
Abstract
Age-related inflammation plays a pivotal role in osteoarthritis (OA) pathogenesis, but the mechanism is not fully understood. Here, we identify decreased IL-36 receptor antagonists (IL-36Ra) in epidermal keratinocytes from a premature-aged skin mice model, aged mice and patients. Decreased IL-36Ra leads to increased secretion of IL-36 agonists to serum and joints, which activates proinflammatory signaling and promotes senescence in chondrocytes and synovial fibroblasts, thereby aggravates OA progression. Deletion of IL-36Ra in keratinocytes exacerbates, whereas intra-articular inhibition of IL-36R signaling effectively attenuates OA progression in male mice. Moreover, we also generate microneedles loaded with mouse recombinant IL-36Ra protein or spesolimab, insert them directly into skin to sustainably inhibit IL-36R signaling, which both clearly attenuate OA progression in male mice. Overall, our results reveal that IL-36 agonists are age-related systemic inflammatory factors released from skin to joints and contribute to OA development, and targeting IL-36R signaling in aged skin with microneedles represents a promising disease-modifying approach.
Similar content being viewed by others

Introduction
Osteoarthritis (OA) is a prevalent, slowly degenerative joint disease that causes joint tenderness, stiffness, dysfunction, and pain in the elderly population, affecting quality of life and increasing mortality, with no available licensed disease-modifying treatments1. OA has long been considered as a “wear and tear” disease affecting articular cartilage, but current opinions suggest that inflammation plays a prominent role in OA pathogenesis, leading to a renewed definition of OA as a complex multifactorial joint pathology2. Inflammation in OA has unique characteristics compared to rheumatoid arthritis and other autoimmune diseases, including that it is chronic and associated with low-grade systemic and local inflammation, with involvement of both the innate and adaptive immune responses2,3,4. Research is increasingly uncovering how age-related systemic and local inflammation can contribute to OA joint damage5,6,7. Thus, given the pivotal role of low-grade inflammation in OA, future work is still needed to gain a deeper mechanistic understanding of the inflammation, identify novel targets, and develop effective disease-modifying drugs to prevent OA progression.
Skin is the largest organ of the human body, serving as the organismal barrier. However, the skin is fragile and possesses a senescent and inflammatory phenotype when continually exposed to external stressors such as ultraviolet rays and pollution8. The impairment of barrier function in the aged epidermis leads to inflammation of the skin and affects other systems throughout the body9. Recent studies have shown that epidermal barrier dysfunction in aged mice increases the levels of proinflammatory cytokines in the skin and serum, whereas correction of epidermal function in aged mice normalizes both10,11,12,13. The joints can be affected by systemic inflammation from skin disease; indeed, 20–30% of patients with psoriasis will develop psoriatic arthritis (PsA), while cytokine inhibition used to control psoriatic skin disease has been shown a protective effect on the risk of PsA, potentially providing a link between the transition of skin-to-joint disease14,15,16. However, how inflammation of the skin modulates the post-injury inflammatory response in OA has yet to be studied.
The IL-36 subfamily belongs to the IL-1 superfamily and consists of three activating ligands (IL-36α/β/γ), a receptor antagonist (named IL-36Ra in mice and IL-36RN in humans), and their common subfamily-specific receptors (IL-36R and IL-1RAcP). IL-36α/β/γ are agonizts that trigger IL-36R/MyD88 to activate proinflammatory signaling, including nuclear factor (NF)-κB, c-Jun N-terminal kinase (JNK), and extracellular signal-regulated kinase (ERK)−1/2 intracellular pathways, leading to the production and secretion of inflammatory cytokines, creating a feedback loop17. IL-36Ra mediates its antagonistic effect by binding IL-36R and inhibiting the recruitment of IL-1RAcP, thus truncating the relative signaling pathway. IL-36α/β/γ and IL-36Ra are physiologically present in skin and are mainly produced by keratinocytes, and all IL-36 agonizts have been found to synergize with IL-17 and TNF-α in keratinocytes18. IL-36 cytokines have been implicated in numerous inflammatory diseases, including psoriasis, inflammatory bowel diseases, allergic rhinitis, rheumatoid arthritis, and OA19,20. Recent studies have revealed that IL-36 blockade fails to ameliorate rheumatoid arthritis21,22; intra-articular injection of IL-36Ra or hydrogel can effectively control the inflammatory response, thereby protecting cartilage and slowing the development of OA in mice23,24,25. However, the origin and targeted cells of IL-36 cytokines involved in OA are not fully elucidated. In this study, we constructed skin aging models in male mice using two approaches: chemical (D-Gal) and genetic (Zmpste24 knockout), and these two specific models can reasonably reflect human skin aging26,27. We confirmed the link between aged skin and OA progression while demonstrated that aged skin keratinocytes are the main source of IL-36 agonizts, inflammatory mediators that promote OA progression. We also showed how best to target the inflammatory response with sustained-release of formulations, which represented a future perspective for clinical anti-inflammatory therapeutic interventions for OA.
Results
Premature skin aging promotes traumatic OA progression in mice
Age is the strongest risk factor for OA. To explore whether skin aging would affect the development of traumatic OA through skin-to-joint crosstalk, we constructed a mouse model of prematurely aged skin combined with DMM surgery-induced OA. First, we induced premature aged skin in C57BL6/J mice by cutaneous application of D-galactose (D-gal), which did not increase the systemic level of D-gal as we reported earlier (Supplementary Fig. 1a)28. DMM surgery was performed in these mice, and in one group of mice, the aged skin was removed at the same time as DMM surgery (Fig. 1a). Premature skin aging was successfully induced in D-gal-treated mice, which displayed a thinner epidermis, decreased cell numbers, and immunofluorescence staining revealed an increased number of p16-positive keratinocytes and a decreased Lamin B1 expression, both of which are markers of cellular senescence (Fig. 1b, c). We also observed more cartilage degradation, synovitis, subchondral bone sclerosis, and osteophyte development in D-gal-treated mice at 2 months after DMM surgery. Remarkably, compared to D-gal-treated mice, aged skin-removed mice displayed only a minor loss in proteoglycan content, with thicker cartilage, decreased synovitis, less osteophyte formation, and lower OARSI scores (Fig. 1d, e). Immunostaining showed increased expression of the catabolic marker metalloproteinase 13 (MMP13) and the fibrosis and lymphatic angiogenesis marker podoplanin (PDPN), along with decreased expression of the anabolic marker Collagen type II (Col2a1) in D-gal-treated mice, while the opposite was observed in skin-removed mice (Fig. 1f, g). Furthermore, the Von Frey filament test verified enhanced knee joint pain in D-gal-treated mice, which was recovered in skin-removed mice (Fig. 1h). Studies in aged mice have claimed that epidermal-barrier dysfunction increases the levels of proinflammatory cytokines in the skin and serum10. To further demonstrate the correlation between skin aging and OA, we established DMM-induced OA in C57BL/6 J mice and administered weekly intra-articular injections of serum from normal mice or mice with D-Gal cutaneous application (Fig. 1i). Two months after DMM surgery, cartilages from mice treated with serum from skin-aged mice displayed more severe cartilage erosion and clefts, whereas those from mice treated with serum from normal mice were comparable to controls (Fig. 1j, k). The Von Frey filament test confirmed consistent levels of pain in both model types (Fig. 1l). These data indicate that mice with premature skin aging may accelerate OA progression through serum components.

a Schematic illustrating the establishment of an OA model in control mice, mice with cutaneous application of D-galactose (D-gal), and mice with D-gal treated skin removed. b, c H&E staining (b) and quantification of epidermal thickness and cell numbers (c). Immunostaining (b) and quantification (c) of p16 and lamin B1 in skin sections from control and D-gal treated mice. Scale bar: 20 μm. d, e Safranin-O staining (d), scoring of OA parameters (e) in sham- or DMM-operated control, D-Gal-treated mice, or mice with D-Gal-treated skin removed at 8 weeks post-DMM surgery. The inset in the images is shown in the magnified images in the bottom row. Scale bar: 50 μm f–h. Immunostaining (f) and quantification (g) of MMP13, Collagen II, PDPN and quantitative analysis of the von Frey threshold (h) in joint sections from sham or DMM-operated control, D-Gal cutaneous applicated mice or mice with D-Gal treated skin removed at 8 weeks post DMM surgery. Scale bar, 50 μm. i Schematic of the experimental design to determine the effects of intra-articular injection serum from control (CS) or mice with D-Gal cutaneous applicated (DS) on OA progression and pain in C57BL/6 J mice. j–l Safranin-O staining (j), scoring of OA parameters (k) and quantitative analysis of von Frey threshold (l) in DMM-operated C57BL/6 J mice with intra-articular injection serum from control or mice with D-Gal cutaneous applicated. Scale bar: 50 μm; n = 6 (b, c) or 8 (d–l) mice per group, and results from one representative replicate are shown. Quantitative data are shown as the mean ± SD. P-values are from two-tailed unpaired t test (c), one-way ANOVA test followed by Tukey’s post hoc test (g, SBP thickness in e or k) and Kruskal–Wallis test followed by Dunn’s post hoc test (OARSI, synovial inflammation scores, osteophyte maturity scores in e or k, h, l). n indicates biological replicates; any technical repeats were averaged within each biological sample. Source data are provided as a Source Data file.
Next, we generated mice with Zmpste24 conditional knockout (Z24-cKO) in epidermal keratinocytes to induce skin premature aging via topical administration of capsid-mutant AAV vectors encoding Cre recombinase (AAV2-Cre) in Zmpste24fl/fl mice26. We confirmed high deletion efficiency of Zmpste24 in epidermal keratinocytes but not in other organs, and compared to control mice, the skin of Z24-cKO mice showed a thinner epidermis, decreased cell numbers, loss of rete ridges, and remarkedly increased senescent keratinocytes, manifested as an increase in p16 and a decrease in Lamin A/C expression, a nuclear envelope stability protein whose processing is impaired due to Zmpste24 knockout (Supplementary Fig. 1b–f). Zmpste24fl/fl and Z24-cKO mice were then subjected to DMM surgery to induce OA, as described in the schematic (Supplementary Fig. 2a). Interestingly, 2 months after DMM surgery, we noticed uneven cartilage surface and loss of proteoglycan content in Zmpste24fl/fl mice, whereas Z24-cKO mice displayed more severe cartilage erosion and even clefts. Z24-cKO mice also exhibited more severe synovitis, subchondral bone sclerosis, osteophyte development and enhanced knee joint pain (Supplementary Fig. 2b–d). The immunostaining results showed enhanced matrix MMP13 and PDPN and decreased Col2a1 expression in Z24-cKO mice, indicating a degenerative process in the cartilage and enhanced synovial inflammation (Supplementary Fig. 2e, f).
Together, our data confirm the connection between aged skin and traumatic OA and suggest that premature skin aging may play a crucial role in promoting the progression of traumatic OA in mice through skin–serum–joint crosstalk.
Decreased IL-36Ra in senescent keratinocytes results in increased systemic IL-36 agonizts and is involved in OA
To elucidate the underlying reasons of aged skin in promoting traumatic OA progression, we performed proteomic screen analysis with epidermal skin tissue from young (3 months old) and aged (18 months old) cutaneous application mice (Supplementary Table 1). As aged skin is fragile and possesses a senescent and inflammatory phenotype, a downregulated protein, IL-36Ra attracted our attention (Fig. 2a). IL-36Ra, a receptor antagonist of the IL-36 subfamily, suppresses inflammatory responses by competitively binding to IL-36R, and is mainly produced by keratinocytes in skin and was reported to be associated with the progression of OA24. In aged mice, the skin displayed a thinner epidermis, decreased cell numbers, and increased senescent keratinocytes (Fig. 2b, c), whereas joint cartilages showed fibrillation and loss of proteoglycan content (Fig. 2d). Immunostaining of the IL-36 family and co-staining with keratin 10 (K10), a type I keratin expressed in suprabasal keratinocytes of the skin, confirmed that IL-36Ra is specifically expressed in epidermal keratinocytes and decreased in senescent keratinocytes of aged skin. In contrast, IL-36 activating ligands (IL-36α/β/γ) are enhanced in senescent keratinocytes, serum, and joint tissues (Fig. 2e–h). We next observed a decrease in the expression of IL-36Ra and an increase in the expression of IL-36 activating ligands in D-Gal-treated mouse skin keratinocytes (Supplementary Fig. 3a–c). IL-36 activating ligands were elevated in the serum of D-Gal mice, but decreased after the removal of D-Gal from the skin (Supplementary Fig. 3d). Moreover, upregulated mRNA expression of IL-36 activating ligands was also detected in left joints from mice treated with D-Gal (Supplementary Fig. 3e). Decreased IL-36Ra in keratinocytes and increased IL-36 agonizts in keratinocytes, serum, and joint tissues were also observed in Z24-cKO mice compared to Zmpste24fl/fl mice (Supplementary Fig. 3f–i). Furthermore, we extracted and cultured primary keratinocytes from young mice and induced senescence by UVB irradiation. Immunostaining and western blot analysis confirmed successfully induced senescence, which was accompanied by decreased IL-36Ra in cultured keratinocytes (Supplementary Fig. 4a–c), and increased IL-36 agonizts in the supernatant liquor (SNL) via ELISA analysis (Supplementary Fig. 4d). Next, we collected epidermal skin from young patients without OA (35 ± 5 years old) and old patients with OA (70 ± 5 years old). Compared to young patients, an increased number of senescent keratinocytes in the skin epidermis and enhanced degradation of proteoglycan content in cartilage were observed in aged patients (Fig. 2i–k). In addition, diminished expression of IL-36RN in keratinocytes and elevated expression of IL-36 agonizts in keratinocytes and serum were detected in aged patients, as we detected previously with cytokine antibody array28 (Fig. 2l–n and Supplementary Fig. 4e). We also found positive correlations of elevated expression of IL-36 agonizts between serum and synovial fluid in aged OA patients, with IL-36α concentrations 359.27 ± 21.78 pg/mL in serum and 337.01 ± 15.36 pg/mL in synovial fluid, IL-36β concentrations 376.32 ± 18.83 pg/mL in serum and 361.26 ± 18.97 pg/mL in synovial fluid, IL-36γ concentrations 546.15 ± 35.58 pg/mL in serum and 535.12 ± 10.25 pg/mL in synovial fluid. The determination coefficients (R²) of IL-36α, β, and γ were 0.4716, 0.4660, and 0.3210, respectively (Fig. 2o). After inducing senescence in human keratinocyte HaCat cells with UVB irradiation, we observed decreased IL-36RN and increased IL-36 agonizts in the SNL (Supplementary Fig. 4f–i). These data verified that decreased IL-36Ra in senescent keratinocytes from aged skin results in increased systemic IL-36 agonizts and may be involved in OA.

a Heatmap of partial differentially present proteins (> 1.5-fold, P < 0.05) identified by proteomic screen analysis in epidermal skin tissue from young (3 month) and aged (18 month) mice. b, c H&E staining, immunostaining of p16 and lamin B1 (b) and quantification of epidermal thickness and numbers of p16- and lamin B1-positive cells (c) in skin sections from young and aged mice. Scale bar: 20 μm. d Safranin-O staining and OARSI scores in joint tissues from young and aged mice. Scale bar: 50 μm. e, f Immunostaining (e) and quantification (f) of K10 with IL-36Ra or IL-36α/β/γ in epidermal skin from young and aged mice. Scale bar: 20 μm. g Quantified ELISA analysis results of IL-36α/β/γ cytokines in serum from young and aged mice. h Quantified RT-qPCR analysis of IL-36α/β/γ mRNA in joint tissues from young and aged mice. i, j H&E staining, immunostaining of p16 and lamin B1 (i), and quantification of epidermal thickness and numbers of p16- and lamin B1-positive cells (j) in skin sections from young and aged male patients. Scale bar: 50 μm. k Safranin-O staining and OARSI scores in joint sections from young and aged male patients. Scale bar: 100 μm. l, m Immunostaining (l) and quantification (m) of K10 with IL-36Ra or IL-36α/β/γ in epidermal skin from young and aged male patients. Scale bar: 50 μm. n Quantified ELISA analysis results of IL-36α/β/γ cytokines in serum from young and aged male patients. o Elisa correlation analysis of serum and synovial fluid. n = 6 (b–g) or 3(h) mice per group, n = 6 (i–j, l–m) patients per group, or n = 6 (Young) or 9 (Aged OA) (k) patients per group, or n = 5 (n) or n = 15 (o) patients per group. Results from one representative replicate are shown. Quantitative data are shown as mean ± SD. P-values are from two-tailed Mann–Whitney U-test (d, k) and two-tailed unpaired t test (c, f, g, h, j, m, n). R² and exact P-values (for the regression slope) are from two-sided simple linear regression (o). n indicates biological replicates; any technical repeats were averaged within each biological sample. Source data are provided as a Source Data file.
IL-36 isoforms are widely expressed in many tissues17. To determine whether skin is the predominant site of IL-36Ra expression and whether the expression is changed with aging, we detected IL-36Ra expression in all reported tissues from young and aged mice. IL-36Ra expression was observed in almost all tissues and organs we analyzed, showing the highest expression in epidermal skin in young mice (Supplementary Fig. 5a). In aged mice, its expression was increased in the spleen but markedly decreased in the epidermal skin (Supplementary Fig. 5b, c). We also noticed lower expression of IL-36Ra in the cartilage and synovium tissues from aged mice, as reported previously. Further analysis with tissues from more age groups revealed that the reduction in IL-36Ra occurred earlier in the skin than in the articular cartilage and synovium (Supplementary Fig. 5d, e). In addition, skin has greater volume and weight than other tissues. We therefore conclude that decreased IL-36Ra in senescent keratinocytes from aged skin is responsible for most systemic increases in IL-36 agonizts and is involved in OA progression.
IL-36Ra deletion in the epidermis keratinocytes activates proinflammatory signaling in vivo
Next, we generated epidermis keratinocyte conditional knockout mice (IL-36Ra-cKO) with topical administration of capsid-mutant Adeno-associated virus 2 (AAV2) vector23 encoding Cre recombinase (AAV2-Cre) to IL-36Rafl/fl mice. Pathological histology staining showed that the epidermis did not differ significantly between young IL-36Rafl/fl and IL-36Ra-cKO mice (Fig. 3a, b). Effective deletion of IL-36Ra was confirmed in epidermal keratinocytes but not in other internal organs of IL-36Ra-cKO mice via immunostaining and western blot analysis, and enhanced IL-36 agonizts were observed in keratinocytes of IL-36Ra-cKO mice (Fig. 3c, d and Supplementary Fig. 6a–c). Moreover, the expression level of IL-36 agonizts in serum and joint sections from IL-36Ra-cKO mice was significantly increased compared to that in IL-36Rafl/fl mice (Fig. 3e, f), confirming that deletion of IL-36Ra in keratinocytes was responsible for systemic increase in IL-36 agonizts. Subsequently, epidermal skin tissues were collected from IL-36Rafl/fl and IL-36Ra-cKO mice for RNA sequencing analysis. KEGG pathway enrichment and gene set enrichment analysis (GSEA) showed that, compared to IL-36Rafl/fl mice, IL-36Ra conditional knockout in keratinocytes resulted in activation of downstream pathways, including NF-κB, MAPK and JAK-STAT signaling, as previously reported (Fig. 3g–i). We noticed elevated expression of key factors in these pathways, including p-p65, p-STAT3, p-p38, p-JNK and p-ERK1/2 in epidermis keratinocytes of IL-36Ra-cKO mice by immunostaining (Fig. 3j, k), confirming that ablation of IL-36Ra results in the activation of proinflammatory signaling in the epidermis. Further, we also detected activated NF-κB, MAPK and JAK-STAT signaling in cartilage and synovium from IL-36Ra-cKO mice, compared with IL-36Rafl/fl mice (Fig. 3l, m). Afterall, these data demonstrate that IL-36Ra deletion in the epidermis keratinocytes increases systemic IL-36 agonizts and results in activation of proinflammatory signaling in skin and joint, including NF-κB/JAK-STAT/MAPK pathways.

a, b H&E staining and quantification of epidermal thickness and cell numbers in skin sections from IL-36Rafl/fl and IL-36Ra-cKO mice at 12 weeks of age. Scale bar: 20 μm. c–f Immunostaining (c) and quantification (d) of K10 with IL-36Ra or IL-36α/β/γ in epidermal skin. Quantified ELISA results of IL-36α/β/γ cytokines in serum (e) and quantified RT-qPCR analysis of IL-36α/β/γ mRNA in joint tissues (f) from IL-36Rafl/fl and IL-36Ra-cKO mice at 12 weeks of age. Scale bar: 20 μm. g–i. Volcano plot showing differentially-expressed genes (DEGs) (g) KEGG pathway (h) and GSEA analysis (i) for DRGs demonstrating NF-κB, JAK-STAT and MAPK signaling pathway enrichment from RNA-sequencing of epidermis skin tissues from IL-36Rafl/fl and IL-36Ra-cKO mice. j, k Immunostaining (j) and quantification (k) of p-p65, p-STAT3, p-p38, p-JNK, p-ERK1/2 in epidermis skin tissues from IL-36Rafl/fl and IL-36Ra-cKO mice at 12 weeks of age. Scale bar: 50 μm. l, m Immunostaining(l) and quantification (m) of p-p65, p-STAT3, p-p38, p-JNK, p-ERK1/2 in cartilages and synovium from IL-36Rafl/fl and IL-36Ra-cKO mice at 12 weeks of age. Scale bar: 50 μm. n = 6 (a–d) or 5 (e) or 3 (f) or 8 (j–m) mice per group. Results from one representative replicate are shown. Quantitative data are shown as the mean ± SD. P-values are from a two-tailed unpaired t test. n indicates biological replicates; any technical repeats were averaged within each biological sample. Source data are provided as a Source Data file.
IL-36Ra ablation in the epidermis keratinocytes aggravates experimental and age-related OA development in mice
IL-36Rafl/fl and IL-36Ra-cKO mice were then subjected to DMM surgery to induce OA at 12-week age, as described in the schematic (Fig. 4a). Importantly, at 2 months after DMM surgery, safranin O staining revealed that IL-36Ra-cKO mice displayed greater cartilage degradation, synovitis, subchondral bone sclerosis, osteophyte maturity, and markedly increased OARSI scores than IL-36Rafl/fl mice. However, no significant differences in SBP thickness were observed between IL-36Ra-cKO and IL-36Rafl/fl mice (Fig. 4b, c). Further analysis showed increased MMP13, PDPN, and p16 expression, decreased Col2a1 expression (Fig. 4d, e), and enhanced knee joint pain (Fig. 4f) in IL-36Ra-cKO mice, indicating that IL-36Ra ablation in the epidermal skin promotes experimental OA pathogenesis in mice. Furthermore, the histopathological changes of the epidermal skin and joints in IL-36Rafl/fl and IL-36Ra-cKO mice without surgical intervention were analyzed at 10 months of age. We did not notice serious psoriasis-like lesions in the epidermal skins of IL-36Ra-cKO mice29, but thinner epidermis, lower cell numbers, and decreased epidermal acanthosis were observed compared to IL-36Rafl/fl mice (Fig. 4g). IL-36Ra-cKO mice exhibited increased cartilage destruction and aggravated synovitis in the joints (Fig. 4h, i). Immunostaining confirmed an imbalance in cartilage metabolism and enhanced chondrocyte senescence (Supplementary Fig. 6d, e), and the Von Frey filament test confirmed enhanced knee joint pain (Fig. 4j) in IL-36Ra-cKO mice. Thus, our data identify the vital role of epidermal skin IL-36R signaling in OA, IL-36Ra ablation in the epidermis keratinocytes aggravates experimental and age-related OA development in mice.

a Schematic illustrating the establishment of a DMM surgery-induced OA model in IL-36Rafl/fl and IL-36Ra-cKO mice. b, c Safranin-O staining (b) and scoring of OA parameters (c) in sham- or DMM-operated IL-36Rafl/fl and IL-36Ra-cKO mice at 8 weeks after DMM surgery. The inset in the images is shown in the magnified images in the bottom row. Scale bar: 50 μm. d–f Immunostaining (d), quantification (e) of MMP13, Collagen II, PDPN, p16 and quantitative analysis of the von Frey threshold (f) in joint sections from sham or DMM-operated IL-36Rafl/fl and IL-36Ra-cKO mice at 8 weeks post DMM surgery. Scale bar: 50μm. g H&E staining and quantification of epidermal thickness and cell numbers in skin sections from sham-operated IL-36Rafl/fl and IL-36Ra-cKO mice at 10 months after AAV-cre injection. Scale bar: 20 μm. h–j Safranin-O staining (h), scoring of OA parameters (i), and quantitative analysis of the von Frey threshold (j) in IL-36Rafl/fl and IL-36Ra-cKO mice 10 months after AAV-cre injection. Scale bar: 50 μm; n = 8 (a–f, h–j) or 10 (g) mice per group. Results from one representative replicate are shown. Quantitative data are shown as the mean ± SD. P-values are from two-tailed Mann–Whitney U-test (f, j, OARSI, synovial inflammation scores, osteophyte maturity scores in c or i), two-tailed unpaired t test (e, g, SBP thickness in c or i). n indicates biological replicates; any technical repeats were averaged within each biological sample. Source data are provided as a Source Data file.
IL-36 agonizts from keratinocytes induce chondrocytes catabolism and inflammation in synovial fibroblasts
To determine which cells are targeted by IL-36 agonizts, we next performed IL-36R immunostaining in various key tissues and cells involved in OA from mice and human joint sections at a young and old age. In the synovium, we observed gradual increases in IL-36R-positive cells with age, and using specific markers, we confirmed that the majority of IL-36R positive cells were synovial fibroblasts, as indicated by PDPN expression, while no significant co-localization was observed with F4/80-positive macrophages (Fig. 5a). We also noticed that IL-36R was positive in articular cartilage chondrocytes, and that the number of positive chondrocytes reached a peak in mice at middle age (10 months), before decreasing at old age (18 months) (Fig. 5b). Moreover, similar changes in IL-36R expression were observed in chondrocytes and synovial fibroblasts from young and aged humans (Supplementary Fig. 7a, b). Furthermore, we detected more blood vessels (CD31) but decreased lymph vessels (PDPN) in the synovium of aged and OA mice (Supplementary Fig. 7c, d), indicating that systemic IL-36 agonizts may affect chondrocytes and synovial fibroblasts via vascular vessels in the synovium. Subsequently, we cultured mouse keratinocytes and induced senescence with UVB irradiation. We next investigated the effects of supernatant liquor from aging keratinocytes (Aging SNL) on chondrocytes or synovial fibroblasts with mIL-36Ra or IL-36R knockdown (Fig. 5c). For synovial fibroblasts, the migration ability, cellular senescence, and release of inflammatory cytokines (e.g., TNF-α, IL-6, IL-1β) were significantly enhanced under the stimulation of Aging SNL, but were restored to levels equal to those of the control group after the addition of mIL-36Ra or knockdown of IL-36R (Fig. 5d, e). Furthermore, in cultured mouse primary chondrocytes, loss of proteoglycan content, increased cellular senescence, inhibited anabolism, and enhanced catabolism were noticed upon stimulation with Aging SNL, all of which were recovered by mIL-36Ra and IL-36R knockdown (Fig. 5f, g). This suggests that the Aging SNL induces catabolic processes in chondrocytes and inflammatory responses in synovial fibroblasts through the IL-36R signaling pathway.

a Immunostaining and quantification of IL-36R (red) with PDPN (green) or F4/80 (green) in synovial tissues from mice aged 3, 10, or 18 months. Scale bar: 20 μm. b Immunostaining and quantification of IL-36R (red) in the articular cartilages of mice aged 3, 10, or 18 months. Scale bar: 20 μm. c Schematic illustrating the induction of mouse keratinocyte senescence with UVB irradiation, and investigation into the effects of Aging SNL on chondrocytes or synovial fibroblasts with mIL-36Ra or IL-36R knockdown. d, e Representative images and quantification of cell wound scratch assay and β-gal staining results. Scale bar, 100 μm (d) qRT–PCR detection of mRNA levels of TNFα, IL-1β, IL-6, IL-8, CCL2, and TGF-β (e) in mice synovial fibroblasts under the indicated stimulation. f, g Representative images and quantification of alcian blue and β-gal staining (f; scale bar, 100 μm for β-gal staining), western blotting results of collagen II, aggrecan, MMP13, MMP3, and IL-36R (g) in mouse chondrocytes under the indicated stimulation conditions. h Schematic illustrating the knockdown of IL-36Ra in mouse keratinocytes, and investigation into the effects of IL-36Ra-KD SNL on chondrocytes or synovial fibroblasts with mIL-36Ra or IL-36R knockdown. i–j Representative images and quantification of cell wound scratch assay and β-gal staining results. Scale bar, 100 μm (i). qRT–PCR detection of the mRNA levels of TNFα, IL-1β, IL-6, IL-8, CCL2, and TGF-β (j) in synovial fibroblasts of mice under the indicated stimulation conditions. k, l Representative images and quantification of alcian blue and β-gal staining (k; scale bar, 100 μm for β-gal staining), and western blotting results of collagen II, aggrecan, MMP13, MMP3, and IL-36R (l) in mouse chondrocytes under indicated stimulation; n = 8 (a, b) mice per group and results from one representative replicate are shown. Experiments were repeated five (d, f, i, k) or three (e, g, j, l) times with similar results. Quantitative data are shown as the mean ± SD. P-values are from two-tailed unpaired t test (a), one-way ANOVA test followed by Tukey’s post hoc test (b, d–f, i–k). n indicates biological replicates; any technical repeats were averaged within each biological sample. Source data are provided as a Source Data file.
RNA-sequencing analysis was then performed in the negative control, and chondrocytes stimulated with the Aging SNL groups. KEGG and GSEA analysis showed that, compared to the control group, IL-36-related signaling, including NF-κB, JAK-STAT and MAPK signaling, were activated in chondrocytes under the stimulation of Aging SNL (Supplementary Fig. 8a). We confirmed the upregulation of p-p38, p-ERK1/2, p-JNK, p-p65, and p-STAT3 in both chondrocytes and synovial fibroblasts following stimulation with Aging SNL, which were recovered after addition of mIL-36Ra (Supplementary Fig. 8b). To confirm that IL-36Ra reduction is critical for the function of senescent keratinocytes, we knocked down IL-36Ra with small interfering RNA in cultured mouse keratinocytes, and used the resulting supernatants to stimulate synovial fibroblasts and chondrocytes (Fig. 5h). Interestingly, we observed similar changes, with increased inflammation in synovial fibroblasts and chondrocyte catabolism with the stimulation of supernatant liquor from IL-36Ra knockdown keratinocytes (IL-36Ra-KD SNL), both of which were recovered by mIL-36Ra and IL-36R knockdown (Fig. 5i–l and Supplementary Fig. 8c). Moreover, activation of these signaling pathways was also verified in joint sections from mice with prematurely aged skin with Immunostaining (Supplementary Fig. 8d–f). Thus, these data suggest that loss of IL-36Ra in keratinocytes leads to increased IL-36 agonist secretion and induces chondrocytes catabolism and synovial inflammation by activating pro-inflammatory signaling, including NF-κB/JAK-STAT/MAPK pathways.
Insertion mIL-36Ra-loaded microneedles into skin targeting epidermal keratinocytes attenuates experimental OA in mice
To further demonstrate the effectiveness of targeted IL-36R signaling in preventing OA in vivo, we established DMM-induced OA and used pharmacological and genetic approaches to inhibit IL-36R in control, Z24-cKO mice (Supplementary Fig. 9a). Mice received intra-articular delivery of mIL-36Ra or IL-36R knockout AAV (shIL-36R) weekly, neither of which affected the normal appearance of the articular cartilage in the knee joints of sham-operated mice (Supplementary Fig. 9b). In Zmpste24fl/fl and Z24-cKO mice with DMM-induced post-traumatic OA, Z24-cKO mice exhibited more severe cartilage destruction, subchondral bone sclerosis, osteophyte development, and synovial inflammation. mIL-36Ra and IL-36R knockout treatment significantly improved the OA-relevant symptoms both in Zmpste24fl/fl and Z24-cKO mice, and mIL-36Ra treatment even restored the degree of OA in Z24-cKO mice to a level comparable to that in Zmpste24fl/fl mice with similar OARSI structure scores (Supplementary Fig. 9c, d). Detection of cartilage metabolism confirmed the upregulated anabolic factors and downregulated catabolic factors, the inhibition of synovial inflammation by mIL-36Ra treatment and IL-36R knockout, and the alleviation in knee joint pain in both groups (Supplementary Fig. 9e–g). We further validated the ameliorating effect of intra-articular delivery mIL-36Ra or IL-36R knockout AAV on changes in OA pathogenesis and pain in IL-36Rafl/fl and IL-36Ra-cKO mice (Supplementary Fig. 10a). Importantly, the use of mIL-36Ra also showed a better effect than IL-36R knockout in IL-36Ra-cKO mice (Supplementary Fig. 10b–g). Collectively, our data suggest that intra-articular knockout of IL-36R or mIL-36Ra delivery to inhibit IL-36R signaling significantly ameliorates OA pathology changes in mice, including wild-type mice, mice with premature aged skin, and mice with IL-36Ra ablation in the epidermis.
In this study, we reached a similar conclusion to previous studies, in that IL-36Ra is a potential therapeutic agent to ameliorate OA23. We also demonstrated that senescent keratinocytes in aged skin represent the major source of IL-36 agonizts and promote OA progression. However, given that weekly or daily injections are impractical for clinical use, we developed MN-mIL-36Ra, microneedles loaded with mIL-36Ra to achieve sustained release of mIL-36Ra, and inserted them directly into the skin to target epidermal keratinocytes (Fig. 6a, b). The images of MN-mIL-36Ra under optical and scanning electron microscopy (SEM) are displayed in Fig. 6c. The in vitro drug release profile showed that the drug-release rate of mIL-36Ra was 41.06% within 24 h, 73.37% within 168 h, indicating that MN-mIL-36Ra had a sustained release effect (Fig. 6d). Next, we established DMM-induced OA in control, Z24-cKO, or IL-36Ra-cKO mice, and covered the skin of mice with MN-mIL-36Ra to examine its effect on OA protection (Fig. 6e). Upon application of MN-mIL-36Ra in mice, the skin exhibited rapid recovery within minutes, skin epidermis and other important organs were not significantly affected after 8 weeks (Supplementary Fig. 11a–d). Reduced IL-36 ligands in keratinocytes and serum were observed in Z24-cKO and IL-36Ra-cKO mice with MN-mIL-36Ra application, while IL-36Ra levels in serum were not affected (Fig. 6f–h). SOFG staining demonstrated that MN-mIL-36Ra treatment effectively improved cartilage histological structural integrity relative to the control group both in Zmpste24fl/fl and Z24-cKO mice, and antagonized the enhanced OA pathology changes and knee joint pain caused by premature skin aging in Z24-cKO mice (Supplementary Fig. 11e and Fig. 6i–k). Moreover, chondrocyte metabolism and synovial inflammation improved and recovered to levels similar to those of Zmpste24fl/fl in Z24-cKO mice (Supplementary Fig. 11f-g). MN-mIL-36Ra treatment also showed comparable alleviation of all OA symptoms in IL-36Rafl/fl and IL-36Ra-cKO mice, including cartilage and subchondral bone structures, synovitis, osteophyte hyperplasia, chondrocyte metabolism, and knee joint pain (Supplementary Fig. 11h–j, Fig. 6l–n). Thus, MN-mIL-36Ra provides an efficient strategy for the treatment of OA, directly targeting IL-36 agonizts derived from skin keratinocytes and provides a promising drug delivery system to broaden the usage of mIL-36Ra.

a, b A schematic and image illustrating the insertion of microneedles loaded with mIL-36Ra (MN-mIL-36Ra) into the mouse epidermis layer. c Image of MN-mIL-36Ra under optical microscope and SEM. Scale bar: 100 μm (c). d Drug release in MN-mIL-36Ra. e Schematic of the experimental design to determine the effects of MN-mIL-36Ra on OA progression and pain in Zmpste24fl/fl and ZM-cKO mice, IL-36Rafl/fl and IL-36Ra-cKO mice with surgically-induced DMM OA. f–g Immunostaining (f) and quantification (g) of K10 with IL-36α/β/γ in epidermal skin from ZM-cKO and IL-36Ra-cKO mice with or without MN-mIL-36Ra insertion. Scale bar: 50 μm. h Quantified ELISA results of IL-36Ra and IL-36α/β/γ cytokines in serum from ZM-cKO and IL-36Ra-cKO mice with or without MN-mIL-36Ra insertion for 8 weeks. i–k Safranin-O staining (i), scoring of OA parameters (j), and quantitative analysis of the von Frey threshold (k) in DMM-operated Zmpste24fl/fl and Z24-cKO mice with or without MN-mIL-36Ra insertion. The inset in the images is shown in the magnified images in the bottom row. Scale bar: 50 μm. l–n Safranin-O staining (l), scoring of OA parameters (m), and quantitative analysis of the von Frey threshold (n) in DMM-operated IL-36Rafl/fl and IL-36Ra-cKO mice with or without MN-mIL-36Ra insertion. The inset in the images is shown in the magnified images in the bottom row. Scale bar: 50 μm; n = 6 (f–h) or n = 8 (i–n) mice per group. Results from one representative replicate are shown. Quantitative data are shown as the mean ± SD. P-values are from two-tailed Mann–Whitney U-test (k, n, OARSI, synovial inflammation scores, osteophyte maturity scores in j or m) and two-tailed unpaired t test (g, h, SBP thickness in j or m). n indicates biological replicates; any technical repeats were averaged within each biological sample. Source data are provided as a Source Data file.
Targeting IL-36R signaling demonstrates potential therapeutic efficiency for human OA
Spesolimab is an IL-36R antagonist approved by the Food and Drug Administration for the treatment of generalized pustular psoriasis flares in adults30,31. We next assessed the potential clinical therapeutic efficacy of spesolimab in human OA using the human C28/I2 chondrocyte cell line, human fibroblast-like synoviocytes, primary human articular chondrocytes, and cartilage tissue explants from patients undergoing total knee arthroplasty. C28/I2 cells were treated with SNL from aging HaCat cells or HaCat cells with IL-36RN knockdown. Loss of chondrocyte metabolism homeostasis and proteoglycan content were determined by immunoblotting and alcian blue staining. The addition of spesolimab to C28/I2 cells showed a good protective effect against the SNL from both HaCat cells (Fig. 7a and Supplementary Fig. 12a). Similar results were obtained in primary human articular chondrocytes from patients with OA, and spesolimab was found to significantly retain the anabolism and proteoglycan content (Fig. 7b). In cartilage tissue explants from patients with OA cultured with spesolimab for 14 days, the degradation of proteoglycan content was inhibited, the expression of degradative enzymes was reduced, and the expression of anabolic factors was increased, indicating the therapeutic efficacy on OA pathogenesis (Fig. 7c, d). In human fibroblast-like synoviocytes, spesolimab treatment effectively reduced cell migration, suppressed cellular senescence, and led to decreased release of inflammatory factors (Fig. 7e, f).

a Western blot analysis results of Collagen II, Aggrecan, MMP13, MMP3 and representative images of alcian blue staining in C28/I2 cells with the stimulation of spesolimab and SNL from HaCat cells upon UVB. b Western blot analysis results of Collagen II, Aggrecan, MMP13, ADAMTS5 and representative images of Collagen II immunostaining in primary human articular chondrocytes from OA patients with or without spesolimab addition. c, d Safranin-O staining, immunostaining of MMP13, MMP3, Collagen II, Aggrecan (c) and quantification scoring of OA parameters and indicated positive cells (d) in cartilage tissue explants from OA patients cultured with or without spesolimab for 14 days. Scale bar, 100 μm. e, f Representative images and quantification of cell wound scratch assay and β-gal staining results. Scale bar, 100 μm (e), qRT-PCR detection of mRNA levels of TNFα, IL-1β, IL-6, IL-8, CCL2 and TGF-β (f) in human fibroblast-like synoviocytes under the indicated stimulation. g, h Representative images and quantification of cell wound scratch assay and β-gal staining results. Scale bar, 100 μm (g), qRT–PCR detection of mRNA levels of TNFα, IL-1β, IL-6, IL-8, CCL2, and TGF-β (h) in mice synovial fibroblasts under the indicated stimulation. i–j Representative images and quantification of Alcian blue and β-gal staining (i; scale bar, 100 μm for β-gal staining), and western blotting of collagen II, aggrecan, MMP13, MMP3, and IL-36R (j) in mouse chondrocytes under indicated stimulation conditions. k Schematic of the experimental design to determine the effects of MN-SPE on OA progression and pain in Zmpste24fl/fl and Z24-cKO, IL-36Rafl/fl and IL-36Ra-cKO mice with surgically-induced DMM OA. l–n Safranin-O staining (l), scoring of OA parameters (m), and quantitative analysis of the von Frey threshold (n) in DMM-operated Zmpste24fl/fl and Z24-cKO mice with or without MN-SPE insertion. Scale bar: 50 μm. o–q. Safranin-O staining (o), scoring of OA parameters (p), and quantitative analysis of the von Frey threshold (q) in DMM-operated IL-36Rafl/fl and IL-36Ra-cKO mice with or without MN-SPE insertion. The inset in the images is shown in the magnified images in the bottom row. Scale bar: 50 μm. n = 6 (Control) or 8 (OA and OA + SPE) (d) patients per group, or n = 8 (l–q) mice per group. Results from one representative replicate are shown. Experiments were repeated three (a, b, f, h, j) or five (e, g, i) times with similar results. Quantitative data are shown as mean ± S.D. P-values are from one-way ANOVA test followed by Tukey’s post hoc test (e–i, MMP13, MMP3, Col2a1, Aggrecan in d), Kruskal–Wallis test followed by Dunn’s post hoc test (OARSI in d), two-tailed unpaired t test (SBP thickness in m or p) and two-tailed Mann–Whitney U-test (n, q, OARSI, synovial inflammation scores, osteophyte maturity scores in m or p). n indicates biological replicates; any technical repeats were averaged within each biological sample. Source data are provided as a Source Data file.
Spesolimab is a humanized IgG1 monoclonal antibody against human IL-36R and previous study claimed that spesolimab has low binding affinity to IL-36R in other species up to 1 μM32. We subsequently performed an SPR assay and confirmed spesolimab had low binding affinity with mouse IL-36R at low concentration, but had higher affinity at higher concentrations, with a KD value of 5.34 × 10⁻⁶ M, a Ka value of 4.92 × 10⁴ 1/Ms, and a Kd value of 2.62 × 10⁻¹ 1/s (Supplementary Fig. 12b). We investigated the effects of Aging SNL on chondrocytes or synovial fibroblasts with spesolimab. Interestingly, we observed similar changes, with increased inflammation in synovial fibroblasts, activation of pro-inflammatory signaling in chondrocytes and synovial fibroblasts, as well as chondrocyte catabolism with the stimulation of Aging SNL, all of which were recovered by spesolimab (Fig. 7g–j and Supplementary Fig. 12c). Next, we established DMM-induced OA in control, Z24-cKO, or IL-36Ra-cKO mice, and covered the skin of mice with MN-SPE (microneedles loaded with spesolimab) to examine its effect on OA protection (Fig. 7k). Upon application of MN-SPE in mice, skin epidermis and other important organs were not significantly affected after 8 weeks (Supplementary Fig. 12d–f). Reduced IL-36 ligands in keratinocytes and serum were observed in Z24-cKO and IL-36Ra-cKO mice with MN-SPE application, while spesolimab levels in serum were not affected (Supplementary Fig. 12g–i). SOFG staining demonstrated that MN-SPE treatment effectively improved cartilage histological structural integrity relative to the control group, both in Zmpste24fl/fl and Z24-cKO mice, and antagonized the enhanced OA pathology changes and knee joint pain caused by premature skin aging in Z24-cKO mice. Moreover, chondrocyte metabolism and synovial inflammation improved and recovered to levels similar to those of Zmpste24fl/fl in Z24-cKO mice (Fig. 7l–m, Supplementary Fig. 12j–l). MN-SPE treatment also showed comparable alleviation of all OA symptoms in IL-36Rafl/fl and IL-36Ra-cKO mice, including cartilage and subchondral bone structures, synovitis, osteophyte hyperplasia, chondrocyte metabolism, and knee joint pain (Fig. 7n, o and Supplementary Fig. 12m–o). Thus, MN-SPE provides an efficient strategy for the treatment of OA, directly targeting IL-36 agonizts derived from skin keratinocytes and provides a promising drug delivery system to broaden the usage of spesolimab.
Discussion
Aging is one of the most prominent risk factors for OA, with the responsible mechanisms considered to be multifactorial, potentially including an age-related low-level inflammatory state that has been termed “inflamm-aging”33. OA has long been recognized as a degenerative disease of cartilage, but accumulating evidence indicates that inflammatory aging plays a central role in driving OA pathogenesis34. Elevated systemic and local inflammatory cytokines induce the expression of enzymes, including MMPs and ADAMTS, in chondrocytes that stimulate the transition of synovial fibroblasts to a proinflammatory state, and contribute to the development of structural damage and joint pain in OA35. The skin is the largest organ of the human body and undergoes disruption with aging, resulting in elevated levels of serum inflammatory mediators, with broad effects on other systems11. Bone and joint tissues are closely connected and can be affected by skin. Studies have revealed that psoriatic patients with higher skin severity are twice as likely to have increased joint involvement. Psoriasis is an autoimmune disease characterized by the formation of inflamed, red, and scaly patches on the skin. Up to 30% of patients with psoriasis develop chronic arthritis, termed PsA, including spondylitis, enthesitis, or peripheral joint arthritis36. Raemdonck et al. reported that intradermal administration of the endogenous TLR7 ligand miR-Let7b intensified joint inflammation in a PsA-like preclinical model, indicating the connection of skin immunopathology with joint inflammation37. In our study, we constructed two prematurely aged skin mouse models using Zmpste24 knockout and D-gal cutaneous application. Mice from both groups displayed accelerated OA pathogenesis in the traumatic OA mouse model, which further verified the connection of skin to joints and emphasized that skin aging is an important driver for traumatic OA progression.
There is evidence that increased inflammation at the time of meniscus injury or meniscectomy accelerates the process of cartilage degradation and further produces inflammation and pain in conjunction with the cycle of altered cartilage loading and subsequent degradation38,39. When analyzing the proteomics results of skin tissue, we focused on inflammatory factors, and the significantly decreased IL-36Ra in aged skin caught our attention. Moreover, several studies have already highlighted the potential therapeutic value of targeting the IL-36 axis in the development of PsA and OA24,40, which further motivated us to analyze whether IL-36Ra is the regulatory molecule from skin to joints. IL-36 cytokines are mostly found in the skin, where their expression is strongly induced by TNF-α and IL-17, and the IL-17 pathway interacts with the IL-36 pathway in a positive feedback system41,42,43,44. We confirmed that keratinocytes are the major cutaneous source of IL-36Ra, and further noticed decreased expression of IL-36Ra in senescent keratinocytes, which resulted in increased secretion of IL-36 agonizts in serum. We also noticed a gradual decrease in the expression of IL-36Ra in articular cartilage and synovium during aging, but the decrease occurred earlier, and the difference was greater in keratinocytes. Although this does not rule out the effect of locally derived IL-36 cytokines on OA, it does suggest that the early increase in serum IL-36 cytokines is a result of keratinocytes. TNF-α and IL-17 signaling was upregulated in chondrocytes treated with Aging SNL, suggesting that the increase in IL-36 agonizts from senescent keratinocytes was induced via TNF-α and IL-17 signaling. We also determined that chondrocytes and synovial fibroblasts are the two major targeted cells of IL-36 agonizts and detected more blood vessels in the aged and OA synovium, indicating that systemic IL-36 agonizts may affect chondrocytes and synovial fibroblasts via the vascular tissues of the synovium.
Inflammation is associated with joint pain and structural damage, and could therefore be a potential target for OA treatment. Recently, Li et al. reported that intra-articular treatment with IL-36Ra attenuated OA development in mice. Our study confirmed that treatment of chondrocytes and synovial fibroblasts with supernatant containing increased IL-36 agonizts from senescent or IL-36Ra knockdown keratinocytes led to senescence and increased expression of catabolic enzymes and SASP, all of which were reversed by mIL-36Ra treatment. Moreover, intra-articular knockout of IL-36R ameliorated OA pathogenesis and joint pain in control and skin aged mice, whereas IL-36Ra replenishment with mIL-36Ra displayed an even better effect against OA. Both MN-mIL-36Ra and MN-SPE showed efficacy in treating OA when applied to the skin. Although we cannot rule out that mIL-36Ra and spesolimab directly reach the joints to treat OA, our data show that they mainly exert their IL-36R blocking effects locally through the skin with only a slight increase observed in serum levels. These results highlight that an increased IL-36 axis originating from aged skin promotes OA and can be targeted to improve OA symptoms. However, Derer et al. demonstrated that the IL-36R-blocking antibody did not significantly affect the phenotype in animal models of RA22. The most striking discrepancy between the levels of inflammation in OA and RA is that the former is low-grade chronic inflammation2. Regarding the distinct role of IL-36 agonizts in OA and RA, together with our findings that IL-36 agonizts increased with aging, we speculate that IL-36 agonizts are systemic inflammatory cytokines that belong to the inflamm-aging group and participate in OA progression.
Blockade of proinflammatory mediators alleviates OA progression in many preclinical models. However, several randomized clinical trials targeting pro-inflammatory cytokines by biologics, especially IL-1, TNFα and IL-6, did not show a clear pain-relieving effect, thus largely reducing the enthusiasm for targeting inflammatory factors as disease-modified OA drugs45,46,47. We concluded that, to achieve a breakthrough, future anti-inflammatory interventions are warranted to focus on the source of inflammatory mediators that target specific cell types and are dependent on the disease stage of OA. Our study demonstrated that targeting enhanced IL-36R signaling in aged skin is more promising for a clinical disease-modifying osteoarthritis drug in early-stage OA because of its ability to inhibit crucial inflammatory signaling, including MAPK and NF-κB. We did not observe any serious adverse events in the local or cutaneous application of mIL-36Ra or spesolimab in mice with OA. We also identified the cells targeted by IL-36 cytokines in the joints and found that intra-articular injection of mIL-36Ra or knockout IL-36R alleviated OA in mice. How best to target the inflammatory response and achieve sustained release of formulations is a critical problem that needs to be solved for clinically therapeutic interventions for OA. Currently, intra-articular injection is one of the most attractive treatments for OA, but the clearance of the drug in the arthritic site, risk of infections, and pain associated with frequent injections make this route highly non-compliant to patients48. In our study, we verified that, together with aging, epidermis keratinocytes are the source of chronic low levels of IL-36 cytokines involved in OA. We generated Microneedles to penetrate the skin, directly targeting keratinocytes to sustain the supply of IL-36Ra and spesolimab, inhibiting the major source of IL-36 cytokines, and achieving a sufficient therapeutic effect for OA. Microneedles provide a prolonged duration of mIL-36Ra and spesolimab compared to the i.a. route, representing a painless means to create hundreds of microchannels in the skin to improve efficiency, which represents a promising drug delivery system in clinical OA treatment.
Spesolimab was recently approved by the FDA as an anti-IL-36 agent to block the IL-36 axis, thereby inhibiting the proinflammatory process, with clinical efficacy and safety profiles in treating generalized pustular psoriasis31. Our study confirmed that spesolimab binds to mouse IL-36R at high concentrations (above 1 μM), protecting against cartilage ECM degradation and reducing synovial inflammation, which significantly expanded the potential of SPE as a therapeutic agent in OA. Therefore, we can conclude that delivery of spesolimab via microneedles penetrating the skin is a promising and translational approach for OA treatment. In summary, our study demonstrated that aged skin intensifies experimental OA progression and that IL-36 agonizts are age-related systemic inflammatory factors released from aged skin to joints that contribute to OA development. Targeting keratinocytes in the epidermis using microneedles loaded with drugs that inhibit IL-36R signaling and inserted into the skin could be a promising option to improve therapeutic efficiency (Fig. 8). Of course, our study still has some limitations. Subchondral bone sclerosis and osteophyte formation did not reach statistical significance in some groups, possibly due to the DMM model, which typically induces milder subchondral bone changes than the ACLT model49,50. Longer treatment durations might be required to observe significant improvements. Future studies with different OA models in larger animals and even sequential phase clinical trials are warranted to demonstrate the ultimate safety and efficacy of IL-36Ra in treating OA.

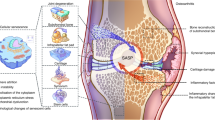
IL-36Ra decreases in senescent epidermal keratinocytes of aged mice and patients, which leads to age-related low-level inflammatory with increased secretion of IL-36 agonizts, activated pro-inflammatory signaling, and promoted senescence in chondrocytes and synovial fibroblasts. Deletion of IL-36Ra in keratinocytes aggravates experimental and age-related OA in mice, whereas mIL-36Ra treatment or deletion of IL-36R effectively attenuates OA progression. Insertion of mIL-36Ra or spesolimab loaded microneedles into skin provides prolonged inhibition of IL-36R signaling directly in skin keratinocytes, representing a promising drug delivery system to broaden the usage of IL-36Ra in clinical OA treatment.
Methods
Human samples
Human skin samples from young (30-40 years old; n = 6; half male and half female) and elderly (70-75 years old; n = 6; half male and half female) individuals were obtained from the back incisions of patients undergoing posterior spinal surgery. Human OA cartilage and synovium were taken from patients with late-stage OA undergoing total knee arthroplasty (n = 8; aged 60–90; half male and half female). Human non-OA cartilage and synovium were from patients with bone tumors undergoing partial knee replacement surgery with no history of OA (n = 6; aged 25–45; half male and half female). Human OA joint synovial fluid and serum were collected from the same patients with late-stage OA undergoing total knee replacement surgery (n = 15; aged 70–90; males). Human serum from young (n = 5; aged 25–35; 2 male and 3 female) and elderly (n = 5; aged 60–80; 2 male and 3 female) individuals was obtained from the hospital health examination center. All clinical samples for this project were collected from the Third Affiliated Hospital of Southern Medical University (Guangzhou, China), with approval from the hospital’s medical ethics committee. Written informed consent was obtained from all subjects or their guardians, including consent to publish potentially identifying information.
Mice
All mice were housed in the Specific Pathogen-Free (SPF) animal facility at Southern Medical University (Guangzhou, China), with no more than five mice per cage. The mice were kept in an environment with adequate ventilation, appropriate temperature and humidity, and had free access to food and water. A 12h light/dark cycle was maintained. All animal experiments conducted in this study were approved by the Animal Care and Use Committee of Southern Medical University and were in compliance with all relevant ethical regulations regarding animal research. Twelve-week-old male C57BL/6 J mice used for the experimental OA and 3-, 10- and 18-month-old male C57BL/6 J mice used for aged-OA studies were all acquired from the Laboratory Animal Center of Southern Medical University. Zmpste24-flox mice and Il1f5 (IL-36Ra)-flox mice were purchased from GemPharmatech Co., Ltd (Nanjing, China). Adeno-associated viruses (AAV2/Kera2-Cre-Null-ZsGreen or AAV2/Kera2-ZsGreen Control) were engineered by Hanbio Biotechnology Co., Ltd (Shanghai, China). We diluted AAV2 (1.4 × 10^12 vg/mL, 20 μl) with PBS and uniformly applied it to the shaved dorsal skin area (4.0 × 2.0 cm²) of mice once a week. Male mice with Zmpste24-flox and IL-36Ra-flox back skin infected by AAV2/Kera2-Cre-Null-ZsGreen were designated as Zmpste24-cKO and IL-36Ra-cKO, respectively, while those infected with AAV2/Kera2-ZsGreen Control on the back skin of Zmpste24fl/fl and IL-36Rafl/fl male mice were designated as controls for Zmpste24-cKO and IL-36Ra-cKO.
Animals model
To induce premature skin aging in mice, D-Galactose (D-Gal; MedChemExpress, Cat. No. HY-N0210) was dissolved in 70% ethanol at a concentration of 20 mg/mL to facilitate dermal absorption. Then, 200 μL of the solution was topically applied once daily onto a shaved dorsal skin area (4.0 × 2.0 cm²) of 8-week-old male mice for two months. The control group was only treated with 70% ethanol. After one month of D-Gal application, we performed a skin excision surgery under sterile conditions. Briefly, after anesthetizing the mice, the shaved dorsal skin was disinfected with iodine. Using sterile scissors and forceps, we carefully excised the D-Gal-treated skin along the edges of the treatment area. The wound edges were then sutured using absorbable sutures (Vicryl, 5-0), followed by a second round of iodine disinfection. After suturing, the wound was covered with sterile gauze and secured with an elastic bandage. For the first week post-surgery, the wound was disinfected daily, and the sterile gauze was replaced. Additionally, enrofloxacin (50–100 mg/L) was administered in the drinking water to prevent infection, continuing for 1-2 weeks post-surgery or until the wound had fully healed. For D-Gal-induced premature skin aging, D-Gal skin removal, and control C57BL/6 J male mice, as well as Zmpste24-cKO, IL-36Ra-cKO male mice, and their respective controls (Zmpste24fl/fl and IL-36Rafl/fl male mice), we induced OA through Destabilization of the Medial Meniscus (DMM). Briefly, after anesthetizing the mice, the right knee was prepared and disinfected. The joint capsule adjacent to the medial side of the patellar tendon was opened to expose the intercondylar area for visualization and transection of the meniscotibial ligament. The joint capsule and skin were then closed. For the sham-operated group, the joint capsule was opened, but the meniscotibial ligament was not transected. At eight weeks post-operation, mice were euthanized by rapid cervical dislocation, and serum, knee joints, and dorsal skin samples were collected. For the transgenic mice (Zmpste24-cKO, IL-36Ra-cKO), we immobilized and exposed their right legs, then administered an 8 μl injection of IL-36R antagonist adenovirus or control virus (provided by Hanbio Biotechnology Co., Ltd.) into the joint capsule to knockout IL-36R, or a 10 μl (0.5 mg/mL) injection of mouse recombinant IL-36Ra protein (mIL-36Ra, TargetMol, Cat No. TMPY-04275) to inhibit IL-36R. A booster dose was administered weekly thereafter to maintain the viral load. For C57BL/6 J male mice, we injected 10 μl of serum (sourced from D-Gal-induced premature skin aging and control mice) into the joint capsule, with injections maintained on a weekly basis. All mice subjected to intra-articular injections commenced treatment one week after DMM surgery and were euthanized at week 8 post-surgery for the collection of knee joint samples. All procedures started one week post-DMM surgery and continued until week 8 post-OA induction, at which point mice were euthanized, and skin and knee joint samples were collected.
Preparation and characterization of MN-mIL-36Ra and MN-SPE
To address the challenges of low oral administration efficiency and poor compliance with injectable medications, we developed a dissolving microneedle (DMN) delivery system loaded with mIL-36Ra (TargetMol, Cat No. TMPY-04275) or spesolimab (MedChemExpress, Cat No. HY-P99396) for the treatment of OA. mIL-36Ra was formulated into a gel-based microneedle (MN) system as MN-mIL-36Ra, with a dosage of 0.05 mg per patch. Briefly, we first uniformly mixed 2% (w/v) Hyaluronic Acid Methacryloyl (HAMA, EACIN BIO; product number YX-HM-231121-01-H), 0.25% (w/v) Lithium Phenyl (2,4,6-trimethylbenzoyl) phosphinate (LAP, EFL; product number 85073-19-4), and mIL-36Ra (<1.5%) in deionized water in a 37 °C water bath to ensure even distribution of mIL-36Ra in the HAMA. The HAMA-mIL-36Ra mixture was used as the aqueous phase, while the oil phase served as the continuous phase. These were injected into microfluidic chip channels (channel size: 20-50μm) to form stable droplets of the aqueous phase in the oil phase. Subsequently, the droplets were solidified into mIL-36Ra-loaded microspheres by UV polymerization. Next, we prepared a 5% (w/v) Gelatin Methacryloyl (GelMA, EFL; model EF-GM-90) solution by dissolving 250 mg of GelMA and 12.5 mg of LAP in 5 ml of deionized water at 37 °C in a water bath. We mixed the GelMA solution with the mIL-36Ra microspheres, then added this mixture to the mold, covering the microneedle tips. The mold was placed in a 60 °C drying oven to remove excess air bubbles, followed by UV polymerization to form the microneedle tips. Afterward, a sufficient amount of GelMA solution without mIL-36Ra was added to form the base of the microneedles, and UV polymerization was performed. The mold was frozen at -20 °C for about 30 min to harden, and after demolding, the MN-mIL-36Ra microneedles were obtained. Spesolimab was formulated into the gel-based microneedle (MN) system as MN-SPE with a dosage of 0.1 mg per patch. Briefly, we first prepared a gel by mixing 15% (w/v) GelMA, 0.25% (w/v) LAP, and 5% (w/v) PEGDA (Sigma Alorich; product number 455008) in deionized water at 37 °C. Spesolimab was then mixed into the gel. Subsequently, the spesolimab-containing gel was poured into the needle-part of a polydimethylsiloxane (PDMS) mold, and the mold was placed in a vacuum oven to remove air bubbles, followed by ultraviolet light polymerization. Additional drug-free mixed gel was added to the mold surface as a base, followed by drying and light polymerization until the MN-SPE was demolded. The surface morphology of both MN-mIL-36Ra and MN-SPE were observed using an optical microscope (Leica) and scanning electron microscope (SEM), and their in vitro drug release profiles were evaluated. Finally, both MN-mIL-36Ra and MN-SPE (microneedle side) were applied to the shaven dorsal skin (4 × 5 cm) of transgenic male mice (Zmpste24-cKO, IL-36Ra-cKO, Zmpste24fl/fl, and IL-36Rafl/fl), covered with moist gauze, secured with tape to prevent movement, and the moist gauze was changed daily.
Surface plasmon resonance (SPR) analysis
SPR analysis was performed by MedChemExpress (Monmouth Junction, NJ, USA) using a Biacore T200 instrument (GE Healthcare) equipped with CM5 sensor chips (GE Healthcare). Briefly, the chip surface was activated by injection of a mixture containing 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and N-hydroxysuccinimide (NHS) at a flow rate of 10 μL/min. Mouse IL-36R (MedChemExpress; Cat No. HY-HY-P70748) was diluted to 50 μg/mL in 10 mM sodium acetate buffer (pH 5.0) and immobilized on the activated surface at 10 μl/min for 420 s. The channel was blocked with 1 M ethanolamine (Sigma, Cat No. E6133) (pH 8.5) at 10 μl/min for 420 s. A neighboring channel was activated and blocked similarly but without protein immobilization to serve as the reference. Both channels were equilibrated with PBS. Spesolimab was diluted to a series of concentrations (0.15625 μM, 0.3125 μM, 0.625 μM, 1.25 μM, 5 μM, 10 μM) in PBS and flowed over the channels at 30 μl/min for 150 s. After each injection, the chip was regenerated with 10 mM glycine-HCl (pH 2.0) at 10 μl/min for 5 min. Data were collected using Biacore T200 Control software, and the results were subtracted by those from the reference channel. By globally fitting the data to a 1:1 Langmuir binding model using Biacore T200 Evaluation software, the binding affinity (KD), association rate constant (Ka), and dissociation rate constant (Kd) of spesolimab with Mouse IL36R were determined. Data were exported to GraphPad Prism 9.3 software for generating the final figures.
Von Fery
To evaluate OA-related pain and the efficacy of treatments in mice, we utilized the von Frey filaments to assess the occurrence of mechanical allodynia. At the eighth week post-DMM surgery, mice were placed in a plexiglass chamber with a metal mesh floor, allowing direct access to the underside of the mice’s heels. After the mice fully acclimated to the setup (at least 15 min) and ceased exploratory behavior, we tested for mechanical allodynia by applying von Frey filaments to the underside of the mice’s heel in ascending force order for up to 5 sec. A positive response was indicated by a sharp retraction of the tested foot, and this value was recorded. Finally, we collected and analyzed the average of three repeated tests for all mice.
Histological analysis
Soft tissues, including human and mouse skin, synovium, mouse heart, brain, spleen, lungs, liver, kidneys, intestines, esophagus, stomach, and pancreas, were fixed in 4% paraformaldehyde for 12 h. Human cartilage and mouse knee joints were fixed in 4% paraformaldehyde for 48 h, followed by decalcification in 10% EDTA (pH 8.0) for 21 days. All tissues were dehydrated in an automatic dehydration machine (Leica; ASP300S) and then embedded in paraffin. Soft tissues and human cartilage were sectioned continuously at 4 μm thickness, while mouse knee joints were sectioned continuously at 4 μm thickness with 20 μm intervals according to a standard protocol. All sections were used for histological and immunohistochemical staining. For human and mouse articular cartilage, Safranin O/Fast green staining is the gold standard for assessing the severity of OA. The severity of articular cartilage OA was blindly scored by two experienced researchers using the Osteoarthritis Research Society International (OARSI) grading system (0–6 points)51,52. To assess the sclerosis of the subchondral bone in mice, we measured the average distance between five evenly distributed measuring points from the subchondral bone margin to the top of the trabecular bone53,54. Synovitis (0 for no inflammation, 1 for mild inflammation, 2 for moderate inflammation, 3 for severe inflammation)55 and osteophyte formation (0 for none, 1 for primarily cartilaginous, 2 for mixed cartilaginous and bony with active vascular invasion and enchondral ossification, 3 for primarily bony)56 were quantified as previously described.
Immunohistochemistry (IHC) and Immunofluorescence (IF)
Representative sections prepared as described above were deparaffinized in xylene, rehydrated through graded ethanol, and then submerged in TE9.0 and incubated overnight at 65 °C to expose antigens. For IHC, sections were treated with 3% hydrogen peroxide for 10 min. Sections were then blocked with 1% sheep serum at 37 °C for 1 h, and incubated overnight at 4 °C with primary antibodies. Subsequently, the sections were incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. Finally, 3, 3-diaminobenzidine was used to observe the chromogen, with hematoxylin or alcian bule for counterstaining. For IF, sections or cells seeded onto confocal dishes were incubated with primary antibodies, then incubated in the dark at room temperature for 1 h with Alexa Fluor 488 (Life Technologies, Cat No. A20000) or Alexa Fluor 594 (Life Technologies, Cat No. A10438) conjugated secondary antibodies. Cell nuclei were stained with DAPI (ThermoFisher Scientific, Cat No. D1306). Images of all stained sections and confocal dishes were acquired using a FluoView FV1000 confocal microscope (Olympus) and FV10-ASW Viewer (v4.2) software. The ratio of the number of positive cells to the total number of cells within multiple standardized regions of interest (ROIs) was assessed using Image Pro Plus (v6.0) and Image J (v1.8) software. For mouse knee joints, we analyzed the ratio of the area/number of positive staining to the total area/total number of cells along the joint surface in the femoral condyle and tibial plateau regions. For the skin, the ratio of the total area to the total length of the epidermal layer was taken as the average thickness of the epidermis. The average intensity of staining was quantified as the integrated optical density (IOD) of the positive cell area. For each group (comprising at least three samples), at least three random fields of view were evaluated from three random sections of each individual sample. Detailed information regarding the primary and secondary antibodies used for IHC and IF staining is provided in Supplementary Table 2.
Human OA cartilage explant culture
As described previously, OA knee cartilage samples from 8 patients undergoing total knee arthroplasty for advanced OA (with no history of medication treatment) and control cartilage from 6 patients with bone tumors undergoing partial knee replacement surgery with no history of OA were obtained. All samples were sourced from the Third Affiliated Hospital of Southern Medical University, with approval from the hospital’s ethics committee and written informed consent from the patients. The cartilage specimens were cut into small cubes of 0.5 cm^3, washed with sterile PBS, and transferred to 12-well plates containing DMEM/F12 medium (Gibco, Cat No. C11330500BT) supplemented with 10% FBS (Gibco, Cat No. 10099141 C). Spesolimab (AtaGenix, Cat. No. ATAD00573) was added to the culture medium at a concentration of 200 ng/mL to evaluate its effects on human OA cartilage cells. These cartilage explants were cultured in media with or without spesolimab at 37 °C in 5% CO2 for 14 days, with cartilage explants from individuals without OA history serving as blank controls. The medium and drug were replaced every other day following the initial steps. After the culture period, cartilage specimens were collected and processed as previously described to prepare sections for histological and immunological staining.
Cell culture
Primary articular chondrocytes (CCs) were isolated from the femoral condyles and tibial plateaus of 3-day-old C57BL/6 J mice. In brief, the chondrocytes were sequentially digested using 0.25% Trypsin-EDTA (Gibco, Cat No. 25200-072) followed by 0.1% collagenase type II (Sigma-Aldrich, Cat No. C-BIOC) at 37 °C. The isolated primary chondrocytes were then cultured in DMEM/F12 medium supplemented with 10% FBS at 37 °C in a 5% CO2 atmosphere to maintain a monolayer. Similarly, the C28/I2 cell line (a Human normal chondrocyte cell line; Shzeye, Cat No. ZY-C6492H) was also cultured under the same conditions of 37 °C and 5% CO2 in DMEM/F12 medium containing 10% FBS.
Primary keratinocytes (KCs) were isolated from the skin of 1–2-day-old C57BL/6 J mice. Briefly, after peeling the skin from the mice, it was rinsed with DPBS (Gibco, Cat No. 14190144) and then treated with 4 mg/mL Dispase II (Roche, Cat No. D4693) at 4 °C for 18 h. Subsequently, the epidermis was separated from the dermis, and the epidermis was incubated with TrypLe™ Express enzyme (ThermoFisher, Cat No. 12604013) for 20 min to dissociate keratinocytes from the epidermis. The cell suspension was then filtered through a 70 µm mesh sieve and centrifuged to pellet the cells. After removing the supernatant, the isolated primary mouse keratinocytes were cultured at 37 °C in 5% CO22 in EpiLife™ keratinocyte growth medium (ThermoFisher, Cat No. MEPI500CA) supplemented with 1% EDGS growth factor (ThermoFisher, Cat No. S0125) and Calcium Chloride (60 µM). The HaCat cell line (a Human immortalized keratinocyte cell line; Procell, Cat No. CL-0090) was maintained at 37 °C in 5% CO2 in HaCat-specific medium (Procell, Cat No. CM-0090) containing 10% FBS.
Mouse primary synovial fibroblasts (SFs; iCell, Cat No. MIC-iCell-s004) and FLS (Human fibroblast-like synoviocytes; iCell, Cat No. iCell-008a) were both cultured at 37 °C in 5% CO2 in the iCell Primary Fibroblast Culture System containing 10% FBS (iCell, Cat No. PriMed-iCell-003).
Cells stimulation
For senescence induction in keratinocytes, we utilized ultraviolet radiation to induce senescence in well-conditioned KCs or HaCat cells. When cell density reached approximately 70%, the supernatant was removed, and a thin layer of DPBS was added. The cells were irradiated with a medium-wave ultraviolet irradiation device (Analytik Jena; Model UVM-28) at 302 nm, delivering a dose of 100mJ/cm^2. Following the irradiation, DPBS was discarded, and complete culture medium was added. The cells were then cultured at 37 °C in a 5% CO2 environment for 24 h. Subsequently, cellular proteins were harvested for the detection of aging-related markers through Western Blot and Immunofluorescence analysis.
For cell transfection, we utilized siRNA (Beijing Tsingke Biotech Co., Ltd.) to silence IL-36Ra in KCs and IL-36R in CCs and SFs. Following the manufacturer’s instructions, siRNA was diluted in Opti-MEM (ThermoFisher, Cat No. 31985070). The diluted siRNA (100 nM) was then transfected into cells using Lipofectamine 3000 (Invitrogen, Cat No. L3000150). After 72 h, cellular proteins were extracted to evaluate the efficiency of the knockdown.
For the application of mIL-36Ra (TargetMol, Cat No. TMPY-04275) or spesolimab (MedChemExpress; Cat No. HY-P99396) to cells, we collected the supernatants from senescent/IL-36Ra knockdown KCs to induce SFs to become OA-SFs. Then, the supernatants from the co-culture of senescent/IL-36Ra knockdown KCs with SFs were collected to induce CCs to become OA-CCs. On this basis, mIL-36Ra (200 ng/mL) or spesolimab (0.5 μM) was added to culture OA-SFs or OA-CCs to evaluate their effects on OA. Then, we treated HaCat cells, FLS, and C28/I2 cells in a similar manner. Spesolimab was added to culture OA-FLS or OA-C28/I2 to evaluate the drug’s effect on OA.
Synovial fibroblasts Migration
We conducted a scratch assay to assess the migration capacity of SFs. Well-conditioned SFs or FLS were seeded in 6-well plates and cultured until they reached nearly 100% confluence. After removing the culture medium, a sterile 200 μl pipette tip was used to create a cross-shaped scratch in each well. Subsequently, a medium containing 1% FBS and the respective intervention was added. After 24 h, the cells were fixed with 4% paraformaldehyde and stained with ammonium oxalate crystal violet for 10 min. The stain was then washed off, and five random fields of view per well were photographed and analyzed to measure the extent of cell migration from the edges of the scratch.
Alcian blue staining of chondrocyte clusters
Well-conditioned CCs or C28/I2 cells were seeded into 6-well plates and cultured until they reached approximately 95% confluence. The cells were then digested with trypsin and resuspended in 100 μl of DMEM/F12 containing 10% FBS. Using a pipette at 20 μl/drop of the high-concentration cell suspension were seeded into 12-well plates to form tightly connected clusters of chondrocytes with a diameter of 0.5 cm. After adhering to the plate for 6 h at 37 °C and 5% CO2, 10% FBS + DMEM/F12 medium, along with the respective intervention, was added. After 24 h, the cells were fixed with 4% paraformaldehyde and stained with Alcian Blue for 15-20 min, followed by washing off the stain. Images were acquired using a scanner, and the staining average intensity was quantified as integrated optical density (IOD) using Image Pro Plus (v6.0) software for assessment.
SA-β-Gal staining
To assess the senescence status of cells, we utilized the Senescence β-Galactosidase Staining Kit (Beyotime, Cat No. C0602) following the manufacturer’s protocol. Briefly, the induced CCs and SFs were fixed for 15 min. The fixative was then washed off, and the cells were incubated with SA-β-Gal staining solution at 37 °C in a sealed, light-protected environment for 16 h. After washing off the staining solution, five random areas were photographed in each culture dish to evaluate the degree of cellular senescence.
Western blot analysis
To isolate tissue proteins, the skin, heart, brain, spleen, lungs, kidneys, and intestines of 3-month-old and 18-month-old mice, and the esophagus, heart, brain, spleen, liver, pancreas, lungs, kidneys, stomach and intestines of Zmpste24fl/fl, Zmpste24-cKO, IL-36Rafl/fl, IL-36Ra-cKO mice were ground in liquid nitrogen. Following experimental induction, proteins from mouse CCs and SFs were extracted for immunoblot analysis. Briefly, tissue fragments or cells were lysed on ice using 2x SDS lysis buffer (1 M Tris-HCl-pH6.8, 10% SDS, glycerol, bromophenol blue, and Roche protease inhibitor cocktail). The lysates were then separated using 10% or 12% SDS-polyacrylamide gel electrophoresis, and the separated proteins were transferred to a 0.22μm PVDF membrane (Sigma; Cat No. ISEQ00010) using a wet transfer method. The PVDF membrane was then blocked with 5% skim milk on a rocking platform at room temperature for 1 h, followed by incubation with the primary antibody at 4 °C for 12-16 h. The next day, the membrane was incubated with the secondary antibody at room temperature for 1 h. Finally, protein bands were visualized and captured on a chemiluminescence imager (Tanon, Model 5200CE) using Ultra High Sensitivity ECL Kit (MedChemExpress, Cat No. HY-K1005). Detailed information regarding the primary and secondary antibodies used for Western blot analysis is provided in Supplementary Table 2.
Quantitative real-time PCR
Total RNA from induced SFs and FLS was extracted using Trizol reagent (TaKaRa; Cat No. 9108). The concentration was measured using a NanoDrop spectrophotometer (ThermoFisher Scientific; Cat No. 840-317500). Following the manufacturer’s instructions, cDNA was synthesized from total mRNA using a cDNA synthesis kit (Vazyme; Cat No. R333-01). RT-PCR was performed using ChamQ SYBR qPCR Master Mix (Vazyme; Cat No. Q311-02) on a LightCycler® 96 system (Roche) with LightCycler® 96 software version 1.1. Relative quantification was conducted using the 2-ΔΔCq method. The expression of each gene was normalized to the housekeeping gene GAPDH. Primer sequences (Beijing Tsingke Biotech Co., Ltd.) used for RT-PCR are listed in Supplementary Table 3. RT-PCR data were analyzed using Microsoft Excel version 16.36, and graphs were generated using GraphPad Prism 9.3.
RNA sequencing
Skin tissue from mice (IL-36Rafl/fl and IL-36Ra-cKO) and chondrocytes from mice (negative control group, senescent keratinocyte supernatant-stimulated group) were subjected to RNA sequencing analysis. In brief, total RNA was extracted using trizol reagent (following the manufacturer’s protocol, with skin tissues initially ground in liquid nitrogen before the addition of trizol), and the quality of RNA was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). mRNA was then enriched and isolated using oligo (dT) magnetic beads, followed by the addition of fragmentation reagent to fragment the mRNA. Subsequently, first-strand and second-strand cDNA synthesis reactions were prepared (using DNA polymerase I, RNase H, dNTPs, and buffer), and reaction programs were set up to synthesize first-strand and second-strand cDNA. The double-stranded cDNA was then end-repaired, A-tailed at the 3’ end, and adapters were ligated to the cDNA. PCR amplification of the product followed, and PCR products were denatured into single-stranded circular DNA molecules, which were then amplified via rolling circle amplification to form DNA nanoballs (DNBs). These DNBs were loaded onto high-density DNA nanoarray chips and sequenced at BGI Genomics Co., Ltd (Shenzhen, China) using the combinatorial Probe-Anchor Synthesis (cPAS) technology. Raw sequencing data were processed using SOAPnuke (v1.5.6) to generate clean data. Differentially expressed genes (DEGs) were identified using DESeq2 (v1.34.0), with significance defined as q‑value < 0.05 calculated by the Benjamini–Hochberg method. KEGG pathway enrichment analysis was performed using a hypergeometric test (Phyper) against the KEGG database (https://www.kegg.jp/), with q‑value < 0.05 as the significance threshold for enriched pathways. Gene Set Enrichment Analysis (GSEA) was performed using GSEA v3.0.
LC-MS/MS mass spectrometry-based proteomics
We collected dorsal skin tissues from 3-month-old and 18-month-old mice, removed hair, and immediately snap-froze the samples in liquid nitrogen. Protein sample preparation and liquid chromatography-tandem mass spectrometry (LC-MS/MS) using label-free quantification (LFQ) were performed at PTM Biolab Co., Ltd. (Hangzhou, Zhejiang, China). Initially, skin tissue proteins were extracted by grinding the skin of each group into powder with liquid nitrogen, followed by the addition of a fourfold volume of lysis buffer and sonication on ice using a high-intensity ultrasonic processor. After sonication, the supernatant was collected by centrifugation (4 °C; 12,000 × g; 10 min) to obtain protein samples, and the protein concentration was determined using a BCA Protein Assay Kit. Subsequently, the protein samples underwent 2-3 rounds of precooled acetone washing, then were dissolved in 200 mM TEAB and disrupted by sonication. Trypsin was added for overnight digestion. The samples were then reduced with 5 mM dithiothreitol at 56 °C for 30 min and alkylated with 11 mM iodoacetamide in the dark at room temperature for 15 min. The trypsin-digested peptides were dissolved in solvent A and loaded onto a homemade reverse-phase analytical column. Peptide separation was carried out using a mobile phase of water with 0.1% formic acid and 2% acetonitrile as solvent A, and water with 0.1% formic acid and 100% acetonitrile as solvent B, with a gradient of 4–22% B from 0 to 70 min, 22–30% B from 70 to 84 min, 30–80% B from 84 to 87 min, and 80% B from 87 to 90 min. Peptides were ionized using a capillary ion source and subsequently analyzed with a tims-TOF Pro mass spectrometer. The ion source voltage was set at 2.0 kV. Both precursor ions and their fragments were detected at high resolution by the TOF analyzer. MS/MS spectra were acquired within the mass range of 100–1700. Data acquisition was performed using the Parallel Accumulation Serial Fragmentation (PASEF) mode. For each full MS scan, 10 PASEF scans were collected for precursor ions with charges ranging from 0 to 5. Dynamic exclusion was enabled for 30 sec to avoid repeated analysis of identical precursor ions. The resulting MS/MS data were analyzed using MaxQuant (version 1.6.6.0). A reverse database was included to estimate the false discovery rate (FDR), and a common contaminant database was used to eliminate contamination proteins from the final identification results. The enzymatic digestion was set to Trypsin/P; the number of missed cleavage sites was set to 2; the minimum peptide length was set at 7 amino acid residues; the maximum number of modifications per peptide was set to 5; the precursor mass tolerance for the first search and the main search were set to 20 ppm and 20 ppm, respectively, and the mass tolerance for secondary fragment ions was set to 20 ppm. Carbamidomethyl (C) was set as a fixed modification for cysteine alkylation, with variable modifications including methionine oxidation, protein N-terminal acetylation, and deamidation (NQ). The FDR for protein identification and peptide-spectrum matches (PSMs) was set to 1%.
We identified significantly differentially abundant proteins using a combination of fold change (FC > 1.5 or < 1/1.5) and adjusted p-values. Specifically, we calculated the fold change (FC) as: FCA/B k = Mean (Rik, i ∈A) / Mean (Rik, i ∈B), where R represents the relative protein quantification value, i represents the sample, and k represents the protein. To assess statistical significance, p-values were calculated using an unpaired two-tailed Student’s t test on log2-transformed values: Pk = T. test (Log2 (Rik, i ∈A), Log2 (Rik, i ∈B)). To correct for multiple testing, the Benjamini-Hochberg procedure was applied to control the false discovery rate (FDR), and q-values were obtained. Proteins with an adjusted q-value < 0.05 and an absolute fold change > 1.5 (i.e., FC > 1.5 or FC < 1/1.5) were considered significantly differentially expressed, ensuring both statistical and biological relevance.
Elisa assays
Serum was collected from both young and aged mice, young and elderly humans, and synovial fluid from elderly OA patients. We also collected serum samples from D-Galactose-treated and their control male mice. Additionally, we collected serum from Zmpste24fl/fl, IL-36Rafl/fl, Zmpste24-cKO, and IL-36Ra-cKO male mice, as well as from Zmpste24-cKO, IL-36Ra-cKO male mice and their control male mice after the application of MN-mIL-36Ra or MN-SPE. Supernatants from UV-induced senescent KCs and HaCat cells were also collected. The levels of IL-36α, IL-36β, IL-36γ, IL-36Ra and D-Gal in these samples were determined using ELISA kits for IL-36α (Cat. No.: MM-45615M2), IL-36β (Cat. No.: MM-45619M2), IL-36γ (Cat. No.: MM-45212M2), IL-36Ra (Cat. No.: MM-48043M2), Spesolimab (Cat. No.: MM-95430O1) and D-Galactose (Cat. No.: MM-45619M2) according to the manufacturer’s instructions (Jiangsu Meimian Industrial Co., Ltd).
D-Galactose content assay
The D-galactose content in serum samples from D-Galactose-induced prematurely aged skin male mice (8 weeks) and their control C57BL/6 J male mice was measured using a D-Galactose Content Assay Kit (ADS-W-TDX046, Jiangsu Meimian Industrial Co., Ltd) according to the manufacturer’s instructions. Briefly, Serum samples were centrifuged at 12,900 × g for 10 min at room temperature, and the supernatant was collected. The assay uses D-galactose reacting with galactose dehydrogenase to form NADH, which produces a yellow compound with an absorbance peak at 450 nm. Supernatants were added to a 96-well plate with the kit reagents, incubated at 25 °C, and absorbance was measured at 450 nm (A1). After adding a second reagent, the plate was incubated for 20 min, and absorbance (A2) was measured again. The change in absorbance (ΔA = A2 - A1) was used to calculate the D-galactose content, expressed as μg/ml.
Statistical analysis
All reported data were derived from at least three independent biological replicates, with the number of biological replicates indicated by “n” in the figures. All data originate from these biological replicates and are presented as mean ± standard deviation (S.D.). We used the Shapiro-Wilk test to assess data normality. The unpaired, two-tailed Student’s t test or Mann-Whitney rank sum test (for non-normally distributed data) was used to compare two independent groups. For comparing multiple values, we used a one-way analysis of variance (ANOVA) with Tukey’s post hoc test or Kruskal-Wallis test (for non-normally distributed data) with Dunn’s multiple comparisons test. All statistical tests used were two-sided. Values of p < 0.05 were considered statistically significant. Finally, we generated the graphs using GraphPad Prism version 9.3. For the construction of all animal and cell models, as well as drug administration, the operators were blinded to the group information. We ensured blinding during the data collection process by assigning random IDs to all samples, preventing access to group information. Histological staining, WB, RT-PCR, IHC, and IF were carried out by individuals other than the experiment designers. For histological analysis, the analysts were blinded to the sample information, including the genotype of the mice, any drug treatments, or surgical conditions.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The original RNA-seq data generated in this study have been deposited in the GEO database under accession code GSE263907 and GSE264103. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD062165. All the data supporting the findings of this study are available within the article and its Supplementary Information file. Source data are provided in this paper.
References
Hunter, D. J. & Bierma-Zeinstra, S. Osteoarthritis. Lancet 393, 1745–1759 (2019).
Robinson, W. H. et al. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 12, 580–592 (2016).
Kloppenburg, M. Inflammation is a relevant treatment target in osteoarthritis. Lancet 402, 1725–1726 (2023).
Scanzello, C. R. Role of low-grade inflammation in osteoarthritis. Curr. Opin. Rheumatol. 29, 79–85 (2017).
De Roover, A., Escribano-Núñez, A., Monteagudo, S. & Lories, R. Fundamentals of osteoarthritis: Inflammatory mediators in osteoarthritis. Osteoarthr. Cartil. 31, 1303–1311 (2023).
Greene, M. A. & Loeser, R. F. Aging-related inflammation in osteoarthritis. Osteoarthr. Cartil. 23, 1966–1971 (2015).
Loeser, R. F. Aging and osteoarthritis. Curr. Opin. Rheumatol. 23, 492–496 (2011).
Lee, Y. I., Choi, S., Roh, W. S., Lee, J. H. & Kim, T.-G. Cellular Senescence and Inflammaging in the Skin Microenvironment. Int. J. Mol. Sci. 22, 3849 (2021).
Franco, A. C., Aveleira, C. & Cavadas, C. Skin senescence: mechanisms and impact on whole-body aging. Trends Mol. Med. 28, 97–109 (2022).
Hu, L. et al. Epidermal dysfunction leads to an age-associated increase in levels of serum inflammatory cytokines. J. Invest. Dermatol. 137, 1277–1285 (2017).
Pilkington, S. M., Bulfone-Paus, S., Griffiths, C. E. M. & Watson, R. E. B. Inflammaging and the skin. J. Invest. Dermatol. 141, 1087–1095 (2021).
Pilkington, S. M., Bulfone-Paus, S., Griffiths, C. E. M. & Watson, R. E. B. Can Skin aging contribute to systemic inflammaging? J. Invest. Dermatol. 142, 484–485 (2022).
Elias, P. M. Barrier function in aging: comments on pilkington et al. “Inflammaging and the Skin”. J. Invest. Dermatol. 142, 483–484 (2022).
Ritchlin, C. Psoriatic disease—from skin to bone. Nat. Clin. Practice Rheumatol. 3, 698–706 (2007).
Schett, G. et al. Psoriatic arthritis from a mechanistic perspective. Nat. Rev. Rheumatol. 18, 311–325 (2022).
Villanova, F., Di Meglio, P. & Nestle, F. O. Biomarkers in psoriasis and psoriatic arthritis. Ann. Rheum. Dis. 72, ii104–ii110 (2013).
Elias, M. et al. IL-36 in chronic inflammation and fibrosis — bridging the gap? J. Clin. Invest. 131, e144336 (2021).
Buhl, A.-L. & Wenzel, J. Interleukin-36 in Infectious and Inflammatory Skin Diseases. Front. Immunol. 10, 1162 (2019).
Frey, S. et al. The novel cytokine interleukin-36α is expressed in psoriatic and rheumatoid arthritis synovium. Ann. Rheum. Dis. 72, 1569–1574 (2013).
Iznardo, H. & Puig, L. Exploring the Role of IL-36 Cytokines as a New Target in Psoriatic Disease. Int. J. Mol. Sci. 22, 4344 (2021).
Lamacchia, C. et al. The severity of experimental arthritis is independent of IL-36 receptor signaling. Arthritis Res. Ther. 15, R38 (2013).
Dietrich, D. & Gabay, C. IL-36 has proinflammatory effects in skin but not in joints. Nat. Rev. Rheumatol. 10, 639–640 (2014).
Li, T. et al. TGF-beta type 2 receptor-mediated modulation of the IL-36 family can be therapeutically targeted in osteoarthritis. Sci. Transl. Med. 11, eaan2585 (2019).
Onuora, S. IL-36 inhibition to treat OA. Nat. Rev. Rheumatol. 15, 386 (2019).
Yi, Y. -h et al. Injectable temperature-sensitive hydrogel loaded with IL-36Ra for the relief of osteoarthritis. ACS Biomater. Sci. Eng. 9, 1672–1681 (2023).
Varela, I. et al. Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature 437, 564–568 (2005).
Umbayev, B. et al. Galactose-induced skin aging: the role of oxidative stress. Oxid. Med. Cell Longev. 2020, 7145656 (2020).
Liang, W. et al. Skin chronological aging drives age-related bone loss via secretion of cystatin-A. Nat. Aging 2, 906–922 (2022).
Watanabe, S. et al. Neutrophil extracellular traps are induced in a psoriasis model of interleukin-36 receptor antagonist-deficient mice. Sci. Rep. 10, 20149 (2020).
Blair, H. A. Spesolimab: first approval. Drugs 82, 1681–1686 (2022).
Ratnarajah, K., Jfri, A., Litvinov, I. V. & Netchiporouk, E. Spesolimab: a novel treatment for pustular psoriasis. J. Cutan. Med. Surg. 24, 199–200 (2020).
Ganesan, R. et al. Generation and functional characterization of anti-human and anti-mouse IL-36R antagonist monoclonal antibodies. mAbs 9, 1143–1154 (2017).
Motta, F., Barone, E., Sica, A. & Selmi, C. Inflammaging and Osteoarthritis. Clin. Rev. Allergy Immunol. 64, 222–238 (2022).
Berenbaum, F. Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis!). Osteoarthritis Cartilage 21, 16–21 (2013).
Knights, A. J., Redding, S. J. & Maerz, T. Inflammation in osteoarthritis: the latest progress and ongoing challenges. Curr. Opin. Rheumatol. 35, 128–134 (2023).
Veale, D. J. & Fearon, U. The pathogenesis of psoriatic arthritis. Lancet 391, 2273–2284 (2018).
Van Raemdonck, K. et al. TLR7 endogenous ligands remodel glycolytic macrophages and trigger skin-to-joint crosstalk in psoriatic arthritis. Eur. J. Immunol. 51, 714–720 (2020).
Lieberthal, J., Sambamurthy, N. & Scanzello, C. R. Inflammation in joint injury and post-traumatic osteoarthritis. Osteoarthritis Cartilage 23, 1825–1834 (2015).
Edd, S. N., Giori, N. J. & Andriacchi, T. P. The role of inflammation in the initiation of osteoarthritis after meniscal damage. J. Biomech. 48, 1420–1426 (2015).
Boutet, M. A. et al. Interleukin-36 family dysregulation drives joint inflammation and therapy response in psoriatic arthritis. Rheumatology 59, 828–838 (2020).
Ni, X. et al. IL-17D-induced inhibition of DDX5 expression in keratinocytes amplifies IL-36R-mediated skin inflammation. Nat. Immunol. 23, 1577–1587 (2022).
Hovhannisyan, Z. et al. Enhanced IL-36R signaling promotes barrier impairment and inflammation in skin and intestine. Sci. Immunol. 5, eaax1686 (2020).
Walsh, P. T. & Fallon, P. G. The emergence of the IL-36 cytokine family as novel targets for inflammatory diseases. Ann. N. Y. Acad. Sci. 1417, 23–34 (2016).
Fukaura, R. & Akiyama, M. Targeting IL-36 in Inflammatory Skin Diseases. BioDrugs 37, 279–293 (2023).
Wood, M. J., Miller, R. E. & Malfait, A.-M. The genesis of pain in osteoarthritis: inflammation as a mediator of osteoarthritis pain. Clin. Geriatr. Med. 38, 221–238 (2022).
Mathieu, S., Tournadre, A., Soubrier, M. & Sellam, J. Effect of disease-modifying anti-rheumatic drugs in osteoarthritis: A meta-analysis. Joint Bone Spine 89, 105444 (2022).
Schieker, M. et al. Effects of Interleukin-1β Inhibition on Incident Hip and Knee Replacement. Annal. Int. Med. 173, 509–515 (2020).
Zhou, P. et al. Strategy for osteoarthritis therapy: Improved the delivery of triptolide using liposome-loaded dissolving microneedle arrays. Int. J. Pharmaceutics 609, 121211 (2021).
Lorenz, J. & Grassel, S. Experimental osteoarthritis models in mice. Methods Mol. Biol. 1194, 401–419 (2014).
Glasson, S. S., Blanchet, T. J. & Morris, E. A. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage 15, 1061–1069 (2007).
Pritzker, K. P. et al. Osteoarthritis cartilage histopathology: grading and staging. Osteoarthritis Cartilage 14, 13–29 (2006).
Glasson, S. S., Chambers, M. G., Van Den Berg, W. B. & Little, C. B. The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage 18, S17–S23 (2010).
Fan, A. et al. Inhibition of fibroblast activation protein ameliorates cartilage matrix degradation and osteoarthritis progression. Bone Res. 11, 3 (2023).
Das Neves Borges, P., Vincent, T. L. & Marenzana, M. Automated assessment of bone changes in cross-sectional micro-CT studies of murine experimental osteoarthritis. PLoS ONE 12, e0174294 (2017).
Krenn, V. et al. Synovitis score: discrimination between chronic low-grade and high-grade synovitis. Histopathology 49, 358–364 (2006).
Little, C. B. et al. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 60, 3723–3733 (2009).
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (NO. 32471180 to K. L.), Basic and Applied Basic Research Foundation of Guangdong Province (2021A1515011004 to E.K., 2023A1515140035 to C.W. and K.L.), Guangzhou Municipal Science and Technology Bureau (2024A04J9995 to K.L.), Grants from Guangxi Key Laboratory of basic and translational research of Bone and Joint Degenerative Diseases (21-220-06 to K.L.). The authors thank Hong Wang, Shuqin Zhang, Wenping Chen and Pinglin Lai for providing secretarial assistance and technical support.
Author information
Authors and Affiliations
Contributions
K.L. and X.B. conceived the ideas for experimental designs and supervised the study. D.C. and C.Y. conducted the majority of the experiments, analyzed data and helped with manuscript preparation. H.L., Y.X., and J.W. conducted cell cultures, immunoblot experiments and qRT-PCR experiments. P.Y., L.C., Q.C., and Z. Z. maintained mice, collected tissue samples, acquired histology, confocal imaging and analysis. C.W. and Z.W. prepared the microneedles loaded with mIL-36Ra and analyzed the characterization. K.L. and E.K. wrote and revised the manuscript. X.G.W., X.B., and B.H. supervised the project and critically reviewed the manuscript. All authors have approved the submitted version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Huilin Li, who co-reviewed with Yue Pan, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, D., Wang, C., Yang, C. et al. Aged skin exacerbates experimental osteoarthritis via enhanced IL-36R signaling. Nat Commun 17, 1695 (2026). https://doi.org/10.1038/s41467-026-68399-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-68399-z
