
Abstract
Age-related obesity is a growing public health concern linked to various metabolic disorders, yet its underlying mechanisms remain incompletely understood. Here we report that S-nitrosoglutathione reductase (GSNOR), a pivotal denitrosation enzyme, increases in adipose tissue of both male mice and humans from middle-age. GSNOR knockout protects against age-related weight gain and enhances metabolism, whereas adipose-specific GSNOR knock-in mice promotes obesity and metabolic decline. Further investigation reveals that aged GSNOR KO mice maintain higher mitochondrial content and more beige adipocytes, whereas adipose-specific GSNOR overexpression promotes adipose tissue whitening. Mechanistically, GSNOR denitrosates Beclin-1 at cysteine 351 and mutation of this site (Beclin-1C351A) increases autophagy by enhancing Beclin-1 and ATG14 interaction, thereby accelerating beige-to-white adipocyte conversion. Together, our findings reveal that GSNOR regulates adipose tissue remodeling during aging through Beclin-1 S-nitrosation, pointing to a potential therapeutic target for age-related obesity.
Similar content being viewed by others


Introduction
Age-related obesity is a major public health problem that occurs in middle-aged individuals1. Obesity exacerbates the age-related decline in physical function and causes a series of diseases2. Excess visceral adipose tissue disrupts systemic homeostasis through chronic inflammation, insulin resistance, and dysregulated adipokine secretion, laying the foundation for multi-organ complications3. Studies have demonstrated that obesity contributes to type 2 diabetes4, hypertension, atherosclerosis, and other cardiovascular conditions5. Therefore, understanding the mechanisms underlying age-related obesity is crucial. Several regulatory factors have been identified to be involved in age-related obesity. One is related to neuromodulation, which weight gain in middle-aged individuals is associated with an increase in appetite due to the degeneration of cells in the brain that control appetite over time. The study showed that the MC4R-expressing primary cilia of hypothalamic neurons progressively shorten with age in rats, resulting in increased appetite and age-dependent metabolic decline6. Another critical regulatory factor is inflammaging, a type of chronic low-grade inflammation associated with aging7. Studies have shown that IgG accumulates in white adipose tissue (WAT) during aging, impairing adipose tissue function and adversely affecting metabolic health8. The suppression of adipocyte lipolysis regulated by adipose tissue macrophages leads to age-related metabolic decline and obesity9. Adipose tissue serves as an energy reservoir and endocrine organ and is critical for maintaining systemic glucose, lipid, and energy homeostasis10; however, the decline in these metabolic functions with aging likely contributes to the development of age-related obesity. It is reported that impaired branched-chain amino acid (BCAA) catabolism in adipose tissues causes age-related obesity11. Metabolic reprogramming has also garnered significant attention as a regulatory factor. Decreased levels of LONP1 in adipose tissue may contribute to the aging-related reduction in white-to-beige adipocyte conversion capacity12. Despite these insights, currently employed treatments based on these mechanisms have not proven entirely effective, indicating a need for a more comprehensive understanding of the mechanisms driving age-related obesity.
Nitric oxide (NO) is a crucial endogenous messenger molecule, and its bioavailability and production decline with age13,14. Increasing evidence has shown that NO plays a pivotal role in regulating mammalian metabolism15. For example, eNOS-derived NO actively participates in regulating adiposity16. L-arginine has been shown to promote adipose tissue lipolysis and improve metabolic function17. Nitrate also increases glucose uptake and oxidative catabolism in primary adipocytes and WAT18. However, the molecular mechanism is not clear. S-nitrosation/S-nitrosylation is an important protein posttranslational modification to convey NO bioactivity, which involves the covalent addition of a nitrosyl group to the reactive thiol group of a cysteine, resulting in the formation of the protein S-nitrosothiol (SNO)19. S-nitrosoglutathione reductase (GSNOR) is essential for maintaining intracellular NO homeostasis and is a key enzyme for denitrosation20,21. It indirectly regulates protein S-nitrosation by modulating GSNO levels within cells22. GSNOR has been implicated in a variety of physiological and pathological processes, such as cardiac dysfunction 23,24,25, tumor growth26, vasculogenesis27, cell senescence28 etc. Our previous research has explored the diverse functions of GSNOR, including its roles in aging-related cognitive impairment29, morphine analgesic tolerance30, immune regulation31 and hematopoietic stem cell regeneration32. Additionally, we have demonstrated that proinflammatory macrophages suppress PPARγ activity in adipocytes via S-nitrosation, underscoring the regulatory influence of S-nitrosation on adipocyte function and metabolism33. However, the associations between GSNOR and NO metabolism and age-related obesity remain poorly understood. Furthermore, the core targets and mechanisms of S-nitrosation in the context of age-related obesity have yet to be elucidated.
During our previous investigation into the role of GSNOR in age-related learning and memory29, we unexpectedly observed that after middle age, there is no significant body weight increasing in GSNOR knockout (KO) mice, as usually increasing in wild-type (WT) controls. These findings led us to hypothesize that GSNOR may be involved in age-related obesity and metabolism. In the present study, we found that age-related metabolic disorders are effectively mitigated in aged GSNOR KO mice, suggesting that GSNOR is a critical factor in age-related obesity and metabolic decline. Moreover, we generated adipose tissue-specific GSNOR-overexpressing mice and observed that these mice exhibited obesity and aberrant metabolic phenotypes. Using quantitative proteomic analysis of S-nitrosation modification, we identified Beclin-1 as an important target of GSNOR. Mechanistically, we demonstrated that GSNOR promotes adipose tissue whitening and contributes to age-related obesity by regulating the S-nitrosation of Beclin-1.
Results
GSNOR KO alleviated age-related obesity and modulated adipose tissue weight and composition in mice
To investigate the role of GSNOR in age-related obesity, we monitored the body weights of WT and GSNOR KO mice used in our previous study during natural aging29. As depicted in Fig. 1a, b, the body weights of WT male and female mice continued to increase from 11 to 20 months of age. In contrast, GSNOR KO mice exhibited resistance to obesity development after reaching middle age (11 months), although weight gain during middle age relative to youth is a more common phenomenon than that during younger ages. In addition, we found that GSNOR KO male mice were obviously thinner than WT mice from 1 month of age to 5 months of age (Fig. 1a), which aligns with previous research34. No difference in survival rates was observed between the WT and GSNOR KO groups (Supplementary Fig. 1a, b). To ascertain whether alterations in fat tissue composition contributed to the observed differences in body weight, we conducted magnetic resonance imaging (MRI). Notably, similar to the observations at 11 months, GSNOR KO mice at 20 months of age maintained a significantly lower body weight than WT mice. MRI analysis revealed that GSNOR KO mice had a lower percentage of fat mass and a higher percentage of lean mass at both 11 and 20 months of age (Fig. 1c, d), although the absolute values of both fat mass and lean mass decreased in GSNOR KO mice (Supplementary Fig. 1c, d). This suggests that the lean body composition of GSNOR KO mice was better maintained with age. To further elucidate this, we isolated inguinal white adipose tissue (iWAT), gonadal white adipose tissue (gWAT), and brown adipose tissue (BAT) from middle-aged mice, when notable variations in body weight began to manifest. Strikingly, the iWAT weights of GSNOR KO mice were significantly lower than those of WT mice, whereas no significant differences in gWAT or BAT were detected between the two groups at this stage (Fig. 1e, f). Notably, the iWAT, gWAT, and BAT weights of GSNOR KO mice were lower than those of WT mice at age 20 months (Fig. 1g, h). This pattern of fat reduction in GSNOR KO mice, particularly in WAT, mirrors the age-related changes observed in young versus middle-aged mice (Supplementary Fig. 1e, f). Hematoxylin and eosin (H&E) staining of iWAT, gWAT, and BAT sections revealed that, compared with WT mice, GSNOR KO mice have significantly more adipocytes per field of view and smaller adipocyte sizes in old age (Fig. 1i). These results indicated that GSNOR KO alleviated age-related obesity in aging mice, suggesting a potential role for GSNOR in the regulation of fat accumulation and body weight during the aging process.

a Body weight of male WT and KO mice during aging (WT n = 19, KO n = 11). b Body weight of female WT and KO mice during aging (WT n = 20, KO n = 16). c Gross view, fat and lean mass percentage of 11-month-old male WT and KO mice, as determined by magnetic resonance imaging (MRI) (n = 8). d Gross view, fat and lean mass percentage of 20-month-old male WT and KO mice, as determined by MRI (n = 5). e, f Gross view and weight percentage of iWAT, gWAT, and BAT relative to body weight from 11-month-old male WT and KO mice (WT n = 9, KO n = 11). g, h Gross view and weight percentage of iWAT, gWAT, and BAT relative to body weight from 20-month-old male WT and KO mice (n = 10). i H&E staining and cell number per view of iWAT, gWAT, and BAT from 20-month-old male WT and KO mice (n = 10). Scale bar, 100 μm. Data are represented as mean ± SEM (a, b) or mean ± SD (c, d, f, h, i); Significance was assessed by Unpaired two-tailed Student’s t test (a, b, c, d, f, h, i) (*p < 0.05, **p < 0.01, ***p < 0.001, ns for not significant). WT wild-type, KO, GSNOR knockout, MRI magnetic resonance imaging, H&E hematoxylin and eosin, iWAT inguinal white adipose tissue, gWAT gonadal white adipose tissue, BAT brown adipose tissue.
We next investigated whether the effects in GSNOR KO mice were related to ectopic lipid deposition. Intriguingly, whole blood analysis (Table 1) and serum biochemical analysis revealed reduced levels of triglyceride (TG) in GSNOR KO mice (Supplementary Fig. 2a–l). Additionally, H&E staining did not reveal fat accumulation in the livers of GSNOR KO mice (Supplementary Fig. 2m), suggesting that GSNOR KO does not lead to hepatic steatosis. Consistent with these findings, we also measured TG level in liver and found a significant reduction in GSNOR KO mice compared to WT mice (Supplementary Fig. 2n).
These findings collectively suggest that the primary effect of GSNOR KO on weight regulation is mediated through the modulation of WAT proportion and composition, with no apparent adverse effects on muscle mass or liver fat accumulation. The reduction in TG levels further indicated a beneficial effect on lipid metabolism in GSNOR KO mice.
GSNOR KO aging mice improved metabolism and increased thermogenesis
To elucidate the mechanisms underlying the protection of GSNOR KO mice from age-related obesity, we investigated various physiological and metabolic parameters. Obesity is commonly influenced by factors such as diet management, physical activity, and metabolic regulation35. Compared with aged WT mice, aged GSNOR KO mice exhibited comparable intake and fecal output (Fig. 2a, b) and similar levels of locomotor activity (Fig. 2c). We investigated the impact of GSNOR deficiency on thermogenesis and cold tolerance in mice and found that GSNOR KO mice improved tolerance to cold exposure than WT mice (Fig. 2d and Supplementary Fig. 3e), which suggests that these mice have enhanced thermogenic capacity, allowing them to maintain body temperature more effectively in cold conditions. To further explore the metabolic implications, we conducted metabolic cage studies on 11-month-old GSNOR KO mice. The results showed that compared to wild-type (WT) mice, aged GSNOR knockout (KO) mice exhibited higher oxygen consumption, as well as higher energy exchange rate (EE) and respiratory quotient (RQ) (Fig. 2e–g). Additionally, we performed metabolic assessments on 20-month-old GSNOR KO mice, which showed higher oxygen consumption, carbon dioxide production, heat production compared to WT mice (Supplementary Fig. 3a–d). These results suggest an enhanced metabolic rate and energy expenditure in GSNOR KO mice. To determine whether the enhanced thermogenesis under cold stimulation was associated with BAT activity, we measured the expression of key thermogenic markers (Ucp1, Pgc1a, Pgc1b, and Cidea) in BAT. At 11 months of age, BAT from GSNOR KO mice showed a significant reduction in Ucp1 mRNA levels compared to WT (Supplementary Fig. 3f). This finding is consistent with a previous report demonstrating that BAT-specific deletion of GSNOR impairs cold-induced UCP1-dependent thermogenesis36. No significant differences in BAT thermogenic marker expression were observed between WT and KO mice at 20 months of age (Supplementary Fig. 3g). Taken together, these results indicated that BAT does not contribute to the improved cold tolerance observed in GSNOR KO mice, prompting us to focus on the role of beige adipocytes in iWAT. Importantly, no significant differences in lipolysis were found between the WT and GSNOR KO mice (Supplementary Fig. 3h, i). These findings suggest that the metabolic changes in GSNOR KO mice may contribute to their resistance to obesity. Furthermore, we performed a glucose tolerance test (GTT) and an insulin tolerance test (ITT) on aged WT and GSNOR KO mice. As expected, aged GSNOR KO mice presented improved metabolic fitness and increased insulin sensitivity compared with aged WT mice (Fig. 2h, i). Taken together, these results suggest that GSNOR KO can both improve metabolism and promote thermogenesis, which likely contribute to resistance to age-related obesity.

a, b Food intake and feces weight of 20-month-old male WT and KO mice (n = 5). c Locomotion activity measurement in an open field for 20-month-old male WT and KO mice (n = 8). d Cold tolerance test of 20-month-old male WT and KO mice (WT n = 10, KO n = 9). e–g Analysis of metabolic performances of 11-month-old male WT and KO mice (n = 6), including oxygen consumption, carbon dioxide production, energy expenditure. The regression plots on the right side of each panel depict the respective metabolic parameter as a function of mouse weight, in the dark and light cycles. h Glucose tolerance test (GTT) performed on 20-month-old male WT and KO mice (WT n = 4, KO n = 5). The area under the curve (AUC) was calculated as a measure of glucose intolerance for each individual mouse. i Insulin tolerance test (ITT) performed on 20-month-old male WT and KO mice (WT n = 3, KO n = 4). The area under the curve (AUC) was calculated as a measure of insulin tolerance for each individual mouse. Data are represented as mean ± SEM (a, b) or mean ± SD (c, d, f, h, i); Significance was assessed by Unpaired two-tailed Student’s t test (a–d, h, i), two-way ANOVA and two-side ANCOVA (e, f, g) (*p < 0.05, **p < 0.01, ***p < 0.001, ns for not significant). WT wild-type, KO, GSNOR knockout.
GSNOR was increased in the iWAT of middle-aged mice and humans
Considering the critical effects of GSNOR on age-related obesity in mice, it is worthwhile to study the changes in GSNOR expression levels during natural aging. Notably, we found that GSNOR protein levels were consistently elevated, whereas UCP1 expression was significantly reduced in the iWAT of middle-aged mice compared with young mice (Fig. 3a), but the protein expression of GSNOR in gWAT and BAT did not differ between young mice and middle-aged mice (Supplementary Fig. 4a, b). This finding was consistent with the obvious decrease in iWAT weight but not in gWAT or BAT in middle-aged GSNOR KO mice (Fig. 1f). To further corroborate these findings, we measured GSNOR expression levels in the adipose tissues of both adult and middle-aged humans. Surprisingly, we found that the level of the GSNOR protein in the subcutaneous adipose tissues (SATs) of middle-aged (38–47 years) men was markedly greater than that in adult (20–30 years) men, which is consistent with the murine data (Fig. 3b). To assess the relevance of GSNOR to human obesity, we analyzed GSNOR gene expression in a dataset obtained from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geoprofiles/). We observed that the mRNA level of GSNOR was greater in the preadipocytes of adults with obesity than in those of lean adults (Fig. 3c). These data collectively suggest that elevated GSNOR levels in SAT may contribute to aging-associated obesity in both mice and humans.

a Immunoblot analysis of GSNOR and UCP1 protein levels in iWAT from young and middle-aged mice (male, young mice n = 6, middle-aged mice n = 7). b Immunoblot analysis of GSNOR protein levels in subcutaneous adipose tissue (SAT) from young (n = 5) and middle-aged men (n = 4). c Gene expression levels of GSNOR in preadipocytes derived from lean and adults with obesity (n = 14). d Gross view and body weight of 3-month-old male control and KI mice (control n = 12, KI n = 10). e, f Gross view and weight percentage of iWAT relative to body weight from 3-month-old male control and KI mice (Control n = 12, KI n = 10). g H&E staining of iWAT, gWAT, and BAT from 3-month-old male control and KI mice (n = 4). Scale bar, 200 μm. h Cold tolerance test of 3-month-old male control and KI mice (control n = 7, KI n = 6). i–k Metabolic performance analysis, including oxygen consumption, carbon dioxide production, and energy expenditure in 3-month-old male control and KI mice (n = 6). The regression plots on the right side of each panel depict the respective metabolic parameter as a function of mouse weight, in the dark and light cycles. Data are represented as mean ± SEM (c, h) or mean ± SD (a, b, d, e, g, h, i, j, k); Significance was assessed by Unpaired two-tailed Student’s t test (a, b, c, d, e, f, h), two-way ANOVA and two-side ANCOVA (i, g, k) (*p < 0.05, **p < 0.01, ***p < 0.001, ns for not significant), KI GSNOR adipocyte knock-in, H&E hematoxylin and eosin, iWAT inguinal white adipose tissue, gWAT gonadal white adipose tissue, BAT brown adipose tissue.
GSNOR overexpression in adipose tissue promoted obesity and suppressed thermogenesis
To determine whether increased GSNOR expression in adipose tissue could promote obesity during aging, adeno-associated viruses (AAVs) expressing GSNOR were injected directly into the iWAT depots of 3-month-old mice (Supplementary Fig. 5a). Four weeks after viral transduction, we observed significant upregulation of GSNOR mRNA in iWAT from the AAV-GSNOR groups compared with the AAV-Ctrl groups (Supplementary Fig. 5b). Strikingly, compared with those from the AAV-Ctrl groups, the iWAT from the AAV-GSNOR groups presented a whiter appearance (Supplementary Fig. 5c), and H&E staining of the iWAT from the AAV-GSNOR groups revealed fewer adipocytes per field of view, suggesting that the adipocyte size in the AAV-GSNOR groups was larger (Supplementary Fig. 5d). Consistent with the increased adipocyte size in the AAV-GSNOR groups, there was a marked decrease in the expression of mitochondrial genes, genes involved in thermogenesis, and genes found in beige adipose tissue (Supplementary Fig. 5e). Moreover, contrary to expectations, there was no appreciable difference in iWAT mass between the AAV-GSNOR and AAV-Ctrl groups (Supplementary Fig. 5f), which might be attributed to the brief duration of GSNOR overexpression induced by the virus. These findings suggest that GSNOR overexpression in iWAT facilitates the conversion of beige adipocytes to white adipocytes.
To circumvent the limitations of transient AAV-mediated overexpression and investigate the chronic effects of GSNOR overexpression on adipose tissue, we developed a fat-specific overexpression mouse model. We subsequently generated conditional overexpression mice (hereby referred to as Rosa26GSNOR-GFP control mice) for robust and sustained genetic manipulation of GSNOR in vivo (Supplementary Fig. 6a). Next, Rosa26GSNOR-GFP mice were bred with Adipoq-Cre (Adp-Cre) mice to generate constitutive GSNOR adipocyte knock-in mice (KI mice) (Supplementary Fig. 6b). We determined that GSNOR was specifically overexpressed in adipose tissue (Supplementary Fig. 6c). On the other hand, we observed that total protein S-nitrosation (PSNO) levels in iWAT from KI mice were significantly lower than those in iWAT from control mice via an irreversible biotin switch assay procedure (IBP) and western blot analysis, which further confirmed the effectiveness of GSNOR overexpression (Supplementary Fig. 6d). In stark contrast to GSNOR KO mice, KI mice exhibited obvious obesity and increased body weight compared with control mice at 3 months of age (Fig. 3d). Moreover, the iWAT, gWAT and BAT weights (Fig. 3e, f) and adipocyte sizes within these tissues (Fig. 3g) were greater in 3-month-old KI mice than in control mice, which is consistent with our lentiviral injection studies. We observed that KI mice exhibited a significantly impaired ability to maintain body temperature compared to control mice during cold exposure (Fig. 3h). Furthermore, lower oxygen consumption, carbon dioxide production, and energy expenditure in 3-month-old KI mice suggested that the metabolic level of the KI mice was decreased (Fig. 3i–k). Meanwhile, there was no significant differences in lipolysis between the WT and GSNOR KI mice (Supplementary Fig. 6e). Together, these data support a critical role for GSNOR as a potential driver in the pathogenesis of age-related obesity.
Aged GSNOR KO mice exhibit enhanced maintenance of beige adipocytes
Brown and beige adipocytes are specialized cells with distinct characteristics compared to white adipocytes, including numerous small lipid droplets and a high number of cristae-rich mitochondria. They are primarily responsible for thermogenesis and metabolic homeostasis37. The maintenance and enhancement of metabolism and thermogenesis is underpinned by the comprehensive remodeling of gene networks38. To elucidate the transcriptional changes underlying the increased fat metabolism observed in aged GSNOR KO mice, we conducted a comparative analysis of gene expression profiles in iWAT from aged WT and GSNOR KO mice via RNA-seq. Our analysis revealed a significant upregulation of genes associated with biological processes related to lipid metabolism and cell components related to mitochondria (Fig. 4a). To deepen our understanding of the temporal dynamics of these metabolic changes, we conducted proteomic analyses of iWAT from both young and middle-aged mice. Our findings revealed that, compared with young mice, middle-aged mice presented a distinct pattern of differentially expressed proteins, which were associated mainly with adipose metabolism and mitochondrial function (Fig. 4b). These data suggest that age-related changes in protein expression are pivotal in the metabolic alterations observed in middle-aged mice. It is possible that GSNOR KO mice mitigate age-related metabolic decline by altering the expression patterns of these critical proteins.

a Gene ontology enrichment analysis of RNA sequencing data from iWAT of 20-month-old male WT and KO mice (n = 3). b Gene ontology enrichment analysis of quantitative proteome from iWAT of young and middle-aged male mice (n = 3). c mRNA expression levels of mitochondrial genes (Mtco1, Mtnd5) in iWAT from 20-month-old male WT and KO mice (WT n = 5, KO n = 4). d mRNA expression levels of beige markers and thermogenic genes in iWAT from 20-month-old male WT and KO mice. Sample sizes per group ranged from n = 3 to 8 biologically independent animals, as detailed in the Source data file. e Immunohistochemical staining for UCP1 protein in iWAT from 20-month-old male WT and KO mice (n = 3 biological replicates). Scale bar, 200 μm. f Immunoblotting for UCP1 protein levels in iWAT from 20-month-old male WT and KO mice (n = 3 biological replicates). Data are represented as mean ± SD; Significance was assessed by unpaired two-tailed Student’s t test (c, d, f) (*p < 0.05, **p < 0.01, ***p < 0.001). WT wild-type, KO GSNOR knockout, iWAT inguinal white adipose tissue.
We hypothesized that GSNOR deficiency alters adipose tissue characteristics. Subsequently, we found that the expression of mitochondrial markers, including Mtco1 and Mtnd5, was elevated in iWAT from aged GSNOR KO mice compared with aged WT mice, suggesting an enhanced presence of beige adipocytes (Fig. 4c). Moreover, the expression levels of genes associated with beige adipocytes and involved in thermogenesis were significantly elevated. (Fig. 4d). These results suggest that the iWAT of GSNOR KO mice may have an enhanced capacity for beige adipocyte formation and increased thermogenic potential. Previous studies have highlighted the potential of beige adipocytes in augmenting energy expenditure39. Uncoupling Protein 1 (UCP1) serves as a key indicator of beige and brown adipocytes, reflecting their thermogenic capabilities40. Notably, the elevated expression of UCP1 in aged GSNOR KO mice compared with aged WT mice was confirmed by immunohistochemistry and western blot analysis of iWAT (Fig. 4e, f). However, no significant differences in PGC1α or UCP1 protein levels were detected in gWATs or BAT between aged WT and KO mice (Supplementary Fig. 7a, b). Together, these results suggest that GSNOR deficiency prevents age-related obesity, possibly by inhibiting the whitening of adipose tissue, thereby maintaining higher levels of beige adipocytes.
GSNOR overexpression accelerated adipose tissue whitening by impairing beige adipocyte maintenance
Studies have shown that a programmed loss of brown adipocytes in the WATs of mice occurs with age41, in addition to a decline in browning markers in human WAT with age42. The maintenance of brown and beige adipocytes is crucial for sustained metabolic health, whereas adipose tissue whitening has detrimental effects39. Given the association between adipose whitening and age-related obesity, we hypothesized that GSNOR overexpression might contribute to obesity by facilitating adipose whitening in iWAT. Indeed, we observed a marked reduction in the protein expression of the mitochondrial marker TIM23 in iWAT from KI mice compared with control mice, suggesting a lower number of beige adipocytes in KI mice (Fig. 5a). To explore our hypothesis further, we treated mice with rosiglitazone or cold exposure to synchronize the stage of beige adipocytes and then subsequently withdrew the treatment to monitor the conversion of beige adipocytes to white adipocytes43. Control and KI mice were treated with rosiglitazone for ten consecutive days to promote beige adipocyte formation (day 0 of treatment withdrawal) and then were subjected to a 15-day rosiglitazone withdrawal period for the conversion of beige adipocytes to white adipocytes. The levels of beige adipocyte and thermogenic markers were detected on day 15 of rosiglitazone withdrawal (Fig. 5b). We found that the levels of UCP1, a marker of beige adipocytes, were significantly elevated in iWAT from both control and KI mice on day 0 of rosiglitazone withdrawal (Fig. 5c), which indicated that their level of browning was consistent and their ability to become beige is comparable. Notably, the level of the beige marker UCP1 in iWAT from KI mice was significantly lower than that in iWAT from control mice on day 15 postrosiglitazone withdrawal (Fig. 5d, e). Additionally, the fluorescence intensity of TOM20, a marker of mitochondrial abundance, was significantly lower in KI mice than in control mice (Fig. 5f). These findings are consistent with significant changes in the beige-to-white-adipose switch. These data demonstrated that the ability to maintain adipose tissue beigeing in KI mice was impaired and that GSNOR overexpression promoted adipose tissue whitening. Taken together, these results emphasized the indispensable role of GSNOR in promoting adipose tissue whitening.

a Immunoblotting for TIM23 protein levels in iWAT from 3-month-old male control and KI mice (n = 4 biological replicates). b Experimental timeline for the collection of iWAT following treatment with rosiglitazone or cold exposure. c Immunoblotting for UCP1 protein levels in iWAT from 3-month-old male control and KI mice at day 0 (DMSO: n = 2, rosiglitazone: Control n = 6, KI n = 7). d, e UCP1 protein levels (n = 5) and Ucp1 mRNA levels (WT n = 3, KI n = 4) in iWAT from 3-month-old male control and KI mice at day 15 after rosiglitazone withdraw. f Immunofluorescence staining for TOM20 protein in iWAT from 3-month-old male control and KI mice at 15 days of rosiglitazone withdrawal (n = 5 biological replicates). Scale bar, 20 μm. Data are represented as mean ± SD; Significance was assessed by Unpaired two-tailed Student’s t test (a, c, d, e, f) (*p < 0.05, ***p < 0.001, ns for not significant). iWAT inguinal white adipose tissue, KI GSNOR adipocyte knock-in. DMSO dimethyl sulfoxide.
GSNOR decreased Beclin-1 S-nitrosation to increase autophagy and promoted adipose tissue whitening
To elucidate the potential mechanisms of GSNOR in age-related obesity, we performed protein quantitative proteomics and quantitative S-nitrosation proteomics44 to investigate the levels and sites of protein S-nitrosation in iWAT from control and KI mice (Fig. 6a). The abundance ratios of proteins and their S-nitrosation states (KI/Control) were assessed, with ratios greater than 1.2 or less than 0.8 considered statistically significant. The results indicated that the KI group exhibited a marked reduction in S-nitrosation modification targets, with approximately half of the identified targets showing significant downregulation (Supplementary Fig. 8a), which aligned with previous results on PSNO levels (Supplementary Fig. 6d), thereby validating the reliability of our quantitative proteomic approach. A clustering analysis of differentially expressed proteins between the two groups highlighted that molecular functions related to fatty acid metabolism and cellular components associated with mitochondria were enriched (Supplementary Fig. 8b, c). We subsequently focused on the top-ranked downregulated (≤0.8) S-nitrosation proteins that were most closely associated with fatty acid metabolism and mitochondria (Fig. 6b). Beclin-1, a core member of autophagy, attracted our attention, and we determined that cysteine 351 of Beclin-1 was S-nitrosated (Fig. 6c). An IBP assay confirmed that the level of Beclin-1 S-nitrosation was lower in iWAT from KI mice than control mice (Fig. 6d), whereas no significant differences in the level of S-nitrosation were detected for mTOR and ATG4B between the two groups (Supplementary Fig. 8d). We further demonstrated that the level of Beclin-1 S-nitrosation was elevated in GSNOR KO mice (Fig. 6e) but reduced in middle-aged mice compared with young mice (Fig. 6f), suggesting that Beclin-1 may be a key target of GSNOR.

a Schematic representation of the experimental approach for quantitative S-nitrosation proteome in iWAT from 3-month-old male control and KI mice (n = 3). b Top-ranked S-nitrosation proteins of quantitative S-nitrosation proteome. c Quantitative S-nitrosation proteomic mass showing the identification of the S-nitrosation site at cysteine 351 in Beclin-1 peptides. d Immunoblotting for SNO-Beclin-1 levels in iWAT from 3-month-old male control and KI mice (n = 3 biological replicates). e Immunoblotting for SNO-Beclin-1 levels in iWAT from 11-month-old male WT and KO mice (n = 6 biological replicates). f Immunoblotting for SNO-Beclin-1 levels in iWAT from young and middle-aged male mice (n = 3 biological replicates). g Immunoblotting for P62 protein levels in iWAT from WT and KO mice (n = 3 biological replicates). h Electron micrographs in iWAT from 3-month-old male WT and KO mice harvested at 2 days of cold exposure withdrawal. Red arrows denote autophagosomes, and the letter L denotes lipid droplets. White scale bar, 1 μm. Black scale bar, 200 nm. Images were acquired from 5 random fields per mouse (n = 4 per group). i Immunoblotting for P62 protein levels in iWAT from control and KI mice at 2 days of cold exposure withdrawal (n = 3 biological replicates). Data are represented as mean ± SD; Significance was assessed by unpaired two-tailed Student’s t test (d, e, f, g, i) (*p < 0.05, **p < 0.01, ***p < 0.001). iWAT inguinal white adipose tissue, WT wild-type, KO GSNOR knockout.
We next determined the functional significance of SNO-Beclin-1 in adipose tissue whitening in the context of obesity. Autophagy-mediated mitochondrial clearance is needed for beige-to-white adipocyte reversal43,45. We hypothesized that Beclin-1 S-nitrosation might modulate autophagy in adipocytes. The accumulation of P62, a marker of reduced autophagic flux, was observed in iWAT but not in gWATs or BATs from aged GSNOR KO mice (Fig. 6g and Supplementary Fig. 7a, b), which is consistent with the specific increase in GSNOR levels in the iWAT of aging mice. Compared with that in WT mice, electron microscopy revealed a decrease in the number of autophagosomes in GSNOR KO mice after cold exposure withdrawal, which further illustrated that the degree of autophagy in GSNOR KO mouse iWAT was reduced during the conversion of beige adipocytes to white adipocytes (Fig. 6h). To delineate the specific roles of GSNO reductase versus formaldehyde dehydrogenase activity, we introduced GSNOR-WT or GSNOR-C174S mutant46 (deficient specifically in GSNO reductase activity) into GSNOR KO primary adipocytes. Following being stimulus withdrawal, C174S-reconstituted cells maintained elevated SNO-Beclin-1 levels, exhibited impaired autophagic flux (indicated by P62 accumulation), and showed increased UCP1 and TOM20 expression - fully recapitulating the GSNOR KO phenotype. In contrast, GSNOR-WT reconstitution significantly reduced SNO-Beclin-1, enhanced autophagic flux, and suppressed UCP1/TOM20 expression (Supplementary Figs. 7c and 8e). These results establish that GSNO reductase activity specifically regulates adipose whitening through control of Beclin-1 S-nitrosation, autophagy, and mitochondrial clearance. We then used KI mice and control mice to analyze autophagic flux during the conversion of beige adipocytes to white adipocytes after cold exposure withdrawal. As expected, the iWAT of KI mice presented decreased levels of P62 compared with those of control mice (Fig. 6i), indicating increased autophagy in iWAT from KI mice and suggesting that GSNOR promotes autophagy. These results highlight the importance of GSNOR deletion in the maintenance of beige adipocytes. Collectively, these data suggest that targeting Beclin-1 S-nitrosation could alleviate age-related obesity by suppressing autophagy.
Beclin-1 S-nitrosation site mutation inhibited the maintenance of beige adipocytes by increasing autophagy
To directly investigate the role of S-nitrosation at the Beclin-1 cysteine 351 residue, we generated a mouse line (Beclin-1C351A) using CRISPR-Cas9 technology, in which the Cys351 residue was substituted with alanine (Fig. 7a). The successful generation of the mutant mice was confirmed by sequencing the mouse genome (Supplementary Fig. 9a). We initially verified that the level of Beclin-1 S-nitrosation was lower in iWAT from Beclin-1C351A mice than WT mice (Fig. 7b). We subsequently observed decreased P62 protein levels in Beclin-1C351A mice compared with WT mice, which was indicative of increased autophagy (Fig. 7c). Moreover, electron microscopy images revealed an increase in the number of autophagosomes in iWAT from Beclin-1C351A mice on day 2 of cold exposure withdrawal compared with that in iWAT from WT mice (Fig. 7d). Collectively, these results revealed that mutation of the Beclin-1 S-nitrosation site increased autophagy in iWAT. We confirmed that Beclin-1 S-nitrosation contributes to the maintenance of beige adipocytes by suppressing autophagy.

a Schematic of Beclin-1 C351A mutant mouse generation. b Immunoblotting for SNO-Beclin-1 levels in iWAT from 3-month-old male WT and Beclin-1C351A mice (n = 5 biological replicates). c Immunoblotting for P62 protein levels in iWAT from 3-month-old male WT and Beclin-1C351A mice (n = 3 biological replicates). d Electron micrographs in iWAT from 3-month-old male WT and Beclin-1C351A mice harvested at 2 days of cold exposure withdrawal. Red arrows denote autophagosomes and the letter L denote lipid droplets. Scale bar, 500 nm. Images were acquired from 5 random fields per mouse (n = 4 per group). e Immunoblot analysis of LC3-I, LC3-II, and P62 levels in WT and Beclin-1C351A primary adipocytes, untreated or treated with 3-MA (10 mM, 24 h) or bafilomycin A1 (BafA1, 10 nM, 24 h), following a 1-day withdrawal period from rosiglitazone treatment. (Representative data from three independent replicates experiments). f Representative fluorescence imaging photographs of primary adipocytes infected with Plv-GFP-RFP-LC3 adenovirus and treated with or without 3-MA (10 mM, 24 h) or bafilomycin A1 (BafA1, 10 nM, 24 h). Scale bar, 10 μm. The numbers of LC3 puncta (including yellow LC3 puncta and red LC3 puncta) per cell in each condition were quantified using Image-J. More than 12 cells were counted in each condition. The n values for WT and Beclin-1C351A in each group are as follows: Control group: n = 21, 21; 3-MA group: n = 12, 23; BafA1 group: n = 14, 13. (Representative data from three independent replicates experiments). g Co-immunoprecipitation of Beclin-1/ATG14 and LC3 immunoblot in primary adipocytes with/without S-nitrosocysteine (SNOC) treatment. (Representative data from three independent replicates experiments). h Mito-Tracker Green staining to visualize mitochondria in primary adipocytes from WT and Beclin-1C351A mice with or without 3-MA treatment, 4 days after rosiglitazone withdrawal. Scale bar, 5 μm. (WT: n = 21, Beclin-1C351A: n = 22, Beclin-1C351A + 3MA: n = 17. Representative data from three independent replicates experiments). i Immunoblotting for UCP1 protein levels in iWAT from 3-month-old male WT and Beclin-1C351A mice at 15 days of rosiglitazone withdrawal (WT: n = 5, Beclin-1C351A: n = 6). j Normalized iWAT mass weight relative to total iWAT weight of WT or Beclin-1C351A mice at 0 day of cold exposure withdrawal (Day 0 n = 4, Day 15 n = 6). Data are represented as mean ± SD; Significance was assessed by Unpaired two-tailed Student’s t test (b, c, f, h, i, j) (*p < 0.05, **p < 0.01, ***p < 0.001, ns for not significant). SNO-beclin-1 beclin-1 S-nitrosation, WT wild-type, SNOC S-nitrosocysteine.
To further validate the effect of Beclin-1 S-nitrosation on the autophagic process, we conducted an autophagic flux analysis utilizing 3-methyladenine (3-MA), an early-stage autophagy inhibitor that suppresses autophagosome formation, and bafilomycin A1 (BafA1), a late-stage autophagy inhibitor that blocks the fusion of autophagosomes with lysosomes. The experimental timeline for collecting primary adipocytes following rosiglitazone treatment is designed to capture critical time points for assessing autophagy levels on Day 1 and associated biological effects on Day 4 (Supplementary Fig. 9b). Interestingly, an analysis of the in primary adipocytes after rosiglitazone withdrawal revealed that the ratio of LC3-Ⅱ/LC3-Ⅰ, a marker of autophagy, increased in Beclin-1C351A adipocytes compared to WT adipocytes (Fig. 7e). LC3-Ⅱ/LC3-Ⅰ ratio decreased both in WT and Beclin-1C351A groups with 3-MA treatment, while LC3-Ⅱand P62 protein levels increased with BafA1 treatment, and more in Beclin-1C351A groups than WT groups (Fig. 7e). Moreover, fluorescence analysis using Plv-GFP-RFP-LC3 showed that mutation of the Beclin-1 S-nitrosation site markedly increased the number of LC3 puncta in primary adipocytes on day 1 of rosiglitazone withdrawal compared with that in WT adipocytes, and this effect was suppressed by 3-MA treatment in both the control and the Beclin-1C351A groups (Fig. 7f). In contrast, more LC3 puncta were observed in BafA1-treated adipocytes than in WT adipocytes, and the number of LC3 puncta in Beclin-1C351A adipocytes was greater (Fig. 7f), suggesting that S-nitrosation of Beclin-1 Cys351 promoted autophagy at the early stage of adipocyte whitening.
Given that protein S-nitrosation regulates protein–protein interactions47, we performed immunoprecipitation (IP) experiments with an anti-Beclin-1 antibody on iWAT from WT and Beclin-1C351A mice on day 2 of rosiglitazone withdrawal. Proteins were then separated via SDS‒PAGE, visualized by silver staining (Supplementary Fig. 9c), and identified by mass spectrometry. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses of the 34 upregulated interacting proteins revealed associations with mitochondrial transport, the cold response and peptide cross-linking (Supplementary Fig. 9d). Among the top four highest scoring proteins (Supplementary Fig. 9e), ATG14 is known to be part of the VPS34 initiation complex (VPS15-VPS34-ATG14-Beclin-1) required for autophagy activation48. On the basis of these results, we hypothesized that S-nitrosation of Beclin-1 Cys351 inhibited its interaction with ATG14. To explore our hypothesis, we further treated primary adipocytes with S-Nitrosocysteine (SNOC) after the withdrawal of rosiglitazone. The immunoprecipitation assay results revealed that the ATG14-Beclin-1 interaction was decreased in the presence of SNOC but increased in the Beclin-1C351A group compared with the WT group (Fig. 7g). An analysis of the autophagic flux in primary adipocytes after rosiglitazone withdrawal revealed that the ratio of LC3-Ⅱ/LC3-Ⅰdecreased in WT adipocytes treated with SNOC but increased when the S-nitrosation of Beclin-1 (Cys351) was abolished (Fig. 7g). These data suggest that Beclin-1 S-nitrosation downregulates autophagy initiation by interfering with the interaction between Beclin-1 and ATG14. In addition, the level of mitochondria, indicative of beigeing levels, was significantly lower in primary adipocytes from the Beclin-1C351A group than in those from the WT group after rosiglitazone withdrawal, which was abrogated by treatment with early-stage autophagy inhibitors (3-MA) (Fig. 7h). Overall, these results demonstrated that Beclin-1C351A promotes autophagy and whitening in mice by interacting with ATG14.
Next, we determined whether alterations in Beclin-1 S-nitrosation affect the rate of adipocyte whitening. We used rosiglitazone or cold exposure to promote the synchronization of beige adipocytes in mice and then ceased rosiglitazone treatment or cold exposure to observe and compare the conversion of beige adipocytes to white adipocytes. At first, by comparing the UCP1 protein levels of WT and Beclin-1C351A mice before and after cold stimulation, we found that the UCP1 levels of the two groups increased after ten days of cold stimulation, and there was no difference before the two groups. Which indicated that the degree of beige of the two groups after cold stimulation was consistent (Supplementary Fig. 9f). Next comes the whitening phase. As expected, we found that the UCP1 protein level in iWAT from Beclin-1C351A mice was lower than that in iWAT from control WT mice on day 15 of rosiglitazone withdrawal (Fig. 7i). Although we did not observe difference in body weight between the WT and Beclin-1C351A mice (Supplementary Fig. 9g), it should be noted that the iWAT weight on day 15 of cold withdrawal normalized to the iWAT weight on day 0 was greater in Beclin-1C351A mice than in WT mice (Fig. 7j), suggesting that, compared with the WT mice, the Beclin-1C351A mice presented impaired beige adipocyte maintenance and increased adipose tissue whitening.
Targeting GSNOR to combat adipose tissue whitening and middle-aged obesity
Considering the systemic mutation in Beclin-1C351A mice, we sought to explore the functional role of Beclin-1 S-nitrosation in the transition from beige to white adipocytes and to determine whether this process is regulated by GSNOR in Beclin-1C351A mice. For this purpose, we isolated primary adipocytes from WT and Beclin-1C351A mice. These adipocytes were treated with rosiglitazone and then subjected to withdrawal to observe the beige-to-white adipocyte conversion. In this system, we investigated the effect of GSNOR knockdown on adipose tissue whitening mediated by Beclin-1 cysteine 351. As anticipated, shRNA-mediated knockdown of GSNOR significantly decreased GSNOR expression in WT adipocytes (Supplementary Fig. 9h). The reduction in GSNOR levels led to an increase in UCP1 expression and smaller lipid droplets, as indicated by oil red O staining, in the WT group (Fig. 8a, b), suggesting maintenance of beige adipocytes. Conversely, in the Beclin-1C351A group, UCP1 levels were markedly reduced, and the lipid droplets were significantly larger than those in the WT group, indicating an accelerated transition to white adipocytes. Moreover, GSNOR knockdown in the Beclin-1C351A group did not increase UCP1 levels or reduce lipid droplet size (Fig. 8a, b), confirming that Beclin-1 S-nitrosation and its function in Beclin-1C351A mice could not be restored by GSNOR depletion. These results underscore the critical role of Beclin-1 S-nitrosation in inhibiting the whitening of adipose tissue. The absence of Beclin-1 S-nitrosation in Beclin-1C351A mice promoted the rapid whitening of adipose tissue and reduced the level of beiging in adipocytes. In conclusion, our results emphasized the importance of Beclin-1 S-nitrosation in preserving beige adipocyte characteristics and preventing the conversion of beige adipocytes to white adipocytes. The data presented here strongly support the notion that manipulating GSNOR to control Beclin-1 S-nitrosation is a promising therapeutic approach for countering age-related obesity and metabolic deterioration.

a Oil Red O staining of primary adipocytes from WT and Beclin-1C351A mice treated with sh-ctrl or shGSNOR adeno-associated virus (AAV) at 4 days of rosiglitazone withdrawal. Scale bar, 50 μm. (n represented the number of lipid droplets. WT untreated (n = 15), WT treated (n = 16), Beclin-1 C351A untreated (n = 17), Beclin-1 C351A treated (n = 14). Representative data from three independent replicates experiments). b Immunoblotting for UCP1 protein levels in primary adipocytes from WT and Beclin-1C351A mice infected with sh-ctrl or shGSNOR AAV. (Representative data from three independent replicates experiments). c Schematic illustration of the site-directed AAV injections used to knockdown GSNOR expression in the iWAT of 14-month-old mice. d Immunoblotting for GSNOR and UCP1 protein levels and densitometric quantification of the bands (n = 7). e Gross view and weight of iWAT injected with control (AAV-sh-ctrl) or AAV-shGSNOR (n = 11). f H&E staining of iWAT from mice injected with control (AAV-sh-ctrl) or AAV-shGSNOR (n = 6). Scale bar, 200 μm. g A graph model depicting the role of GSNOR in age-related obesity. Data are represented as mean ± SD; Significance was assessed by Unpaired two-tailed Student’s t test (a, b), Paired two-tailed Student’s t test (d–f) (*p < 0.05, ***p < 0.001, ns for not significant). AAV adeno-associated virus, H&E iWAT, H&E hematoxylin and eosin, iWAT inguinal white adipose tissue.
To further explore the therapeutic potential of targeting GSNOR, we investigated the effects of iWAT-specific GSNOR intervention on obesity in middle-aged mice. We injected an AAV carrying shRNA targeting GSNOR (AAV-shGSNOR) or control (AAV-sh-ctrl) into the iWAT of middle-aged (14-month-old) mice (Fig. 8c). AAV-shGSNOR successfully knocked down GSNOR expression in the iWAT (Fig. 8d). Compared with the sh-ctrl group, this knockdown led to increased UCP1 protein levels, indicating that GSNOR ablation suppressed the conversion of beige adipocytes to white adipocytes (Fig. 8d). Consequently, compared with those in the sh-ctrl group, the fat mass and adipocyte size in the shGSNOR group were reduced (Fig. 8e, f). These findings collectively demonstrated that Beclin-1 S-nitrosation plays a critical role in preserving beige adipocyte characteristics and inhibiting adipose tissue whitening and that targeting GSNOR to maintain Beclin-1 S-nitrosation is a potential therapeutic strategy to combat adipose tissue whitening and middle-aged obesity.
Discussion
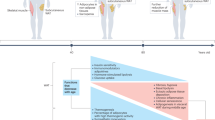
Age-related obesity and metabolic decline are significant high-risk factors for age-related diseases, and effective management of middle-aged obesity is crucial in controlling age-related metabolic disorders. In this study, we discovered that GSNOR, whose expression in iWAT increases significantly in naturally aged mice and humans, is a novel key molecule that regulates age-related obesity and metabolic decline. Denitrosation of Beclin-1 by GSNOR promotes autophagy initiation in iWAT by increasing the interaction between Beclin-1 and ATG14, which exacerbates the loss of beige adipocyte characteristics, contributing to adipose tissue whitening and metabolic deterioration in age-related obesity. Conversely, specific knockdown of GSNOR in the iWAT of old mice abrogates the age-related increase of iWAT mass (Fig. 8g). This study revealed that increased adipose whitening mediated by GSNOR-denitrosation of Beclin-1 is a new mechanism for age-related obesity apart from the previously reported mechanisms regarding neuromodulation, inflammaging and the reduction in white-to-beige conversion capacity, providing new insights for the prevention of age-related obesity.
GSNOR is a new age-related obesity gene
We reported for the first time that GSNOR expression in WAT was increased in middle-aged mice and humans, and its expression was strongly correlated with weight gain during aging. This correlation underscores its potential pivotal role in age-related obesity and metabolic decline. The organ-specific temporal signatures of aging, as identified by Schaum et al.49, indicate that WAT is among the first tissues to exhibit aging hallmarks. Moreover, several genes have been associated with severe monogenic and syndromic forms of obesity50. Additionally, genome-wide association studies (GWASs) have revealed numerous single nucleotide polymorphisms (SNPs) associated with obesity. The FTO gene, for example, is the first locus identified with common variants that significantly influence obesity predisposition and fat mass at the population level51. Other genes critical for appetite regulation and fat metabolism, such as PR12052, MC4R53, Mrap254,55, and PCSK156, are also related to obesity. Despite various strategies for body weight management, including caloric restriction57, exercise58, targeting the glucagon-like peptide-1 receptor agonist (GLP-1RA) semaglutide59,60, and lipectomy-induced compensatory recovery of body fat61, satisfactory results have not been consistently achieved. This highlights the need for further research into new targets and mechanisms. Our study revealed that GSNOR overexpression in adipocytes resulted in beige cell whitening, metabolic decline, and obesity. Elevated GSNOR levels in preadipocytes from adults with obesity further support these findings. Conversely, GSNOR KO alleviated age-related obesity by increasing fat metabolism and promoting thermogenesis, suggesting that GSNOR is indeed an obesity-related gene. Therefore, we propose that GSNOR may be a new intervention target for age-related obesity and the development of specific GSNOR inhibitors may have promising effect inmitigating the middle-aged onset of obesity.
Adipose tissue whitening or beiging matters in age-related obesity and autophagy accounts
In our study, a higher proportion of beige adipocytes in adipose tissue was associated with reduced age-related obesity, as observed in GSNOR KO mice. Conversely, an increased abundance of white adipocytes correlated with exacerbated age-related obesity, particularly evident in GSNOR KI mice. The characteristics of whitening or beiging determine whether or not of the occurrence of age-related obesity. While the state of whitening or beiging is up to the level of autophagy in the adipose tissue. Our study revealed that the increased GSNOR in adipose tissue during aging leads to an increase in autophagy levels, which contributes to the alterations in adipocyte characteristics, leading to phenotypic transition from beige to white adipocytes. Conversely, GSNOR knockdown abrogates all of these effects. Previous studies also provide consistent evidence. For example, it has been reported that autophagy increases in adipose tissue with age and promotes metabolic disorders62. This phenomenon is also observed in the adipose tissues of individuals with obesity (Kovsan et al.)63. Adipocyte-specific deletion of the autophagy-related genes Atg5 or Atg12 maintains high thermogenic capacity and protects against diet-induced obesity by preventing beige adipocyte loss43. Similarly, BAT-specific deletion of the autophagy-related gene Atg7 suppressed mitochondrial clearance and maintained brown adipocytes45. Additionally, Parkin deletion specifically in adipose tissue protects mice against high-fat diet- and aging-induced obesity, which is partially due to a mild reduction in autophagy levels64. In together, our study clearly elucidates the correlation of the state of adipose tissue (white or beige), the levels of autophagy and the obesity. Adipose tissue whitening or beiging state matters in age-related obesity and autophagy accounts for the state of adipose tissue.
S-nitrosation of Beclin-1 is a brake of autophagy
Autophagy is crucial for mitochondrial clearance during the beige-to-white adipocyte transition. Therefore, understanding the mechanism of autophagy regulation during this process is important. Beclin-1 is a key protein in the autophagy process65. In this study, we found that Beclin-1 S-nitrosation status is integral to regulating autophagy. With increasing GSNOR levels in WAT during aging, the S-nitrosation of Beclin-1 at Cys351 decreases. This reduction increases the Beclin-1-ATG14 interaction in adipocytes, thereby initiating autophagy, reducing the number of mitochondria in beige adipocytes, and promoting adipose tissue whitening, ultimately leading to the development of age-related obesity. These findings were confirmed in Beclin-1C351A S-nitrosation mutant mice (Fig. 7). Conversely, GSNOR knockdown mitigated these effects. Therefore, the S-nitrosation state of Beclin-1 acts as a switch for autophagy, the beige-to-white adipocyte transition and middle-aged obesity. A decrease in Beclin-1 S-nitrosation signals facilitates the activation of autophagy and adipose tissue whitening and the onset of middle-aged obesity; while S-nitrosation of Beclin-1 is a brake of autophagy. In this study, we present novel findings indicating that Beclin-1 modulates autophagy initiation via S-nitrosation, providing a comprehensive understanding of the underlying mechanism of age-related obesity. Our previous study demonstrated that S-nitrosation of ATG4B, another crucial autophagy-related protein, impairs autophagy and leads to neurotoxicity in response to hyperglycemia66, highlighting S-nitrosation plays an important role in regulating autophagy.
The reason why GSNOR levels increase in iWAT during aging also deserves further exploration. In addition, specific mutation of Beclin-1 S-nitrosation in adipose tissue will provide more precision mechanism. Furthermore, systematic screening for GSNOR inhibitors is immediate follow-up work for our group with the long-term goal of testing GSNOR inhibitors in clinical obesity studies. The approach to specifically increasing the S-nitrosation of Beclin-1 in adipose tissue represents a new challenge and precision intervention of age-related obesity in the future.
In conclusion, our study demonstrates a new mechanism for age-related obesity and provides a comprehensive understanding of the role of GSNOR and Beclin-1 S- nitrosation in age-related obesity and adipose tissue whitening. GSNOR is found as a key target in age-related obesity. The denitrosation of Beclin-1, driven by increased GSNOR levels, promotes autophagy, thereby promoting iWAT whitening and metabolic decline during aging. Targeting GSNOR in iWAT during aging can prevent the conversion of beige adipocytes to white adipocytes. Our study provides a new avenue for preventing and treating middle-aged obesity.
Methods
Statement of ethical regulations
This study was conducted in accordance with all relevant ethical regulations for animal research. The experimental protocols were reviewed and approved by the ethics committee of animal experiments of the Institute of Biophysics, Chinese Academy of Sciences (No: SYXK2021097). The use of adipose tissue from humans was approved by the Medical Ethics Committee of Zhengzhou Central Hospital (Approval Number: ZXYY2024135).
Animals
All animal experiments were performed in accordance with procedures approved by the ethics committee of animal experiments of the Institute of Biophysics, Chinese Academy of Sciences. The mice were randomly allocated to experimental groups. The mice were housed in an enriched environment with ad libitum access to a standard diet and tap water, unless otherwise stated. The light in the facility was set to a 12-h light/dark cycle. The mice were housed at room temperature (22 ± 2 °C) during breeding. A standard normal chow diet (SiPeiFu, SPF-F02) and sterile water were given. All in vivo studies were performed in male animals. C57BL/6J mice were the WT control strain used for this study.
GSNOR KO mice
GSNOR KO mice were generated as previously described67 and were compared with age-matched littermates (C57BL/6J mice).
GSNOR knock-in mice
The Rosa26GSNOR-GFP mice were generated by Beijing VIEWSOLID Biotechnology (Beijing, China). This strain was constructed on the C57BL/6J background. In brief, the human GSNOR gene was cloned and fused with the GFP reporter gene. A termination sequence was inserted between exon 1 and exon 2 of the mouse Rosa26 locus to ensure that the GSNOR-GFP fusion gene was expressed only upon Cre-mediated recombination. LoxP sites flanking the termination sequence were added to allow Cre recombinase to excise the sequence and activate GSNOR-GFP expression. The targeting vector was assembled to contain the GSNOR-GFP fusion gene, LoxP sites, and selection markers. This was followed by microinjection into embryonic stem cells and selection, microinjection into blastocysts to generate chimeric mice, and breeding to produce heterozygous Rosa26GSNOR-GFP mice. GSNOR KI mice were generated by crossing Rosa26GSNOR-GFP mice with Adipoq-Cre mice, resulting in GSNOR overexpression specifically in adipose tissue.
Beclin-1C351A mice
Beclin-1C351A mice were produced through CRISPR-Cas9 technology, which involved replacing cysteine 351 with alanine in the Beclin-1 protein, thereby preventing S-nitrosation at this specific residue. The generation of Beclin-1C351A mice was achieved via CRISPR/Cas9 genome editing in a C57BL/6J mouse background. We designed an sgRNA sequence that targets the region upstream of the 11th exon of the mouse Beclin-1 gene (5′-TTGCCGTTATACTGTTCTGGGGG-3′). Additionally, we constructed a donor plasmid harboring homology arms and the sgRNA target site. For the microinjection process, a cocktail consisting of sgRNA, in vitro transcribed SaCas9 mRNA, and the donor plasmid was injected into the cytoplasm of zygotes derived from superovulated C57BL/6J mice. The microinjected zygotes were then implanted into pseudopregnant female mice to yield viable offspring. To confirm Beclin-1 mutation, genomic DNA from the tail tissue of each mouse was sequenced.
Diet intake monitoring
Food intake was measured weekly throughout the study. Two mice were housed per cage in metabolic cages, and the amount of food and water consumed was recorded.
Fecal collection and analysis
Feces were collected from the mice housed in metabolic cages. The metabolic cages were fitted with feces collection trays. Urine and feces were collected at designated time points and stored at −80 °C until analysis.
Open field test
The open field test is a common behavioral test used to assess exploratory activity in mice. The open field test was conducted as previously described68. Each mouse was gently placed in the center of a square chamber (50 × 50 × 40 cm), and they were allowed to freely explore the environment for 10 min. The movement of each mouse was meticulously recorded using a high-resolution video camera, and subsequent analysis was performed using the EthoVision XT software.
Survival curve
The survival of the mice was monitored daily, and the data were analyzed via the statistical software GraphPad Prism 8. Survival probability was calculated at each time point, and the curves were compared between groups via the log-rank test.
Mouse fat mass analysis
Mouse fat mass analysis was conducted using the Echo-MRITM-4in1 Body Composition Analyzer. The mice were fasted and anesthetized before being positioned in the prone position for noninvasive, rapid fat mass measurement. After measurement, the data were recorded and statistically analyzed, and the recovery of the mice was closely monitored after anesthesia.
Glucose tolerance test and insulin tolerance test
The mice were housed under a 12-h light/dark cycle with ad libitum access to food and water.
In the glucose tolerance test (GTT), the mice were fasted overnight for 14 h. The mice were subsequently injected intraperitoneally with a glucose solution (1 g/kg body weight). Blood glucose levels were measured using a glucometer at various time points (e.g., 0, 15, 30, 45, 60, 75, 90, 120, and 180 min) after glucose injection. In the insulin tolerance test (ITT), the mice were fasted for 4 h and injected intraperitoneally with insulin solution (1.5 U/kg body weight). Blood glucose levels were measured at various time points (e.g., 0, 15, 30, 45, 60, 75, and 90 min) after insulin injection. The areas under the curve (AUCs) were computed to evaluate glucose tolerance and insulin sensitivity.
Core temperature measurement during cold exposure
To assess thermogenic function, cold stimulation was performed by exposing the mice to 6 °C, and rectal temperature measurements were taken at the indicated times via a digital thermometer, as previously described by Altshuler-Keylin et al.43. The change in body temperature was calculated from the initial and final measurements.
Metabolic cage studies
Mouse metabolic cage studies were conducted using the PhenoMaster system (TSE Systems) to assess energy expenditure in WT and GSNOR KO mice or control and GSNOR KI mice. Male mice were individually housed in TSE metabolic chambers for an initial 24-h acclimation period, followed immediately by a 24-h experimental period with data collection. The room temperature for all metabolic studies was maintained at 22 °C with a 12-h light/dark cycle. Air flow into the chamber was maintained at 0.6 L/min, and exhaust air from each chamber was measured for 2 min every 18 min. The body weight of each mouse was measured and entered into the system prior to the initiation of monitoring. Oxygen consumption and carbon dioxide production were measured and normalized to body weight. Data were collected continuously and analyzed using the Phenomaster software to calculate energy expenditure in accordance with established guidelines for metabolic phenotyping69.
Human biospecimen acquisition
Human adipose tissue samples were obtained from participants undergoing elective abdominal surgery at Zhengzhou Central Hospital, following the obtainment of informed consent and Institutional Review Board (IRB) approval (Ethical review approval from the medical ethics reference number: ZXYY2024135). Subcutaneous adipose tissue (SATs) was collected, immediately snap-frozen in liquid nitrogen and stored at −80 °C for subsequent protein analysis.
Murine primary preadipocyte culture and differentiation
Primary preadipocytes were isolated from the iWAT of 2–4-week-old mice and digested with collagenase to obtain a single-cell suspension. The suspension was filtered through a 70 µm cell strainer and centrifuged to pellet the cells. The cell pellet was resuspended in DMEM/F12 containing 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 µg/ml streptomycin. The cell suspension was filtered again with a 40 µm cell strainer. The cells were cultured in a cell culture dish.
Differentiation of adipocytes
At 90% confluence, the medium was replaced with DMEM containing 10% FBS, 0.5 mM IBMX, 1 µM dexamethasone, and 1 µM insulin. The mixture was incubated for 48 h, after which the medium was replaced with DMEM containing 10% FBS and 1 µM insulin. The mixture was incubated for an additional 48 hours, after which the medium was replaced with DMEM containing 10% FBS. The cells were maintained in differentiation medium for 6–8 days until mature adipocytes formed. Oil red O staining of the cells was performed using a standard protocol.
PPAR agonist administration
Rosiglitazone, a PPARγ agonist, was used to promote beige (brown-like) adipocyte differentiation in this study. Rosiglitazone treatment of control and KI mice (or Beclin-1C351A mice) were treated with rosiglitazone (10 mg/kg body weight) by intraperitoneal injection for ten consecutive days to promote beige adipocyte differentiation. Rosiglitazone was then withdrawn for 15 days to allow the beige adipocytes to convert to white adipocytes. The levels of beige and thermogenic markers were detected on the 15th day after the withdrawal of rosiglitazone.
Rosiglitazone treatment of primary adipocytes: Primary adipocytes were treated with rosiglitazone (5 µM) for 4 days to induce beige adipocyte differentiation. Markers of autophagy were then analyzed on the first day after rosiglitazone withdrawal.
Site-directed AAV delivery
pAAV-CBG-EGFP-3xFLAG-WPRE-H1-shRNA (Adh5), pAAV-CBG-EGFP-3xFLAG-WPRE-H1-shRNA (NC), pAAV-CMV-3xFLAG-Becn1 (p. C351A)-HA-WPRE and pAAV-CMV-3xFLAG-Becn1-HA-WPRE were custom constructed and packaged by Biology (Shanghai, China). All virus titers were >1012 genomic copies/ml. Gene-overexpressing and gene-knockdown adeno-associated viruses (AAVs) were produced by HeYuan Biology. To directly transfer genes into the iWAT of mice, AAV-mediated gene transfer was used. In brief, the mice were anesthetized with isoflurane and placed on a heating pad to maintain body temperature. The injection site (inguinal region) was shaved and sterilized with iodophor. A small incision (approximately 0.5 cm) was made in the skin above the iWAT. A Hamilton syringe equipped with a 30-gauge needle was used to inject AAV vectors into the iWAT depot at three different sites. The total volume of each injection was 60 μl, and the AAV dose was 1 × 1011 viral genomes. After 6 weeks, the animals were euthanized, and iWAT samples were collected. The overexpression or knockdown efficiency was assessed using western blotting or quantitative polymerase chain reaction (qPCR).
Biochemical analysis of whole blood and serum from aged WT and GSNOR KO mice
Whole blood and serum samples were collected from aged WT and GSNOR KO mice (20 months old) using standard procedures. Briefly, the mice were anesthetized, and blood was drawn via enucleation. Whole blood samples were immediately processed for hematology analysis. Serum was obtained by centrifugation of the collected blood at 1000 × g for 15 min at -4 °C and stored at −80 °C until further analysis by the Pony Testing International Group. Hematology analysis: Whole-blood samples were analyzed using an automated hematology analyzer to measure various parameters, including red blood cell count (RBC), hemoglobin (Hb) levels, hematocrit (Hct), mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH) levels, mean corpuscular hemoglobin concentration (MCHC), white blood cell count (WBC), and neutrophil, lymphocyte, monocyte, eosinophil, and basophil counts. Serum biochemistry analysis: Serum samples were analyzed using an automated biochemical analyzer to measure the levels of various parameters, including alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), gamma-glutamyltransferase (GGT), total protein (TP), urea, creatinine (Cr), uric acid (UA), cholesterol (CHO), high-density lipoprotein (HDL), low-density lipoprotein (LDL), and TGs.
Adipocyte lipolytic activity ex vivo
Adipose tissue was harvested from 20-month-old mice, 100 mg of tissue per sample was minced with scissors, and the samples were incubated with 1 mL of Krebs buffer (Sigma, K4002) at 37 °C for 120 min. The supernatants (100 μL) were collected at 0, 0.5, 1, 1.5, and 2 . To inactivate the lipase, the samples were heated at 70 °C for 10 min and then centrifuged at 3000 × g for 3 min. The supernatants were transferred to new EP tubes. The glycerol content in the incubation medium served as an indicator of lipolysis and was measured via the GPO-Trinder reagent (Sigma, FG0100) according to the manufacturer’s instructions. The protein concentration of the adipose tissue was determined using a BCA kit.
Histology and immunohistochemical analysis
Histologic analysis of adipose tissue was conducted as previously described70. Briefly, adipose tissue or muscle tissue samples were fixed in 4% neutral-buffered formalin for 24 h at room temperature. After fixation, the samples were dehydrated through a series of ethanol concentrations, cleared in xylene, and embedded in paraffin. Five-micron-thick sections were cut and mounted on glass slides. The sections were then stained with H&E to assess the general morphology and structure of the adipose tissue.
Immunohistochemical staining was performed as described by Vamvini et al.71. Briefly, adipose tissue slides were deparaffinized in xylene and rehydrated through an alcohol series. Antigen retrieval was performed using a pressure cooker in citrate buffer (10 mM, pH 6.0), followed by blocking with 5% normal goat serum. The slides were incubated with primary antibodies overnight at 4 °C and then with secondary antibodies (goat anti-rabbit IgG-FITC) for 1 h each at room temperature. The nuclei were stained with Hoechst, and the slides were subsequently sealed and observed via confocal laser microscopy.
Electron microscopy
Adipose tissue samples were harvested and fixed in 2.5% glutaraldehyde in PBS at 4 °C overnight. After washing with PBS twice and with distilled water once, the adipose tissues were postfixed with 1% OsO4 and 1.5% potassium ferricyanide (K3Fe (CN)6) for 50 min at room temperature. Then, the adipose tissues were washed with distilled water and placed in thiocarbohydrazide TCH (thiocarbohydrazide) solution for 30 min at room temperature. Then, the adipose tissues were washed in distilled water again and incubated with 1% OsO4 for an additional 45 min at room temperature. After being rinsed with water, the samples were incubated in ice-cold 2% aqueous uranyl acetate for 1 h at room temperature. The samples were then rinsed once more with distilled water, gradually dehydrated through a series of increasing concentrations of ethanol, and embedded in epoxy EMBED-812 resin. Imaging was conducted using a 120-kV electron microscope (Tecnai Spirit, FEI) equipped with a CCD camera (Morada G3; EMSIS) and RADIUS software.
Mouse iWAT quantitative S-nitrosation proteomics and LC‒MS/MS analysis
For quantitative S-nitrosation proteomic analysis, total SNO-modified proteins were prepared according to the above IBP procedures, and the biotin labeling reagent was replaced with iodoTMT reagent. The protein sample was subsequently alkylated by iodoacetamide incubation. The iWAT protein was digested with sequencing-grade modified trypsin (25 μg of trypsin to digest 1 mg of protein). After incubation at 37 °C overnight, the protein digests were desalted using C18 SPE columns and dried in a centrifugal vacuum concentrator, and 200 μl of 1X TBS buffer was used to suspend the freeze-dried peptide. Two hundred microliters of settled resin was added to enrich the iodo-TMT-labeled proteins. The peptide was analyzed by mass spectrometry. All nanoLC-MS/MS experiments were performed on a Orbitrap Exploris 480 (Thermo Scientific) equipped with an Easy n-LC 1200 HPLC system (Thermo Scientific). The peptides were loaded onto a 100 μm id×2 cm fused silica trap column packed in-house with reversed phase silica (Reprosil-Pur C18 AQ, 5 μm, Dr. Maisch GmbH) and then separated on an a 75 μm id×25 cm C18 column packed with reversed phase silica (Reprosil-Pur C18 AQ, 1.9 μm, Dr. Maisch GmbH). The peptides bounded on the column were eluted with a 73-min linear gradient. The solvent A consisted of 0.1% formic acid in water solution and the solvent B consisted of 80% acetonitrile and 0.1% formic acid. The segmented gradient was 4–9% B, 3 min; 9–20% B, 22 min; 20–30% B, 20 min; 30-40% B, 15 min; 40-95% B, 5 min; 95% B, 8 min at a flow rate of 300 nl/min.
The MS analysis was performed with Orbitrap Exploris 480 mass spectrometer with the FAIMS Pro interface (Thermo Scientific). FAIMS separations were performed with two compensation voltage (−45 and −65). With the data-dependent acquisition mode, the MS data were acquired at a high resolution 60,000 (m/z 200) across the mass range of 350–1500 m/z. The target value was 3.00E + 06 with a maximum injection time of 22 ms. Data dependent mode was selected as cycle time mode which was set as 2 s. The precursor ions were selected from each MS full scan with isolation width of 1.6 m/z for fragmentation in the Ion Routing Multipole with normalized collision energy of 28%. Subsequently, MS/MS spectra were acquired at resolution 15,000 at m/z 200. The target value was 7.50E + 04 with a maximum injection time of 22 ms. The dynamic exclusion time was 40 s. For nano electrospray ion source setting, the spray voltage was 2.0 kV; no sheath gas flow; the heated capillary temperature was 320 °C.
Protein Identification and quantification analysis
The raw data from Orbitrap Exploris 480 were analyzed with Proteome Discovery version 2.4.1.15 using Sequest HT search engine for protein identification. The Uniprot mouse protein database (updated on 07-2022) was used for searching the data from samples. Some important searching parameters were set as following: trypsin was selected as an enzyme and two missed cleavages were allowed for searching; the mass tolerance of precursor was set as 10 ppm and the product ions tolerance was 0.02 Da.; TMT 6plex (lysine and N-terminus of peptides) and cysteine carbamidomethylation were specified as fixed modifications; iodoTMT6plex of cysteine and methionine oxidation were set as variable modifications; The methionine oxidation was chosen as variable modifications. FDR analysis was performed with Percolator and FDR <1% was set for protein identification. The peptides confidence was set as high for peptides filter. Proteins quantification was also performed on Proteome Discovery 2.4.1.15 using the ratio of the intensity of reporter ions from the MS/MS spectra. Only unique and razor peptides of proteins were selected for protein relative quantification. The co-isolation threshold was specified as 50% and average reporters S/N value should be above 10. The normalization mode was selected as total peptide amount to corrected experimental bias.
Mouse iWAT gene expression analysis (RT‒qPCR and RNA sequencing)
TRIzol reagent was used to extract total RNA from mouse iWAT tissue. The RNA was reverse transcribed into cDNA via the M-MLV reverse transcriptase system. The manufacturer’s instructions were followed, and oligo(dT) primers were used to specifically prime for the synthesis of cDNA from mRNA.
The specific primer sequences for genes of interest are listed in Oligonucleotides. A real-time PCR system (Applied Biosystems) and SYBR Green master mix were used to perform qPCR, and 36B4 was used as the reference gene. The Ct values were analyzed, and the relative expression levels were calculated via the ΔΔCt method. The Q-PCR primer sequences used in this study are detailed in Supplementary Table 1 within the Supplementary Information.
RNA sequencing was conducted by Biomarker Technologies (BMK) in Beijing. RNA samples were isolated from the iWAT of the mice. The indexing and clustering of the samples were carried out on a cBot cluster generation system with the Hieff NGS Ultima Dual-mode mRNA Library Prep Kit for Illumina, following the manufacturer’s protocol. After cluster formation, the prepared libraries were sequenced on an Illumina platform. The RNA-seq data were analyzed via bioinformatics tools to identify differentially expressed genes between WT and GSNOR KO old mouse iWAT samples. A functional enrichment analysis was performed to understand the biological processes and pathways associated with the gene expression changes.
Western blotting
Western blotting was performed as previously described44. Briefly, cell or tissue lysates were prepared using radioimmunoprecipitation assay (RIPA) buffer (comprising 50 mM Tris-HCl at pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, and 0.1% SDS) supplemented with a protease inhibitor cocktail (Roche). Protein concentrations were determined using BCA assays. Each sample was then mixed with 5× SDS-reducing sample loading buffer and heated at 100 °C for 10 min. A total of 10–20 μg of protein was separated via SDS‒PAGE and transferred onto a polyvinylidene fluoride (PVDF) membrane (Millipore). The membranes were subsequently blocked with 5% nonfat dry milk and probed with primary antibodies, followed by incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies (diluted 1:5000). Imaging was performed using an e-blot imaging system with enhanced chemiluminescence (Thermo Fisher). The antibodies and reagents used in this study are detailed in Supplementary Table 1 within the Supplementary Information.
Irreversible biotin switch assay procedure (IBP)
An IBP to detect S-nitrosation was carried out according to the method described by Qiao et al.44. Mouse cells or adipose tissue were lysed using a HEN buffer (comprising 100 mM HEPES, 1 mM EDTA, and 0.1 mM neocuproine, pH 7.7) supplemented with 1% Nonidet P-40 and cocktail. The resulting supernatant was then incubated with S-methyl methanethiosulfonate (MMTS) at 50 °C for 30 min, with frequent vortexing, to remove excess MMTS via ice-cold acetone precipitation, followed by centrifugation at 3500 × g for 10 min. This precipitation step was repeated three times. The precipitated material was resuspended in HEN buffer with 2.5% SDS, 0.2 mM biotin-maleimide, and 10 mM ascorbate and incubated at 37 °C for 1 h. Any remaining biotin-maleimide was removed using ice-cold acetone precipitation, as previously described. The pellet was then resuspended in HEN buffer with 200 mM DTT and heated at 100 °C for 15 min before the addition of neutralization buffer (HEN buffer supplemented with 100 mM NaCl) and streptavidin-agarose (50–100 μl per sample) to isolate the biotinylated proteins. The mixture was incubated at 4 °C overnight. The streptavidin-agarose mixture was subsequently washed three times with neutralization buffer (1000 × g for 1 min), and the proteins were eluted with HEN buffer containing 2.5% SDS at 100 °C for 15 min. The eluted proteins were then analyzed by SDS‒PAGE and subjected to immunoblotting with the appropriate antibody.
Co-immunoprecipitation (Co-IP)
Adipocytes were collected and lysed in ice-cold RIPA buffer (mild). The obtained samples were coimmunoprecipitated with an anti-Beclin-11 antibody immobilized on the DYNABEADS PROTEIN according to the standard procedure. Upon elution, the samples were separated by 10% SDS‒PAGE. The samples were subsequently subjected to gelatolytic hydrolysis after silver staining and analyzed by MS or western blotting using a standard protocol.
Statistics and reproducibility
All the statistical analyses were performed using GraphPad Prism 8. The results were presented as the mean ± standard error of the mean (SEM) or the mean ± standard deviation (SD) as specified in the figure legends. Statistical significance was determined using the appropriate test (Paired/Unpaired two-tailed Student’s t-test, two-way ANOVA, two-sided ANCOVA, or log-rank test). A p-value < 0.05 was considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001; ns, not significant). The specific p values and n values were given are indicated in the figures or corresponding legends. In animal studies, “n” represented the number of biologically independent animals; in cellular assays, “n” referred to the number of independent biological replicates. Results were presented based on at least three independent experiments with consistent results. Mouse experiments were conducted under blinded conditions with random group allocation based on genotype or ear tag number. No formal power calculation was used to predetermine sample size, and no animals or data points were excluded from the analyses.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of this study are available within the paper and Supplementary Information. RNA-seq data: The RNA sequencing data generated in this study have been deposited in the Gene Expression Omnibus (GEO) database under accession code GSE317377. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the iProX partner repository72,73 with the dataset identifier PXD072412 [https://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD072412]. Source data are provided with this paper.
Change history
11 May 2026
A Correction to this paper has been published: https://doi.org/10.1038/s41467-026-73041-z
References
Nguyen, T. T. & Corvera, S. Adipose tissue as a linchpin of organismal ageing. Nat. Metab. 6, 793–807 (2024).
Chia, C. W., Egan, J. M. & Ferrucci, L. Age-related changes in glucose metabolism, hyperglycemia, and cardiovascular risk. Circ. Res. 123, 886–904 (2018).
Zatterale, F. et al. Chronic adipose tissue inflammation linking obesity to insulin resistance and type 2 diabetes. Front. Physiol. 10, 1607 (2019).
Pillon, N. J., Loos, R. J. F., Marshall, S. M. & Zierath, J. R. Metabolic consequences of obesity and type 2 diabetes: balancing genes and environment for personalized care. Cell 184, 1530–1544 (2021).
Mouton, A. J., Li, X., Hall, M. E. & Hall, J. E. Obesity, hypertension, and cardiac dysfunction: novel roles of immunometabolism in macrophage activation and inflammation. Circ. Res. 126, 789–806 (2020).
Oya, M. et al. Age-related ciliopathy: obesogenic shortening of melanocortin-4 receptor-bearing neuronal primary cilia. Cell Metab. 36, 1044–1058.e1010 (2024).
Franceschi, C., Garagnani, P., Parini, P., Giuliani, C. & Santoro, A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 14, 576–590 (2018).
Yu, L. et al. IgG is an aging factor that drives adipose tissue fibrosis and metabolic decline. Cell Metab. 36, 793–807.e795 (2024).
Camell, C. D. et al. Inflammasome-driven catecholamine catabolism in macrophages blunts lipolysis during ageing. Nature 550, 119–123 (2017).
Marcelin, G., Gautier, E. L. & Clément, K. Adipose tissue fibrosis in obesity: etiology and challenges. Annu. Rev. Physiol. 84, 135–155 (2022).
Han, H. S. et al. Impaired BCAA catabolism in adipose tissues promotes age-associated metabolic derangement. Nat. Aging 3, 982–1000 (2023).
Fu, T. et al. Proteolytic rewiring of mitochondria by LONP1 directs cell identity switching of adipocytes. Nat. Cell Biol. 25, 848–864 (2023).
Herrera, M. D., Mingorance, C., Rodríguez-Rodríguez, R. & Alvarez de Sotomayor, M. Endothelial dysfunction and aging: an update. Ageing Res. Rev. 9, 142–152 (2010).
Baylis, C. Changes in renal hemodynamics and structure in the aging kidney; sexual dimorphism and the nitric oxide system. Exp. Gerontol. 40, 271–278 (2005).
Dai, Z. et al. Nitric oxide and energy metabolism in mammals. BioFactors 39, 383–391 (2013).
Sessa, W. C. A new approach to weight loss: just activate endothelial NO synthase!. Circ. Res. 111, 1111–1112 (2012).
Wu, Z., Satterfield, M. C., Bazer, F. W. & Wu, G. Regulation of brown adipose tissue development and white fat reduction by L-arginine. Curr. Opin. Clin. Nutr. Metab. Care 15, 529–538 (2012).
McNally, B. D. et al. Inorganic nitrate promotes glucose uptake and oxidative catabolism in white adipose tissue through the XOR-catalyzed nitric oxide pathway. Diabetes 69, 893–901 (2020).
Huang, B. & Chen, C. Detection of protein S-nitrosation using irreversible biotinylation procedures (IBP). Free Radic. Biol. Med 49, 447–456 (2010).
Zhou, H. L., Premont, R. T. & Stamler, J. S. The manifold roles of protein S-nitrosylation in the life of insulin. Nat. Rev. Endocrinol. 18, 111–128 (2022).
Ovadia, H. et al. Increased adipocyte S-nitrosylation targets anti-lipolytic action of insulin: relevance to adipose tissue dysfunction in obesity. J. Biol. Chem. 286, 30433 (2011).
Barnett, S. D. & Buxton, I. L. O. The role of S-nitrosoglutathione reductase (GSNOR) in human disease and therapy. Crit. Rev. Biochem. Mol. Biol. 52, 340–354 (2017).
Tang, X. et al. Mitochondrial GSNOR alleviates cardiac dysfunction via ANT1 denitrosylation. Circ. Res. 133, 220–236 (2023).
Tang, X. et al. NEDD4-mediated GSNOR degradation aggravates cardiac hypertrophy and dysfunction. Circ. Res. 136, 422–438 (2025).
Hayashida, K. et al. Improvement in outcomes after cardiac arrest and resuscitation by inhibition of S-nitrosoglutathione reductase. Circulation 139, 815–827 (2019).
Rizza, S. et al. GSNOR deficiency promotes tumor growth via FAK1 S-nitrosylation. Cell Rep. 42, 111997 (2023).
Gomes, S. A. et al. S-nitrosoglutathione reductase (GSNOR) enhances vasculogenesis by mesenchymal stem cells. Proc. Natl. Acad. Sci. USA 110, 2834–2839 (2013).
Rizza, S. et al. S-nitrosylation drives cell senescence and aging in mammals by controlling mitochondrial dynamics and mitophagy. Proc. Natl. Acad. Sci. USA 115, E3388–e3397 (2018).
Zhang, Y. et al. Increased GSNOR expression during aging impairs cognitive function and decreases S-nitrosation of CaMKIIalpha. J. Neurosci. 37, 9741–9758 (2017).
Su, L. Y. et al. S-nitrosoglutathione reductase alleviates morphine analgesic tolerance by restricting PKCα S-nitrosation. Redox Biol. 75, 103239 (2024).
Liu, Q. et al. GSNOR facilitates antiviral innate immunity by restricting TBK1 cysteine S-nitrosation. Redox Biol. 47, 102172 (2021).
Yi, W. et al. Protein S-nitrosylation regulates proteostasis and viability of hematopoietic stem cell during regeneration. Cell Rep. 34, 108922 (2021).
Yin, R. et al. Pro-inflammatory macrophages suppress PPARgamma activity in adipocytes via S-nitrosylation. Free Radic. Biol. Med 89, 895–905 (2015).
Cao, Y. et al. S-nitrosoglutathione reductase-dependent PPARγ denitrosylation participates in MSC-derived adipogenesis and osteogenesis. J. Clin. Investig. 125, 1679–1691 (2015).
González-Muniesa, P. et al. Obesity. Nat. Rev. Dis. Prim. 3, 17034 (2017).
Sebag, S. C. et al. ADH5-mediated NO bioactivity maintains metabolic homeostasis in brown adipose tissue. Cell Rep. 37, 110003 (2021).
Cohen, P. & Kajimura, S. The cellular and functional complexity of thermogenic fat. Nat. Rev. Mol. Cell Biol. 22, 393–409 (2021).
Kaartinen, M. T. et al. Matrisome alterations in obesity - Adipose tissue transcriptome study on monozygotic weight-discordant twins. Matrix Biol.: J. Int. Soc. Matrix Biol. 108, 1–19 (2022).
Rabiee, A. Beige fat maintenance; toward a sustained metabolic health. Front. Endocrinol. 11, 634 (2020).
Chouchani, E. T., Kazak, L. & Spiegelman, B. M. New advances in adaptive thermogenesis: UCP1 and beyond. Cell Metab. 29, 27–37 (2019).
Rogers, N. H., Landa, A., Park, S. & Smith, R. G. Aging leads to a programmed loss of brown adipocytes in murine subcutaneous white adipose tissue. Aging cell 11, 1074–1083 (2012).
Gu, P. et al. Compromised browning in white adipose tissue of ageing people. Eur. J. Endocrinol. 188, lvad014 (2023).
Altshuler-Keylin, S. et al. Beige adipocyte maintenance is regulated by autophagy-induced mitochondrial clearance. Cell Metab. 24, 402–419 (2016).
Qiao, X. et al. ER reductive stress caused by Ero1alpha S-nitrosation accelerates senescence. Free Radic. Biol. Med 180, 165–178 (2022).
Kim, D. et al. Suppression of brown adipocyte autophagy improves energy metabolism by regulating mitochondrial turnover. Int. J. Mol. Sci. 20, 3520 (2019).
Kasamatsu, S. et al. Supersulfide catalysis for nitric oxide and aldehyde metabolism. Sci. Adv. 9, eadg8631 (2023).
Ye, H., Wu, J., Liang, Z., Zhang, Y. & Huang, Z. Protein S-nitrosation: biochemistry, identification, molecular mechanisms, and therapeutic applications. J. Med. Chem. 65, 5902–5925 (2022).
Pavlinov, I., Salkovski, M. & Aldrich, L. N. Beclin 1-ATG14L protein-protein interaction inhibitor selectively inhibits autophagy through disruption of VPS34 complex I. J. Am. Chem. Soc. 142, 8174–8182 (2020).
Schaum, N. et al. Ageing hallmarks exhibit organ-specific temporal signatures. Nature 583, 596–602 (2020).
Hinney, A., Körner, A. & Fischer-Posovszky, P. The promise of new anti-obesity therapies arising from knowledge of genetic obesity traits. Nat. Rev. Endocrinol. 18, 623–637 (2022).
Huang, C., Chen, W. & Wang, X. Studies on the fat mass and obesity-associated (FTO) gene and its impact on obesity-associated diseases. Genes Dis. 10, 2351–2365 (2023).
Ichimura, A. et al. Dysfunction of lipid sensor GPR120 leads to obesity in both mouse and human. Nature 483, 350–354 (2012).
Farooqi, I. S. et al. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N. Engl. J. Med. 348, 1085–1095 (2003).
Sebag, J. A., Zhang, C., Hinkle, P. M., Bradshaw, A. M. & Cone, R. D. Developmental control of the melanocortin-4 receptor by MRAP2 proteins in zebrafish. Science 341, 278–281 (2013).
Asai, M. et al. Loss of function of the melanocortin 2 receptor accessory protein 2 is associated with mammalian obesity. Science 341, 275–278 (2013).
Jackson, R. S. et al. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat. Genet. 16, 303–306 (1997).
Das, M., Gabriely, I. & Barzilai, N. Caloric restriction, body fat and ageing in experimental models. Obes. Rev. Off. J. Int. Assoc. Study Obes. 5, 13–19 (2004).
Villareal, D. T. et al. Aerobic or resistance exercise, or both, in dieting obese older adults. N. Engl. J. Med. 376, 1943–1955 (2017).
Weghuber, D. et al. Once-weekly semaglutide in adolescents with obesity. N. Engl. J. Med. 387, 2245–2257 (2022).
Wilding, J. P. H. et al. Once-weekly semaglutide in adults with overweight or obesity. N. Engl. J. Med. 384, 989–1002 (2021).
Mauer, M. M., Harris, R. B. & Bartness, T. J. The regulation of total body fat: lessons learned from lipectomy studies. Neurosci. Biobehav. Rev. 25, 15–28 (2001).
Yamamuro, T. et al. Age-dependent loss of adipose Rubicon promotes metabolic disorders via excess autophagy. Nat. Commun. 11, 4150 (2020).
Kovsan, J. et al. Altered autophagy in human adipose tissues in obesity. J. Clin. Endocrinol. Metab. 96, E268–E277 (2011).
Moore, T. M. et al. Parkin regulates adiposity by coordinating mitophagy with mitochondrial biogenesis in white adipocytes. Nat. Commun. 13, 6661 (2022).
Fernández, ÁF. et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature 558, 136–140 (2018).
Li, Y. et al. Autophagy impairment mediated by S-nitrosation of ATG4B leads to neurotoxicity in response to hyperglycemia. Autophagy 13, 1145–1160 (2017).
Liu, L. et al. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell 116, 617–628 (2004).
Zhang, Y. et al. Increased GSNOR expression during aging impairs cognitive function and decreases S-nitrosation of CaMKIIα. J. Neurosci. 37, 9741–9758 (2017).
Müller, T. D., Klingenspor, M. & Tschöp, M. H. Revisiting energy expenditure: how to correct mouse metabolic rate for body mass. Nat. Metab. 3, 1134–1136 (2021).
Ou, Q. et al. Apoptosis releases hydrogen sulfide to inhibit Th17 cell differentiation. Cell Metab. 36, 78–89.e75 (2024).
Vamvini, M. et al. Exercise training and cold exposure trigger distinct molecular adaptations to inguinal white adipose tissue. Cell Rep. 43, 114481 (2024).
Ma, J. et al. iProX: an integrated proteome resource. Nucleic Acids Res. 47, D1211–d1217 (2019).
Chen, T. et al. iProX in 2021: connecting proteomics data sharing with big data. Nucleic Acids Res. 50, D1522–d1527 (2022).
Acknowledgements
We are grateful to Professor Guangjun Nie of the National Center for Nanoscience and Technology for his support with mouse MRI experiments, and to Professor Changtao Jiang of the Basic Medical College, Peking University, for his help with metabolic cage experiments. We also thank the staff of the Scientific Research Platform at the Institute of Biophysics, Chinese Academy of Sciences: Jifeng Wang for LC-MS/MS analysis, Yun Feng for image data analysis using Imairs software, and Xiaoxiao Zhu, Dongdong Fan, and Songge Hou for animal care and maintenance. Special thanks to Professor Hong Zhang of the Institute of Biophysics, Chinese Academy of Sciences for his valuable discussions and suggestions, which greatly improved our study. This work was supported by the National Key R&D Program of China (2022YFA1303000 to C.C.), the National Natural Science Foundation of China (32371297 to X.Q. and 91849203 to C.C.), the Chinese Academy of Sciences Strategic Priority Research Program B grants (XDB39000000 to C.C.), the National Key R&D Program of China (2022YFA1305100 to M.J.), and the Beijing Natural Science Foundation (5254040 to C.S.).
Author information
Authors and Affiliations
Contributions
C.C. and X.Q. designed the project. X.Q., T.X., and C.C. wrote the manuscript. X.Q., T.X., Y.Z., C.S., X.W., Y.C., Q.Y., H.Z., T.W., J.M., and S.L. conducted the experiments. H.S. and S.L. helped to analyze the GEO database and the metabolic cage data. L.Z., M.N., Y.H., and H.L. helped to collect human adipose tissue samples, and C.C. revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Zhenji Gan, who co-reviewed with Tingting Fu, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Qiao, X., Xie, T., Zhang, Y. et al. S-nitrosoglutathione reductase GSNOR drives age-related obesity by promoting adipose tissue whitening through de-nitrosation of Beclin-1. Nat Commun 17, 3059 (2026). https://doi.org/10.1038/s41467-026-69793-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-026-69793-3
