
Abstract
Four, eight or twenty C3 symmetric protein trimers can be arranged with tetrahedral, octahedral or icosahedral point group symmetry to generate closed cage-like structures1,2. Viruses access more complex higher triangulation number icosahedral architectures by breaking perfect point group symmetry3,4,5,6,7,8,9, but nature appears not to have explored similar symmetry breaking for tetrahedral or octahedral symmetries. Here we describe a general design strategy for building higher triangulation number architectures starting from regular polyhedra through pseudosymmetrization of trimeric building blocks. Electron microscopy confirms the structures of T = 4 cages with 48 (tetrahedral), 96 (octahedral) and 240 (icosahedral) subunits, each with 4 distinct chains and 6 different protein–protein interfaces, and diameters of 33 nm, 43 nm and 75 nm, respectively. Higher triangulation number viruses possess very sophisticated functionalities; our general route to higher triangulation number nanocages should similarly enable a next generation of multiple antigen-displaying vaccine candidates10,11 and targeted delivery vehicles12,13.
Similar content being viewed by others

Main
Natural and designed protein nanocages consist of one or multiple unique components arranged with tetrahedral, octahedral and icosahedral point group symmetry1,3,4,5,6,7,14,15,16. Although there are no other point group symmetries, in the icosahedral case, viruses access larger and more complex higher triangulation (T) number architectures by interspersing varying numbers of hexagons between 12 pentagonal substructures (pentons), which expand the structure without changing the curvature. Viruses achieve this symmetry breaking by placing at symmetrically non-equivalent positions either the same subunit in different conformations (quasisymmetry)3,4,5,6,7,8, or closely related but distinct subunits (pseudosymmetry)9. The accessing of higher T number icosahedral structures by such symmetry breaking is critical to the remarkable functionality of viruses, such as the ability to package and deliver large nucleic acid cargos. Similarly, the de novo design of higher T number protein assemblies could enable new approaches to nucleic acid delivery and, as the potency of nanoparticle immunogens can increase with increasing valency of display, could lead to more potent vaccines. However, although protein design has had considerable success in designing symmetric assemblies that assemble from identical interacting subunits1,14,15,16, the design of assemblies with multiple identical or nearly identical chains in non-symmetry equivalent positions is an outstanding challenge.
We set out to develop a systematic approach to design higher T-number protein assemblies that could generate not only icosahedral (I-sym) architectures but also tetrahedral (T-sym) and octahedral (O-sym) higher T-number nanostructures, which, to our knowledge, have not been found in nature. Of the two routes to breaking symmetry described above, we reasoned that using closely related but distinct subunits (pseudosymmetry) would have the advantage over quasisymmetry as it avoids the complexity of designing single subunits with multiple distinct states and sets of interactions. We reasoned further that a design strategy that started with creating T = 1 architectures from homotrimers, and then extended these to T = 4 by inserting pseudosymmetric heterotrimers, would be more robust than directly designing for higher T-number cages, as subsets of the cage forming interfaces could be experimentally validated independently.
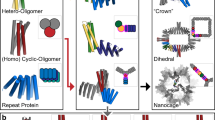
Our design strategy is illustrated in Fig. 1a for the icosahedral case. A T = 1 cage with icosahedral point group symmetry can be built from 20 homotrimers aligned along the icosahedral threefold symmetry axes (Fig. 1a). The centres of the homotrimers can be visualized as the vertices of 12 pentagonal faces that form the sides of a dodecahedron (Fig. 1a). Higher T-number cages conserve the number of pentagons (pentons), which are always aligned along the 12 icosahedral fivefold symmetry axes. T = 4 cages with hexagons bridging the 12 pentons could be generated if it were possible to extract pentons from the T = 1 cages and insert homotrimers along the icosahedral threefold symmetry axes connecting three pentons (Fig. 1b,c). Extracting pentons from T = 1 cages requires that one of the three interfaces joining each trimer with its nearest neighbours be eliminated. This can be accomplished by substituting the homotrimers with geometrically identical (that is, pseudosymmetric) heterotrimers composed of three distinct chains with different amino acid sequences. This symmetry breaking allows the interface facing outwards from the penton to be specifically eliminated, isolating the penton as a free-standing structure that can be used to build larger structures.

a–c, General design route to T = 4 icosahedral cages using substructures extracted from a T = 1 cage. Twelve pentagonal substructures (pentons) from a T = 1 cage (a) are docked with 20 homotrimers to form a closed cage structure (b), which creates hexagonal local structures (shaded red) placed between pentons (shaded yellow; c). d–f, Schematic of the route to T = 4 tetrahedral (d), octahedral (e) and icosahedral (f) cages. Step 1: T = 1 cages are designed starting from C3 symmetric trimeric building blocks. Step 2: the trimers constituting each face of the T = 1 cages are displaced away from the origin along the symmetry axis orthogonal to the face (left) and replaced by ABC-type pseudosymmetric heterotrimers to produce crowns in which one of the three components (yellow) is free to be designed to dock to other building blocks (right). Step 3: the crowns are docked with a new set of homotrimers aligned along the threefold symmetry axis of each architecture, which produces T = 4 cages.
We reasoned that this approach could be used to design T = 4 tetrahedral, octahedral and icosahedral nanostructures using pseudosymmetric heterotrimers to precisely programme the six distinct interfaces in such structures. We adopted the three-step hierarchical approach outlined in Fig. 1d–f. We first designed T = 1 cages by arranging C3 symmetric homotrimers along the threefold symmetry axis of each architecture1,2 (Fig. 1d–f, step 1). Following experimental validation, we next extracted cyclic ‘crown’-like substructures from these cages (pentons in the icosahedral case) by replacing the symmetric homotrimers with ABC-type heterotrimers that lack the cage-forming trimer–trimer interface on one of the subunits, and redesigning the crown-forming trimer–trimer interface to be heterodimeric (Fig. 1d–f, step 2). Following experimental validation, these crown-like structures are arranged along one of the threefold symmetry axes of T-sym, the fourfold symmetry axis of O-sym or the fivefold symmetry axis of I-sym architectures, and docked and designed to interact with an additional symmetric homotrimer placed on the remaining threefold axis (Fig. 1d–f, step 3). This generates four component T = 4 cages for each cage symmetry, with a hexameric motif between edges of the crowns (h = 2 and k = 0 in the Caspar and Klug nomenclature8).
Design of pseudosymmetric heterotrimers
Our design strategy requires homotrimeric building blocks that can be replaced by pseudosymmetric heterotrimeric blocks with distinct intra-trimer interfaces but identical outwards-facing interfaces that mediate nanocage assembly. We chose to utilize a previously designed family of ring-shaped homotrimers and heterotrimers17, which have the same structure but different amino acid sequences at the protomer–protomer interfaces. The heterotrimer designs contain all three chains in equal stoichiometries, and both homotrimer and heterotrimer designs assemble into symmetric cyclic rings very close to the computational design models17. To generate a wider diversity of geometries to enable docking of the rings into closed architectures through helix–helix interfaces (Fig. 2a), we built on previous work17,18,19 and rigidly fused various helical repeat protein extensions onto the termini19 (referred to as ‘arms’ below), yielding homotrimers of approximately 10 nm in diameter (Fig. 2b–e, Supplementary Table 2, Supplementary Fig. 1 and Supplementary information 1). Of the designs, 37 of 39 were soluble following expression in Escherichia coli and purification from immobilized metal affinity chromatography, 33 of 39 had size-exclusion chromatography (SEC) retention volumes consistent with the design models and the SEC profiles were monodisperse for 22 of the designs (Supplementary Figs. 2 and 3). The homotrimeric state was confirmed by native mass spectrometry (nMS) for 27 of 39 designs (Supplementary Figs. 4–6), and small-angle X-ray scattering profiles were closely consistent with profiles computed from the design models for 19 (Supplementary Figs. 7 and 8). Negative-stain electron microscopy (nsEM) characterization of the overall shapes of the designs was again consistent with the design models, with curved, straight, wide and narrow arm arrangements evident in 2D class averages and 3D reconstructions (Fig. 2b,d and Supplementary Fig. 9). We obtained a crystal structure of one of the designs, BGL17_A31 (see Supplementary information 1.3 for protein naming), which was very close to the design model and AlphaFold2 (AF2) prediction (Fig. 2c and Extended Data Table 2).

a, Schematic of the design strategy for homotrimers with arms and pseudosymmetric heterotrimers. b, nsEM micrograph of the BGL17_A31 homotrimer and a 2D class average (inset) along a threefold symmetry axis. c, Structure of BGL17_A31 solved by X-ray crystallography (white) compared with its design model (light blue) and AF2 prediction (orange). d, Superpositions of the 3D-reconstructed nsEM map (transparent cloud) to the structure model (blue) of BGL17_A32 (no map; top left), BGL18_A35 (top middle) and BGL19_A39 (top right). 2D class averages along threefold symmetry axis are also shown (bottom). e, SEC results of homotrimers. f, ABC-type pseudosymmetric heterotrimer (hetBGL0-18-17_A32) with arms of different lengths extending from each component. Top, the 3D-reconstructed nsEM map (transparent cloud) superimposed on the design model (colours indicate the three different chains). Bottom, a 2D class average along the pseudo-threefold symmetry axis. g, nMS analysis of hetBGL0-18-17_A32; only the heterotrimeric ABC species is observed. Scale bars, 100 nm (b) and 10 nm (b (inset),d,f). See Extended Data Table 1 for the amino acid sequences of the oligomers.
To design pseudosymmetric heterotrimers, we used an interface transplantation approach (Supplementary Figs. 10 and 11), preserving the overall structural C3 symmetry (Supplementary information 2). A homotrimer was selected as the host scaffold and three different protomer–protomer interfaces from different homo-oligomers (guests) were transplanted onto the subunit–subunit interfaces (Fig. 2a and Supplementary Table 1), conserving the residues at the arm junction. We first checked the compatibility between a host and a guest in the homo-oligomer context by symmetrically transplanting the guest interface into the host. We selected highly expressed and homogenous homotrimers for hybridization: seven with arms (BGL0_A10, BGL0_A11, BGL17_A31, BGL17_A32, BGL18_A35, BGL19_A38 and BGL19_A39) as host scaffolds and four without arms (BGL0, BGL17, BGL18 and BGL19)17 as guest interfaces. We experimentally characterized 18 of the 28 combinations (Supplementary Table 3); 14 of 18 had SEC peaks at the correct oligomeric size (Supplementary Fig. 12) and 11 of 18 had strong nMS signals at the correct masses (Supplementary Fig. 13). The BGL17_A32 host backbone was found to be compatible with multiple guest interfaces; we constructed heterotrimers by splicing interfaces from different guests together in different combinations17 (Supplementary Table 4). To enable assignment of chain type by electron microscopy, we varied the number of repeat units on the arms protruding from each chain (−1 repeat for the A component and +1 to the C component). Three of five heterotrimers (hetBGL0-17-19_A32, hetBGL0-18-17_A32 and hetBGL0-19-17_A32; see Supplementary information 2 for protein naming) formed ABC-type heterotrimers as shown by SEC, SDS–PAGE and nMS (Fig. 2f and Supplementary Figs. 14–17). For two of these (hetBGL0-18-17_A32 and hetBGL0-19-17_A32), the expected differences in arm lengths were clearly evident in 2D nsEM averages and 3D reconstructions (Fig. 2f and Supplementary Fig. 18). This approach to creating pseudosymmetric hetero-oligomers should be readily applicable to symmetric protein oligomers quite generally, if the amino acid sequence of the protomer–protomer interface can be diversified.
Cage design
We generated base T = 1 tetrahedral, octahedral and icosahedral cages from the BGL17_A32 homotrimer using RPXdock20,21 to sample rotational and translational displacements of the trimer C3 axis along the threefold cage axes (Supplementary information 3). We found that for the different symmetries, different numbers of repeat units of arms (1.5, 2.5 and 3.5 repeat units for tetrahedral, octahedral and icosahedral cages, respectively) were optimal for docking of the trimers with good shape complementarity (Fig. 3a,b,e,f,i,j and see Supplementary information 3.1). The newly generated cage interfaces were designed using ProteinMPNN22, and designs for which the AF2 prediction of the arm–arm interface was less than 2.0 Å root-mean-square deviation (RMSD) from the design model were selected for experimental characterization; these comprise seven tetrahedral cages (TetT = 1-1 to TetT = 1-7; Supplementary Table 5) with 12 subunits and diameters of approximately 13 nm, 8 octahedral cages (OctT = 1-1 to OctT = 1-8; Supplementary Table 6) with 24 subunits and diameters of approximately 20 nm, and 4 icosahedral cages (IcoT = 1-1 to IcoT = 1-4; Supplementary Table 7) with 60 subunits and diameters of approximately 40 nm. All of the tetrahedral designs had peaks at the expected retention volume (Supplementary Fig. 20) on SEC, and four (TetT = 1-1, TetT = 1-2, TetT = 1-4 and TetT = 1-6) were structurally homogeneous by nsEM (Extended Data Fig. 1 and Supplementary Fig. 21), with 2D class averages and 3D-reconstructed nsEM maps (Fig. 3c and Supplementary Fig. 21) matching the design models (Fig. 3b,c). Seven of the octahedral cages had single peaks on SEC, and two (OctT = 1-2 and OctT = 1-4) showed homogenous structures matching the design models by nsEM (Fig. 3f,g and Supplementary Figs. 22 and 23). One of the icosahedral cages, IcoT = 1-1, had a single peak on SEC and was close to the design model by nsEM (Fig. 3j,k), although imperfectly formed cages were also observed (Supplementary Fig. 24).

a–c,e–g,i–k, BGL17_A32 with 1.5 (a), 2.5 (e) and 3.5 (i) repeat unit arms docked into tetrahedral (TetT = 1-4; b,c), octahedral (OctT = 1-2; f,g) and icosahedral (IcoT = 1-1; j,k) T = 1 cages. Superpositions of the 3D-reconstructed nsEM map (transparent cloud) on the cage design model (colours) are shown (b,f,j). nsEM micrographs (left) and characteristic 2D class averages (right) of the cages are also displayed (c,g,k). d,h,l, C3 (crownC3-3), C4 (crownC4-2) and C5 (crownC5-1) crowns made from pseudosymmetric heterotrimers. Left, superpositions of the 3D-reconstructed nsEM map on the crown design model (ch_A (green), ch_B (blue) and ch_C (orange)). Right, 2D class averages along threefold, fourfold and fivefold symmetry axes. The diameters of the crowns are 11 nm (C3), 20 nm (C4) and 35 nm (C5). Scale bars, 100 nm (c,g,k (left)) and 10 nm (c,g,k (right),d,h,l). See Extended Data Tables 3 and 4 for the amino acid sequences of the T = 1 cages and crowns.
We next extracted C3, C4 and C5 symmetric cyclic oligomers (which we refer to as crowns because of their shape) from the structurally confirmed T = 1 cages by substituting in the structurally identical pseudosymmetric hetBGL0-18-17_A32 heterotrimer in place of the BGL17_A32 homotrimer (Fig. 1, third and fourth columns). The chain A (ch_A) and ch_B interfaces were redesigned using ProteinMPNN to interact at the crown trimer–trimer interface; this was necessary to avoid a potential off-target structure possible with the original C2 interface (Supplementary Fig. 25). The surface of the arm of ch_C, which points outwards from the crown, was redesigned to be entirely polar. This isolates the crowns as free-standing building blocks that can be used to build T = 4 cages in the next step. We selected designs for which AF2 predicted the ch_A–ch_B interface with RMSD < 2 Å and did not predict the ch_A–ch_A or ch_B–ch_B homodimer interfaces to form (Supplementary Fig. 19). We obtained genes encoding 19 sets of crowns that passed these filters (crownC3-1 to crownC3-5 for C3 crowns, crownC4-1 to crownC4-7 for C4 crowns and crownC5-1 to crownC5-7 for C5 crowns; Supplementary Tables 8–10), and the three chains for each crown were expressed separately in independent E. coli cultures. Following expression, the amount of each protein was estimated by SDS–PAGE gel densitometry, and appropriate amounts of culture were combined to achieve mixtures with stoichiometric amounts of the three chains, which were co-lysed and co-purified. By SEC, four of five C3, five of seven C4 and two of seven C5 crowns had peaks at the expected elution volumes (Supplementary Figs. 26–31) containing three distinct bands by SDS–PAGE (Supplementary Fig. 32), indicating that the complexes were heterotrimers. nsEM 2D class averages and 3D-reconstructed nsEM maps matched well with the crown design models (Fig. 3d,h,l and Extended Data Fig. 1). Thus, symmetric substructures can be extracted from larger symmetric assemblies by substituting homotrimers with pseudosymmetric heterotrimers.
In the final step of our hierarchical design approach, we designed T = 4 cages by combining the experimentally confirmed crowns with the BGL17_A32 homotrimer (Fig. 1, last column, and Supplementary information 3). The C3, C4 and C5 crowns were aligned with the threefold, fourfold and fivefold axes of tetrahedral, octahedral and icosahedral architectures, and BGL17_A32 was aligned to the remaining threefold axis. This generated assemblies in which the heterotrimer arms pointing outwards from the crowns (ch_C of heterotrimer) interact with the arms of the homotrimer (ch_ho). To find optimal docking interfaces, we used RPXdock, sampling the lengths of the interacting arms and the rotations and translations along the common axis, and designed sequences using proteinMPNN for the highest RPX scoring models for each symmetry. Designs were filtered based on the formation of the designed interface, and lack of formation of the self interfaces, in AF2 predictions (Supplementary Fig. 19). Very few initial designs for the octahedral architecture did not form predicted self interfaces; thus, to decrease the probability of self-interaction, we performed explicit negative design using proteinMPNN22 against the predicted self interfaces. We experimentally tested 14 sets of T = 4 cage designs (TetT = 4-1 to TetT = 4-5, OctT = 4-1 to OctT=4-4 and IcoT = 4-1 to IcoT = 4-5; Supplementary Tables 11–13) that passed the AF2 filters. The four components were expressed independently in different E. coli. cultures, mixed with 1:1:1:1 stoichiometry and co-lysed. The lysed samples were purified using immobilized metal affinity chromatography and SEC, and the cage structures were characterized by nsEM (Extended Data Fig. 1 and Supplementary Figs. 35–47). As described in the following paragraphs, the major species in each case was the designed T = 4 structure; we also observed minor species of smaller off-target T = 1-like cages (Supplementary Figs. 37, 42 and 45).
The T = 4 tetrahedral cage (TetT = 4-2) has a tetrapod shape (diameter of 33 nm) with the four C3 crowns pointing outwards, and the homotrimers bridging the crowns closer to the centre of the cage and facing inwards (Fig. 4a,d,g and Supplementary Fig. 36). The homotrimer–heterotrimer distance (11.5 nm) is almost twice the heterotrimer–heterotrimer distance (6.0 nm) due to the arm length difference between components (1.5 repeat units for ch_A and ch_B, and 3 repeat units for ch_C and ch_ho), and the interior volume is a tetrahedral channel 6.0 nm in width (Fig. 4d). Overall, the structure maps to a T = 4 Goldberg polyhedra with tetrahedral symmetry23, in which the hexagonal motifs between triangles are highly elongated (Fig. 4a). These structural features are evident in the nsEM map (Fig. 4d), micrographs (Fig. 4e) and 2D average classes (Fig. 4f), and the design model is closely consistent with the reconstructed nsEM map (Fig. 4g). The design model could be readily relaxed to fit the nsEM 3D map with all four components clearly within density (Supplementary Fig. 36); overall, the relaxed model matches well with the design model, with the exception of a slight twist of the overall structure resulting from curvature in the arm near the homotrimer–heterotrimer interface (Fig. 4d and Supplementary Fig. 36).

a–c, Ball-and-stick models of T = 1 (left) and T = 4 (right) cages for each symmetry, defined by the Caspar and Klug nomenclature8. d–o, Four component T = 4 tetrahedral cage with 48 subunits (d–g), octahedral cage with 96 subunits (h–k) and icosahedral cage with 240 subunits (l–o). Design models of the T = 4 cages (left) and a 3D-reconstructed nsEM map (right) are shown (d,h,l). In the design models, each chain is shown in a distinct colour (ch_A (green), ch_B (blue), ch_C (orange) and ch_ho (purple)). nsEM micrographs (e,i,m), characteristic 2D class averages of nsEM (f,j,n) and superpositions of the 3D-reconstructed nsEM map (grey density) on the cage design model (colours; g,k,o) are also displayed. Scale bars, 100 nm (e,i,m) and 20 nm (f,j,n). See Extended Data Table 5 for the amino acid sequences of the T = 4 cages.
The T = 4 octahedral cage (OctT = 4-3) has a 3D cross shape structure (diameter of 43 nm) with the original cubic shape of the T = 1 structure repeated six times and shifted away from the origin to positive and negative values of x, y and z along the three fourfold symmetry axes (Fig. 4b,h). Six C4 crowns form the outward faces of the structure along the x, y and z axes, which are connected by eight homotrimers placed in a cubic arrangement closer to the centre of the cage; as for the tetrahedral cage, the homotrimers and heterotrimers face in opposite directions (Supplementary Fig. 40). The overall architecture is that of a T = 4 Goldberg polyhedra with octahedral symmetry23, with an elongated hexagon bridging the square faces (Fig. 4b) and a 10-nm cavity in the centre. Homogeneous populations of cages are observed in nsEM micrographs (Fig. 4i), and 2D class averages along the twofold, threefold and fourfold symmetry axes are closely consistent with the design model (Fig. 4j). The nsEM 3D reconstruction is very close to the design model (Fig. 4k), but a slightly curved connection between crowns and homotrimers leading to a slight twist of the overall structure (Supplementary Fig. 40). To characterize the structure of OctT = 4-3 at higher resolution, we collected cryo-electron microscopy (cryo-EM) data and generated a 3D reconstruction, which, following refinement, resulted in a 3D cryo-EM map with 6.87 Å resolution with clear secondary structure features (Fig. 5a–d and Supplementary Fig. 51). Following relaxation via molecular dynamics24, the design model fits well into the cryo-EM map (Fig. 5e–g), with the crown and homotrimer substructures and the individual chains clearly defined (Supplementary Fig. 41). Around the fourfold symmetry axes, ch_A and ch_B of the hetBGL0-18-17_A32 heterotrimers (Fig. 5b,e, green and blue) form square motifs; the five helices in each arm in both chains are clearly evident in the cryo-EM map (Fig. 5e). BGL17_A32 homotrimers are placed along the threefold symmetry axes (Fig. 5d); the arm of each subunit has six helices and forms an interface with ch_C of the heterotrimer at the end (Fig. 5f,g, purple and orange). The slight twist of the C4 crown substructures relative to the design model arises from shifts at the homotrimer–heterotrimer interface (Supplementary Fig. 41).

a–d, 3D cryo-EM map of OctT = 4-3 from different views. e–g, Overlay between the cryo-EM map (grey transparent) and the design model relaxed into the map (colours) for each substructure: C4 crown (e), the homotrimer–heterotrimer interface (f) and the homotrimer (g). h–k, 3D cryo-EM map of IcoT = 4-4 from different views. The inset (h) is an adeno-associated virus (AAV) capsid shown at the same scale for size comparison. l–n, Overlay between the cryo-EM map (continuous density) and the relaxed design model (colours) for each substructure: C5 crown (l), the homotrimer–heterotrimer interface (m) and the homotrimer (n). Both the cryo-EM map and the protein model are coloured by chain (ch_A (green), ch_B (blue), ch_C (orange) and ch_ho (purple)). See Extended Data Table 7 for cryo-EM data collection information.
The T = 4 icosahedral cage (IcoT = 4-4) consists of 12 C5 crowns (pentons) connected by 20 outwards-facing homotrimers (Fig. 4c,l). Largely homogeneous 75-nm-sized cages were identified by nsEM and dynamic light scattering (DLS; Fig. 4m and Supplementary Fig. 44). SDS–PAGE showed clear bands corresponding to each component, suggesting that all four chains are present (Supplementary Fig. 43). The 2D class averages (Fig. 4n) and 3D nsEM reconstructions (Fig. 4l,o) have the overall designed shape but the orientations of the C5 crowns, and homotrimers appeared inverted from the design model (Supplementary Figs. 34 and 47). We collected cryo-EM images, and 3D reconstruction and refinement of the cryo-EM data yielded a 3D cryo-EM map with 13.15 Å resolution (Fig. 5h–k and Supplementary Fig. 51), in which the holes at the centre of trimers and the orientations of trimers are clearly identified. The design model with inverted components fits well into the cryo-EM density following relaxation (Fig. 5l–n and Supplementary Fig. 48). The overall structure has the architecture of a T = 4 Goldberg polyhedron with icosahedral symmetry23, with boat-type hexagonal motifs placed between pentagons (Fig. 4c). Surrounding the fivefold symmetry axes are pentons formed from hetBGL0-18-17_A32 heterotrimers (Fig. 5h,i, green, blue and orange). The pentons are bridged by BGL17_A32 homotrimers, which form tripod-like protrusions on the threefold symmetry axis (Fig. 5h,k, purple). On the twofold axes are boat-type distorted hexagons with two homotrimers and four heterotrimers on the vertices; two of the edges are formed by interacting heterotrimer subunits, and four edges are formed by interacting homotrimer and heterotrimer subunits (Fig. 5j). The outer diameter of the IcoT = 4-4 cage is about three times larger than that of the adeno-associated virus capsid (Fig. 5h), and the inner diameter of the empty pore at the centre of the IcoT = 4-4 cage is approximately 50 nm (volume of approximately 6.55 × 104 nm3), which can be used to package diverse cargos such as nucleic acids and enzymes.
We tested the thermal stability and pH tolerance of the OctT = 4-3 and IcoT = 4-4 cages using DLS (Supplementary information 4.5). The Z-average diameter (dZ) of the two cages was consistent with the expected diameters of the intact cages (approximately 80 nm for IcoT = 4-4 and approximately 50 nm for OctT = 4-3) at 25 °C (the smaller T = 1 contaminants observed by electron microscopy were not resolved by DLS probably due to the screening effect of the larger of two similarly sized particles25), was unchanged from 25–70 °C and then decreased above 75 °C (Fig. 6a,b). To test pH tolerance of the cages, we measured dZ while decreasing the pH of the buffer. The IcoT = 4-4 dZ of around 80 nm was unchanged from pH 8.0 to pH 5.3, but increased at pH 4.7, probably due to aggregation (Fig. 6c), whereas the OctT = 4-3 of approximately 50 nm was unchanged between pH 8.0 and pH 6.4, but increased at pH 5.9 or lower (Fig. 6d). Thus, the cages are quite thermostable (melting temperatures higher than 70 °C), which should facilitate their development as delivery vehicles, and undergo pH-dependent transitions at the pH sampled in the endocytic pathway, which may be exploitable for endosomal escape.

a–d, Z-average diameter of IcoT = 4-4 and OctT = 4-3 at varying temperature (a,b) and pH (c,d), measured by DLS. e, An ASGPR binding cage was generated by fusing the ch_ho chain of IcoT = 4-4 to an ASGPR-binding protein. f,g, Schematic of the ASGPR cage (f) and internalization of the cage in HEP3B cells (g). h,i, Confocal microscope images of HEP3B cells treated without (h) and with (i) the cages. Nuclei were stained with DAPI (blue), and nanocages with anti-His–Alexa-488 are shown in green. Scale bars, 30 µm (h,i).
As a first step towards exploring the use of the cages for delivery applications, we evaluated internalization by fusing one of the four components of IcoT = 4-4 (ch_ho) to a designed binder26 of asialoglycoprotein receptor (ASGPR) (Fig. 6e and Extended Data Table 6). ASGPR is an endocytic internalizing receptor that is exclusively expressed on the surface of liver cells. This receptor has been utilized for delivery of therapeutic oligonucleotides such as small interfering RNA27 and liver-specific LYTAC-mediated protein degradation26,28. Sixty ASGPR mini-binders were displayed on the outer surface of the IcoT = 4-4 cage through genetic fusion (Fig. 6f). We labelled the ASGPR cages with anti-His tag–Alexa-488 antibody (the subunits each have His tags) and incubated them with the hepatocellular carcinoma cell line HEP3B. By confocal imaging, we observed a substantial intracellular accumulation of the cage in a punctate localization pattern, probably indicating vesicle localization, whereas treatment with only anti-His–Alexa-488 antibody without the cage resulted in no intracellular signal (Fig. 6h,i). This result suggests that these large designed T = 4 cages can be internalized by cells for delivery or LYTAC applications.
Conclusion
Our T = 4 assemblies are a considerable advance in compositional and structural complexity over previous nanocage designs, which have one or two distinct components and up to three distinct interfaces; the designs described here have four distinct core structural components and six distinct interfaces (Supplementary Fig. 33). Our design approach is not limited to T = 4 cages but can be extended to higher T; for example, T = 7 cages with 420 subunits can be constructed by incorporating a second heterotrimer, based on either another BGL homodimer or using a different set of designed heterotrimers29 (see Supplementary Figs. 49 and 50 for routes to T = 7 and T = 9). Our design approach can be applied to any set of protein oligomers that can be pseudosymmetrized and have dockable interfaces for the target point group symmetry. To our knowledge, pseudosymmetric tetrahedral and octahedral assemblies have not been observed in nature; with four distinct chains and eight termini available for functionalization, these provide starting points for sophisticated multicomponent materials. With a 75-nm diameter, our T = 4 icosahedral design has considerably more interior volume available for packaging nucleic acid and other cargoes than previously designed cages; the accompanying paper by Dowling et al.30 (see ref. 31 for a high-level comparison of approaches) describes a route to even larger, albeit less homogeneous, structures using two-component heterotrimers. For vaccine and delivery applications, the four distinct structural components of our T = 4 assemblies provide many more opportunities for further functional elaboration than the one-component and two-component nanoparticles designed to date; for vaccines, presentation of multiple antigens and immunomodulators; and for targeted delivery, incorporation of distinct designed modules for targeting and internalization.
Methods
Computational design
All computational design methods are described in Supplementary information 1–3.
Protein expression
Plasmids (100 ng) were transformed into a chemically competent E. coli expression strain, BL21(DE3) or BL21(DE3)Star, for protein expression following the manufacturer’s protocol, with the exception of using 10 µl competent cells per reaction. Following transformation and recovery, the entire transformation products were used to inoculate 1 ml LB medium containing 100 µg ml−1 kanamycin and grown at 37 °C with shaking at 225 rpm overnight. Of overnight cultures, 500 µl was diluted into 50 ml TBM-5052 supplemented with 100 µg ml−1 kanamycin in 250 ml baffled flasks and incubated at 37 °C with shaking at 225 rpm for 18–24 h.
For systems using hetero-oligomers (pseudosymmetric hetero-oligomers, crowns and T = 4 cages), we used two different protocols: protocol 1 and protocol 2.
Protocol 1
Each component was transformed and incubated independently. After the incubation, we first checked relative expression level between components using SDS–PAGE (Supplementary information 4.5) based on the intensity of bands. Then, the incubated E. coli cultures of each component were mixed with a correct stoichiometry for target assemblies (1:1:1 for heterotrimers and crowns; 1:1:1:1 for T = 4 cages). After mixing the E. coli cultures, we harvested proteins following Supplementary information 4.4.
Protocol 2
Plasmids of all components were transformed into E. coli together. For example, in heterotrimer expression (three components: A, B and C), 300 ng of plasmids (100 ng of A + 100 ng of B + 100 ng of C) were transformed into 10 µl E. coli cells and incubated as described in this section. Then, proteins were harvested following Supplementary information 4.4.
Protein purification
Immobilized metal affinity chromatography
Cultures were harvested by centrifugation at 4,000g for 10 min, culture supernatant was decanted, and pellets were resuspended to 30 ml in lysis buffer. Of PMSF, 300 µl (100 mM in 100% ethanol) was added immediately before sonication at 70% power for 5 min. ‘Lysate’ fractions were saved, and then lysates were clarified by ultracentrifugation at 18,000g at 12 °C for at least 30 min and applied to 1.5-ml Ni-NTA resin (Qiagen) pre-equilibrated with lysis buffer and packed into Econo-Pac columns (Bio-Rad) for gravity chromatography. The columns were washed twice with 15 ml wash buffer and eluted with 10 ml elution buffer. Hetero-oligomers were purified according to a similar procedure, except they used the ‘Het’ variants of the lysis, wash and elution buffers, as this was found to improve the yield of single-His-tagged complexes over nonspecific multiple-His-tagged complexes, which can arise at high concentrations and probably dominate binding to the Ni-NTA resin. Samples prepared for crystallization were treated similarly, except 500-ml cultures were used and lysate was divided among six gravity columns.
SEC
Immobilized metal affinity chromatography (IMAC)-eluted samples were concentrated using 10k MWCO spin concentrators and were purified using a Superdex 200 10/300 increase (for oligomers) or Superdex 6 10/300 increase (for crowns and cages) columns (Cytiva) in SEC buffer using an ÄKTA pure system (Cytiva). SEC traces were also used to qualitatively determine homogeneity and quantitatively measure total yield by A280 absorbance integrated over the collected fractions using Unicorn (Cytiva).
Note that, for T = 4 icosahedral cages (IcoT = 4-4), we kept the IMAC-eluted sample at room temperature for 24 h before concentrating and running SEC. The 24-h equilibration was important to generate homogeneous samples, and we think that an even longer equilibration time could be helpful to further increase the level of homogeneity. Given the complexity of this sample (4 components, 6 different interfaces and 240 subunits), a relatively slow assembly kinetics seems reasonable; thus, giving an enough equilibration time (more than 24 h) is critical to get homogeneous samples.
For all other systems, the equilibration time is not a critical issue, so we concentrated the elution sample right after IMAC and ran SEC within a few hours. However, giving enough equilibration time (more than 24 h) may not harm the sample quality, but rather possibly increase the homogeneity.
TEVp cleavage
Purification and mass tags were buffer exchanged into TEV buffer and cleaved with TEVp at a ratio of 1 mg TEV per 100 mg substrate for 24–72 h at room temperature. After TEV cleavage, samples were exchanged into lysis buffer and passed over a Ni-NTA gravity column and washed with 10 ml lysis buffer. Flowthrough was collected, concentrated using 10k MWCO spin concentrators and purified once again by SEC.
Sample analysis
SDS–PAGE
Samples were diluted 1:1 with 2× Laemmli sample buffer (Bio-Rad) without β-mercaptoethanol, and 15 µl was loaded onto AnykD Criterion TGX Precast Midi Protein Gels (Bio-Rad). The ladder was 10 µl of Precision Plus Protein Kaleidoscope Prestained Protein Standards (Bio-Rad). Gels were run at 300 V for 18 min, then stained using the eStain L1 Protein Staining System (Genscript). Stained gels were imaged using Chemidoc XRS+ (Bio-Rad).
Liquid chromatography–mass spectrometry
To identify the molecular mass of each protein and thus verify sample identity and integrity, intact mass spectra were obtained via reverse-phase light chromatography–mass spectrometry on an Agilent G6230B TOF on an AdvanceBio RP-Desalting column, and subsequently deconvoluted by way of Bioconfirm using a total entropy algorithm.
nMS
The oligomeric state of SEC-purified and light chromatography–mass spectrometry-verified samples was analysed by on-line buffer exchange mass spectrometry in 200 mM ammonium acetate using a Vanquish ultra-high performance light chromatography system coupled to a Q Exactive ultra-high mass range Orbitrap mass spectrometer (Thermo Fisher Scientific). A self-packed buffer-exchange column was used (P6 polyacrylamide gel; Bio-Rad). The recorded mass spectra were deconvolved with UniDec version 4.2+.
nsEM
SEC-purified samples were diluted (0.005 mg ml−1 for oligomers and 0.05–0.1 mg ml−1 for crowns and cages) using SEC buffer immediately before application for 45 s to glow-discharged thick carbon film-coated 400 mesh copper grids (CF400-CU TH, Electron Microscopy Sciences). Grids were then stained and dried immediately twice using 2% uranyl formate. Dried grids were screened on a 120 kV Talos L120C transmission electron microscope. The E. Pluribus Unum (EPU; FEI Thermo Scientific) software was used for automated data collection. Data processing was carried out in CryoSPARC (Structura Biotechnology Inc).
Cryo-EM sample preparation
To prepare cryo-EM sample grids, 3 μl of 0.5–1.0 mg ml−1 of protein in 25 mM Tris and 300 mM NaCl at pH 8.0 was applied to glow-discharged Quantifoil R 2/2 300 mesh copper grids overlaid with a thin layer of carbon. Vitrification was performed on a Mark IV Vitrobot with a wait time of either 5 s or 7.5 s, a blot time of 0.5 s and a blot force of either 0 or −1 before being immediately plunged frozen into liquid ethane. The sample grids were clipped following standard protocols before being loaded into the microscope for imaging.
Note that we were not able to conduct cryo-EM for T = 4 tetrahedral cages, because the cages were aggregated when they are concentrated higher than 0.3 mg ml−1. They were stable below 0.15 mg ml−1, which was too dilute for conducting cryo-EM.
Cryo-EM data collection
Data collection was performed automatically using either Leginon32 or SerialEM to control either a Thermo Fisher Titan Krios 300 kV TEM equipped with a standalone K3 Summit direct electron detector with an energy filter or a Thermo Fisher Glacios 200 kV equipped with a standalone K3 Summit direct electron detector33. Both samples were collected using counting mode, with random defocus ranges spanning between −0.7 µm and −1.8 μm using image shift, with one shot per hole on the Glacios for OctT = 4-3 or multiple shots per hole on the Titan Krios for IcoT = 4-4. Movies (n = 1,320 and 1,048) were collected with a pixel size of 0.44 Å for OctT = 4-3 with a total dose of approximately 50 e− Å−2, and 6,678 movies were collected with a pixel size of 0.84 Å for IcoT = 4-4 with a total dose of approximately 61 e− Å−2.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Code availability
The source code from RPXdock for the protein docking is freely available at https://github.com/willsheffler/rpxdock. The source code from ProteinMPNN for the sequence design is freely available at https://github.com/dauparas/ProteinMPNN.
References
King, N. P. et al. Computational design of self-assembling protein nanomaterials with atomic level accuracy. Science 336, 1171–1174 (2012).
Hsia, Y. et al. Design of a hyperstable 60-subunit protein icosahedron. Nature 535, 136–139 (2016).
Rayment, I., Baker, T. S., Caspar, D. L. D. & Murakami, W. T. Polyoma virus capsid structure at 22.5 Å resolution. Nature 295, 110–115 (1982).
Liddington, R. C. et al. Structure of simian virus 40 at 3.8-Å resolution. Nature 354, 278–284 (1991).
Johnson, J. E. Functional implications of protein–protein interactions in icosahedral viruses. Proc. Natl Acad. Sci. USA 93, 27–33 (1996).
Prasad, B. V. V. et al. X-ray crystallographic structure of the Norwalk virus capsid. Science 286, 287–290 (1999).
Baker, T. S., Olson, N. H. & Fuller, S. D. Adding the third dimension to virus life cycles: three-dimensional reconstruction of icosahedral viruses from cryo-electron micrographs. Microbiol. Mol. Biol. Rev. 63, 862–922 (1999).
Caspar, D. L. D. & Klug, A. Physical principles in the construction of regular viruses. Cold Spring Harb. Symp. Quant. Biol. 27, 1–24 (1962).
De Colibus, L. et al. Assembly of complex viruses exemplified by a halophilic euryarchaeal virus. Nat. Commun. 10, 1456 (2019).
Marcandalli, J. et al. Induction of potent neutralizing antibody responses by a designed protein nanoparticle vaccine for respiratory syncytial virus. Cell 176, 1420–1431.e17 (2019).
Boyoglu-Barnum, S. et al. Quadrivalent influenza nanoparticle vaccines induce broad protection. Nature 592, 623–628 (2021).
Tong, G. J., Hsiao, S. C., Carrico, Z. M. & Francis, M. B. Viral capsid DNA aptamer conjugates as multivalent cell-targeting vehicles. J. Am. Chem. Soc. 131, 11174–11178 (2009).
Banskota, S. et al. Engineered virus-like particles for efficient in vivo delivery of therapeutic proteins. Cell 185, 250–265.e16 (2022).
Lai, Y.-T., Cascio, D. & Yeates, T. O. Structure of a 16-nm cage designed by using protein oligomers. Science 336, 1129 (2012).
King, N. P. et al. Accurate design of co-assembling multi-component protein nanomaterials. Nature 510, 103–108 (2014).
Lai, Y.-T. et al. Structure of a designed protein cage that self-assembles into a highly porous cube. Nat. Chem. 6, 1065–1071 (2014).
Kibler, R. D. et al. Stepwise design of pseudosymmetric protein hetero-oligomers. Preprint at bioRxiv https://doi.org/10.1101/2023.04.07.535760 (2023).
Brunette, T. et al. Exploring the repeat protein universe through computational protein design. Nature 528, 580–584 (2015).
Hsia, Y. et al. Design of multi-scale protein complexes by hierarchical building block fusion. Nat. Commun. 12, 2294 (2021).
Fallas, J. A. et al. Computational design of self-assembling cyclic protein homo-oligomers. Nat. Chem. 9, 353–360 (2017).
Sheffler, W. et al. Fast and versatile sequence-independent protein docking for nanomaterials design using RPXDock. PLoS Comput. Biol. 19, e1010680 (2023).
Dauparas, J. et al. Robust deep learning–based protein sequence design using ProteinMPNN. Science 378, 49–56 (2022).
Schein, S. & Gayed, J. M. Fourth class of convex equilateral polyhedron with polyhedral symmetry related to fullerenes and viruses. Proc. Natl Acad. Sci. USA 111, 2920–2925 (2014).
Kidmose, R. T. et al. Namdinator — automatic molecular dynamics flexible fitting of structural models into cryo-EM and crystallography experimental maps. IUCrJ 6, 526–531 (2019).
Tomaszewska, E. et al. Detection limits of DLS and UV-Vis spectroscopy in characterization of polydisperse nanoparticles colloids. J. Nanomater. 2013, 313081 (2013).
Huang, B. et al. Designed endocytosis-inducing proteins degrade targets and amplify signals. Nature https://doi.org/10.1038/s41586-024-07948-2 (2024).
Springer, A. D. & Dowdy, S. F. GalNAc–siRNA conjugates: leading the way for delivery of RNAi therapeutics. Nucleic Acid Ther. 28, 109–118 (2018).
Ahn, G. et al. LYTACs that engage the asialoglycoprotein receptor for targeted protein degradation. Nat. Chem. Biol. 17, 937–946 (2021).
Bermeo, S. et al. De novo design of obligate ABC-type heterotrimeric proteins. Nat. Struct. Mol. Biol. 29, 1266–1276 (2022).
Dowling, Q. et al. Hierarchical design of pseudosymmetric protein nanocages. Nature https://doi.org/10.1038/s41586-024-08360-6 (2024).
Lee, S. et al. Expanding protein nanocages through designed symmetry-breaking. Baker Lab https://www.bakerlab.org/wp-content/uploads/2024/04/Expanding-protein-nanocages-through-designed-symmetry-breaking.pdf (2024).
Suloway, C. et al. Automated molecular microscopy: the new Leginon system. J. Struct. Biol. 151, 41–60 (2005).
Sun, M. et al. Practical considerations for using K3 cameras in CDS mode for high-resolution and high-throughput single particle cryo-EM. J. Struct. Biol. 213, 107745 (2021).
Acknowledgements
We thank E. C. Yang, Q. M. Dowling and N. P. King for helpful discussions; H. Eisenach for advice on explicit negative design; X. Li for help with mass spectrometry analysis of proteins; and C. Liu for advice on cryo-EM data processing. nMS measurements were provided by F. Busch, A. Norris, N. Horvath and S. Cleary of the NIH-funded Resource for Native Mass Spectrometry Guided Structural Biology at The Ohio State University (NIH P41 GM128577 awarded to V. Wysocki). Small-angle X-ray scattering was conducted at the Advanced Light Source, a national user facility operated by Lawrence Berkeley National Laboratory on behalf of the US Department of Energy (DOE), Office of Basic Energy Sciences, through the Integrated Diffraction Analysis Technologies program, supported by the DOE Office of Biological and Environmental Research. Additional support comes from the NIH project ALS-ENABLE (P30 GM124169) and the High-End Instrumentation grant S10OD018483. This research used resources of the National Energy Research Scientific Computing Center (NERSC), a US DOE Office of Science User Facility located at Lawrence Berkeley National Laboratory, operated under contract no. DE-AC02-05CH11231 using the NERSC award BER-ERCAP0022018. We acknowledge funding from the Howard Hughes Medical Institute (S.L. and D.B.); the Audacious Project at the Institute for Protein Design (S.L., R.D.K., A.J.B., A.P. and D.B.); the Open Philanthropy Project Improving Protein Design Fund (Y.H. and D.B.); the Open Philanthropy Project Universal Flu Vaccine Fund (Y.H. and D.B.); the Bill and Melinda Gates Foundation (OPP1156262 to A.J.B.); the Defense Advanced Research Projects Agency (DARPA) Biostasis (Y.H.); and the NIH’s National Institute on Aging (R01AG063845 to A.J.B.). This work was also supported by Outstanding Young Scientist grants (RS-2024-00345974) and the Nano & Material Technology Development Program (RS-2024-00409405) through the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT)(no. RS-2024-00411809). This work was supported by the Institute for Protein Design Breakthrough Fund, for “De novo design of 100 nm scale protein assemblies” (S.L., R.D.K., Y.H. and D.B.).
Author information
Authors and Affiliations
Contributions
S.L., R.D.K., Y.H. and D.B. conceptualized the study and designed the protocol. R.D.K., S.L. and Y.H. curated the computational design of oligomers. S.L. and R.D.K. came up with the computational design of cages. S.L. and R.D.K. performed protein synthesis and characterizations. G.A., S.L., R.D.K. and B.H. performed the functionalization of protein cages and cell experiments. A.J.B., A.P., R.D.K. and S.L. undertook cryo-EM. M.A.K. and B.S. performed X-ray crystallography. D.B. supervised the study. S.L., R.D.K. and D.B. wrote the original draft of the manuscript. All authors read and contributed to the manuscript.
Corresponding author
Ethics declarations
Competing interests
D.B., S.L., R.D.K., Y.H. and G.A. are inventors on a provisional patent application (#49728.01US1) submitted by the University of Washington for the design, composition and function of the proteins created in this study.
Peer review
Peer review information
Nature thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Zoomed-in view of nsEM images.
T = 1 cages (top), crowns (middle), T = 4 cages (bottom).
Supplementary information
Supplementary Information (download PDF )
This file contains Sections 1–4; Supplementary Figs. S1–S51; Supplementary Table S1–S13 and Supplementary References
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lee, S., Kibler, R.D., Ahn, G. et al. Four-component protein nanocages designed by programmed symmetry breaking. Nature 638, 546–552 (2025). https://doi.org/10.1038/s41586-024-07814-1
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-024-07814-1
This article is cited by
-
Of condensates and coats - reciprocal regulation of clathrin assembly and the growth of protein networks
Nature Communications (2025)
-
Nonviral protein cages as tools to decipher and combat viral threats
npj Viruses (2025)
-
Hierarchical design of pseudosymmetric protein nanocages
Nature (2025)
-
Computational design of bifaceted protein nanomaterials
Nature Materials (2025)
-
Engineering complexity into protein-based biomaterials for biomedical applications
Nature Reviews Materials (2025)
