
Abstract
The tissue origin(s) and the earliest viral dynamics of HIV rebound after antiretroviral therapy (ART) remain unclear. Here, using barcoded SIVmac239 in rhesus macaques (n = 24), we defined the distribution of barcode-specific viral RNA expression in tissues during ART (n = 6) and then assessed initial clonal rebound 5 and 7 days after ART cessation by identifying barcodes in individual tissues that exceeded the 99th percentile of the on-ART distribution (‘outliers’). In 4 of 11 aviraemic and 6 of 7 viraemic animals, 32 such outlier barcodes were identified. Sixteen of these barcodes were also identified in rebound viraemia, confirming specific tissues as rebound origin and early amplification sites. Overall, 27 of the 32 outlier barcodes were determined to reflect rebound origins, of which 96% were in the gastrointestinal tract (26%) or gastrointestinal tract-associated lymphoid tissues (70%). These results indicate that distinct tissue sites differentially support post-ART viral rebound, with potential therapeutic implications for interventions designed to prevent or control these events.
Similar content being viewed by others


Main
Antiretroviral therapy (ART) has the capacity to drastically suppress or eliminate ongoing replication of human immunodeficiency virus (HIV) and simian immunodeficiency virus (SIV), yet residual virus-infected cells persist long term in both humans and macaques and will, with few exceptions, reignite spreading and progressive infection when ART is discontinued1,2,3,4,5. The long-term, stable viral population that can reignite infection after ART—the rebound competent viral reservoir (RCVR)—is complex and includes multiple subsets of cluster of differentiation 4+ (CD4+) T cells particularly including clonal expansions of CD4+ memory T cells, and possibly macrophages that are widely distributed throughout the lymphoid tissues of the body2,4,6,7,8,9,10. To mediate post-ART viraemic rebound, RCVR virus must be genetically intact, replication competent and inducible in vivo8,11,12; however, viral rebound is not a simultaneous universal resumption of virus replication from the entire RCVR but rather begins with an initial oligoclonal outgrowth of virus from a limited number of infected cells that subsequently spreads throughout the body, reseeding systemic infection4,13,14. Virus expression, including viral RNA (vRNA) and protein production and even release of intact virions from infected cells, is an ongoing process during ART15 because ART acts not by blocking virus expression by already infected cells but by preventing de novo infection of susceptible target cells1,16. Together, these observations suggest that local cellular, anatomic and immunologic tissue milieus that support the persistence and outgrowth of the RCVR might also contribute to regulation of post-ART rebound and that this local regulation restricting and/or promoting viral reactivation and initial viral spread after ART discontinuation represents a potential point of intervention for therapies designed to interfere with the rebound process. Despite the potential importance of understanding the origins of post-ART viral rebound, little is known about these early local events in tissues, with previous work primarily focused on matching on-ART/pre-rebound sequence- or barcode-defined tissue viral lineages with those lineages contributing to overt post-ART viraemia, with variable success13,14,17,18,19.
Identifying rebound origin sites in vivo is a daunting objective, given that the RCVR may be found in hundreds of different tissues throughout the body10,13,20, any of which potentially represent sites of origin and early amplification for rebound viraemia. Moreover, the initial events leading to rebound are temporally inconsistent, with the time between ART washout and systemic viral spread varying between individuals, making the task of observing initial viral rebound processes even more challenging. Despite recent advances, imaging technology is currently unable to rigorously distinguish the earliest focal clonal viral outgrowth that would reflect origin sites leading to viraemic rebound21,22,23. To identify the initial tissue origins leading to rebound plasma viraemia, we used barcoded SIV24,25 in a calibrated rhesus macaque model of SIV infection4, including through extended ART suppression and post-ART rebound, along with strategically timed, on- and off-ART necropsies with comprehensive tissue analysis. We first developed and validated criteria for recognizing initial viral replication and spread in tissues and then used this approach to identify the earliest sites of local post-ART viral replication leading to viraemic rebound and subsequent viral dissemination. Our results indicate that with our approach it is possible to robustly identify the early stages of local viral spread leading to post-ART viral rebound and that these early processes preferentially occur in the gastrointestinal (GI) tract and GI tract-associated lymphoid tissues.
Use of barcoded SIV to characterize processes leading to post-ART viral rebound in rhesus macaques
This study was designed to identify the anatomic sites of early viral replication and spread that lead to viraemic rebound after ART cessation using a calibrated rhesus macaque model with barcoded SIV. We have previously shown that intravenous administration of SIVmac239M (a virus stock containing ~10,000 viral variants of a SIVmac239 infectious molecular clone, each of which contains a unique 34-base ‘barcode’ between the vpx and vpr genes), allows deep sequencing-based genetic tracking in vivo over time of distinct individual, phenotypically equivalent viral lineages (each lineage referred to as a viral clonotype) varying only in their barcode sequences4,24,25. When SIVmac239M-infected macaques are started on ART at day 9 after infection, high-level acute-phase viraemia is consistently observed, followed by rapid and complete viral control, with establishment of a saturated RCVR, and predictable rebound dynamics with 1–2 distinct barcode-defined reactivation events per day leading to measurable rebound plasma viraemia at a median 7 days after ART (plasma viral load (PVL) > 15 copies per ml)4. We hypothesized that individual viral clonotypes undergoing initial post-ART viral replication and spread would be identifiable by manifesting a detectable increase in local tissue barcode-specific cell-associated vRNA levels relative to viral DNA (vDNA) because in spreading SIV infections, productively infected cells can express up to thousands of RNA copies for every DNA provirus26.
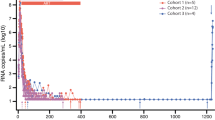
To test this hypothesis, we established a cohort of 24 macaques started on ART 9 days after intravenous infection with 5,000 infectious units of SIVmac239M (Fig. 1a and Supplementary Table 1). Measured PVLs peaked at 9 or 12 days post infection (d.p.i.) with a geometric mean of 7 log10 RNA copies per ml containing a median of 422 distinct barcodes per macaque (Fig. 1b,c). After ART initiation, PVLs declined to <15 copies per ml in almost all macaques by 15 weeks post infection (w.p.i.) and were below 1 copy per ml at most time points throughout the remainder of the 70 week ART treatment period (Fig. 1b). Peripheral blood mononuclear cell (PBMC)-associated and biopsy tissue cell-associated vRNA and vDNA (including peripheral lymph node (pLN), mesenteric lymph node (mLN), spleen, liver, bone marrow, duodenal and rectal mucosa), mirrored PVL with high vRNA and vDNA levels at day 12, with a subsequent on-ART decline of 3–6 log10 copies per 106 cells for vRNA, compared with only 1–2 log10 copies per 106 cells for vDNA (Fig. 1d,e), consistent with previous observations of SIV on-ART dynamics4,5. Despite this early and complete control of SIV replication, this level of infection was sufficient to induce the development and maintenance of both CD4+ and CD8+ T cell responses to SIV in the blood and representative effector site (lung airspace) and lymphoid tissues (pLN, mLN, spleen) of all study macaques (Extended Data Fig. 1). At the end of the 70-week ART phase, the 24 macaques were divided into 3 balanced groups based on peak acute phase PVL, time to viral suppression, number of viral blips and overall virus levels in PBMCs and on-ART biopsies (Extended Data Figs. 2 and 3). Six macaques were necropsied on ART (to establish a baseline for virologic parameters during ART-suppressed infection), and 9 macaques each were necropsied at day 5 and day 7 after ART cessation to capture the predicted earliest rebound events (Fig. 1a). Among the macaques necropsied at days 5 and 7 after ART, 2 of 9 and 5 of 9, respectively, manifested measurable rebound viraemia (≥5 copies per ml) at necropsy (Fig. 1f). In the macaques with measurable plasma viral rebound, PVLs were less than 30 copies per ml at necropsy in 6 of 7 animals with the remaining macaque showing more advanced plasma viral rebound at 4,700 copies per ml. Barcode sequencing showed 2–5 rebound barcodes in plasma of low-PVL macaques, whereas 9 barcodes were identified in the plasma of the higher-PVL macaque (Extended Data Fig. 4).

a, Twenty-four rhesus macaques were intravenously (IV) infected with barcoded SIVmac239M with daily ART (TDF, tenofovir disoproxil fumarate; FTC, 2′,3′-dideoxy-5-fluoro-3′-thiacytidine (emtricitabine); DTG, dolutegravir) beginning at 9 d.p.i. Macaques were necropsied (Nx) either on ART (n = 6), at 5 days after ART (n = 9) or at 7 days after ART (n = 9). b, Blood PVL (assay threshold 15 copies per ml through 36 w.p.i., then 1 copy per ml thereafter; indicated by a dotted grey line) reveals the characteristic SIVmac239 initial viral growth rate (average 1.6, range 1.3–1.8) and on-ART viral first-phase decay rates (0.75, range 0.59–0.91). Bold lines indicate log mean PVL for all macaques. c, Peak PVL plasma samples (12 d.p.i.) were used to identify the number of unique barcodes initiating infection per macaque (median 422, range 111–649). d, vDNA and vRNA were quantified from PBMCs from all 24 study macaques at 12 d.p.i., 21 w.p.i. and 66 w.p.i. and were compared between the three time points (two-sided Wilcoxon signed-rank test; with P values indicated). e, Both vDNA and vRNA from biopsy tissues taken from all 24 macaques at 12 d.p.i., 21 w.p.i. and 66 w.p.i. (pLN, mLN, spleen, bone marrow (BM), duodenum (Duo), rectum and liver) showed typical declines over time. f, After 72 w.p.i., 18 macaques were released from ART with daily PVLs obtained, and the detectable rebounding barcode lineages (BCs) identified; 7 of 18 macaques were viraemic (≥5 copies per ml) at the time of necropsy (2 of 9 from animals necropsied 5 days off-ART and 5 of 9 from animals necropsied 7 days off-ART). There was a total of 27 barcodes detectable in blood at necropsy (mean 3.86; range 2–9).
In the 24 macaques studied here, an average of 93 tissue samples (range 86–123; Supplementary Table 1) across ~60 distinct tissue sites were collected and analysed from each macaque, including the GI tract, GI tract-associated lymphoid tissues (lymphoid tissues draining or associated with non-sterile mucosal surfaces of the GI tract), non-GI tract-associated lymphoid tissues (other lymph nodes (LNs), as well as spleen, thymus, bone marrow) and non-lymphoid tissues, using a consistent tissue collection protocol across all study animals (Supplementary Table 2). Our goal was extensive sampling of both major and minor sites known to harbour residual virus on ART10,13,20. Each collected sample was examined for overall SIV cell-associated vRNA and vDNA with the vast majority of GI tract, and GI tract- and non-GI tract-associated lymphoid tissues showing detectable vDNA and most with detectable vRNA (Extended Data Fig. 5) at levels that were quite similar to those observed in on-ART biopsies (Extended Data Fig. 3). Both cell-associated vDNA and vRNA were detected less frequently in non-lymphoid tissues, likely reflecting the lower proportion of potential viral target cells in these sites.
Tissue viral barcode analysis on ART defines the vRNA expression landscape during full viral suppression
Barcode sequencing was performed on vRNA- and vDNA-positive tissue specimens, with barcode-specific analyses focused initially on the 6 macaques that were necropsied on ART. This sequencing showed that the majority of detected barcodes across all types of tissue were vDNA+ only (that is, without detection of matching barcode vRNA expression) (Fig. 2a,b). The percentage of barcodes expressing vRNA was higher in GI tract and in GI tract-associated lymphoid tissues (16–17%), compared with non-GI tract lymphoid tissues (9%) and non-lymphoid tissues (3%). Part of this difference can be attributed to the observation that barcodes that are present at higher vDNA levels (more cells infected with a given barcode variant in the population), even with a fixed proportion expressing vRNA, are more likely to have sufficient numbers of these cells being transcriptionally active and therefore detected as vDNA+/vRNA+. Reflecting this, between 2 and 3 log10 copies per barcode for vDNA, there is a notable shift in the population from vRNA expression likely originating from single cells (each with a distinct barcode) to apparent production of vRNA from multiple cells expressing the same barcode. This is most notable in the GI tract-associated lymphoid tissues, where proportionately more barcodes have vDNA levels exceeding 1,000 copies compared with all other tissue groups (347 out of 15,632 versus 101 out of 16,412; two-sided P < 2 × 10−16; two-proportion z-test; Fig. 2a). Mixed-effects logistic regression indicates that while barcode vDNA level is the strongest predictor of vRNA expression (P < 0.001; two-sided Wald test), tissue type is also a significant explanatory variable, with barcodes in GI tract and GI tract-associated lymphoid tissues having significantly higher odds of being vRNA+ for a given vDNA level than barcodes in non-GI tract-associated lymphoid tissues (P < 0.001; two-sided Wald test; Fig. 2c and Supplementary Table 3). Taken together, these observations suggest that at the level of barcode-defined clonotypic populations, the vDNA+ infected cell pool is larger or more active or both in GI tract and GI tract-associated lymphoid compared with other sites.

a, vRNA (cDNA) and vDNA from individual samples with >50 available viral templates were sequenced to determine the viral barcode. Each barcode from each individual tissue sample is plotted per tissue group with barcodes expressing vRNA in only 3–17% of the total barcodes per tissue. b, Histogram (across all tissue groups) of the number of vDNA+-only counts compared with the vDNA+/vRNA+ counts based on the total vDNA count per viral barcode lineage showing that the more frequently a barcode is found in the vDNA, the more likely it is also to be expressed as vRNA. c, The marginal effect of the vDNA level (log10) stratified by type of tissue on the predicted probability of vRNA expression for a logistic regression model including animal-level random effects. For a given vDNA level, barcodes in GI tract tissue have the highest probability of being vRNA+, followed by GI tract-associated lymphoid tissues, non-GI LN and non-lymphoid tissues. The shaded coloured bands around each coloured data line represent the 95% confidence intervals, indicating uncertainty around the estimated marginal effects, with odd ratios for types of tissue relative to non-GI LNs shown in the figure inset with P values computed using a two-sided Wald test (GI tract, ***P < 2 × 10−16; GI LN, ***P < 2 × 10−16; non-lymphoid (non-lymph), P = 0.172, non-significant (NS)).
To develop criteria for detection of rare barcode-defined viral lineages contributing to initial viral outgrowth in tissues after ART cessation (rebounding viral clonotypes that are the progeny of presumptively unique initial infection events from a given barcode present in the viral inoculum and identified in each animal’s primary peak plasma), the distribution of vRNA and vDNA expression levels per barcode measured across all tissues in the 6 macaques necropsied on fully suppressive ART was plotted (Fig. 3a). A piecewise linear regression was used to define the relationship between the levels of vRNA expression and levels of vDNA per barcode. A 99% prediction interval (99% PI) was then determined for this distribution, defining the expected range of barcode vRNA expression for a given barcode vDNA level in macaques on fully suppressive ART (that is, the levels of vRNA expressed without active viral replication/spread). As expected, the barcode-specific vRNA levels from the late-phase, on-ART (pre-necropsy) tissue biopsies for all 24 macaques in the study also fell within this 99% PI (Extended Data Fig. 6). As indicated above, the earliest post-ART stages leading to viraemic rebound are pauci-clonal, and thus the transition from an ART-suppressed, non-replicative infection to active local viral replication with tissue spread would be expected to involve one or more individual barcode clonotypes with a measured increase in vRNA expression with initially little change in vDNA for those barcodes, given the potential for a single proviral vDNA to give rise to multiple logs of vRNA copies. Barcodes showing local viral replication with spread would therefore be detected as outliers—individual barcodes with RNA expression exceeding the 99% PI determined from the on-ART tissue samples.

a, The per barcode vDNA and vRNA levels across all tissues for each on-ART macaque (n = 6) reveal a piecewise linear regression (purple line) with a breakpoint at 2.77 log10 copies per barcode of DNA which was used to generate a 99% PI (blue lines with shaded area) for how much total vRNA is expected for a given vDNA level under conditions of full ART suppression. b, The vRNA and vDNA values show 7 of 11 off-ART, aviraemic macaques were within the 99% PI. c, One off-ART, aviraemic animal (AV1) had evidence of three distinct viral barcodes (blue, BC.93; red, BC.68; purple, BC.194) outside the prediction interval in one of two distinct tissue pieces (lower jejunum and submandibular LN). Inset: barcode proportional distribution at peak primary viraemia. Barcode 93 is the dominant peak plasma barcode which is also highly enriched in necropsy tissues. Barcode 68 was the second highest peak plasma barcode lineage and shows a highly enriched vDNA+ population in necropsy tissues with ~66% of tissues positive for vRNA. By contrast, barcode 194 is not prominently represented in the peak plasma and shows outlier vRNA expression in a single necropsy tissue (submandibular LN). d, For the remaining 3 off-ART, aviraemic macaques (AV2–AV4), the percentage of vDNA+, vRNA+ positive barcodes ranged from 12% to 17% with evidence of 1–3 outlier barcode lineages present at high peak plasma proportions (right) and total necropsy tissue vDNA+ population (BC.2022, BC.4152, BC.452 and BC.2) while BC.268 in AV4 is an outlier barcode despite a modest to low level of overall viral burden at peak and at necropsy.
Identification of presumptive tissue origin sites of initial clonotypic post-ART viral rebound
We next analysed tissue samples obtained using the same necropsy/tissue sampling protocol from the 11 aviraemic macaques that were necropsied at 5 days (n = 7) or 7 days (n = 4) off-ART, each with PVL < 1 copy per ml at necropsy. As expected, given the limited time off ART and aviraemic status, the per barcode vRNA and vDNA distribution from 7 of 11 aviraemic macaques was indistinguishable from the distribution from the macaques necropsied on ART (Fig. 3b), suggesting that in the tissues examined in these 7 macaques, there had not yet been any increase in expression of the vRNA of any individual barcode above the 99% PI determined for the on-ART group. However, in the other 4 of these aviraemic, off-ART macaques, 8 individual barcodes in 6 distinct tissue samples were identified as outliers that exceeded the 99% PI (Fig. 3c,d). The number of outlier barcodes detected per animal was 3, 3, 1 and 1 (AV1, AV2, AV3 and AV4, respectively) all originating from the GI tract or GI tract-associated lymphoid tissues. Interestingly, 3 of these outlier barcodes (BC.93, BC.68, BC.2022) were identified near the top of the primary infection peak plasma virus barcode distribution. On the other end of the spectrum, barcodes BC.194 and BC.268 were not dominant barcodes with no other vRNA+ tissue sites except for the noted outlier. Despite these differences in the distribution within barcode hierarchies for individual macaques, only 8 barcodes (of the over 4,500 barcodes detected in these 11 macaques) and 6 tissue sites (of the over 1,000 tissues analysed) show evidence of vRNA levels higher than expected for viral expression without viral replication (that is, greater than the 99% PI), likely reflecting early off-ART viral replication with only local, within-tissue spread and therefore constituting potential rebound origin and/or early amplification sites. However, as these macaques were aviraemic at necropsy, the increased barcode vRNA detected at these sites cannot be explicitly linked to systemic, viraemic rebound.
However, if outlier barcodes in specific tissue sites reflect local replication and spread as a precursor to post-ART viraemic rebound, we predicted that the outlier barcodes in such tissues should overlap with the barcodes detected in peripheral blood in the macaques with early post-ART viraemia. Indeed, among the 6 macaques with low-level post-ART viraemia (LV1–LV6; <30 copies per ml rebound PVL), we detected 16 outlier barcodes in one or more tissues in 5 macaques (LV2–LV6), of which 9 were also identified in the low-level early rebound viraemia for 4 of these animals (LV3–LV6) (Fig. 4). Conversely, of the 18 barcodes detected in rebound plasma of LV1–LV6, half were also identified as outlier barcodes in one or more tissues examined. All but 1 of the 16 outlier barcodes detected in these low-viraemic macaques were found in either GI tract or GI tract-associated lymphoid tissues. Furthermore, 12 of these 16 outlier barcodes were found in only one tissue site, consistent with an initial rebound origin site, while the other 4 outlier barcodes were found in more than one site that were anatomically linked within the GI tract or LNs draining these sites (dotted circles; Fig. 4d,f), consistent with rebound origin plus local contiguous spread. Overall, in these 6 low-viraemic macaques, only 16 distinct tissues out of the nearly 600 examined (2.7%) had evidence of barcode clonotypic viral replication and spread, highlighting the rarity of early tissue viral spread and showing that early, limited off-ART viral replication with local within-tissue spread can produce low-level rebound viraemia.

a,b, Two low-viraemia macaques (LV1 (a) and LV2 (b)) each had evidence of two rebounding barcode lineages in plasma, but none were identified as outliers in any analysed necropsy tissue. Right panels show barcode proportional distribution in peak primary and rebound plasma. LV2 showed evidence of two outlier barcodes in ascending colon LN (BC.2557) and an intercostal LN (BC.1012) but not in rebound viraemia. Asc. colon, ascending colon. c, LV3 had two distinct viral plasma rebound barcode lineages (BC.2431 and BC.1136), of which BC.2431 was also identified as a vRNA expression outlier in a mesenteric LN, linking this distinct tissue site with rebound plasma viraemia. d, From LV4, four detectable barcode lineages were observed in rebound viraemia (BC.5385, BC.1872, BC.213 and BC.834), three of which were also vRNA expression outliers in one or two tissues. Trans. colon, transverse colon; Desc. colon, descending colon. e, LV5 had three detectable plasma rebound barcode lineages; of these, two (BC.3338 and BC.1078) were also found as vRNA expression outliers in a descending colon LN and in an ileocaecal LN, respectively. Although barcode 401 (purple) was not identified in rebound plasma, it was enriched within the same ileocaecal LN sample that gave rise to BC.1078. f, VL6 had five detectable rebounding viral lineages in plasma; two of these rebounding lineages (BC.8128 (orange) and BC.4406 (green)) were also found as vRNA expression outliers within a transverse colon LN and in a mesocolic LN, respectively. One additional rebounding lineage (BC.3963; blue) was the dominant lineage in rebound plasma and was found enriched in four distinct GI tract and GI tract-draining LNs. Two additional barcode lineages (BC.662 (purple) and BC.2567 (red)) were not found in rebound plasma but were identified as vRNA expression outliers in transverse colon and in three sites in the GI tract and GI tract-draining LNs, respectively. The frequency of all tissue barcodes that were vDNA+/vRNA+ ranged from 10% to 18%. Blue lines with shaded area delineate the 99% PI.
Characterization of tissue origins and spread with subsequent clonal viral rebound
These observations of the earliest stages of viral rebound are consistent with the hypothesis that the outlier barcodes, enriched for vRNA expression, reflect local viral replication and tissue spread upon ART discontinuation, with subsequent spillover into the peripheral blood and local lymphatic spread, all of which would be expected to be a prelude to exponentially increasing viraemia and distal (systemic) viral spread. Indeed, in the one macaque (HV1) that manifested more advanced rebound with a PVL of 4,700 copies per ml at necropsy (Fig. 1 and Extended Data Fig. 4b) there were overt signs of replication consistent with both local and distal viral dissemination (Fig. 5a). In this animal, a total of 9 barcodes were detected in rebound plasma, 7 of which were detected as outlier barcodes in 1 or more of the sampled tissues (BC.4116, BC.994, BC.1322, BC.3139, BC.7069, BC.1547 and BC.457), while only two rebounding lineages were not found as tissue outlier barcodes (BC.5833 and BC.1767) (Fig. 5a,b). Among rebounding barcodes, there was a direct relationship between the level of the rebounding barcode vRNA in blood and the number of tissues in which that barcode was found as an outlier. Note that the two barcodes with the highest representation in blood (BC.457 and BC.1547) showed outlier status in 33 and 18 diverse tissues, respectively, with a broad range of vRNA detected, with some tissues being far from the 99% PI boundary and others closer to this boundary. Intermediate viraemic barcodes (BC.7069, BC.3139, BC.1322, BC.994) manifested outlier status in fewer but still disparate tissues, whereas the lowest viraemic barcode (BC.4116) was found as an outlier in only 1 tissue. Finally, one outlier barcode (BC.2440) was found in 2 tissues, but not in rebound plasma, consistent with a more recent viral reactivation and corresponding limited spread.

a, Macaque HV1 had a rebound PVL of 4,700 copies per ml with 9 detectable rebounding barcode lineages in plasma and 8 barcodes showing outlier vRNA expression levels in 1 or more tissues (7 of these barcodes were also present in rebound plasma); 14% of all tissue barcodes were vDNA+/vRNA+. Barcodes identified in rebound plasma but not showing outlier vRNA expression in any analysed tissue (BC.5833, yellow; BC.1767, brown, only vDNA+) are shown in the top left panel along with the only barcode lineage with outlier vRNA expression that is not detected in rebound plasma (BC.2440, teal). Abd., abdominal; Sup. Mes., superior mesenteric. b, The peak primary infection plasma barcode distribution compared with the rebounding, off-ART barcode hierarchy. Blue lines with shaded area delineate the 99% PI.
To identify the likely presumptive origin/initial amplification site in 3 macaques (LV4, LV6, HV1) with outlier barcodes found in more than one tissue (n = 11 outlier barcodes), we hypothesized that ‘origin’ site viruses with a particular barcode would have a longer period of active viral replication in that site than viruses with that same barcode in sites of (subsequent) secondary spread, which would be reflected by the putative origin site barcode having significantly higher vRNA levels than the same barcode in secondary spread sites. Thus, for these 11 barcodes, the adjusted vRNA level, for example, the difference in measured vRNA compared with the on-ART predicted value (based on the corresponding vDNA level), was determined for each tissue in which it was an outlier. For each barcode, the tissues were rank ordered based on their adjusted vRNA, and the differences in adjusted vRNA levels between consecutively ranked tissues were computed and used to determine a threshold difference level (99th quantile) to distinguish presumptive origin sites from sites of secondary spread. A presumptive origin site (for which the adjusted vRNA distance to the next-highest site was above the threshold) could be identified for 6 of 11 barcodes, including 3 from HV1 (Extended Data Fig. 7). Thus, this single macaque shows the entire spectrum of sequential reactivation and spread of individual barcode lineages ranging from clonotypes that have replicated and spread locally in tissue but not yet reached detectable levels in blood, clonotypes that replicated and spread in a single tissue site and have spilled into blood at low levels, and clonotypes that have extensively replicated and spread from initial sites to diverse distant tissues (consistent with secondary reseeding of tissues) with high levels in blood.
These observations are consistent with sequential post-ART viral replication and spread, initially of just a few viral lineages with local spread within the origin site, followed by local (lymphatic) spread, and eventually to distal (hematogenous) spread with exponentially increasing rebound viraemia. In this regard, it is interesting to note that when outlier barcodes are found in a single site, the majority (67%) are not detected in rebound viraemia with the remaining 33% observed in plasma at levels of 1–18 RNA copies per ml (Fig. 6a). Locally spreading barcodes are more likely to become viraemic but still at very low levels (1–21 copies per ml). By contrast, barcodes with demonstrated distal spread show PVL levels above 150 copies per ml.

a, The rebound PVL for each outlier barcode found at a single site (n = 7) or with only local replication (n = 3) was significantly different from outlier barcodes found to have to spread to anatomically distal sites (n = 6) (two-sided Wilcoxon test; Benjamini–Hochberg correction with P values indicated). b, The peak barcode levels in primary viraemia are plotted against the total vDNA per barcode count in tissues (n = 9,100) with outlier and rebounding barcodes of interest highlighted and colour-coded by animal. The peak barcode viral load was separated into quartiles (for the boxplots, the midline represents the median, the boxes represent the interquartile range (IQR) and the whiskers represent the largest or lowest value within 1.5 × IQR), with the majority of outlier and rebounding barcodes of interest found mostly in the top quartile (33 of 43 lineages, 77%) with only one lineage originating from the bottom quartile (2%). c, Heat map shows all outlier barcodes above the 99% PI, plotted by tissue sample type with the colour values based on the level of vRNA above the expected value given its vDNA concentration. The tissues that represent rebound origin sites are outlined in cyan. There are seven origin sites found within five GI tract tissues (upper and lower jejunum, cecum, and ascending and transverse colon). For the GI tract-associated lymphoid tissues, 19 origin sites were detected from 10 different tissues (mesenteric LN, ileocaecal LN, ascending colon LN, transverse colon LN, descending colon LN, mesocolic LN, abdominal para-aortic LN, tonsil, submandibular LN and retropharyngeal LN). The non-GI LNs had evidence of a single origin site (intercostal LN). No origin sites were detected in non-lymphoid organs. Asterisk in column name indicates that the specific, outlier barcode was also found in rebound plasma.
Initial viral rebound preferentially occurs in GI tract-associated lymphoid tissues
Barcode RNA proportional representation at peak PVL during primary infection reflects levels of acute phase viral replication for individual barcode viral clonotypes and correlates with extent of total tissue seeding as measured by vDNA levels for all barcodes across all macaques (r = 0.88, P < 2 × 10−16; Pearson correlation). Each of the tissue outlier and/or plasma rebounding barcodes shown in Figs. 3–5 are highlighted within this overall distribution (Fig. 6b). It is worth noting that 77% of outlier or rebounding barcodes detected derive from the top quartile of these barcode hierarchies (Fig. 6b). In fact, for macaques necropsied off-ART, mixed-effects logistic regression indicates that a 10-fold increase in the total tissue vDNA reservoir size for an individual barcode increases its odds of contributing to rebound nearly 12-fold (P < 0.001; two-sided Wald test) (Supplementary Table 4). However, barcode vDNA levels explain only about half of the variation (marginal R2 = 0.51), with 23% of outlier barcodes found much lower in the distribution (Fig. 6b), suggesting that tissue location-specific factors could influence the activation of such rarer clonotypes.
To visualize the tissue origins of rebound, a heat map of all 32 outlier barcodes in the aviraemic, low-viraemic and high-viraemic macaques was generated with the colour scale indicating the individual barcode vRNA level above predicted baseline expression (based on the amount of vDNA for that barcode in each tissue), with samples meeting the criteria for rebound origin sites indicated by a cyan outline (27 of 32 outlier barcodes) in Fig. 6c. It is striking that with one exception (an intercostal LN in LV2), all other presumptive rebound origin sites were in the GI tract (7 of 27; 26%) or GI tract-associated lymphoid tissues (19 of 27; 70%). Logistic regression indicates that across the tissues in which these 27 lineages were found, the odds of rebound originating in GI tract-associated lymphoid tissues was 10-fold higher compared with non-GI tract-associated lymphoid tissues (P = 0.022, two-sided Wald test), whereas in this comparison the barcode vDNA level in tissues was not a significant predictor of where rebound originated (Supplementary Table 5 and Extended Data Fig. 8). Even when oropharyngeal sites were considered separately from GI-tract associated lymphoid tissues, the increased likelihood of rebound from the latter was not substantially altered (odds ratio (OR) = 9, P = 0.033, two-sided Wald test), and oropharyngeal sites were also independently associated with rebound (OR = 18, P = 0.008, two-sided Wald test) (Supplementary Table 6).
The barcode itself is simply a marker to quantify distinct rebound events in tissues, allowing us to investigate predictors of rebound at the level of tissues. To this end, we extended our analysis to the 697 distinct tissue samples from which we obtained both RNA and DNA barcode sequences from the 18 off-ART animals using a mixed-effects Poisson regression, which revealed a 10-fold higher incidence of rebound (that is, the number of distinct barcodes for which a given tissue is the presumptive rebound origin site) in GI tract-associated lymphoid tissues compared with non-GI tract-associated lymphoid tissues (P = 0.025; two-sided Wald test) even when oropharyngeal sites are considered separately (Supplementary Tables 7 and 8), while the total level of barcoded vDNA in individual tissues was not associated with rebound (Extended Data Fig. 9a). Across the total 1,717 sampled tissues from the 18 off-ART macaques, only 21 (1.2%) were identified as presumptive rebound origin sites. Remarkably, 5 of these 21 rebound tissue origin sites had more than 1 rebounding barcode (Figs. 3–5 and Extended Data Fig. 9a), suggesting that local conditions in specific tissue sites likely also contribute to a milieu that favours initiation and/or amplification of viral rebound. Moreover, within these 21 rebound origin sites, the ranking of barcode DNA levels was predictive overall of which lineages rebounded (P < 0.001; Supplementary Table 9). However, in contrast to this overall pattern, several barcodes with low levels of DNA also contributed to rebound (Extended Data Fig. 9b), further supporting a possible role for tissue micro-environments or cell-specific factors (for example, integration site, activation state) contributing to off-ART rebound.
Discussion
The processes that connect the RCVR on ART to viraemic rebound are not well understood. Yet, the fact that post-ART viral rebound is oligoclonal and temporally dispersed despite the widespread distribution of the RCVR indicates that microenvironmental mechanisms in tissues govern the rebound process, mechanisms that might be targetable for new approaches to achieve durable post-ART viral remission or cure. To study these potential mechanisms, the sites hosting the earliest phase of viral rebound, those first viral clonotypes emerging after ART discontinuation and spreading to new target cells, must be identified. Here, we developed a statistically based criterion for detecting these initial sites of post-ART clonotypic viral spread and validated this criterion by demonstrating that barcodes of viruses showing evidence of local or regional replication after ART (‘vRNA outliers’) contribute to the earliest measurable post-ART viraemia.
Not all barcoded viruses found in post-ART plasma were identified in sampled tissues, which is not surprising as even our extensive tissue sampling still represented only a relatively minor component of each animal’s total body lymphoid tissues. The implication is that the viraemic barcodes not identified in sampled tissues are derived from sites that were not analysed, including tissues/sites that were not collected, or in larger organs, regions that were not included in the analysed material. It is also not surprising that we identified locally spreading barcodes in individual tissues from some post-ART macaques that were either aviraemic or viraemic with different barcoded viruses. In the former situation, these locally spreading barcoded viruses likely reflect presumptive early rebound founder sites that had not achieved sufficient replication to disseminate into blood by the time of necropsy, and in the latter situation, we would interpret these barcodes as reflecting later reactivating and spreading viral lineages (for example, preceded by other barcoded viruses present in the rebound viraemia) for which replication and spread was again interrupted by necropsy, before achieving viraemia. On the other end of the spectrum, in a macaque with more advanced viraemic rebound (4,700 copies per ml), 7 of 9 rebounding barcodes in plasma were also found in tissues, with the barcodes showing the highest plasma levels showing disseminated replication and spread and those lineages at the lowest levels showing local replication and spread in one or two sites. These results are consistent with a temporal succession of viral clonotype local replication and spread: first in a single local tissue without viraemia, continued spread in that tissue and possibly draining LNs with low viraemia, and finally dissemination to multiple non-contiguous tissues with exponentially increasing viraemia. This successive temporal process would be expected to continue for many viral lineages. Indeed, in a separate parallel study with necropsy of barcoded SIV-infected macaques at day 12 after ART, successive secondary rebounding lineages can be visualized, even as the initial lineages approach peak viraemia27. Although this process has been previously shown using barcoded clonotype proportions in plasma rebound4,24, these current studies advance our understanding by demonstrating this temporal progression in specifically identified tissues, culminating in rebound viraemia and recrudescent systemic infection with individual lineages reactivating at various times off-ART.
Overall, we identified 32 outlier tissue barcodes in study macaques, including 27 barcodes that met statistical criteria for presence in a presumptive (barcode found in plasma) or potential (barcode not detected in plasma) rebound origin site for that lineage (Fig. 6). We acknowledge that, given the sampling limitations discussed above, we cannot rule out that the actual first site of viral reactivation and spread occurred in an unsampled or unanalysed site in the near vicinity of the identified site (adjacent or directly connected via lymphatics), such that the identified sites represent early local spread. Even with this caveat, the statistically based analyses of these potential/presumptive origin sites provide key insights into the characteristics of post-ART viral rebound biology. First, early rebound is typically detected in a single tissue in the absence of measurable viraemia, but in some instances, replication in a single tissue or in a few anatomically contiguous tissues can be sufficient to achieve low but measurable levels of viraemia (<30 copies per ml). In the single, higher viraemic macaque, distant spread (non-anatomically contiguous tissues) was associated with viraemia at ≥150 copies per ml. If these associations hold true for HIV rebound in humans, the implication would be that by the time rebounding plasma virus is detected with clinically used assays, between 50 and 200 copies per ml, the rebounding infection has already disseminated across the body. Second, almost 80% of the barcodes participating in initial tissue rebound derive from the top quartile of the total body barcode hierarchy. These highly represented barcodes are more likely to express vRNA on ART, possibly increasing the probability of their participation in initial events leading to viral rebound. Moreover, viruses with highly represented barcodes are more broadly distributed across the body and are therefore more likely to reside in tissues or tissue microenvironments that support viral rebound. However, our results also emphasize that 23% of the time, viruses containing less represented barcodes, including lineages with little to no vRNA expression, can initiate tissue rebound, suggesting that tissue microenvironmental factors contribute to reactivation and spread of less extensively distributed proviral barcodes. Indeed, the striking observation that 5 of the 21 identified rebound tissue origin sites had more than one distinct rebounding barcode is consistent with specific tissue conditions favouring rebound initiation and amplification in these sites.
Finally, and most importantly, we found that, independent of sampling, initial rebound was almost exclusively found in tissues associated with the GI tract, in particular GI tract-associated lymphoid tissues such as intestine-draining mesenteric LNs. Indeed, our data suggest that initial viral rebound occurred 10-fold more frequently in GI tract-associated lymphoid tissues than non-GI tract-associated lymphoid tissues. The immunologic importance of the gut microbiome has been repeatedly demonstrated28,29, including specifically in the setting of HIV infection where intestinal dysbiosis and microbial translocation have been documented, processes to which GI tract-associated lymphoid tissues are preferentially exposed and which have been implicated as major drivers of systemic inflammation and immune activation, even on ART30,31,32. The innate immune stimulation resulting from microbial translocation could have multiple, non-mutually exclusive relevant effects on post-ART rebound, including creating immunologic conditions that (1) support the maintenance of a larger and/or more transcriptionally active virally infected cell pool, (2) decrease the innate and/or adaptive immune barriers to viral outgrowth and/or (3) as suggested above, provide specific triggers that promote virus expression leading to productive viral replication and spread in the absence of ART31,33,34,35. Indeed, in the cohort of macaques studied here, barcode lineages in GI tract and GI tract-associated lymphoid tissues manifest significantly higher odds of being vRNA+ for a given vDNA level than barcodes in non-GI tract lymphoid tissues, suggesting that the RCVR in these GI tract-associated lymphoid tissues is constitutively more active than in their non-GI tract counterparts. Interestingly, in the above-mentioned parallel study of rebounding macaques necropsied at day 12 the founder sites for both later primary and secondary rebounding lineages (lineages well down in the temporal rebounding hierarchy) were still preferentially located in GI tract and GI tract-associated lymphoid tissues27, even after the onset of systemic, post-rebound immune activation. These observations suggest that the mechanisms that underlie preferential initial rebound in GI tract-associated tissues may persist through peak viral replication.
While further work will be required to ascertain the nature of these mechanisms, this observation has three important near-term implications for development of HIV cure therapies. First, given that these sites are difficult to access in humans, this observation should spur development of imaging or specialized collection techniques focused on these lymphoid tissues. Second, these data suggest that against usual assumptions post-ART rebound is, at least initially, a process that predominantly occurs in GI tract and GI tract-associated lymphoid tissues and that interventions designed to intercept and interrupt post-ART rebound must be designed, formulated and delivered in a way that ensures activity in these sites. Finally, delineation of the mechanism(s) underlying preferential initial rebound in GI tract-associated tissues would provide potential points of therapeutic intervention, possibly leading to novel approaches for HIV cure therapy. This is not to say that non-GI tract-associated sites are irrelevant for rebound, but clearly, interventions lacking activity in GI tract-associated sites would not be expected to be effective. Moreover, even a specific suppression of initial tissue rebound in GI tract-associated sites, delaying viraemia for days to weeks, might facilitate immune control of infection, potentially leading to long-term remission36,37.
We acknowledge that this study uses a reductionist animal model and study design that does not capture all of the subtleties and complexities of human infection with HIV. While SIV infection of rhesus macaques authentically recapitulates many key aspects of the pathogenesis of HIV in humans38, host species differences do exist. Moreover, this study used SIVmac239M, a virus stock that uses a unique barcode tag in an otherwise isogenic and phenotypically equivalent virus to enable tracking of chains of viral infection events originating from individual virus variants, while obviating viral phenotypic differences as a potential confounding variable. While this critical feature does not capture the genotypic and phenotypic diversity of the complex swarms typically present in individuals with HIV infection, particularly if they started ART later in the course of their infection, this model does allow for the ultra-sensitive and unambiguous identification of each individual viral barcode lineage within an animal and for each tissue sampled. Similarly, initiation of ART in this study at a precise uniform time in acute infection allowed the establishment of well-calibrated, saturated RCVR levels, based on characterization in previous studies in the model4,24, and although this does not reflect the typical timing of ART initiation in most humans, immediate ART treatment is the recommended treatment guideline39,40. A consequence of early ART initiation and just over a year of treatment before ART discontinuation is that the RCVR may still be undergoing decay4,24,41 and remains genetically intact42. This is in contrast to the accumulation of defective proviral genomes in individuals after extended periods of ART—often decades43,44. In addition, early ART initiation and SIVmac239 resistance to antibody neutralization45 obviate the development of autologous neutralizing antibody as a potential confounding factor influencing viral rebound dynamics, while such antibodies have been shown to be a potentially important consideration in people living with HIV46. Finally, while we have shown that expanded clones of infected CD4+ T cells can be established with early initiation of ART following SIV infection47, we did not characterize this aspect of the RCVR for this study, and it is likely that clonal expansion is more limited in this model than seen in people with HIV infection on long-duration ART48,49,50.
The virology and immunobiology underlying local tissue-level processes and events leading to post-ART rebound in SIV-infected rhesus macaques is likely similar for people living with HIV, in particular our fundamental finding that a few individual lineages begin actively replicating, most often in GI tract-associated lymphoid tissues with tissue-specific factors influencing the probability for a given viral clonotype to contribute to post-ART rebound. This general biology might be confirmed in people with imaging or other non-invasive means in study participants undergoing analytical treatment interruption. Moreover, the rhesus macaque model itself, the barcode data and the approach reported here will allow for a tissue-based, detailed analysis of initial rebound sites, providing insight into the mechanisms promoting or restricting post-ART viral rebound and thereby informing the development of therapeutics that can exploit these mechanisms to achieve off-ART viral remission.
Methods
Ethical statement
All use of animals, including the experimental protocols, procedures, administered reagents and housing/care plan, was approved by the Oregon National Primate Research Center (ONPRC) Institutional Animal Care and Use Committee (IACUC) under protocol number IP00001053. ONPRC is a Category I facility. The Laboratory Animal Care and Use Program are fully accredited by the American Association for Accreditation of Laboratory Animal Care, with an approved Assurance (A3304-01) for the use and care of animals on file with the National Institutes of Health (NIH) Office of Laboratory Animal Welfare. The ONPRC IACUC adheres to national guidelines established in the Animal Welfare Act (7 USC Sections 2131–2159) and the Guide for the Care and Use of Laboratory Animals (8th Edition) as mandated by the US Public Health Service Policy, as well as using the standards of the US NIH Guide for the Care and Use of Laboratory Animals (National Academies Press, 2011; https://www.nap.edu).
Animals
A total of 24 purpose-bred male rhesus macaques (Macaca mulatta) of Indian genetic background, aged 2.5–4.7 years at study start, were used for these experiments (see Supplementary Table 1 for detailed animal demographics). These animals were born and housed at the ONPRC, following standards established by the centre’s IACUC and the NIH Guide for the Care and Use of Laboratory Animals. After project assignment, study macaques were housed in Animal Biosafety level (ABSL)-2+ rooms with autonomously controlled temperature, humidity and lighting and with automatic water systems that provided fresh, potable water. Macaques were fed Purina LabDiet 5000 (Purina Mills International) twice daily and received daily food enrichment (for example, fresh fruit, vegetables). Ketamine HCl (Ketathesia, Henry Schein Animal Health) with or without dexmedetomidine (Dexmedesed, Dechra) was used to sedate macaques for procedures. At the study end-point, macaques were euthanized with sodium pentobarbital overdose (>50 mg kg−1) and exsanguinated via the distal aorta. A board-certified veterinary pathologist performed and collected tissues at necropsy.
All macaques were determined to be specific pathogen free, as defined by being free of cercopithecine herpesvirus 1, D-type simian retrovirus, simian T-lymphotropic virus type 1 and Mycobacterium tuberculosis. MHC-1 genotyping for protective MHC alleles (Mamu-A*01, Mamu-B*08 and Mamu-B*17; Supplementary Table 1) was performed by sequence-specific PCR as described previously51. Prior to study initiation, all macaques received a multimodal therapeutic regimen to eliminate common gastrointestinal pathogens as previously described52. Macaques were inoculated intravenously with 5,000 infectious units (IU) of barcoded SIVmac239M4,24 before starting daily ART as previously described53 (subcutaneous injections of 5.1 mg kg−1 d−1 tenofovir disoproxil, 40 mg kg−1 d−1 emtricitabine and 2.5 mg kg−1 d−1 dolutegravir in a solution containing 15% (v/v) kleptose at pH 4.2) 9 d.p.i. through at least 70 w.p.i. The viral dose and timing of ART start was calibrated based on past data in the model to provide a saturated reservoir as well as high resolving power for identifying individual barcode reactivation events, while not impacting acute infection viral dynamics4,24.
Tissue sampling
Tissue biopsies included bone marrow (collected from iliac crest or humerus), peripheral LNs (collected from axillary or inguinal sites), mesenteric LNs (collected from colonic mesentery), spleen and liver (collected as laparoscopic pinch biopsies) and duodenum and rectum (collected as endoscopic pinch or direct forceps biopsies)54,55. At necropsy, all efforts were made to collect lymphoid tissues and intestines comprehensively while representative samples were collected from other tissues of interest (Supplementary Table 2). All detectable LNs (greater than ~2 mm in diameter) were collected from at least 20 pre-defined sites, with each LN collected individually. The spleen was serially divided into 3 mm sections orthogonal to the major axis. Bone marrow was collected from each femur. The thymus was divided into 4 equal-sized sections. The small intestine was divided into the duodenum (rostral-most 8 cm), jejunum and ileum (caudal-most 8 cm), while the large intestine was divided into the cecum, ascending colon, transverse colon, descending colon and rectum based on macroscopic morphological features. Each section of the intestines was further divided into 4-cm-long segments. Representative samples were collected from other tissues, including the brain, spinal cord, heart, kidney, liver, lung, urinary bladder, prostate, testes, seminal vesicles, visceral fat, nasal mucosa and oral mucosa. Tissue samples for virological analysis were collected in either Lysing Matrix D (MP Biomedicals; PBMC, tonsil, LNs, bone marrow, spleen, thymus, adenoids, bronchoalveolar lavage and spinal cord) or Precellys lysing kit ‘Tissue Homogenizing CK28-15 ml’ (Bertin Technologies; intestines, oral and nasal mucosa, lung, male reproductive tract, brain, liver, kidney, heart, fat) and flash-frozen immediately after collection.
Viral nucleic acid quantitation assays
Levels of plasma SIV RNA and vDNA and vRNA from cell pellets/tissues were determined using assays targeting a gag amplicon56,57,58. For PBMC cell pellet and tissue cell-associated SIV RNA and DNA measurements, snap-frozen dry cell pellets were lysed on ice with Trizol reagent and sonicated using a Branson cup sonifier. Snap-frozen tissue specimens were disrupted and lysed in 2 ml microfuge tubes containing 1.4 mm zirconium beads using TRIzol and a Precellys homogenizer (Bertin Technologies). Following the addition of 1-bromo-3-chloropropane, vortexing and centrifugation, RNA was extracted by careful aspiration of the entire RNA-containing aqueous phase, taking care not to disturb the proteinaceous interphase and transferring it to a separate tube containing glycogen. The remaining organic layer contains the DNA from the sample and was extracted as described below. The RNA was purified by the addition of isopropanol, vortexing, and centrifugation (room temperature, 21,000 × g, 10 min) to pellet the RNA. The supernatant was removed, and the pellet was resuspended in 70% ethanol. After overnight incubation at −20 °C followed by pelleting, a second 70% ethanol wash, pelleting by centrifugation and decanting of the supernatant, the pellet was briefly air dried. The dried pellet was dissolved in 5 mM Tris (pH 8.0), vortexed and gently warmed to dissolve for assay.
DNA was extracted from the organic phase from the RNA extraction by the addition of Back Extract Solution (4 M GuSCN, 1 M Tris base, 50 mM sodium citrate), followed by vortexing and centrifugation (4 °C, 14,000 × g, 15 min). The resulting aqueous phase was carefully aspirated as above and transferred to a fresh tube with glycogen, followed by addition of isopropanol, vortexing and pelleting of the DNA by centrifugation (room temperature, 21,000 × g, 10 min). After decanting the supernatant, 70% ethanol was added, and the pellet was dislodged and stored overnight at 4 °C. Following centrifugation (room temperature, 21,000 × g, 10 min) and decanting, the pellet was washed again with 70% ethanol, resuspended in 5 mM Tris, pH 9.0 and heated in a dry bath (45 °C) to fully dissolve the pellet. After brief sonication, 5 M NaCl and 100% ethanol were added, followed by vortexing, centrifugation (4 °C, 21,000 × g, 10 min) and decanting, then a 70% ethanol wash, pelleting and decanting. Any residual ethanol was removed, and the pellet was briefly air dried, followed by thorough resuspension of the pellet in 5 mM Tris, pH 9.0 with heating in a dry bath (45 °C) to fully dissolve the pellet. Samples were then sonicated, followed by a flash spin, heated (120 °C in a bead dry bath, 15 min) and flash frozen.
Quantitation of SIV RNA and SIV DNA was performed using RNA and DNA extracted from the same specimen, as described above, with quantitation of vRNA and vDNA determined by quantitative PCR with reverse transcription/PCR assays targeting a conserved sequence in gag, with results for both vRNA and vDNA normalized relative to 106 diploid genome cell equivalents of co-analysed genomic DNA, as determined by quantitation of a single copy sequence from the CCR5 gene (two copies per cell)—based on empirically demonstrated consistent, essentially quantitative recovery of DNA and RNA in the extraction process used. To cover the broad range of target amplicon values characteristic of the specimens typically analysed (up to 6 logs dynamic range), while providing sufficient assay throughput, the hybrid assay approach combines and takes advantage of features of both traditional real-time PCR methods and a statistical frequency-based approach, with multiple replicate reactions run on aliquots of the same extracted nucleic acid samples. For each specimen the extracted DNA or complementary DNA (following reverse transcription, for all specimens analysed for vRNA quantitation,see below) was used to set up 12 replicate reactions. Two of the 12 wells were spiked with a known quantity of control standard nucleic acid to monitor for any reaction inhibition. If all replicate reactions yield a positive result, the mean value of the threshold cycle (CT) for all of the positive reactions was interpolated onto a real-time PCR standard curve to determine the per reaction copy number, which was then normalized based on any relevant specimen division, dilution factor and resuspension volume to provide a copy number per 106 diploid genome cell equivalents in the source specimen. If all replicate reactions did not yield a positive result, copy number was estimated using a statistically imputed value based on the frequency of positive reactions (copies per well = −ln(1 − f), where f is the fraction of positive wells), independent of the specific above-threshold CT value of the positive wells, based on nominal single-copy detection sensitivity for the PCR reaction, with normalization based on any relevant specimen division, dilution factors and resuspension volume to provide a copy number per 106 diploid genome cell equivalents in the source specimen. If no wells gave a positive reaction, results were reported as less than the value that would have been statistically imputed if a single well had been positive, normalized based on any relevant specimen division, dilution factors and resuspension volume to provide a copy number per 106 diploid genome cell equivalents in the source specimen.
As different specimens can contain very different numbers of cells, yielding different amounts of DNA, to maximize sensitivity, the DNA assay uses as much extracted DNA as can be included without causing reaction inhibition, based on spectrophotometric quantitation of DNA before assay set-up, using a Biomek FXp lab robot and Biotek Epoch spectrophotometer. If reaction inhibition was documented for a given specimen based on spiked internal control reactions giving results below quality control specifications, analysis was repeated with dilution to relieve reaction inhibition. All results were normalized back to vDNA copies per 106 diploid genome equivalents of DNA, taking into account the amount of specimen analysed and all relevant dilution factors.
To perform reverse transcription for qRT PCR, the isolated and resuspended RNA was aliquoted into 12 wells of a 384-well plate as noted above (including 2 reactions with quantified positive control vRNA template spikes to confirm reverse transcription efficiency) and reverse transcribed with MuLV Reverse transcriptase (37 °C for 60 min and 90 °C for 30 min). A known quantity of CCR5 DNA standard was included in the master mix as a positive control for each well to confirm specimen transfer from the completed reverse transcription reactions to the quantitative PCR (qPCR) reactions. Following reverse transcription, the qPCR assays for vRNA (viral cDNA) and vDNA were identical and used a limited nested pre-amplification reaction to maximize sensitivity. An initial round of 12 cycles of PCR (95 °C for 10 min activation; 95 °C for 30 s, 60 °C for 1 min and 72 °C for 10 s) was performed with primers (gag Nest Forward 5′– GATTTGGATTAGCAGAAAGCCTG TTG –3′ and gag Nest Reverse 5′–GTTGGTCTACTTGTTTTTGGCATAGTTTC –3′) designed to amplify a conserved 247-base pair region in gag. Following the nested PCR, 10% of the resulting reaction from each individual well was transferred to a well in a new 384-well plate with a master mix that contains primers that amplify a conserved 84-base pair region within the nested sequence (gag21 5′–GTCTGCGTCAT-dP-TGGTGCATTC–3′ and gag22 5′–CACTAG-dK-TGTCTCTGCACTAT-dP-TGTTTTG–3′), a FAM labelled Taqman probe (psgag23 5′–FAM-CTTC-dP-TCAGT-dK-TGTTTCACTTTCTCTTCTGCG –BHQ1–3′) (where special base dP binds to A or G and dK binds to T or C, both from Biosearch Technologies and used to avoid potential amplification bias at sites of sequence divergence among experimentally used SIV/SHIV variants) and primers for CCR-5 (rHR5F01 5′– CCAGAAGAGCTGCGACATCC –3′, rHR5R03 5′– GTTAAGGCTTTTACTCATCTCAGAAGCTAAC –3′) with the associated Red610 labelled Taqman probe (rHR5P01 5′– Red610 –TTCCCCTACAAGAAACTCTCCCCGG TAAGTA –BHQ2 –3′). qPCR was performed using AptaTaq (Roche; 95 °C for 2 min activation; 95 °C for 30 s and 60 °C for 60 s) with results determined after 35 amplification cycles. After quality control review, the raw data were exported to a Filemaker-Pro database for calculation of final normalized results as described above. In addition to the control template titration reactions used for the real-time PCR standard curve, each assay run included ‘no template’ negative controls to detect potential false positive reactions and quantified standardized control template standards to control for amplification efficiency.
Barcode sequencing
RNA and DNA for barcode sequencing was obtained from samples extracted for vRNA and vDNA quantitation56,57,58. Although viral quantification is performed using a gag amplicon and viral barcode is found between vpr and vpx, we have previously reported that early ART and short-term treatment cause the retention of intact viral genomes that would be expected to have both gag and vpr/vpx at nearly identical proportions42,59. For sequencing vRNA, cDNA was generated with Superscript III reverse transcriptase (Invitrogen) and an SIV-specific reverse primer (Vpr.cDNA3: 5′-CAG GTT GGC CGA TTC TGG AGT GGA TGC-3′). Before sequencing, single-round PCR was performed on cDNA or DNA with VpxF1 (5′-CTA GGG GAA GGA CAT GGG GCA GG-3′) and VprR1 (5′-CCA GAA CCT CCA CTA CCC ATT CATC-3′) primers containing either the F5 or F7 Illumina adaptors with a unique 8-nucleotide index sequence for multiplexing and the High Fidelity Platinum Taq (ThermoFisher) as previously described4,24. For cDNA samples, 2–6 replicate PCR reactions were performed, and for DNA 12–24 replicate PCR reactions were performed before pooling and sequencing on a MiSeq instrument (Illumina). Sequences were analysed with our custom Barcode Analysis Tool (https://github.com/KeeleLab) as previously described4,24. As part of this analysis, a 1/input lower limit of detection (LOD) is used as a threshold for valid barcodes, and only barcode sequences from tissues that matched exactly those found in each macaque’s peak plasma (day 12 after infection) distribution were retained for analysis. While the total amount of viral templates used for sequencing varied between samples depending on dilutions and repeats required during quantification, at least 50% of the extracted material was used for barcode sequencing, which is reported as the relative proportion of each barcode lineage multiplied by the total nucleic acid templates quantified. Furthermore, as a final quality assurance measure to avoid false positives resulting from under-quantification of vDNA, we manually compared the SIV DNA quantified value for each sample by the estimated input based on sequencing depth. In rare cases, where there was clear evidence of deeper sequencing capacity than would be expected for the quantified calculated vDNA input, vDNA quantity was adjusted upward accordingly thereby reducing the RNA to DNA ratio and minimizing the risk of false positive enriched barcodes.
Statistical analysis
The Friedman rank sum test was used to compare PBMC vDNA and vRNA levels across all sampled time points, with the paired Wilcoxon signed-rank test used for pairwise comparisons between consecutive time points (12 d.p.i. versus 21 w.p.i. and 21 w.p.i. versus 66 w.p.i.), with values below the LOD set to the LOD value (0.004 cp 10−6 cell equivalents). The two-sided Kruskal–Wallis test was used to compare macaques between the three different experimental groups (on-ART, off-ART 5 days and off-ART 7 days) in terms of peak PVL, frequency of viral blips and PBMC vDNA and vRNA at each sampled time point. In addition, the log-rank test was used to compare the Kaplan Meier survival curves of time to suppression between the experimental groups. Finally, the two-sided Kruskal–Wallis test was used to compare the cell-associated vDNA and vRNA obtained from different tissue biopsies between the three experimental groups at each biopsy time point. All statistical analyses were performed in R version 4.3.1 and Prism 10.4.1.
Identifying active viral replication based on higher-than-expected on-ART RNA expression
To determine how on-ART baseline RNA expression (per barcode per sample) varied as a function of DNA quantity, we fit a piecewise linear regression using the R package ‘segmented’ with a single unknown breakpoint to the set of RNA+ DNA+ barcodes (greater than 1 SIV RNA and 1 SIV DNA copy) across all samples from all 6 animals necropsied on-ART. The data were log10-transformed, and the regressions were weighted based on the DNA quantity due to increased precision of larger values. Davies test was performed to confirm a non-zero difference in slope between the two segments. We estimated the expected range of RNA expression for a given DNA level on-ART by constructing a 99% PI for the fitted model. For all animals necropsied after ART discontinuation, we then identified all outlier data points with RNA expression above the on-ART 99% PI as putative sites of active viral replication.
Identifying rebound origin sites
Barcodes identified in rebound plasma that had a single outlier site indicative of active viral replication were deemed to have also rebounded from that site. To identify the putative rebound origin site for barcodes that were RNA outliers in more than one tissue sample, we looked for a signal of delayed viral replication consistent with spread from the site of predominant RNA expression to other outlier sites. Specifically, we assumed that after rebound occurs at a single site, the virus will replicate at that site for some time before spreading to related tissues or disseminating broadly, resulting in bigger differences in the level of replication between the origin site and the secondary site of replication than between distinct secondary sites, which may have started replicating at around the same time. For each outlier, we first determined how much vRNA was expressed above the expected level based on the on-ART linear piecewise model fit, that is, adjusted vRNA = observed vRNA (log10) − predicted vRNA (given the level of vDNA). For each barcode, the outlier sites were rank ordered based on their adjusted vRNA levels, and the difference in adjusted vRNA was computed between consecutive sites. We used the 99th quantile of adjusted vRNA differences between secondary sites (rank >1) as the threshold level for discriminating delayed replication consistent with replication from the origin site.
Regression analyses
Regression analyses were performed in R using the lme4 package (version 1.1.37) for mixed effects and the geepack package (version 1.3.13) for generalized estimating equations. All models investigated the effect of vDNA level on the outcome (probability of RNA expression or probability or incidence of rebound), with a subset of models additionally including tissue type. For models with more than one predictor of interest, we first investigated the effect of each covariate on the dependent variable independently and compared the models against each other based on the Akaike information criterion score and against the null model (containing just the intercept) by the likelihood ratio test. If both covariates were significantly associated with the outcome (based on the asymptotic Wald test), we investigated whether the full model containing both covariates explained the data better than the best-fitting single covariate model using the likelihood ratio test. Within the generalized estimating equations (GEE) framework, nested models were compared using the Wald test.
Probability of RNA expression per barcode per sample (model 1)
We performed mixed effects logistic regression analyses to investigate the effect of (1) the DNA level (log10) per barcode per tissue sample, (2) the tissue type of each sample and (3) both covariates together, on the probability of RNA expression in the 6 on-ART animals. Clustering of observations within individual animals was accounted for by including a random effect on the intercept (assumed normally distributed with a zero mean).
Probability of rebound per barcode (model 2)
We performed mixed effects logistic regression analyses to investigate for the 18 off-ART animals whether a barcode’s total DNA across all sampled tissues was predictive of whether it rebounded (that is, the barcode was an outlier based on the on-ART 99% PI in any tissue sample and/or was detected in rebound plasma viraemia). Clustering of observations within individual animals was accounted for by including a random effect on the intercept (assumed normally distributed with a zero mean).
Probability of rebound per sample within barcodes with identified origin sites (model 3)
For the 27 barcoded lineages across the 18 off-ART animals for which we could identify presumptive rebound origin sites, we investigated whether tissue type or the barcode’s vDNA level (log10) in individual tissue samples were predictive of whether they were rebound origin sites. We again performed logistic regression analysis using GEE, accounting for clustering of observations within distinct barcoded lineages using an independent correlation structure.
Incidence of rebound per tissue sample (model 4)
We performed mixed effects Poisson regression analysis to investigate for the 18 off-ART animals whether either the total vDNA level of a tissue or tissue type were predictive of the rebound incidence rate per tissue. Clustering of observations within individual animals was accounted for by including a random effect on the intercept (assumed normally distributed with a zero mean).
Probability of rebound per barcode within origin sites (model 5)
For the 21 presumptive tissue origin sites across the off-ART animals, we investigated whether the vDNA level of individual barcodes were predictive of their probability of rebounding using logistic regression in the GEE framework, accounting for clustering of observations within distinct tissue origin sites. We used independent correlation structures in the models because more complex working correlation matrices did not improve quasi-likelihood under independence model criterion scores.
T cell response assays
SIV-specific CD4+ and CD8+ T cell responses were detected and quantified by intracellular cytokine staining analysis using mononuclear cells isolated from peripheral blood (PBMCs), bronchoalveolar lavage, LN cells (mesenteric and peripheral) and spleen, as previously described58,60,61. These T cell responses were measured by stimulating these cells with mixes of sequential 15-mer peptides (with an 11 amino acid overlap) that encompass the SIVmac239 Gag, Pol, Nef, Rev, Tat, Vpr, Vpx and Vif proteins (sequence based on GenBank accession number M33262; from Intavis AG) in the presence of the co-stimulatory antibodies anti-CD28 (CD28.2, Purified 500 ng per test; Life Tech, CUST03277) and anti-CD49d (9F10, Purified 500 ng per test; Life Tech, CUST03278). They were then incubated in RPMI medium supplemented with 10% newborn calf serum (Hyclone, SH30401.01) at 37 °C in a humidified incubator with 5% CO2, along with individual peptides or peptide mixes and antibodies for 1 h, followed by an additional 8 h incubation in the presence of brefeldin A (5 μg ml−1; BioLegend, 91850). Stimulation without peptides served as background control. After incubation, the stimulated cells were stored at 4 °C until staining with combinations of fluorochrome-conjugated monoclonal antibodies including anti-CD3 (SP34-2: PacBlue, BD Biosciences, catalogue number 24034; 100 ng per test), anti-CD4 (L200; BV510, BD Biosciences, catalogue number 624340; 220 ng per test), anti-CD8α (SK-1: PerCP-eFluor710, Life Technologies, CUST04424; 8 ng per test), anti-CD69 (FN50: PE/Dazzle594, BioLegend, 93437; 50 ng per test), anti-IFNγ (B27: APC, BioLegend, 96018; 80 ng per test), and anti-TNF (Mab11: PE, BioLegend, 96019; 20 ng per test). For memory phenotyping, the following antibodies were used on PBMCs: anti-CD28 (CD28.2: PE/Dazzle594, BioLegend, 93924; 40 ng per test), anti-CD3 (SP34-2, BUV395, BD Biosciences, catalogue number 624310; 120 ng per test), anti-CD20 (2H7: APC-Fire 750, BioLegend, 93924; 30 ng per test), anti-CD95 (DX2: BV605, BioLegend, 93384; 110 ng per test), anti-CD8α (RPA-T8: BV711, BioLegend, 900006277; 20 ng per test) and anti-CD4 (L200: BV510, BD Biosciences, catalogue number 624340; 220 ng per test). Stained samples were analysed on an FACSymphony A5 flow cytometer (BD Biosciences). Data analysis was performed using FlowJo software (BD Biosciences). Data were collected using FACSDiva software v6.1.3, and Flow Cytometry Standard (FCS) files were analysed using Flowjo v10.10. In all analyses, gating on the lymphocyte population was followed by the separation of the CD3+ T cell subset and progressive gating on CD4+ and CD8+ T cell subsets. Antigen-responding cells in both CD4+ and CD8+ T cell populations were determined by their intracellular expression of CD69, and one or both of the cytokines IFNγ and TNF (gating/analysis procedures are shown in Supplementary Fig. 1)61. After background subtraction, the raw response frequencies above the assay LOD were ‘memory-corrected’ (for example, percentage responding out of the memory CD4+ or CD8+ T cell population), as described58,60,61. CD4+ and CD8+ T cells were subdivided into the memory subsets of interest based on surface phenotype (CD28 versus CD95), with memory defined as CD28+/− and CD95+ (ref. 62).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All of the data that support the findings of this study are available within the article and Supplementary Information. Source data are provided with this paper.
Code availability
The Barcode Analysis Tool is available via GitHub at https://github.com/KeeleLab.
References
Landovitz, R. J., Scott, H. & Deeks, S. G. Prevention, treatment and cure of HIV infection. Nat. Rev. Microbiol. 21, 657–670 (2023).
Chou, T. C., Maggirwar, N. S. & Marsden, M. D. HIV persistence, latency, and cure approaches: where are we now? Viruses 16, 1163 (2024).
Li, J. Z. et al. Time to viral rebound after interruption of modern antiretroviral therapies. Clin. Infect. Dis. 74, 865–870 (2022).
Keele, B. F. et al. Early antiretroviral therapy in SIV-infected rhesus macaques reveals a multiphasic, saturable dynamic accumulation of the rebound competent viral reservoir. PLoS Pathog. 20, e1012135 (2024).
Okoye, A. A. et al. Early antiretroviral therapy limits SIV reservoir establishment to delay or prevent post-treatment viral rebound. Nat. Med. 24, 1430–1440 (2018).
Churchill, M. J., Deeks, S. G., Margolis, D. M., Siliciano, R. F. & Swanstrom, R. HIV reservoirs: what, where and how to target them. Nat. Rev. Microbiol. 14, 55–60 (2016).
Cohn, L. B., Chomont, N. & Deeks, S. G. The biology of the HIV-1 latent reservoir and implications for cure strategies. Cell Host Microbe 27, 519–530 (2020).
Siliciano, J. D. & Siliciano, R. F. In vivo dynamics of the latent reservoir for HIV-1: new insights and implications for cure. Annu. Rev. Pathol. 17, 271–294 (2022).
Cole, B. et al. Extensive characterization of HIV-1 reservoirs reveals links to plasma viremia before and during analytical treatment interruption. Cell Rep. 39, 110739 (2022).
Estes, J. D. et al. Defining total-body AIDS-virus burden with implications for curative strategies. Nat. Med. 23, 1271–1276 (2017).
Roux, H. & Chomont, N. Measuring human immunodeficiency virus reservoirs: do we need to choose between quantity and quality? J. Infect. Dis. 229, 635–643 (2024).
Bone, B. & Lichterfeld, M. “Block and lock” viral integration sites in persons with drug-free control of HIV-1 infection. Curr. Opin. HIV AIDS 19, 110–115 (2024).
Chaillon, A. et al. HIV persists throughout deep tissues with repopulation from multiple anatomical sources. J. Clin. Invest. 130, 1699–1712 (2020).
De Scheerder, M. A. et al. HIV rebound is predominantly fueled by genetically identical viral expansions from diverse reservoirs. Cell Host Microbe 26, 347–358, e347 (2019).
Kearney, M. F. et al. Origin of rebound plasma HIV includes cells with identical proviruses that are transcriptionally active before stopping of antiretroviral therapy. J. Virol. 90, 1369–1376 (2016).
Bandera, A., Gori, A., Clerici, M. & Sironi, M. Phylogenies in ART: HIV reservoirs, HIV latency and drug resistance. Curr. Opin. Pharmacol. 48, 24–32 (2019).
Vibholm, L. K. et al. Characterization of intact proviruses in blood and lymph Nnde from HIV-infected individuals undergoing analytical treatment interruption. J. Virol. 93, e01920-18 (2019).
Liu, P. T. et al. Origin of rebound virus in chronically SIV-infected rhesus monkeys following treatment discontinuation. Nat. Commun. 11, 5412 (2020).
Solis-Leal, A. et al. Lymphoid tissues contribute to plasma viral clonotypes early after antiretroviral therapy interruption in SIV-infected rhesus macaques. Sci. Transl. Med. 15, eadi9867 (2023).
Wong, J. K. & Yukl, S. A. Tissue reservoirs of HIV. Curr. Opin. HIV AIDS 11, 362–370 (2016).
Obregon-Perko, V. et al. Dynamics and origin of rebound viremia in SHIV-infected infant macaques following interruption of long-term ART. J. JCI Insight 6, e152526 (2021).
Srinivasula, S. et al. Whole-body PET imaging of simian immunodeficiency virus using gp120-targeting probes fails to reveal regions of specific uptake in rhesus macaques. Eur. J. Nucl. Med. Mol. Imaging 52, 2645–2657 (2025).
Song, J. et al. Visualization and quantification of simian immunodeficiency virus-infected cells using non-invasive molecular imaging. J. Gen. Virol. 96, 3131–3142 (2015).
Fennessey, C. M. et al. Genetically-barcoded SIV facilitates enumeration of rebound variants and estimation of reactivation rates in nonhuman primates following interruption of suppressive antiretroviral therapy. PLoS Pathog. 13, e1006359 (2017).
Khanal, S. et al. In vivo validation of the viral barcoding of simian immunodeficiency virus SIVmac239 and the development of new barcoded SIV and subtype B and C simian-human immunodeficiency viruses. J. Virol. 94, e01420-19 (2019).
Chen, H. Y., Di Mascio, M., Perelson, A. S., Ho, D. D. & Zhang, L. Determination of virus burst size in vivo using a single-cycle SIV in rhesus macaques. Proc. Natl Acad. Sci. USA 104, 19079–19084 (2007).
King, I. V. et al. Origin and correlates of viral rebound in SIV-infected rhesus macaques following ART discontinuation. Preprint at bioRxiv https://doi.org/10.1101/2025.08.30.673277 (2025).
Medzhitov, R. Recognition of microorganisms and activation of the immune response. Nature 449, 819–826 (2007).
Slack, E. et al. Innate and adaptive immunity cooperate flexibly to maintain host-microbiota mutualism. Science 325, 617–620 (2009).
Brenchley, J. M. et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 12, 1365–1371 (2006).
Estes, J. D. et al. Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS Pathog. 6, e1001052 (2010).
Nganou-Makamdop, K. et al. Translocated microbiome composition determines immunological outcome in treated HIV infection. Cell 184, 3899–3914, e3816 (2021).
Hataye, J. M. et al. Principles governing establishment versus collapse of HIV-1 cellular spread. Cell Host Microbe 26, 748–763, e720 (2019).
Siddiqui, S. et al. Persistent viral reservoirs in lymphoid tissues in SIV-infected rhesus macaques of Chinese-origin on suppressive antiretroviral therapy. Viruses 11, 105 (2019).
Rabezanahary, H. et al. Despite early antiretroviral therapy effector memory and follicular helper CD4 T cells are major reservoirs in visceral lymphoid tissues of SIV-infected macaques. Mucosal Immunol. 13, 149–160 (2020).
Rutishauser, R. L. & Trautmann, L. CD8 + T-cell responses in HIV controllers: potential implications for novel HIV remission strategies. Curr. Opin. HIV AIDS 17, 315–324 (2022).
Sandel, D. A., Rutishauser, R. L. & Peluso, M. J. Post-intervention control in HIV immunotherapy trials. Curr. Opin. HIV AIDS 20, 70–79 (2025).
Van Rompay, K. K. A. Tackling HIV and AIDS: contributions by non-human primate models. Lab Anim. 46, 259–270 (2017).
Rutstein, S. E., Limarzi-Klyn, L., Miller, W. C. & Powers, K. A. Public health implications of diagnosing and treating acute HIV. Curr. Opin. HIV AIDS 20, 236–246 (2025).
Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV (Clinical info HIV.gov, 2019); https://clinicalinfo.hiv.gov/en/guidelines/hiv-clinical-guidelines-adult-and-adolescent-arv/initiation-antiretroviral-therapy?view=full
Fray, E. J. et al. Antiretroviral therapy reveals triphasic decay of intact SIV genomes and persistence of ancestral variants. Cell Host Microbe 31, 356–372.e5 (2023).
Long, S. et al. Evaluating the intactness of persistent viral genomes in simian immunodeficiency virus-infected rhesus macaques after initiating antiretroviral therapy within one year of infection. J. Virol. 94, e01308-19 (2019).
Gandhi, R. T. et al. Selective decay of intact HIV-1 proviral DNA on antiretroviral therapy. J. Infect. Dis. 223, 225–233 (2021).
Ho, Y. C. et al. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 155, 540–551 (2013).
Means, R. E., Greenough, T. & Desrosiers, R. C. Neutralization sensitivity of cell culture-passaged simian immunodeficiency virus. J. Virol. 71, 7895–7902 (1997).
Bertagnolli, L. N. et al. Autologous IgG antibodies block outgrowth of a substantial but variable fraction of viruses in the latent reservoir for HIV-1. Proc. Natl Acad. Sci. USA 117, 32066–32077 (2020).
Ferris, A. L. et al. Clonal expansion of SIV-infected cells in macaques on antiretroviral therapy is similar to that of HIV-infected cells in humans. PLoS Pathog. 15, e1007869 (2019).
McMyn, N. F. et al. The latent reservoir of inducible, infectious HIV-1 does not decrease despite decades of antiretroviral therapy. J. Clin. Invest. 133, e171554 (2023).
Maldarelli, F. et al. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 345, 179–183 (2014).
Cohn, L. B. et al. HIV-1 integration landscape during latent and active infection. Cell 160, 420–432 (2015).
Loffredo, J. T. et al. Mamu-B*08-positive macaques control simian immunodeficiency virus replication. J. Virol. 81, 8827–8832 (2007).
Bochart, R. M. et al. Mitigation of endemic GI-tract pathogen-mediated inflammation through development of multimodal treatment regimen and its impact on SIV acquisition in rhesus macaques. PLoS Pathog. 17, e1009565 (2021).
Del Prete, G. Q. et al. Comparative evaluation of coformulated injectable combination antiretroviral therapy regimens in simian immunodeficiency virus-infected rhesus macaques. AIDS Res. Hum. Retroviruses 32, 163–168 (2016).
Smedley, J. et al. Laparoscopic technique for serial collection of para-colonic, left colic, and inferior mesenteric lymph nodes in macaques. PLoS ONE 11, e0157535 (2016).
Zevin, A. S. et al. Laparoscopic technique for serial collection of liver and mesenteric lymph nodes in macaques. J. Vis. Exp. 123, e55617 (2017).
Okoye, A. A. et al. CD8+ T cells fail to limit SIV reactivation following ART withdrawal until after viral amplification. J. Clin. Invest. 131, e141677 (2021).
Hansen, S. G. et al. Addendum: immune clearance of highly pathogenic SIV infection. Nature 547, 123–124 (2017).
Hansen, S. G. et al. Immune clearance of highly pathogenic SIV infection. Nature 502, 100–104 (2013).
Immonen, T. T. et al. No evidence for ongoing replication on ART in SIV-infected macaques. Nat. Commun. 15, 5093 (2024).
Hansen, S. G. et al. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 473, 523–527 (2011).
Hansen, S. G. et al. Myeloid cell tropism enables MHC-E-restricted CD8(+) T cell priming and vaccine efficacy by the RhCMV/SIV vaccine. Sci. Immunol. 7, eabn9301 (2022).
Pitcher, C. J. et al. Development and homeostasis of T cell memory in rhesus macaque. J. Immunol. 168, 29–43 (2002).
Acknowledgements
The work was funded by the Bill and Melinda Gates Foundation and in part with Federal funds from the National Institute of Allergy and Infectious Disease, National Institutes of Health grant UM1AI164560 (to L.J.P.), the Oregon National Primate Research Center Core grant P51OD011092 and the National Cancer Institute, National Institutes of Health, currently under contract number 75N91019D00024 (B.F.K. and J.D.L.). These funders had no role in the conceptualization, design, data collection, analysis, decision to publish or preparation of this manuscript. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services; nor does mention of trade names, commercial products or organizations imply endorsement by the US Government. Funding agency websites are https://www.niaid.nih.gov/, https://orip.nih.gov/ and https://www.cancer.gov/. We thank the Quantitative Molecular Diagnostics Core and the Viral Evolution Core of the AIDS and Cancer Virus Program at the Frederick National Laboratory for Cancer Research for viral load analyses and barcode sequencing. We thank the Infectious Disease Resource of the Oregon National Primate Research Center for animal protocol management. ART drugs were provided by Gilead Sciences Inc. We also thank A. Sylwester, S. Hagen, M. Fischer, A. Selseth, O. Fadeyi, M. Medina, M. Chaunzwa, W. Omange, R. MacAllister, A. Marenco, I. Espinoza, R. Butler and Z. Ettaki for technical assistance or animal care.
Author information
Authors and Affiliations
Contributions
L.J.P., J.D.L. and D.H.B. conceived and obtained funding for the study. B.F.K., A.A.O., B.V.-M., J.D.E., J.D.L. and L.J.P. designed and supervised the study. D.D., C.N., W.G. and S.H. collected, processed and analysed samples from study animals. J.S. supervised animal care and collected biopsy samples. C.S.L. and M.K.A. performed necropsies on study animals with anatomic/pathologic analysis. C.M. Hughes and S.G.H. performed T cell response analysis on blood and tissue samples. W.J.B., R.F., C.M. Homick, M.H., K.O., R.S., L.S. and J.L.W. performed quantitative virologic analysis on study samples. C.M.F. and A.M. performed barcode sequencing. B.F.K., T.T.I., E.K., A.C. and C.A.G. analysed barcode data. T.T.I. performed all statistical tests. B.F.K., T.T.I., J.D.L. and L.J.P. wrote the original and revised paper. All authors reviewed and edited the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Microbiology thanks Namita Rout and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Despite early ART, all study macaques develop SIV-specific CD4+ and CD8+ T cell responses.
a, Longitudinal analysis of SIV-specific CD4+ and CD8+ T cell responses in blood and bronchoalveolar lavage fluid (BAL). Responses were determined by intracellular cytokine (ICS) analysis (IFN-γ and TNF-α) using whole open reading frame (ORF) mixes of overlapping 15mer peptides (including SIVmac239 Gag; Rev, Nef, Tat, Pol, Vif, Vpr, and Vpx) to stimulate PBMCs. The net frequencies of CD69+, IFN-γ+ and/or TNF+ positive memory T cell responses to each ORF peptide mix after stimulation were summed to get the overall responses shown in the figure. The black line indicates the mean frequencies for each time point, with the grey area indicating the 95% confidence limit. b, Analysis of CD4+ and CD8+ T cell responses in blood, peripheral (p) LN, mesenteric (m) LN, and spleen at 60 wpi. Responses were determined by intracellular cytokine (ICS) analysis as described in part a. The box plots show the median and 25th/75th percentiles with the whiskers showing the minimal and maximal values.
Extended Data Fig. 2 Blood viral burden and dynamic viral parameters are balanced between experimental groups.
a, Peak pVL was not significantly different between macaques in the 3 experimental groups [on-ART (n = 6), off-ART d5 (n = 9), or off-ART d7(n = 9), Kruskal-Wallis test]. b, Time to viral suppression (first time point with less than 15 copies/ml of plasma) was not significantly different between groups using a Log-rank Mantel-Cox test. c, The number of detectable blips in pVL (>1 copy/ml) was assessed between 36-66 wpi showing no difference between experimental groups (Kruskal-Wallis test). d, e, The amount of vDNA (d) and vRNA (e) in PBMCs at 12 dpi, 21 and 66 wpi was not significantly different between macaques in the assigned experimental groups (Kruskal-Wallis test was used for each individual time point assessed). [horizontal black lines indicate median value in all panels; open circle symbols represent samples below the limit of detection, which varied depending on specimen input].
Extended Data Fig. 3 Tissue biopsy viral loads are comparable between experimental groups during ART.
a, b, The amount of vDNA (a) and vRNA (b) from on-ART biopsy samples from peripheral LN (pLN), mesenteric LN (mLN), spleen, bone marrow, duodenum, rectum, and liver at 12 dpi, 21 wpi, and 66 wpi was not significantly different between macaques based on the assigned experimental groups (on-ART necropsy or with necropsy after 5 or 7 days off ART). Kruskal-Wallis test was used for each individual time point assessed. Open symbols plotted below the dotted line represent samples below the limit of detection, which varied depending on specimen input. [horizontal black lines indicate median value in all panels with the n values reflecting the number of samples with DNA or RNA levels above the limit of detection].
Extended Data Fig. 4 Off-ART plasma viral rebound dynamics and barcode proportions.
a, Of the 9 macaques that were necropsied 5 days off ART, 7 remained aviraemic (<1 copy/ml) and 2 (LV1 and LV4) were viraemic with 13 and 21 copies/ml, respectively. b, Of the 9 macaques that were necropsied 7 days off ART, 4 remained aviraemic (<1 copy/ml), 4 (VL2, LV3, LV5, LV6) had low-level viraemia (5-28 copies/ml) and 1 (HV1) had a much higher rebound at 4,700 copies/ml. For low viraemic macaques, the number of unique barcode lineages detected in rebound plasma ranged from 2-5, while HV1 had a total of 9 detectable lineages. Each barcoded lineage is shown as a proportion of the total PVL at the time of sampling.
Extended Data Fig. 5 Necropsy viral loads across various tissue types.
a, Viral DNA and RNA from on-ART (n = 6) and off-ART aviraemic macaques (n = 11). b, Viral DNA and RNA from off-ART viraemic macaques (n = 7). Tissue samples were grouped anatomically into 4 groups: GI tract, GI tract-associated lymphoid tissues, non-GI-tract-associated lymphoid tissues, and non-lymphoid tissues. For some tissues, multiple separate samples were analyzed and plotted individually. Symbols plotted below the black line represent samples below the limit of detection, which varied depending on specimen input.
Extended Data Fig. 6 On-ART biopsy barcode data do not exceed the on-ART tissue-based 99% prediction interval.
Extensive biopsies were performed on all 24 study rhesus macaques at 66 wpi while still on ART, collecting samples from duodenum and rectum (GI tract; blue circles), mesenteric LNs (GI tract LN; red diamonds), liver, spleen, bone marrow, peripheral LN, and PBMCs (non-GI tract tissues; green triangle). Individual barcode vDNA and vRNA levels are plotted with only 14% of all barcodes in all tissues expressing viral RNA. There was no vRNA barcode quantity that exceeded the 99% prediction interval (blue lines with shaded area) that was generated using the on-ART necropsy data (see Fig. 2a).
Extended Data Fig. 7 Identification of presumptive rebound origin sites in macaques with distal spread of rebounding lineages.
a, An example barcode (LV6, BC.3963) is shown with 4 distinct tissues each with vRNA expression above the 99% prediction interval (blue lines with shaded area). The above expected vRNA value was calculated from the prediction interval regression line to the measured value of each site (dotted lines). b, The difference in vRNA levels above expected (pink line) between the first and second highest barcode was calculated, followed by the same comparison between the second and third highest barcode, and so forth for all remaining tissue sites. c, The difference in vRNA levels above expected was calculated for each 11 outlier barcodes found in more than one tissue site. d, The difference in the adjusted RNA levels between all tissues with the same outlier barcode was plotted with the difference between the 1st and 2nd barcode shown in pink and the difference between 2nd and 3rd shown in green with the remaining comparisons shown in grey. We determined the 99th quantile of the adjusted RNA difference distribution using all sites with rank >1 (for example, not potential origin sites). The adjusted-RNA difference between the 1st and 2nd tissue sites was higher than the 99% quantile in 6 of the 11 outlier barcodes consistent with them being rebound origin sites.
Extended Data Fig. 8 Barcode DNA distributions comparing rebound origin and non-0rigin sites across all tissues in which the rebound origin (outlier) barcodes were detected.
For each outlier barcode with a defined origin site (n = 27), the DNA values for that specific barcode across all tissues where it was identified is shown. Non-GI-associated lymphoid tissues are shown on the left with origin sites designated by green triangles. GI tract samples are shown in the middle with origin sites designated by blue circles, GI tract-associated lymphoid tissues are shown on the right with origin sites designated by red diamonds.
Extended Data Fig. 9 Relationship between the overall tissue viral DNA levels and the individual barcode DNA levels in origin sites with the incidence of early post-ART viral rebound.
a, The overall viral DNA copies from all 697 individual tissues from the 18 off-ART macaques, from which we obtained both RNA and DNA barcode sequences, are plotted by tissue type with the non-rebound origin tissues coloured grey and the 21 origin site tissues coloured based on whether they include 1 (blue), 2 (green) or 3 (red) outlier barcodes. b, Each of the 21 origin site tissues is shown separately with the tissue-level barcode DNA distribution for each tissue plotted. Non-outlier barcodes are grey; outlier barcodes are coloured based on whether there were 1, 2 or 3 in that tissue, colours as described in panel a.
Supplementary information
Supplementary Information (download PDF )
Gating strategy for intracellular cytokine analysis.
Supplementary Tables 1–9 (download XLSX )
Table 1. Animal demographics. Table 2. Tissues analysed and anatomic group assignments. Tables 3–9. Various regression tables.
Source data
Source Data Fig. 1 (download XLSX )
All numerical data in Excel.
Source Data Fig. 2 (download XLSX )
All numerical data in Excel.
Source Data Fig. 3 (download XLSX )
All numerical data in Excel.
Source Data Fig. 4 (download XLSX )
All numerical data in Excel.
Source Data Fig. 5 (download XLSX )
All numerical data in Excel.
Source Data Fig. 6 (download XLSX )
All numerical data in Excel.
Source Data Extended Data Fig. 1 (download XLSX )
All numerical data in Excel.
Source Data Extended Data Fig. 2 (download XLSX )
All numerical data in Excel.
Source Data Extended Data Fig. 3 (download XLSX )
All numerical data in Excel.
Source Data Extended Data Fig. 4 (download XLSX )
All numerical data in Excel.
Source Data Extended Data Fig. 5 (download XLSX )
All numerical data in Excel.
Source Data Extended Data Fig. 6 (download XLSX )
All numerical data in Excel.
Source Data Extended Data Fig. 7 (download XLSX )
All numerical data in Excel.
Source Data Extended Data Fig. 8 (download XLSX )
All numerical data in Excel.
Source Data Extended Data Fig. 9 (download XLSX )
All numerical data in Excel.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Keele, B.F., Okoye, A.A., Immonen, T.T. et al. Initial sites of SIV rebound after antiretroviral treatment cessation in rhesus macaques. Nat Microbiol 11, 648–663 (2026). https://doi.org/10.1038/s41564-025-02258-3
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41564-025-02258-3
