
Abstract
Bacterial cellulose possesses excellent biocompatibility and mechanical strength but lacks the bioactivity needed for many biomedical and healthcare applications. To address this limitation, we develop a metabolic glycoengineering–click chemistry strategy that enables in situ incorporation of azide groups into bacterial cellulose, followed by mild and selective conjugation of alkyne-bearing functional molecules. This approach avoids harsh chemical treatments, preserves the native properties of bacterial cellulose, and supports stable attachment of diverse bioactive agents, including antibacterial porphyrins, arginine-glycine-aspartic acid peptides, and recombinant proteins with fluorescent or enzymatic functions. As a proof-of-concept, a cascade catalytic system comprising glucose oxidase and superoxide dismutase is immobilized onto azide-modified bacterial cellulose, yielding a multifunctional wound dressing designed to address hyperglycemia and oxidative stress—key barriers to chronic wound healing. In male diabetic mice, this glucose oxidase/superoxide dismutase-integrated bacterial cellulose dressing (low endotoxin <0.1 EU/mL) accelerates wound closure to 92.1% by day 14, significantly outperforming the controls. Our strategy highlights a scalable and bio-orthogonal route for enhancing bacterial cellulose with user-defined bioactivities, thereby expanding its utility in advanced biomaterials development.
Similar content being viewed by others


Introduction
Bacterial cellulose (BC), produced by various Acetobacteraceae species, has emerged as a versatile biomaterial due to its unique physicochemical properties, including high purity, mechanical strength, and biocompatibility1,2,3,4. Unlike plant-derived cellulose, BC’s three-dimensional nanofiber network, free of lignin and hemicellulose, provides exceptional porosity, water retention, and structural robustness5. These attributes have spurred its use in applications ranging from biodegradable composites, flexible electronics, to engineered living materials6,7,8,9. However, BC inherently lacks bioactive functionalities, such as antimicrobial, anti-inflammatory, anti-oxidative, and cell-adhesive properties, critical for advanced biomedical and healthcare applications10. While strategies to functionalize BC exist, they often compromise its structural integrity, biocompatibility, or scalability, highlighting the need for innovative modification approaches11.
Current functionalization methods rely on physical adsorption or chemical grafting. Physical adsorption, though mild, suffers from rapid agent leaching due to weak non-covalent interactions12,13. Chemical modifications, while more stable, typically require harsh conditions (e.g., strong acids, toxic solvents) to activate BC’s hydroxyl groups, risking material degradation and biocompatibility loss14,15,16. Moreover, the dense hydrogen-bonding network in BC restricts its solubility in water and most organic solvents, posing challenges for efficient modification and limiting scalability17.
Recent advancements in metabolic glycoengineering have opened new possibilities for modifying the features of BC materials18,19. This approach enables the biosynthetic incorporation of noncanonical monosaccharide derivatives bearing tailored chemical moieties, such as fluorescent molecules20,21 or antimicrobial agents22, directly into the growing polysaccharide chains. Following the paradigm of non-natural amino acid incorporation in recombinant proteins23, researchers have successfully fed synthesized sugar precursors to cellulose-producing bacteria (e.g., Komagataeibacter sucrofermentans), yielding ‘built-in’ functionalities within BC products24. For instance, Liu et al. developed a photosensitizer-modified sugar derivative (TPEPy) to generate a light-activated antimicrobial BC, which effectively killed pathogens and supported wound healing with minimal antibiotic use22. They then extended this concept to engineer a two-layered BC structure embedded with photosensitizers and VEGF-secreting cells, thereby combining antibacterial and tissue-regenerative functions in chronic wound healing25. However, despite progress in employing metabolic glycoengineering for tailoring BC materials, several critical challenges remain unresolved. Firstly, chemical engineering of biocompatible monosaccharide derivatives remains labor-intensive and inefficient, with typical synthesis routes requiring at least three reaction steps to achieve desired substitutions with suboptimal yields20,22,25. Secondly, even after the derivatives have been successfully synthesized, they often face stringent metabolic barriers, which either impair Komagataeibacter growth or reduce BC yields26. Last but not least, the compatibility, stability, and functionality of each derivative must be evaluated through case-specific studies, requiring extensive time and resources22,25,27. These limitations collectively constrain the broader application of metabolic glycoengineering for BC functionalization, particularly in scenarios demanding high versatility, bioactivity, and scalability.
To address these challenges, we develop a bioengineering platform that combines metabolic glycoengineering with click chemistry, for robust and bio-orthogonal functionalization of BC28. Our strategy leverages the previously unrecognized metabolic adaptability of Komagataeibacter to incorporate GlcNAz (an azide-modified N-acetylglucosamine analog) into BC during biosynthesis. Remarkably, GlcNAz supplementation at concentrations up to 2 mM does not inhibit BC growth, while efficiently enabling the incorporation of azide groups. These azide moieties facilitate subsequent copper-catalyzed azide-alkyne cycloaddition (CuAAC) and strain-promoted azide-alkyne cycloaddition (SPAAC) with alkyne-tagged molecules under mild, aqueous conditions29. This strategy bypasses toxic reagents, preserves BC’s native properties, and supports stable conjugation of diverse functionalities, including antimicrobial porphyrins, RGD peptides, and biocatalytic enzymes (Fig. 1). To demonstrate the therapeutic potential, we covalently immobilize a glucose oxidase (GOx)/superoxide dismutase (SOD) enzymatic cascade onto azide-functionalized BC, generating a dual-enzyme hydrogel designed to target hyperglycemia and oxidative stress in chronic wounds. In male diabetic (db/db) mice, this enzyme-bearing BC dressing accelerates wound closure to 92.12% by day 14, significantly outperforming pure BC (77.05%), gauze (59.93%), and untreated wounds (44.78%). Our platform offers a scalable, modular route to engineer BC with user-defined bioactivities, addressing longstanding challenges in bioactive BC biomaterial design.

(i) Biosynthesis of GlcNAz-BC: Komagataeibacter rhaeticus (K. rhaeticus) is co-fed with glucose and N-azidoacetylglucosamine (GlcNAz), enabling metabolic incorporation of azide groups into the growing cellulose matrix. (ii) Bio-orthogonal functionalization: Azide-modified BC (i.e., GlcNAz-BC) serves as a versatile scaffold for covalent conjugation with alkyne-tagged molecules (e.g., small compounds, peptides, and proteins) via copper-catalyzed azide-alkyne cycloaddition (CuAAC), yielding BC with tailored bioactivities under mild, aqueous conditions.
Results
Design and characterization of GlcNAz-functionalized BC (GlcNAz-BC)
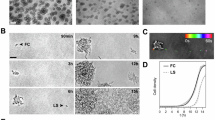
To engineer bioactive BC, we introduced azide moieties via metabolic incorporation of N-azidoacetylglucosamine (GlcNAz). GlcNAz was synthesized through amidation of glucosamine’s amino group with azidoacetic acid30. Its chemical structure was confirmed via Nuclear Magnetic Resonance (NMR) and Electrospray Ionization Mass Spectrometry (ESI-MS) (Supplementary Figs. 1, 2). Supplementation of GlcNAz together with glucose into K. rhaeticus cultures during fermentation results in BC with metabolically integrated azide groups (GlcNAz-BC). To confirm azide reactivity, we performed CuAAC with di(butynyl) bispyridinium salt (VA), a fluorescent alkyne probe31 (Fig. 2a and Supplementary Fig. 3). Post-reaction analysis using confocal laser scanning microscopy (CLSM) revealed a nearly 5-fold increase in fluorescence intensity in GlcNAz-modified BC (GlcNAz-BC), compared to BC controls mixed with VA, GlcNAz, or both. These results confirm the successful covalent incorporation of azide groups on BC during fermentation and exclude non-specific adsorption of GlcNAz or VA as the source of fluorescence (Fig. 2a and Supplementary Fig. 4). Unlike 2-azido-2-deoxy-D-glucose (deO), a previously established azide-functionalization agent for microbial polysaccharide25, feeding 1 mM GlcNAz to cellulose-producing strains resulted in BC with a 4.61-fold higher fluorescence intensity after click-labeling with VA (Fig. 2b), indicating superior azide incorporation efficiency. We hypothesized that GlcNAz’s structural configuration enhances compatibility with metabolic glycoengineering pathways, based on analogous mechanisms observed in sialic acid metabolic pathways32. Moreover, molecular modeling revealed that the azide moiety in GlcNAz extends farther from the cellulose backbone (7.43 Å) compared to that in deO-modified cellulose (4.11 Å), where it remains sterically constrained. This enhanced spatial projection likely facilitates improved accessibility for cycloaddition reactions, consistent with both the lower calculated energy barrier and the stronger fluorescence intensity observed upon click labeling (Fig. 2b and Supplementary Fig. 5).

a Schematic of fluorescence labeling of BC samples using di(butynyl) bispyridinium salt (VA) molecules, and quantification of mean fluorescence intensity (arbitrary units, AU) by CLSM. Groups include: BC, BC + VA (natural BC incubated with VA), BC + GlcNAz + VA (natural BC incubated with free VA and GlcNAz molecules), and GlcNAz-BC + VA (GlcNAz-modified BC incubated with VA). Fluorescence intensity was quantified using ImageJ (n = 3 per group; statistical significance was determined using one-way ANOVA; ***p < 0.001). All reactions were catalyzed by copper(I). b Comparative analysis of mean fluorescence intensity for labeled BC materials under various GlcNAz or 2-azido-2-deoxy-D-glucose (deO) feeding concentrations and CLSM images of deO-BC + VA and GlcNAz-BC + VA (scale bar: 100 μm) (n = 3 per group). c Comparative analysis of the macro- and micro-scale structure features of BC and GlcNAz-BC (scale bar: 1 μm). The experiment was repeated three times independently and yielded comparable results. d Thickness measurements of BC and GlcNAz-BC membranes over a 5-day culturing period at a GlcNAz concentration of 1 mM (n = 3 per group). e FT-IR spectra highlighting the chemical differences between BC and GlcNAz-BC. The experiment was repeated three times independently and yielded comparable results. f Tensile stress-strain curves comparing the mechanical properties of BC and GlcNAz-BC. The experiment was repeated three times independently and yielded comparable results. g Thermogravimetric analysis (TGA) profiles showing the thermal stability of BC and GlcNAz-BC. The experiment was repeated three times independently and yielded comparable results. h Enzymatic degradation rates of BC and GlcNAz-BC in cellulase solution (n = 3 per group; statistical significance was determined using unpaired Student’s t-tests; n.s., not significant, p = 0.518). i Cell viability assessment of BC and GlcNAz-BC using the CCK-8 assay (n = 5 per group; statistical significance was determined using one-way ANOVA; n.s., not significant, p = 0.878, p = 0.997). Data are presented as mean ± SD. Source data for this figure is available in the Source Data file.
Concentration optimization experiments with varying GlcNAz during fermentation, followed by click reaction with VA, revealed peak fluorescence at 1 mM (Fig. 2b), indicating saturation of azide group incorporation via metabolic labeling. No further enhancement occurred at higher GlcNAz concentrations, likely due to substrate competition or cytotoxicity limitation28,33,34. Spatial uniformity of azide distribution was confirmed via cross-sectional CLSM imaging, which showed consistent fluorescence signals throughout all layers of the cellulose network, indicating uniform grafting of azide groups across the entire BC matrix (Supplementary Fig. 6).
Post fermentation at 30 °C for 5 days, GlcNAz-BC and BC were ultrasonically cleaned and freeze-dried. Both materials exhibited similar appearances, characterized by white, loose, and porous sheets (Fig. 2c). However, quantitative analysis of scanning electron microscopy (SEM) images revealed that the average pore diameter in GlcNAz-BC was 0.1 µm smaller than that of unmodified BC (Fig. 2c and Supplementary Fig. 7), presumably due to enhanced intermolecular interactions and denser packing of the nanofiber network introduced by the azide modification. While this compact structure is evident in the dry state, the swelling of the cellulose matrix in the reaction medium ensures that the accessibility of azide groups for the subsequent CuAAC reaction is maintained.
Introducing GlcNAz did not affect BC pellicle thickness or yield at concentrations below 2 mM, whereas BC production declined at ≥5 mM, indicating that only excessive GlcNAz supplementation impairs cellulose biosynthesis (Fig. 2d and Supplementary Fig. 8). Fourier-transform infrared spectroscopy (FTIR) analysis further verified successful azide incorporation, as evidenced by distinct peaks at 1540.6 cm−1 and 1647.7 cm−1 in the GlcNAz-BC spectra, corresponding to the C = O stretching vibrations of acylamino groups (Fig. 2e). Mechanical testing demonstrated comparable Young’s moduli for GlcNAz-BC and native BC, but GlcNAz-BC exhibited a 120% increase in fatigue strain (Fig. 2f), consistent with azide-mediated hydrogen-bond reinforcement. Thermogravimetric (TG) analysis, biodegradation assays, and cytocompatibility tests showed no significant differences between GlcNAz-BC and unmodified BC (Fig. 2g–i), indicating that azide functionalization did not compromise thermal stability, degradability, or biocompatibility.
Finally, to evaluate the generalizability of our metabolic glycoengineering strategy, we expanded testing to Komagataeibacter xylinus and Komagataeibacter sucrofermentans. Together with the previously characterized K. rhaeticus, all three species tolerated 1 mM GlcNAz supplementation without detectable effects on growth or BC production, and the fluorescent VA probe consistently and robustly labeled the resulting BC materials across strains after CuAAC (Supplementary Fig. 9). These results demonstrate that the metabolic glycoengineering approach is broadly applicable to multiple Komagataeibacter species, enabling reliable and modular functionalization of BC in a strain-independent manner.
Functionalization of azide-modified bacterial cellulose (GlcNAz-BC) for tailored applications
To demonstrate the versatility of GlcNAz-BC as a functionalization platform, we systematically explored its application in incorporating small chemical molecules, peptides, and proteins with distinct biological functionalities (Fig. 3a). This investigation covers three main functionalization modules, each highlighting specific advantages and potential applications of the modified BC materials.

a Schematic illustration depicting the synthesis of multifunctional BC materials through click chemistry and the alkynylation of bioactive proteins. b SEM images of C. albicans and S. aureus following incubation on Por-BC and BC surfaces (scale bar: 1 μm). The experiment was repeated three times independently and yielded comparable results. c Colony-forming unit (CFU) counts of C. albicans on Por-BC versus BC, determined by plate counting (n = 3 per group, Statistical significance was determined using unpaired Student’s t-tests; **p = 0.00573, ***p = 2.79E-06). d Colony-forming unit (CFU) counts of S. aureus on Por-BC versus BC, determined by plate counting (n = 3 per group; statistical significance was determined using unpaired Student’s t-tests; ***p < 0.001). e CLSM images of L929 cells cultured on RGD-BC and BC. Actin (red) and cell nuclei (blue) were fluorescently labeled (scale bar = 100 µm). The experiment was repeated three times independently and yielded comparable results. f Quantification of the averaged cell area on each membrane (n = 6 per group, Statistical significance was determined using unpaired Student’s t-tests. ***p = 1.60E-10.) using ImageJ. g The number of attached cells (per 1 mm²) on RGD-BC and BC (n = 3 per group, Statistical significance was determined using unpaired Student’s t-tests. ***p = 2.28E-03). h Fluorescence images illustrating the conjugation of mCherry proteins onto BC materials. Control groups include Alkyne (−): GlcNAz-BC incubated with non-alkyne-modified mCherry, and Azide (−): alkyne-tagged mCherry incubated with natural BC lacking azide groups (scale bar = 200 μm). The experiment was repeated three times independently and yielded comparable results. i Representative chemiluminescence images of BC samples treated with luminol (scale bar = 1 cm). The experiment was repeated three times independently and yielded comparable results. j Mean fluorescence intensity of mCherry proteins on BC materials (n = 3 per group; statistical significance was determined using one-way ANOVA; ***p < 0.001). k Measured chemiluminescence lifetime of BC samples incubated with luminol (n = 3 per group; statistical significance was determined using one-way ANOVA; ***p < 0.001). Data are presented as mean ± SD. Source data for this figure is available in the Source Data file.
Beginning with small molecule functionalization, we developed an antibacterial BC material by covalently attaching alkyne-tagged photo-bactericidal porphyrin (Zn-Epor-PS)35 to GlcNAz-BC through CuAAC reaction, yielding porphyrin-modified BC (Por-BC) (Fig. 3a and Supplementary Fig. 10). The bactericidal efficacy of Por-BC was evaluated against Candida albicans and Staphylococcus aureus (1×108 CFUs each) under calibrated white light irradiation. SEM analysis revealed that light exposure induced notable deformation and collapse in microorganisms on Por-BC surfaces, while control samples (e.g., BC and GlcNAz-BC) maintained intact cellular structures (Fig. 3b and Supplementary Fig. 11). Quantitative assessment through colony-forming unit (CFU) counting demonstrated Por-BC’s potent antibacterial activity, achieving 90% reduction of C. albicans after 30 min and over 99% reduction of S. aureus within 10 minutes of light exposure (Fig. 3c-d and Supplementary Figs. 11, 12). These results validate the successful integration of photo-bactericidal properties into BC through porphyrin functionalization.
Extending our approach to peptide incorporation, we functionalized the azide-bearing BC with Pra-RGD peptide using the CuAAC method under mild aqueous conditions (Fig. 3a). The resulting RGD-modified BC (RGD-BC) was evaluated for its biological activity using L929 fibroblast cells36. Phalloidin/DAPI staining revealed well-developed cytoskeletal structures on the RGD-BC surface, indicating effective cell spreading and cytoskeleton organization. Conversely, cells on BC and GlcNAz-BC remained round, showing limited attachment and spreading (Fig. 3e and Supplementary Fig. 13). The spreading area of a single cell on the surface of RGD-BC is 6.5 times that of the BC (Fig. 3f). Comparative studies between native BC and RGD-BC revealed dramatic improvements in cellular response, with RGD-BC demonstrating a 12.8-fold increase in viable cell count compared to native BC (Fig. 3g and Supplementary Fig. 14). Similarly, BC materials grafted with RGD via SPAAC showed comparable enhancements in cellular adhesion behavior (Supplementary Fig. 13), underscoring the generality and robustness of our conjugation strategy. Although PAR (4-(2-pyridylazo) resorcinol) assay-based measurements confirmed that GlcNAz-BC adsorbs only minimal copper under CuAAC conditions (Supplementary Fig. 15), and that thorough washing reduces residual copper to trace levels (Supplementary Fig. 16), the metal-free SPAAC approach eliminates the need for Cu(I) catalysts entirely and may therefore be preferable for applications requiring heightened biocompatibility or regulatory compliance.
While small molecules and peptides could be seamlessly grafted onto BC, protein conjugation posed hurdles, mainly attributed to their fragile molecular structures and lack of inherent alkyne groups necessary for click reactions. Many conventional methods rely on harsh reagents and conditions that can compromise protein integrity and bioactivity, making their application tricky. Aiming to overcome these challenges, we adopted a site-selective functionalization technique that modifies methionine residues in proteins, using But-3-yn-1-yl 4-(vinylsulfonyl)benzoate (BVSB) to introduce alkyne groups while preserving the native protein structure (Fig. 3a and Supplementary Fig. 17)37. This strategy employs photoexcited lumiflavin as a catalyst under mild alkaline aqueous conditions, offering a gentle yet effective way to prepare proteins for subsequent conjugation (Fig. 3a).
As a proof of concept, we modified recombinant mCherry using this method (Supplementary Fig. 18) and subsequently conjugated it onto GlcNAz-BC via click chemistry, resulting in the formation of mCherry-BC. Fluorescence microscopy confirmed successful protein grafting, as mCherry-BC samples exhibited 4-6 times higher fluorescence intensity compared to two control groups: the natural BC (lacking azide groups) reacted with alkyne-mCherry, and the azide-functionalized BC reacted with mCherry (lacking alkyne groups) (Fig. 3h, j). Recognizing the broader potential of this methodology, we extended our strategy to the immobilization of enzymes such as horseradish peroxidase (HRP) on GlcNAz-BC. Using the same click chemistry method, we conjugated HRP onto GlcNAz-BC. Following incubation with 5 mM luminol and 0.3% (v/v) H2O2, the chemiluminescence lifetime was immediately measured. The HRP-grafted BC (HRP-BC) exhibited a notably longer emission duration ( > 30 s) compared to pure BC (non-luminous) and BC carrying physically adsorbed HRP (HRP/BC) (short quenching time ≈3 s), confirming successful enzyme functionalization and enhanced stability via covalent attachment (Fig. 3i, k).
Our method’s flexibility is underscored by its applicability to a wide range of proteins and enzymes, thus establishing that biomaterials can be functionally different depending on the specific therapeutic requirements. To demonstrate the practical utility of our protein-functionalized BC platform, we then focused on a clinically relevant and therapeutically demanding application—chronic wound healing.
Functional wound dressings enabled by enzymatic functionalization of BC with GOx and SOD
When faced with complicated wound environments, particularly in cases like diabetic wounds, single functional wound dressings often fail to meet diverse requirements. The healing of diabetic wounds remains a clinical challenge due to the complexity of the wound microenvironment, which features hyperglycemia, hypoxia, and excess oxidative stress38. Addressing this, we planned to orchestrate an intricate therapeutic response through a multifunctional dressing, employing click chemistry. We aimed to incorporate vital enzymes central to wound repair, such as GOx and SOD, into our GlcNAz-BC scaffold (Fig. 4a).

a Schematic illustration of the GOx/SOD-BC dressing and its proposed mechanism of reaction. b DPPH assay absorbance curves showing the free radical scavenging activity of BC, physically adsorbed SOD (SOD/BC), covalently bound SOD (SOD-BC), and dual-enzyme system (GOx/SOD-BC). Milli-Q water served as a blank control. The experiment was repeated three times independently and yielded comparable results. c Corresponding DPPH radical scavenging rates calculated at 519 nm for BC, SOD/BC, SOD-BC, and GOx/SOD-BC (n = 3 per group; statistical significance was determined using one-way ANOVA; ***p < 0.001). d Representative fluorescence images showing intracellular ROS levels in RAW264.7 cells treated with BC or GOx/SOD-BC. Endo indicates exogenously added endotoxin ( − Endo, absence; +Endo, presence). Images were obtained using an inverted fluorescence microscope (scale bar: 200 μm). The experiment was repeated three times independently and yielded comparable results. e Absorbance curves from the GOx kit assay for BC, GOx/BC, GOx-BC, and GOx/SOD-BC. Milli-Q water served as a blank control. The experiment was repeated three times independently and yielded comparable results. f Calculated GOx enzyme activity of BC, GOx/BC, GOx-BC, and GOx/SOD-BC (n = 3 per group; statistical significance was determined using one-way ANOVA; ***p < 0.001). Data are presented as mean ± SD. Source data for this figure is available in the Source Data file.
GOx reduces glucose levels by catalyzing its oxidation into gluconic acid and hydrogen peroxide39. However, the generated hydrogen peroxide can lead to the production of reactive oxygen species (ROS), which may exacerbate inflammation and tissue injury. To counteract this harmful effect, SOD is typically incorporated into engineered materials to convert ROS (specifically, superoxide anions, O2⁻) into less harmful molecules, such as oxygen (O2) and hydrogen peroxide. Subsequently, hydrogen peroxide is further converted into non-toxic oxygen and water, effectively reducing oxidative stress and alleviating hypoxia40 (Fig. 4a). Thus, the BC composite bearing the GOx and SOD enzymes would provide a coordinated, enzymatic antioxidant defense system that mitigates oxidative tissue damage and promotes wound healing41,42,43,44. Consistent with this design principle, FRET analysis using alkyne-modified GFP and mCherry confirmed nanoscale co-localization of the two proteins on GlcNAz-BC (Supplementary Fig. 19), indicating that GOx and SOD are likewise co-localized under our conjugation conditions.
To evaluate the enzymatic activities of SOD and GOx immobilized on BC, individual control samples (SOD-BC and GOx-BC) were prepared in addition to the dual-functionalized composite (GOx/SOD-BC). Moreover, to verify the efficiency of covalent immobilization achieved by click chemistry, control samples with physically adsorbed enzymes (SOD/BC and GOx/BC) were prepared by passive incubation of BC substrates in enzyme solutions, serving as benchmarks for comparison. Our analyses showed that SOD-BC and GOx/SOD-BC possessed remarkable radical-scavenging prowess, neutralizing both charged and uncharged nitrogen radicals and oxygen radicals effectively (Fig. 4b). Compared with physically adsorbed SOD/BC samples, click-conjugated SOD-BC and GOx/SOD-BC retained much higher enzymatic activity after extensive washing, consistent with stable covalent attachment rather than easily removable surface adsorption (Fig. 4b). The DPPH (2,2-diphenyl-1-picrylhydrazyl) assay revealed about a 30% increase in antioxidative activity with SOD-BC compared to BC and SOD/BC samples, attributing to the limited capacity of BC through its terminal aldehyde groups. (Fig. 4c). The oxygen radical-neutralizing effect of GOx/SOD-BC was further corroborated at the cellular level, where it suppressed lipopolysaccharide (LPS)-induced ROS elevation in cultured cells (Fig. 4d).
Concurrently, GOx-BC and GOx/SOD-BC exhibited efficient glucose consumption, as quantified by a colorimetric assay (absorbance at 500 nm) (Fig. 4e, f), confirming preserved enzymatic activity post-immobilization. Noted that immobilized GOx does not exhibit higher intrinsic specific activity than the free enzyme at equal loading (Supplementary Fig. 20). Bradford assays showed that our click-conjugation strategy yields a retained enzyme density of 3.20 μg GOx per mg BC after washing, whereas physically adsorbed GOx/BC samples display markedly lower post-wash activity under the same conditions (Fig. 4f and Supplementary Fig. 20), indicating that the superior catalytic output of GOx-BC and GOx/SOD-BC primarily arises from the increased retained enzyme density on the BC matrix. Biocompatibility assessments via CCK-8 assays revealed no significant cytotoxicity of GOx/SOD-BC, with cell viability comparable to the pristine culture medium (DMEM) after 24-hour incubation (Supplementary Fig. 21). Hemocompatibility evaluation further demonstrated materials’ great blood compatibility: both BC and GOx/SOD-BC displayed hemolysis rates below 5%, starkly contrasting with the severe hemolysis induced by 1% Triton X-100 (positive control), which generated intensely red lysate solutions (Supplementary Fig. 22). Moreover, both BC and GOx/SOD-BC exhibited consistently low endotoxin levels (<0.1 EU/mL) (Supplementary Fig. 23), indicating their compliance with international safety standards (ISO 10993-1) for biomaterials and supporting their potential utility in biomedical applications.
Our results underscore the utility of our platform in fabricating advanced wound dressings that integrate multiple bioactive proteins, thereby facilitating a cascade of therapeutic actions essential for managing the multifaceted challenges of diabetic wound healing.
Evaluating the in vivo therapeutic efficacy of protein-integrated BC dressings
The in vivo efficacy of our developed GOx/SOD-BC dual-enzyme dressing was investigated using a diabetic mouse model with excisional cutaneous wounds (Fig. 5a). This model was established by creating round, 10 mm diameter full-thickness wounds on the backs of db/db mice, a well-recognized strain for simulating human-type diabetes due to their impaired glucose tolerance, obesity, and delayed wound-healing capacity45,46,47. After surgery, all mice were randomly assigned to four groups to evaluate the wound-healing performance of different treatment modalities. One group received no treatment and served as the control (blank), while the other three groups were treated with standard gauze, plain BC, or GOx/SOD-BC, respectively (Fig. 5b).

a Schematic illustration of the GOx/SOD-BC dressing, its application process, and the timeline for creating and treating wounds in the animal model. b Representative photographs of wound progression at days 0, 3, 5, 7, and 14 (scale bar: 1 cm). The experiment was repeated three times independently and yielded comparable results. c, d Quantitative analysis of wound area changes among the control, gauze, BC, and GOx/SOD-BC groups (n = 3 per group). e H&E-stained histological sections of wound tissue in each group on day 14 (Red arrows: new vessels; yellow arrows: sebaceous gland (SG); blue arrows: hair follicle (HF). Scale bar: 1 mm and 200 μm). The experiment was repeated three times independently and yielded comparable results. f Measured dermal thickness in the different treatment groups (n = 3 per group; statistical significance was determined using one-way ANOVA; **p < 0.01). g Masson’s trichrome-stained sections of wound tissue on day 14, highlighting collagen distribution (scale bar: 1 mm and 200 μm). The experiment was repeated three times independently and yielded comparable results. h Percentage of collagen deposition in each group (n = 3 per group; statistical significance was determined using one-way ANOVA; **p < 0.01). Data are presented as mean ± SD. Source data for this figure is available in the Source Data file.
In both the untreated control and gauze-treated groups, wounds showed persistent inflammation and progressive deterioration over the first five days. In contrast, wounds treated with BC or GOx/SOD-BC dressings remained relatively dry, exhibiting notable reduced inflammation and exudate by day 5 (Fig. 5b). Correspondingly, wound closure was markedly accelerated in the BC and GOx/SOD-BC groups compared to the control and gauze groups by day 7 (Fig. 5c). Approximately 83.91% and 74.19% of the initial wound areas remained unhealed in the control and gauze groups, whereas only 45.16% and 26.40% remained unhealed in the BC and GOx/SOD-BC groups, respectively. These findings were consistent with the angiogenesis assessment, where the GOx/SOD-BC treatment showed the strongest CD31 upregulation and a 29.25% increase in CD31-positive area relative to the blank control (Supplementary Fig. 24), indicating markedly enhanced neovascularization at the wound site. By day 14, GOx/SOD-BC treatment demonstrated superior wound healing outcomes compared to all other groups (Fig. 5d), achieving 92.12% wound closure. In comparison, wound closure reached 77.05% in the BC group, 59.93% in the gauze group, and only 44.78% in untreated controls.
Further histological examinations were conducted to verify the wound healing effects at the tissue level. On day 14 post-surgery, wound tissues were harvested from the mice in each group for hematoxylin and eosin (H&E) staining (Fig. 5e) and Masson trichrome staining (Fig. 5g). H&E staining showed that the dermis tissues of the control and gauze groups healed on the 12th day were still defective, with incomplete structures, few sebaceous glands, and an uneven epithelium. In contrast, wounds treated with BC or GOx/SOD-BC displayed a relatively continuous and uniform epithelial layer, a well-organized dermis, newly formed blood vessels, and the presence of hair follicles and sebaceous glands. Besides, the dermal thickness of the GOx/SOD-BC group exceeded 189.3 μm, surpassing other groups (Fig. 5f). Masson staining of the wounds revealed that the blue collagen fibers in the wounds of the BC and GOx/SOD-BC groups were interwoven. Conversely, the collagen fibers in the wounds of the control and gauze groups were disorganized, and their area percentage was about 5% lower than that of the BC and GOx/SOD-BC groups (Fig. 5h). During the wound healing process, excessive inflammation can lead to sustained secretion of TNF-α and IL-6, thereby delaying wound repair. In contrast, IL-4, as an anti-inflammatory factor, promotes tissue repair and accelerates healing by inhibiting inflammatory responses and balancing immune reactions48. Immunofluorescence staining revealed that GOx/SOD-BC enhances healing by reducing TNF-α/IL-6 levels and elevating IL-4 expression (Supplementary Fig. 25), demonstrating their dual role in modulating the inflammatory microenvironment.
These results indicate that wound healing in the GOx/SOD-BC group progresses well beyond simple surface closure and involves deeper regenerative processes. The presence of newly formed appendages reflects more robust tissue remodeling, which is typically associated with more complete and functionally superior healing outcomes. In line with these findings, 21-day subcutaneous implantation studies showed normal blood routine parameters (Supplementary Fig. 26), together with no detectable pathological changes in major organs (Supplementary Fig. 27), further supporting the excellent in vivo biosafety of the GOx/SOD-BC materials.
Discussion
In this work, we have demonstrated a versatile strategy for functionalizing BC by coupling metabolic glycoengineering with click chemistry. Through in situ fermentation with azide-modified monosaccharides, we introduced reactive azide groups into the BC matrix under mild and environmentally friendly conditions, thereby enabling efficient covalent conjugation of diverse functional agents via azide-alkyne click reactions. A key feature of this strategy is that the azide handle remains genuinely bioorthogonal during BC biosynthesis. The reaction partners required for CuAAC or SPAAC are absent under Komagataeibacter fermentation conditions, and the robust ex vivo labeling of purified GlcNAz-BC confirms that azide groups are not consumed intracellularly and are retained throughout cellulose secretion. This approach not only preserves the native properties of BC, such as mechanical stability and biocompatibility, but also provides a bioorthogonal scaffold for integrating multi-scale, multifunctional components. We showcased this capability by successfully imparting fluorescent, antibacterial, cell-adhesive, and enzymatic activities onto BC. Notably, the immobilization of dual enzymes (GOx and SOD) led to a multifunctional dressing for enhanced diabetic wound healing, illustrating the platform’s potential in advanced biomedical applications.
Despite these achievements, several challenges warrant further investigation. Quantifying the azide grafting rate remains difficult, primarily due to the high crystallinity of the BC membrane21,49 and the complex, incompletely characterized processing of synthetic monosaccharide derivatives in Komagataeibacter metabolism18. While we attempted to measure relative azide concentrations, high-resolution analytical methods proved insufficient, pointing to the need for improved techniques or alternative approaches that can accurately determine grafting efficiency. In addition, although this clickable platform shows promise for biomedical applications, further optimization is needed to broaden the repertoire of functional cargos and to better control their integration into the BC network. A major practical limitation is that many clinically used small molecules and therapeutic proteins do not inherently possess alkyne groups and may be sensitive to alkynylation strategies such as BVSB modification. We propose that this challenge can be mitigated by using multifunctional linkers or universal binding modules (such as streptavidin) that anchor to GlcNAz-BC via click chemistry and subsequently capture unmodified biologics through conventional conjugation or affinity interactions.
Building on these promising findings, ongoing work will seek to manipulate the metabolic pathways of cellulose-producing bacteria to increase the grafting efficiency and further customize the properties of BC. Future research will also explore the synergistic interplay of multiple enzyme systems, advanced sensing molecules, and innovative polymerization methods. By broadening the range of clickable moieties and optimizing in situ fermentation processes, we expect this platform to transform BC into advanced materials, thereby fostering innovations in diverse fields such as biosensing, biocatalysis, and plastic degradation.
Method
Animals
All animal experiments were approved by the Animal Committee of Changzheng Hospital, the Second Affiliated Hospital of Naval Medical University (IACUC Issue No.CX052403108), and were performed according to relevant guidelines and regulations (including ARRIVE guidelines). Eight-week-old male C57BLKS (db/db) mice and eight-week-old male C57BL/6 J mice were purchased from Leagene Biotechnology Co., Ltd. (Shanghai, China). Animals were housed in specific pathogen-free (SPF) grade animal facilities at the Second Affiliated Hospital of Naval Medical University, under controlled conditions (23 ± 1 °C, 50% ± 10% relative humidity, 12-hour light-dark cycle, with ad libitum access to food and water).
Materials and equipment
Glucosamine hydrochloride, 2-azidoacetic acid, triethylamine (TEA), 1-hydroxybenzotriazole (1-HOBt), 1-ethyl-3-(3-dimethyl aminopropyl) carbodiimide hydrochloride (EDC), triphenylphosphine (Ph3P), ninhydrin, 2-azido-2-deoxy-D-glucose (deO), 4,4′-bipyridine, 4-bromo-1-butyne, 4-pyrindinecarboxaldehyde, potassium carbonate, zinc acetate, iodomethane, 4-(vinylsulfonyl)benzoic acid, 3-butyn-1-ol, N, N-diisopropylethylamine (i-Pr2NEt), lumiflavin, PAR (4-(2-pyridylazo)resorcinol), and nitric acid were purchased from Aladdin and used without further purification. 4-[(trimethylsilyl)ethynyl]benzaldehyde was purchased from Leyan (Shanghai) Ltd. Pra-RGD was purchased from GL Biochem (Shanghai) Ltd. HRP, Lipase, GOx, and SOD were purchased from Sigma-Aldrich. FITC-phalloidin was purchased from Share-Bio. DAPI and Bradford Protein Assay Kit were purchased from Beyotime. DBCO-PEG-RGD was purchased from Ruixibio (Xi’an) Co., Ltd.
Nuclear magnetic resonance spectra (NMR) were recorded on a Bruker 400 MHz instrument at ambient temperature using either CDCl3, DMSO-d6, or deuterium oxide as solvent and tetramethylsilane as internal standard.
Electrospray ionization mass spectrometry (ESI-MS) was performed on an Ultimate 3000-Q Exactive Focus LC-HRMS system (Thermo, USA). A single sample of each compound, GlcNAz and Zn-Por-PS, was analyzed. The sample (1 μL) was injected and separated using a mobile phase of (A) water with 0.1% acetic acid and (B) acetonitrile at a flow rate of 0.3 mL/min. The elution gradient was as follows: initial 50% B; increased linearly to 100% B over 1.5 min; maintained at 100% B for an additional 1.5 min. Mass detection was conducted in negative ion mode for GlcNAz and positive ion mode for Zn-Por-PS. Data were processed using Xcalibur Qual Browser software (version 4.3.73.11, Thermo).
Matrix-assisted laser desorption/ionization time-of-flight mass spectra (MALDI-TOF-MS) were acquired on a Bruker Autoflex Speed mass spectrometer (Bruker). 1 μL of sample was mixed with 1 μL of matrix (sinapic acid, SA) and loaded onto the polished target board. After evaporation, mass weight measurement was conducted in linear positive mode. Data were analyzed using FlexAnalysis software (version 3.4, Bruker). A total of 6 independent samples were analyzed. For each sample, three technical replicates were acquired by spotting the sample onto three separate positions on the MALDI target plate, while 3 independent biological replicates were performed. Appropriate controls, including native mCherry, were included in each experiment.
A field emission scanning electron microscope (FESEM, JEOL JSM-IT500HR/LA) was applied to observe bacterial cellulose structure and bacterial morphology. Fourier Transform Infrared Spectroscopy (FT-IR) was tested on the Bruker VERTEX 70. Confocal images were obtained by a Leica SP8 confocal laser scanning microscope (CLSM). The Shimadzu DTG-60AH instrument was utilized to perform thermogravimetric analysis (TGA). A Universal Mechanical Testing Machine (68SC-2) was used to test the material’s strength and toughness.
Infinite® E Plex was used to measure absorbance at different wavelengths. All calculations were carried out using the xtb package and Gaussian 09, visualization and inspection of molecular structures, vibrational analyses, and solvent-accessible surfaces were performed using GaussView 5 and VMD 1.9.3.
Cell lines
NCTC clone 929 (L929) fibroblasts (ATCC CCL-1) and RAW264.7 mouse mononuclear macrophage leukemia cells (ATCC TIB-71), both obtained from the American Type Culture Collection (ATCC, USA), were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 10 µg/mL gentamicin. Cells were maintained at 37°C in a humidified incubator with 5% CO2.
Microorganism
Komagataeibacter rhaeticus (ATCC BAA-2831) used for BC production, was purchased from ATCC. Komagataeibacter sucrofermentans (DSM 15973) and Komagataeibacter xylinus (CGMCC 2955) used for BC production, were purchased from Bio-sci Co., Ltd. The culture medium contained 20 g/L glucose, 10 g/L yeast extract, 20 g/L peptone, 2.7 g/L disodium hydrogen phosphate, and 1.5 g/L citric acid, with the pH adjusted to ~7.0. Candida albicans (ATCC 18804), and Staphylococcus aureus (ATCC 25923) were obtained from ATCC. S. aureus and E. coli were cultured in LB broth at 37 °C and 220 rpm, while C. albicans was grown in potato dextrose broth (PDB) at 30 °C and 220 rpm.
Chemical synthesis of GlcNAz
Glucosamine hydrochloride (3 g, 20 mmol) and 2-azidoacetic acid (1.26 g, 30 mmol) were dissolved in 150 mL of methanol in a 250 mL round-bottom flask equipped with a condenser. TEA (5.6 mL, 40 mmol), 1-HOBt (1.72 g, 14 mmol), and EDC (5.36 g, 14 mmol) were added. After the mixture was cooled in an ice bath, it was stirred overnight at room temperature. Completion was verified by TLC (9:1 dichloromethane/methanol). The azido sugar on TLC plates was detected via a Staudinger reaction with 0.05 M triphenylphosphine (Ph3P), followed by ninhydrin staining. After solvent removal, the residue was purified using preparative thin-layer chromatography (methanol/dichloromethane = 1:10, v/v) to yield GlcNAz (1.73 g, 53% yield) as a pale yellow solid30.
1H NMR of GlcNAz (400 MHz, D2O, ppm): δ 5.14 (d, NH), 3.99 (s, 2H, CH2N3), 3.38-3.88 (m, H-2, H-3, H-4, H-5, H-6, H-6′).
13C NMR of GlcNAz (mixture of α- and β-anomers) (101 MHz, D2O) δ 94.64, 90.75, 75.96, 73.63, 71.57, 70.64, 70.02, 69.86, 60.73, 60.56, 56.76, 54.09, 51.98, 51.73.
ESI-MS, m/z: [M-H] calcd 261.0841, found 261.0837.
Synthesis of di(butynyl) bispyridinium salt (VA)
4,4′-bipyridine (0.26 g, 1.67 mmol) and 4-bromo-1-butyne (625 µL, 6.7 mmol) were heated at 85 °C for 48 h in 10 mL dry DMF. After the reaction mixture had cooled to room temperature, the resulting precipitate was collected by filtration and washed sequentially with DMF and CH₃CN, yielding a yellow solid with a 94% yield31.
1H NMR (400 MHz, D2O, ppm): δ 9.09 (d, J = 6.8 Hz, 4H), 8.47 (d, J = 6.7 Hz, 4H), 4.78 (t, J = 6.2 Hz, 4H), 2.93 (tt, J = 8.1,4.0 Hz, 4H), 2.41 (t, J = 2.6 Hz, 2H).
13C NMR of VA (101 MHz, D2O) δ 150.56, 145.81, 126.97, 78.41, 74.16, 59.90, 20.70.
Synthesis of Zn-Epor-PS
4-pyridinecarboxaldehyde, 4-[(trimethylsilyl)ethynyl]benzaldehyde, and pyrrole were refluxed in propionic acid to yield an A3B porphyrin. Deprotection of the ethynyl group was accomplished with potassium carbonate in MeOH/THF, followed by zinc metalation of the porphyrin macrocycle using zinc acetate in THF/MeOH. The nitrogen atoms on the pyridine groups were subsequently quaternized with iodomethane in DMF at elevated temperatures35,50.
1H NMR of Zn-Epor-PS (400 MHz, DMSO-d6): δ 9.47 (d, 6H), 9.14 (m, 4H), 9.06 (d, 2H), 8.98 (m, 8H), 8.25 (d, 2H), 7.99 (d, 2H), 4.77 (s, 9H), 4.55 (s, 1H).
13C NMR (101 MHz, DMSO-d6) δ 162.80, 158.57, 158.54, 150.35, 148.91, 148.69, 148.50, 144.24, 144.20, 142.85, 134.88, 133.75, 133.20, 132.96, 132.58, 132.50, 130.73, 122.73, 121.96, 116.22, 115.56, 83.95, 82.63.
FTMS(ESI): (m/z) calcd for Zn-Por-PS [C46H34N7Zn]3+ 249.4050, found 249.4048.
Synthesis of But-3-yn-1-yl 4-(vinylsulfonyl)benzoate
4-(vinylsulfonyl)benzoic acid (200 mg, 0.94 mmol), 3-butyn-1-ol (73 mg, 1.04 mmol), EDC (217 mg, 1.13 mmol), 1-HOBt (173 mg, 1.13 mmol), i-Pr2NEt (0.33 mL, 1.88 mmol), and CH2Cl2 (4 mL) were stirred at room temperature for 15 h. After workup and flash column chromatography (30–100% EtOAc in hexanes), the product (111 mg, 0.42 mmol, 45% yield) was obtained as a clear oil37.
1H NMR (400 MHz, CDCl3): δ 8.24 (d, J = 8.4 Hz, 2H), 8.00 (d, J = 8.4 Hz, 2H), 6.68 (dd, J = 16.5, 9.7 Hz, 1H), 6.55 (d, J = 16.5 Hz, 1H), 6.14 (d, J = 9.7 Hz, 1H), 4.48 (t, J = 6.7 Hz, 2H), 2.71 (td, J = 6.7, 2.7 Hz, 2H), 2.06 (t, J = 2.6 Hz, 1H) ppm.
13C NMR of BVSB (101 MHz, CDCl3) δ 164.73, 143.65, 137.89, 134.58, 130.61, 129.09, 128.04, 79.66, 70.29, 63.31, 19.06.
Microbial fermentation for BC
Komagataeibacter rhaeticus was inoculated from a glycerol stock in 5 mL YPD medium containing 1% (v/v) cellulase and grown for 3 days under shaking. The bacterial culture was resuspended and inoculated into fresh YPD medium at 1:100 (v/v). BC membranes were produced under static conditions at 30 °C for 2 days, then purified with 1 wt% NaOH solution at 50 °C for 20 minutes to remove residual cells and proteins. The samples were washed with deionized water until the pH reached 7.0 and stored at 4 °C for further characterization. For cell culture and animal studies, all BC samples were sterilized by gamma irradiation and stored in PBS at 4 °C until use.
Preparation of GlcNAz-BC
Komagataeibacter rhaeticus was grown in YPD medium supplemented with 1 mM GlcNAz at 30 °C for 2 days, then the resulting GlcNAz-BC membranes were processed using the same preparation and sterilization protocols as BC. For Fig. 2b, varying concentrations of GlcNAz (0.1, 0.2, 0.3, 0.5, 0.8, 1.0, 1.5 and 2.0 mM) were tested. For Supplementary Fig. 8, additional 5.0 and 10.0 mM GlcNAz concentrations were tested.
Reactions for methionine alkylation on proteins
The used protein (mCherry, GFP, HRP, lipase, GOx, and SOD) was dissolved in degassed Milli-Q water to a final concentration of 5 mg/mL. Lumiflavin (10 mM in H₂O), Michael acceptor (0.2 M in DMF), and phosphate-buffered saline (PBS, 10×, pH 7.4) solutions were degassed separately. In a 2 mL vial equipped with a stir bar, 100 µL protein solution, 100 µL 10× PBS, 50 µL lumiflavin solution, 50 µL Michael acceptor solution (10 µmol), and 700 µL water were sequentially added. The sealed vial was placed in an ice bath and stirred under blue LED irradiation (440 nm, 40 W, 4 cm distance) for 30 min37. Reaction mixtures were purified using spin desalting columns (Zeba, Thermo Fisher; 0.5 mL, 3 kDa MWCO), and the products were stored at 4 °C.
Synthesis of functional molecules-grafting BC materials
Dehydrated GlcNAz-BC (or unmodified BC) membranes were incubated with VA (10 mg/mL, 23.7 mM) in 24-well plates. A freshly mixed catalyst (CuSO4 (2 mg/mL) + sodium ascorbate (10 mg/mL, 50.5 mM), 1:1 v/v) was then added at 1:10 (v/v) to initiate the click reaction under gentle shaking overnight. VA-BC materials were repeatedly washed until no fluorescence was detected in the wash solution. Other click-based modifications (Por-BC, RGD-BC, mCherry-BC, GFP-BC, mCherry/GFP-BC, HRP-BC, Lipase-BC, SOD-BC, GOx-BC, GOx/SOD-BC) followed the same procedure, replacing VA with each target molecule. Protein reactions were carried out at 4 °C in the dark to preserve enzyme activity.
Synthesis of DBCO-PEG-RGD-BC
Dehydrated GlcNAz-BC membranes were incubated with DBCO-PEG-RGD (10 mg/mL) in 24-well plates. No catalyst was added. The click reaction was performed at 4 °C under gentle shaking overnight, and the membranes were washed repeatedly to acquire the DBCO-PEG-RGD-BC materials.
Theoretical modeling of azido-sugar-modified cellulose
The investigation of the reaction pathway began with a transition-state (TS) search performed using the semi-empirical GFN2-xTB method as implemented in the xtb package. This approach allowed for efficient identification of reasonable initial TS geometries for the CuAAC reaction under solvated conditions. These preliminary TS structures were subsequently reoptimized at the density functional theory (DFT) level using Gaussian 09, employing the PBE1PBE (PBE0) functional together with the def2-SVP basis set and Grimme’s D3(BJ) dispersion correction (empirical dispersion = gd3bj). Vibrational frequency analyses were conducted to confirm the nature of the transition states, each of which exhibited a single imaginary frequency corresponding to the expected cycloaddition reaction coordinate.
Characterization of GlcNAz-BC via FT-IR, TGA, and lap-shear tests
BC and GlcNAz-BC membranes were cultured in YPD medium for 48, 72, 96, or 120 h, rinsed with Milli-Q water, and then measured for thickness using a Vernier caliper (n ≥ 3). Dried BC and GlcNAz-BC samples were next subjected to Fourier Transform Infrared (FT-IR) analysis, recorded in the 400–4000 cm⁻1 range at 25 °C and 30% relative humidity (n ≥ 3). Thermogravimetric analysis (TGA) was conducted on a Shimadzu DTG-60AH instrument: 2–3 mg of each dried sample was sealed in aluminum pans and heated from 30 °C to 600 °C at a rate of 10 °C min⁻1 under a nitrogen atmosphere. For mechanical testing, rectangular BC and GlcNAz-BC specimens (100 × 10 × 20 mm3) were prepared in molded grooves, thoroughly washed, and compressed to a thickness of 1 mm. Pure shear tests were carried out on an Instron machine at a constant stretch rate of 10 mm min⁻1, allowing stress-strain curves to be plotted to evaluate the materials’ toughness.
Degradation rate of GlcNAz-BC
BC and GlcNAz-BC fragments (2–3 mg, mass m) were incubated in 2 mL of 0.1% cellulase-containing PBS buffer at 37 °C for 24 h under agitation. The reaction mixtures were centrifuged (8000 × g, 10 min, 25 °C), and supernatants were collected. For reducing sugar quantification, 50 μL of supernatant was mixed with 100 μL DNS reagent in a 96-well plate, heated at 99 °C for 5 min, cooled to 4 °C for 1 min, and absorbance at 540 nm was measured using a microplate reader. In parallel, a glucose standard curve (0.125–4 g/L) was prepared with anhydrous glucose under identical DNS reaction conditions. Degradation-derived glucose content was calculated based on the standard curve.
Acid digestion of BC membranes
Precisely weigh 10 mg of BC or GlcNAz-BC membrane and transfer it into a dry glass test tube. Add 5 mL of concentrated nitric acid (HNO3, ≥65%) and heat the tube at 120 °C for 30 minutes. After the initial digestion, carefully add 3 mL of 30% hydrogen peroxide (H2O2) to the mixture and continue heating at 120 °C for an additional 10 minutes, or until the solution becomes clear and no solid residue remains. Allow the digested solution to cool naturally to room temperature. Neutralize the solution by slowly adding sodium hydroxide solution under gentle mixing until the pH reaches 6-7. Quantitatively transfer the neutralized digestate to a 10 mL with deionized water.
PAR-based colorimetric copper assay
Prepare a series of copper ion standard solutions from a stock copper sulfate (CuSO₄) solution, covering a concentration range of 0 to 15 µg/mL. Piper 100 µL of each diluted digestate sample or copper standard solution into individual wells of a 96-well microplate. Perform all measurements in triplicate. To each well, add 100 µL of the PAR reagent solution (0.2 mg/mL). Securely seal the microplate and incubate with shaking for 5 min to ensure uniform mixing and complete color development. Measure the absorbance of each well at a wavelength of 499 nm using a microplate reader. Generate a standard curve by plotting the average absorbance at 499 nm against the known concentration of the copper standard solutions. Determine the copper ion concentration in the original digestate samples by interpolating their measured absorbance values from the standard curve.
Cytotoxicity evaluation in vitro
BC, GlcNAz-BC, RGD-BC, and GOx/SOD-BC materials (13 mm diameter) were assessed using both leaching and direct contact assays. In the leaching assay, samples were incubated in the serum-containing medium at 37 °C for 24 h (6 cm2/mL). L929 cells (1 × 105 cells/mL) were seeded in 24-well plates for 24 h, and the medium was replaced with material extracts (experimental) or fresh DMEM (control). After 24 h, cell viability was measured by CCK-8 at 450 nm. Cell viability was calculated using the following formula:
Hemolysis ratio assay
To evaluate the hemocompatibility of the materials, BC, GlcNAz-BC, RGD-BC, and GOx/SOD-BC samples (13 mm diameter) were incubated with 2 mL of 5% (v/v) red blood cell (RBC) suspension at 37 °C for 1 h. Hemolysis was quantified by measuring the OD540 of the supernatant after centrifugation (845 × g, 10 min). The hemolysis ratio was then calculated using the following formula:
Endotoxin evaluation in vitro
The residual endotoxin levels in sterilized BC, GlcNAz-BC, and GOx/SOD-BC composites were quantified using a chromogenic Limulus amebocyte lysate (LAL) assay. Briefly, samples were subjected to endotoxin extraction by incubating in endotoxin-free water (6 cm²/mL surface-to-volume ratio) at 37 °C for 24 h. Subsequently, 10 μL aliquots of the extracts were analyzed in triplicate alongside a calibration series (0 ~ 0.5 EU/mL) prepared from endotoxin standards. Absorbance at 545 nm was measured using a microplate reader.
Bactericidal assays on Por-BC in vitro
A 50 µL aliquot of S. aureus or C. albicans (1 × 106 CFU/mL) was placed on BC, GlcNAz-BC, or Por-BC films (14 mm diameter). Samples were irradiated for 0, 5, 10, or 30 min, then washed with 5 mL PBS. Suspensions were serially diluted and plated on LB or PDB agar to determine CFU counts. For SEM imaging, bacteria on each film were fixed with 2 wt% glutaraldehyde, progressively dehydrated with ethanol solutions (30 ~ 100%), then sputter-coated with platinum before FE-SEM observation.
Characterization of cells on RGD-BC
To evaluate cell adhesion and viability, BC, GlcNAz-BC, RGD-BC and DBCO-RGD-BC membranes were rinsed in PBS using five cycles of 2-hour washes each, then incubated in culture medium for 6 hours before being transferred to 24-well plates. L929 or GFP-MSC cells (1 × 106 cells/well) were seeded onto each membrane for 6 h. Cells were then fixed in 4% paraformaldehyde and permeabilized in 0.1% Triton X-100, followed by staining with FITC-phalloidin (30 min) and DAPI (5 min). CLSM was used to visualize the cytoskeleton and nuclei. FDA/PI assays were also employed to assess cell viability over 3 days in culture.
DPPH radical scavenging activity assay
To evaluate the antioxidant activity of the samples, a stock DPPH solution was prepared by dissolving 1 mg of DPPH in 24 mL of anhydrous ethanol. An additional 0.5 mL ethanol was added to 1 mL of this solution to achieve an absorbance of 0.6–1.0 at 519 nm. BC and SOD-BC samples were incubated with the DPPH solution for 30 min in the dark. Scavenging (%) = [(A0 − A)/A0] × 100%, (A0 is the absorbance value without sample addition; A is the absorbance value after sample addition).
GOx enzyme activity measurement
The GOx-BC and BC samples were lyophilized and weighed. A working solution was prepared according to the manufacturer’s protocol for the GOx activity assay kit and pre-incubated at 37 °C for 10 minutes. Subsequently, the working solution was added to each sample at a ratio of 1 mg per 200 μL. The mixtures were incubated in a 37 °C orbital shaker at 220 rpm for 1 hour. Absorbance measurements at 500 nm were taken before (A₁) and after (A₂) incubation. Enzyme activity was calculated using the following formula:
(V = total reaction volume; ε = molar extinction coefficient;d = light path length of the cuvette; m = dry weight of the sample; T = reaction time.)
GOx protein binding quantification
A GOx reaction mixture containing click chemistry reagents (copper sulfate and sodium ascorbate) was prepared as the standard solution. Aliquots (200 μL each) of this standard solution were applied separately onto the surfaces of both BC and GlcNAz-BC membranes, followed by incubation for 60 minutes at room temperature. After incubation, 100 μL of the supernatant from each membrane reaction mixture was transferred to a 96-well plate. For the standard curve, 100 μL of serially diluted GOx reaction mixtures with known concentrations were also added to separate wells. Then, 100 μL of Coomassie Brilliant Blue G-250 dye was added to each well, and the absorbance at 595 nm was measured using a microplate reader. The protein concentration in each sample was determined by interpolating from the standard curve.
In vitro assessment of ROS scavenging activity
RAW264.7 cells were cultured on BC and SOD-BC membranes overnight, followed by stimulation with endotoxin (10 µg/mL, 24 h) to increase intracellular ROS. Cells were then incubated with a DCFH-DA probe for 30 min at 37 °C in the dark, and fluorescence images were captured (Ex = 488 nm, Em = 525 nm) using an inverted fluorescence microscope or CLSM.
Wound healing in vivo
A total of 12 eight-week-old male db/db mice were employed to investigate wound healing. They were allowed to acclimatize to the environment by being fed for one week before the experiment. Animals were randomly assigned to four groups (GOx/SOD-BC, BC, medical gauze, and untreated control), with three mice per group. After anesthetization with pentobarbital sodium (40 mg/kg), four circular full-thickness wounds, each with a diameter of 10 mm, were created on the dorsal area using a sterile biopsy punch. The wounds were then treated with either sterilized GOx/SOD-BC, BC, gauze, or left untreated. Wound images were captured on days 0, 3, 7, and 14 post-surgery. The wound area was measured using ImageJ software, and the wound healing rate was calculated according to the following formula:
(W0 = the area of the wound at the beginning in cm2; Wt = the area of the wound at day t in cm2; here t = 3, 7, and 14.)
Histological analysis
On day 14, db/db mice were anesthetized and euthanized. The tissues surrounding the wound area were excised, fixed in 4% paraformaldehyde, and subsequently embedded in paraffin. The tissue samples were sectioned at a thickness of 4 µm. The resulting sections were stained with hematoxylin and eosin (H&E) and Masson’s trichrome for histopathological analysis. The expression levels of inflammatory cytokines TNF-α, IL-6, and IL-4 were analyzed through immunofluorescence staining.
Neovascularization analysis
A total of 16 eight-week-old male db/db mice were used to investigate the effect of angiogenesis. Before the experiment, the mice were acclimatized for one week to adapt to the environment. The animals were randomly divided into four groups (GOx/SOD-BC group, BC group, gauze group, and blank group), with 4 mice per group. After anesthesia, one circular full-thickness wound with a diameter of 10 mm was created on the dorsal region of the mice using a sterile biopsy punch. Subsequently, the wounds were then treated with either sterilized GOx/SOD-BC, BC, gauze, or left untreated. On day 7 post-surgery, the db/db mice were anesthetized and euthanized. The tissues surrounding the wound area were harvested, fixed in 4% paraformaldehyde, and then embedded in paraffin. The expression level of the angiogenesis factor CD31 was detected by immunohistochemistry.
In vivo toxicity evaluation
A total of 20 eight-week-old male C57BL/6 mice were employed to investigate toxicity evaluation. Twenty C57BL/6 mice were randomly divided into four groups, three of which served as experimental groups. The same size of gauze, BC, and GOx/SOD-BC modified materials were subcutaneously implanted into the dorsal region of the mice in the three experimental groups, respectively. On the 21st day, the rats were anesthetized and sacrificed, and blood was collected from the eyeballs for blood routine examination. Subsequently, major organs, including the heart, liver, spleen, lungs, and kidneys, were harvested. Hematoxylin-eosin (H&E)-stained sections were used to observe whether there were pathological abnormalities in the tissues of each organ.
Statistics and reproducibility
Plate reader data were acquired with BioTek Gen5 (version 3.11). Fluorescent images from Nikon AX/AX R confocal microscopy were collected using NIS-Elements (version 5.21). Data analysis, statistical evaluation, and graph generation were conducted primarily in Excel 2019 and Origin 2021. NMR spectra were processed with MestReNova (v16.0.0), while mass spectrometry data were analyzed using Xcalibur Qual Browser (v4.3.73.11, Thermo) and FlexAnalysis (v3.4, Bruker). Chemical structures were drawn with ChemDraw (version 21.0.0). Image-based quantification, including mean fluorescence intensity and single-cell area measurements, was performed using ImageJ (1.54p). Figures and diagrams were assembled and illustrated with Adobe Illustrator 2022 and Autodesk 3ds Max 2020. Molecular visualization and structural analyses were carried out with GaussView 5 and VMD 1.9.3. All software was used according to standard protocols to maintain consistency and support the reproducibility of experimental results.
Comparisons between two groups were conducted using two-tailed Student’s t-tests. For comparisons across three or more groups, one‑way ANOVA was applied. Data are presented as mean ± standard deviation (SD). Representative images are derived from at least three independent samples unless otherwise indicated.
No statistical method was used to predetermine sample size. No data were excluded from the analyses. All experiments were performed in triplicate or greater, and all attempts at replication were successful (sample size indicated in figure legend). All experiments were not randomized, and investigators were not blinded to group allocation during experiments and outcome assessment. Mice were randomly assigned to experimental groups. Randomization procedures applicable to human or animal studies are not relevant to this study. Experimental groups consisted of the developed BC materials, and sample allocation was determined by the experimental design rather than by random assignment. The study does not contain experiments where blinding would be applicable.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data are available within the Article, the Supplementary Information, and the Source Data. Source data are provided with this paper.
References
Wu, S. et al. All-in-one porous membrane enables full protection in guided bone regeneration. Nat. Commun. 15, 119 (2024).
Caro-Astorga, J., Walker, K. T., Herrera, N., Lee, K.-Y. & Ellis, T. Bacterial cellulose spheroids as building blocks for 3D and patterned living materials and for regeneration. Nat. Commun. 12, 5027 (2021).
Janmohammadi, M. et al. Cellulose-based composite scaffolds for bone tissue engineering and localized drug delivery. Bioact. Mater. 20, 137–163 (2023).
Mikaeeli Kangarshahi, B., Naghib, S. M., Younesian, D. & Rabiee, N. Unlocking the rhythmic power of bacterial cellulose: a comprehensive review on green energy harvesting and sustainable applications. Adv. Funct. Mater. 35, 2413760 (2025).
Moon, R. J., Martini, A., Nairn, J., Simonsen, J. & Youngblood, J. Cellulose nanomaterials review: structure, properties and nanocomposites. Chem. Soc. Rev. 40, 3941 (2011).
Swingler, S. et al. Recent advances and applications of bacterial cellulose in biomedicine. Polymers 13, 412 (2021).
Gilbert, C. et al. Living materials with programmable functionalities grown from engineered microbial co-cultures. Nat. Mater. 20, 691–700 (2021).
Walker, K. T. et al. Self-pigmenting textiles grown from cellulose-producing bacteria with engineered tyrosinase expression. Nat. Biotechnol. 43, 345–354 (2025).
An, B. et al. Engineered living materials for sustainability. Chem. Rev. 123, 2349–2419 (2023).
Liu, W. et al. Bacterial cellulose-based composite scaffolds for biomedical applications: a review. ACS Sustain. Chem. Eng. 8, 7536–7562 (2020).
Samyn, P. et al. Opportunities for bacterial nanocellulose in biomedical applications: review on biosynthesis, modification and challenges. Int. J. Biol. Macromol. 231, 123316 (2023).
Zheng, Z., Guo, S., Yan, M., Luo, Y. & Cao, F. A functional Janus Ag nanowires/bacterial cellulose separator for high-performance dendrite-free zinc anode under harsh conditions. Adv. Mater. 35, 2304667 (2023).
Zhang, X. et al. Uniformly aligned flexible magnetic films from bacterial nanocelluloses for fast actuating optical materials. Nat. Commun. 13, 5804 (2022).
Subbotina, E., Ram, F., Dvinskikh, S. V., Berglund, L. A. & Olsén, P. Aqueous synthesis of highly functional, hydrophobic, and chemically recyclable cellulose nanomaterials through oxime ligation. Nat. Commun. 13, 6924 (2022).
Li, J. et al. Molecular engineering of renewable cellulose biopolymers for solid-state battery electrolytes. Nat. Sustain. 7, 1481–1491 (2024).
Habibi, Y. Key advances in the chemical modification of nanocelluloses. Chem. Soc. Rev. 43, 1519–1542 (2014).
Kaldéus, T., Larsson, P. T., Boujemaoui, A. & Malmström, E. One-pot preparation of bi-functional cellulose nanofibrils. Cellulose 25, 7031–7042 (2018).
Thongsomboon, W. et al. Phosphoethanolamine cellulose: a naturally produced chemically modified cellulose. Science 359, 334–338 (2018).
Verma, P., Ho, R., Chambers, S. A., Cegelski, L. & Zimmer, J. Insights into phosphoethanolamine cellulose synthesis and secretion across the Gram-negative cell envelope. Nat. Commun. 15, 7798 (2024).
Gao, M. et al. A natural in situ fabrication method of functional bacterial cellulose using a microorganism. Nat. Commun. 10, 437 (2019).
Sun, Y. et al. Microbe-assisted fabrication of circularly polarized luminescent bacterial cellulosic hybrids. Nat. Commun. 16, 1115 (2025).
Liu, X. et al. Direct synthesis of photosensitizable bacterial cellulose as engineered living material for skin wound repair. Adv. Mater. 34, 2109010 (2022).
Niu, W. & Guo, J. Cellular site-specific incorporation of noncanonical amino acids in synthetic biology. Chem. Rev. 124, 10577–10617 (2024).
Yin, S. et al. Advanced composite solid electrolyte architecture constructed with amino-modified cellulose and carbon nitride via biosynthetic avenue. Adv. Funct. Mater. 34, 2314976 (2024).
Liu, X. et al. Living artificial skin: photosensitizer and cell sandwiched bacterial cellulose for chronic wound healing. Adv. Mater. 36, 2403355 (2024).
Teh, M. Y. et al. An expanded synthetic biology toolkit for gene expression control in Acetobacteraceae. ACS Synth. Biol. 8, 708–723 (2019).
Natalio, F. et al. Biological fabrication of cellulose fibers with tailored properties. Science 357, 1118–1122 (2017).
Lavrador, P., Moura, B. S., Almeida-Pinto, J., Gaspar, V. M. & Mano, J. F. Engineered nascent living human tissues with unit programmability. Nat. Mater. 24, 143–154 (2025).
Till, N. A., Ramanathan, M. & Bertozzi, C. R. Induced proximity at the cell surface. Nat Biotechnol. https://doi.org/10.1038/s41587-025-02592-1 (2025).
Laughlin, S. T. & Bertozzi, C. R. Metabolic labeling of glycans with azido sugars and subsequent glycan-profiling and visualization via Staudinger ligation. Nat. Protoc. 2, 2930–2944 (2007).
Zhang, Y.-M. et al. Molecular binding behavior of a bispyridinium-containing bis(β-cyclodextrin) and its corresponding [2]rotaxane towards bile salts. Org. Biomol. Chem. 12, 2559 (2014).
Wratil, P. R., Horstkorte, R. & Reutter, W. Metabolic glycoengineering with N -Acyl side chain modified mannosamines. Angew. Chem. Int Ed. 55, 9482–9512 (2016).
Del Bino, L. et al. Synthetic glycans to improve current glycoconjugate vaccines and fight antimicrobial resistance. Chem. Rev. 122, 15672–15716 (2022).
Wang, H. et al. Selective in vivo metabolic cell-labeling-mediated cancer targeting. Nat. Chem. Biol. 13, 415–424 (2017).
Feese, E., Sadeghifar, H., Gracz, H. S., Argyropoulos, D. S. & Ghiladi, R. A. Photobactericidal porphyrin-cellulose nanocrystals: synthesis, characterization, and antimicrobial properties. Biomacromolecules 12, 3528–3539 (2011).
Weishaupt, R. et al. Antibacterial, cytocompatible, sustainably sourced: cellulose membranes with bifunctional peptides for advanced wound dressings. Adv. Healthc. Mater. 9, 1901850 (2020).
Kim, J. et al. Site-selective functionalization of methionine residues via photoredox catalysis. J. Am. Chem. Soc. 142, 21260–21266 (2020).
Yin, M. et al. Multifunctional magnesium organic framework-based microneedle patch for accelerating diabetic wound healing. ACS Nano 15, 17842–17853 (2021).
Fu, L., Qi, C., Hu, Y., Lin, J. & Huang, P. Glucose oxidase-instructed multimodal synergistic cancer therapy. Adv. Mater. 31, 1808325 (2019).
Zhang, L. et al. A composite hydrogel of chitosan/heparin/poly (γ-glutamic acid) loaded with superoxide dismutase for wound healing. Carbohydr. Polym. 180, 168–174 (2018).
Du, X. et al. pH-switchable nanozyme cascade catalysis: a strategy for spatial–temporal modulation of pathological wound microenvironment to rescue stalled healing in diabetic ulcer. J. Nanobiotechnol. 20, 12 (2022).
Meng, X. et al. Ultrasmall metal alloy nanozymes mimicking neutrophil enzymatic cascades for tumor catalytic therapy. Nat. Commun. 15, 1626 (2024).
Gao, W. et al. Deciphering the catalytic mechanism of superoxide dismutase activity of carbon dot nanozyme. Nat. Commun. 14, 160 (2023).
Shang, L. et al. Ultrasound-augmented multienzyme-like nanozyme hydrogel spray for promoting diabetic wound healing. ACS Nano 17, 15962–15977 (2023).
Wei, S. et al. A composite hydrogel with co-delivery of antimicrobial peptides and platelet-rich plasma to enhance healing of infected wounds in diabetes. Acta Biomaterialia 124, 205–218 (2021).
Li, G. et al. A small molecule HIF-1α stabilizer that accelerates diabetic wound healing. Nat. Commun. 12, 3363 (2021).
Song, J. W. et al. Bioresorbable, wireless, and battery-free system for electrotherapy and impedance sensing at wound sites. Sci. Adv. 9, eade4687 (2023).
Wang, H. et al. Carbon dots with integrated photothermal antibacterial and heat-enhanced antioxidant properties for diabetic wound healing. Small 20, 2403160 (2024).
Jiang, W., Jiang, Z., Zhu, M. & Fan, X. Oriented bacterial cellulose for achieving high carbon yield through pre-stretching. Cellulose 29, 4323–4333 (2022).
Sathyapalan, A. et al. Preparation, characterization, and electrical properties of a self-assembled meso-pyridyl porphyrin monolayer on gold surfaces. Aust. J. Chem. 58, 810 (2005).
Acknowledgements
This work was jointly sponsored by the National Key R&D Program of China (2024YFA0920100 to B.A.), the Shenzhen Medical Research Fund (A2403048 to B.A.), the National Natural Science Foundation of China (Grant No. 52473115 to Y.Z., and U24A20478 to X.D.), the National Science Fund for Distinguished Young Scholars (32125023 to C.Z.), the National Natural Science Foundation of Shanghai (23ZR1442700 to Y.Z.), and Shenzhen Science and Technology Program (Grant No. KJZD20230923114217034 to Y.Y.W.). The authors thank the HPC Platform of ShanghaiTech University and Shanghai Supercomputer Center for computing time, and Mr. Zhongchuan Yin for his assistance in the preparation of the Fig. 1. The authors also gratefully acknowledge the Center for Instrumental Analysis at the Materials Synthetic Biology Center and the Shenzhen Synthetic Biology Infrastructure at the Shenzhen Institute of Synthetic Biology.
Author information
Authors and Affiliations
Contributions
C.Z., Y.Z., T.Y., and B.A. conceived the concept and directed the research. S.C. systematically designed and conducted experiments on diverse BC materials synthesis and characterization with the assistance of B.L., X.D., and X.F. in FT-IR, SEM, and cell imaging. H.T. designed and performed all animal experiments. Y.M.W. and Y.Y.W. provided the purified recombinant proteins. Z.W., X.J., and P.Z. assisted in cell and material characterization. S.C. and B.A. drafted the paper with help from all authors.
Corresponding authors
Ethics declarations
Competing interests
C.Z. is a cofounder and equity holder of Shenzhen PAM2L Biotechnologies Co., Ltd. C.Z., B.A., S.C., and Y.Y.W. are co-inventors on a patent application (PCT-CN2025-129902) filed by Shenzhen Institutes of Advanced Technology based on the bacterial cellulose modification methods covered in this article. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yong-Qiang Li, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, S., Tang, H., Fan, X. et al. Bio-orthogonal functionalization of bacterial cellulose combining metabolic glycoengineering and click chemistry. Nat Commun 17, 2304 (2026). https://doi.org/10.1038/s41467-026-69130-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69130-8
