
Abstract
Thromboinflammation occurs in various diseases, leading to life-threatening microvascular occlusion with resulting end-organ failure1,2,3,4. Importantly, how microvascular thromboinflammation resolves remains poorly understood due to the small size-scale of microvasculature and the long duration (weeks to months) of this process. Here we introduce a hydrogel-based thromboinflammation-on-a-chip model with long-term culture capabilities to model microvascular thromboinflammation and monitor clot resolution over clinically and physiologically relevant timescales (up to months). Using this system, we mapped out the distinct temporal phases of clot resolution in microvascular thromboinflammation. Using multiplexed RNA fluorescence in situ hybridization in combination with our thromboinflammation-on-a-chip model, we observed that inflammation shifts the endothelium fibrinolytic balance to favour thrombosis and pinpointed neutrophil elastase as a double-edged sword that induces clot resolution but also tissue damage. We then investigated the mechanisms of potential therapeutic agents that either prevent microvascular thrombosis or accelerate clot resolution. Specifically, we observed that, in thromboinflammation, (1) early tissue plasminogen activator administration within 3 h directly improves endothelial barrier function; (2) prophylactic defibrotide and enoxaparin suppress microvascular thromboinflammation through endothelium-mediated mechanisms; and (3) combining enoxaparin with crizanlizumab reduces microvascular occlusion and protects endothelial function in sickle cell disease. These data introduce a paradigm in investigating the underlying mechanisms of thromboinflammatory clot resolution and conducting drug discovery thereof.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others

Data availability
The data supporting the findings of this study are available from the corresponding authors on request. Source data are provided with this paper.
References
Bray, M. A., Sartain, S. E., Gollamudi, J. & Rumbaut, R. E. Microvascular thrombosis: experimental and clinical implications. Transl. Res. 225, 105–130 (2020).
Conran, N. & De Paula, E. V. Thromboinflammatory mechanisms in sickle cell disease—challenging the hemostatic balance. Haematologica 105, 2380–2390 (2020).
Gu, S. X. et al. Thrombocytopathy and endotheliopathy: crucial contributors to COVID-19 thromboinflammation. Nat. Rev. Cardiol. 18, 194–209 (2021).
Jackson, S. P., Darbousset, R. & Schoenwaelder, S. M. Thromboinflammation: challenges of therapeutically targeting coagulation and other host defense mechanisms. Blood 133, 906–918 (2019).
Needleman, L. et al. Ultrasound for lower extremity deep venous thrombosis: multidisciplinary recommendations from the Society of Radiologists in Ultrasound Consensus Conference. Circulation 137, 1505–1515 (2018).
Canedo-Antelo, M. et al. Radiologic clues to cerebral venous thrombosis. Radiographics 39, 1611–1628 (2019).
Schuijf, J. D. et al. CT imaging with ultra-high-resolution: opportunities for cardiovascular imaging in clinical practice. J. Cardiovasc. Comput. Tomogr. 16, 388–396 (2022).
Choe, K. et al. Intravital three-photon microscopy allows visualization over the entire depth of mouse lymph nodes. Nat. Immunol. 23, 330–340 (2022).
Whyte, C. S. & Mutch, N. J. “Going with the flow” in modeling fibrinolysis. Front. Cardiovasc. Med. 9, 1054541 (2022).
Bonnard, T., Law, L. S., Tennant, Z. & Hagemeyer, C. E. Development and validation of a high throughput whole blood thrombolysis plate assay. Sci. Rep. 7, 2346 (2017).
Kuiper, G. J. et al. Validation of a modified thromboelastometry approach to detect changes in fibrinolytic activity. Thromb. J. 14, 1 (2016).
Mutch, N. J. et al. The use of the Chandler loop to examine the interaction potential of NXY-059 on the thrombolytic properties of rtPA on human thrombi in vitro. Br. J. Pharmacol. 153, 124–131 (2008).
Pandian, N. K. R., Mannino, R. G., Lam, W. A. & Jain, A. Thrombosis-on-a-chip: prospective impact of microphysiological models of vascular thrombosis. Curr. Opin. Biomed. Eng. 5, 29–34 (2018).
Zhang, Y. S. et al. Bioprinted thrombosis-on-a-chip. Lab Chip 16, 4097–4105 (2016).
Qiu, Y. et al. Microvasculature-on-a-chip for the long-term study of endothelial barrier dysfunction and microvascular obstruction in disease. Nat. Biomed. Eng. 2, 453–463 (2018).
Pober, J. S. & Sessa, W. C. Inflammation and the blood microvascular system. Cold Spring Harb. Perspect. Biol. 7, a016345 (2014).
Suzuki, Y., Yasui, H., Brzoska, T., Mogami, H. & Urano, T. Surface-retained tPA is essential for effective fibrinolysis on vascular endothelial cells. Blood 118, 3182–3185 (2011).
Chapin, J. C. & Hajjar, K. A. Fibrinolysis and the control of blood coagulation. Blood Rev. 29, 17–24 (2015).
Adams, S. A., Kelly, S. L., Kirsch, R. E., Robson, S. C. & Shephard, E. G. Role of neutrophil membrane proteases in fibrin degradation. Blood Coagul. Fibrinolysis 6, 693–702 (1995).
Nicklas, J. M., Gordon, A. E. & Henke, P. K. Resolution of deep venous thrombosis: proposed immune paradigms. Int. J. Mol. Sci. 21, 2080 (2020).
Varma, M. R. et al. Neutropenia impairs venous thrombosis resolution in the rat. J. Vasc. Surg. 38, 1090–1098 (2003).
Ali, M. R. et al. Aspect of thrombolytic therapy: a review. ScientificWorldJournal 2014, 586510 (2014).
The NINDS t-PA Stroke Study Group. Intracerebral hemorrhage after intravenous t-PA therapy for ischemic stroke. Stroke 28, 2109–2118 (1997).
Adams, H. P. Jr et al. Guidelines for thrombolytic therapy for acute stroke: a supplement to the guidelines for the management of patients with acute ischemic stroke. A statement for healthcare professionals from a Special Writing Group of the Stroke Council, American Heart Association. Circulation 94, 1167–1174 (1996).
Richardson, P. G. et al. The importance of endothelial protection: the emerging role of defibrotide in reversing endothelial injury and its sequelae. Bone Marrow Transplant 56, 2889–2896 (2021).
Mohty, M. et al. Prophylactic, preemptive, and curative treatment for sinusoidal obstruction syndrome/veno-occlusive disease in adult patients: a position statement from an international expert group. Bone Marrow Transplant 55, 485–495 (2020).
Tekgunduz, E. et al. Does defibrotide prophylaxis decrease the risk of acute graft versus host disease following allogeneic hematopoietic cell transplantation? Transfus. Apher. Sci. 54, 30–34 (2016).
Richardson, P. G., Carreras, E., Iacobelli, M. & Nejadnik, B. The use of defibrotide in blood and marrow transplantation. Blood Adv. 2, 1495–1509 (2018).
Buijsers, B., Yanginlar, C., Maciej-Hulme, M. L., de Mast, Q. & van der Vlag, J. Beneficial non-anticoagulant mechanisms underlying heparin treatment of COVID-19 patients. eBioMed. 59, 102969 (2020).
McLaughlin, K. et al. Low molecular weight heparin improves endothelial function in pregnant women at high risk of preeclampsia. Hypertension 69, 180–188 (2017).
Shet, A. S., Lizarralde-Iragorri, M. A. & Naik, R. P. The molecular basis for the prothrombotic state in sickle cell disease. Haematologica 105, 2368–2379 (2020).
Fredman, G. Resolving inflammation and pain of sickle cell. Blood 133, 190–191 (2019).
Zhang, D., Xu, C., Manwani, D. & Frenette, P. S. Neutrophils, platelets, and inflammatory pathways at the nexus of sickle cell disease pathophysiology. Blood 127, 801–809 (2016).
Ataga, K. I. et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N. Engl. J. Med. 376, 429–439 (2017).
Welsh, J. D. et al. A systems approach to hemostasis: 1. The interdependence of thrombus architecture and agonist movements in the gaps between platelets. Blood 124, 1808–1815 (2014).
Henke, P. K. et al. Interleukin-8 administration enhances venous thrombosis resolution in a rat model. J. Surg. Res. 99, 84–91 (2001).
Sahoo, M., del Barrio, L., Miller, M. A. & Re, F. Neutrophil elastase causes tissue damage that decreases host tolerance to lung infection with species. PLoS Pathog. 10, e1004327 (2014).
Szepanowski, R. D. et al. Thromboinflammatory challenges in stroke pathophysiology. Semin. Immunopathol. 45, 389–410 (2023).
Noubouossie, D. F., Reeves, B. N., Strahl, B. D. & Key, N. S. Neutrophils: back in the thrombosis spotlight. Blood 133, 2186–2197 (2019).
Peiseler, M. & Kubes, P. More friend than foe: the emerging role of neutrophils in tissue repair. J. Clin. Invest. 129, 2629–2639 (2019).
Qi, H., Yang, S. & Zhang, L. Neutrophil extracellular traps and endothelial dysfunction in atherosclerosis and thrombosis. Front. Immunol. 8, 928 (2017).
Wang, R. et al. Neutrophil extracellular traps promote tPA-induced brain hemorrhage via cGAS in mice with stroke. Blood 138, 91–103 (2021).
Richardson, E. et al. Defibrotide: potential for treating endothelial dysfunction related to viral and post-infectious syndromes. Expert Opin. Ther. Targets 25, 423–433 (2021).
Li, G., Hilgenfeld, R., Whitley, R. & De Clercq, E. Therapeutic strategies for COVID-19: progress and lessons learned. Nat. Rev. Drug Discov. 22, 449–475 (2023).
Corbacioglu, S. et al. Defibrotide for prophylaxis of hepatic veno-occlusive disease in paediatric haemopoietic stem-cell transplantation: an open-label, phase 3, randomised controlled trial. Lancet 379, 1301–1309 (2012).
Richardson, P. G. et al. Multi-institutional use of defibrotide in 88 patients after stem cell transplantation with severe veno-occlusive disease and multisystem organ failure: response without significant toxicity in a high-risk population and factors predictive of outcome. Blood 100, 4337–4343 (2002).
Richardson, P. G. et al. Phase 3 trial of defibrotide for the treatment of severe veno-occlusive disease and multi-organ failure. Blood 127, 1656–1665 (2016).
Frame, D. et al. Defibrotide therapy for SARS-CoV-2 ARDS. Chest 162, 346–355 (2022).
Ruggeri, A. et al. Use of defibrotide in patients with COVID-19 pneumonia: comparison of a phase II study and a matched real-world corhort control. Haematologia 109, 3261–3268 (2024).
Morici, N. et al. Enoxaparin for thromboprophylaxis in hospitalized COVID-19 patients: the X-COVID-19 randomized trial. Eur. J. Clin. Invest. 52, e13735 (2022).
Price, G. M. & Tien, J. Methods for forming human microvascular tubes in vitro and measuring their macromolecular permeability. Methods Mol. Biol. 671, 281–293 (2011).
Wang, C., Lu, H. & Schwartz, M. A. A novel in vitro flow system for changing flow direction on endothelial cells. J. Biomech. 45, 1212–1218 (2012).
Acknowledgements
We thank A. Shaw and S. Hsieh (Parker H. Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology) for technical assistance; and T. Byun and S. Panicker from Star Therapeutics who provided Cablivi as a gift. This work was supported by National Institutes of Health, Institute of Heart, Lung and Blood grants R01HL130918 (W.A.L.), R01HL140589 (W.A.L.), R35HL145000 (W.A.L.) and U54HL141981 (Y.Q.), National Institute of Biomedical Imaging and Bioengineering grant R21EB028519 (Y.Q.) and Health Resources and Services Administration of the US Department of Health and Human Services Maternal and Child Health Bureau 340B Program (5H30MC24049-08-00). This work was performed in part at the Georgia Tech Institute for Matter and Systems, a member of the National Nanotechnology Coordinated Infrastructure, which is supported by National Science Foundation, division of Electrical, Communications, and Cyber Systems grant 1542174.
Author information
Authors and Affiliations
Contributions
Conception and design: Y.Q. and W.A.L. Development of the methodology: Y.Q., J.L., A.W., Z.F., Y.S., H.C., E.K.W., E.T.H., K.M., A.F.C., G.W. and W.A.L. On-chip experiments: Y.Q. Fibrin gel degradation experiments: J.L. and H.C. RT–qPCR analysis: Y.Q., J.L. and A.W. Silicon master mould: E.T.H. and E.K.W. Cell maintenance and sample preparation: Y.Q. and Y.S. RNA-FISH: Z.F. and A.F.C. Formal analyses: Y.Q., J.L., A.W. and Z.F. Writing the original draft: Y.Q. and W.A.L. Writing, reviewing and/or revising the manuscripts: all of the authors. All of the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
W.A.L. and Y.Q. receive research funding from Novo Nordisk and Star Therapeutics that is unrelated to this Article. W.A.L. also receives research funding from Roche for work that is unrelated to this Article. The other authors declare no competing interests.
Peer review
Peer review information
Nature thanks Paul Richardson and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
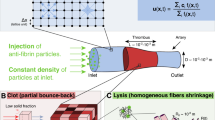
Extended Data Fig. 1 The hydrogel-based thromboinflammation-on-a-chip model was maintained under microcirculatory flow conditions for at least two months and exhibited physiologically relevant microvascular barrier function.
a, Computational fluid dynamics (CFD) simulation of the microchannels indicating the wall shear stress within the microchannels. b, A representative phase-contrast microscopy image of the microvasculature-on-a-chip during Step 1 in Fig. 1. c, A representative epifluorescence image of the engineered microvasculature after a 15-min perfusion of 70 kDa dextran-FITC demonstrates that the system is impermeable to dextran-FITC under physiological flow conditions during Step 1. in Fig. 1. d, Representative stitched-composite of epifluorescence images after a 15-min perfusion of 70 kDa dextran-FITC into the microvasculature inflamed by either 1 or 10 ng ml−1 TNF, demonstrate that TNF increases permeability in a dose-dependent manner. e, Quantitative analysis of the apparent permeability of the microvasculature after 16 h of stimulation with different doses of TNF. f, Representative confocal microscopy images show that endothelial-specific proteins VE-cadherin and PECAM−1 remain primarily located at cell-cell junction after two months of culture in the microvasculature-on-a-chip model. g, Representative confocal microscopy images show that endothelial cells continue to express VE-cadherin and PECAM-1 after 16 h of exposure to 10 ng ml−1 TNF, although these proteins become more diffusively distributed compared to the resting endothelium in (f). [Scale bar = 20 μm (f,g). n = 4 biologically independent replicates. Data are presented as mean values +/− SD. p values were calculated using one-way ANOVA with Tukey correction (e)].
Extended Data Fig. 2 IL-6 leads to less fibrin formation and faster fibrin resolution post-thromboinflammation than TNF in the thromboinflammation-on-a-chip model.
a, Timeline for studying IL-6-induced thromboinflammation formation and resolution. b-c, Representative 3D confocal microscopy Z-stack images of thromboinflammation formation and resolution after 16 h of exposure to either a lower-dose (1 ng ml−1) (b) or a higher-dose (10 ng ml−1) (c) of IL-6. d, Quantitative analysis of fibrin formation and resolution post-thromboinflammation indicates that, regardless of the concentration, IL-6 induces a similar level of fibrin formation as 1 ng ml−1 TNF. However, IL-6-induced fibrin resolves faster than fibrin induced by 1 ng ml−1 TNF on Day1 post-thromboinflammation. [Scale bar = 20 μm (b-c). Data are presented as mean values +/− SD. n = 4 biologically independent replicates. p values were calculated using one-way ANOVA with Tukey correction].
Extended Data Fig. 3 A 16-hour exposure to TNF induces vWF release by the endothelium, facilitating platelet aggregation in the thromboinflammation-on-a-chip model.
a, A representative confocal microscopy image shows vWF strings attached to the endothelial surface at the bifurcation area after a 16-hour exposure to 10 ng ml−1 TNF. b, A representative confocal microscopy image shows a vWF string exceeding 100 μm in length after a 16-hour exposure to 10 ng ml−1 TNF. c, Representative confocal microscopy images of the bifurcation, and d, Representative confocal microscopy images of the channels downstream the bifurcation show that P-selectin positive, activated platelets are located at the core of the aggregated platelets, with only a small number of platelets being phosphatidylserine (PS)-positive after healthy whole blood is perfused over the inflamed endothelium (16-hour exposure to 10 ng ml−1 TNF). e, Representative confocal microscopy images show that the addition of Cablivi to whole blood inhibits platelet aggregation in the inflamed microvasculature (16-hour exposure to 10 ng ml−1 TNF).
Extended Data Fig. 4 Characterization of apoptotic cells in the thromboinflammation-on-a-chip model.
a, Timeline for studying cell apoptosis during thromboinflammation formation and resolution under the medium-dose (1 ng ml−1) TNF condition. b, Representative 3D confocal microscopy Z-stack images showing cell apoptosis over time under the medium-dose (1 ng ml−1) TNF condition. c-h, Confocal images of the boxes in b at higher magnification show apoptotic phosphatidylserine (PS) fluorescence signals (red) colocalize with neutrophils (green) and platelets (blue), indicating that the recruited neutrophils and platelets, but not endothelial cells, undergo apoptosis during microvascular thromboinflammation under the medium-dose (1 ng ml−1) TNF condition. i, Timeline for studying cell apoptosis during thromboinflammation formation and resolution under the high-dose (10 ng ml−1) TNF condition. j, Representative confocal microscopy images showing cell apoptosis after thromboinflammation formation. Phosphatidylserine (PS) is positively stained in some of the recruited neutrophils and aggregated platelets under the high-dose (10 ng ml−1) TNF condition. k, Representative confocal microscopy images on Day 7 post-thromboinflammation show that endothelial cells become apoptotic under the high-dose (10 ng ml−1) TNF condition. [Scale bar = 20 μm (b-h,j-k)].
Extended Data Fig. 5 D-dimer staining confirms fibrin degradation post-thromboinflammation in the thromboinflammation-on-a-chip model.
a, Timeline for studying fibrin degradation post-thromboinflammation using D-dimer staining. b, Representative confocal microscopy images showing fibrin formation after perfusing with whole blood (top row). D-dimer, the fibrin degradation by-product, is not detectable on Day 1 post-thrombosis (middle row) but becomes detectable on Day 3 post-thromboinflammation (bottom row).
Extended Data Fig. 6 Elevated TNF affects the expression of fibrinolysis-related genes in endothelial cells.
RT-qPCR results show exposure to 10 ng ml−1 TNF significantly reduces the expression of PLAT (encoding tPA), PLAU (encoding uPA), and/or PLAUR (encoding uPAR), while the expression of SERPINE1 (encoding PAI-1) and SERPINB2 (encoding PAI-2) is either less affected or increased in four types of primary human endothelial cells cultured under static condition. (n = 4 biological replicates for HUVECs. n = 3 biological replicates for cardiac, dermal, and glomerular microvascular endothelial cells. Data are shown as fold change compared to gene expression in control endothelial cells without TNF stimulation and are presented as mean values +/− SD. p values were calculated using a two-sided t-test between TNF-treated and control endothelial cells).
Extended Data Fig. 7 Recruited neutrophils gradually release elastase during and post-thromboinflammation in the thromboinflammation-on-a-chip model, and both recombinant human neutrophil elastase and isolated neutrophils induce fibrin degradation in vitro.
a, Timeline for studying the release of neutrophil elastase in the model, and representative 3D confocal microscopy Z-stack images show that neutrophils recruited to the microvasculature gradually become positive for elastase staining. b-e, Confocal microscopy images of the boxes in (a) at higher magnification showing neutrophils positive (arrowheads) and negative (arrows) for elastase staining during and post thromboinflammation. f, Recombinant human neutrophil elastase results in fibrin degradation in a dose-dependent manner. Compared to the condition with PBS, 690 nM elastase shows a significant difference (p < 0.0001) after 3 h, 9.4 ug ml−1 plasmin shows a significant difference (p < 0.0001) after 1 day, 345 nM elastase shows a significant difference (p = 0.0002) after 1 day, 138 nM elastase shows a significant difference (p < 0.0001) after 2 days, and 69 nM elastase shows a significant difference (p < 0.0309) also after 2 days. (n = 3 biological replicates. Data are presented as mean values +/− SD. p values were calculated using two-way ANOVA with Tukey correction.) g, Isolated neutrophils result in fibrin degradation in a dose-dependent manner, and neutrophil activation accelerate fibrin degradation in vitro. (n = 4 biological replicates for 1 M ml−1 neutrophils and n = 3 biological replicates for other conditions. Data are presented as mean values +/− SD. p values were calculated using one-way ANOVA with Tukey correction).
Extended Data Fig. 8 Recombinant human neutrophil elastase induces endothelial detachment and apoptosis in a dose-dependent manner.
a, Representative epi-fluorescence images with or without overlaying with phase-contrast images demonstrate that 690 nM recombinant human neutrophil elastase results in HUVEC detachment and apoptosis after a 3-hour culture. b, High magnification images of the boxes in (a). c, Representative epifluorescence images with or without overlaying with and phase-contrast images demonstrate that a 1-day culture with 690 nM human neutrophil elastase results in complete HUVEC detachment and cell fragmentation, while a 1-day culture with 345 nM elastase results in HUVEC detachment and apoptosis, similar as that observed after a 3-hour culture with 690 nM elastase. d-e, Higher magnification images of the boxes in (c). [(scale bar = 100 μm (a-d)].
Extended Data Fig. 9 Neutrophil elastase induces fibrin degradation and endothelial damage during thromboinflammation resolution in the thromboinflammation-on-a-chip model.
a, Timeline for studying the role of neutrophil elastase in thromboinflammation under the 10 ng ml−1 TNF condition. Representative 3D confocal microscopy Z-stack images and a bright field image demonstrate that the human neutrophil elastase inhibitor BAY 85-8501 blocks fibrin degradation and prevents endothelial detachment. b, Timeline for studying the role of neutrophil elastase and endogenous tPA in thromboinflammation under the 1 ng ml−1 TNF condition. Representative 3D confocal microscopy Z-stack images show that the human neutrophil elastase inhibitor BAY 85-8501 and antifibrinolytic drug 6-aminocaproic acid together block fibrin degradation. c, Timeline for studying the role of endogenous tPA in thromboinflammation under the 1 ng ml−1 TNF condition. Representative 3D confocal microscopy Z-stack images show that antifibrinolytic drug 6-aminocaproic acid alone does not block fibrin degradation. d, Quantitative analysis of fibrin shows that inhibition of human neutrophil elastase by BAY85-8501 significantly reduces fibrin degradation on Day 7 post-thromboinflammation compared to the control without inhibitor under the 10 ng ml−1 TNF condition. e, Quantitative analysis of fibrin shows that inhibition of human neutrophil elastase significantly reduces fibrin degradation post-thromboinflammation under the 1 ng ml−1 TNF condition, whereas the antifibrinolytic drug 6-aminocaproic acid alone does not. [n = 4 biologically independent replicates. Data are presented as mean values +/− SD. p values were calculated using two-way ANOVA with Šídák correction (d), or with Dunnett correction (e)].
Extended Data Fig. 10 Crizanlizumab and enoxaparin treatment mitigates “in vitro vaso-occlusive episodes” in sickle cell disease in the thromboinflammation-on-a-chip model.
a, Timeline for studying thrombus resolution with sickle cell disease patient blood, and representative 3D confocal microscopy Z-stack images show that whole blood from sickle cell disease patients, in and of itself, is pro-thrombotic and sufficient to induce fibrin production in resting, non-inflamed microvasculature. Fibrin degradation gradually occurs over time only after perfusion with healthy serum. b, Quantitative analysis of fibrin fluorescence signals indicates fibrin degrades over the 5 days post-thrombosis following perfusion with healthy serum. c, A representative epifluorescence microscopy image and fluorescence intensity analysis along the linescan demonstrate that perfusion of healthy whole blood does not increase permeability on Day 1 post-thrombosis. d, A representative epifluorescence image and fluorescence intensity analysis along the linescan demonstrate that perfusion of sickle cell disease whole blood increases permeability on Day 1 post-thrombosis. e, Quantitative analysis of localized endothelial permeability confirms that perfusion with sickle cell disease whole blood alone increases endothelial permeability compared to perfusion of healthy whole blood. f, Timeline of enoxaparin and crizanlizumab treatment during thrombosis formation. g, Representative confocal images show that without crizanlizumab, neutrophils (red) and platelets (green) form stable aggregates, leading to complete microvascular occlusion (e.g., >740 s in this case). h, Representative confocal microscopy images show that crizanlizumab prevents the formation of neutrophil-platelet aggregates in sickle cell disease patient blood, thereby reducing occlusion duration in the microvasculature (e.g., ~240 s in this case). i, Representative epifluorescence images after a 15-min perfusion of 70 kDa dextran-FITC show that enoxaparin alone does not prevent endothelial permeability following perfusion with sickle cell disease whole blood. j, Representative epifluorescence images after a 15-min perfusion of 70 kDa dextran-FITC show that enoxaparin combined with crizanlizumab reduces permeability following perfusion with sickle cell disease whole blood. k, Quantitative analysis of occlusion duration shows that crizanlizumab significantly reduces occlusion duration. (Occlusive events were recorded from three biologically independent replicates for each condition.) l, Quantitative analysis of localized endothelial cell permeability shows that the combination of enoxaparin and crizanlizumab preserves endothelial barrier function, whereas enoxaparin alone does not prevent occlusion-induced permeability increases. [Scale bar = 20 μm (a,c-d,g-h); 100 μm (i,j). n = 4 biologically independent replicates (b,e). n = 3 biologically independent replicates (l). Data are presented as mean values +/− SD. p values were calculated using one-way ANOVA with Tukey correction (b), or using two-sided t-test (e,k), or two-way ANOVA with Tukey correction (l)].
Supplementary information
Supplementary Information (download PDF )
Supplementary Figs. 1–12.
Supplementary Video 1 (download MP4 )
Thromboinflammation forms in the thromboinflammation-on-a-chip model-A. A representative video showing all imaging channels (bright field; green, platelets; red, neutrophils; blue, fibrin), capturing a 30 min perfusion of healthy whole blood into the microvasculature pre-exposed to 10 ng ml−1 TNF for 16 h.
Supplementary Video 2 (download MP4 )
Thromboinflammation forms in the thromboinflammation-on-a-chip model-B. A representative video showing only fluorescence channels (green, platelets; red, neutrophils; blue, fibrin), capturing a 30 min perfusion of healthy whole blood into the microvasculature pre-exposed to 10 ng ml−1 TNF for 16 h.
Supplementary Video 3 (download MP4 )
Resting microvasculature-on-a-chip is antithrombotic-A. A representative video showing all imaging channels (bright field, green: platelets; red, neutrophils; blue, fibrin) shows that a 30 min perfusion of healthy whole blood into the resting microvasculature does not induce thrombosis.
Supplementary Video 4 (download MP4 )
Resting microvasculature-on-a-chip is antithrombotic-B. A representative video showing only fluorescence channels (green, platelets; red, neutrophils; blue, fibrin) shows that a 30 min perfusion of healthy whole blood into the resting microvasculature does not induce thrombosis.
Source data
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Qiu, Y., Lin, J., Wang, A. et al. Clinically relevant clot resolution via a thromboinflammation-on-a-chip. Nature 641, 1298–1308 (2025). https://doi.org/10.1038/s41586-025-08804-7
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-025-08804-7
This article is cited by
-
Heart-on-a-chip and vasculature-on-a-chip platforms as models of cardiovascular disease
Nature Reviews Cardiology (2026)
-
Advanced technologies of artery-on-a-chip: a review of construction strategies and disease models
Angiogenesis (2026)
