
Abstract
Parkinson’s disease (PD) is a neurodegenerative disorder characterized by the aggregation of α-synuclein (α-syn) and the nigrostriatal dopaminergic neuronal degeneration. Depression is one of the most common non-motor symptoms of PD patients. However, the pathogenic connection between PD and depression is not well understood. Herein, we report that chronic stress upregulates the expression of α-syn in the mouse brain. Overexpression of α-syn in the hippocampus replicates depressive-like phenotypes, whereas the genetic deletion of α-syn enhances resistance to chronic stress. Furthermore, chronic stress in early life promoted the deposition of α-syn aggregates in a transgenic mouse model that overexpresses human A53T mutant α-syn (A53T mice). Chronic stress also exacerbated dopaminergic degeneration and motor impairments in A53T mice. Strikingly, α-syn inclusions were also observed in the brains of some aged non-transgenic mice subjected to chronic stress. Together, our findings suggest that chronic stress upregulates α-synuclein expression, resulting in depression-like behaviors and parkinsonism.
Similar content being viewed by others


Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease and is characterized by the loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) and the presence of Lewy body (LB) aggregates1. PD patients suffer from motor symptoms, such as bradykinesia, rigidity, and resting tremor, and non-motor symptoms, such as depression, sleep disorders, cognitive decline, and autonomic dysfunction2. Depression is one of the most common non-motor symptoms and is considered clinically significant in 40–50% of PD patients3,4. Depression may predate the onset of motor symptoms and accelerate the progression of motor symptoms and cognitive impairments5. Thus, depression has been identified as one of the major determinants of quality of life in PD patients. However, the pathogenic relationship between PD and depression is poorly understood.
Two hypotheses have been proposed regarding the relationship between PD and depression. One possibility is that depression is an early manifestation of PD, which is supported by the fact that depression in PD patients is related to neuroanatomical alterations, neurotransmitter disturbances, and neuroinflammation, similar to individuals with naive depression6. The other hypothesis presumes that premorbid depression may increase susceptibility to PD. Some studies have reported that depression is associated with an increased risk of developing PD in late life7,8,9. To better dissect these two hypotheses, studies on the pathophysiological integration between PD and depression are needed.
α-Synuclein (α-syn), encoded by the SNCA gene, is highly enriched in presynaptic terminals. α-Syn is believed to play roles in neurotransmission, synaptic plasticity, and vesicle dynamics under physiological conditions10. Pathological deposits of α-syn in LBs and Lewy neurites (LNs) are the hallmarks of PD and underlie the development and progression of PD11. Missense mutation of SNCA (A53T, A30P, E46K) and multiplication of SNCA cause familial PD12,13. Even in sporadic PD, SNCA polymorphisms are genetic risk factors14. Interestingly, several studies have reported that α-syn mRNA levels are increased in patients with major depressive disorder15,16. Additionally, two autopsy studies revealed that depressive symptoms are associated with the presence of α-syn aggregates in subcortical structures17,18. These results suggest that α-syn pathology may underlie both PD and depression.
In the present study, we found both increased protein and mRNA levels of α-syn in different brain regions of depressive mice induced by chronic unpredicted mild stress (CUMS). Overexpression of α-syn in the hippocampus leads to spontaneous depression-like phenotypes, whereas its genetic deletion enhances resistance to pathological changes under chronic stress. Moreover, CUMS promoted α-syn aggregation and compromised motor ability in a genetic PD mouse model. We further demonstrated that CUMS could increase the likelihood of pathological α-syn in aged non-transgenic mice. These observations indicate that α-syn may bridge chronic stress to both depression and PD.
Results
α-Syn expression is upregulated in different brain regions after CUMS
The experimental design and timeline of our study are shown in Supplementary Fig. 1. We first determined the effects of chronic stress on phenotypes in a CUMS-induced mouse model. After 4 weeks of CUMS treatment, the sucrose preference of the CUMS group was lower than that of the WT group (Supplementary Fig. 2a). The body weight curve revealed that the mice in the CUMS group gained less body weight than the mice in the control group did (Supplementary Fig. 2b). In the behavioral tests, the immobility times in both the FST and TST of the mice in the CUMS group were longer than those of the mice in the control group (Supplementary Fig. 2c, d). Furthermore, the EPM test revealed that the mice in the CUMS group had fewer entries into the open arms and less time spent in the open arms (Supplementary Fig. 2e). In the OFT, the time spent in the center area, number of central entries, and total distance traveled were significantly lower in the CUMS group than in the control group (Supplementary Fig. 2f). These results indicate that the CUMS procedure successfully increases depression-like behaviors in mice.
Next, TEM was performed to evaluate the extent of synaptic loss after CUMS exposure. We found that the synaptic density in the hippocampus of the mice in the CUMS group was lower than that in the control group (Fig. 1a, b). In addition, Golgi staining revealed that CUMS caused the loss of dendritic spines in the hippocampus (Fig. 1c, d). Morphological analysis of spines revealed that CUMS decreased the number of thin and mushroom spines (Supplementary Fig. 2g). To test whether CUMS influences synaptic plasticity, we recorded LTP induced by HFS in the Schaffer collateral-CA1 region of hippocampal slices and observed that LTP was diminished in the CUMS group (Supplementary Fig. 2h, i). WB analysis (Fig. 1e, f) revealed that the abundances of postsynaptic density protein 95 (PSD95), synapsin I, and synaptophysin were lower in the hippocampal lysates of the CUMS group than those of the control group, indicating impaired synaptic plasticity. The levels of syntaxin, vesicle-associated membrane protein 2 (VAMP2) and synaptosomal associated protein 25 (SNAP25) in the CUMS group were higher than those in the control group. Since syntaxin, VAMP2 and SNAP25 are core components of the soluble N-ethylmaleimide–sensitive factor attachment protein receptor (SNARE) complex, we also determined the levels of these proteins in the striatum and substantia nigra. The results revealed increased levels of syntaxin in the striatum and substantia nigra after CUMS treatment. There was no difference in the level of VAMP2 in the striatum or substantia nigra. The level of SNAP25 was elevated only in the striatum (Supplementary Fig. 2j, k). We further tested the effect of stress on α-syn expression. Interestingly, CUMS exposure increased the protein levels of α-syn in the hippocampus, striatum, and substantia nigra (Fig. 1g, h). Consistently, qPCR revealed that chronic stress increased the level of Snca mRNA in these brain regions (Fig. 1i). Thus, the CUMS procedure could cause depression-like phenotypes and the upregulation of α-syn.

a Representative electron microscopy images of synapses in the hippocampus. The red arrows indicate synapses. Scale bar, 1 μm. b Quantification of synapses. n = 45 fields from 9 sections from 3 mice per group. c Representative Golgi staining images of spines in the hippocampus. Scale bar, 10 μm. d Quantification of spines. n = 45 branches from 3 mice per group. e Representative immunoblot images of PSD95, synapsin I, synaptophysin, syntaxin, VAMP2 and SNAP25 and (f) quantification. n = 6 mice per group. g Representative immunoblot images of α-syn in the HIP, CPU and SN and (h) quantification. n = 6 mice per group. i Quantification of the mRNA levels of α-syn in different brain regions. n = 6 mice per group. The box-and-whisker plots depict the median, quartiles and range. Other data are shown as mean ± SEM. Student’s t-test WAS used. The P values are labeled in the diagram. CUMS chronic unpredictable mild stress.
Overexpression of α-syn induces depression-like phenotypes
To test whether α-syn plays a role in depression, WT mice were injected with AAV particles encoding the human SNCA gene in the bilateral hippocampus. Two months after injection, WB analysis confirmed that exogenous α-syn was expressed in the hippocampus (Fig. 2a, b). Compared with the control mice, the α-syn-overexpression mice consumed less sucrose (Fig. 2c). The body weight differed between control AAV-injected mice and α-syn-overexpression mice (Supplementary Fig. 3a). Compared with the control groups, the α-syn-overexpression group displayed increased immobility time in both the FST and TST (Fig. 2d, e). The EPM test results revealed a significant decrease in the time spent in the open arms and the number of entries into the open arms in the α-syn-overexpression group (Supplementary Fig. 3b). The OFT yielded similar results, with the α-syn-overexpression group exhibiting fewer central entries and fewer total distances moved, although there was no difference in the time spent in the center area among the groups (Supplementary Fig. 3c). These findings indicate that α-syn overexpression in the hippocampus leads to depression-like behaviors in the absence of stress.

a Representative immunoblot images of PSD95, synapsin I, synaptophysin, syntaxin, VAMP2 and SNAP25 and (b) quantification. n = 6 mice per group. Results of the body weight (c), FST (d) and TST (e) tests. n = 10 mice per group. f Representative electron microscopy images of synapses in the hippocampus. The red arrows indicate synapses. Scale bar, 1 μm. g Quantification of synapses. n = 45 fields from 9 sections from 3 mice per group. h Representative Golgi staining images of spines in the hippocampus. Scale bar, 10 μm. i Quantification of spines. n = 45 branches from 3 mice per group. The box-and-whisker plots depict the median, quartiles and range. Other data are shown as mean ± SEM. The date were analyzed using one-way ANOVA followed by Tukey’s post hoc test. The P values are labeled in the diagram. ns not significant, FST forced swim test, TST tail suspension test.
We further investigated whether α-syn overexpression impacts synapses. TEM analysis revealed a decrease in the synaptic density in the hippocampus of the mice in the α-syn overexpression group compared with those in the control groups (Fig. 2f, g). Additionally, the α-syn overexpression group presented a pronounced loss of dendritic spines compared with the control groups (Fig. 2h, i). Although the overexpression of α-syn significantly decreased the number of thin and mushroom spines, more stubby spines were present in the α-syn overexpression group (Supplementary Fig. 3d). Compared with that in the control group, the induction of LTP in the α-syn-overexpressing group was impaired (Supplementary Fig. 3e, f). WB analysis revealed that the levels of PSD95, synapsin I, synaptophysin, syntaxin and VAMP2 decreased, whereas the level of SNAP25 increased in the hippocampus of α-syn-overexpression mice (Fig. 2a, b). These results suggest that α-syn triggers synaptic dysfunction in the hippocampus.
Genetic deletion of α-syn attenuates depression-like phenotypes induced by CUMS
To further explore the role of α-syn in depression-like phenotypes induced by chronic stress, WT and Snca-/- mice were subjected to the CUMS paradigm. Sucrose preferences were similar in WT and Snca-/- mice before CUMS, whereas Snca-/- mice presented a lower reduction in sucrose preference than WT mice did after CUMS (Fig. 3a). Although CUMS treatment resulted in less body weight gain in both WT and Snca-/- mice, the reduction was partially attenuated in Snca-/- mice (Supplementary Fig. 4a). Compared with the WT-CUMS group, the Snca-/--CUMS group had a shorter immobility time in both the FST and TST. There was no difference in immobility time in the TST between the Snca-/--control group and the Snca-/--CUMS group (Fig. 3b, c). Knockout of α-syn also partially improved performance in the EPM and OFT tests after CUMS treatment (Supplementary Fig. 4b, c). Hence, Snca-/- mice are resistant to CUMS-induced depression-related behaviors.

Results of the body weight (a), FST (b) and TST (c) tests. n = 10 mice per group. d Representative electron microscopy images of synapses in the hippocampus. The red arrows indicate synapses. Scale bar, 1 μm. e Quantification of synapses. n = 45 fields from 9 sections from 3 mice per group. f Representative Golgi staining images of spines in the hippocampus. Scale bar, 10 μm. g Quantification of spines. n = 45 branches from 3 mice per group. h Representative immunoblot images of PSD95, synapsin I, synaptophysin, syntaxin, VAMP2 and SNAP25 and (i) quantification. n = 6 mice per group. The box-and-whisker plots depict the median, quartiles and range. Other data are shown as mean ± SEM. The data were analyzed by two-way ANOVA followed by Tukey’s post hoc test. The P values are labeled in the diagram. ns not significant, FST forced swim test, TST tail suspension test.
In addition, TEM analysis revealed that the Snca-/--CUMS mice exhibited less synaptic loss than the WT-CUMS mice did (Fig. 3d, e). We also found that the loss of dendritic spines was less severe in Snca-/--CUMS mice than in WT-CUMS mice (Fig. 3f, g). The detrimental effects of CUMS treatment on thin and mushroom spines were alleviated in Snca-/--CUMS mice (Supplementary Fig. 4d). Knockout of α-syn partially attenuated the impairment of LTP caused by CUMS (Supplementary Fig. 4e, f). Consistently, the knockdown of α-syn also attenuated the decreases in PSD95, synapsin I and synaptophysin and the increases in syntaxin, VAMP2 and SNAP25 (Fig. 3h, i). These results indicate that the knockout of α-syn partially alleviates the synaptic impairments caused by chronic stress.
CUMS exacerbates α-syn pathology in A53T mice
Given that α-syn mediates depression-related changes caused by chronic stress, we next determined whether chronic stress affects α-syn deposition in α-syn A53T transgenic mice. These mice express human A53T mutant α-syn and are used as a mouse model of synucleinopathy. After 4 weeks of CUMS exposure, we found that sucrose preferences were decreased in both WT and A53T mice subjected to CUMS, and this effect was more prominent in A53T mice (Supplementary Fig. 5a). Consistently, CUMS treatment impaired body weight gain in both WT and A53T mice, with A53T-CUMS mice gaining the lowest body weight among the groups (Supplementary Fig. 5b). Compared with WT-CUMS mice, A53T-CUMS mice presented increased immobility time in both the FST and TST (Supplementary Fig. 5c, d). Both WT and A53T mice exposed to CUMS exhibited worse performance in the EPM and OFT tests. There was no difference between WT-CUMS mice and A53T-CUMS mice (Supplementary Fig. 5e, f).
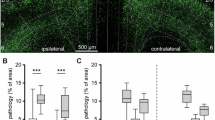
We further tested whether CUMS accelerates the pathological aggregation of α-syn. Five months after CUMS treatment, no phosphorylated α-syn (Ser129) was detected in any of the tested areas in WT mice with or without CUMS (Fig. 4a, b). Immunostaining revealed weak phosphorylated α-syn signals, which appeared as dots and neurites, in unstressed A53T mice. CUMS exposure augmented the formation of neuronal phosphorylated α-syn in A53T mice. Obvious LB-like aggregates were detected in the striatum, hippocampus, amygdala and substantia nigra (Fig. 4a, b). We also applied anti-aggregated α-syn (5G4) antibody and anti-misfolded α-syn (Syn303) antibody. Both the 5G4 antibody and Syn303 antibody detected no signal in WT-control and WT-CUMS mice, while more cytoplasmic staining was found in A53T-CUMS mice than in A53T-control mice (Supplementary Fig. 6a–d). These results were validated by WB analysis. There was no difference in the level of total α-syn in the soluble fraction between the control and CUMS groups in either genotype. No α-syn was detected in the insoluble fraction of WT mice with or without CUMS. Compared with A53T-control mice, A53T-CUMS mice presented higher levels of α-syn in the insoluble fraction, as detected by total α-syn antibodies, phosphorylated α-syn antibodies and 5G4 antibodies (Fig. 4c, d).

a Representative immunohistochemical results of phosphorylated α-syn from WT and A53T mice with and without CUMS treatment in the striatum, hippocampus, amygdala and substantia nigra and quantification b. n = 6 mice per group. Scale bar, 20 μm. c Representative immunoblot images of α-syn, phosphorylated α-syn, aggregated α-syn (5G4) and misfolded α-syn (Syn303) in the soluble and insoluble fractions of the striatum of WT and A53T mice with and without CUMS treatment and quantification d. n = 6 mice per group. Data are shown as mean ± SEM. The data were analyzed by two-way ANOVA followed by Tukey’s post hoc test. The P values are labeled in the diagram. ns not significant, CUMS chronic unpredictable mild stress.
Finally, we investigated degeneration of the nigrostriatal system and motor function in WT and A53T mice after CUMS. The surviving dopaminergic cells in the substantia nigra were quantified by stereological counts after immunostaining with tyrosine hydroxylase (TH), a marker of dopaminergic neurons. The number of TH-positive neurons in the substantia nigra and the density of TH-positive fibers in the striatum were not altered in WT-control mice, WT-CUMS mice, or A53T-control mice. However, after CUMS, extensive loss of dopaminergic neurons and dopaminergic fibers was detected in A53T mice (Fig. 5a–d). The rotarod test and pole test were used to assess the effects of CUMS on locomotor activity. In both tests, no obvious motor impairment was detected in WT-control or A53T-control mice, suggesting normal motor ability in eight-month-old A53T mice (Fig. 5e, f). After exposure to CUMS, A53T mice exhibited impaired motor performance (Fig. 5e, f). In summary, these results indicate that CUMS accelerates the onset of PD-like pathology and motor symptoms in A53T mice.

a Representative images of SNpc TH-positive neurons and (b) quantification. Scale bar, 100 μm. n = 6 mice per group. c Representative images of striatum TH-positive fibers and (d) quantification. Scale bar, 200 μm. n = 6 mice per group. Performance in the (e) rotarod test and (f) pole test. n = 10 mice per group. Data are shown as mean ± SEM. The data were analyzed by two-way ANOVA followed by Tukey’s post hoc test. The P values are labeled in the diagram. ns not significant, SNpc substantia nigra pars compacta, AOD average optical density.
CUMS induces α-syn deposition in aged WT mice
Finally, we tested whether WT mice subjected to CUMS could spontaneously develop α-syn pathology with aging. WT mice were subjected to CUMS at 2 months of age. α-Syn deposition was detected by immunohistochemistry at 15 months of age. Interestingly, phosphorylated α-syn was detected in the striatum, hippocampus and substantia nigra in nine out of the fifteen WT-CUMS mice, whereas no phosphorylated α-syn signaling was detected in the WT-control mice (Fig. 6a). In these nine mice, phosphorylated α-syn staining was mild. WB analysis revealed that no pathological α-syn was detected by the phosphorylated α-syn antibody, 5G4 antibody or Syn303 antibody in WT-control mice, whereas pathological α-syn was detected in WT-CUMS mice (Fig. 6b). Double immunofluorescence staining revealed that phosphorylated α-syn colocalized with p62 in the striatum and substantia nigra of WT-CUMS mice at 15 months of age (Fig. 6c).

WT mice were subjected to 4-week CUMS treatment at 2 months of age. a Representative immunohistochemical results of phosphorylated α-syn in the striatum, hippocampus and substantia nigra at 15 months of age. Scale bar, 20 μm. b Representative immunoblot images of α-syn, phosphorylated α-syn, aggregated α-syn (5G4) and misfolded α-syn (Syn303) in the soluble and insoluble fractions of the striatum. c Representative images showing phosphorylated α-syn (red), P62 (green) and DAPI (blue) staining in the striatum and substantia nigra. Scale bar, 10 μm. CUMS chronic unpredictable mild stress.
Discussion
In the present study, we explored the role of α-syn in depression induced by chronic stress. The α-syn mRNA and protein levels were elevated in multiple brain regions of stressed mice. The change in α-syn in stressed mice is similar to that observed in patients with depression19,20. Several studies have shown that Lewy bodies in the hippocampus are associated with cognitive impairment in PD patients21,22. According to Braak staging theory, the hippocampus is affected by α-syn pathology at neuropathological stage IV23. However, different PD patients present distinct patterns of α-syn pathology11.
Next, we determined the effects of α-syn overexpression on the development of depression. To avoid the effects of motor deficits, which may be induced by α-syn overexpression in the striatum or substantia nigra, on performance in depression-related tests, we chose to overexpress α-syn in the hippocampus24. We found that hippocampal α-syn overexpression caused depression-like behaviors and synaptic loss at 8 weeks after AAV injection. These results are supported by two other studies showing that the overexpression of α-syn (one in raphe serotonin neurons and the other in midbrain dopaminergic neurons) also triggers depressive-like phenotypes at 8 weeks after AAV injection25,26. This time pattern is different from that of CUMS treatment, since a 4-week period results in minimal heterogeneity in C57BL/6 J mice27.
In addition, α-syn can regulate the fusion and clustering of synaptic vesicles through VAMP2, which ultimately leads to the recruitment of SNAP-25 and syntaxin28. Interestingly, our results demonstrated that the levels of VAMP2, SNAP25 and syntaxin increased in the hippocampus after chronic stress, whereas α-syn overexpression resulted in increased levels of SNAP25. The increased levels of these three proteins are associated with glutamate neurotransmission, which is one of the core features of stress-related mental disorders29,30,31. Antidepressant drugs reduce hippocampal glutamate by inhibiting the levels of syntaxin, SNAP25 and VAMP232,33.
Excessive accumulation of α-syn in neurons is toxic and may be involved in the complex pathological mechanisms of depression. First, α-syn may regulate the synthesis of monoamine neurotransmitters34. The overexpression of α-syn disrupts the function of vesicular monoamine transporter 2 to affect neurotransmitter storage35. α-Syn also modulates the trafficking of monoamine transporters to the cell surface, thus influencing neurotransmitter reuptake36,37,38. A recent study reports brain area-dependent changes in α-syn, β-syn, and γ-syn. An imbalance in the levels of synucleins may affect serotonin transporter levels and function in patients with depression39. In addition, hypothalamic pituitary adrenal axis dysregulation is detected in transgenic rats overexpressing α-syn40. Moreover, α-syn has been reported to regulate neuroinflammation and the complement system41,42, which contributes to depression43,44.
Here, we showed that α-syn overexpression caused a decrease in dendritic spine density, which is consistent with previous results45,46. However, reports indicate that α-syn overexpression in the prefrontal cortex leads to increased dendritic spine density47,48. The changes in spines secondary to α-syn may be partly attributed to cell vulnerability. In addition, two studies reported the detrimental effects of α-syn preformed fibrils on the dendritic spines in layer V of the somatosensory cortex and on striatal spiny projection neurons49,50.
Additionally, our results revealed that the depression-like phenotypes induced by chronic stress in Snca-/- mice were not as apparent as those in WT mice. The potential of targeting α-syn in the treatment of depression needs further evaluation. Two independent clinical studies have shown that the efficacy of tricyclic antidepressants (desipramine and nortriptyline) is more favorable than that of selective serotonin reuptake inhibitors in managing depression in PD patients51,52. Nortriptyline directly binds to soluble α-syn to inhibit its pathological aggregation53. Desipramine affects the expression of α-syn38,54, whereas fluoxetine fails to do so55. It has also been reported that, in an A53T mouse model, α-syn affects serotonergic projections in a space-dependent pattern within the hippocampus, thereby influencing the response to fluoxetine treatment56.
Given that α-syn plays a vital role in both PD and depression, it would be interesting to dissect the relationship between these two clinical entities. Depression may occur prior to PD symptoms and indicate a worse prognosis for PD patients57,58. Chronic stress and stress hormones can affect the function of the central nervous system by affecting transcriptional regulation, cellular signaling, neurotransmission release and so on59. Previous studies have reported that immobilization stress leads to increased dopamine levels and oxidative damage to the nigrostriatal system60 and that chronic restraint stress promotes dopaminergic neuronal degeneration via microglial activation and oxidative stress61. However, only extremely severe and prolonged stress can lead to permanent dopaminergic neuronal death.
Furthermore, several studies have shown that chronic stress can exaggerate neural degeneration and compromise motor function in neurotoxin-induced PD models62,63,64. Considering that neurotoxic models rarely reflect age-dependent neurodegeneration, we used A53T mice to examine neuropathological and behavioral changes at 5 months after CUMS. This model is a late-onset transgenic model of PD. We did not find loss of dopaminergic neurons in 8-month-old A53T mice without CUMS. This finding is consistent with previous reports65,66,67,68. Moreover, chronic stress exacerbated pathological α-syn aggregation, the depletion of TH-positive neurons and motor impairments in A53T mice. Other studies reported similar results in transgenic PD mouse models40,69. Chronic restraint stress exacerbated PD-like motor disorders in A53T mice, whereas chronic corticosterone administration aggravated nigrostriatal degeneration and phosphorylated α-syn in transgenic rats overexpressing α-syn. In an α-syn fibril model, chronic corticosterone administration was also associated with increased dopaminergic cell loss and aggravated α-syn pathology70. In addition, we found few phosphorylated α-syn and P62 double-positive aggregates in some 15-month-old WT mice that were exposed to chronic stress at 2 months of age, which is supported by Ren’s report that phosphorylated α-syn inclusions were detected in aged rats subjected to maternal separation71. Nevertheless, future studies are warranted to determine the effects of stress/depression on pathological α-syn in nontransgenic mice. Together, these results indicate a connection between stress and PD and the role of α-syn as a potential focal point of these two diseases. Together, the findings of the present study highlight the role of α-syn in mediating depressive phenotypes and connecting depression and Parkinsonism.
There are limitations in this study. We used only male mice for two reasons. First, experiments on rodent models have shown that there is sex difference in the onset of depression. Many studies use male rodents because more depression-like behaviors are observed in males than in females72,73. Second, it has been demonstrated that estrogen exerts neuroprotective effects against PD74. Female rodents exhibit fluctuating estrogen levels75,76, which may affect the experimental results. Notably, the prevalence of depression is greater in females than in males, and women with PD are more vulnerable to depressive symptoms than men with PD77,78. It would be helpful to include female rodents in future research.
Methods
Animals
Wild-type (WT) C57BL/6 J mice (strain #000664), homozygous human A53T mutant α-syn transgenic mice (B6;C3-Tg(Prnp-SNCA*A53T)83Vle/J, A53T mice, strain #004479) and homozygous Snca knockdown (Snca-/-) mice (B6;129×1-Sncatm1Rosl/J, strain #003692) were obtained from Jackson Laboratory65,79. The mice were maintained in a specific pathogen-free environment with suitable humidity (50–60%) and temperature (23 °C, daily temperature difference <3 °C) on a 14–10-h light–dark cycle (lights on at 07:00) and were fed commercial rodent food and sterilized water ad libitum. The sample size was determined via Power and Precision software (GPower 3.1)80. The protocol was reviewed and approved by the Animal Care and Use Committee of Renmin Hospital of Wuhan University, with ethical approval number 20220803. Only male mice of 8 to 9 weeks were used in this study.
CUMS procedure
The CUMS procedure was adopted from previous studies81,82. In brief, the mice were exposed to randomized mild stressors once a day for 4 weeks, including food deprivation (24 h), water deprivation (24 h), cage tilt (30°, 24 h), reversed light/dark cycle, overnight illumination, restraint stress (2 h), soiled cage (24 h), cage shaking (40 r.p.m, 5 min), tail pinch (1 min) and forced swimming (4 °C, 5 min). The same stressor was not applied in succession. The control mice were held under the same conditions without the stressors and were only switched to individual housing during the behavioral test phase.
Viral construction and stereotaxic injection
Adeno-associated virus (AAV) particles encoding human SNCA with the human synapsin I (hSyn1) promoter (AAV2/9-hSyn1-hSNCA, 1.2 × 1012 vector genome/ml) and EGFP control (AAV2/9-hSyn1-EGFP, 1.3 × 1012 vector genome/ml) were prepared by BrainVTA (BrainVTA Co., Ltd.). The mice were anesthetized via the intraperitoneal injection of ketamine (100 mg/kg) and xylazine (10 mg/kg) in saline, and the heads were fixed in a stereotaxic frame (RWD). The stereotaxical injection of AAVs was performed bilaterally into the hippocampus at the coordinates anteroposterior −2.5 mm and mediolateral ±2 mm relative to the bregma and dorsoventral 1.8 mm from the dural surface. A total of 300 nl of viral suspension containing 4 × 109 vector genomes was injected into each site with a 10 μl glass syringe (Hamilton) with custom glass needle syringes (50 nl/min). The needle was left in place for an additional 5 min before being withdrawn. After surgery, the mice were subcutaneously injected with ketoprofen (2 mg/kg) and placed on a heating pad for recovery. Two months later, depressive-like behavioral tests were performed.
Transmission electron microscopy
Tissue preparation for transmission electron microscopy (TEM) was performed according to a previous report83. First, the mice were anesthetized and perfused transcardially with precooled fixation buffer (G1124, Servicebio). Then, the hippocampal tissues were separated and postfixed in fixation buffer at 4 °C for 2 h. After fixation, the samples were washed with phosphate-buffered saline (PBS), postfixed with 1% osmium tetroxide for 1.5 h, again washed in PBS, dehydrated in an acetone series, and then embedded in epoxy resin. For ultrathin sections, the cut surface was first trimmed, and semithin sections were obtained at a thickness of 1.5 μm, stained with 1% toluidine blue, and examined via light microscopy. After the desired area was determined, ultrathin sections (~70 nm thick) were cut with an ultramicrotome (EM UC7, Leica) and picked up on 100 mesh copper grids. The sections were stained with 2% uranyl acetate for 3 min and then with 0.5% lead citrate for 5 min. Finally, the samples were viewed via transmission electron microscopy (1400plus, JEOL). Micrographs were taken at 20,000× magnification.
Quantitative analyses of synaptic density were performed by investigators who were blinded to the treatment conditions84. For each of three animals from the experimental groups, three samples of the hippocampus were obtained for the preparation of embedding blocks, totaling n = 9 blocks per group. Synapses were quantified in areas totaling ~3000 μm2 for each experimental group. Synapses were identified by the presence of synaptic vesicles and postsynaptic densities.
Golgi staining
Golgi staining was performed via the FD Rapid GolgiStai Kit (PK401, FD NeuroTechnologies) according to the manufacturer’s instructions. The brain tissue was removed from the skull quickly and immersed in impregnation solution at room temperature under dark conditions for 14 days (the impregnation solution was replaced on the second day). Then, the brain was sectioned serially into 150-μm thick coronal sections via a vibratome (VT1200S, Leica), and the slices were mounted on gelatin-coated slides. All the sections were placed in the staining solution for 20 min and rinsed with double distilled water. The sections were placed in sequential solutions of 50%, 75%, and 95% ethanol, then in 100% ethanol 4 times, and finally in xylene 3 times. The sections were subsequently mounted with cover glasses. The dendritic spines in the cornu ammonis 1 (CA1) and CA3 regions were evaluated morphologically via light microscopy (Eclipse ci, Nikon). The density of the dendritic spines was analyzed with ImageJ software (v.1.54, US National Institutes of Health).
The selected pyramidal neurons in the CA1 and CA3 regions fulfilled the following criteria: (1) the dendrites were dark and had consistent Golgi staining across their entire length, (2) the dendrites were visibly in-tact, and (3) each neuron had enough space between them to prevent interference during the analysis85,86. To count the dendritic spines, straight branches that provided a clear resolution of spines and were longer than 15 μm were preferred. The spines are defined as thin if the length is greater than the diameter of the neck with the same diameter. The mushroom type is defined as when the diameter of the head of the spine is greater than that of the neck. Spines are considered stubby if the length and width of a spine are equal. We analyzed three mice in each group, five neurons were randomly selected per hemisphere and fifteen branches per mouse were included for statistical analysis.
Electrophysiology
The protocols were adapted from previous literature87,88. After the mice were anesthetized by isoflurane, the brains were carefully removed and placed in ice-cold oxygenated sucrose slicing buffer for approximately 1 min. Appropriate portions of the brain were trimmed away, and the brain was then glued to an ice-cold stage and placed in a vibrating blade microtome (VT1200S, Leica Biosystems) filled with chilled oxygenated sucrose slicing solution. The coronal 300 μm hippocampal slices were collected and transferred to a recovery chamber (maintained at 32 °C) containing oxygenated artificial cerebrospinal fluid (aCSF) for 60 min. The recipes of sucrose slicing solution and aCSF are listed in Supplementary Tables 1 and 2. For recording, the slices were transferred to an 8 × 8 array of planar microelectrodes, each 50 × 50 μm in size, with an interpolar distance of 150 μm (MED-P515A, Alpha MED Scientific Inc.) and carefully positioned to ensure that CA1 and CA3 were within the array. The array chamber was perfused with oxygenated aCSF at a rate of 3 ml/min at 32 °C.
Field excitatory postsynaptic potentials (fEPSPs) were recorded in the CA1 region by stimulating electrodes in the CA1 and CA3 regions along the Schaffer collateral pathway with the MED64 system (Alpha MED Scientific, Inc.). Input–output (I/O) curves were obtained by giving stimuli with increasing amplitudes from 10 to 100 μA. The stimulus intensity for further recording was determined as the strength needed to generate 40% of the maximum fEPSP amplitude during the I/O curve measurement. The slice was stimulated once every 30 s until a stable baseline was reached for at least 20 min. Long-term potentiation (LTP) was induced through 3 traces of high-frequency stimulation (HFS) pulses at 100 Hz with 1 s intervals. After that, the baseline stimulation was resumed, and fEPSP amplitudes were recorded for at least 60 min. LTP magnitude was quantified as the percentage change in the mean fEPSP amplitude (45–60 min) after HFS and normalized to the mean fEPSPs amplitude of baseline recordings 10 min prior to tetanus. The data were filtered at 1 kHz, digitized at 20 kHz, and analyzed with Mobius software (Alpha MED Scientific Inc.).
Western blots
For general procedure, brain tissues were lysed in precooled radioimmunoprecipitation assay buffer (P0013B, Beyotime) containing protease and phosphatase inhibitor cocktail (78442, Thermo Fisher Scientific) in a grinder (KZ-III-FP, Servicebio) for 5 min at −10 °C. The lysates were then sonicated briefly on ice and centrifuged at 15,000 r.p.m. for 20 min at 4 °C. The protein concentrations of the supernatants were determined with a Pierce BCA protein assay kit (23227, Thermo Fisher Scientific). For sequential extraction, the tissues were homogenized into soluble and insoluble fractions with TBS+ (50 mM Tris-HCl (pH 7.4), 175 mM NaCl, 5 mM EDTA) and a protease inhibitor mixture. After ultracentrifugation at 120,000 g at 4 °C for 30 min, the supernatants representing soluble fractions were collected, and the pellets were further rinsed with TBS+ containing 1% Triton X-100, TBS+ containing 1 M sucrose and radioimmunoprecipitation assay buffer, followed by ultracentrifugation at 120,000 g at 4 °C for 20 min. The final pellets representing Triton X-100 insoluble fractions were solubilized in 8 M urea/5% SDS.
The proteins were electrophoresed on 10% polyacrylamide gel and transferred to nitrocellulose membranes (1620097, Bio-Rad). After incubation with 5% nonfat milk in Tris-buffered saline containing 0.1% Tween-20 for 1 h at room temperature, the membranes were incubated with primary antibodies overnight at 4 °C. The membranes were washed 5 times, incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature and visualized with enhanced chemiluminescence (1705061, Bio-Rad). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal reference. The commercially available antibodies used are listed in Supplementary Table 3. Unprocessed western blot (WB) images are provided in the source data.
Quantitative real-time PCR
The total RNA of the mouse brain was extracted via TRIzol reagent (RK30129, ABclonal). The concentration and quality of the RNA were determined via spectrophotometer measurements at 260 and 280 nm absorbance wavelengths, with values between 1.8 and 2.0 considered to be used. Next, 1 µg of total RNA from each sample was reverse transcribed to cDNA via ABScript III RT Master Mix (RK20428, ABclonal). Quantitative real-time PCR (qPCR) was performed with 2X Universal SYBR Green Fast qPCR Mix (RK21203, ABclonal) and the CFX96TM Real-Time PCR Detection system (Bio-Rad). All genes were analyzed in triplicate. The primers used are listed in Supplementary Table 4. The comparative Ct (2-ΔΔCt) method was used to quantify the expression of genes. Gapdh was used as an internal control.
Immunohistochemistry and immunofluorescence
The mice were anesthetized and transcardially perfused with ice-cold PBS and 4% paraformaldehyde (PFA). The brains were incubated in 4% PFA for 48 h, embedded in paraffin and cut into 4-μm-thick sections. After deparaffinization, the sections were subjected to heat-induced antigen retrieval in 10 mM citrate buffer for 20 min. For immunohistochemistry, the sections were treated with peroxidase blocking solution and 3% bovine serum albumin (BSA) blocking buffer. The sections were subsequently incubated with primary antibodies overnight at 4 °C. After rinsing with PBS, the sections were incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature and stained with 3,3’-diaminobenzidine. For immunofluorescence, the sections were treated with 3% BSA blocking buffer, followed by incubation with primary antibodies overnight at 4 °C. Fluorochrome-conjugated secondary antibodies and 4’,6-diamidino-2-phenylindole (DAPI) were used. For stereological quantification of tyrosine hydroxylase (TH)-positive cells, fixed brains were washed with PBS, cryoprotected with 30% sucrose and embedded in Tissue-Plus OCT compound (Thermo Fisher Scientific). Frozen brains were sliced (25-μm-thick sections) with a cryostat (Leica) and kept in PBS at 4 °C. A set of eight sections for TH immunohistochemistry was obtained from every fourth 25-μm-thick section through the entire SNpc. MicroBrightField stereological software (StereoInvestigator v.9.14, MBF Bioscience) was used to assess the cell counts. The commercially available antibodies used are listed in Supplementary Table 3.
Behavioral analysis
All tests were executed by an expert blinded to group assignment between 09:00 and 15:00. The mice were placed in the test room 1 h before the tests, and the equipment was cleaned with 70% ethanol between the animals to minimize odor interference.
Sucrose preference test
The sucrose preference test (SPT) was used to assess anhedonia, which is considered a key feature of depression, and was performed before and after the administration of CUMS89,90. In brief, the mice were first individually housed and then habituated to two bottles filled with tap water for 2 days, followed by two bottles filled with 1% sucrose solution (w/v) for 2 days. After that, the water was deprived for 24 h. Finally, the mice were given ad libitum access to two bottles, one containing 1% sucrose solution and the other containing tap water, for 24 h. The position of the bottles was switched after 12 h to avoid the side preference of the mice in drinking behavior. The consumption of the sucrose solution and water was then recorded, and the percentage preference for sucrose consumption was calculated via the following formula: sucrose preference (%) = (sucrose solution consumption/total liquid intake) × 100%. A lower sucrose preference indicates depression-like behavior.
Tail suspension test
The tail suspension test (TST) was conducted as previously reported91,92. In brief, each mouse was individually suspended by the tail from a rod fixed 20 cm above the ground with adhesive tape for 6 min. A white background was used. The mice were filmed with a high-definition video camera for 6 min, and the immobility time of the mice during the last 4 min was recorded. Small movements confined to only the front limbs and oscillations and pendulums that followed previous mobility were not considered mobility.
Forced swim test
The forced swim test (FST) was adapted from previous reports93. Briefly, the mice were individually placed into a glass cylinder (35 cm in height and 20 cm in diameter) filled with 15 cm of warm water (25 °C). The mice were forced to swim for 6 min, and the process was complete. After each trial, the water was renewed, and the mice were treated with drying paper, heat lamps and heat pads. The immobility time during the final 4 min was assessed. Immobility was defined as floating without any movement except for those necessary for keeping the head above the water.
Open-field test
General locomotor activity and anxiety were measured via the open-field test (OFT) according to previously described methods94,95. The ground area of a rectangular chamber (50 × 50 × 50 cm) was divided into two parts: a 25 × 25 cm central zone and the surrounding zone. The mice were gently placed in the corner and permitted unrestricted exploration for 5 min. A video camera was fixed over the center of the apparatus, and movements were digitally tracked via the ANY-maze Video Tracking System (Stoelting Co.). The time spent in the central zone, the number of entries into the central zone, and the total distance traveled in the whole field were calculated for each mouse via ANY-maze behavioral analysis software (v.4.3, Stoelting Co.).
Elevated plus maze
The anxiety-like behavior of the mice was also evaluated through the elevated plus maze (EPM) test96. The apparatus consists of two open arms (30 × 5 × 0.5 cm), two closed arms (30 × 5 × 15 cm), and a central zone (5 × 5 cm), elevated 50 cm above the ground. At the beginning of each test, each mouse was gently placed in the central zone, facing an open arm. The movements of the mice during a 5 min trial were tracked by the ANY-maze Video Tracking System (Stoelting Co.). Arm entry was defined as the center point of the mouse across the border of the arm. The number of entries into each arm and the amount of time spent in each arm were recorded via ANY-maze behavioral analysis software (v.4.3, Stoelting Co.).
Rotarod test
The mice were placed on a spinning rod with a gradually accelerating spinning rate from 5 to 40 r.p.m. within 3 min. The mice were trained 3 times a day for 3 consecutive days before being subjected to the formal experiment. A 30-min break was given between trial intervals. The latency time to fall off was documented, and the time longer than 180 s was recorded as 180 s.
Pole test
The pole was made up of a long wooden rod (75 cm long, 1 cm diameter) that was wrapped with bandage gauze. The mice were placed head-upward on the top of the pole and waited for autonomous descent. The formal test started after the mice were subjected to three trials for training. The total time taken to reach the base was recorded.
Statistical analysis
GraphPad Prism 10.0 software (GraphPad Software) was used to perform the statistical analysis. Normal tests and homogeneity tests of variance for all continuous variables were performed before the analysis. All data were presented as mean ± standard error of the mean (SEM) unless otherwise stated. Differences between two groups were established via an unpaired two-sided Student’s t-test. One-way analysis of variance (ANOVA) or two-way ANOVA followed by Tukey’s post hoc test was applied for multiple-group comparisons. Fisher’s exact test was used for count data. P < 0.05 was considered statistically significant. The P values are labeled in the diagram. The test statistics can be found in the Source Data.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available on request from the corresponding author.
References
Tanner, C. M. & Ostrem, J. L. Parkinson’s Disease. N. Engl. J. Med. 391, 442–452 (2024).
Tolosa, E., Garrido, A., Scholz, S. W. & Poewe, W. Challenges in the diagnosis of Parkinson’s disease. Lancet Neurol. 20, 385–397 (2021).
Schapira, A. H. V., Chaudhuri, K. R. & Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 18, 509 (2017).
Weintraub, D. et al. The neuropsychiatry of Parkinson’s disease: advances and challenges. Lancet Neurol. 21, 89–102 (2022).
Galts, C. P. C. et al. Depression in neurodegenerative diseases: common mechanisms and current treatment options. Neurosci. Biobehav. Rev. 102, 56–84 (2019).
Assogna, F. et al. Drug choices and advancements for managing depression in Parkinson’s disease. Curr. Neuropharmacol. 18, 277–287 (2020).
Schuurman, A. G. et al. Increased risk of Parkinson’s disease after depression: a retrospective cohort study. Neurology 58, 1501–1504 (2002).
Shen, C.-C., Tsai, S.-J., Perng, C.-L., Kuo, B. I.-T. & Yang, A. C. Risk of Parkinson disease after depression: a nationwide population-based study. Neurology 81, 1538–1544 (2013).
Gustafsson, H., Nordström, A. & Nordström, P. Depression and subsequent risk of Parkinson disease: a nationwide cohort study. Neurology 84, 2422–2429 (2015).
Burré, J., Sharma, M. & Südhof, T. C. Cell biology and pathophysiology of α-Synuclein. Cold Spring Harb. Perspect. Med. 8, a024091 (2018).
Morris, H. R., Spillantini, M. G., Sue, C. M. & Williams-Gray, C. H. The pathogenesis of Parkinson’s disease. Lancet Lond. Engl. 403, 293–304 (2024).
Sahay, S., Ghosh, D., Singh, P. K. & Maji, S. K. Alteration of structure and aggregation of α-Synuclein by familial Parkinson’s disease associated mutations. Curr. Protein Pept. Sci. 18, 656–676 (2017).
Alefanti, I. et al. The novel p.A30G SNCA pathogenic variant in Greek patients with familial and sporadic Parkinson’s disease. Eur. J. Neurol. 32, e16562 (2025).
Park, K. W. et al. Ethnicity- and sex-specific genome wide association study on Parkinson’s disease. NPJ Park. Dis. 9, 141 (2023).
Ishiguro, M. et al. Increased serum levels of α-Synuclein in patients with major depressive disorder. Am. J. Geriatr. Psychiatry.27, 280–286 (2019).
Rotter, A. et al. Alpha-Synuclein RNA expression is increased in major depression. Int. J. Mol. Sci. 20, 2029 (2019).
Wilson, R. S. et al. Brainstem aminergic nuclei and late-life depressive symptoms. JAMA Psychiatry 70, 1320–1328 (2013).
Tsopelas, C. et al. Neuropathological correlates of late-life depression in older people. Br. J. Psychiatry J. Ment. Sci. 198, 109–114 (2011).
Frieling, H. et al. Alpha-synuclein mRNA levels correspond to beck depression inventory scores in females with eating disorders. Neuropsychobiology 58, 48–52 (2008).
Brazdis, R.-M. et al. Peripheral Upregulation of Parkinson’s Disease-Associated Genes Encoding α-Synuclein, β-Glucocerebrosidase, and Ceramide Glucosyltransferase in Major Depression. Int. J. Mol. Sci. 25, 3219 (2024).
Harding, A. J. & Halliday, G. M. Cortical Lewy body pathology in the diagnosis of dementia. Acta Neuropathol.102, 355–363 (2001).
Armstrong, R. A. et al. A quantitative study of α-synuclein pathology in fifteen cases of dementia associated with Parkinson disease. J. Neural Transm.121, 171–181 (2014).
Braak, H. et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211 (2003).
Huntington, T. E. & Srinivasan, R. Adeno-associated virus expression of α-Synuclein as a tool to model Parkinson’s disease: current understanding and knowledge gaps. Aging Dis. 12, 1120–1137 (2021).
Miquel-Rio, L. et al. Human α-synuclein overexpression in mouse serotonin neurons triggers a depressive-like phenotype. Rescue by oligonucleotide therapy. Transl. Psychiatry 12, 79 (2022).
Caudal, D., Alvarsson, A., Björklund, A. & Svenningsson, P. Depressive-like phenotype induced by AAV-mediated overexpression of human α-synuclein in midbrain dopaminergic neurons. Exp. Neurol. 273, 243–252 (2015).
Antoniuk, S., Bijata, M., Ponimaskin, E. & Wlodarczyk, J. Chronic unpredictable mild stress for modeling depression in rodents: Meta-analysis of model reliability. Neurosci. Biobehav. Rev. 99, 101–116 (2019).
Burré, J. et al. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667 (2010).
Müller, H. K., Wegener, G., Popoli, M. & Elfving, B. Differential expression of synaptic proteins after chronic restraint stress in rat prefrontal cortex and hippocampus. Brain Res. 1385, 26–37 (2011).
Cao, Y. J., Wang, Q., Zheng, X. X., Cheng, Y. & Zhang, Y. Involvement of SNARE complex in the hippocampus and prefrontal cortex of offspring with depression induced by prenatal stress. J. Affect. Disord. 235, 374–383 (2018).
Fan, L. et al. Soluble N-ethylmaleimide-sensitive Factor Attachment Receptor (SNARE) protein involved in the remission of depression by acupuncture in rats. J. Acupunct. Meridian Stud. 9, 242–249 (2016).
Bonanno, G. et al. Chronic antidepressants reduce depolarization-evoked glutamate release and protein interactions favoring formation of SNARE complex in hippocampus. J. Neurosci.25, 3270–3279 (2005).
Li, Y. C. et al. Paeoniflorin ameliorates depressive-like behavior in prenatally stressed offspring by restoring the HPA axis- and glucocorticoid receptor- associated dysfunction. J. Affect. Disord. 274, 471–481 (2020).
Perez, R. G. et al. A role for alpha-synuclein in the regulation of dopamine biosynthesis. J. Neurosci.22, 3090–3099 (2002).
Surguchov, A. Molecular and cellular biology of synucleins. Int. Rev. Cell Mol. Biol. 270, 225–317 (2008).
Sidhu, A., Wersinger, C. & Vernier, P. alpha-Synuclein regulation of the dopaminergic transporter: a possible role in the pathogenesis of Parkinson’s disease. FEBS Lett. 565, 1–5 (2004).
Wersinger, C., Rusnak, M. & Sidhu, A. Modulation of the trafficking of the human serotonin transporter by human alpha-synuclein. Eur. J. Neurosci. 24, 55–64 (2006).
Jeannotte, A. M., McCarthy, J. G., Redei, E. E. & Sidhu, A. Desipramine modulation of alpha-, gamma-synuclein, and the norepinephrine transporter in an animal model of depression. Neuropsychopharmacology 34, 987–998 (2009).
Sarriés-Serrano, U. et al. Impaired unfolded protein response, BDNF and synuclein markers in postmortem dorsolateral prefrontal cortex and caudate nucleus of patients with depression and Parkinson’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 138, 111299 (2025).
Nakos Bimpos, M. et al. Alpha-synuclein-induced stress sensitivity renders the Parkinson’s disease brain susceptible to neurodegeneration. Acta Neuropathol. Commun. 12, 100 (2024).
Hentrich, T., Wassouf, Z., Riess, O. & Schulze-Hentrich, J. M. SNCA overexpression disturbs hippocampal gene expression trajectories in midlife. Aging 10, 4024–4041 (2018).
Gregersen, E. et al. Alpha-synuclein activates the classical complement pathway and mediates complement-dependent cell toxicity. J. Neuroinflamm.18, 177 (2021).
Yao, Q. & Li, Y. Increased serum levels of complement C1q in major depressive disorder. J. Psychosom. Res. 133, 110105 (2020).
Crider, A. et al. Complement component 3a receptor deficiency attenuates chronic stress-induced monocyte infiltration and depressive-like behavior. Brain. Behav. Immun. 70, 246–256 (2018).
Winner, B. et al. Role of α-synuclein in adult neurogenesis and neuronal maturation in the dentate gyrus. J. Neurosci.32, 16906–16916 (2012).
Oliveira da Silva, M. I. et al. α-Synuclein triggers cofilin pathology and dendritic spine impairment via a PrPC-CCR5 dependent pathway. Cell Death Dis. 15, 264 (2024).
Bosch, P. J. et al. Enhanced spine stability and survival lead to increases in dendritic spine density as an early response to local alpha-synuclein overexpression in mouse prefrontal cortex. Cell. Mol. Neurobiol. 44, 42 (2024).
Wagner, L. M., Nathwani, S. M., Ten Eyck, P. P. & Aldridge, G. M. Local cortical overexpression of human wild-type alpha-synuclein leads to increased dendritic spine density in mouse. Neurosci. Lett. 733, 135051 (2020).
Pérez-Acuña, D., Shin, S. J., Rhee, K. H., Kim, S. J. & Lee, S.-J. α-Synuclein propagation leads to synaptic abnormalities in the cortex through microglial synapse phagocytosis. Mol. Brain 16, 72 (2023).
Marino, G. et al. Intensive exercise ameliorates motor and cognitive symptoms in experimental Parkinson’s disease restoring striatal synaptic plasticity. Sci. Adv. 9, eadh1403 (2023).
Devos, D. et al. Comparison of desipramine and citalopram treatments for depression in Parkinson’s disease: a double-blind, randomized, placebo-controlled study. Mov. Disord. 23, 850–857 (2008).
Menza, M. et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology 72, 886–892 (2009).
Collier, T. J. et al. Nortriptyline inhibits aggregation and neurotoxicity of alpha-synuclein by enhancing reconfiguration of the monomeric form. Neurobiol. Dis. 106, 191–204 (2017).
Jeannotte, A. M., McCarthy, J. G. & Sidhu, A. Desipramine induced changes in the norepinephrine transporter, alpha- and gamma-synuclein in the hippocampus, amygdala and striatum. Neurosci. Lett. 467, 86–89 (2009).
Kohl, Z. et al. Fluoxetine rescues impaired hippocampal neurogenesis in a transgenic A53T synuclein mouse model. Eur. J. Neurosci. 35, 10–19 (2012).
Deusser, J. et al. Serotonergic dysfunction in the A53T alpha-synuclein mouse model of Parkinson’s disease. J. Neurochem. 135, 589–597 (2015).
Leentjens, A. F. G., Van den Akker, M., Metsemakers, J. F. M., Lousberg, R. & Verhey, F. R. J. Higher incidence of depression preceding the onset of Parkinson’s disease: a register study. Mov. Disord. 18, 414–418 (2003).
Pålhagen, S. E., Carlsson, M., Curman, E., Wålinder, J. & Granérus, A.-K. Depressive illness in Parkinson’s disease-indication of a more advanced and widespread neurodegenerative process?. Acta Neurol. Scand. 117, 295–304 (2008).
Nowacka-Chmielewska, M. et al. Running from stress: neurobiological mechanisms of exercise-induced stress resilience. Int. J. Mol. Sci. 23, 13348 (2022).
Kim, S. T., Choi, J. H., Chang, J. W., Kim, S. W. & Hwang, O. Immobilization stress causes increases in tetrahydrobiopterin, dopamine, and neuromelanin and oxidative damage in the nigrostriatal system. J. Neurochem. 95, 89–98 (2005).
Sugama, S. et al. Chronic restraint stress triggers dopaminergic and noradrenergic neurodegeneration: Possible role of chronic stress in the onset of Parkinson’s disease. Brain. Behav. Immun. 51, 39–46 (2016).
Lauretti, E., Di Meco, A., Merali, S. & Praticò, D. Chronic behavioral stress exaggerates motor deficit and neuroinflammation in the MPTP mouse model of Parkinson’s disease. Transl. Psychiatry 6, e733 (2016).
Hemmerle, A. M., Dickerson, J. W., Herman, J. P. & Seroogy, K. B. Stress exacerbates experimental Parkinson’s disease. Mol. Psychiatry 19, 638–640 (2014).
Janakiraman, U. et al. Chronic mild stress augments MPTP induced neurotoxicity in a murine model of Parkinson’s disease. Physiol. Behav. 173, 132–143 (2017).
Giasson, B. I. et al. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 34, 521–533 (2002).
Tian, Y. et al. SYNJ1 rescues motor functions in hereditary and sporadic Parkinson’s disease mice by upregulating TSP-1 expression. Behav. Brain Res. 452, 114569 (2023).
Oaks, A. W., Frankfurt, M., Finkelstein, D. I. & Sidhu, A. Age-dependent effects of A53T alpha-synuclein on behavior and dopaminergic function. PloS ONE 8, e60378 (2013).
Ramsey, C. P., Tsika, E., Ischiropoulos, H. & Giasson, B. I. DJ-1 deficient mice demonstrate similar vulnerability to pathogenic Ala53Thr human alpha-syn toxicity. Hum. Mol. Genet. 19, 1425–1437 (2010).
Lin, X.-M. et al. Membrane phospholipid peroxidation promotes loss of dopaminergic neurons in psychological stress-induced Parkinson’s disease susceptibility. Aging Cell 22, e13970 (2023).
Burtscher, J. et al. Chronic corticosterone aggravates behavioral and neuronal symptomatology in a mouse model of alpha-synuclein pathology. Neurobiol. Aging 83, 11–20 (2019).
Ren, C. et al. Early-life stress induces prodromal features of Parkinsonism in ageing rats. J. Gerontol. A. Biol. Sci. Med. Sci. 77, 705–716 (2022).
Pitzer, C., Kurpiers, B. & Eltokhi, A. Sex differences in depression-like behaviors in adult mice depend on endophenotype and strain. Front. Behav. Neurosci. 16, 838122 (2022).
Harro, J. Animal models of depression: pros and cons. Cell Tissue Res. 377, 5–20 (2019).
Smith, K. M. & Dahodwala, N. Sex differences in Parkinson’s disease and other movement disorders. Exp. Neurol. 259, 44–56 (2014).
Lliberos, C. et al. Evaluation of inflammation and follicle depletion during ovarian ageing in mice. Sci. Rep. 11, 278 (2021).
Brooks, H. L., Pollow, D. P. & Hoyer, P. B. The VCD mouse model of menopause and perimenopause for the study of sex differences in cardiovascular disease and the metabolic syndrome. Physiology 31, 250–257 (2016).
Silveira, P. P., Pokhvisneva, I., Howard, D. M. & Meaney, M. J. A sex-specific genome-wide association study of depression phenotypes in UK Biobank. Mol. Psychiatry 28, 2469–2479 (2023).
Santos-García, D. et al. Sex differences in motor and non-motor symptoms among spanish patients with Parkinson’s Disease. J. Clin. Med. 12, 1329 (2023).
Abeliovich, A. et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25, 239–252 (2000).
Faul, F., Erdfelder, E., Lang, A.-G. & Buchner, A. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods 39, 175–191 (2007).
Ma, H. et al. Amygdala-hippocampal innervation modulates stress-induced depressive-like behaviors through AMPA receptors. Proc. Natl. Acad. Sci. USA 118, e2019409118 (2021).
Li, Y. et al. The Eph receptor A4 plays a role in demyelination and depression-related behavior. J. Clin. Investig. 132, e152187 (2022).
Heidari, M. H. et al. Effect of chronic morphine consumption on synaptic plasticity of Rat’s hippocampus: a transmission electron microscopy study. Neurol. Res. Int. 2013, 290414 (2013).
Lecca, S. et al. Rescue of GABAB and GIRK function in the lateral habenula by protein phosphatase 2A inhibition ameliorates depression-like phenotypes in mice. Nat. Med. 22, 254–261 (2016).
Groves, T. R., Wang, J., Boerma, M. & Allen, A. R. Assessment of hippocampal dendritic complexity in aged mice using the golgi-cox method. J. Vis. Exp. JoVE 55696 https://doi.org/10.3791/55696 (2017).
Yan, Y. et al. Down-regulation of MST1 in hippocampus protects against stress-induced depression-like behaviours and synaptic plasticity impairments. Brain. Behav. Immun. 94, 196–209 (2021).
Martin, E. et al. New role of P2X7 receptor in an Alzheimer’s disease mouse model. Mol. Psychiatry 24, 108–125 (2019).
Orock, A., Logan, S. & Deak, F. Age-related cognitive impairment: role of reduced Synaptobrevin-2 levels in deficits of memory and synaptic plasticity. J. Gerontol. A. Biol. Sci. Med. Sci. 75, 1624–1632 (2020).
Yan, Z.-Y., Li, X.-J., Ding, X.-F., Liu, Y.-Y. & Chen, J.-X. Evaluating the Anti-depression Effect of Xiaoyaosan on Chronically-stressed Mice. J. Vis. Exp. JoVE https://doi.org/10.3791/58276 (2019).
Li, Y. et al. Inflammation-activated C/EBPβ mediates high-fat diet-induced depression-like behaviors in mice. Front. Mol. Neurosci. 15, 1068164 (2022).
Tang, M., Chen, M. & Li, Q. Paeoniflorin ameliorates chronic stress-induced depression-like behavior in mice model by affecting ERK1/2 pathway. Bioengineered 12, 11329–11341 (2021).
Xu, X., Xiao, X., Yan, Y. & Zhang, T. Activation of liver X receptors prevents emotional and cognitive dysfunction by suppressing microglial M1-polarization and restoring synaptic plasticity in the hippocampus of mice. Brain. Behav. Immun. 94, 111–124 (2021).
Yankelevitch-Yahav, R., Franko, M., Huly, A. & Doron, R. The forced swim test as a model of depressive-like behavior. J. Vis. Exp. JoVE 52587 https://doi.org/10.3791/52587 (2015).
Jiang, P.-E. et al. Behavioral Assessments of Spontaneous Locomotion in a Murine MPTP-induced Parkinson’s Disease Model. J. Vis. Exp. JoVE https://doi.org/10.3791/58653 (2019).
Wu, F. et al. Genetic and pharmacological inhibition of two-pore domain potassium channel TREK-1 alters depression-related behaviors and neuronal plasticity in the hippocampus in mice. CNS Neurosci. Ther. 27, 220–232 (2021).
Ari, C. et al. Elevated plus maze test combined with video tracking software to investigate the anxiolytic effect of exogenous ketogenic supplements. J. Vis. Exp. JoVE https://doi.org/10.3791/58396 (2019).
Acknowledgements
This work was supported by grants from the National Key Research and Development Program of China (2019YFE0115900), the National Natural Science Foundation of China (82271447, 81822016), and the Innovative Research Groups of Hubei Province (2022CFA026), and the “New 20 Terms of Universities in Jinan” grant (No. 202228022, to Z.Z.).
Author information
Authors and Affiliations
Contributions
Z.Z. conceived and designed the experiments. Z.Z. and L.M. coordinated the whole project. D.X. and M.X. conducted most of the experiments with help from Y.Y. and X.W. Q.C. and S.L. provided statistical analysis and technical support. Z.Z., D.X., and M.X. participated in final data analysis and interpretation. Z.Z. and X.D. carried out most of the writing with input from other authors. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xia, D., Xiong, M., Yang, Y. et al. Chronic stress induces depression-like behaviors and Parkinsonism via upregulating α-synuclein. npj Parkinsons Dis. 11, 139 (2025). https://doi.org/10.1038/s41531-025-00998-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41531-025-00998-x
This article is cited by
-
GDNF attenuates a-synuclein aggregation-induced damage to VTA-NAc dopaminergic transmission and alleviates depression-like behaviors in mice
Scientific Reports (2025)
-
Chronic unpredictable mild Stress- induced neurobehavioral and metabolic alteration: insights into molecular mechanisms and emerging therapeutic Strategies”
Psychopharmacology (2025)
