p = 0.0033. C tsh-GAL80 was used to suppress Crz neurons’ expression in the VNC. Scale bars represent 100 μm. Green, GFP antibody; magenta, nc82 antibody. D Crz neurons in the brain were not required for the emission of 20E from the male reproductive system. Crz-Gal4/+ naive, N = 4; Crz-Gal4/+ primed, N = 4; tsh-Gal80/+;UAS-Kir2.1/+ naive, N = 4; tsh-Gal80/+;UAS-Kir2.1/+ primed, N = 4; Crz-Gal4,tsh-Gal80;UAS-Kir2.1 naive, N = 6; Crz-Gal4,tsh-Gal80;UAS-Kir2.1 primed, N = 6. MRS, male reproductive system. Each data point represents 10 flies. Error bars represent ± SEM. Two-tailed unpaired t test, column A vs column B, p = 0.0194; column C vs column D, p = 0.0411; column E vs column F, p = 0.0390. E The brain-gonad axis is composed of P1 neurons, Crz neurons, and the male reproductive system. F VNC Crz neurons responded to the P1 neurons’ activation (left) and the traces of fluorescence intensity after ATP or AHL adding (right). The dotted line indicates the time when ATP or AHL was added to the imaging chamber. The calibration bar ranges from 0–255. The quantitation of the fluorescence changes in Crz neurons (middle). N = 10. Error bars represent ± SEM. Two-tailed unpaired t test, p < 0.0001. ns, not significant. *p < 0.05. **p < 0.01, ***p < 0.001, ****p < 0.0001.
We then investigated the neural pathway that links P1 neurons and the male reproductive system in the flies. Four male-specific Crz neurons and their downstream CrzR neurons located in the ventral nerve cord (VNC) are responsible for male ejaculation28. CrzR neurons projected to the male reproductive system with elaborate arborizations28, suggesting that Crz and CrzR neurons may trigger 20E emission during sexual arousal. To test this notion, we blocked Crz neurons using the inwardly rectifying potassium channel kir2.1 and measured the levels of 20E in the male reproductive system after priming. In this case, we found that priming-induced 20E emission was blocked (Fig. 4B). To rule out the involvement of the Crz neurons in the brain, we specifically targeted brain Crz neurons by suppressing Crz-GAL4 in the VNC with tsh-GAL80 (Fig. 4C). Blocking brain-specific Crz neurons did not interfere with 20E emission during courtship (Fig. 4D), suggesting that Crz neurons in the VNC exclusively control 20E emission. To further confirm the functional connection between P1 neurons and Crz neurons, we performed Ca2+ imaging on VNC Crz neurons with the calcium sensor GCaMP7s. The Ca2+ level in Crz neurons was largely increased when we used ATP to activate P1 neurons bearing P2X2 (Fig. 4F), suggesting that VNC Crz neurons are downstream of P1 neurons.
A neuropeptide pathway mediates the descending control of 20E emission
How is the activation from the P1 neuron during priming relayed to VNC Crz neurons? Drosulfakinin (DSK) neurons, which receive direct synaptic transmission from P1 neurons, are known to act downstream of P1 neurons to regulate mating behaviors29,30. Two putative receptors of Dsk peptides, CCKLR-17D1 and CCKLR-17D3 have been identified. We found that selectively knocking down CCKLR-17D3 but not CCKLR-17D1 with RNAi in Crz neurons significantly suppressed 20E emission during courtship behavior (Fig. 5A), indicating that DSK and its receptor CCKLR-17D3 are indispensable for 20E emission. We then examined the CCKLR-17D3 expression in Crz neurons with a knock-in GAL4 into the CCKLR-17D3 locus (17D3GAL4)29 and the Crz antibody. Two out of the four male-specific Crz neurons in the abdominal ganglion (AbG) of the VNC expressed CCKLR-17D3 (Fig. 3B), consistent with the single-cell RNA sequencing results from FCA (FLY CELL ATLAS). Furthermore, stimulation of DSK neurons with ATP elicited a Ca2+ response in all four Crz neurons in the AbG (Fig. 3C). While only two Crz neurons express CCKLR-17D3 and are the direct downstream of DSK neurons, mutual activation between the four Crz neurons could trigger the synchronized activation of all four Crz neurons31. Thus, the DSK/CCKLR pathway bridges P1 neurons and Crz neurons to control the emission of 20E. However, we did not rule out the possibility that the activation of the DSK/CCKLR pathway could result in the biosynthesis of 20E in EB. This scenario is likely, considering that the CCKLR may elicit a downstream cascade to boost the biosynthesis of 20E. Up to this point, we have identified the two-directional communication along the brain-gonad axis during sexual priming. During sexual priming, this axis controls the release of 20E from the male reproductive system. As feedback, 20E activates P1 neurons to promote the males’ mating drive, as reflected by the fact that males became less picky about mates.

A Knocking down CCKLR-17D3 but not CCKLR-17D1 in Crz neurons inhibited the emission of 20E from the male reproductive system. Crz-Gal4/+ naive, N = 4; Crz-Gal4/+ primed, N = 4; UAS-CCKLR-17D1-RNAi/+ naive, N = 4; UAS-CCKLR-17D1-RNAi/+ primed, N = 6; Crz-Gal4 > UAS-CCKLR-17D1-RNAi naive, N = 4; Crz-Gal4 > UAS-CCKLR-17D1-RNAi primed, N = 4; UAS-CCKLR-17D3-RNAi/+ naive, N = 4; UAS-CCKLR-17D3-RNAi/+ primed, N = 6; Crz-Gal4 > UAS-CCKLR-17D3-RNAi naive, N = 4; Crz-Gal4 > UAS-CCKLR-17D3-RNAi primed, N = 6. Each data point represents 10 flies. MRS, male reproductive system. Error bars represent ± SEM. Column A and column C didn’t pass the normality test (Shapiro-Wilk test). Column A vs column B and column C vs column D used a nonparametric test (two-tailed Mann-Whitney test). Other tests used a two-tailed unpaired t test, column A vs column B, p = 0.0286; column C vs column D, p = 0.019; column E vs column F, p = 0.0023; column G vs column H, p = 0.054. B The brain-gonad axis contributed to the emission of 20E and is composed of P1 neurons, Dsk neurons, CCKLR-17D3 in Crz neurons, and the male reproductive system. C Two of four VNC Crz neurons expressed CCKLR-17D3 (indicated by white arrows). Scale bars represent 100 μm. Green, GFP antibody; magenta, nc82 antibody; red, Crz antibody. D VNC Crz neurons responded to the DSK-expressed neurons’ activation (left). The calibration bar ranges from 0–177. Traces of fluorescence intensity changes after ATP or AHL adding (right). The dotted line indicates the time when we added ATP to the chamber. The quantitation of the fluorescence changes in Crz neurons (middle). Scale bars represent 50 μm. + AHL, N = 7; + ATP, N = 9. Error bars represent ± SEM. Column A didn’t pass the normality test (Shapiro-Wilk test). Column A vs column B used a nonparametric test (two-tailed Mann-Whitney test), p = 0.0002. ns, not significant. *p < 0.05. **p < 0.01, ***p < 0.001, ****p < 0.0001.
Males’ sexual arousal is correlated with their reproductive strategy
What’s the biological significance of the priming-induced mating drive elevation? Male animals display lower mating intensity toward aged females, probably due to the lower fecundity of those mates. It is conceivable that the mating drive levels of males are determined by the expectation for the reproductive outcome of their mates. It has been well demonstrated that aged females laid significantly fewer eggs and produced markedly fewer progeny than optimal mates32. Thus, we speculated that the increased mating drive of males to sub-optimal mates (aged females) may result from an increased anticipation of the reproductive outcome when mated with aged females. Indeed, we found that aged females mated with males who experienced 10 min priming laid more eggs on the first day after mating than those mated with naïve males (Fig. 6A and Supplementary Fig. S3A). Specifically, it primarily affects the initial phase of egg-laying after mating. This result suggests that multiple factors may influence female fecundity, and the increased mating plug from males induced by priming is just one of them. Nevertheless, as females lay the most eggs in the first day after mating and are more likely to mate with other males later on, this initial increase may be important for the number of this males’ offspring. Additionally, the larger mating plug may be beneficial for the males in a competitive environment where tandem matings usually occur.

A The total number of eggs laid by aged females was fewer than optimal females after mating. This defect could be partly rescued after they mated with primed males. Optimal, N = 11; Aged, N = 10; Aged (♂ primed), N = 11. Error bars represent ± SEM. Two-tailed unpaired t test, column A vs column B, p < 0.0001; column B vs column C, p = 0.0002. B Male flies transferred a similar number of sperms to optimal and aged females indicated by sperm counts in their uterus right after mating. N = 10. Error bars represent ± SEM. Two-tailed unpaired t test. C Sperms in aged females could not be stored at the maximal level 8 h after mating. N = 12. Error bars represent ± SEM. Two-tailed unpaired t test, p = 0.0095. D Confocal images revealed a smaller mating plug size in the uterus of aged females compared to optimal females. Scale bars represent 50 μm. Optimal, N = 23; Aged, N = 17. Error bars represent ± SEM. Two-tailed unpaired t test. E Aged females expelled the mating plug faster than optimal females. Optimal, N = 20; Aged, N = 18. Log-rank (mantel-Cox) test, p = 0.0150. F The mating plug size in aged females was increased after they mated with primed males. Optimal, N = 13; Aged, N = 15; Aged (Primed), N = 15. Error bars represent ± SEM. Two-tailed unpaired t test, column A vs column B, p < 0.0001; column B vs column C, p = 0.0014. G Mating plug size in aged female mice was smaller than in optimal females. However, the mating plug size was increased in aged female mice after their mates experienced priming. The scale bar represents 2.5 mm. H The weight of the mating plug was decreased in aged female mice compared with optimal female mice and was restored in aged female mice after their mates experienced priming. Optimal, N = 11; Aged, N = 10; Aged (Primed), N = 7. Error bars represent ± SEM. Two-tailed unpaired t test, column A vs column B, p = 0.0117; column B vs column C, p = 0.0906. ns, not significant. *p < 0.05. **p < 0.01, ***p < 0.001, ****p < 0.0001.
To investigate how primed males enhanced the reproductive outcomes of the sub-optimal mates, we quantified the different reproductive strategies of males between optimal and sub-optimal mates. We first examined the number of sperms transferred to the females during mating. By visualizing sperms using protamine-GFP, we observed that both optimal and aged females processed a similar number of sperms in their uterus immediately after ejaculation (Fig. 6B and Supplementary Fig. S3B), indicating that males transferred a similar number of sperms regardless of the states of their mates. Interestingly, aged females stored fewer sperms in their seminal receptacle compared with optimal females 8 h after mating, which may account for the reduced egg number (Fig. 6C and Supplementary Fig. S3C).
Males with priming invest more mating plug in sub-optimal mates
Despite around a thousand sperms being transferred by the males during mating, as few as 30-40% of them are stored in mated females33. The mating plug from the ejaculates acts as a barrier to prevent sperm loss and facilitate sperm storage in mated females34,35. Mating plugs undergo a phase transition in the female reproductive tract to form a barrier against sperm loss35. In addition, mating plugs enhance sperm competitiveness by subsequent competing males36,37. The mating plug’s comprehensive functionality prompts questions about its impact on sperm movement and its activation of reproductive tract stretch receptors.
Given that the sperm number transferred to the aged females was not reduced, we reasoned that a defective mating plug may cause sperm loss after mating in the aged females. The mating plug can be visualized with PEBme, a major protein component with autofluorescence under UV light34. We observed that the mating plug in the uterus of aged females is small after ejaculation (Fig. 6D), suggesting that males reduce their allocation of the mating plug to the aged females. Mated females expel both the mating plug and the majority of sperm from their uterus several hours after mating33. We found that the aged females ejected the mating plug faster than the optimal females (Fig. 6E), likely due to the small size of the mating plug. Thus, males make different reproductive investment strategies between optimal and sub-optimal mates by adjusting mating plug allocation. We then found that males after priming 10 min allocated more mating plug to the aged females (Fig. 6F), indicating that males increase their reproductive investment to aged females when they are at high mating drive state.
In parallel, we observed a smaller size mating plug in aged female mice after mating with a naïve male (Fig. 6G). Quantifying mating plug weight, the mating plug in aged female mice is lighter than in optimal female mice (Fig. 6H). In addition, the weight of the mating plug is not correlated with the weight of the female or the male, or the female receptivity index (Fig. S3D–F). Then, we observed that male mice experienced priming increased their mating plug allocation to aged females (Fig. 6G, H). Taken together, we can conclude that males with priming invest more mating plug in sub-optimal mates across taxa, making males less picky.
Ejaculatory bulb emission contributes to mating plug allocation
To study how the male mating drive affects mating plug allocation, we sought to elucidate the neural basis underlying mating plug emission during sexual priming. The mating plug is mainly composed of proteins secreted by the ejaculatory bulb (EB) of the male reproductive system. We proposed that sexual priming triggers the emission of EB, e.g., proteins secreted to the lumen, through the brain-gonad axis, and the increase of mating plug allocation on sub-optimal mates avoids the waste of prepared reproductive materials. To test it, we dissected the male flies and examined the EB size after they were primed for 10 min (Fig. 7A). The EB size of the primed males was larger than the naive males (Fig. 7B), due to the accumulation of mating plug proteins in the EB lumen during courtship. These results suggest that mating plug proteins are secreted to the EB lumen before copulation, a process resembling the emission of mammals, e.g., the preparation of the contents in the seminal vesicles38. Thus, primed males increase their mating plug allocation to aged females to avoid wasting prepared reproductive materials.

A The ejaculatory bulb (EB) size was measured after males experienced 10 min priming. B The projected area of EB in w1118 males who experienced 10 min priming was larger than that in unmated w1118 males. N = 20. Error bars represent ± SEM. Two-tailed unpaired t test, p < 0.0001. Scale bars represent 100 μm. C Activation of P1 neurons using dTRPA1 could induce EB enlargement. R71G01-Gal4/+, 22 °C, N = 10; UAS-dTrpA1/+, 22 °C, N = 11; R71G01-Gal4 > UAS-dTrpA1, 22 °C, N = 10; R71G01-Gal4/+, 30 °C, N = 10; UAS-dTrpA1/+, 30 °C, N = 13; R71G01-Gal4 > UAS-dTrpA1, 30 °C, N = 14. Error bars represent ± SEM. One-way ANOVA, p < 0.0001. D Silencing Crz neurons blocked the EB enlargement. Crz-Gal4/+ naive, N = 9; Crz-Gal4/+ primed, N = 10; UAS-Kir2.1/+ naive, N = 12; UAS-Kir2.1/+ primed, N = 10; Crz>Kir2.1 naive, N = 10; Crz>Kir2.1 primed, N = 11. Error bars represent ± SEM. Two-tailed unpaired t test, column A vs column B, p < 0.0001; column C vs column D, p < 0.0001. E The Crz neurons in the brain were dispensable for EB enlargement during priming. Crz-Gal4/+ naive, N = 10; Crz-Gal4/+ primed, N = 7; tsh-Gal80/+;UAS-Kir2.1/+ naive N = 10; tsh-Gal80/+;UAS-Kir2.1/+ primed N = 8; Crz-Gal4,tsh-Gal80;UAS-Kir2.1 naive N = 10; Crz-Gal4,tsh-Gal80;UAS-Kir2.1 primed N = 10. Error bars represent ± SEM. Two-tailed unpaired t test, column A vs column B, p < 0.0001; column C vs column D, p < 0.0001; column E vs column F, p < 0.0001. F Knocking down CCKLR-17D3 but not CCKLR-17D1 in Crz neurons inhibited the mating plug allocation of males. Crz-Gal4/+ naive N = 20; UAS-CCKLR-17D1-RNAi/+ naive, N = 9; Crz-Gal4 > UAS-CCKLR-17D1-RNAi primed N = 10; Crz-Gal4 > UAS-CCKLR-17D3-RNAi naive, N = 10; Crz-Gal4 > UAS-CCKLR-17D3-RNAi primed, N = 10. Error bars represent ± SEM. Two-tailed unpaired t test, column D vs column E, p < 0.0001. ns, not significant. *p < 0.05. **p < 0.01, ***p < 0.001, ****p < 0.0001.
On the other hand, a prepared mating plug stored in males for a long time may be potentially harmful for males’ reproductive organs. To test this notion, males who experienced priming were single-housed, and their EB was examined. We observed significant protein precipitation and aggregation in EB 11 days after priming, compared with 1 day (Supplementary Fig. S4A, B). The major component of the mating plug is PEBme, which is transferred from a liquid state in males to a gel-like state in mated females39. We then observed that purified PEBme precipitates and aggregates in vitro with increasing concentrations ranging from 5 μM to 60 μM (Supplementary Fig. S4C), indicating that PEBme may form aggregation when stored in EB for a long time. We thus proposed that high concentration PEBme may form precipitation and aggregation inside the lumen of males’ EB, which would block emission and ejaculation. By expelling the prepared mating plug, primed male flies and mice may be able to maintain the normal function of the EB.
The brain-gonad axis coordinates male reproductive investment
We then investigated whether the P1-DSK-Crz pathway also controls EB emission. Males with P1 neurons blocked failed to expand their EB after they were primed with optimal females (Supplementary Fig. S5A). Furthermore, activating P1 neurons with dTPRA1 for 10 min could induce EB enlargement without any sexual experience (Fig. 7C). These results indicate that P1 neurons control the EB emission to modulate males’ reproductive material allocation.
We found that CrzR neurons projected to the male reproductive system with elaborate arborizations (Supplementary Fig. S5B), suggesting that Crz neurons may also trigger EB enlargement during sexual priming. We found that silencing Crz neurons in the VNC blocked EB enlargement during priming (Fig. 7D). To exclude the effect of Crz neurons in EB enlargement, we used tsh-gal80 specifically labeled Crz neurons in the brain and we found these neurons are not involved in EB enlargement during courtship behavior (Fig. 7C). Meanwhile, CCKLR-17D3 but not CCKLR-17D1 in Crz neurons regulates males’ mating plug allocation (Fig. 7F). The reduced mating plug size of ΔCCKLR-17D3 mutant males could be rescued by expressing CCKLR-17D3 with Crz-GAL4 (Supplementary Fig. S5C). Thus, the brain-gonad axis controls EB enlargement during sexual priming via the P1-DSK-Crz pathway. This pathway conveys prior sexual experience into a stronger mating drive to boost reproductive output and probably also to avoid the waste of the prepared mating plug materials.
Discussion
In this study, we show that sex hormones released during sexual arousal/priming regulate male mate preference and make males less picky about mates, likely due to the elevated mating drive state. This highlights the crucial roles of hormones in shaping male mating behavior. In Drosophila, priming-induced modification of mate preference was recapitulated by selective activation of P1 neurons through 20E. 20E is a steroid hormone which can freely diffuse through the membrane of their target cells (Nielsen & Herrera, 2017) and function in the transmembrane-spanning G-protein coupled receptor DopEcR, rapidly activating neurons via intracellular Ca2+ and cAMP signaling (H. Ishimoto et al., 2013). This mechanism suffices a rapid regulation of male sexual behavior and is crucial for males to immediately adjust their behavior after they enter into an elevated mating drive state. Notably, in both primed male flies and mice, neither the courtship latency index nor mating intensity toward optimal females appears to increase. This may be because the one‑on‑one mating assay fails to detect the elevated mating drive toward optimal females. This limitation can be overcome through a more detailed analysis of mating behaviors or alternative experimental approaches.
This study unveils the neural mechanisms of top-down regulation from the nervous system to the reproductive system. In male flies, the nervous system integrates female cues, prompting the release of hormones from the reproductive system and the preparation of reproductive materials. This communication is mediated by both neural circuits and neuropeptides. However, the circuitry underlying brain-gonad communication in mammals remains unclear. In male mice, the medial preoptic area (MPOA) within the hypothalamus emerges as a central hub for controlling mating behavior, akin to the role of P1 neurons in fruit flies6,11. This integrative role of the MPOA or other sexual-related brain areas emphasizes the conserved logic in the control of sexual behaviors across species.
Additionally, the spinal nucleus of the bulbocavernosus (SNB) neuromuscular system in male mice is responsible for mediating ejaculation reflexes40, which likely serves a conserved function in the vertebrate central nervous system. Its functional analogy to the Crz/CrzR system in control of emission and ejaculation in flies, suggests a shared evolutionary origin. Thus, we propose there likely exist MPOA-SNB neural circuits, playing a crucial role not only in controlling testosterone emission but also in orchestrating the preparation of reproductive materials.
Owing to the limited reproductive materials and opportunities, a suitable reproductive strategy is crucial for polygamous males. Our observations suggest that males tend to deposit larger mating plug to optimal females with high fecundity, which raises a question: why do animals use mating plug as investment substances instead of sperms? We can envision three possible reasons. 1, The mating plug plays an equally important role with sperms in reproduction, including facilitating sperm storage and preventing immediate mating from competitors. These functions have been well demonstrated in Drosophila and mice33,34,37. In addition, the semen of humans becomes thick and gelatinous after ejaculation and eventually liquefies to enable the sperms to swim better41. This gel-like structure resembles the same function as the mating plug to promote pregnancy42. 2, The stored sperms in a male are often sufficient to support multiple mating in a short period, while the mating plug depletes rapidly and requires a longer time to replenish43. 3, The mating plug is energetically expensive. The mating plug in a single ejaculation of a mouse is around 0.03 ~ 0.05 g, roughly 0.1% of the body weight of an adult male. Similarly, the depleted EB contents can take 3-4 days to recover in fruit flies. Thus, the mating plug is rich in protein content, which could be a heavy burden for male animals engaged in active mating activity. Interestingly, a study in red-sided garter snakes (Thamnophis sirtalis parietalis) quantified the energetic expense of size-dependent ejaculate strategies and found that a single mating plug without sperm constitutes 5–18% of daily energy expenditure44. Taken together, these parallel studies across species suggest that the non-sperm components of an ejaculate, especially the mating plug, can be essential to male reproductive success and must be deliberately invested in mating.
Notably, the increased mating plug investment to old females induced by priming primarily affects the initial phase of egg-laying after mating, suggesting that female fecundity is likely influenced by multiple factors, with the priming-induced increase in mating plug size representing just one of them. Nevertheless, since females lay the majority of their eggs on the first day after mating and are more likely to remate thereafter, this early boost in egg output could significantly contribute to the reproductive success of the primed male. Moreover, a larger mating plug may offer a competitive advantage in environments where tandem matings are common.
In conclusion, our study not only sheds light on the intricate regulation of sexual states by hormones but also unravels the neural basis dialog between the nervous and reproductive systems in adult males. This multidimensional perspective contributes to a more nuanced understanding of male mate behavior by emphasizing the dynamic and adaptive nature of reproductive strategies.
Methods
We confirm that our research complies with all relevant ethical regulations. All experimental procedures in this study were performed in accordance with the Tsinghua University guidelines and animal protocols, which were approved by Tsinghua University Institutional Animal Care and Use Committee (IACUC).
Fly experiments
Animals
Fly strains utilized in this study were procured from the Bloomington Drosophila Stock Center (BDSC) or other reputable laboratories. Flies were maintained on a standard medium under controlled environmental conditions, including a temperature of 25 °C, 60% relative humidity, and a 12 h light–dark cycle. Exceptions were made for thermo-activation and thermo-inactivation experiments, during which flies were subjected to an 18 °C environment. w1118 was used as a wild-type control and aged female (35–45 d old). All assays were completed with 3–7 days flies except the RNAi experiment and rescue experiment, which used flies aged 10–16 days. The sub-optimal female refers to the aged female if not otherwise mentioned. All male flies were raised single-housed. For a comprehensive list of fly strains used in this study, please refer to the Supplementary Materials (see KRT).
Courtship and copulation assays
Courtship assays were conducted using cylindrical courtship chambers with dimensions of 10 mm in diameter and 3 mm in height. These chambers provided a controlled environment for observing courtship behaviors. The experiments were carried out at a temperature of 25 °C with 60% relative humidity. Unless otherwise noted, a male fly was paired with a w1118 virgin female fly in each chamber. The courtship assays were videotaped to allow for detailed analysis and manual scoring of courtship behaviors. The courtship index was manually scored as the fraction of time during which the male fly exhibited courtship behaviors. These behaviors included orientation, following, single-wing vibration, attempted copulation, and copulation. The scoring was performed within a 5 min window, initiated once the male fly commenced courtship activities.
Priming assays
Before the assays, the female flies were anesthetized using CO2 to facilitate handling. The genitalia of the female flies were carefully sealed using nail polish. This procedure aimed to prevent copulation during the primed-courtship assays. Following genital sealing, the female flies were allowed a recovery period of 2 h and this recovery time took place in individual food tubes. After the recovery period, the prepared female flies were utilized in 10 min priming assay. During these assays, individual male flies were introduced to the chambers.
Courtship competition assay
Courtship competition assay was carried out in a chamber with dimensions of 10 mm in diameter and 3 mm in height. All w1118 female flies were collected within 2 h after eclosion and housed in groups. Male flies were collected and singly housed right after eclosion. Female flies aged for 4–6 days were briefly anesthetized on ice and introduced into the recording chamber to recover for at least 2 h. Two competing male flies aged for 4-13 days were introduced into the chamber simultaneously. To identify each genotype, the flies in either the control group or experimental group were marked with a dot on their wings.
20E ELISA
The hemolymph samples for each experiment were collected from 20 adult flies. 20E are extracted by adding 3 volumes of chilled methanol (75% of the total volume) and vortexing for 30 s. The solution was centrifuged at 9391 × g for 10 min at 4 °C. Supernatant was carefully aspirated without disturbing the pellet, and the pellet was dried down completely using a centrifugal concentrator at 30 °C for 2-3 hrs. The dried powder/pellet can be stored at − 20 °C until assayed or can be dissolved in a minimum of 125 μL ELISA buffer to immediate quantification. A 50 μL sample was used to detect the levels of 20E using an enzyme immunoassay kit (ACE Enzyme Immunoassay).
The central neural system, gut, and reproductive system samples were collected from 10 adult flies. The dissection was performed in a dissection buffer (PBST: 0.015% Triton X-100 in 1x PBS. The collected samples were ground in methanol at 4 °C to extract 20-hydroxyecdysone (20E), a hormone. The grinding process was facilitated by sonication, performed five times for five seconds each at a frequency of 20 KHz. Following sonication, the samples were centrifuged at 10,000 × g for 20 min at 4 °C. The resulting supernatant, containing the extracted 20E, was carefully collected. The collected supernatant was subjected to evaporation until complete dryness. The dried pellet was reconstituted in 125 μL of ELISA buffer. This step aimed to ensure the solubility of the extracted 20E for subsequent analysis. The pellet was dissolved in 125 μL ELISA buffer. A 50 μL sample was used to detect the levels of 20E using an enzyme immunoassay kit (ACE Enzyme Immunoassay).
20E administration
For 20E supply, flies first underwent a starvation period lasting 4 to 6 h. A 25 mM stock solution of 20E in ethanol was prepared for use. The final concentration used for administration was 3 mM. This concentration was achieved by diluting the 25 mM stock solution appropriately. The ethanol served as a solvent for 20E. Following the starvation period, flies were provided with the 3 mM 20E solution for 5 h.
Fecundity (egg-laying) assays
Each female fly was paired with a male in a food vial. The mating pairs were allowed to interact for a duration of 2 h. Mated females were subsequently identified by the presence of a mating plug. Confirmed mated females were then transferred onto a grape juice plate. The grape juice medium was prepared by dissolving 0.25% agarose in grape juice, resulting in a final concentration of 15% (v/v) agarose. This grape juice plate served as the substrate for egg laying. The mated females were given 24 h for egg laying on the grape juice plate. After this period, the total number of eggs laid by five mated females was counted. This count provided a measure of reproductive output within the given timeframe.
Each female fly was paired with a male in a food vial for a duration of 2 h. Mated females were identified by the presence of a mating plug. Mated females were then transferred onto a new food vial. This food vial served as the substrate for egg laying. The food vial was changed every day. The number of eggs laid by each mated female was counted for 10 days.
Sperm counts
Female flies were allowed to mate with protamine-GFP males. The use of protamine-GFP males facilitated the visualization and identification of sperm cells through green fluorescent protein (GFP) labeling. Post-mating, the female reproductive tract and seminal receptacle were carefully dissected in a dissection buffer (PBST: 0.015% Triton X-100 in 1x PBS). The dissected female reproductive tract and seminal receptacle were mounted between two pieces of cover glass with two spacers of 0.18 mm. Confocal image stacks were acquired to visualize and manually count GFP-positive sperm heads. Two specific time points were considered for analysis: immediately after ejaculation to assess the number of sperms transported to the uterus, and 8 h post-mating to examine sperm storage in the seminal receptacle. It was noted that confocal imaging might not capture every sperm, particularly in deeper optical Z-sections, due to the thickness of the uterus. As a result, the sperm counts obtained from this procedure were consistent between samples but may be underestimated compared to previous reports.
Mating plug size and retention time measurement
To assess the size of the mating plug in the female uterus, the following steps were taken. Female reproductive tracts were dissected 2 h after ejaculation in a dissection buffer (PBST: 0.015% Triton X-100 in 1x PBS). The dissected female reproductive tracts were mounted between two pieces of cover glass with two spacers of 0.18 mm. Confocal images were acquired at 405 nm/440 nm excitation/emission to detect the auto-fluorescence of PEBme in the mating plug. This choice of wavelength allowed for the visualization of specific components within the mating plug. ImageJ, a widely used image analysis tool, was employed to analyze the projected area of fluorescence particles. This analysis provided quantitative data on the size of the mating plug.
For mating plug retention time measurement, UV light was used to detect the presence of mating plugs every half hour after ejaculation.
EB size measurement
EB also includes PEBme and auto-fluorescence, the secretions transferred to the female are stored in the lumen of EB. We used the same method described in the mating plug size measurement to dissect EB and analyze the projected area of the lumen in EB.
Thermo-genetics
Flies utilized in heat activation or inactivation experiments were raised in a controlled incubator set to a temperature of 18 °C. All experiments, specifically heat activation or inactivation studies, were conducted at 30 °C. This elevated temperature was chosen to induce specific physiological responses or conditions in the experimental flies.
Tissue dissection, staining, and imaging
Brains and VNC of 5–7 d old flies were dissected in dissection buffer (PBST: 0.015% triton X-100 in 1x PBS) and fixed in 4% PFA at room temperature on a shaker for 30 min, then washed for 4 × 15 min in wash buffer (0.3% triton in 1x PBS). After this, the tissues were blocked in block buffer (1x heat-inactivated normal goat serum with 0.3% triton in 1x PBS) for 30 min at room temperature. The samples were then incubated with the primary antibody at 4 °C overnight. The primary antibodies used were: Rabbit-GFP (1:500 dilution), Mouse-GFP (1:500 dilution), Chicken-GFP (1:500 dilution), and Mouse-nc82 (1:50 dilution). On the second day, tissues were washed for 4 × 15 min and then incubated in secondary antibodies. The secondary antibodies were all from Invitrogen and used at 1:200 dilution: Alexa Fluor 555 anti-Rabbit (1:200 dilution), Alexa Fluor 488 anti-Mouse (1:200 dilution), Alexa Fluor 647 anti-Mouse (1:200 dilution). After incubation, tissues were washed for 4 × 15 min. VNC and brains were mounted on a slide for imaging. An Olympus FV1000 microscope with 10X air lens or 20X air lens was used for confocal imaging.
Calcium imaging
The male flies were raised single-housed for 7 to 11 days, and the entire brain and VNC were dissected in a recording chamber filled with the AHL (103 mM NaCl, 3 mM KCl, 5 mM TES, 10 mM trehalose, 10 mM glucose, 26 mM NaHCO3, 1 mM NaH2PO4, and 4 mM MgCl2, pH 7.25, 310 mOsm). 2 mM CaCl2 was added to the saline before use. Calcium imaging was performed using an Olympus BX51WI microscope with a 40 × water immersion objective, an Andor Zyla camera, and a Uniblitz shutter. The Ca2+ indicator GCaMP7s was used to measure the Ca2+ signal. GCaMP7s was excited with 488 nm light, and the fluorescent signals were collected at 1 Hz. For P2X2 stimulation, 10 μl of 0.2 M ATP solution or AHL was loaded into the recording chamber to reach the final concentration of 1 mM. ROIs were manually selected with ImageJ. The fluorescent change was calculated as %Peak ΔF/F0 = (Fpeak − F0)/F0 × 100, where F0 corresponds to the average intensity of 20 frames of background-subtracted baseline fluorescence before ATP application and Fpeak corresponds to the highest fluorescence after ATP application.
For monitoring the activity of P1 neurons, brains were prepared as described above. Images were acquired on an Olympus FV1000 confocal microscope using a 20X Air lens. GCaMP7s signals were excited with 488 nm light, and green fluorescent signals were collected. All operation was automatically controlled by the FV10-ASW 3.0 software. Selected regions of interest (ROIs) of P1 neurons using the tdTomato channel and the GCaMP7s channel. ROI sizes were kept roughly the same between samples. The average fluorescence in each ROI was calculated with ImageJ. Normalized fluorescence was calculated as GCaMP7s/tdTomato.
For 20E stimulation, 20E dissolved in DMSO or AHL was loaded into the recording chamber to reach the final concentration of 10−1 nM to 10−3 nM. ROIs of P1 neurons were manually selected with ImageJ. The fluorescent change was calculated as %Peak ΔF/F0 = (Fpeak − F0)/F0 × 100, where F0 corresponds to the average intensity of 20 frames of background-subtracted baseline fluorescence before 20E application, and Fpeak corresponds to the highest fluorescence after 20E application.
PEBme expression and purification
The coding sequence of PEBme was amplified from Drosophila melanogaster and cloned into an expression vector pET-28b with 6 × His tag. BL21 cells were transformed with the expression plasmids and plated on Ampicillin LB plates. Single clones were selected, sub-cultured in median volume flasks, and transferred to larger ones in LB medium with agitation on an incubated shaker (180 rpm at 37 °C) until they reached an OD600 of 0.8. Then 0.5 M IPTG was added to induce protein expression, and culture continued at 220 rpm for 4 h.
Bacteria were centrifuged at 1503 × g and lysed in Native Lysis Buffer (150 mM NaCl, 20 mM Tris-HCl, pH = 7.5, 1 mM PMSF, Protein Inhibitor Cocktail 1:1000) and sonicated for 30 min (10 s on/ 10 s off, 180 W). After centrifugation at 20,000 × g for 40 min, the pellet was collected and resuspended in Denaturing Lysis Buffer (6 M Urea, 150 mM NaCl, 20 mM Tris-HCl, pH = 7.5, 1 mM PMSF, Protein Inhibitor Cocktail 1:1000). Purification of His-tagged PEBme was performed by Ni-affinity chromatography. After centrifugation at 20,000 × g, the supernatant was collected. Purification of His-tagged PEBme was then performed by Ni-affinity chromatography. The purified proteins were further concentrated to 300 μM using centrifugal filters.
In vitro phase transition assays
The PEBme was diluted in a pH 7.5 buffer solution, with concentrations ranging from 5 μM to 60 μM. The phase transition products were added to glass-bottom 384-well plates and settled at room temperature for 30 min. Images were captured using an Olympus IXplore spinning disk confocal microscope.
Mice experiments
Animals
C57BL/6 J mice were used for all studies. All care and experimental procedures involving mice were conducted following the approval of the Laboratory Animal Resources Center of Tsinghua University and Beijing Vital River Laboratory Animal Technology Co., Ltd. Ethical considerations were prioritized to ensure the well-being and humane treatment of the animals. Male and female mice of 8 weeks old were used in the experiments. Aged female mice between 25 to 27 weeks were used. Mice were housed in a controlled environment with a 12-h light. and 12 h darkness cycle. The temperature was maintained between 20–22 °C, and humidity levels were kept within the range of 30–70%. Standard mouse chow (Experimental mouse breeding feed with 6% fat content. Other ingredients are similar to those of 5K52 used by JAX) and water (Reverse Osmosis water, sterile) were provided ad libitum unless otherwise specified. Male mice were single-housed in their home cage, while female mice were group-housed, accommodating up to five individuals. Mice were euthanized by carbon dioxide inhalation in accordance with institutional guidelines.
Mice behavioral experiments and data analysis
All behavioral experiments were conducted 1 h after the onset of the dark cycle in a new cage. Before tests, mice were habituated in the behavior recording room for 1-2 h. Behavioral experiments were video-recorded using a Logitech camera for 30 min at a frame rate of 30 per second.
Male-female mating assays
Male mice were single-housed for at least 1 week. Female mice were estrous females, typically housed in groups, and were selected for high receptivity. Before the experiment, female mice were introduced into a new cage, and male mice were introduced into the cage after approximately one hour.
Sexual priming behavior assays
Individually housed male mice were primed by an estrous female before the assay. In the priming-mating assay, males were allowed to interact with females for up to 5 times intromission, and were separated before ejaculation. Intromissions longer than 15 s were interrupted to prevent early ejaculation. Behaviors of the primed male mice with another estrous female were recorded.
Testosterone injection mating behavior assays
Each male mouse was subcutaneously injected with 500 ng testosterone (dissolved in ethanol and diluted with corn oil) in the back, with a 30 min rest. Behaviors of the testosterone-injected mice when exposed to estrous female mice were recorded.
Data analysis
We used BORIS to analyze male mice mating behavior, including sniffing, mounting, intromission, and ejaculation. The onset of a bout of sniffing was defined as the moment when the male’s nose touched any part of the female’s body and culminated when the male stopped making contact with the female. The onset of a bout of mounting was defined as the moment when the male climbed over a female and began rapid, shallow lower-body thrusting, and terminated when the male was no longer over the female. During a bout of mounting, the male could gain intromission, which was evident from the male’s thrusting movements becoming deeper and slower. The ejaculation was marked by a specific series of events in which an intromitted male began to shake for a few seconds, then fell on his side, then became temporarily immobilized even when the female moved away, and then stood up again to clean himself.
To measure a male’s latency to mounting, we calculated the time period between male entry and the first attempt to mounting.
To measure a male’s mating intensity, we calculated the fraction of time that a male spent sniffing, mounting, and intromission behavior in the ejaculation latency.
To measure a male’s female receptivity index, we calculated the fraction of time that a male spent intromission behavior in mounting behavior.
Weighing mice mating plug
After each mating session, which lasted for 3–8 h, female mice were selected for further analysis. Female mice were anesthetized using avertin at a dosage of 0.15 mL per gram of body weight. Avertin, a commonly used anesthetic, was administered to ensure the mice were under anesthesia during the mating plug retrieval process. The mating plug was obtained from the vagina of anesthetized female mice. To facilitate the retrieval, a blunt wire was used to gently remove the mating plug from the reproductive tract. The retrieved mating plug was weighed using a precise scale. After weighing, the mating plug was stored at 4 °C. This storage condition helps maintain the integrity of the mating plug for further analysis.
Testosterone ELISA
The testosterone from each mouse was extracted from 200 μl of serum. Firstly, 5 times the sample volume of diethyl ether was added and thoroughly mixed with a vortex. After allowing the layers to separate, 90% of the ether (upper layer) was transferred into a clean test tube using a pipette. This extraction process was repeated three more times. The combined ether extracts were then evaporated under a gentle stream of nitrogen. The resulting extract was dissolved in 500 μl of ELISA buffer. A 50 μL sample was used to measure the levels of testosterone using an enzyme immunoassay kit (ACE Enzyme Immunoassay).
Quantification and statistical analysis
GraphPad Prism 8 software was employed for statistical analysis. Error bars in all figures represent the Standard Error of the Mean (SEM). For all analyses, statistical notations are as follows: *p < 0.05. **p < 0.01, ***p < 0.001, ****p < 0.0001. No sample size estimation, inclusion and exclusion of any data or subjects were conducted in this study.
Normality Tests, Small Sample (n < 8): Shapiro-Wilk test was utilized for normality testing. Large Sample (n ≥ 8): D’Agostino & Pearson test was used for normality testing.
Comparison Tests, Parametric Datasets (Two Groups): Two-tailed unpaired t test was employed to evaluate significance. Nonparametric Datasets (Two Groups): The Mann-Whitney test was used for evaluating significance. Parametric Datasets (Three Groups): One-way ANOVA was employed for evaluating significance. Correlation Test: A correlation test was performed to assess the correlated degree between the weight of the mating plug and the weight of the female, male, or female receptivity index.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
We declare that all data supporting the findings are provided in the Source Data file. All data supporting the findings described in this manuscript are available in the article and in the Supplementary Information and from the corresponding author upon request. All figures were generated by the authors. No external image databases or third-party graphical resources were used. Source data are provided in this paper.
References
Bayless, D. W. et al. A neural circuit for male sexual behavior and reward. Cell 186, 3862–3881 (2023).
Yamamoto, D. & Koganezawa, M. Genes and circuits of courtship behavior in Drosophila males. Nat. Rev. Neurosci. 14, 681–692 (2013).
Ågmo, A. Neuroendocrinology of sexual behavior. Int. J. Impot. Res. 36, 305–311 (2024).
Maras, P. M. & Petrulis, A. Chemosensory and steroid-responsive regions of the medial amygdala regulate distinct aspects of opposite-sex odor preference in male Syrian hamsters. Eur. J. Neurosci. 24, 3541–3552 (2006).
Veening, J. G. & Coolen, L. M. Neural activation following sexual behavior in the male and female rat brain. Behav. Brain Res. 92, 181–193 (1998).
Hull, E. M. & Dominguez, J. M. Sexual behavior in male rodents. Horm. Behav. 52, 45–55 (2007).
Kwon, J.-T. et al. An amygdala circuit that suppresses social engagement. Nature 593, 114–118 (2021).
Pan, Y., Meissner, G. W. & Baker, B. S. Joint control of Drosophila male courtship behavior by motion cues and activation of male-specific P1 neurons. Proc. Natl. Acad. Sci. USA 109, 10065–10070 (2012).
Zhang, L., Guo, X. & Zhang, W. Nutrients and pheromones promote insulin release to inhibit courtship drive. Sci. Adv. 8, eabl6121 (2022).
Lin, H. H. et al. A nutrient-specific gut hormone arbitrates between courtship and feeding. Nature 602, 632–638 (2022).
Zhang, S. X. et al. Hypothalamic dopamine neurons motivate mating through persistent cAMP signalling. Nature 597, 245–249 (2021).
Fu, O. et al. Hypothalamic neuronal circuits regulating hunger-induced taste modification. Nat. Commun. 10, 4560 (2019).
Nielsen, S. E. & Herrera, A. Y. Sex steroids, learning and memory. In Hormones, Brain and Behavior (eds Pfaff, D. W. & Joëls, M.) 399–422 (Academic Press, 2017).
Borella, M. I. et al. The brain–pituitary–gonad axis and gametogenesis. In Biology and Physiology of Freshwater Neotropical Fish (eds Baldisserotto, B., Urbinati, E. C. & Cyrino, J. E. P.) 315–341 (Academic Press, London, 2020).
Maruska, K. P. & Fernald, R. D. Social regulation of gene expression in the hypothalamic–pituitary–gonadal axis. Physiology 26, 412–423 (2011).
Nyby, J. G. Reflexive testosterone release: a model system for studying the nongenomic effects of testosterone upon male behavior. Front. Neuroendocrinol. 29, 199–210 (2008).
van Anders, S. M. & Watson, N. V. Social neuroendocrinology: effects of social contexts and behaviors on sex steroids in humans. Hum. Nat. 17, 212–237 (2006).
Ishimoto, H., Sakai, T. & Kitamoto, T. Ecdysone signaling regulates the formation of long-term courtship memory in adult Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 106, 6381–6386 (2009).
Genazzani, A. R. et al. Androgens and the brain. Maturitas 57, 27–30 (2007).
Contreras-Zarate, M. J. & Cittelly, D. M. Sex steroid hormone function in the brain niche: implications for brain metastatic colonization and progression. Cancer Rep. 5, e1241 (2022).
Ortiz, J. B. et al. Puberty and traumatic brain injury. in Cellular, Molecular, Physiological, and Behavioral Aspects of Traumatic Brain Injury 351–362 (Elsevier, 2022).
Li, H. et al. Fly Cell Atlas: a single-nucleus transcriptomic atlas of the adult fruit fly. Science 375, eabk2432 (2022).
Kimura, K. -i. et al. Fruitless and Doublesex coordinate to generate male-specific neurons that can initiate courtship. Neuron 59, 759–769 (2008).
Andersson, M. & Iwasa, Y. Sexual selection. Trends Ecol. Evol. 11, 53–58 (1996).
Bond, N. D., Hoshizaki, D. K. & Gibbs, A. G. The role of 20-hydroxyecdysone signaling in Drosophila pupal metabolism. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 157, 398–404 (2010).
Freeman, M. R. et al. The dare gene: steroid hormone production, olfactory behavior, and neural degeneration in Drosophila. Development 126, 4591–4602 (1999).
Abrieux, A. et al. The GPCR membrane receptor DopEcR mediates the actions of both dopamine and ecdysone to control sex pheromone perception in an insect. Front. Behav. Neurosci. 8, 312 (2014).
Tayler, T. D. et al. A neuropeptide circuit that coordinates sperm transfer and copulation duration in Drosophila. Proc. Natl. Acad. Sci. USA 109, 20697–20702 (2012).
Wu, S. et al. Drosulfakinin signaling in fruitless circuitry antagonizes P1 neurons to regulate sexual arousal in Drosophila. Nat. Commun. 10, 4770 (2019).
Wu, F. et al. A neuropeptide regulates fighting behavior in Drosophila melanogaster. ELife 9, e54229 (2020).
Thornquist, S. C. et al. CaMKII measures the passage of time to coordinate behavior and motivational state. Neuron 105, 334–345 (2020).
Mossman, J. A. et al. Age of both parents influences reproduction and egg dumping behavior in Drosophila melanogaster. J. Hered. 110, 300–309 (2019).
Lee, K. M. et al. A neuronal pathway that controls sperm ejection and storage in female Drosophila. Curr. Biol. 25, 790–797 (2015).
Avila, F. W. et al. Retention of ejaculate by Drosophila melanogaster females requires the male-derived mating plug protein PEBme. Genetics 200, 1171–1179 (2015).
Avila, F. W. et al. Don’t pull the plug! The Drosophila mating plug preserves fertility. Fly 9, 62–67 (2015).
Datta, S. R. et al. The Drosophila pheromone cVA activates a sexually dimorphic neural circuit. Nature 452, 473–477 (2008).
Stockley, P. et al. Revealing mechanisms of mating plug function under sexual selection. Proc. Natl. Acad. Sci. USA 117, 27465–27473 (2020).
Mulhall, J. P. & Müller, A. Erection, emission, and ejaculation: mechanisms of control. In Infertility in the Male (eds Howards, S. S., Lipshultz, L. I. & Niederberger, C. S.) 132–152 (Cambridge Univ. Press, Cambridge, 2009).
Lung, O. & Wolfner, M. F. Identification and characterization of the major Drosophila melanogaster mating plug protein. Insect Biochem. Mol. Biol. 31, 543–551 (2001).
Raskin, K. et al. Characterization of the spinal nucleus of the bulbocavernosus neuromuscular system in male mice lacking androgen receptor in the nervous system. Endocrinology 153, 3376–3385 (2012).
Dorus, S. et al. Rate of molecular evolution of the seminal protein gene SEMG2 correlates with levels of female promiscuity. Nat. Genet. 36, 1326–1329 (2004).
Anamthathmakula, P. & Winuthayanon, W. Mechanism of semen liquefaction and its potential for a novel non-hormonal contraception. Biol. Reprod. 103, 411–426 (2020).
Zhang, S. X., Rogulja, D. & Crickmore, M. A. Dopaminergic circuitry underlying mating drive. Neuron 91, 168–181 (2016).
Friesen, C. R. et al. Size dependence in non-sperm ejaculate production is reflected in daily energy expenditure and resting metabolic rate. J. Exp. Biol. 218, 1410–1418 (2015).
Acknowledgements
We thank members of the Zhang lab for the discussions. This work was supported by grant 2021ZD0203303 from Ministry of Science and Technology, China, grants 31871059 and 32022029 from the National Natural Science Foundation of China, grant Z181100001518001 from the Beijing Municipal Science & Technology Commission, and a ‘Brain + X’ Seeds grant from the IDG/McGovern Institute for Brain Research at Tsinghua to W.Z. W.Z. is supported by the Chinese Institute for Brain Research, Beijing.
Author information
Authors and Affiliations
Contributions
K.L., Y.J., T.L., and H.S. performed the experiments and analyzed the data. Y.L. and W.Z. supervised the project. K.L. and W.Z. wrote the manuscript. All authors discussed and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Young Joon Kim, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, K., Jin, Y., Liu, T. et al. Brain-gonad axes harmonize male mating drive and reproductive strategy via steroid hormones. Nat Commun 17, 1553 (2026). https://doi.org/10.1038/s41467-025-68275-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-68275-2
