
Abstract
Tumour-associated macrophages (TAMs) contribute to immune checkpoint blockade resistance, but their impact on intratumoural CD8⁺ T cell distribution remains unclear. Here we show that the expression of the glucose transporter SLC2A1 is spatially negatively correlated with CD8⁺ T cell distribution in both non-small-cell lung cancer (NSCLC) biopsies and murine tumour models. Tumour cell-specific Slc2a1 knockdown fails to reproduce the therapeutic benefit of SLC2A1 inhibition, whereas TAM-specific deletion of Slc2a1 suppresses tumour growth by enhancing the spatial homogeneity and effector function of intratumoural CD8⁺ T cells, thereby improving αPD-L1 efficacy. Spatial profiling of NSCLC specimens further revealed that SLC2A1⁺ TAM-enriched regions exhibit reduced CD8⁺ T cell density, and spatial proximity between these populations predicts resistance to αPD-(L)1 therapy. These findings identify SLC2A1⁺ TAMs as drivers of spatial CD8⁺ T cell exclusion and highlight TAM-specific SLC2A1 as a therapeutic target to overcome immune checkpoint blockade resistance in NSCLC.
Similar content being viewed by others


Main
Immune checkpoint blockade (ICB) therapies, particularly agents targeting PD-(L)1 and CTLA-4, have revolutionized the therapeutic paradigm for non-small-cell lung cancer (NSCLC)1,2,3, yet 5-year survival remains low4,5,6,7 and predictive biomarkers are lacking8. Both tumour-intrinsic factors and an immunosuppressive tumour microenvironment (TME) orchestrate ICB resistance9, with tumour-associated macrophages (TAMs) emerging as central mediators through checkpoint expression10,11, induction of T cell exhaustion12,13 and recruitment or polarization of regulatory T cells14,15. Notably, specific immunosuppressive TAM subsets have been identified16, highlighting the importance of deciphering TAM-intrinsic programmes for therapeutic targets and predictive insights.
Beyond these canonical functions, TAMs are metabolically active17 and can surpass tumour cells in glucose uptake18, thereby limiting glycolysis and IFN-γ production in CD8⁺ T cells19, while driving the IL-10 and PD-L1 expression within themselves in a lactate-dependent manner20,21. These findings underscore glucose metabolism as a key determinant of TAM immunoregulatory function. In addition, TAM heterogeneity is shaped by spatial localization22: hypoxic niches preferentially recruit and reprogramme TAMs towards immunosuppressive phenotypes23,24,25, and the spatial colocalization of specific TAM subsets with CD8⁺ T cells has been linked to ICB outcomes26. However, it remains unclear how TAM glucose metabolism regulates their spatial organization relative to CD8⁺ T cells and whether their spatial proximity modulates responses to ICB therapies in NSCLC.
Given that glucose transporters of the SLC2A family critically shape cellular competition for glucose in the TME, here we identify SLC2A1 as a determinant of TAM-mediated spatial distribution of CD8⁺ T cells in NSCLC. Pharmacological inhibition or TAM-specific genetic ablation of Slc2a1 significantly enhanced CD8⁺ T cell infiltration, spatial homogeneity and effector function, thereby suppressing tumour growth and improving response to αPD-L1 therapy. Spatial analysis of human NSCLC specimens revealed that regions enriched in SLC2A1⁺ TAMs were associated with reduced CD8⁺ T cell density and that close spatial proximity between these populations predicted poor outcomes to αPD-(L)1 therapies. These findings establish SLC2A1⁺ TAMs as mediators of spatially dependent immunosuppression and identify their proximity to CD8⁺ T cells as a prognostic factor for ICB resistance in NSCLC.
Results
Negative spatial correlation between SLC2A1 expression and CD8+ T cells in NSCLC
To investigate whether and how the spatial distribution of glucose transporter expression affects CD8+ T cell immune responses, we performed analysis on in-house spatial transcriptomics data of three NSCLC surgical samples focused on possible associations between the SLC2A family and the activity of the IFN-γ-related pathway, a well-established surrogate of responsiveness to ICB therapies27. Local indicators of spatial association (LISA) and bivariate Moran’s I analysis identified SLC2A1 as the transporter most negatively correlated with ICB-responsive signature genes (Fig. 1a,b), confirmed by contour plots (Fig. 1c). Validation across The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) bulk RNA sequencing (RNA-seq) datasets further showed SLC2A1’s consistent negative correlation with both ICB-responsive and IMvigor-210 signatures28 (Extended Data Fig. 1a,b).

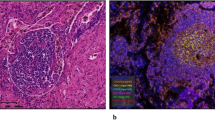
a, A heatmap visualization of spatial associations between 14 SLC2A family members and 18 genes in the ICB-responsive signature. Spatial associations with glycolysis activity are presented separately. The colour gradient represents the strength of significance (P values) in Pearson correlations. n.s., P value ≥ 0.05. b, The stacked barplot of statistically significant correlations for each SLC2A family member, sorted by the number of positive and negative correlations. The colour gradient represents P values in Pearson correlation. n.s., P value ≥ 0.05. c, Contour map of localized SLC2A1 (middle) and ICB-responsive signature (right) expression in three NSCLC spatial transcriptome datasets. d, A schematic of the workflow for spatial colocalization analysis between SLC2A1 and CD8+ T cells in clinical NSCLC specimens from the XQ cohort. The left panel shows representative images of NSCLC tissues with haematoxylin and eosin (HE) staining; the three panels to the right show representative images with immunohistochemistry (IHC). Scale bars, 5 mm (left), 2.5 mm (the middle two images) and 250 μm (right). e, A representative image of IHC shows the distribution of SLC2A1 and CD8A. Red, SLC2A1; brown, CD8A. Scale bars, 250 μm. f, A representative LISA plot of SLC2A1 and infiltrating CD8+ T cell spatial distribution patterns. g, A scatter plot of spatial associations between SLC2A1 and CD8+ T cells in patients with NSCLC (n = 38). h, Proportional distribution of positive versus negative spatial associations (n = 38). Red, positive associations (bivariate Moran’s I > 0); blue, negative associations (bivariate Moran’s I < 0). i,j, LISA mapping (i) and statistical summary (j) of SLC2A1–CD8 spatial relationships in LLC allograft tumours (n = 5). k,l, LISA mapping (k) and statistical summary (l) of SLC2A1–CD8 spatial relationships in the LLC orthotopic lung cancer model (n = 9). Scale bar, 2.5 mm. The strength and P values of the correlation in a and b were assessed using a two-sided Pearson’s correlation coefficient. P values for the bivariate Moran’s I in g–l were determined using a one-sided test.
CD8⁺ T cells serve as the primary source of IFN-γ in the TME and are central to the activity of the IFN-γ-related pathway. In silico analyses revealed SLC2A1’s negative correlation with overall T cell infiltration and activated CD8+ T cell abundance (Extended Data Fig. 1c,d), whereas no significant spatial link was found with CD4+ T cells, PD-1 or PD-L1 (Extended Data Fig. 2), underscoring CD8+ T cells’ role in defining tumour immunophenotypes29. Furthermore, we performed dual immunohistochemical staining for SLC2A1 and CD8A on surgically resected specimens from 38 patients with treatment-naive NSCLC from the XQ cohort (Supplementary Table 1). A grid-based quantitative spatial analysis, dividing whole-tissue sections into 1-mm2 regions, revealed a mutually exclusive distribution wherein regions with high SLC2A1 expression had significantly lower CD8+ T cell density compared with regions with low SLC2A1 signal (Fig. 1d,e and Extended Data Fig. 3). Bivariate Moran’s I confirmed this correlation in 78.9% (30/38) of cases, with statistical significance in 50% (19/38) (Fig. 1f–h). Cross-species validation in murine models reinforced that SLC2A1 shared a robust negative correlation with CD8+ T cell localization, with obvious spatial segregation observed in all subcutaneously (Fig. 1i,j) and intravenously (Fig. 1k,l) implanted Lewis lung carcinoma (LLC) tumours.
Collectively, our data identified SLC2A1 expression level as a factor in spatial exclusion of CD8⁺ T cells in NSCLC tumours, potentially contributing to an immunosuppressive microenvironment.
Pharmacological inhibition of SLC2A1 enhances CD8⁺ T cell responses
Given the negative spatial association between SLC2A1 expression and CD8⁺ T cell infiltration, we hypothesized that SLC2A1 inhibition could promote CD8⁺ T cell responses, enhancing antitumour immunity. We tested this hypothesis using BAY-876, a selective SLC2A1 inhibitor30,31, in immunocompetent and immunocompromised LLC and TC-1 (lung adenocarcinoma cell line) tumour-bearing mice. BAY-876 significantly delayed tumour growth in immunocompetent mice (Fig. 2a) but not in immunodeficient hosts (Fig. 2b), confirming an immune-dependent mechanism. To further dissect the contribution of immune responses, we comprehensively analysed the cellular TME phenotype resulting from BAY-876 treatment in the LLC tumour model (Fig. 2c and Extended Data Fig. 4a). Flow cytometry (Extended Data Fig. 4b) revealed increased CD4⁺ and CD8⁺ T cell infiltration (Fig. 2d) and elevated IFN-γ and TNF expression (Fig. 2e) in BAY-876-treated LLC tumours, with no changes in regulatory T (Treg) cells, natural killer (NK) cells, myeloid-derived suppressor cells (MDSCs), TAMs or dendritic cells (Extended Data Fig. 4c–g). Likewise, a similar response was observed in B16-F10 melanoma models (Extended Data Fig. 4h,i).

a, Tumour growth curves of subcutaneously implanted LLC (n = 7) and TC-1 (n = 12) tumours in immunocompetent mice treated with BAY-876 or vehicle control. b, Tumour growth curves of subcutaneously implanted LLC (control group (n = 6) and BAY-876-treated group (n = 7)) and TC-1 (n = 7) tumours in immunodeficient mice treated with BAY-876 or vehicle control. c,d, Treatment timeline (c) and flow cytometric analysis of CD4+ and CD8+ T cells in CD45+ cells (d) from LLC tumours (control group (n = 8) and BAY-876-treated group (n = 7)). e, CD8⁺ T cell populations expressing IFN-γ and TNF in LLC tumours (control group (n = 8) and BAY-876-treated group (n = 7)). f–h, Schematic of the experimental design (f), tumour growth curves following αCD8 intervention in the LLC tumour model (g) (BAY-876 group (n = 7) and the other groups (n = 6 each)), and tumour growth curves following αCD4 intervention in the LLC tumour model (h) (BAY-876 group (n = 7), BAY-876 + αCD4 group (n = 7) and the other groups (n = 6 each)). i,j, LLC allograft tumour growth curves (i) and weights (j) in C57BL/6J mice following BAY-876 treatment with or without αPD-L1 antibody therapy (combination group (n = 6) and the other groups (n = 5 each)). k, CD8+ T cell proportion and number (combination group (n = 6) and the other groups (n = 5 each)) in LLC tumours after treatment. l,m, Scatter plots (left) and proportion (middle) and number (right) of CD8⁺ T cell populations expressing IFN-γ (l) and TNF (m) (combination group (n = 6) and the other groups (n = 5 each)). n,o, Outline of the orthotopic lung tumour model and therapeutic regimen (n). Representative lung histology (left) and the quantitative analysis of tumour burden (right) (o) (control group (n = 5), αPD-L1 group (n = 6) and the other groups (n = 7 each)). Scale bars, 2.5 mm. Data are reported as the mean ± s.e.m., and group differences were assessed by a two-tailed unpaired Student’s t-test.
Importantly, CD8⁺ T cell depletion abolished BAY-876’s antitumour effects (Fig. 2f,g and Extended Data Fig. 4j), whereas CD4⁺ T cell depletion had no impact (Fig. 2h and Extended Data Fig. 4j), demonstrating CD8⁺ T cell dependence. This CD8⁺ T cell-dependent mechanism of action was similarly observed in the B16-F10 tumour-bearing mice (Extended Data Fig. 4k).
Pharmacological inhibition of SLC2A1 potentiates αPD-L1 therapy
We next examined whether SLC2A1 inhibition could augment ICB therapies using subcutaneous LLC tumour models. While αPD-L1 monotherapy showed no efficacy (Fig. 2i,j), combining BAY-876 with αPD-L1 restored sensitivity, significantly inhibiting tumour growth (Fig. 2i,j). Flow cytometry revealed that the combination regimen promoted the infiltration of CD8⁺ T cells (Fig. 2k) and boosted IFN-γ and TNF production (Fig. 2l,m) compared with monotherapies. In parallel, TME analysis confirmed this effect was specific to CD8⁺ T cells, with no significant changes in other immune populations (Extended Data Fig. 5a–f). The synergy was replicated in an orthotopic lung tumour model (Fig. 2n,o) and extended to MC38 colon adenocarcinoma and B16-F10 melanoma models (Extended Data Fig. 5g,h), where SLC2A1 inhibition consistently enhanced αPD-L1 efficacy. These results demonstrate that SLC2A1 inhibition could not only promote CD8⁺ T cell-mediated tumour control, but also sensitize tumours to αPD-L1 therapy, suggesting this combinatorial strategy holds promise for improving ICB therapeutic efficacy in solid tumours.
Genetic targeting of SLC2A1 in TAMs suppresses tumour growth
Given that multiple cell populations within the TME rely on SLC2A1-mediated glucose uptake, we sought to identify the key cellular mediators of antitumour immune responses following SLC2A1 inhibition. Through analysis of public single-cell data from NSCLC32, we found that among SLC2A1+ components, tumour cells exhibited the predominant expression, while among immune cells, TAMs showed the highest proportion (Extended Data Fig. 6a). Using 2-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) amino)-2-deoxyglucose (2-NBDG) glucose uptake assays together with flow cytometry, we confirmed that BAY-876 selectively reduced glucose uptake in tumour cells and TAMs (Fig. 3a), with minimal impact on other immune populations (Extended Data Fig. 6b–e). These findings suggest that BAY-876 might preferentially disrupt glucose uptake in tumour cells and TAMs.

a, Flow cytometry assays and summarized data of 2-NBDG (glucose analogue) uptake by tumour cells (left) and TAMs (right) (control group (n = 5) and BAY-876-treated group (n = 4)). b, Growth curves (by volume) of LLC-Slc2a1KD or LLC-vector control allograft tumours in C57BL/6 mice (BAY-876-treated groups (n = 4 each) and the other groups (n = 5 each)) treated with BAY-876 or vehicle. c, Growth curves of LLC allograft tumours in C57BL/6J following BAY-876 treatment with or without αCSF1R antibody (n = 6). d–f, Percentage (left) and number (right) of CD8+ T cells (d) and CD8⁺ T cell populations expressing IFN-γ (e) and TNF (f) (n = 6). g, Growth curves of subcutaneous LLC tumours in Slc2a1fl/fl (n = 6) and Slc2a1cko (n = 7) mice. h, Growth curves of subcutaneous LLC tumours in Slc2a1cko mice treated with BAY-876 or dimethyl sulfoxide (DMSO) control (n = 4). i–k, Schematic of the experimental design for co-implantation of TC-1 cells with TAMs from Slc2a1fl/fl or Slc2a1cko LLC tumour-bearing mice (i), tumour growth curves (j) and weights (k) for groups from i (Slc2a1fl/fl group (n = 6) and Slc2a1cko group (n = 5)). Data are reported as the mean ± s.e.m., and group differences were assessed by a two-tailed unpaired Student’s t-test.
To determine whether the antitumour effects of SLC2A1 inhibition were tumour cell dependent, we generated LLC-Slc2a1 knockdown (KD) cells (Extended Data Fig. 6f,g). In immunodeficient mice, LLC-Slc2a1KD and control (LLC-vector) tumours grew similarly (Extended Data Fig. 6h). While BAY-876 suppressed tumour growth in immunocompetent mice, this effect was independent of tumour-intrinsic SLC2A1 expression, as LLC-Slc2a1KD and control tumours responded equally to BAY-876 (Fig. 3b). Consistent with this notion, we found that LLC-Slc2a1 KD did not affect glucose uptake in T cells, further supporting the tumour cell-independent activity (Extended Data Fig. 6i). However, TAM depletion via αCSF1R antibodies (Extended Data Fig. 6j) abolished BAY-876’s tumour suppression (Fig. 3c) and negated its effects on CD8⁺ T cell abundance and function (Fig. 3d–f), supporting a central role of SLC2A1+ TAMs in mediating this therapeutic response.
To directly test whether SLC2A1 expression in TAMs was functionally required for tumour progression, we generated Slc2a1fl/flLyz2cre (Slc2a1cko) mice (Extended Data Fig. 6k,l). Tumours in Slc2a1cko mice grew significantly slower than in controls (Fig. 3g), and BAY-876 provided no additional benefit (Fig. 3h), confirming SLC2A1+ TAMs as the primary therapeutic target of SLC2A1 inhibition. To functionally validate this observation, we sorted TAMs (CD11b⁺GR1⁻F4/80⁺) from LLC-bearing Slc2a1cko or Slc2a1fl/fl mice and then co-implanted them with TC-1 tumour cells into wild-type recipient mice (Fig. 3i). Co-injection of Slc2a1-deficient TAMs significantly suppressed tumour progression compared with that in mice injected with Slc2a1fl/fl TAMs (Fig. 3j,k), confirming their suppressive role.
Together, these findings indicate that SLC2A1+ TAMs are critical for tumour progression and that genetic ablation of Slc2a1 in TAMs is sufficient to recapitulate the antitumour effects of pharmacological SLC2A1 inhibition.
TAM-specific Slc2a1 deletion enhances CD8⁺ T cell effector function and sensitizes tumours to αPD-L1 therapy
We aimed to explore the phenotype resulting from Slc2a1 deficiency in TAMs. Fluorescence-activated cell sorting (FACS) was used to isolate TAMs from LLC-bearing Slc2a1cko and Slc2a1fl/fl mice, and subsequent bulk RNA-seq analysis revealed extensive reprogramming in Slc2a1-deficient TAMs (Fig. 4a). Transcriptomic studies have defined immunosuppressive TAM subpopulations (C1QC-TAMs, SPP1-TAMs, TREM2-TAMs and so on16), whose characteristic genes were broadly downregulated in Slc2a1-deficient TAMs (Fig. 4b), confirming the prevalent immunosuppressive identity of SLC2A1+ TAMs. The Myeloid immunosuppressive score analysis further showed Slc2a1 deficiency significantly reduced this score33 (Fig. 4c), quantitatively linking SLC2A1 to TAM immunosuppression. Focusing on immunosuppressive pathways, Slc2a1 deficiency significantly dampened the activity of major TAM-related immunosuppressive pathways, including arginine metabolism, IL-10 and TGF-β signalling (Extended Data Fig. 7a–c). These transcriptional changes were corroborated at the protein level through flow cytometry analysis, in either Slc2a1-deficient (Fig. 4d,e) or BAY-876-treated TAMs (Extended Data Fig. 7d).

a, PCA of TAMs from LLC tumours of Slc2a1fl/fl and Slc2a1cko mice (n = 4). b, Heatmap of macrophage subset scores in Slc2a1fl/fl and Slc2a1cko TAMs (n = 4). c, Immunosuppressive gene signature scores in Slc2a1fl/fl and Slc2a1cko TAMs (n = 4). d,e, ARG1 expression (d) and immunosuppressive cytokines (TGF-β, IL-10 and IL-1β) (e) in Slc2a1fl/fl and Slc2a1cko TAMs (Slc2a1fl/fl group (n = 6) and Slc2a1cko group (n = 7)). MFI, mean fluorescence intensity. f, Schematic diagram (left) depicting the coculture system. CD8⁺ T cells were cocultured with either SLC2A1⁺ or SLC2A1⁻ TAMs, and their functional state was subsequently evaluated by flow cytometry (n = 3). g,h, Flow cytometric analysis of the proportion (g) and total numbers (h) of CD8⁺ T cell populations expressing IFN-γ, TNF or Ki-67 in TC-1 tumours co-implanted with TAMs derived from Slc2a1fl/fl or Slc2a1cko mice (Slc2a1fl/fl group (n = 6) and Slc2a1cko group (n = 5)). i–k, Proportion and numbers of CD8⁺ T cells (i) and the frequency and absolute number of IFN-γ⁺, TNF⁺ or Ki-67⁺ subsets among them, with the frequency shown in (j) and the absolute number shown in (k), in LLC tumour-bearing Slc2a1fl/fl or Slc2a1cko mice (Slc2a1fl/fl group (n = 6) and Slc2a1cko group (n = 7)). l, Growth of LLC tumours of Slc2a1fl/fl and Slc2a1cko mice treated with isotype control or αPD-L1 antibody therapy (Slc2a1cko group (n = 3) and the other groups (n = 4 each)). m, Schematic and representative images of CD8+ T cell spatial distribution in LLC tumours from Slc2a1fl/fl and Slc2a1cko mice. Scale bar, 100 μm. n, Inter-CD8+ T cell spatial analysis in LLC tumours from Slc2a1fl/fl and Slc2a1cko mice (n = 6). o, Representative flow cytometry scatter plots (left) and summarized data (right) of the percentage of Ki-67+ and active Caspase-3+ cells among CD8+ T cells (n = 3). Data are shown as the mean ± s.e.m. Statistical analysis was performed using the two-tailed Wilcoxon rank-sum test (c) and unpaired Student’s t-test (d–k, l, n and o). For the violin and box plots, the centre line denotes the median, the box encompasses the first and third quartiles and whiskers denote range. The diagram in f was created using BioRender.com.
To directly assess the functional impacts of SLC2A1+ TAM on CD8+ T cells, we cocultured FACS-isolated SLC2A1+ and SLC2A1− TAMs from LLC tumours with splenic naive CD8⁺ T cells under CD3/CD28 stimulation. This ex vivo coculture assay demonstrated SLC2A1+ TAMs suppressed CD8⁺ T cell cytokine release (Fig. 4f). To validate these findings in vivo, we co-implanted wild-type recipient mice with TC-1 tumour cells and TAMs isolated from LLC-bearing Slc2a1cko or Slc2a1fl/fl mice. This assay revealed Slc2a1-deficient TAMs enhanced intratumoural CD8⁺ T cell cytokine production (IFN-γ and TNF) and proliferation (Ki-67) (Fig. 4g,h). We next investigated the functional phenotype of CD8+ T cells in LLC tumour-bearing Slc2a1cko or Slc2a1fl/fl mice. LLC tumour-bearing Slc2a1cko mice displayed a higher CD8⁺ T cell frequency and absolute count (Fig. 4i), with elevated IFN-γ, TNF and Ki-67 expression (Fig. 4j,k), indicating enhanced effector function. Critically, in the αPD-L1-resistant LLC tumour model, αPD-L1 therapy significantly inhibited tumour growth in Slc2a1cko but not Slc2a1fl/fl mice (Fig. 4l), demonstrating that TAM-specific Slc2a1 deletion sensitizes tumours to αPD-L1.
These findings suggest that SLC2A1 could dictate TAMs towards a phenotype that impairs the antitumour immune function of CD8⁺ T cells and confers resistance to αPD-L1 therapies.
TAM-specific Slc2a1 deletion promotes the spatial homogenization of CD8⁺ T cells
In terms of spatial phenotype, immunofluorescent staining revealed CD8⁺ T cell infiltration was significantly increased in LLC-bearing Slc2a1cko mice, characterized by obviously greater homogeneity in the spatial distribution of CD8⁺ T cells compared with that in LLC-bearing Slc2a1fl/fl control mice (Fig. 4m,n). To explore the mechanisms underlying this spatial homogeneity upon Slc2a1 deletion, we measured the uptake of the fluorescent glucose tracer 2-NBDG by CD8⁺ T cells cocultured with SLC2A1⁺ or SLC2A1− TAMs. Coculture experiments revealed no significant alteration in glucose import by CD8⁺ T cells (Extended Data Fig. 7e). In vivo studies further confirmed comparable 2-NBDG uptake among total T cell populations, as well as in the CD4⁺ and CD8⁺ subsets from both Slc2a1fl/fl and Slc2a1cko mice (Extended Data Fig. 7f), indicating that altered glucose availability does not account for the observed functional changes. We next evaluated whether SLC2A1 expression in TAMs influences CD8⁺ T cell migration. Transwell assays indicated that the migratory capacity of CD8⁺ T cells was not altered by the presence of SLC2A1⁺ or SLC2A1⁻ TAMs (Extended Data Fig. 7g). Finally, in coculture assays of CD8⁺ T cell with TAMs, we observed that SLC2A1⁺ TAMs significantly suppressed CD8⁺ T cell proliferation and enhanced apoptosis compared with SLC2A1⁻ TAMs (Fig. 4o). This reduction in viable CD8⁺ T cells near SLC2A1⁺ TAMs provides a mechanistic explanation for the decreased intratumoural CD8⁺ T cell abundance and loss of spatial homogeneity.
SLC2A1⁺ TAMs shape CD8⁺ T cell spatial distribution in NSCLC
We then investigated whether SLC2A1⁺ TAMs influence the spatial distribution of CD8⁺ T cells in human NSCLC. To this end, multiplex immunofluorescence staining for SLC2A1, CD68 and CD8A was conducted in tumour samples from two independent tissue microarrays, including TJ cohort 1 (treatment-naive resected NSCLC; Supplementary Table 1) and TJ cohort 2 (treatment-naive biopsies from patients with NSCLC before αPD-(L)1 therapy; Supplementary Table 1). Regions enriched with SLC2A1⁺ TAMs showed markedly reduced CD8⁺ T cell infiltration, while SLC2A1⁻ TAMs low areas exhibited robust CD8⁺ T cell accumulation (Fig. 5a). Quantitative image analysis confirmed the negative correlation between the SLC2A1⁺/SLC2A1⁻ TAM ratio and CD8⁺ T cell density in TJ cohort 1 (Fig. 5b). Spatial mapping revealed that SLC2A1⁺ TAMHigh tumours had dispersed CD8⁺ T cells with increased nearest-neighbour distances, whereas SLC2A1⁻ TAMHigh tumours showed tight CD8⁺ clustering (Fig. 5c–e), suggesting SLC2A1⁺ TAMs spatially restrict CD8⁺ access, contributing to immune-excluded TME.

a, Representative multiplex immunofluorescence (mIF) images of CD8+ T cells and SLC2A1⁺ TAMs in tissue microarray dots from TJ cohort 1 tumours with high or low SLC2A1⁺ TAM infiltration. Blue, DAPI; white, CD8A; red, SLC2A1; green, CD68. Scale bar, 50 μm. b, Scatter plots of CD8⁺ T cell proportions in total cells by the SLC2A1⁺ TAM/SLC2A1⁻ TAM ratio with Pearson correlation analysis in tumours from TJ cohort 1 (n = 110). c,d, Spatial analysis of CD8⁺ T cells in TJ cohort 1 (n = 110), including nearest-neighbour distances between CD8⁺ T cells (c) and mean distance to five nearest CD8⁺ T cells (d). Pearson correlation analysis demonstrated associations between the SLC2A1⁺/SLC2A1⁻ TAM ratio and these two spatial indices. e, Representative multiplex immunofluorescence images and spatial analysis of SLC2A1⁺ TAM/SLC2A1− TAM versus CD8+ T cells in the tumour samples from TJ cohort 2. Scale bar, 200 μm. f, Contour maps of CD8+ T cell and SLC2A1+ TAM spatial distribution patterns in tumours from TJ cohort 1. Scale bar, 200 μm. g, Schematic of the experimental workflow for colocalization analysis between SLC2A1⁺ TAMs and CD8+ T cells. h,i, Median CD8+ T cell density plotted by distance to SLC2A1+ TAMs (left) and SLC2A1− TAMs (right) in TJ cohort 1 (h) (n = 110) and TJ cohort 2 (i) (n = 127). Error bars indicated the 33rd and 67th percentile. The strength and P values of the correlation in b–d were assessed using a two-sided Pearson’s correlation coefficient. Statistical analysis was performed using repeated measures analysis of variance in h and i.
This spatial antagonism is visually demonstrated by the segregated distribution of SLC2A1⁺ TAMs and CD8⁺ T cells in tumour regions (Fig. 5f). To quantitatively analyse this spatial interaction, we measured the CD8⁺ T cell density at defined distances (0–500 μm, in 100-μm increments) from individual SLC2A1⁺ or SLC2A1− TAMs (Fig. 5g). This proximity analysis (0–500 μm) demonstrated that CD8⁺ T cell density decreased near SLC2A1⁺ TAMs but enriched near SLC2A1⁻ TAMs in both TJ cohort 1 (Fig. 5h) and TJ cohort 2 (Fig. 5i), indicating SLC2A1⁺ TAMs actively disrupt CD8⁺ T cell organization.
Spatial proximity between SLC2A1⁺ TAMs and CD8⁺ T cells confer αPD-(L)1 resistance in NSCLC
The negative spatial correlation between SLC2A1⁺ TAMs and CD8⁺ T cells prompted investigation of its clinical significance in αPD-(L)1-treated NSCLC patients. A transcriptome-based approach34 was used to estimate the abundance of SLC2A1⁺ and SLC2A1⁻ TAMs from bulk RNA-seq data from two independent αPD-(L)1-treated NSCLC cohorts (SYS cohort and TUSM cohort; Supplementary Table 1). We found that SLC2A1⁺ TAM infiltration was correlated with worse progression-free survival (PFS), while SLC2A1⁻ TAMs were associated with improved PFS in both cohorts (Fig. 6a,b). To further dissect this relationship, patients were categorized into three groups according to the median infiltration of SLC2A1+ and SLC2A1⁻ TAMs: SLC2A1+ TAMLow SLC2A1⁻ TAMHigh, SLC2A1+ TAMHigh SLC2A1⁻ TAMLow, and otherwise. Pathway analysis uncovered significant enrichment of CD8⁺ T cell-related pathways (for example, activated CD8⁺ T cells and interferon gamma production) in the SLC2A1+ TAMLow SLC2A1⁻ TAMHigh group relative to the SLC2A1+ TAMHigh SLC2A1⁻ TAMLow group in both cohorts (Fig. 6c,d), suggesting various responsiveness to ICB therapies. Clinically, these patients also experienced significantly longer PFS under αPD-(L)1 therapy (Fig. 6e,f), supporting a detrimental effect of SLC2A1⁺ TAMs.

a,b, Forest plots of SLC2A1+ TAM signature influence on PFS in the SYS (a) (n = 109) and TUSM (b) (n = 18) immunotherapy cohorts. Error bars indicate the 95% confidence interval (CI). The hazard ratio for SLC2A1+ TAM was 1.914 (95% CI, 1.042–3.518) in the SYS cohort and 2.151 (95% CI, 0.693–6.671) in the TUSM cohort. c,d, Patients were stratified into three subgroups based on combined infiltration levels of SLC2A1⁺ and SLC2A1⁻ TAMs: SLC2A1+ TAMLow SLC2A1⁻ TAMHigh, SLC2A1+ TAMHigh SLC2A1⁻ TAMLow and otherwise. Box plots comparing activated CD8+ T cell (left) or interferon gamma production (right) across groups in the SYS (c) (n = 109) and TUSM (d) (n = 18) immunotherapy cohorts. e,f, Kaplan–Meier survival curves of PFS stratified according to the above groups in the SYS (e) (n = 109) and TUSM (f) (n = 18) cohorts. g, Clinical nomogram integrating key variables to predict 1-, 2- and 3-year survival probabilities. Points from individual predictors are summed to estimate total survival risk. h, ROC curves evaluating 3-year survival prediction in the discovery (n = 76) and validation cohorts (n = 51) derived from TJ cohort 2. i, Area under the ROC curve analysis for the discovery (n = 76) and validation cohorts (n = 51). j,k, Kaplan–Meier curves of OS in the discovery (j) (n = 76) and validation cohorts (k) (n = 51). l, Schematics highlighting the major findings of this study. Statistical analysis was performed using the two-sided Cox proportional regression P values (a and b), two-tailed unpaired Student’s t-test (c and d), and log-rank (Mantel–Cox) test (e, f, j and k). For the box plots, the centre line denotes the median, the box encompasses the first and third quartiles and whiskers denote 1.5 times the interquartile range. The schematic in l was created using BioRender.com.
Because CD8⁺ T cell infiltration alone poorly predicts ICB response, we integrated spatial features into a predictive model. A nomogram model demonstrated that, as expected, higher CD8⁺ T cell density was correlated with improved overall survival (OS) rates, whereas elevated SLC2A1⁺ TAM density was associated with poorer outcomes (Fig. 6g). Notably, mean distance between CD8⁺ T cells and SLC2A1⁺ TAMs emerged as the most weighted negative survival predictor (Fig. 6g). This risk scoring algorithm demonstrated relatively strong performance, with an area under the curve (AUC) of 0.80 in the discovery set and 0.71 in the validation set (Fig. 6h,i). Stratification by risk scores revealed that patients in the low-risk group showed a significant association with prolonged OS following αPD-(L)1 therapy in both the discovery (Fig. 6j) and validation set (Fig. 6k) from TJ cohort 2.
Collectively, these findings demonstrate that spatial proximity between SLC2A1⁺ TAMs and CD8⁺ T cells emerges as an informative, independent predictor of patient resistance to αPD-(L)1 therapy in patients with NSCLC (Fig. 6l).
Discussion
In this study, we found that glucose uptake by TAMs drove immunosuppression by creating spatial exclusion zones that hinder CD8⁺ T cell infiltration, thereby promoting resistance to αPD-(L)1 therapy in NSCLC. Notably, our analyses of clinical samples identified spatial proximity between SLC2A1⁺ TAMs and CD8⁺ T cells as a strong predictor of ICB therapeutic response in NSCLC, highlighting a clinically relevant factor contributing to immune resistance in the TME.
While tumour cells are known for aberrant glucose uptake35, TAMs consume even more glucose in the TME18. Targeting glucose uptake, either in tumour cells or TAMs, has emerged as a promising metabolic therapeutic strategy. Among glucose transporters, SLC2A1 is critical, and its inhibitor BAY-876 has shown antitumour efficacy in preclinical models30,36,37,38,39. However, these studies have largely overlooked the specific cellular targets of BAY-876 within the TME. We found that BAY-876 primarily targets TAMs and tumour cells. More importantly, genetic approaches revealed that its antitumour effect is mediated mainly through SLC2A1 inhibition in TAMs, establishing TAMs as the key regulators of therapeutic response to SLC2A1 inhibition.
In addition to direct tumour suppression, targeting SLC2A1 has been shown to enhance immunotherapeutic efficacy40,41,42. However, the clinical relevance of SLC2A1 inhibition remains largely unexplored. In the present study, spatial transcriptomics and multiplex immunofluorescence revealed an inverse correlation between SLC2A1 expression and CD8⁺ T cells in both NSCLC clinical samples and preclinical tumour models. This suggests that targeting SLC2A1 could improve CD8⁺ T cell distribution, a notion confirmed in mice where BAY-876 treatment increased CD8⁺ T cell infiltration and effector function.
The metabolic microenvironment within tumours is a key determinant of TAM plasticity and immunosuppressive function43,44. Contrary to the paradigm that glycolytic inhibition affects immunity primarily by modulating glucose availability, we found that inhibiting SLC2A1 in TAMs does not increase glucose for CD8+ T cells or attract CD8+ T cells. Instead, SLC2A1 in TAMs directly impairs proliferation and survival of CD8+ T cells, reducing CD8+ T cell numbers near SLC2A1+ TAMs. This mechanism is clinically relevant, as close proximity between SLC2A1+ TAMs and CD8+ T cells correlates with resistance to ICB therapy.
Previous studies have found that spatial heterogeneity of intratumoural T cells is associated with diminished antitumour immune responses45,46,47,48. More recently, reducing local heterogeneity among tumour cell populations might facilitate restoration of antitumour immunity49,50,51. In our study, genetic ablation of Slc2a1 in TAMs potentiated the infiltration of CD8⁺ T cells into LLC tumours and reduced spacing between neighbouring CD8⁺ T cells. This finding was mirrored in human NSCLC samples, where SLC2A1⁻ TAMs correlated with decreased intercellular distances among CD8⁺ T cells. These findings suggest that targeting SLC2A1 in TAMs may reverse spatial heterogeneity, transforming a dispersed T cell distribution into a cohesive one that is effective against tumours.
Local cell communities within the TME were recently shown to play a role in determining CD8⁺ T cell fate, such as survival, stemness, cytotoxicity and restimulation52,53,54,55,56,57. The structural organization of these microanatomical niches governs the spatial positioning of CD8⁺ T cells, thereby holding potential as a predictor of responses to immunotherapy58,59,60,61,62. The spatial heterogeneity of TAMs, where immunosuppressive subsets accumulate in hypoxic regions24,25, is probably linked to their metabolic activity, which also underlies their exclusion of effector T cells. In the present study, we found that SLC2A1⁺ TAMs and CD8⁺ T cells are spatially segregated, while SLC2A1⁻ TAMs colocalize with CD8⁺ T cells in NSCLC tissues. A high SLC2A1⁺/SLC2A1⁻ TAM ratio correlated with reduced IFN-γ production and poorer ICB outcomes, indicating that SLC2A1⁺ TAMs form immunosuppressive niches. Critically, the spatial proximity between SLC2A1⁺ TAMs and CD8⁺ T cells was a better predictor of ICB response than cell abundance alone, highlighting the translational value of spatial metrics in guiding immunotherapy decisions.
In summary, our study uncovers a previously unappreciated mechanism by which SLC2A1⁺ TAMs mediate spatial immune exclusion of CD8⁺ T cells, consequently impairing their effector functions, and conferring resistance to αPD-(L)1 therapy in NSCLC. Our integrated data establish TAM-specific glucose metabolism as a key driver and predictive biomarker of tumour immune evasion, providing a rationale for immunometabolic combination therapies.
Methods
Ethical statement
The protocols of this study were approved by the institutional review boards of Tongji Hospital (Huazhong University of Science and Technology), Xinqiao Hospital (Third Military Medical University) and Shanghai Pulmonary Hospital (Tongji University School of Medicine). Informed consent was obtained from all participants. All animal experiments were conducted in full compliance with the guidelines and under the approval of the Laboratory Animal Welfare and Ethics Committee of Third Military Medical University.
Study cohorts
Patients for all cohorts were recruited from the following collaborating institutions: the XQ cohort from the Department of Oncology, Xinqiao Hospital; TJ cohort 1 and TJ cohort 2 from the Department of Oncology, Tongji Hospital; TUSM cohort from the Department of Medical Oncology, Shanghai Pulmonary Hospital; SYS cohort from the Department of Medical Oncology, State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, Sun Yat-sen University Cancer Center.
The XQ cohort comprises patients with resectable NSCLC tumours at the Department of Thoracic Surgery (Xinqiao Hospital) from 2020 to 2022. Selected sections were stained with haematoxylin and eosin to confirm tumour mass. Tumour samples were used for dual immunohistochemistry (IHC) to validate the spatial distribution of SLC2A1 and CD8.
To validate the spatial distribution of SLC2A1+ TAMs and CD8+ T cells, we performed multiplex immunofluorescence analysis on TJ cohort 1 (microarray), which consisted of NSCLC tumour tissues obtained between 2015 and 2018. To validate the association between SLC2A1+ TAMs and immunotherapeutic responses, TJ cohort 2 (microarray) was collected, including patients with metastatic NSCLC who received an ICB-containing regimen from 2018 to 2022. For TJ cohort 2, OS data following ICB administration were extracted from medical records.
To investigate the association between SLC2A1+ TAMs and responses to immunotherapy, we analysed two independent cohorts. The TUSM cohort (RNA-seq data) comprised patients with metastatic NSCLC from Shanghai Pulmonary Hospital (2018–2020), treated with ICB-containing regimens. The SYS cohort, described previously63, included similar patients from Sun Yat-sen University Cancer Center (2018–2021) who received either ICB combined with platinum-based chemotherapy or chemotherapy alone. For both cohorts, transcriptomic data were generated via RNA sequencing of treatment-naive formalin-fixed, paraffin-embedded tumour specimens, with clinical data and outcomes provided by the respective institutions. OS was defined as the time from the initiation of ICB treatment.
Spatial transcriptomics analysis
In-house raw sequencing data of three NSCLC surgical specimens underwent primary quality control using FastQC, followed by alignment and feature-barcode matrix generation via Space Ranger software (v.2.1.0). Downstream analyses were performed using Seurat (v.4.0.5)64 with SCTransform (v.0.3.3)65 for normalization. Single-sample gene set enrichment analysis (ssGSEA)66 was implemented in R to calculate an enrichment score for glycolytic signature67. Subsequent Pearson correlation analysis was conducted between expression of SLC2A family molecules and glycolytic signature versus the aforementioned 18 genes of ICB-responsive signature27, visualized through heatmap representation.
Bulk RNA-seq data analysis of public datasets
The following gene sets were interrogated in this study: ICB-responsive signature, IMvigor210-derived signature68, T cell-associated genes69, glycolytic signature. ssGSEA was implemented in R to calculate enrichment scores for these gene signatures across TCGA and GEO datasets. Subsequently, the association between SLC2A1 expression levels and the computed gene set scores was assessed by Pearson correlation analysis.
The abundance of SLC2A1+ TAMs and SLC2A1− TAMs in bulk RNA-seq data was estimated using BayesPrism. This tool uses a Bayesian framework to deconvolve bulk RNA-seq data, predicting both cellular composition and cell type-specific expression by leveraging single-cell RNA-seq data derived from patients with lung cancer as prior information34. Genes located on chromosomes X and Y, along with low-expressed genes and non-protein-coding genes, were excluded from the raw count matrix. SLC2A1+ TAMs and SLC2A1− TAMs were defined on the basis of SLC2A1 expression levels. All software parameters were retained as default settings unless otherwise specified. Samples were stratified into three groups according to median abundance values: (1) SLC2A1+ TAMhigh and SLC2A1− TAMlow group, (2) SLC2A1+ TAMlow and SLC2A1− TAMhigh group and (3) other intermediate group. The R packages survival and survminer were applied for survival analysis. Specifically, Kaplan–Meier estimates were used to visualize survival probability, and the log-rank test was used to compare survival curves. The prognostic value of variables was quantified via univariable Cox regression, which served to calculate hazard ratios.
Cell culture
The murine cell lines used in this study—including colon adenocarcinoma MC38 (a gift from Dr Liufu Deng, Shanghai Jiao Tong University), melanoma B16-F10 (ATCC CRL-6475), LLC (ATCC CRL-1642) and TC-1 (BeNa Culture Collectin BNCC341334)—were routinely screened and confirmed to be free of mycoplasma contamination. In addition, all cell lines were authenticated. All cells were cultured under standard conditions (37 °C, 5% CO2) in Dulbecco’s modified Eagle medium (DMEM) containing 10% foetal bovine serum (Gibco) and 1% penicillin–streptomycin (Gibco).
Cell line construction
To knock down Slc2a1 in the murine LLC carcinoma cell line, we used lentiviral transduction. Recombinant lentiviral vectors containing GFP were commercially obtained from Genechem. At 72 h post-transduction, GFP-positive cells were purified using FACS to establish a stably transfected population. To confirm the KD efficacy, we performed real-time quantitative reverse transcription PCR (qRT–PCR) and flow cytometry to measure its efficacy at the mRNA and protein levels, respectively. qRT–PCR was performed with a Bio-Rad CFX384 system (primers listed in Supplementary Table 2). For protein detection, we used an APC-conjugated GLUT1 antibody (Novus Biologicals; 1:200 dilution). The cell line exhibiting the highest KD efficiency was chosen for all subsequent functional assays.
qRT–PCR
Total RNA was extracted from tumour cells using TRIzol reagent (Thermo Fisher Scientific) and subsequently reverse-transcribed into cDNA with PrimeScript RT Master Mix following the manufacturer’s protocol (37 °C for 15 min, 85 °C for 5 s). Gene expression was then quantified by qRT–PCR on a CFX384 system (Bio-Rad) using SYBR Green Premix Ex Taq II (Takara) following the manufacturer’s protocol.
Mice
To generate myeloid-specific Slc2a1 knockout mice (Slc2a1cko), we crossed Slc2a1fl/fl mice with Lyz2Cre mice, both on a C57BL/6 background and obtained from Shanghai Model Organisms. Genotyping primers are listed in Supplementary Table 2. Wild-type C57BL/6J and BALB/c-nude mice were acquired from HFK Bioscience. All experiments utilized 6- to 8-week-old mice. Mice were housed in specific pathogen-free facilities at Third Military Medical University and under strictly controlled environmental conditions. The light/dark cycle was set at 12 h each (lights on from 06:00 to 18:00). The ambient temperature was maintained at 23 ± 2 °C with approximately 50% relative humidity. All animals had free access to food and water.
Tumour models and treatments
Female mice (6–8 weeks old) received subcutaneous injections of tumour cells into the right flank: LLC (1 × 10⁶), TC-1 (5 × 10⁵), B16-F10 (5 × 10⁵ or 2.5 × 10⁵) or MC38 (5 × 10⁵) cells. An orthotopic lung model was established via intravenous injection of 5 × 10⁵ LLC cells in 200 μl phosphate-buffered saline. Tumour volumes, measured by calipers and calculated as 0.5 × length × width2, were monitored. To ensure ethical compliance, mice were euthanized when tumours reached the predefined limit of 2,000 mm3, which was not exceeded in any experiment.
For BAY-876 (MCE) treatment, mice were treated by gavage feeding 7 days after tumour inoculation at a dose of 3 mg kg−1. For CD8+ T cell and CD4+ T cell depletion, mice received intraperitoneal injections of 200 μg αCD8 (BE0061, clone 2.43, Bio X Cell) or αCD4 (BE0003-1, clone GK1.5, Bio X Cell) 1 day before BAY-876 treatment, repeated every 3 days. Macrophages were depleted using 600 μg CSF1R antibody (BE0213, AFS98, Bio X Cell) administered intraperitoneally 1 day before BAY-876 treatment, with doses repeated every 7 days. For αPD-L1 immunotherapy treatment, 200 μg of αPD-L1 antibody (BE0101, clone 10F.9G2, Bio X Cell) was intraperitoneally injected 4 days post-inoculation every 3 days with or without BAY-876 in LLC orthotopic tumour models, 5 days post-inoculation every 3 days with or without BAY-876 in LLC/MC38 subcutaneous tumour models and 7 days post-inoculation every 3 days with or without BAY-876 in B16-F10 tumour models.
For in vivo macrophage coinjection experiments, TAMs isolated from LLC tumour-bearing Slc2a1cko or Slc2a1fl/fl mice were mixed with TC-1 tumour cells at a 1:1 ratio. A total of 5 × 10⁵ cells from this mixture, or 5 × 10⁵ TC-1 cells alone as a control, were subcutaneously injected into the flanks of C57BL/6J mice. Tumour volume was tracked by volume measurements every 3 days.
Flow cytometry and cell sorting
Single-cell suspensions were prepared from tumour tissues with enzymatic digestion using 1 mg ml−1 collagenase I (Sigma) and 50 μg ml−1 DNase I (Roche). Flow cytometric analysis and cell sorting were conducted using a Gallios flow cytometer (BECKMAN COULTER) and a FACS Aria II sorter (BD Biosciences), respectively, with data processed in FlowJo V10. The following antibodies were used for cell surface and intracellular staining: Fixable Viability Dye (eBioscience; 1:1,000), CD45 (30-F11, BioLegend; 1:100), CD3 (17A2, BioLegend; 1:100), CD4 (GK1.5, BioLegend; 1:100), CD8 (53-6.7, BioLegend; 1:100), NK1.1 (PK136, BioLegend; 1:100), CD11b (M1/70, BioLegend; 1:100), CD11c (N418, BioLegend; 1:100), F4/80 (BM8, BioLegend; 1:100), I-A/I-E (M5/114.15.2, BioLegend; 1:100), Gr-1 (RB6-8C5, BioLegend; 1:100), GLUT1 (NB110-39113APC, Novus Biologicals; 1:200), Arg1 (A1exF5, eBioscience; 1:100), IL-1β (Pro-form) (NJTEN3, eBioscience; 1:100), TGF-β1 (TW7-16B4, BioLegend; 1:100), IL-10 (JES5-16E3, eBioscience; 1:100), IFN-γ (XMG1.2, BioLegend; 1:100), TNF (MP6-XT22, BioLegend; 1:100), GZMB (GB11, BioLegend; 1:100), Perforin (eBioOMAK-D, BioLegend; 1:100), Ki-67 (16A8, BioLegend; 1:100) and active Caspase-3 (C92-605.rMAb, BD Biosciences; 1:100). To assess T cell function, single-cell suspensions were stimulated for 5 h with a Cell Activation Cocktail containing Brefeldin A (BioLegend; 1:250) before surface staining. Following surface marker staining, cells were fixed and permeabilized using the BD Fixation/Permeabilization Solution Kit for intracellular effector function cytokine detection, or the Foxp3/Transcription Factor Staining Buffer Set (eBioscience) for Ki-67 staining. TAMs (CD11b+ GR1−F4/80+) were isolated from LLC tumours at day 20 post-inoculation using FACS.
RNA sequencing
Freshly isolated TAMs from LLC tumours of Slc2a1fl/fl or Slc2a1cko mice were collected. Following RNA extraction with TRIzol reagent, the integrity and quality of the isolated RNA were assessed using an Agilent Bioanalyzer, and samples with an RNA integrity number (RIN) greater than 5.0 were selected for further analysis. mRNA sequencing libraries were prepared by OE Biotech and sequenced on the Illumina platform using the HiSeq system with a 2 × 150–bp read configuration in accordance with the manufacturer’s guidelines. Raw reads were quality controlled and filtered (adapter and low-quality bases removal) using Fastp (v.0.23.4). The cleaned reads were then aligned to the mouse reference genome (GRCm39) using STAR (v.2.7.10a). Gene-level read counts were generated using FeatureCounts (v.2.0.1). Sample similarity was assessed by principal component analysis (PCA) using top two principal components derived from transcripts per million (TPM) matrices. A heatmap of the normalized expression of selected macrophage signatures in Slc2a1fl/fl or Slc2a1cko TAMs was visualized using the Pheatmap (v.1.0.8) package.
Glucose uptake assay
To assess glucose uptake in the TME, single-cell suspensions were prepared from freshly collected LLC tumours. These cells were adjusted to a density of 1 × 10⁶ cells ml−1 and incubated with 100 μM 2-NBDG (Thermo Fisher Scientific) in a glucose-free medium at 37 °C for 30 min. Following this incubation, cells were stained with specific surface antibodies to enable cell type discrimination, and 2-NBDG fluorescence intensity was immediately quantified via flow cytometry. For the coculture experiments, CD8⁺ T cells were first cultured with SLC2A1⁺ or SLC2A1⁻ TAMs for 72 h. The resulting cell mixtures were then subjected to the same 2-NBDG labelling protocol, followed by antibody staining to detect 2-NBDG uptake in CD8+ T cells.
T cell migration assay
A Transwell migration assay was performed to assess CD8⁺ T cell chemotaxis. CD8⁺ T cells (5 × 10⁵) isolated from wild-type mouse spleens were placed in the upper chamber of a 24-well transwell insert (LABSELECT, China; 8.0 μm pore size). Concurrently, SLC2A1+ or SLC2A1− TAMs freshly isolated from LLC tumours were seeded in the lower chamber at a density of 2.5 × 10⁵ cells in 800 μl of RPMI 1640 complete medium. Following a 24-h incubation at 37 °C with 5% CO2, the CD8⁺ T cells that had migrated to the lower chamber were collected and quantified.
In vitro coculture with T cells
CD8⁺ T cells were purified from the spleens of tumour-free C57BL/6 mice using a CD8αβ T-Cell Isolation Kit (Stemcells) and subsequently labelled with 5 μmol l−1 CellTrace Violet (Invitrogen). The labelled T cells (5 × 10⁵ per well) were then seeded in a 96-well plate and activated with αCD3/CD28 beads. To assess the impact of different macrophage subsets, SLC2A1⁺ or SLC2A1⁻ TAMs (isolated from LLC tumours as described earlier) were introduced into the culture at a 1:2 ratio (TAMs:T cells). Following a 3-day coculture, the CD8⁺ T cells were collected and analysed by flow cytometry to evaluate their effector functions, proliferative capacity and apoptotic status.
IHC and immunofluorescence of human and mouse samples
Surgically resected NSCLC samples from the XQ cohort were stained with SLC2A1 and CD8A using a DoubleStain IHC kit (ZSBIO), and the spatial distribution of SLC2A1 and CD8+ T cells was analysed with bivariate Moran’s I algorithm and LISA—methodologies originally established in geospatial sciences and subsequently validated for spatial transcriptomics applications70. The spatial distributions of SLC2A1 and CD8⁺ T cells in mouse subcutaneous and orthotopic tumour tissues were further validated using multiplex immunofluorescence staining and analysed with the same spatial analytical approach.
To further evaluate and visualize the spatial distribution of SLC2A1+ TAMs and CD8+ T cells in TMEs, multiplex fluorescence imaging was performed on tissue microarrays of TJ cohort 1 and TJ cohort 2. Tissue microarrays were processed with commercial multiplex IHC reagents (PANO IHC kit, Panovue) following antigen retrieval and blocking procedures. Primary antibody cocktails contained: CD68 (1:1,500; 76437, Cell Signaling Technology), SLC2A1 (1:500; 73015, Cell Signaling Technology) and CD8α (1:500; 70306, Cell Signaling Technology). Fluorophore-conjugated secondary antibodies (PPD520/650/570, Panovue) were applied through sequential incubation, followed by 4′,6-diamidino-2-phenylindole (DAPI) nuclear counterstaining. Whole-section imaging was conducted on an Olympus VS200 microscope.
For murine tumour analysis, six tissue samples per experimental group underwent standard histological processing, including fixation, paraffin embedding and sectioning (4 μm thickness). Tissue sections were processed in accordance with the above procedure. The primary antibody contained CD8α (1:100; 98941, Cell Signaling Technology). Fluorophore-conjugated secondary antibodies (PPD570, Panovue) were applied through sequential incubation, followed by DAPI nuclear counterstaining.
Spatial correlation analysis
For each CD8+ T cell, the distance to its nearest neighbouring CD8+ T cell was calculated, along with the mean distance to its five nearest neighbouring CD8+ T cells. These spatial proximity metrics were utilized to quantify the intratumoural infiltration extent of CD8+ T cells.
To evaluate and visualize spatial correlations between two variables, we implemented the bivariate Moran’s I algorithm and LISA methodologies. For multiplex immunofluorescence staining data, cellular identities were determined using predefined marker thresholds: SLC2A1+ TAMs (dual positivity for SLC2A1 and CD68) and CD8+ T cells (CD8A positivity). Spatial co-aggregation patterns were visualized through two-dimensional density heatmaps. To quantify proximity-dependent interactions, concentric annular zones (<100 μm, 100–200 μm, 200–300 μm, 300–400 μm and 400–500 μm) were defined around each SLC2A1+ TAM and SLC2A1− TAM. For each sample, we calculated the mean CD8+ T cell density (number of CD8+ T cells/total cells) within each annular region for all SLC2A1+ TAMs.
Construction of prognostic nomogram
Patients from the TJ cohort 2 were randomly assigned to discovery and validation sets in a 6:4 ratio. Leveraging our previous findings, we developed a multivariable Cox proportional hazards model that integrated three pivotal parameters: CD8+ T cell abundance, SLC2A1+ TAM abundance and their spatial proximity. The predictive performance was assessed by receiver operating characteristic (ROC) analysis. Subsequently, we constructed a prognostic nomogram to visualize and weight the contribution of each variable. Using the composite scores generated from this nomogram, we stratified patients into distinct high-risk and low-risk categories. Finally, Kaplan–Meier survival curves were plotted, and the statistical difference in outcomes between the risk groups was evaluated with the log-rank test.
Statistics and reproducibility
The data were presented as the mean ± standard error of the mean (s.e.m.). Statistical analyses were performed utilizing GraphPad Prism (v.9.0) and RStudio (v.4.2.2). Statistical analyses are specified in the figure legends and Methods. Throughout all statistical evaluations, a threshold of P < 0.05 was established for determining statistical significance. No a priori power calculations were performed to determine sample sizes. For in vivo experiments, mice were randomly assigned to different treatment groups. No data were excluded from the analyses. Data collection and analysis were not performed blind to the conditions of the experiments. Data distribution was assumed to be normal, but this was not formally tested. In addition, representative images of haematoxylin–eosin, immunofluorescence and genotyping are from one experiment with at least three independent biological replicates.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Spatial transcriptomic data that support the findings of this study have been deposited in the Genome Sequence Archive in National Genomics Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences, under accession number HRA010548 (https://ngdc.cncb.ac.cn/gsa-human/browse/HRA010548). RNA-seq data of isolated TAMs from LLC tumour tissue are available via Zenodo at https://doi.org/10.5281/zenodo.15605493 (ref. 71). The human lung cancer data are available via the TCGA Research Network at http://cancergenome.nih.gov/. The datasets derived from GEO (https://www.ncbi.nlm.nih.gov/geo/) that support the findings of this study are available under accession numbers GSE24551, GSE14814, GSE86166, GSE72968, GSE157009, GSE58812, GSE57495, GSE85916, GSE16446, GSE157010 and GSE157011. All other data supporting the findings of this study are available from the corresponding author on reasonable request. Source data are provided with this paper.
Code availability
All data analyses in this study were performed exclusively with publicly available tools, and the corresponding parameters have been elaborated in the respective Methods sections.
Change history
19 January 2026
A Correction to this paper has been published: https://doi.org/10.1038/s41556-026-01874-3
References
Reck, M. et al. Updated analysis of KEYNOTE-024: pembrolizumab versus platinum-based chemotherapy for advanced non-small-cell lung cancer with PD-L1 tumor proportion score of 50% or greater. J. Clin. Oncol. 37, 537–546 (2019).
Gandhi, L. et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N. Engl. J. Med. 378, 2078–2092 (2018).
Mok, T. S. K. et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. Lancet 393, 1819–1830 (2019).
Reck, M. et al. Five-year outcomes with pembrolizumab versus chemotherapy for metastatic non-small-cell lung cancer with PD-L1 tumor proportion score >/= 50. J. Clin. Oncol. 39, 2339–2349 (2021).
Garassino, M. C. et al. Pembrolizumab plus pemetrexed and platinum in nonsquamous non-small-cell lung cancer: 5-year outcomes from the phase 3 KEYNOTE-189 Study. J. Clin. Oncol. 41, 1992–1998 (2023).
de Castro, G. Jr. et al. Five-year outcomes with pembrolizumab versus chemotherapy as first-line therapy in patients with non-small-cell lung cancer and programmed death ligand-1 tumor proportion score >/= 1% in the KEYNOTE-042 study. J. Clin. Oncol. 41, 1986–1991 (2023).
Novello, S. et al. Pembrolizumab plus chemotherapy in squamous non-small-cell lung cancer: 5-year update of the phase III KEYNOTE-407 study. J. Clin. Oncol. 41, 1999–2006 (2023).
Mino-Kenudson, M. et al. Predictive biomarkers for immunotherapy in lung cancer: perspective from the International Association for the Study of Lung Cancer Pathology Committee. J. Thorac. Oncol. 17, 1335–1354 (2022).
Sharma, P., Hu-Lieskovan, S., Wargo, J. A. & Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723 (2017).
Bader, J. E. et al. Obesity induces PD-1 on macrophages to suppress anti-tumour immunity. Nature 630, 968–975 (2024).
Gordon, S. R. et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 545, 495–499 (2017).
Kersten, K. et al. Spatiotemporal co-dependency between macrophages and exhausted CD8+ T cells in cancer. Cancer Cell 40, 624–638 (2022).
Waibl Polania, J. et al. Antigen presentation by tumor-associated macrophages drives T cells from a progenitor exhaustion state to terminal exhaustion. Immunity 58, 232–246 (2025).
Kuratani, A. et al. Platelet factor 4-induced TH1-Treg polarization suppresses antitumor immunity. Science 386, eadn8608 (2024).
Mulder, K. et al. Cross-tissue single-cell landscape of human monocytes and macrophages in health and disease. Immunity 54, 1883–1900 (2021).
Ma, R. Y., Black, A. & Qian, B. Z. Macrophage diversity in cancer revisited in the era of single-cell omics. Trends Immunol. 43, 546–563 (2022).
Jin, R., Neufeld, L. & McGaha, T. L. Linking macrophage metabolism to function in the tumor microenvironment. Nat. Cancer 6, 239–252 (2025).
Reinfeld, B. I. et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature 593, 282–288 (2021).
Chang, C. H. et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 162, 1229–1241 (2015).
De Leo, A. et al. Glucose-driven histone lactylation promotes the immunosuppressive activity of monocyte-derived macrophages in glioblastoma. Immunity 57, 1105–1123 (2024).
Morrissey, S. M. et al. Tumor-derived exosomes drive immunosuppressive macrophages in a pre-metastatic niche through glycolytic dominant metabolic reprogramming. Cell Metab. 33, 2040–2058 (2021).
Chu, X., Tian, Y. & Lv, C. Decoding the spatiotemporal heterogeneity of tumor-associated macrophages. Mol. Cancer 23, 150 (2024).
Zheng, X. et al. Spatial density and distribution of tumor-associated macrophages predict survival in non-small cell lung carcinoma. Cancer Res. 80, 4414–4425 (2020).
Sattiraju, A. et al. Hypoxic niches attract and sequester tumor-associated macrophages and cytotoxic T cells and reprogram them for immunosuppression. Immunity 56, 1825–1843 (2023).
Wang, W. et al. Identification of hypoxic macrophages in glioblastoma with therapeutic potential for vasculature normalization. Cancer Cell 42, 815–832 (2024).
van Elsas, M. J. et al. Immunotherapy-activated T cells recruit and skew late-stage activated M1-like macrophages that are critical for therapeutic efficacy. Cancer Cell 42, 1032–1050 (2024).
Ayers, M. et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest. 127, 2930–2940 (2017).
Mariathasan, S. et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548 (2018).
Giles, J. R., Globig, A. M., Kaech, S. M. & Wherry, E. J. CD8+ T cells in the cancer-immunity cycle. Immunity 56, 2231–2253 (2023).
Siebeneicher, H. et al. Identification and optimization of the first highly selective GLUT1 inhibitor BAY-876. ChemMedChem 11, 2261–2271 (2016).
Cao, S., Chen, Y., Ren, Y., Feng, Y. & Long, S. GLUT1 biological function and inhibition: research advances. Future Med. Chem. 13, 1227–1243 (2021).
Salcher, S. et al. High-resolution single-cell atlas reveals diversity and plasticity of tissue-resident neutrophils in non-small cell lung cancer. Cancer Cell 40, 1503–1520 (2022).
Lyu, A. et al. Evolution of myeloid-mediated immunotherapy resistance in prostate cancer. Nature 637, 1207–1217 (2025).
Chu, T., Wang, Z., Pe’er, D. & Danko, C. G. Cell type and gene expression deconvolution with BayesPrism enables Bayesian integrative analysis across bulk and single-cell RNA sequencing in oncology. Nat. Cancer 3, 505–517 (2022).
Pavlova, N. N., Zhu, J. & Thompson, C. B. The hallmarks of cancer metabolism: still emerging. Cell Metab. 34, 355–377 (2022).
Wu, Q. et al. GLUT1 inhibition blocks growth of RB1-positive triple negative breast cancer. Nat. Commun. 11, 4205 (2020).
Miller, Z. A. et al. GLUT1 inhibitor BAY-876 induces apoptosis and enhances anti-cancer effects of bitter receptor agonists in head and neck squamous carcinoma cells. Cell Death Discov. 10, 339 (2024).
Guo, L. et al. Inhibition of carbohydrate metabolism potentiated by the therapeutic effects of oxidative phosphorylation inhibitors in colon cancer cells. Cancers 16, 1399 (2024).
Sawayama, H. et al. Glucose transporter 1 regulates the proliferation and cisplatin sensitivity of esophageal cancer. Cancer Sci. 110, 1705–1714 (2019).
Wang, K. et al. Sonodynamic activated nanoparticles with Glut1 inhibitor and cystine-containing polymer stimulate disulfidptosis for improved immunotherapy in bladder cancer. Biomaterials 319, 123178 (2025).
Li, T. et al. Metabolism/immunity dual-regulation thermogels potentiating immunotherapy of glioblastoma through lactate-excretion inhibition and PD-1/PD-L1 blockade. Adv. Sci. 11, e2310163 (2024).
Ren, X. et al. Inhibition of glycolysis-driven immunosuppression with a nano-assembly enhances response to immune checkpoint blockade therapy in triple negative breast cancer. Nat. Commun. 14, 7021 (2023).
Vitale, I., Manic, G., Coussens, L. M., Kroemer, G. & Galluzzi, L. Macrophages and metabolism in the tumor microenvironment. Cell Metab. 30, 36–50 (2019).
Chen, D., Zhang, X., Li, Z. & Zhu, B. Metabolic regulatory crosstalk between tumor microenvironment and tumor-associated macrophages. Theranostics 11, 1016–1030 (2021).
Jia, Q. et al. Local mutational diversity drives intratumoral immune heterogeneity in non-small cell lung cancer. Nat. Commun. 9, 5361 (2018).
Liu, S. et al. Spatial maps of T cell receptors and transcriptomes reveal distinct immune niches and interactions in the adaptive immune response. Immunity 55, 1940–1952 (2022).
Yang, B. et al. Spatial heterogeneity of infiltrating T cells in high-grade serous ovarian cancer revealed by multi-omics analysis. Cell Rep. Med. 3, 100856 (2022).
Joshi, K. et al. Spatial heterogeneity of the T cell receptor repertoire reflects the mutational landscape in lung cancer. Nat. Med. 25, 1549–1559 (2019).
Tanaka, M. et al. Tumor cell heterogeneity drives spatial organization of the intratumoral immune response. J. Exp. Med. 222, e20242282 (2025).
Li, J. et al. Tumor cell-intrinsic factors underlie heterogeneity of immune cell infiltration and response to immunotherapy. Immunity 49, 178–193 e7 (2018).
Klein, L. et al. Spatial tumor immune heterogeneity facilitates subtype co-existence and therapy response in pancreatic cancer. Nat. Commun. 16, 335 (2025).
Di Pilato, M. et al. CXCR6 positions cytotoxic T cells to receive critical survival signals in the tumor microenvironment. Cell 184, 4512–4530 (2021).
Jansen, C. S. et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 576, 465–470 (2019).
Meiser, P. et al. A distinct stimulatory cDC1 subpopulation amplifies CD8+ T cell responses in tumors for protective anti-cancer immunity. Cancer Cell 41, 1498–1515 (2023).
Elewaut, A. et al. Cancer cells impair monocyte-mediated T cell stimulation to evade immunity. Nature 637, 716–725 (2025).
Baldominos, P. et al. Quiescent cancer cells resist T cell attack by forming an immunosuppressive niche. Cell 185, 1694–1708 (2022).
Chen, D. et al. DNASE1L3-expressing dendritic cells promote CD8+ T cell function and anti-PD-(L)1 therapy efficacy by degrading neutrophil extracellular traps. Cancer Cell 43, 1758–1775 (2025).
Magen, A. et al. Intratumoral dendritic cell-CD4+ T helper cell niches enable CD8+ T cell differentiation following PD-1 blockade in hepatocellular carcinoma. Nat. Med. 29, 1389–1399 (2023).
Chen, J. H. et al. Human lung cancer harbors spatially organized stem-immunity hubs associated with response to immunotherapy. Nat. Immunol. 25, 644–658 (2024).
Duraiswamy, J. et al. Myeloid antigen-presenting cell niches sustain antitumor T cells and license PD-1 blockade via CD28 costimulation. Cancer Cell 39, 1623–1642 (2021).
Helmink, B. A. et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577, 549–555 (2020).
Vanhersecke, L. et al. Mature tertiary lymphoid structures predict immune checkpoint inhibitor efficacy in solid tumors independently of PD-L1 expression. Nat. Cancer 2, 794–802 (2021).
Yang, Y. et al. Updated overall survival data and predictive biomarkers of sintilimab plus pemetrexed and platinum as first-line treatment for locally advanced or metastatic nonsquamous NSCLC in the phase 3 ORIENT-11 study. J. Thorac. Oncol. 16, 2109–2120 (2021).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Hafemeister, C. & Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296 (2019).
Barbie, D. A. et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108–112 (2009).
Zappasodi, R. et al. CTLA-4 blockade drives loss of Treg stability in glycolysis-low tumours. Nature 591, 652–658 (2021).
Balar, A. V. et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. Lancet 389, 67–76 (2017).
Charoentong, P. et al. Pan-cancer immunogenomic analyses reveal genotype–immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 18, 248–262 (2017).
Zormpas, E., Queen, R., Comber, A. & Cockell, S. J. Mapping the transcriptome: realizing the full potential of spatial data analysis. Cell 186, 5677–5689 (2023).
Wang, L. & Chu, H. Data for ‘SLC2A1+ tumour-associated macrophages spatially control CD8+ T cell function and drive immunotherapy resistance in non-small cell lung cancer’. Zenodo https://doi.org/10.5281/zenodo.15605493 (2025).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (grant numbers 82472969 and 62131009 to Q.C.; 82525107 to B.Z.; and 82241233 to Q.J.) and Natural Science Foundation of Chongqing (grant number CSTB2023NSCQ-JQX0026 to Q.J.).
Author information
Authors and Affiliations
Contributions
Q.C., B.Z. and Q.J. designed the study and provided overall supervision. Experimental work and subsequent data analysis were carried out by L.W., D.C., Y.W., J.J., L.P., S.X., L.H. and X.D. Omics data analysis was performed by H.C. Clinical data provision and analysis were managed by L.L., F.L., S.H., Z.J., D.Z., W.F.F. and T.J. Technical support was contributed by H.Y.C. and X.L. The paper was drafted by L.W., H.C., D.C., Q.J., B.Z. and Q.C, and all authors participated in reviewing the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Cell Biology thanks Sergei Grivennikov and the other, anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Validation analyses for SLC2A1 correlations with ICB therapy responsiveness.
a,b, Association patterns of SLC2A1 with the ICB-responsive and IMvigor210 gene signatures in TCGA (a) and GEO datasets (b). c,d, Analysis of immune infiltration demonstrating the correlation between SLC2A1 and total T cell or CD8+ T cell-specific infiltration levels across TCGA (c) and GEO datasets (d). p-values were calculated using a two-sided Pearson’s correlation coefficient.
Extended Data Fig. 2 SLC2A1 expression is exclusively correlated with CD8+ T cells in human NSCLC tumors.
a,b,c, Spatial correlations between SLC2A1 expression and CD274 expression (a), PDCD1 expression (b), and CD4⁺ T cell transcript signature scores (c) across three NSCLC spatial transcriptome datasets. p-values were calculated using a two-sided Pearson’s correlation coefficient.
Extended Data Fig. 3 Spatial correlation of SLC2A1 and CD8+ T cells validated by immunohistochemistry.
Local Indicators of Spatial Association (LISA) cluster maps showing spatial distribution patterns between SLC2A1+ regions and CD8+ T cell infiltration in individual NSCLC samples from the XQ cohort (n = 38). p-values for the bivariate Moran’s I were determined using a one-sided test.
Extended Data Fig. 4 The therapeutic efficacy of SLC2A1 inhibition is dependent on CD8+ T cells.
a, LLC tumor weights from BAY-876- or vehicle-treated tumor-bearing mice (Control group (n = 8) and BAY-876-treated group (n = 7)). b, Gating strategy used to identify tumor-infiltrating immune cells. c,d,e,f,g, Flow cytometric analysis was performed to evaluate the proportion and number of Treg cells (c), NK cells (d), MDSCs (e), TAMs (f) and DCs (g) from BAY-876- or vehicle-treated LLC tumor-bearing mice (Control group (n = 8) and BAY-876-treated group (n = 7)). h, Flow cytometry analysis of the CD8+ T cell proportion among CD45+ T cells from BAY-876- or vehicle-treated B16-F10 tumor-bearing mice (Control group (n = 8) and BAY-876-treated group (n = 11)). i, CD8⁺ T cell populations expressing IFN-γ, Granzyme B, or Perforin from BAY-876- or vehicle-treated B16-F10 tumor-bearing mice (Control group (n = 8) and BAY-876-treated group (n = 11)). j, Validation of the depletion efficacy of αCD8 and αCD4 antibodies. k, Tumor growth curves following αCD8 intervention in B16-F10 tumor model (BAY-876 group (n = 5) and the other groups (n = 4 each)). Data are reported as mean ± s.e.m., and group differences were assessed by a two-tailed unpaired Student’s t test.
Extended Data Fig. 5 BAY-876 administration enhances efficacy of αPD-L1 treatment.
a,b,c,d,e,f, Summarized data of the proportion (left) and total numbers (right) of CD45+ cells (a), CD4+ T cells (b), NK cells (c), MDSCs (d), TAMs (e), and DCs (f) in each indicated group from LLC tumors following BAY-876 treatment with or without αPD-L1 antibody therapy (Combination group (n = 6) and the other groups (n = 5 each)). g, MC38 allograft tumor growth kinetics (left) and the endpoint measurement of tumor mass (right) in C57BL/6 J mice following BAY-876 treatment with or without αPD-L1 antibody therapy (αPD-L1 group (n = 6) and the other groups (n = 8 each)). h, B16-F10 allograft tumor growth kinetics (left) and the endpoint measurement of tumor mass (right) in C57BL/6 J mice following BAY-876 treatment with or without αPD-L1 antibody therapy (n = 5). Data are reported as mean ± s.e.m., and group differences were assessed by a two-tailed unpaired Student’s t test.
Extended Data Fig. 6 Tumor-associated macrophages determine the efficacy of BAY-876.
a, The pie chart shows the proportion of SLC2A1-positive cells within tumor, immune, and stromal cell populations. b,c,d,e, Flow cytometry assays of glucose uptake (using 2-NBDG) by CD3+ T cells (b), CD4+ and CD8+ T cells (c), myeloid cells (d), myeloid-derived suppressive cells and dendritic cells (e) (Control group (n = 5) and BAY-876-treated group (n = 4)). f,g, Validation of Slc2a1 knockdown efficiency in LLC cells at the mRNA (f) and protein (g) levels (n = 4). The sh2 cell line, which had the highest knockdown efficiency, was subsequently selected to establish in vivo tumor models. h, Growth curves (by volume) of LLC-Slc2a1KD or LLC-vector tumors in nude mice (n = 7). i, Flow cytometry assays and summarized data of 2-NBDG (glucose analog) uptake by CD3+ T cells, CD4+ T cells, and CD8+ T cells in LLC Slc2a1-knockdown tumor models (n = 7). j, Percentage (left) and number (right) of TAMs in each indicated group (n = 6). k, The verification of myeloid-specific Slc2a1-knockout in mice by genotyping. l, Flow cytometry analysis of SLC2A1 expression on TAMs from allograft LLC tumors in Slc2a1fl/fl and Slc2a1cko mice (Slc2a1fl/fl group (n = 3) and Slc2a1cko group (n = 4)). MFI, mean fluorescence intensity. Data are reported as mean ± s.e.m., and group differences were assessed by a two-tailed unpaired Student’s t test.
Extended Data Fig. 7 Slc2a1 conditional knockout in macrophages impairs the immunosuppressive function of TAMs.
a,b,c, Quantification of Arginine metabolism (a), IL-10 pathway (b) and TGF-β pathway (c) of TAMs derived from LLC tumors of Slc2a1fl/fl or Slc2a1cko mice (n = 4). d, Comparison of metabolic immunosuppressive ARG1 expression, and immunosuppressive cytokines expression (TGF-β, IL-10, IL-1β) in TAMs from BAY-876- or vehicle-treated LLC tumor-bearing mice (Control group (n = 8) and BAY-876-treated group (n = 7)). MFI, mean fluorescent intensity. e, CD8+ T cells were co-cultured with SLC2A1+ and SLC2A1− TAMs. Flow cytometry was conducted to measure the glucose uptake by sorted CD8+ T cells (n = 3). f, Analysis of glucose uptake in T cell subsets from TAM-specific Slc2a1-knockout LLC models (Slc2a1fl/fl group (n = 6) and Slc2a1cko group (n = 5)). Flow cytometry was used to evaluate 2-NBDG incorporation in CD3⁺, CD4⁺, and CD8⁺ T cells, with summarized data. g, Flow cytometry analysis compared the migration of CD8+ T cells in transwell assay co-cultured with SLC2A1+ and SLC2A1− TAMs (n = 3). Data are reported as mean ± s.e.m. The two-tailed Wilcoxon Rank-Sum test was applied in (a,b,c), and the two-tailed unpaired Student’s t test was used for (d-g). In the box plots, the center line represents the median, the box spans are the first and third quartiles, and whiskers denote 1.5 times interquartile range.
Supplementary information
Supplementary Table 1
Summary of in-house and publicly available cohorts.
Supplementary Table 2
Sequences of primers used in this study.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Statistical source data.
Source Data Extended Data Fig.6
Unprocessed gels.
Source Data Extended Data Fig.2
Statistical source data.
Source Data Extended Data Fig.4
Statistical source data.
Source Data Extended Data Fig.5
Statistical source data.
Source Data Extended Data Fig.6
Statistical source data.
Source Data Extended Data Fig.7
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, L., Chu, H., Chen, D. et al. SLC2A1+ tumour-associated macrophages spatially control CD8+ T cell function and drive resistance to immunotherapy in non-small-cell lung cancer. Nat Cell Biol (2026). https://doi.org/10.1038/s41556-025-01840-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41556-025-01840-5
This article is cited by
-
Metabolic borders shape immune resistance
Nature Cell Biology (2026)
