
Abstract
Treatment options for Staphylococcus aureus infections are increasingly limited, particularly in livestock, where S. aureus causes mastitis requiring prolonged antibiotic therapy. This study engineered Phage Inducible Chromosomal Islands (ePICIs) to deliver CRISPR-Cas9 modules targeting small RNA genes. ePICIs exhibit bactericidal activity without chromosomal integration, an expanded host range compared to their parental phages, and biofilm-dependent efficacy influenced by the extracellular matrix composition. Biofilms mediated by the Bap protein strongly protect bacteria from ePICIs, whereas PIA/PNAG-based biofilms do not. Despite Bap-mediated protection in vitro, ePICIs achieved bactericidal effects comparable to vancomycin in a mouse mastitis model caused by Bap-producing strains. These findings reveal key factors affecting phage-delivered CRISPR-Cas efficacy and highlight that antibiofilm therapies should not be dismissed based solely on in vitro performance. Non-replicative ePICIs thus represent a promising alternative for treating localized infections such as mastitis.
Similar content being viewed by others

Introduction
Phage-inducible chromosomal islands (PICIs) are phage satellites that typically lack phage-like structural or lysis genes but retain the ability to replicate their genetic material independently after induction of their life cycle by a helper phage. To move from one cell to another, PICIs hijack the capsid from a helper phage. This helper phage may be an induced prophage present in the same cell as the PICI, or it may be a phage that infects the cell in which the PICI resides. In either case, as the phage multiplies in the cell, it replicates its genome, synthesizes the proteins of the capsid, and the proteins needed to lyse the bacterium. The PICI synchronizes its excision and replication with active phage replication as phage proteins sequester the PICI repressor. During the lysogenic state, this repressor suppresses the expression of genes required for genome excision and replication. At this point, copies of the phage genome and PICI genomes, as well as phage capsids, coexist in the bacterial cytoplasm. PICIs hijack the phage capsids using different strategies and package their genome inside. After cell lysis, both the PICIs and the phage particles are released, free to infect new bacteria1,2,3,4,5,6,7,8,9.
Like phages, PICIs have a modular structure, with genes involved in related functions—such as cleavage-integration, replication, and packaging—grouped together. This modular organization facilitates genetic manipulation, allowing the addition of beneficial modules that confer new traits of interest while enabling the removal of undesirable ones, such as toxin-coding genes. The addition of new modules to PICIs is possible because the phage capsids are prepared to accommodate the larger phage genome, leaving sufficient space for the smaller PICI elements. Taking advantage of this, Ram et al.10 conducted a pioneering study in which they engineered PICIs carrying the Cas9 protein and guide RNAs (gRNAs) targeting the agr two-component system as a strategy to combat Staphylococcus aureus infections. Although the use of PICIs for bacterial elimination is conceptually similar to phage therapy, a key distinction is that phages replicate during infection. This replication can facilitate the spread of virulence or antibiotic resistance genes through generalized transduction. In contrast, PICIs do not replicate in the absence of their cognate helper phage, meaning their numbers remain constant throughout treatment. This has two important implications: it reduces the risk of horizontal gene transfer between bacteria; however, it also means that a higher initial concentration of PICI particles than bacteria must be administered to achieve effective therapeutic outcomes.
Chronic infections caused by S. aureus are closely linked to the bacterium’s ability to adhere to host tissue and form biofilms, in which bacterial cells grow embedded in an extracellular matrix. This matrix can be either of proteinaceous or polysaccharidic nature11,12,13,14. The exopolysaccharidic matrix consists of a polymer composed of poly-N-acetyl-b-(1-6)-glucosamine (PIA/PNAG)15,16,17, while the proteinaceous matrix consists of surface proteins of the MSCRAMM family (microbial surface components that recognize adhesive matrix molecules)18,19,20,21,22. A notable example of a proteinaceous biofilm is that produced by the Bap protein, which is found exclusively in some S. aureus isolates from bovine mastitis. This strong association suggests that Bap plays a key role in the pathogenesis of S. aureus mastitis23,24,25. Bovine mastitis, an inflammatory disease of the mammary gland, is a major health concern in the dairy and livestock industries, and its treatment accounts for the largest use of antibiotics in the dairy production26,27. Among the various causative agents of mastitis, S. aureus is one of the most prevalent, responsible for both subclinical (asymptomatic) and clinical infections, with reported prevalence rates ranging from 5% to 50% across different countries28,29,30.
In this study, we developed non-replicative ePICIs to deliver CRISPR-Cas9 modules designed to target S. aureus small RNAs (sRNAs). Our findings indicate that ePICIs do not need to integrate into the genome of the recipient cell to exert their bactericidal activity and have a broader host range than phages, likely due to having fewer components recognizable by the bacterial immune system. The bactericidal efficacy of ePICIs in vitro is significantly affected by the biofilm matrix composition. Specifically, the biofilm matrix formed by Bap confers strong protection against ePICIs, whereas that composed of PIA/PNAG exopolysaccharide matrix does not. Importantly, and in contrast to in vitro results, ePICIs demonstrated bactericidal activity comparable to vancomycin against Bap-producing bacteria in a murine model of mastitis, highlighting their potential as a safe and effective alternative for the treatment of S. aureus infections.
Results
Assembly and production of engineered ePICIs
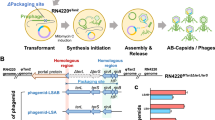
To create recombinant PICIs with the capability to deactivate S. aureus, we employed a previously established rebooting procedure31. The engineered PICIs (ePICIs) incorporated the gene encoding the Cas9 protein along with gRNAs targeting three DNA regions (rsaE, rsaH, and rsaI) that encode small regulatory RNA (sRNA) and a tetracycline resistance cassette (tetM), which facilitates our studies (Fig. 1). Targeting the coding DNA sequence of sRNA with Cas9, was chosen due to its high conservation at the sequence level32. The PICI genome was amplified in four 30-nucleotide overlapping fragments, enabling assembly through recombination in yeast (Supplementary Fig. 1).

Left panel: Diagram of the S. aureus RN4220 chromosome engineered to generate the ePICI. The chromosome contains the ePICI genome, which encodes an anti-Staphylococcus CRISPR-Cas module and a tetracycline resistance cassette. In addition, the strain also carries a lysogenic phage 80α (helper) with a deletion in the terS gene, which prevents self-packaging of the phage genome. To avoid CRISPR-Cas self-targeting, the rsaE, rsaH, and rsaI genes were deleted from the host chromosome. Right panel: Upon exposure to stress conditions (e.g., mitomycin C), phage 80α ΔterS enters the lytic cycle, excising its genome and producing capsid components. Concurrently, 80α PICI antirepressor induces the excision, circularization, and replication of the ePICI genome, which then hijacks the phage-derived capsids to package its own genome.
After their assembly in yeast, the four YAC derivatives, each containing a recombinant ePICI, were isolated and introduced into S. aureus RN4220 ΔrsaE rsaH rsaI. This strain lacked the rsaE, rsaH, and rsaI target sequences and harbored a lysogenic 80α phage derivative with a deletion in the terS gene, which encodes the small terminase subunit (Fig. 1). The terS mutation disrupts the packaging of phage DNA into capsids but does not affect the packaging of ePICI DNA, thus preventing the presence of phage contaminants in ePICI samples10. After transformation, the ePICIs integration into the chromosomal attC site was monitored through the acquisition of tetracycline resistance and confirmed by PCR, using one oligonucleotide from the flanking region and another from the ePICI sequence (Table 3). To generate ePICI particles, S. aureus RN4220 80α-terS ePICI strains were treated with mitomycin C, and ePICIs were purified from culture supernatants as described in “Methods” section (Fig. 1B).
To confirm their functionality, a purified solution of ePICI particles was used to infect S. aureus RN4220 and the triple mutant S. aureus RN4220 ΔrsaE rsaH rsaI. Infections with ePICIrsaE, ePICIrsaI, and ePICIrsaH resulted in transductant particles exclusively in the S. aureus RN4220 ΔrsaE rsaH rsaI strain (Fig. 2A). Infection with ePICI-gRNA2 resulted in tetracycline-resistant transductants in both strains, as its gRNA does not target any region of the bacterial chromosome. These findings confirm the functionality of the constructed ePICIs, demonstrating their ability to transfer into other strains as long as those strains lack the gRNA-targeted sequence. Additionally, the results show that ePICIs carrying the Cas9-gRNA module effectively kill S. aureus. To investigate whether the bactericidal activity of ePICIs extend beyond a specific strain, we assessed the ability of the three ePICI variants to reduce the growth of three genetically unrelated S. aureus strains: S. aureus HG001 (RN1 derivative with restored rsbU), S. aureus V329 (a bovine mastitis isolate producing a strong Bap protein-mediated biofilm matrix) and S. aureus 15981 (a clinical isolate with a robust PIA/PNAG exopolysaccharide-mediated biofilm matrix) (Fig. 2B). The results showed strain-dependent bactericidal efficacy of the ePICIs: ePICIrsaE and ePICIrsaH inhibited HG001 strain growth, only ePICIrsaE significantly reduced 15981 strain growth, and none of the three ePICIs affected the growth of V329 strain. Notably, combining all three ePICIs did not enhance their bactericidal effect. In all cases, ePICIrsaE exhibited the strongest bactericidal activity across strains and was therefore selected for further studies.

A Bacterial spot assay evaluating the bactericidal activity of ePICIs. Genetically unrelated S. aureus RN4220 and S. aureus V329 (109 cfu) were treated with increasing concentrations of ePICIs and spotted in tetracycline-containing agar plates. In the absence of a chromosomal target for the sgRNA, ePICI-infected bacteria gain tetracycline resistance. Conversely, when the ePICI-encoded sgRNA targets the bacterial chromosome, only CRISPR-escape mutants are able to grow in the presence of tetracycline. B Growth kinetics of S. aureus HG001, S. aureus V329, and S. aureus 15981 exposed to different ePICIs. ePICIs were applied at titers exceeding the bacterial inoculum, and bactericidal activity was inferred from growth inhibition. Data are representative of three independent biological replicates.
Influence of biofilm matrix composition on susceptibility to ePICIrsaE
Discrepancies in the bactericidal efficacy of ePICIrsaE across diverse S. aureus strains may be influenced by the composition of the extracellular matrix. To explore the impact of the matrix on susceptibility to ePICIrsaE infection, we investigated its antibacterial effects on both S. aureus strain V329 and its isogenic mutant lacking the bap gene. Additionally, we examined S. aureus strain 15981 and its isogenic mutant deficient in the icaB gene. As a control, the S. aureus strain V329 ΔrsaE, lacking the recognition site for the island-carrying gRNA, was included. While the growth of S. aureus V329 was minimally affected by the presence of ePICIrsaE, the growth of its isogenic bap-deficient mutant was severely affected in the presence of ePICIrsaE. On the other hand, the susceptibility of S. aureus 15981 was lower than that of its isogenic mutant in the icaB gene (Fig. 3A). These results indicate that the Bap-mediated proteinaceous matrix confers protection against infection by ePICIrsaE, while the PIA/PNAG exopolysaccharide matrix benefits infection by ePICIrsaE. The variation in ePICIrsaE bactericidal efficacy depending on biofilm matrix composition raises the question of whether this phenomenon is specific to ePICIrsaE or part of a broader mechanism affecting phage susceptibility. To explore this possibility, we decided to analyze the lytic effect of phage 80α, the capsid donor of ePICI, on S. aureus V329 and S. aureus 15981 strains, along with their respective bap or icaB mutants. Similar to ePICIrsaE, the Bap-mediated protein matrix provided protection against phage infection, whereas the PIA/PNAG exopolysaccharide matrix increased phage lysis (Fig. 3B).

A Growth curves of S. aureus V329, its isogenic mutants deficient in bap (V329 Δbap) and rsaE (V329 ΔrsaE), as well as S. aureus 15981 and its isogenic mutant deficient in icaB (15981 ΔicaB), in the absence (gray curves) and presence of ePICIrsaE (blue curves). Red dots indicate the time points at which samples were taken for bacterial enumeration, as shown in the corresponding bar graph. B Growth curves of S. aureus V329 and its isogenic mutant deficient in bap (V329 Δbap), along with S. aureus 15981 and its isogenic mutant deficient in icaB (15981 ΔicaB), in the presence of phage 80α. Data are representative of three independent biological replicates. S. aureus V329 was cultured in DMEM to induce Bap expression, while S. aureus 15981 was grown in TSB with 0.25% glucose, as this medium optimally promotes PIA/PNAG expression. Statistical significance was determined using the Mann–Whitney U-test (ns no significant; *P < 0.05).
The differences in susceptibility to the bactericidal effect of ePICIrsaE observed between S. aureus strains V329 and 15981 could be due to intrinsic characteristics of each strain. To evaluate this possibility, we analyzed the effect of the Bap protein and the PIA/PNAG exopolysaccharide matrices on the same genetic background. To do this, the S. aureus V329 Δbap strain was complemented with the icaADBC genes (S. aureus V329 Δbap + ica). The S. aureus V329 WT strain produced a protein biofilm matrix sensitive to proteinase K treatment, whereas the S. aureus V329 Δbap + ica strain produced an exopolysaccharide matrix sensitive to dispersin B treatment, an enzyme that hydrolyzes the glycosidic linkages in PIA/PNAG (Fig. 4A, B). Infection with ePICIrsaE confirmed that the presence of the Bap protein confers protection against infection, whereas the presence of the PIA/PNAG exopolysaccharide increased killing of strain V329 (Fig. 4C). Finally, to demonstrate that the protective effect of Bap is also present in other S. aureus strains, the effect of ePICIrsaE was analyzed on the S. aureus Newman strain containing the bap gene in the chromosome (Fig. 4D). Again, S. aureus Newman expressing Bap was more resistant to the bactericidal effect of ePICIrsaE than the isogenic wild-type strain. These results demonstrate that the Bap protein matrix protects against infection by ePICI and phages, whereas the PIA/PNAG exopolysaccharide matrix increases the susceptibility of S. aureus to infection by ePICI or to phage-mediated killing.

A Characterization of the biofilm matrix composition in S. aureus V329, V329 Δbap, and V329 Δbap + ica. Biofilms formed by these strains were treated with proteinase K (protease) or dispersin B (an enzyme that selectively degrades the exopolysaccharide PIA/PNAG). The Bap-dependent biofilm is sensitive to proteinase K but resistant to dispersin B, whereas the biofilm produced by V329 Δbap + ica is resistant to proteinase K but susceptible to dispersin B treatment. B Dot-blot analysis using specific antibodies against PIA/PNAG to reveal the presence of PIA/PNAG in S. aureus V329, V329 Δbap, and V329 Δbap + ica. C Growth curves of S. aureus V329, V329 Δbap, and V329 Δbap + ica in the absence (gray curves) and in the presence of ePICIrsaE (blue curves). Data are representative of three independent biological replicates. D Growth curves of S. aureus Newman and its isogenic derivative carrying the bap gene integrated into the chromosome (S. aureus Newman + bap). All growth assays were performed in TSB supplemented with 0.25% glucose to promote optimal PIA/PNAG expression. Data are representative of three independent biological replicates.
ePICI kills bacteria without chromosomal integration
After infection, ePICI introduces its circular DNA into the recipient bacterium, where it integrates into the attC site of the bacterial chromosome. Cas9 and gRNA can theoretically be expressed from the circular form before or after integration into the genome of the recipient cell. To determine whether ePICIrsaE requires chromosomal integration for bactericidal activity, we generated a derivative, ePICIrsaE Δint, in which the integrase was mutated by the insertion of two stop codons. Since integrase mutants exhibit inefficient excision33 we complemented the strain producing ePICIrsaE Δint with plasmid pCN51::SaPIbov2 integrase, which carries the integrase gene. Our rationale was that ePICIrsaE Δint could form infectious particles using integrase expressed in trans from the plasmid, but would remain unable to integrate into the chromosome of the recipient cell. To confirm the excision and circularization of ePICIrsaE Δint, we performed PCR using PICI-specific oligonucleotides before and after mitomycin C treatment (Fig. 5A). The E18 and E19 oligonucleotides amplified an internal PICI fragment, confirming the presence of ePICI regardless of its integration status (Fig. 5B). The amplification product of divergent oligonucleotides OL17 and OL18, corresponding to the circular form of ePICIrsaE Δint, was only detected in mitomycin C-induced bacteria, regardless of the presence of integrase (Fig. 5B). Next, we evaluated the bactericidal efficacy of ePICIrsaE and ePICIrsaE Δint against S. aureus. To determine whether the presence of the ePICI integrase was necessary for bactericidal activity in the recipient strain, we infected S. aureus HG001 carrying either an empty plasmid (pCN51) or a plasmid containing the ePICI integrase gene (pCN51::SaPIbov2 integrase). Our results showed that when ePICIrsaE and ePICIrsaE Δint were produced from a donor strain expressing integrase in trans, their bactericidal efficacy was comparable (Fig. 5C). Furthermore, the presence of integrase in the recipient strain had no effect on the bactericidal activity of ePICIrsaE Δint. The only condition in which ePICIrsaE Δint exhibited reduced bactericidal efficacy was when it was produced from a donor strain lacking integrase. This reduction is likely due to lower ePICI titers in the absence of integrase, leading to diminished efficacy. However, its activity remained independent of integrase presence in the recipient strain. These findings indicate that ePICI-mediated bactericidal activity does not require integrase function and occurs independently of chromosomal integration in the recipient cell. In contrast, integrase appears important in the donor strain to achieve high ePICI titers. This integration-independent activity of ePICIs provides an additional advantage by minimizing the potential for horizontal gene transfer or inadvertently increasing the expression of virulence factors.

A Schematic representation of the oligonucleotide design used to detect the linear and circular forms of ePICIrsaE and its integrase-deficient mutant (ePICIrsaE ∆int). Oligonucleotides E18 and E19 amplify a fragment of ePICI (PCRC) regardless of whether it is integrated into the genome (linear form) or excised (circular form). Oligonucleotides OL17 and OL18 produce an amplification product (PCRD) only when ePICI is excised from the chromosome and forms the circular intermediate. B Agarose gel electrophoresis showing PCR amplification products obtained using oligonucleotides E18 and E19 (top panel) and the divergent primers OL17 and OL18 (bottom panel). PCR was conducted on bacteria with either ePICIrsaE (Int ePICI +) or ePICIrsaE ∆int (Int ePICI −), along with an empty plasmid or one carrying the ePICI integrase, both before and after mitomycin C treatment. Lanes 1–8 from the top gel show a 4 kb band corresponding to ePICIrsaE integrated into the chromosome or its excised form following mitomycin treatment. Lanes 1–8 from the bottom gel show an amplification product only in mitomycin-treated samples, confirming the circular form of ePICIrsaE after excision from the bacterial chromosome. C Growth curves of the S. aureus HG001 strain and HG001 complemented with a plasmid expressing the ePICI integrase, in the presence of ePICIrsaE or ePICIrsaE ∆int purified from bacteria complemented with an empty plasmid or the plasmid producing ePICI integrase in trans. Data are representative of three independent biological replicates.
ePICIrsaE exhibits a wider infection spectrum than phage 80α
Phages tend to infect a limited range of strains, either because the receptor that the phage recognizes is missing on the surface of the bacterium, or because the bacterium has defense mechanisms against the phage (CRISPR, restriction enzymes, etc.)34. Because ePICIs are packaged in phage capsids, they are likely to face similar limitations in recognition of the host receptors. However, their smaller genome and lack of many phage-associated proteins allow them to evade the bacterial immune systems that typically defend against phages. To assess the infection spectrum of ePICIrsaE in comparison to phage 80α, we evaluated the susceptibility to infection in a collection of 20 S. aureus isolates obtained from bovine mastitis. The growth inhibitory effect of ePICIrsaE and 80α phage was determined using growth kinetic curves (iAUC)35. Sensitivity of the strains to any of the treatments was established for an iAUC ≥0.1. On this basis, results showed that out of the 20 strains, 17 exhibited sensitivity to ePICIrsaE, while only nine strains were susceptible to the phage (Fig. 6). Among the strains sensitive to both phage and ePICIrsaE, four displayed increased sensitivity to the island. These findings underscore that ePICIrsaE has a broader spectrum of infection than phage 80α.

Heat map depicting the bactericidal activity of ePICIrsaE and phage 80α, determined from bacterial growth curves over 12 h in DMEM medium. The infection area under the curve (iAUC) was calculated as the difference between the growth curve areas of treated and untreated cultures, reflecting the growth inhibition. Values were normalized on a scale from 0 to 1. Data are representative of three independent biological replicates.
Bactericidal efficacy of ePICIrsaE in a murine model of staphylococcal mastitis
Given that our results indicated that Bap-mediated protein biofilms protect S. aureus from the bactericidal activity of ePICIs, and that the Bap protein is enriched among S. aureus strains associated with mastitis, we next evaluated the therapeutic potential of ePICIs using a murine mastitis model. Prior to in vivo testing, the activity of ePICIrsaE was assessed under planktonic and biofilm conditions in milk. The results showed that the Bap protein conferred protection against ePICIrsaE under both planktonic (Fig. 7A) and biofilm-forming conditions (Fig. 7B), with the protective effect being significantly more pronounced in biofilms. In contrast, production of the exopolysaccharide PIA/PNAG did not provide protection against ePICIrsaE under either condition.

A S. aureus V329, V329 Δbap, and V329 Δbap + ica were grown in pasteurized milk and treated with phage ePICIrsaE or buffer (control). After two treatments, viable bacteria were quantified by CFU counting on TSA. Data represent mean ± SD (n = 6); statistical significance was determined using the Mann–Whitney U-test (**P < 0.01). B S. aureus V329 and 15981 were cultured in milk under static conditions for 18 h with two ePICIrsaE applications. Adherent biofilm-associated cells were recovered and enumerated by CFU. Representative images are shown from biological triplicates. Graphs display mean ± SD (n = 6); Mann–Whitney U-test (ns not significant; **P < 0.01).
To evaluate the bactericidal efficacy of ePICIrsaE in vivo, a murine model of mastitis was used. CD1 lactating mice were separated from their 7-day-old offspring 2 h prior to infection and randomly assigned to three groups. The L4 (left) and R4 (right) mammary glands were intraductally injected with 100 µl of a bacterial solution containing 103 CFU ml−1 of S. aureus V329, V329 Δbap, and V329 Δbap + ica (Fig. 8A). The negative control group received 100 µl of PBS, the positive control group received 100 µl of vancomycin solution (500 µg ml−1) and the experimental group received 100 µl of phage buffer containing 108 ePICIrsaE particles ml−1. Each animal received two doses of ePICIrsaE, administered 4 and 16 h after infection. At twenty-four hours post-infection, mammary glands were aseptically excised, homogenized, and bacterial loads were quantified. Unlike the results observed in vitro, ePICIrsaE significantly reduced the bacterial load in mammary glands infected with any of the three strains, indicating that Bap production does not protect against the bactericidal activity of ePICIrsaE in vivo. Regarding PIA/PNAG production, and consistent with in vitro findings, results showed that its presence does not confer protection against the bactericidal activity of ePICI. Taken together, in vivo results suggest that ePICIrsaE exhibits potent bactericidal activity within the confined space of the mammary gland.

A Schematic representation of the experimental design for the mouse mastitis model and the treatment regimen with PBS (negative control), vancomycin (positive control), and ePICIrsaE. B S. aureus V329, V329 Δbap, and V329 Δbap + ica bacterial counts from individual mammary glands following treatment with PBS, vancomycin, and ePICIrsaE. Each point represents one mammary gland; horizontal bars indicate mean values. Statistical significance was assessed using the Kruskal–Wallis test (ns no significant; *P < 0.05, **P < 0.01; ****P < 0.0001).
Discussion
In this study, we engineered CRISPR-Cas9-containing pathogenicity islands (ePICIs) with guide RNAs targeting S. aureus sRNA-encoding genes, using a rebooting system in S. cerevisiae. The ePICIs function as Trojan horses, delivering the Cas9 and gRNA to cleave the chromosome of the targeted S. aureus cell. The antibacterial efficacy of the three ePICIs varied significantly; ePICIrsaE exhibited the strongest bactericidal activity, ePICIrsaH showed a moderate effect, and ePICIrsaI had the weakest impact. The effectiveness of guide RNAs is influenced by multiple factors, including nucleotide composition near the protospacer adjacent motif, methylation patterns that may limit target accessibility, and the binding affinity of the guide RNA to the chromosomal target36. Optimal guide RNAs typically have balanced GC and AU content, low self-hybridization potential, and stronger binding at the 3′ end37,38. Both rsaE and rsaH guide RNAs have a GC content of 33.3%, whereas rsaI guide RNA has a lower GC content of 22%, which may explain its reduced efficacy in all strains tested. Notably, the combination of all three ePICIs did not increase bactericidal activity and was even less effective than ePICIrsaE alone. This suggests that the high efficacy of RsaE may be diluted by the moderate performance of the other two guide RNAs, as the total PICI concentration remains constant. A potential strategy to improve the efficacy of ePICI could be to integrate all three gRNAs into a single ePICI construct, allowing simultaneous expression within the infected cell.
The ePICI design incorporates the strong constitutive promoters blaZ and SP01 upstream of the Cas9 gene and the guide RNA, respectively. This configuration ensures constitutive expression of the Cas9 protein after ePICI infection of a host39. Our results indicate that these expression levels are sufficient for ePICI to exert its bactericidal effect without requiring genomic integration into the recipient bacterium. This is a critical advantage, as integration poses a higher risk of enabling the transfer of virulence or antibiotic resistance genes, especially when the recipient strain carries a helper phage capable of packaging ePICIs into capsids. Although ePICIs and phage 80α share the same tail structure, ePICIs infect a wider range of hosts, likely because they interact differently with bacterial immune systems34,40,41. Phages inject a larger genome, whereas ePICIs deliver a smaller genetic payload, minimizing detection and susceptibility to bacterial immune defenses. For instance, degradation of ePICI DNA by bacterial restriction-modification systems does not necessarily impair CRISPR module expression and its bactericidal activity. In contrast, restriction-mediated digestion of phage genomes disrupts their ability to complete the lytic cycle. Additionally, ePICIs maintain bactericidal efficacy in non-replicating bacteria, whereas most phages depend on host replication to achieve lysis, although a phage capable of killing dormant bacteria has recently been reported42.
The analysis of the bactericidal efficacy of ePICIs revealed significant differences depending on the type of extracellular matrix formed by the target strain. The biofilm matrix serves as a protective shield against phage infection, hindering the access of phages to the embedded bacteria43,44,45. Our results show that the protein matrix mediated by Bap acts as an effective shield against ePICI infection under in vitro conditions. However, the matrix formed by the exopolysaccharide PIA/PNAG does not protect against the action of ePICI under identical environmental conditions. These differences in protection between the protein and the exopolysaccharide matrices are also consistent against phage 80α infection, suggesting that the protective effect of the Bap matrix is likely to be related to the ability of ePICI or the phage to infect bacteria. The lack of protection by the PIA/PNAG matrix may be due to the action of exopolysaccharide depolymerase enzymes present in the capsid of ePICI and phage 80α. These enzymes degrade bacterial polysaccharides associated with the cell surface, facilitating phage adsorption46,47. Poly-N-acetylglucosamine depolymerases produced by S. aureus phages have been described as capable of degrading the PIA/PNAG polysaccharide48. An analysis of the phage 80α genome does not identify any protein homologous to an enzyme with depolymerase activity, but considering that 23% of 80α phage proteins have no known function, we cannot rule out that some of these proteins have this activity. It would be interesting to develop a platform to package ePICIs into capsids of different phages to see if any type of capsid can evade the protective action of the Bap protein matrix. Another possibility is that the endolysin of phage 80 alpha may degrade PIA/PNAG. Several bacteriophage-derived endolysins, particularly those from phages infecting Staphylococcus spp., have been shown to exhibit activity against PIA/PNAG in addition to their ability to degrade peptidoglycan, such as φ11, CF-301, and LysH5 lysins49,50,51,52. Notably, similar to ePICIs, endolysins also exhibit a broader spectrum of activity compared to whole phages53.
Mastitis caused by S. aureus is a major challenge to dairy production, resulting in significant economic losses due to reduced milk yield, reproductive problems, veterinary costs, and food safety concerns54,55. Current control measures rely on improved hygiene practices and antibiotic therapy. However, the persistence of S. aureus strains that are either antibiotic-resistant or reside in biofilms or intracellular compartments significantly reduces treatment efficacy, highlighting the need for alternative therapeutic approaches. One strategy to enhance the effectiveness of antibiotics involves nanoparticle-based formulations that improve drug stability, bioavailability, and targeted delivery to the mammary gland56. Other promising alternatives include bacteriophage therapy and bacteriophage-encoded peptidoglycan hydrolases (endolysins)57, both of which have demonstrated efficacy against S. aureus mastitis in vitro and in vivo58,59,60,61,62,63. Treatment with ePICIs is a form of phage therapy, but with important differences. Unlike phages, PICI do not replicate when they infect new bacterial cells, meaning that the initial dose must exceed the bacterial burden. However, this non-replicative nature provides a major advantage; ePICIs do not persist in milk, minimizing concerns for downstream dairy processes such as raw milk fermentation64. During our experiments, we encountered technical challenges in obtaining functional ePICI particle titers higher than 108 ml−1. Although repeated treatments can be administered safely without observable toxicity, excessively high bacterial loads may exceed the available number of ePICIs, reducing treatment efficacy. Nevertheless, even partial bacterial reduction may facilitate immune clearance, similar to the action of bacteriostatic antibiotics. Secondly, the inability of ePICIs to replicate in recipient bacteria also serves as a biosafety advantage, as it prevents unintended horizontal gene transfer. Moreover, ePICIs retain bactericidal activity against non-dividing bacterial populations, a condition that limits the efficacy of most antibiotics and phages, providing them with a unique therapeutic edge. In addition, unlike phages, ePICIs do not cause bacterial lysis, thereby avoiding the release of peptidoglycan fragments that can stimulate biofilm formation and enhance bacterial resistance65.
Our findings demonstrate that ePICIs are bactericidal in a murine model of staphylococcal mastitis and thus represent a promising therapeutic alternative. Interestingly, ePICIs exhibited greater bactericidal efficacy in vivo than in vitro. Although the underlying reasons remain unclear, possible explanations include reduced Bap expression in the mammary gland compared with culture media, inactivation of planktonic bacteria that have detached from the biofilm, or the presence of milk or mammary components that enhance ePICI activity. Calcium, a natural constituent of milk, may play a role, as phage 80α, from which the ePICI capsid is derived, requires calcium for infection. Understanding the mechanisms underlying the superior in vivo efficacy of ePICIs is essential, as this observation highlights that limited in vitro activity does not necessarily predict reduced effectiveness during infection. However, caution is warranted when extrapolating these findings from murine models to bovine intramammary infections. The bovine mammary gland is significantly larger and more structurally complex, which can impede the uniform distribution of intramammary treatments. Thus, while mouse models provide valuable proof-of-concept data, they may not fully replicate the therapeutic challenges encountered in cattle.
We cannot rule out the possibility that other combinations of ePICIs with capsids from different phages may offer a broader spectrum of action and enhanced bactericidal properties. Therefore, developing a platform of ePICIs with novel gRNAs and capsids from various phages is essential. Moreover, the use of phages, phage-derived enzymes, or antibiotics on the one hand, and ePICIs on the other, does not have to be mutually exclusive66. Investigating whether their simultaneous application might produce a synergistic effect is another promising avenue that warrants further study.
Methods
Bacterial strains, oligonucleotides, and culture conditions
Bacterial strains, plasmids, and oligonucleotides used in this work are listed in Tables 1–3, respectively. Escherichia coli strains were grown in Luria-Bertani medium (LB; Conda-Pronadisa) at 37 °C. S. aureus strains were routinely incubated at 37 °C in trypticase soy broth (TSB; Conda-Pronadisa) and Gibco Dulbecco’s Modified Eagle Medium (DMEM, TermoFisher scientific) or trypticase soy broth glucose 0,25% (w/v) when synergy tests were conducted. Saccharomyces cerevisiae was grown either in yeast extract – peptone – dextrose (YPD; Sigma-Aldrich), 2x YPD, or synthetic defined (SD) medium at 30 °C. A stock solution of 10× SD was prepared (6.8 g of Yeast Nitrogen Base Without Amino Acids (Sigma-Aldrich), 50 g of glucose (VWR), and 1.92 g of Yeast Synthetic Drop-out Medium Supplements (Sigma-Aldrich) in 100 ml of distilled water), filter-sterilized, and stored at 2–8 °C. Bacteriological agar was used as a gelling agent (VWR).
DNA manipulations
Routine DNA manipulations were performed using standard procedures unless otherwise indicated. Oligonucleotides were synthesized by StabVida (Caparica, Portugal). FastDigest restriction enzymes, Phusion DNA polymerase, Rapid DNA ligation kit (Thermo Scientific), and KAPA High Fidelity DNA polymerase (Roche) were used according to the manufacturer’s instructions. Plasmids were purified using a Macherey-Nagel plasmid purification kit according to the manufacturer’s protocol. To extract plasmids from S. cerevisiae, an established protocol was modified as follows. After being grown in 15 ml of SD medium for 48 h, yeasts were collected (5000 × g, 1 min), resuspended in 520 µl of Yeast Lysis Solution (500 µl of Lyticase Buffer: 0.1 M Na2EDTA (pH 7.5) (AppliChem), 1 M sorbitol (Sigma-Aldrich); 20 µl of Lyticase Solution: 10 mM sodium phosphate (Na2HPO4, pH 7.5) (Merk), 1.2 M sorbitol, 500 U Lyticase from Arthrobacter luteus (Sigma-Aldrich)) and incubated at 37 °C for 1.5 h. After Lyticase treatment, protoplasts were collected (5000 × g, 2 min), and from this step onward, samples were treated according to the manufacturer’s protocol.
Plasmids were transformed in E. coli by electroporation (1 mm cuvette; 200 Ω, 25 µF, 1,250 V; Gene Pulser X-Cell electroporator). S. aureus competent cells were generated as previously described in ref. 67. Plasmids were transformed in S. aureus by electroporation (1 mm cuvette; 100 Ω, 25 µF, 1250 V; Gene Pulser X-Cell electroporator). To transform S. cerevisiae, a 20 ml overnight culture grown in 2× YPD, 30 °C, and 200 rpm was diluted 1.5:100 in fresh medium and incubated under shaking conditions until an OD595nm of 1.0. 3 ml of the culture was collected, washed twice with 1 ml of 0.1 M Lithium acetate (LiAc) (pH 7.5), and used for transformation. The aforementioned pellets were resuspended in the transformation mixture (containing 260 µl of 50% (w/v) polyethylene glycol 4000 (PEG; Merck), 36 µl of 1.0 M LiAc (Sigma-Aldrich), 50 µl of 2 mg ml−1 single-stranded carrier DNA (Sigma-Aldrich), and 14 µl of the DNA to be transformed) and incubated at 42 °C for 45 min under shaking conditions. After the incubation period, cells were collected (13,000 × g, 30 s), washed twice with 1 ml of water, and resuspended in 200 µl of water. Yeast suspension was plated on SD solid medium and incubated at 30 °C for 48–72 h.
Construction of pEMPTY0 derivatives
To ensure the absence of sgRNA off-target activity and to generate the templates for the PCRs needed in the yeast-mediated ePICI assembly, we constructed four plasmids based on a non-thermosensitive predecessor of pEMPTY68, pEMPTY0. We used oligonucleotides 247, 294, E7, and E15, containing the strong constitutive SP01 promoter from Bacillus subtilis bacteriophage SP0169 and the gRNA sequence that allows targeting either a highly conserved plasmid sequence or the RsaE, RsaH, and RsaI coding sequences (Table 4). These oligonucleotides were used in combination with primer 245 to amplify the Cas9 gene from plasmid pFREE-Amp70. The resulting fragments were cloned into the pJET1.2/blunt plasmid and then sub-cloned into pEMPTY0 digested with PstI and SmaI enzymes, leading to plasmids pEMPTY0::sgRNA2, pEMPTY0::sgRNArsaE, pEMPTY0::sgRNArsaH, and pEMPTY0::sgRNArsaI.
Removal of chromosomal genes
To generate deletional mutants, two fragments of at least 500 bp, which flanked the left and right sequences of the region targeted for deletion, were PCR amplified. As a PCR template, chromosomal DNA from S. aureus RN4220 was used. To amplify rsaE, rsaH, and rsaI flanking regions, primers 330-332 and 334-336, E1-E2 and E3-E4, and E9-E10 and E11-E12 were respectively used. An overlapping PCR of the aforementioned fragments was performed using primers 330-336 (rsaE), E1-E4 (rsaH), and E9-E12 (rsaI), and overlapped products were gel-purified with the silica bead DNA gel extraction kit (Thermo Scientific). rsaH and rsaI related fragments were cloned into pJET1.2/blunt and then, pJET1.2/blunt::rsaH and pJET1.2/blunt::rsaI were digested with NcoI and BamHI or NcoI and EcoRI enzymes, respectively, and inserts were ligated into the shuttle vector pMAD71, leading to plasmids pMAD::rsaH and pMAD::rsaI. An ApaI linearized pMAD_lic plasmid72, and the amplified rsaE-related PCR fragment were treated with T4 DNA polymerase (Novagen) in the presence of dTTP and dATP (Novagen), respectively, for 30 min at 22 °C. After the incubation period, the enzyme was inactivated. A mix of vector and insert was incubated for 5 min at 22 °C, and then, 6.25 mM EDTA was added, and an additional incubation of 5 min at 22 °C was applied. The annealed species were transformed into chemically competent E. coli NovaBlue GigaSingles™ cells. pMAD::rsaH, pMAD::rsaI, and pMAD_lic::rsaE were checked by PCR, purified from E. coli, and transformed into S. aureus by electroporation. Homologous recombination experiments were performed as previously described in73. Erythromycin-sensitive white colonies, which did not further contain the pMAD plasmid, were tested by PCR using primers located outside the amplified-flanking regions (346-348 for rsaE mutants; E5-E6 for rsaH mutants; and E13-E14 for rsaI mutants). All constructed plasmids were confirmed by Sanger sequencing.
Plasmid construction in yeast and ePICIs rebooting
ePICIs assembly in yeast was performed as described elsewhere31. Briefly, KAPA High Fidelity DNA polymerase was used to amplify the fragments of the plasmid to be constructed. Overlapping PCR fragments (≥30 bp) were generated mirroring the structure of the integrated PICIs in the chromosome. Fragments 1 and 7 were designed with 30 nucleotides complementary to the ends of the YAC plasmid (pAUR112). Fragment 4 represented the cas9 gene under the control of a constitutive promoter, fragment 5 contained the sequence for gRNA cloning, and fragment 6 carried the tetM gene, providing tetracycline resistance. These three fragments also included 30 nucleotides complementary to their adjacent fragments to facilitate yeast assembly by recombination. All seven SaPIbov2-CRISPR_Cas9-tetM fragments were transformed into S. cerevisiae BJ5464 cells to assemble the engineered PICI genome (ePICI). The S. cerevisiae colonies were subsequently restreaked and analyzed through colony PCR to confirm the presence of ePICI. Four distinct ePICIs were generated, with three islands each harboring a gRNA targeting an sRNA (RsaE, RsaH, and RsaI), and a fourth island featuring a gRNA directed against a conserved DNA region in cryptic plasmids of S. aureus MW268. This latter sequence, absent in the S. aureus chromosome, served as a negative control for the activity of the CRISPR-Cas9 system. The set of primers used for the amplification of the different fragments were: 404-410 (product obtained: linearized plasmid pAUR112; template: pAUR112), E50-E17 (product obtained: Fragment 1, partial SaPIbov2 sequence; template: genomic DNA RN4220 SaPIbov2 bap::tetM), E18-E19 (product obtained: Fragment 2, partial SaPIbov2 sequence; template: genomic DNA RN4220 SaPIbov2 bap::tetM), E20-452 (product obtained: Fragment 3, partial SaPIbov2 sequence; template: genomic DNA RN4220 SaPIbov2 bap::tetM), 422-424 (product obtained: Fragment 4.1, strong constitutive PblaZ promoter; template: pCN40), 382-129 (product obtained: Fragment 4.2, 5′ UTR and partial cas9 sequence; template: pEMPTY0), 131-E49 (product obtained: Fragment 5, partial cas9 sequence and module containing the constitutive promoter SP01 promoter-sgRNA-tracrRNA; template: pEMPTY0::sgRNA2/ pEMPTY0::sgRNArsaE/ pEMPTY0::sgRNArsaH/ pEMPTY0::sgRNArsaI), E22-E23 (product obtained: Fragment 6, tetM resistance cassette; template: pRN6680), and E24-E51 (product obtained: Fragment 7, partial SaPIbov2 sequence; template: genomic DNA RN4220 SaPIbov2 bap::tetM). Fragments 4.1 and 4.2 were fused using primers 422-129, generating Fragment 4. The first and last fragments included 500 bp outside the attL and attR sites, respectively. For clarity, the whole amplification and assembly process is depicted in Supplementary Fig. 1.
PCR products were purified using the QIAquick PCR Purification Kit (Qiagen) and quantified (Eppendorf BioPhotometer model 6131). The amplified pAUR112 fragment was treated with DpnI, and then 250 ng of DpnI-treated pAUR112 vector and 500 ng of each PCR product suspended in a maximum volume of 14 µl were transformed into the yeast as described above. Fragments used for transformation were not shorter than 1000 bp, as several reports have highlighted poor cloning efficiency when assembling small DNA fragments74. Colonies obtained after transformation were checked by colony PCR, plasmid extraction was performed, and the presence of the different cloned fragments was confirmed by PCR amplification.
S. aureus competent cells were transformed with >2 μg of the yeast-assembled plasmids (pAUR112 derivatives), incubated for 2–4 h at 37 °C and 200 rpm, plated on medium containing 3 µg ml−1 tetracycline and grown for 24–48 h at 37 °C. Tetracycline-resistant colonies obtained after the rebooting process were checked by colony PCR.
Disruption of integrase production in ePICIs
To minimize potential polar effects and abolish integrase production, two stop codons were introduced into the gene encoding the integrase in the SaPIbov2 derivative. This approach reduces the likelihood of generating leaky stop codons while simultaneously creating an EcoRI restriction site to facilitate the screening of positive clones. The mutation was introduced via double-crossover recombination using the pMAD plasmid in a strain of S. aureus RN4220, which harbors a modified version of the PICI integrated into its chromosome.
Construction of pCN47::icaADBC plasmid
AU56 and AU57 primers were employed for the amplification of the complete icaADBC operon from S. aureus 15981. Subsequently, the PCR product was inserted into the pCN47 plasmid, which had been cleaved using SalI and KpnI restriction enzymes, utilizing the In-fusion cloning technique.
Marking and moving phage 80α ΔterS
Phage 80α ΔterS was marked with an erythromycin resistance cassette by lateral transduction75 using a lysate from RN450 80α::ermC on a RN4220 80α ΔterS strain. Phage 80α ΔterS::ermC was mobilized by generalized transduction after trans-complementing the lysogenic strain with the gene encoding the terminase small-subunit (pCN51(CmR)::Pcad-terS80α-TT).
Phage and ePICI induction and titration
An S. aureus overnight culture was diluted (1:50) in fresh TSB medium supplemented with chloramphenicol when required and incubated at 37 °C and 200 rpm until an OD595nm of 0.4. The culture was treated with 2 μg ml−1 of mitomycin C and incubated at 32 °C and 80 rpm for the next 4 h, and then left overnight at room temperature under static conditions. Phage/ePICI stocks were filtered (0.2 μm) and stocked at 4 °C. To remove the presence of mitomycin C and perform a buffer exchange to phage buffer (1 mM NaCl; 0.05 M Tris, pH 7.8; 1 mM MgSO4; 4 mM CaCl2), lysates were dialyzed using Slide-A-LyzerTM 10 K MWCO (ThermoFisher).
Phages and ePICIs used in this study are marked with erythromycin and tetracycline resistance markers, respectively, that allow the selection of transductants on media containing the aforementioned drugs. To titrate both phages and ePICIs, an overnight culture of the S. aureus laboratory strain RN4220 ΔrsaE ΔrsaH ΔrsaI was diluted (1:50) in fresh TSB medium and incubated at 37 °C and 200 rpm until an OD595nm of 1.4. Serial dilutions of the lysate were made in phage buffer and added to the recipient bacteria in a proportion of 100 μl of lysate per ml of bacterial culture. The mixture was homogenized and incubated at 37 °C for 20 min without shaking. 3 ml of TTA (TSB Top Agar: TSB + 0.75% (w/v) agar) at 55 °C was added to the tube containing the lysate-cell combination and immediately poured over the surface of a plate containing the selective drug and 1.7 mM of sodium citrate to prevent bacterial infection after plating. Once solidified, plates were incubated at 37 °C for 24–48 h.
CRISPR-Cas9 efficiency and gRNA specificity
To illustrate the sequence-dependent activity of the different sgRNAs, overnight cultures of S. aureus RN4220 and the triple mutant S. aureus RN4220 ΔrsaE ΔrsaH ΔrsaI were diluted (1:50) in fresh TSB medium and incubated at 37 °C and 200 rpm until an OD600nm of 1.4. Serial dilutions of the lysates were made in phage buffer and added to the recipient bacteria in a proportion of 100 μl of lysate per ml of bacterial culture. The mixture was homogenized and incubated at 37 °C for 20 min, then 20 µl drops were plated in a TSA tetracycline 3 µg ml−1 plate and incubated at 37 °C for 24 h.
Growth kinetic assays
Overnight S. aureus cultures were washed twice with fresh TSB medium, and the cellular density was adjusted to an OD600nm of 0.1. 5 μl of this suspension was mixed with 175 μl of fresh TSB 0.25% glucose medium or Gibco Dulbecco’s Modified Eagle Medium (DMEM, TermoFisher scientific) and 20 μl of phage buffer containing 108 ePICI particles or PFU of 80α phage ml−1. Growth assays were carried out using sterile 96-well polystyrene microtiter plates (Thermo Code: 130188) with 200 µl of volume per well. When required, media was supplemented with chloramphenicol (10 µg ml−1) for plasmid retention. Growth kinetics were obtained using a Synergy H1 Hybrid Multi-Mode Microplate Reader (Biotek). Growth data (OD600nm) was collected every 15 min for up to 24 h at 37 °C and 425 cpm. All the experiments were repeated at least three times.
Bacterial counts during growth kinetics assay
Six hours after the start of the growth kinetics assay, samples were collected from two wells per strain and condition (treated and untreated). Serial dilutions were prepared, followed by duplicate cell counts.
Calculation of mastitis isolates growth inhibition through iAUC
In the growth kinetic assays, OD600nm measurements were collected over a 12-h period for both treated (with ePICIrsaE) and non-treated conditions. These measurements were utilized to construct growth curves for each condition. The area under the curve (AUC) for each growth curve was computed using the trapezoidal rule, a numerical integration method that involved partitioning the time interval into segments, approximating each segment with a trapezoid, calculating the area of each trapezoid, and summing these areas to estimate the total area under the curve. Subsequently, the integrated AUC (iAUC) was determined by subtracting the baseline AUC of the non-treated control from the AUC of the treated condition. This calculation was expressed as iAUC = 1 − AUCTreatment/AUCControl, where AUCTreatment represents the AUC of the spline generated by a specific bacteria and ePICIrsaE, and AUC Control denotes the AUC of the spline created with the same bacteria without any treatment. As a result, iAUC values ranged between 0 and 1, where 0 indicated no growth inhibition, and 1 indicated complete growth inhibition. Susceptibility was defined at iAUC values ≥ 0.1.
Biofilm matrix disgregation susceptibility test
Strains were cultured overnight in TSB with 0.25% glucose at 37 °C. The culture was adjusted to an OD600nm of 1 and diluted 1:40 in TSB-glucose. Subsequently, 200 µl of this diluted cell suspension was inoculated into sterile 96-well plates and incubated for 24 h at 37 °C. Wells were emptied and gently washed three times with water. Biofilm was treated with 100 µl of either proteinase K at 100 µg ml−1, dispersin B76 at 40 µg ml−1 or PBS (negative control treatment) and incubated at 37 °C for 2 h. Wells were emptied and washed three times by immersion in water. Remaining surface-adsorbed cells in each well were stained with 200 µl of crystal violet (VWR) for 5 min. Wells were washed three times and air-dried. Crystal violet-stained cells were revealed by solubilizing the dye with 200 µl of ethanol-acetone (80:20, vol/vol) for 5 min.
Dot-blot PNAG analysis
Surface-anchored PNAG was detected as described elsewhere16. Overnight cultures were grown in Tryptic Soy Broth supplemented with 0.25% glucose, their optical density at 600 nm was measured, and equivalent cell numbers from each culture were suspended in 50 ml of 0.5 M EDTA (pH 8.0). Then, cells were heat-treated for 5 min at 100 °C and centrifuged at 12,000 rpm for 10 min to collect the cell pellets. A 40 µl volume of each supernatant was incubated with 10 µl proteinase K (20 mg ml−1 Sigma-Aldrich) for 30 min at 37 °C. Following this, 10 µl of Tris-buffered saline [20 mM Tris/HCl, 150 mM NaCl (pH 7.4)] with 0.01% bromophenol blue was introduced, and 5 µl of serial decimal dilutions were spotted onto a nitrocellulose filter using a Bio-Dot microfiltration apparatus (Bio-Rad). The filter was blocked overnight with 5% skimmed milk in PBS with 0.1% Tween 20, and incubated for 2 h with rabbit antibodies raised against S. aureus deacetylated PNAG conjugated to diphtheria toxoid diluted 1:10,00077. Detection of bound antibodies was achieved using peroxidase-conjugated goat anti-rabbit IgG antibodies (Jackson ImmunoResearch Laboratories; diluted 1:10,000) and the Amersham ECL Western blotting system.
Bactericidal activity of ePICI-rsaE against planktonic and biofilm S. aureus in milk
Bactericidal activity of ePICIS against planktonic S. aureus was performed as follows. Overnight cultures of S. aureus strains V329 (wild type), V329 Δbap, and V329 Δbap + ica (S. aureus V329 Δbap complemented with icaADBC genes in the pCN47 plasmid) were adjusted to a final concentration of 103 CFU ml−1 in pasteurized milk. Six replicate wells per strain and treatment were filled with 200 µl of bacterial suspension, and the plate was incubated at 37 °C with shaking for 1 h. Subsequently, 40 µl of phage buffer containing 108 ePICI-rsaE ml⁻¹ were added to the treatment wells, while negative controls received phage buffer alone. Plates were incubated at 37 °C for 3 h, followed by a second identical treatment and an additional 4 h of incubation. Cultures of each well were then collected, serially diluted in PBS, and plated on tryptic soy agar (TSA). Plates were incubated at 37 °C, and bacterial counts were determined by colony enumeration. For biofilm assays, overnight cultures of S. aureus strains V329 and 15981 were diluted 1:40 in pasteurized milk. Aliquots of 200 µl of the bacterial suspension were added to sterile 96-well polystyrene plates and incubated statically at 37 °C for 4 h to allow biofilm formation. Treatment conditions were applied as described above for planktonic bacteria. Plates were incubated statically at 37 °C for 10 h, followed by a second identical treatment and an additional 4 h of static incubation. Supernatants were discarded, and wells were gently rinsed with sterile phosphate-buffered saline (PBS) to remove non-adherent cells. Remaining biofilms were detached by adding 100 µl of PBS and sonicating for 2 min. The resulting biofilm suspensions were serially diluted, plated on TSA, and incubated at 37 °C for colony enumeration.
In vivo murine model of induced staphylococcal mastitis
A modified version of the mastitis intramammary infection model described in ref. 23 was followed (Fig. 8A). Briefly, lactating CD1 mice were detached from their 7-day-old offspring 2 h prior to infection and randomly divided into 3 groups. Based on Breyne et al.78, 100 µl of a bacterial suspension containing 103 CFU ml−1 of S. aureus strains V329, V329 Δbap, and V329 Δbap + ica were used to inoculate the L4 (left) and R4 (right) mammary glands by intraductal injection. Treatment doses were 100 µl of PBS for the negative control group, 100 µl of vancomycin solution 500 µg ml−1 for the positive control group, and 100 µl of phage buffer containing 108 ePICIrsaE ml−1 particles for the last group. At 4 h post-infection, the first dose of treatment was inoculated, followed by a second dose 12 h after the first one. 24 h post-infection, the mammary glands involved in the experiment were aseptically extracted and homogenized. Serial dilutions of the homogenates were plated in TSA and incubated at 37 °C.
Statistical analysis
All statistical analyses were performed using GraphPad Prism v5.01 (GraphPad Software, San Diego, CA, USA) and the R programming language. Significant differences between treated and untreated groups were determined using the non-parametric two-tailed Mann–Whitney U-test. For comparisons involving more than two groups, the non-parametric Kruskal–Wallis test was applied. Non-parametric tests were chosen because the limited number of data points per group precluded verification of normality.
Ethics statement
Animal procedures were reviewed and approved by “Comité de Ética para la Experimentación Animal (CEEA) de la Universidad de Navarra” (protocol 012-22). Described animal work was carried out at the “Centro de Investigación Médica Aplicada” building (ES312010000132) under the principles and guidelines established in the European Directive 2010/63/EU for the protection of animals used for experimental purposes.
Data availability
No datasets were generated or analyzed during the current study.
References
Novick, R. P., Christie, G. E. & Penadés, J. R. The phage-related chromosomal islands of Gram-positive bacteria. Nat. Rev. Microbiol. 8, 541–551 (2010).
Penadés, J. R. & Christie, G. E. The phage-inducible chromosomal islands: a family of highly evolved molecular parasites. Annu. Rev. Virol. 2, 181–201 (2015).
Ram, G. et al. Staphylococcal pathogenicity island interference with helper phage reproduction is a paradigm of molecular parasitism. Proc. Natl. Acad. Sci. USA 109, 16300–16305 (2012).
Fillol-Salom, A., Miguel-Romero, L., Marina, A., Chen, J. & Penadés, J. R. Beyond the CRISPR-Cas safeguard: PICI-encoded innate immune systems protect bacteria from bacteriophage predation. Curr. Opin. Microbiol. 56, 52–58 (2020).
Fillol-Salom, A. et al. Phage-inducible chromosomal islands are ubiquitous within the bacterial universe. ISME J. 12, 2114–2128 (2017).
Úbeda, C. et al. Antibiotic-induced SOS response promotes horizontal dissemination of pathogenicity island-encoded virulence factors in staphylococci. Mol. Microbiol. 56, 836–844 (2005).
Úbeda, C. et al. SaPI mutations affecting replication and transfer and enabling autonomous replication in the absence of helper phage. Mol. Microbiol. 67, 493–503 (2008).
Lindqvist, B. H., Dehò, G. & Calendar, R. Mechanisms of genome propagation and helper exploitation by satellite phage P4. Microbiol. Rev. 57, 683–702 (1993).
Bowring, J. et al. Pirating conserved phage mechanisms promotes promiscuous staphylococcal pathogenicity island transfer. eLife 6, e26487 (2017).
Ram, G., Ross, H. F., Novick, R. P., Rodriguez-Pagan, I. & Jiang, D. Conversion of staphylococcal pathogenicity islands to CRISPR-Cas9-based antibacterial drones that cure staph infections in mice. Nat. Biotechnol. 36, 971–976 (2018).
Aguilar, B., Amorena, B. & Iturralde, M. Effect of slime on adherence of Staphylococcus aureus isolated from bovine and ovine mastitis. Vet. Microbiol. 78, 183–191 (2001).
Otto, M. Staphylococcal Biofilms. Microbiol Spectr. 6, https://doi.org/10.1128/microbiolspec.gpp3-0023-2018 (2018).
Schilcher, K. & Horswill, A. R. Staphylococcal biofilm development: structure, regulation, and treatment strategies. Microbiol. Mol. Biol. Rev. 84, e00026–19 (2020).
Gotz, F. Staphylococcus and biofilms. Mol. Microbiol. 43, 1367–1378 (2002).
Mack, D. et al. The intercellular adhesin involved in biofilm accumulation of Staphylococcus epidermidis is a linear beta-1,6-linked glucosaminoglycan: purification and structural analysis. J. Bacteriol. 178, 175–183 (1996).
Cramton, S. E., Gerke, C., Schnell, N. F., Nichols, W. W. & Gotz, F. The intercellular adhesion (ica) locus is present in Staphylococcus aureus and is required for biofilm formation. Infect. Immun. 67, 5427–5433 (1999).
Heilmann, C. et al. Molecular basis of intercellular adhesion in the biofilm-forming Staphylococcus epidermidis. Mol. Microbiol. 20, 1083–1091 (1996).
Vergara-Irigaray, M. et al. Relevant role of fibronectin-binding proteins in Staphylococcus aureus biofilm-associated foreign-body infections. Infect. Immun. 77, 3978–3991 (2009).
Merino, N. et al. Protein A-mediated multicellular behavior in Staphylococcus aureus. J. Bacteriol. 191, 832–843 (2009).
Foster, T. J., Geoghegan, J. A., Ganesh, V. K. & Höök, M. Adhesion, invasion and evasion: the many functions of the surface proteins of Staphylococcus aureus. Nat. Rev. Microbiol. 12, 49–62 (2014).
Corrigan, R. M., Rigby, D., Handley, P. & Foster, T. J. The role of Staphylococcus aureus surface protein SasG in adherence and biofilm formation. Microbiology 153, 2435–2446 (2007).
Patti, J. M., Allen, B. L., McGavin, M. J. & Hook, M. MSCRAMM-mediated adherence of microorganisms to host tissues. Annu. Rev. Microbiol. 48, 585–617 (2003).
Valle, J. et al. Bap, a biofilm matrix protein of Staphylococcus aureus prevents cellular internalization through binding to GP96 host receptor. PLoS Pathog. 8, e1002843 (2012).
Cucarella, C. et al. Bap, a Staphylococcus aureus surface protein involved in biofilm formation. J. Bacteriol. 183, 2888–2896 (2001).
Taglialegna, A. et al. Staphylococcal Bap proteins build amyloid scaffold biofilm matrices in response to environmental signals. PLoS Pathog. 12, e1005711 (2016).
Ruegg, P. L. A 100-year review: mastitis detection, management, and prevention. J. Dairy Sci. 100, 10381–10397 (2017).
Rodriguez, Z., Cabrera, V. E., Hogeveen, H. & Ruegg, P. L. Economic impact of treatment of subclinical mastitis in early lactation using intramammary nisin. J. Dairy Sci. 107, 4634–4645 (2024).
Leitner, G., Lubashevsky, E. & Trainin, Z. Staphylococcus aureus vaccine against mastitis in dairy cows, composition and evaluation of its immunogenicity in a mouse model. Vet. Immunol. Immunopathol. 93, 159–167 (2003).
Zadoks, R. N., Middleton, J. R., McDougall, S., Katholm, J. & Schukken, Y. H. Molecular epidemiology of mastitis pathogens of dairy cattle and comparative relevance to humans. J. Mammary Gland Biol. Neoplasia 16, 357 (2011).
Lin, W. C. et al. Octanoic acid promotes clearance of antibiotic-tolerant cells and eradicates biofilms of Staphylococcus aureus isolated from recurrent bovine mastitis. Biofilm 6, 100149 (2023).
Ibarra-Chávez, R., Haag, A. F., Dorado-Morales, P., Lasa, I. & Penadés, J. R. Rebooting synthetic phage-inducible chromosomal islands: one method to forge them all. Biodes. Res. 2020, 1–14 (2020).
Geissmann, T. et al. A search for small noncoding RNAs in Staphylococcus aureus reveals a conserved sequence motif for regulation. Nucleic Acids Res. 37, 7239–7257 (2009).
Brady, A. et al. Molecular basis of lysis–lysogeny decisions in Gram-positive phages. Annu Rev. Microbiol. 75, 1–19 (2021).
Bernheim, A. & Sorek, R. The pan-immune system of bacteria: antiviral defence as a community resource. Nat. Rev. Microbiol. 18, 113–119 (2020).
Gencay, Y. E. et al. Engineered phage with antibacterial CRISPR–Cas selectively reduce E. coli burden in mice. Nat. Biotechnol. 42, 265–274 (2024).
Konstantakos, V., Nentidis, A., Krithara, A. & Paliouras, G. CRISPR–Cas9 gRNA efficiency prediction: an overview of predictive tools and the role of deep learning. Nucleic Acids Res. 50, 3616–3637 (2022).
Doench, J. G. et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 34, 184–191 (2016).
Labun, K. et al. CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 47, W171–W174 (2019).
Novick, R. P. & Ram, G. Staphylococcal pathogenicity islands—movers and shakers in the genomic firmament. Curr. Opin. Microbiol. 38, 197–204 (2017).
Millman, A. et al. An expanded arsenal of immune systems that protect bacteria from phages. Cell Host Microbe 30, 1556–1569.e5 (2022).
Rostøl, J. T. & Marraffini, L. (Ph)ighting phages: how bacteria resist their parasites. Cell Host Microbe 25, 184–194 (2019).
Maffei, E. et al. Phage Paride can kill dormant, antibiotic-tolerant cells of Pseudomonas aeruginosa by direct lytic replication. Nat. Commun. 15, 175 (2024).
Melo, L. D. R. et al. The protective effect of Staphylococcus epidermidis biofilm matrix against phage predation. Viruses 12, 1076 (2020).
Pires, D., Melo, L., Boas, D. V., Sillankorva, S. & Azeredo, J. Phage therapy as an alternative or complementary strategy to prevent and control biofilm-related infections. Curr. Opin. Microbiol. 39, 48–56 (2017).
Azeredo, J. & Sutherland, I. The use of phages for the removal of infectious biofilms. Curr. Pharm. Biotechnol. 9, 261–266 (2008).
Pires, D. P., Oliveira, H., Melo, L. D. R., Sillankorva, S. & Azeredo, J. Bacteriophage-encoded depolymerases: their diversity and biotechnological applications. Appl. Microbiol. Biotechnol. 100, 2141–2151 (2016).
Gutiérrez, D. et al. Real-Time assessment of Staphylococcus aureus biofilm disruption by phage-derived proteins. Front. Microbiol. 8, 1632 (2017).
Gutiérrez, D. et al. Role of the pre-neck appendage protein (Dpo7) from phage vB_SepiS-phiIPLA7 as an anti-biofilm agent in staphylococcal species. Front. Microbiol. 6, 1315 (2015).
Schuch, R., Khan, B. K., Raz, A., Rotolo, J. A. & Wittekind, M. Bacteriophage lysin CF-301, a potent antistaphylococcal biofilm agent. Antimicrob. Agents Chemother. https://doi.org/10.1128/aac.02666-16 (2017).
Liu, B., Guo, Q., Li, Z., Guo, X. & Liu, X. Bacteriophage endolysin: a powerful weapon to control bacterial biofilms. Protein J. 42, 463–476 (2023).
Gutiérrez, D., Ruas-Madiedo, P., Martínez, B., Rodríguez, A. & García, P. Effective removal of Staphylococcal biofilms by the endolysin LysH5. PLoS ONE 9, e107307 (2014).
Sass, P. & Bierbaum, G. Lytic activity of recombinant bacteriophage φ11 and φ12 endolysins on whole cells and biofilms of Staphylococcus aureus. Appl. Environ. Microbiol. 73, 347–352 (2007).
Elst, N. V. Bacteriophage-derived endolysins as innovative antimicrobials against bovine mastitis-causing streptococci and staphylococci: a state-of-the-art review. Acta Vet. Scand. 66, 20 (2024).
Bergonier, D., Crémoux, R. de, Rupp, R., Lagriffoul, G. & Berthelot, X. Mastitis of dairy small ruminants. Vet. Res. 34, 689–716 (2003).
Vasileiou, N. G. C. et al. Role of staphylococci in mastitis in sheep. J. Dairy Res. 86, 254–266 (2019).
Algharib, S. A., Dawood, A. & Xie, S. Nanoparticles for treatment of bovine Staphylococcus aureus mastitis. Drug Deliv. 27, 292–308 (2020).
Schmelcher, M. & Loessner, M. J. Bacteriophage endolysins, extending their application to tissues and the bloodstream. Curr. Opin. Biotech. 68, 51–59 (2021).
Titze, I., Lehnherr, T., Lehnherr, H. & Krömker, V. Efficacy of bacteriophages against Staphylococcus aureus isolates from bovine mastitis. Pharmaceuticals 13, 35 (2020).
Dias, R. S. et al. Use of phages against antibiotic-resistant Staphylococcus aureus isolated from bovine mastitis. J. Anim. Sci. 91, 3930–3939 (2013).
Iwano, H. et al. Bacteriophage ΦSA012 has a broad host range against Staphylococcus aureus and effective lytic capacity in a mouse mastitis model. Biology 7, 8 (2018).
Guo, M. et al. Development and mouse model evaluation of a new phage cocktail intended as an alternative to antibiotics for treatment of Staphylococcus aureus-induced bovine mastitis. J. Dairy Sci. 107, 5974–5987 (2024).
Geng, H. et al. Evaluation of phage therapy in the treatment of Staphylococcus aureus-induced mastitis in mice. Folia Microbiol. 65, 339–351 (2020).
Elst, N. V. et al. Development of engineered endolysins with in vitro intracellular activity against streptococcal bovine mastitis-causing pathogens. Microb. Biotechnol. 16, 2367–2386 (2023).
Fernández, L. et al. Bacteriophages in the dairy environment: from enemies to allies. Antibiotics 6, 27 (2017).
Vaidya, S. et al. Bacteria use exogenous peptidoglycan as a danger signal to trigger biofilm formation. Nat Microbiol. 10, 144–157 (2025).
Elst, N. V., Farmen, K., Knörr, L., Merlijn, L. & Iovino, F. Bacteriophage-derived endolysins restore antibiotic susceptibility in β-lactam- and macrolide-resistant Streptococcus pneumoniae infections. Mol. Med. 31, 170 (2025).
Valle, J. et al. SarA and not sigmaB is essential for biofilm development by Staphylococcus aureus. Mol. Microbiol. 48, 1075–1087 (2003).
Dorado-Morales, P., Garcillán-Barcia, M. P., Lasa, I. & Solano, C. Fitness cost evolution of natural plasmids of Staphylococcus aureus. mBio 12, e03094–20 (2021).
Stewart, C. R. et al. Genes and regulatory sites of the “Host-Takeover Module” in the terminal redundancy of Bacillus subtilis bacteriophage SPO1. Virology 246, 329–340 (1998).
Lauritsen, I., Porse, A., Sommer, M. O. A. & Nørholm, M. H. H. A versatile one-step CRISPR-Cas9 based approach to plasmid-curing. Micro. Cell Fact. 16, 135 (2017).
Arnaud, M., Chastanet, A. & Débarbouillé, M. New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl. Environ. Microbiol. 70, 6887–6891 (2004).
Burgui, S., Gil, C., Solano, C., Lasa, I. & Valle, J. A systematic evaluation of the two-component systems network reveals that ArlRS is a key regulator of catheter colonization by Staphylococcus aureus. Front. Microbiol. 9, 342 (2018).
Valle, J. et al. SarA and not σB is essential for biofilm development by Staphylococcus aureus. Mol. Microbiol. 48, 1075–1087 (2003).
Watson, J. F. & García-Nafría, J. In vivo DNA assembly using common laboratory bacteria: A re-emerging tool to simplify molecular cloning. J. Biol. Chem. 294, 15271–15281 (2019).
Chen, J. et al. Genome hypermobility by lateral transduction. Science 362, 207–212 (2018).
Kaplan, J. et al. Genes involved in the synthesis and degradation of matrix polysaccharide in Actinobacillus actinomycetemcomitans and Actinobacillus pleuropneumoniae biofilms. J. Bacteriol. 186, 8213–8220 (2004).
Maira-Litrán, T., Kropec, A., Goldmann, D. A. & Pier, G. B. Comparative opsonic and protective activities of Staphylococcus aureus conjugate vaccines containing native or deacetylated staphylococcal Poly-N-Acetyl-β-(1-6)-glucosamine. Infect. Immun. 73, 6752–6762 (2005).
Breyne, K. et al. Efficacy and safety of a bovine-associated Staphylococcus aureus phage cocktail in a murine model of mastitis. Front. Microbiol. 8, 2348 (2017).
Acknowledgements
N.G.-A. was supported by an FPI contract (PRE2021-097385), and P.D.-M. was supported by an FPI contract (BES-2015-07285) from the Spanish Ministry of Science, Innovation, and Universities. This work was financially supported by the Spanish Ministry of Science, Innovation and Universities grant PID2020-113494RB-I00/AEI (Agencia Española de Investigación/Fondo Europeo de Desarrollo Regional, European Union) to I.L. The funders had no role in the study design, data collection, and interpretation, or the decision to submit the work for publication.
Author information
Authors and Affiliations
Contributions
Conceptualization: I.L. and J.P. Data curation: N.G.-A. and P.D.-M. Formal analysis: N.G.-A. and P.D.-M. Funding acquisition: I.L. Investigation: N.G.-A., P.D.-M., C.G., B.G., M.E., and C.S. Methodology: N.G.-A., P.D.-M., and C.G. Project administration: I.L. Validation: N.G.-A., P.D.-M., and C.G. Visualization: N.G.-A. and P.D.-M. Writing – original draft: I.L. Writing – review & editing: I.L., N.G.-A., P.D.-M., J.P., and C.S.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Garmendia-Antoñana, N., Dorado-Morales, P., Gil, C. et al. Targeted elimination of Staphylococcus aureus mastitis infections with synthetic phage-based CRISPR-Cas delivery systems. npj Biofilms Microbiomes 12, 67 (2026). https://doi.org/10.1038/s41522-026-00931-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41522-026-00931-x
